

Abstract
颅骨锁骨发育不全是一种罕见的常染色体显性遗传,主要影响全身骨骼和牙齿发育,发病率约为1∶1 000 000。本文对1例颅骨锁骨发育不全病例进行报道及文献回顾,基因检测证实RUNX2 6p21.1 NM_001024630.3 Exon4 c.534dupA p.(Val179fs)移码突变,为一个新的突变位点。
Keywords: 颅骨锁骨发育不全, 常染色体显性遗传, 移码突变
Abstract
Cleidocranial dysplasia is a rare autosomal dominant hereditary disease that mainly affects the skeletal and dental development and has an incidence rate of about 1∶1 000 000. In this study, a case of cranio-clavicular dysplasia was reported, and related literature was reviewed. RUNX2 6p21.1 NM_001024630.3 Exon4 c.534dupAp.(Val179fs) was identified to be a new frameshift mutation by gene analysis.
Keywords: cleidocranial dysplasia, autosomal dominant inheritance, frame shift mutation
颅骨锁骨发育不全(cleidocranial dysplasia,CCD)(在线人类孟德尔遗传数据库编号:119600)是一种罕见的常染色体显性遗传,主要影响以软骨内成骨方式发育的躯干四肢骨和以膜内成骨方式发育的颅颌面骨[1],发病率约为1∶1 000 000[2]–[3],可以散发也可以家族聚集性发病,其中散发病例约为1/3,男女无显著差异[4]。本文对1例颅骨锁骨发育不全病例进行报道及文献回顾,并经基因检测证实为一个新的移码突变。
1. 病例报告
患者,男,12岁,主诉为乳牙滞留、恒牙未萌多年。患者个头矮小,智力正常,特殊面貌,前额突出,眼距宽,鼻梁塌陷,面中部凹陷,前倾鼻,鼻上翘,下唇外翻,面下1/3短。肩部窄小,胸廓呈圆锥形,锁骨短小,仅在胸骨处可扪及,肩关节活动度过大,双肩下垂可在胸前并拢,末端指节较膨隆(图1)。父母及其妹妹表型无特殊。
图 1. 先证者外观.

Fig 1 Appearance of the proband
A:正面观;B:侧面观;C:手掌面。
口腔检查:双侧颜面部对称,凹面型,前牙反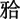 ;混合牙列,16、22、26、31、32、36、41、42、46牙已萌出,其余均为乳牙;51、52、54、55、16、61、65、26、74、75、84、85、46牙龋坏,64牙缺失(图2)。
;混合牙列,16、22、26、31、32、36、41、42、46牙已萌出,其余均为乳牙;51、52、54、55、16、61、65、26、74、75、84、85、46牙龋坏,64牙缺失(图2)。
图 2. 口内照.

Fig 2 Pictures inside the mouth
A:上颌观;B:下颌观;C:右侧观;D:正面观;E:左侧观。
锥形束CT(cone beam CT,CBCT)检查示:11—15、21、23—25、33—35、43—45牙埋伏阻生,多颗多生牙埋伏阻生;上颌骨发育不足、下颌前突,骨缝增宽,颧骨发育不全;额窦未见气化,乳突气化不良(图3~5)。
图 3. CBCT-重组全景片.

Fig 3 CBCT-reconstructed panoramic view
图 5. CBCT断层扫描.

Fig 5 Computed tomography scan
A:冠状面;B:矢状面;C:水平面。
图 4. CBCT三维重建.

Fig 4 CBCT-three-dimensional reconstruction
A:右侧观;B:正面观;C:左侧观。
头颅正侧位片示:脑颅骨大于面颅骨,前囟门未闭合,颅缝增宽;凹面型,安氏Ⅲ类骨性错畸形,测得SNA角80.03°,SNB角94.87°,ANB角-14.84°(图6、7)。胸片示:胸廓呈锥形,双侧肩胛骨较小,右侧锁骨中外2/3、左侧1/3缺如(图8)。
图 6. 头颅正位片.

Fig 6 Posteroanterior cephalogram
图 7. 头颅侧位片.
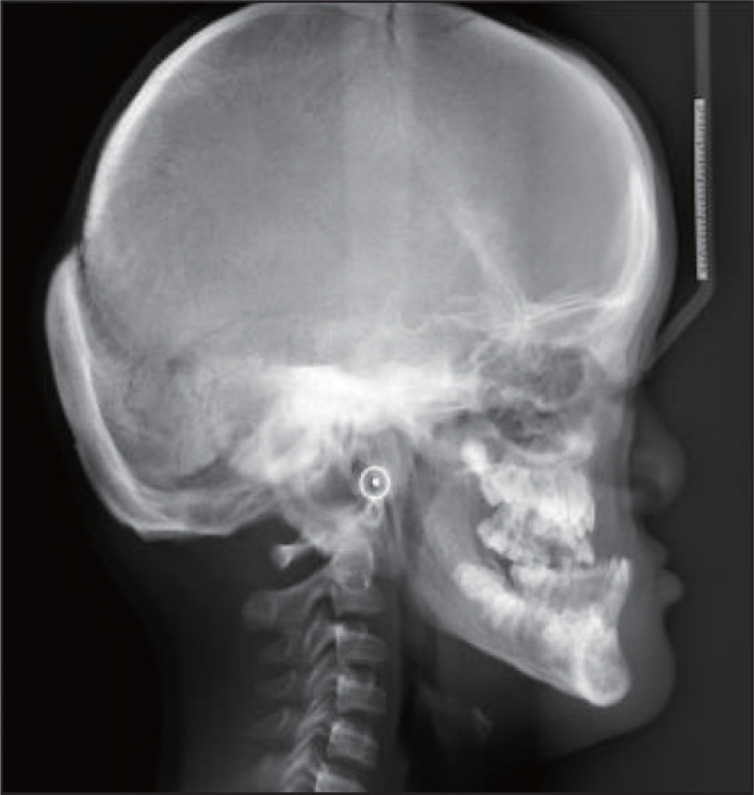
Fig 7 Lateral cephalometric radiograph
图 8. 胸片.
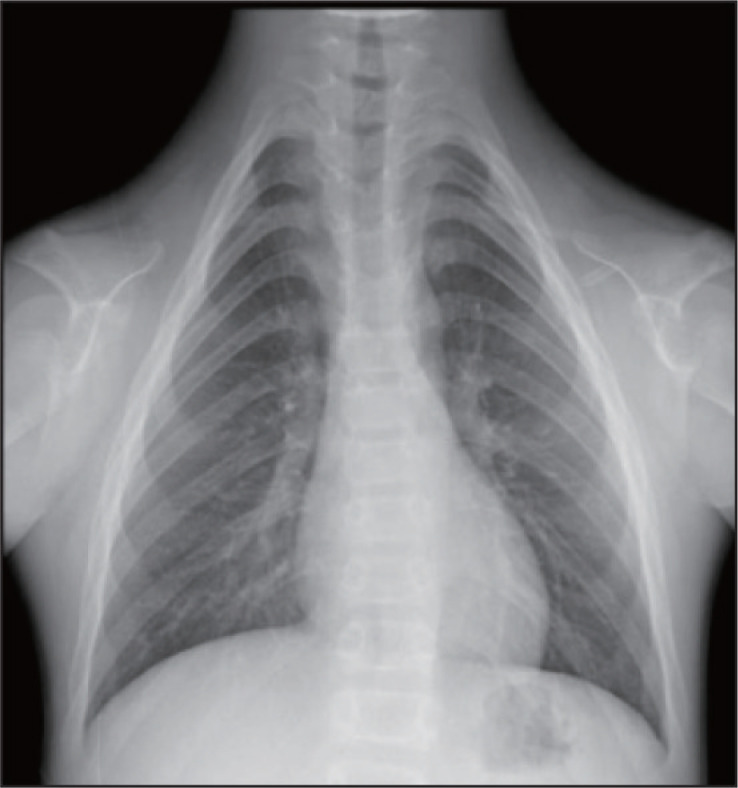
Fig 8 Chest radiography
基因检测显示:患者RUNX2 6p21.1 NM_001-024630.3 Exon4 c.534dupA发生移码突变,导致所编码的蛋白质自第179位开始发生移码,从而丧失其正常功能,该突变位点来自于其父亲(图9)。
图 9. 基因测序图谱.

Fig 9 Gene sequencing graph
上:先证者,箭头为突变位点;中:先证者父亲,为杂合携带者;下:先证者母亲,为健康成员。
本例患者经多科室联合会诊,入院拔除松动的乳牙及多生牙(图10),后期拟正畸牵引助恒牙萌出。若助萌成功,成年之后行正颌治疗;若恒牙无法牵出或效果差,成年之后行种植或修复体修复,恢复咬合功能。
图 10. 拔除松动的乳牙和大部分多生牙.

Fig 10 Extracted loose deciduous teeth and most supernumerary teeth
2. 讨论
2.1. 发病机制
CCD主要是由位于常染色体6p21上的成骨细胞特异性转录因子RUNX2基因突变所致[3],RUNX2可通过调节成骨细胞和软骨细胞的分化以及分化过程中细胞外基质蛋白基因的表达来控制骨骼发育[5]–[6],而敲除RUNX2基因可直接导致成骨细胞和骨生成的完全缺失[7]。RUNX2基因还通过参与成釉器的形成、牙本质细胞的分化及牙板的增殖,对牙齿的正常发育起了重要的作用[6],[8]–[9]。RUNX2基因的突变形式超过90种[10],其插入、缺失、错意表达、移码等等都可造成CCD发生。目前对CCD的研究主要集中在分子生物学领域。
本例先证者RUNX2基因测序结果显示,在Exon4上第c.534位点发生移码突变,导致所编码的蛋白质自第179位缬氨酸开始发生移码,导致翻译提前终止,从而丧失其正常功能。查阅相关文献,该位点突变尚未见相关报道,人类基因突变数据库也未见收录,是一个新的杂合突变,可用于该家系后代的产前诊断。
2.2. 临床表现及影像学特征
CCD患者的临床表现差异较大,可以从单纯的牙齿发育异常到影响全身多处骨骼,即使是同一家系表现也不尽相同[11]。本例患者具有CCD典型的锁骨发育不全、囟门闭合延迟、牙齿萌出异常“三联征”,还伴有身材矮小、前额突出、眼距增宽、颌面部发育不足、短指等,影像学表现为脑颅骨大于面颅骨、骨缝增宽,前囟门未闭合,上颌骨发育不足、下颌前突,气腔气化不全,颌骨内多数恒牙及多生牙的埋伏阻生,主要集中在双侧上下颌前磨牙区,胸廓呈锥形,双侧肩胛骨翼状改变,双侧锁骨发育异常等。部分患者还可伴有腭裂、手足指/趾异常、脊椎侧弯、漏斗胸、膝外翻/骨盆发育不良、牙瘤和融合牙、釉质发育不全、含牙囊肿、传导性听力损害、复发性中耳炎等[12]–[15]。
2.3. 诊断及鉴别诊断
CCD患者,根据其典型面容、咬合关系、全身情况及影像学检查即可诊断,但由于其典型症状在青春期才显著,早期轻微的表现容易被忽视。临床上可通过第二磨牙与乳牙共存,下切牙间隙、骨缝存在以及下颌骨发育特点等对早期患者做出诊断[14],选择最佳的治疗时机。对于高危家庭应进行产前咨询,而产前诊断可选择超声波或基因检查。超声检查应在怀孕12~15周进行,定期检查颅骨锁骨等的发育情况[16],改善妊娠结局。
目前CCD还需与佝偻病、软骨发育不全、克汀病、成骨不全及与外伤有关的锁骨部分缺如等进行鉴别。对于轻症特别是仅有牙齿萌出异常、临床诊断困难的患者,检测RUNX2基因可以进行鉴别诊断。
2.4. 治疗
目前CCD患者尚并无确切有效的治疗方法,有学者[17]认为应该根据患者的年龄、恒牙的发育及萌出情况、颌面部畸形的严重程度采取个性化、多学科综合治疗方案。Park等[13]也给出了具体的治疗原则:1)适时去除滞留的乳牙及多生牙,去除萌出阻力;2)外科手术暴露阻生恒牙;3)正畸牵引排齐;4)Lefort Ⅰ术改善颌面部畸形,恢复美观和功能;5)成年患者采用种植体或修复体恢复咬合功能。因为整个治疗周期长,效果不确定,因此要关注患者及家属的心理健康,进行必要的心理疏导。
Funding Statement
[基金项目] 国家自然科学基金(82160194,81960492);江西省自然科学基金(20181ACB20022);江西省重点研发计划(20212BBG73022)
Supported by: The National Natural Science Foundation of China (82160194, 81960492); Natural Science Foundation of Jiangxi Province (20181ACB20022); Key Research and Development Plan of Jiangxi Province (20212BBG73022).
Footnotes
利益冲突声明:作者声明本文无利益冲突。
References
- 1.Ryoo HM, Kang HY, Lee SK, et al. RUNX2 mutations in cleidocranial dysplasia patients[J] Oral Dis. 2010;16(1):55–60. doi: 10.1111/j.1601-0825.2009.01623.x. [DOI] [PubMed] [Google Scholar]
- 2.Cooper SC, Flaitz CM, Johnston DA, et al. A natural history of cleidocranial dysplasia[J] Am J Med Genet. 2001;104(1):1–6. doi: 10.1002/ajmg.10024. [DOI] [PubMed] [Google Scholar]
- 3.Mundlos S, Otto F, Mundlos C, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia[J] Cell. 1997;89(5):773–779. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- 4.Otto F, Kanegane H, Mundlos S. Mutations in the RUNX2 gene in patients with cleidocranial dysplasia[J] Hum Mutat. 2002;19(3):209–216. doi: 10.1002/humu.10043. [DOI] [PubMed] [Google Scholar]
- 5.Chen HY, Ghori-Javed Farah Y, Harunur R, et al. Runx2 regulates endochondral ossification through control of chondrocyte proliferation and differentiation[J] J Bone Miner Res. 2014;29(12):2653–2665. doi: 10.1002/jbmr.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toshihisa K. Regulation of bone development and extracellular matrix protein genes by RUNX2[J] Cell Tissue Res. 2010;339(1):189–195. doi: 10.1007/s00441-009-0832-8. [DOI] [PubMed] [Google Scholar]
- 7.Dalle Carbonare L, Innamorati G, Valenti MT. Transcription factor Runx2 and its application to bone tissue engineering[J] Stem Cell Rev Rep. 2012;8(3):891–897. doi: 10.1007/s12015-011-9337-4. [DOI] [PubMed] [Google Scholar]
- 8.Dorotheou D, Gkantidis N, Karamolegkou M, et al. Tooth eruption: altered gene expression in the dental follicle of patients with cleidocranial dysplasia[J] Orthod Craniofac Res. 2013;16(1):20–27. doi: 10.1111/ocr.12000. [DOI] [PubMed] [Google Scholar]
- 9.王 小娟, 轩 东英, 董 绍忠, et al. 颅骨锁骨发育不良患者牙齿矿化不全超微结构的研究[J] 实用口腔医学杂志. 2009;25(3):356–360. [Google Scholar]; Wang XJ, Xuan DY, Dong SZ, et al. A study on the ultrastructure of hypocalcification in patient with cleidocranial dysostosis[J] J Pract Stomatol. 2009;25(3):356–360. [Google Scholar]
- 10.郭 凌燕, 徐 珮琼, 陈 林林. 颅锁骨发育不全1例及基因突变分析[J] 华西口腔医学杂志. 2019;37(6):677–680. doi: 10.7518/hxkq.2019.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; Guo LY, Xu PQ, Chen LL. Cleidocranial dysplasia: a case report and gene mutation analysis[J] West China J Stomatol. 2019;37(6):677–680. doi: 10.7518/hxkq.2019.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang CY, Zheng SG, Wang YX, et al. Mutational analysis of RUNX2 gene in Chinese patients with cleidocranial dysplasia[J] Mutagenesis. 2010;25(6):589–594. doi: 10.1093/mutage/geq044. [DOI] [PubMed] [Google Scholar]
- 12.Lu H, Zeng BH, Yu DS, et al. Complex dental anomalies in a belatedly diagnosed cleidocranial dysplasia patient[J] Imaging Sci Dent. 2015;45(3):187–192. doi: 10.5624/isd.2015.45.3.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park TKN, Vargervik K, Oberoi S. Orthodontic and surgical management of cleidocranial dysplasia[J] Korean J Orthod. 2013;43(5):248–260. doi: 10.4041/kjod.2013.43.5.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paul SA, Simon SS, Karthik AK, et al. A review of clinical and radiological features of cleidocranial dysplasia with a report of two cases and a dental treatment protocol[J] J Pharm Bioallied Sci. 2015;7(Suppl 2):S428–S432. doi: 10.4103/0975-7406.163490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.段 小红. 口腔罕见病名录(第一版)[J] 中华口腔医学杂志. 2020;55(7):494–500. doi: 10.3760/cma.j.cn112144-20200226-00092. [DOI] [PubMed] [Google Scholar]; Duan XH. The first edition of oral rare diseases list[J] Chin J Stomatol. 2020;55(7):494–500. doi: 10.3760/cma.j.cn112144-20200226-00092. [DOI] [PubMed] [Google Scholar]
- 16.Hermann NV, Hove HD, Jørgensen C, et al. Prenatal 3D ultrasound diagnostics in cleidocranial dysplasia[J] Fetal Diagn Ther. 2009;25(1):36–39. doi: 10.1159/000195634. [DOI] [PubMed] [Google Scholar]
- 17.Daskalogiannakis J, Piedade L, Lindholm TC, et al. Cleidocranial dysplasia: 2 generations of management[J] J Can Dent Assoc. 2006;72(4):337–342. [PubMed] [Google Scholar]