

Abstract
Non‐alcoholic steatohepatitis (NASH) results from the accumulation of excessive liver lipids leading to hepatocellular injury, inflammation, and fibrosis that greatly increase the risk for hepatocellular carcinoma (HCC). Despite the well‐characterized clinical and histological pathology for NASH‐driven HCC in humans, its etiology remains unclear and there is a deficiency in pre‐clinical models that recapitulate the progression of the human disease. Therefore, we developed a new mouse model amenable to genetic manipulations and gene targeting that mimics the gradual NASH to HCC progression observed in humans. C57BL/6NJ mice were fed a Western high‐fat diet and sugar water (HFD/SW) and monitored for effects on metabolism, liver histology, tumor development, and liver transcriptome for up to 54 weeks. Chronic HFD/SW feeding led to significantly increased weight gain, serum and liver lipid levels, liver injury, and glucose intolerance. Hepatic pathology progressed and mice developed hepatocellular ballooning, inflammation, and worse fibrosis was apparent at 16 weeks, greatly increased through 32 weeks, and remained elevated at 54 weeks. Importantly, hepatocellular cancer spontaneously developed in 75% of mice on HFD/SW, half of which were HCC, whereas none of the mice on the chow diet developed HCC. Chronic HFD/SW induced molecular markers of de novo lipogenesis, endoplasmic reticulum stress, inflammation, and accumulation of p62, all of which also participate in the human pathology. Moreover, transcriptome analysis revealed activation of HCC‐related genes and signatures associated with poor prognosis of human HCC. Overall, we have identified a new preclinical model that recapitulates known hallmarks of NASH‐driven HCC that can be utilized for future molecular mechanistic studies of this disease.
Keywords: hepatocellular carcinoma, liver cancer, liver fibrosis, non‐alcoholic fatty liver disease, non‐alcoholic steatohepatitis
Abbreviations
- AFP
α‐fetoprotein
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CC3
cleaved caspase‐3
- CK‐18
cytokeratin‐18
- CPM
counts per million
- DEGs
differentially expressed genes
- ER
endoplasmic reticulum
- FASN
fatty acid synthase
- FDR
false discovery rate
- GTT
glucose tolerance test
- HCC
hepatocellular carcinoma
- HFD
high‐fat diet
- JNK
c‐Jun N‐terminal kinase
- NAFLD
non‐alcoholic fatty liver disease
- NASH
non‐alcoholic steatohepatitis
- NEFAs
non‐esterified fatty acids
- NES
normalized enrichment score
- SW
sugar water
- α‐SMA
α‐smooth muscle actin
1. INTRODUCTION
Non‐alcoholic fatty liver disease (NAFLD) is a significant cause of chronic liver disease globally and is often associated with central obesity, adipose dysfunction, inflammation, and insulin resistance. As a spectrum of liver disease states, NAFLD ranges from simple steatosis to non‐alcoholic steatohepatitis (NASH) and pericellular fibrosis that can gradually progress to cirrhosis. 1 , 2 NASH is also characterized by hepatocellular injury and ballooning, lobular inflammation, and varying levels of fibrosis. 3 Approximately 25% of NAFLD patients are diagnosed with NASH, which can further progress to end‐stage liver disease and hepatocellular carcinoma (HCC), a major cause of cancer‐related mortality. 4 , 5 Epidemiological studies show that NAFLD is a prominent etiology in HCC. 6
The onset of obesity and insulin resistance has been associated with an increased risk for NASH and advanced fibrosis. 7 , 8 Although the mechanisms are unclear, this is accompanied by additional events that drive the disease, such as liver oxidative stress, endoplasmic reticulum (ER) stress, and inflammation that further progresses to NASH with the manifestation of hepatocellular injury/ballooning and fibrosis. 4 , 5 , 6 Both NASH development and HCC progression depend on elevated proinflammatory cytokines, such as TNF‐α, and hepatic accumulation of p62, an autophagy receptor required for premalignant progression of hepatocytes to HCC. 9 , 10 Nevertheless, while much is known about the clinical pathology of this disease, there remains a lack of reliable biomarkers as well as effective therapies for NASH‐driven HCC. 5
Mouse models that mimic the gradual progression of NASH toward HCC observed in humans are imperative to designing better treatments. Thus far, mouse models of NASH and HCC have relied on carcinogen administration, transgenic/isogenic mice, and/or dietary manipulations, most of which do not fully recapitulate all hallmarks of this disease. 11 A major trigger associated with the development of NASH in humans is the intake of excessive calories, particularly with the consumption of food high in fat and sugar. Specifically, diets high in saturated fat, trans‐fat, carbohydrates in the form of fructose, and cholesterol increase the risk of developing NAFLD/NASH. 12 , 13 , 14 Diet‐induced obese mice fed a more physiologically relevant ‘Western’ diet designed to promote NASH pathology exhibit similar clinical and histological features of human NASH. 12 , 15 However, the progression of diet‐induced NASH to liver cancer has only been observed in transgenic or specific isogenic hybrid strains of mice. 9 , 11 , 15 While this is a significant advancement in preclinical models, these models are not amenable to genetic manipulations and cannot be used for gene‐targeted mice. Preclinical models that can use gene‐targeted knockout mice will provide the field with the foundation for further research as it is highly suitable for gaining new knowledge of how NASH progresses to HCC and the development of new targets for treatment. It will enable exploration of the phenotypes of gene‐targeted mice required for the genetic elucidation of factors and/or signaling pathways involved in NASH to HCC progression.
Guided by a diet‐induced mouse model in isogenic 129S1/C57Bl/6 hybrid mice 15 and to overcome the problem of the complex genetic background, here we present a new preclinical model of Western diet‐induced NAFLD/NASH and progression to HCC. To this end, commonly used C57BL/6NJ mice were fed a high‐fat diet (HFD) with ad libitum access to high fructose and glucose sugar water (SW) and monitored for changes in metabolism, liver histology, tumor development, and the liver transcriptome. We demonstrated that this model recapitulates the changes found in human NASH and progression to HCC. Moreover, genes and pathways altered in HFD/SW‐fed mice that spontaneously develop HCC are similarly regulated in human NASH and HCC. Thus, this model reliably mimics key pathophysiological, metabolic, histologic changes, and transcriptomic gene signature and clinical outcomes observed in the progression of the human disease, emphasizing its applicability to future mechanistic studies of NASH‐driven HCC.
2. MATERIALS AND METHODS
2.1. Animals
C57BL/6NJ mice were purchased from Jackson Laboratory (Bar Harbor) and maintained by inbreeding. Mice were housed in a 12 h light, 12 h dark cycle in a 21–23°C facility. The Institutional Animal Care and Use Committee of Virginia Commonwealth University approved all animal procedures.
2.2. Dietary treatments
Male mice were maintained on a standard chow diet (Harlan TD.7912; Envigo). At 8–9 weeks of age, randomly assigned mice were fed ad libitum a western high‐fat diet (HFD), high in fat (42% kcal from milk fat), cholesterol (0.2%), and sucrose‐enriched carbohydrates (42.7% kcal; Harlan TD.88137; Envigo). Drinking water was supplemented with a high d‐fructose (23.1 g/L) and D‐glucose (18.9 g/L). Control mice were fed ad libitum chow diet with access to normal drinking water.
2.3. Histology
At the indicated timepoints of dietary treatment, mice were weighed and euthanized by inhaled isoflurane and cervical dislocation. Livers were extracted and portions were either fixed in 10% formalin followed by histological processing, snap‐frozen in liquid nitrogen, or frozen in Tissue‐Tek optimal cutting temperature solution. Formalin‐fixed, paraffin‐embedded liver sections were stained with H&E. Liver fibrosis was assessed by staining with Masson's trichrome and Sirius Red as described 16 and collagen content with the Sirius Red/Fast Green collagen kit (Chondrex, Inc.). Liver histology was assessed by an expert liver pathologist in a treatment‐blinded manner based on the NASH‐Clinical Research Network scoring system to score steatosis, lobular inflammation, and hepatocellular ballooning as previously described. 17 , 18 Steatosis (percentage hepatocytes with fat droplets) was scored as 0 (<0.5%), 1 (5%–33%), 2 (>33%–66%) or 3 (>66%). Hepatocyte ballooning was scored as 0 (none), 1 (few cells), or 2 (many cells). Lobular inflammation was scored as 0 (no foci), 1 (<2 foci per field), 2 (2–4 foci), or 3 (>4 foci). NAFLD activity score was from the combined steatosis, inflammation, and ballooning scores. Fibrosis was determined as stage F0 (none), F1 (mild), F2 (perisinusoidal and portal/periportal fibrosis), F3 (bridging fibrosis), or F4 (obvious cirrhosis). Liver steatosis was confirmed with Oil‐Red‐O staining of frozen sections as described. 19 Tumor histology was classified as defined previously. 15 Adenoma tumors were characterized as a nodular proliferation of normal hepatocytes while retaining a trabecular organization. Well‐differentiated HCC was defined as cytological and architectural abnormalities such as major trabecular thickening, loss of sinusoidal barrier, pseudo‐acinar formation, and isolated arteries. Poorly differentiated HCC was defined as major cytological abnormalities, including significantly increased nuclear/cytoplasmic ratio and architectural disorganization with loss of trabecular organization.
2.4. Liver and serum biochemistry
For liver triglyceride analysis, 40 mg of tissue was ground to powder and saponified in six volumes of 2:1 ethanol and 30% KOH at 60°C for 5 h. Next, 1.08 volumes of 1 M MgCl2 were added, incubated on ice for 10 min, centrifuged, and the supernatant analyzed with the Stanbio Triglyceride LiquiColor Test (Mono, 2200‐225). For serum measurements, serum was first obtained from whole blood after coagulation and centrifugation using BD Microtaine Capillary Blood Collector tubes (#BD 365967). Alanine and Aspartate Aminotransferase (ALT/AST) measurements were performed using the Cobas ALTL and ASTL kits with a Cobas c311 Analyzer (Roche Diagnostics). Serum levels of triglycerides, cholesterol, and non‐esterified fatty acids (NEFAs) were measured by the University of Cincinnati Mouse Metabolic Phenotyping Center.
2.5. Glucose tolerance test
Mice were fasted overnight, and baseline blood glucose levels were measured from tail‐vein blood using a TRUEtrack Blood Glucose meter (Nipro Diagnostics). Glucose (2 mg dextrose/g body weight) was dissolved in sterile PBS and injected intraperitoneally. After glucose injection, blood glucose levels were measured at 15, 30, 60, 90, and 120 min, and areas under the curve were determined.
2.6. Immunohistochemistry
Sections from formalin‐fixed and paraffin‐embedded livers were stained for α‐smooth muscle actin (α‐SMA; 1:200; ab5694), cleaved caspase‐3 (CC3; 1:500; CST9664), and Ki67 (1:400; CST12202) by the Virginia Commonwealth University Cancer Mouse Models Core using the Leica Biosystems Bond RX staining system. Positive cells and the percentage of area stained were quantitated with ImageJ. For α‐fetoprotein (AFP) staining, formalin‐fixed/paraffin‐embedded sections were deparaffinized with xylene and rehydrated with ethanol (100%, 90%, 80%, and 70%) and then in 100% PBS. Next, sections were immersed in antigen retrieval buffer (10 mM sodium citrate, 0.05% Tween‐20, pH 6.0) for 20 min at 95°C, rinsed in PBS, and quenched with 2% hydrogen peroxide for 20 min. Sections were rinsed in PBS, blocked in 1.5% goat serum in PBS plus 0.1% Tween‐20 (PBST) for 1 h at room temperature, and incubated overnight with anti‐AFP antibody (1:300, Proteintech 14550‐1‐AP) in blocking buffer. After washing five times with PBST, the anti‐rabbit biotinylated secondary antibody was applied, followed by incubation with avidin and biotinylated‐HRP according to the Vectastain Elite ABC kit (Vector Laboratories). The sections were then rinsed in PBST, incubated with DAB peroxidase substrate for 2 min, and rinsed in tap water for 10 s. Nuclei were stained with hematoxylin (Vector Laboratories).
2.7. Immunofluorescence
Paraffin‐embedded liver sections were dewaxed with Histo‐Clear (twice for 5 min) and Histo‐Clear 90% with 10% ethanol for 5 min, followed by 5 min incubations two times with ethanol 100%, ethanol 95%, ethanol 80%, ethanol 70%, and finally two baths in ddH2O for at least 20 min. According to the manufacturer's instructions, antigen retrieval was performed with the Universal HIER antigen retrieval reagent (Abcam). Afterward, the sections were washed three times with PBS for 10 min each. Sections were blocked and permeabilized with PBS containing 5% Normal Serum Blocking Solution (Biolegend), 1% BSA, and 0.4% Triton‐X100 for 1 h at room temperature in the dark. Subsequently, sections were incubated with rabbit anti‐cytokeratin‐18 (CK‐18; Invitrogen) at 1:200 in PBS containing 5% Normal Serum Blocking Solution, 1% BSA, and 0.4% Triton‐X100 overnight in a dark‐wet chamber at 4°C. After washing three times for 5 min with PBS, sections were stained with anti‐rabbit AlexaFluor488 1:300 in PBS containing 5% Normal Serum Blocking Solution, 1% BSA, and 0.4% Triton‐X100 for 2 h in a dark‐wet chamber at room temperature. Sections were washed three times for 5 min with PBS and stained with 1 µg/ml DAPI for 15 min at room temperature in the dark. Afterward, sections were washed with PBS three times for 5 min and mounted with CC/Mount on coverslips. After drying for at least 6 h, sections were imaged with a BZ‐X810 Keyence fluorescence microscope.
2.8. Immunoblotting
Frozen liver tissues (20 mg) were ground to powders and proteins extracted with a modified RIPA buffer (50 mM Tris base, 1 mM EDTA, 150 mM NaCl, 0.1% SDS, 1% Triton X‐100, 1% sodium deoxycholate). Proteins were separated by SDS‐PAGE, transferred to nitrocellulose, and immunoblotted with antibodies for fatty acid synthase (FASN) (#CST3189), CHOP (#CST2895), phospho‐JNK (#CST4668), total JNK (#CST9252), p62 (#CST5114), and β‐actin (#CST3700).
2.9. RNA extraction and sequencing
Frozen liver tissues were homogenized in Trizol (Invitrogen), and total RNA was isolated with the RNeasy mini kit (Qiagen). RNA‐sequencing was performed by the Genomics, Epigenomics, and Sequencing Core (GES Core) at the University of Cincinnati. The quality of RNA was determined by Bioanalyzer (Agilent), and 1 µg of total RNA was used to isolate polyA RNA with the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England BioLabs). PolyA RNA was enriched with the SMARTer Apollo NGS library prep system (Takara Bio USA). The library was prepared with the dUTP‐based stranded library NEBNext Ultra II Directional RNA Library Prep Kit (New England BioLabs), indexed, and amplified with 8 PCR cycles. Next, individually indexed libraries were analyzed by Bioanalyzer, proportionally pooled and sequenced using the Nextseq 550 sequencer (Illumina) with the single read 1 × 85 bp setting, which generated about 25 million pass filter reads per sample.
Fastq files were generated using Illumina BaseSpace. The reference genome used was mouse GRCm38/mm10 from the UCSC Genome Browser Gateway (http://hgdownload.soe.ucsc.edu/goldenPath/mm10/bigZips/chromFa.tar.gz). Reads were aligned using STAR (Aligner) version 2.6.1a. 20 Gene counts were quantified for each sample based on the number of aligned reads that match each annotated gene using the RnaReadCounter tool within the BaseSpace RNA‐Seq Alignment app v2.0.2 to generate counts per million (CPM). Gene counts were preprocessed and analyzed for differential expression using the R package edgeR v.3.30.0. 21 p‐Values obtained for differentially expressed genes (DEGs) were corrected using a false discovery rate multiple testing correction method. Gene Ontology functional enrichment analysis for biological processes was conducted using Metascape with default settings and p‐value cutoff of 0.01. Gene Set Enrichment Analysis was done with R package clusterProfiler v.4.2.0 22 with gene signatures for human NASH liver samples, 23 human liver disease prognosis, 24 and human HCC subclasses. 25 Expression profiles for selected genes were visualized using log2(CPM) values with Heatmapper. 26
2.10. Statistical analysis
Statistical significances were calculated with Student's t‐test for comparison of two groups, or with ANOVA followed by post hoc tests for multiple comparisons using GraphPad Prism 7.0 software. All bioinformatics and statistical calculations were conducted within the R/Bioconductor environment v4.0.0.
3. RESULTS
3.1. C57BL/6NJ mice fed HFD/SW exhibit obesity and metabolic dysfunction
To develop a mouse model of NASH and HCC, C57BL/6NJ mice were fed a Western high fat, high carbohydrate with cholesterol diet (HFD) and high fructose‐glucose drinking water (SW) for up to 54 weeks (Figure 1A). Feeding HFD/SW led to obesity, as evident in increased body weights at 16, 32, and 54 weeks, compared to chow‐fed mice (Figure 1B). Liver weights also increased at each time point (Figure 1C), which paralleled elevated serum ALT and AST enzymes, indicative of liver damage (Figure 1D). Liver lipid accumulation was apparent as triglycerides were markedly increased at 16 and 54 weeks by 11.4‐ and 4.9‐fold, respectively (Figure 1E). HFD/SW‐fed mice had hypercholesterolemia and also significantly elevated serum NEFAs at both time points with a trend for increased serum triglycerides at 16 weeks and no change after 54 weeks, similar to previous results in mice fed a Western diet 15 , 27 (Figure 1F). To determine calorie consumption, mice fed HFD/SW for 15 weeks were housed individually in metabolic chambers and average food consumption was measured for 5 days. Calorie intake was 10 ± 0.7 and 4.9 ± 0.5 kcal/mouse/day during dark and light cycles, respectively. In agreement with previous studies, 15 , 27 metabolic parameters in HFD/SW fed mice at 16 weeks showed minimal variation and were consistent with little variation in calorie consumption of less than 10%. Importantly, HFD/SW led to glucose intolerance, which is strongly associated with human NAFLD and risk for other adverse effects 28 (Figure 1G). Therefore, chronic HFD/SW fed C57BL/6NJ mice exhibit obesity, liver injury, dyslipidemia, and glucose intolerance as observed in obesity‐associated NASH in humans. 28 , 29
FIGURE 1.
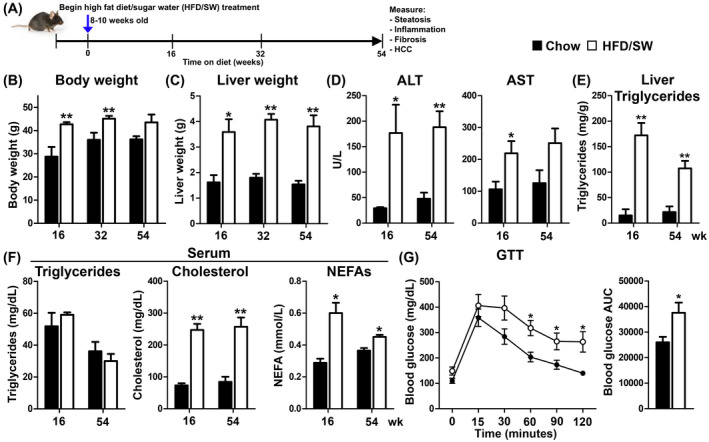
C57BL/6NJ mice fed HFD/SW induces weight gains and metabolic alterations consistent with the development of NASH. (A) Schematic representation of chronic chow or high fat/carbohydrate with cholesterol diet (HFD) with high fructose/glucose (SW) feeding, and timepoints for monitoring fatty liver, NASH, and HCC. (B) Body weights, (C) liver weights, (D) ALT and AST serum enzyme levels, (E) liver triglycerides, (F) and serum triglycerides, cholesterol, and NEFAs measured at the indicated time points (n = 3–9 mice per group). (G) GTT and calculated area under the curve (AUC) at 32 weeks (n = 6–7 mice per group). Data are expressed as means ± SEMs. *p < .05, **p < .01 compared to chow diet at the respective timepoints. ALT, alanine aminotransferase; AST, aspartate aminotransferase; GTT, glucose tolerance test; HCC, hepatocellular carcinoma; NEFAs, non‐esterified fatty acids; NASH, non‐alcoholic steatohepatitis; SW, sugar water.
3.2. Chronic HFD/SW feeding leads to histological signs of NASH
Determination of NAFLD and NASH in humans largely relies on quantitative assessment of histopathology from liver biopsies. 30 In response to HFD/SW, steatosis was clearly observed as macrovesicular and small well‐defined hepatic fat droplets (Figure 2A,B). Steatosis was corroborated by increased neutral lipid Oil‐Red‐O staining in livers from mice fed HFD/SW compared to chow (Figure 2C), consistent with increased liver triglycerides. Liver histology also indicated that hepatocellular ballooning and inflammation were increased by HFD/SW diet. Hepatic ballooning, a key determinant of steatohepatitis, 31 was significantly apparent at 16 weeks, increased at 32, and remained elevated at 54 weeks (Figure 2A,E). This was confirmed by the lack of CK‐18 staining in ballooned hepatocytes, which is present in normal hepatocytes (Figure 2E), as described in humans. 32 Lobular inflammation was elevated at 32 weeks and persisted through 54 weeks (Figure 2A,E). Likewise, the NAFLD activity score increased by week 16 and remained considerably higher thereafter (Figure 2A).
FIGURE 2.
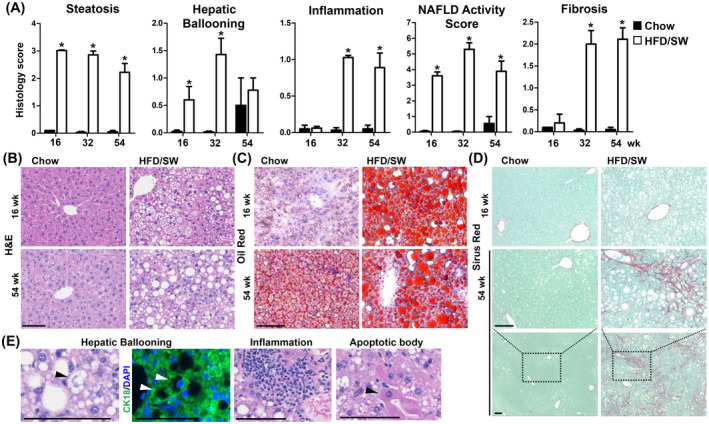
Chronic HFD/SW feeding leads to liver steatosis, steatohepatitis, and fibrosis. (A) Histology scores for steatosis, hepatic ballooning, inflammation, fibrosis, and NAFLD activity score at 16, 32, and 54 weeks for mice fed chow diet (n = 3) or HFD/SW (n = 7–10). Data are expressed as means ± SEM. *p < .05 compared to the chow diet at the respective timepoints. (B–D) Representative liver sections stained with H&E, Oil‐Red‐O, and Sirius Red, respectively. (E) Representative H&E images of hepatic ballooning, inflammation, and apoptotic bodies. Hepatic ballooning is also depicted as loss of cytoplasmic CK18 staining (white arrow). Scale bar: 100 µm. CK18, cytokeratin 18; NAFLD, non‐alcoholic fatty liver disease; SW, sugar water.
Liver fibrosis, a cardinal sign of human NASH, 6 began at 16 weeks and progressed to stages 2 and 3, after 34 and 54 weeks on HFD/SW based on Sirius red and Masson's trichrome staining displaying pericellular and bridging fibrosis (Figure 2A,D), similar to the patterns in human NASH. Moreover, this correlated with elevated α‐SMA staining (3.8‐fold increase), a marker of hepatic stellate cell activation (Figure 3A).
FIGURE 3.
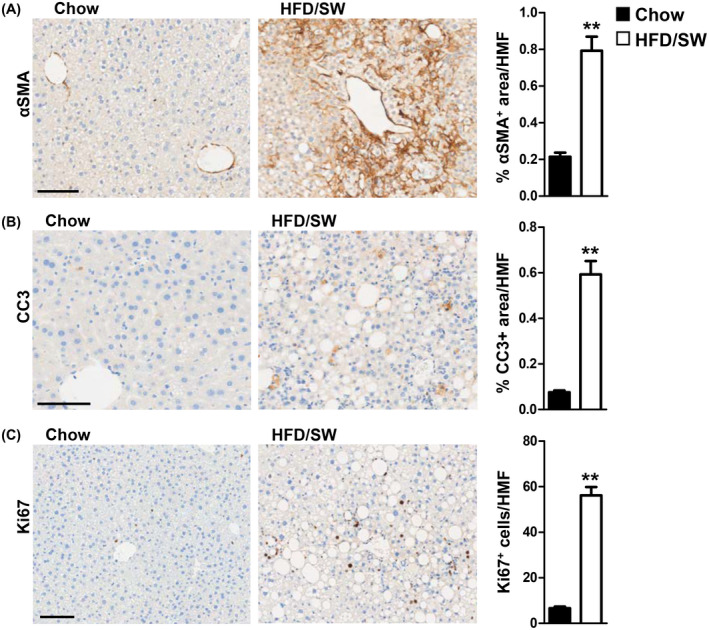
Myofibroblast activation, hepatocyte apoptosis, and proliferation are increased in livers of HFD/SW‐fed mice. (A–C) Representative liver sections from mice fed chow diet or HFD/SW stained for α‐SMA, CC3, and Ki67, respectively. Scale bar: 100 µm. (A) Myofibroblast activation is determined by the percentage of α‐SMA positive area per high magnification field (HMF). (B) Hepatic apoptosis measured as the percentage of CC3 positive area per HMF. (C) Hepatocyte proliferation measured as Ki67 positive cells per HMF. Data are expressed as means ± SEMs, n = 3–4 mice per group. *p < .05 compared to chow diet. α‐SMA, α‐smooth muscle actin; CC3, cleaved caspase‐3; SW, sugar water.
Apoptotic bodies were also observed in the livers of HFD/SW fed mice (Figure 2E). This was associated with significant cell death revealed by increased CC3 staining, which was accompanied by increased Ki67‐positive proliferating hepatocytes at 54 weeks (Figure 3B,C). Thus, chronic HFD/SW induces constant hepatocellular cell death and compensatory proliferation, an important characteristic for the development of hepatocarcinogenesis. 33
3.3. HFD/SW induces spontaneous HCC in C57BL/6NJ mice
Hepatocellular cancer was detected as macroscopic nodules in 75% of mice (12 of 16) after 54 weeks of HFD/SW (Figure 4A,B). Histological examination found that 50% of tumors were HCC, with well‐differentiated and steatohepatitic features and loss of trabecular organization, while the rest were adenomas (Figure 4B). None of the mice on the chow diet developed HCC, and only one mouse on the chow diet presented with an adenoma (1 of 9 mice; Figure 4B). Importantly, similar to human females with reduced susceptibility to HCC, 34 , 35 female mice on HFD/SW for 54 weeks did not develop liver cancer (data not shown). Circulating AFP is often used in clinical diagnosis of HCC, as it is expressed by hepatocytes during development and absent in adults, but it is re‐expressed from hepatocellular tumors. 36 Therefore, we stained for AFP in HFD/SW fed livers. AFP was expressed in tumor tissue, but not in the surrounding non‐tumor area, indicative of HCC (Figure 4C). Moreover, assessment of proliferation showed tumors contain numerous Ki67‐positive proliferating hepatocytes (Figure 4D). Together, these findings demonstrate that C57BL/6NJ mice fed HFD/SW develop pathological features of NASH leading to spontaneous development of a high incidence of adenomas and HCC.
FIGURE 4.
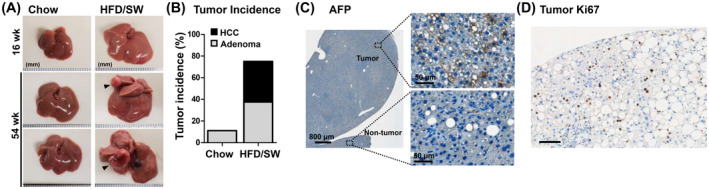
Mice fed HFD/SW for 54 weeks develop HCC. (A) Representative liver images of mice fed chow diet or HFD/SW for 16 and 54 weeks. Black arrows indicate liver tumors. (B) Liver tumor incidence for mice fed chow diet (n = 9) and HFD/SW for 54 weeks (n = 16) represented as the percentage of mice with HCCs and adenomas. (C) Representative immunohistochemical staining for tumor marker AFP in tumor and non‐tumor tissue. Scale bars: 800 and 50 µm (image insets). (D) Intratumor hepatocyte proliferation was shown as Ki67 staining. Scale bar: 100 µm. AFP, α‐fetoprotein; HCC, hepatocellular carcinoma; SW, sugar water.
3.4. NASH‐associated molecular pathways are activated by HFD/SW
The molecular pathways implicated in NASH include activation of de novo lipogenesis, ER stress, inflammation, and inhibition of autophagy. 6 , 8 Indeed, protein levels for key genes involved in fatty acid synthesis, including FASN, and ER stress markers C/EBP homologous protein (CHOP), and phospho‐c‐Jun N‐terminal kinase (JNK) were elevated in livers of mice fed HFD/SW (Figure 5A). The ubiquitin‐binding autophagy receptor p62 involved in impaired autophagy, which accumulates in premalignant hepatocytes and is necessary and sufficient for HCC induction in mice and humans, 10 , 37 was also increased by HFD/SW (Figure 5A).
FIGURE 5.
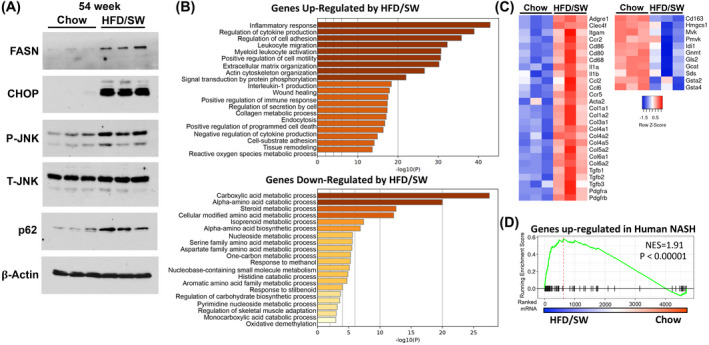
Molecular pathways associated with NASH are activated by HFD/SW. (A) Hepatic protein levels from mice fed chow diet or HFD/SW for 54 weeks were analyzed by immunoblotting with the indicated antibodies. β‐Actin was used as a loading control. (B–D) Liver transcriptome analysis of chow and HFD/SW mice at 54 weeks. (B) GO biological processes pathway enrichment analysis of significantly differentially expressed genes upregulated or downregulated in HFD/SW compared to chow diet. (C) mRNA expression heatmap for genes associated with fibrosis, stellate cell activation, and inflammation from livers of chow and HFD/SW fed mice. (D) Gene Set Enrichment Analysis of hepatic genes upregulated in human NASH patients compared to those from mice fed chow and HFD/SW. NES, normalized enrichment score; NASH, non‐alcoholic steatohepatitis; GO, gene ontology; SW, sugar water.
Global liver transcriptome analysis was used to identify HFD/SW‐induced DEGs. Out of 1800 DEGs (|fold‐change| >2, p < .05), 1565 genes were upregulated and 235 genes downregulated by HFD/SW compared to chow‐fed mice. Analysis of genes upregulated by HFD/SW showed significant enrichment in biological processes related to multiple inflammatory responses, including cytokine production, interleukin‐1 production, leukocyte migration, and activation of myeloid leukocytes, as well as extracellular matrix organization, adhesion, and collagen metabolic processes (Figure 5B). Markers previously shown to detect and differentiate Kupffer cell and monocyte‐derived macrophage populations (Adgre1, Clec4f, Itgam, Ccr2) in diet‐induced NASH 38 were among the genes upregulated by HFD/SW. In addition, genes related to proinflammatory macrophage phenotypes (Cd86, Cd80, Cd68), interleukins (Il1a, Il1b), chemokines (Ccl2, Ccl6), and hepatic stellate cell activation and fibrosis (Acta2/α‐Sma, collagens, Tgfb1‐3, Pdgfra, Pdgfrb) were also elevated (Figure 5C).
Genes downregulated by HFD/SW feeding are enriched in biological processes related to the metabolism of carboxylic acids, amino acids, and steroids (Figure 5B). Cd163, a marker of immunosuppressive macrophages, was repressed the most among all downregulated genes (Figure 5C). These also include genes involved in amino acid degradation (Gls2, Gcat, Sds), the synthesis of sterols (Hmgcs1, Mvk, Pmvk, Idi1), detoxification by glutathione S‐transferases (Gsta2, Gsta4), and S‐adenosylmethionine catabolism (Gnmt; Figure 5C). Next, we compared the overlap of genes activated in human NASH liver samples 23 with genes regulated in livers from HFD/SW fed mice. Importantly, genes known to be upregulated in human NASH were significantly enriched in the mouse genes induced by HFD/SW (normalized enrichment score [NES] = 1.91, p < 1e−5; Figure 5D). Taken together, these results demonstrate that chronic HFD/SW alters the expression of molecular pathways in mice related to inflammation, fibrosis, and metabolism that are consistent with the development of NASH in humans.
3.5. Chronic HFD/SW feeding activates HCC‐related genes and signatures associated with human disease prognosis and HCC subclasses
Genes known to be expressed in HCC progenitors as well as HCC 39 , 40 were significantly increased in the livers of mice fed HFD/SW for 54 weeks compared to the chow diet, whereas liver‐specific genes were not changed or repressed, suggesting a shift toward a tumor microenvironment more prone to liver cancer (Figure 6A). We next assessed the significance of HFD/SW‐regulated genes with respect to human disease prognosis using a human 186 gene signature from non‐tumor tissue that predicts prognosis of patients with HCC as well as patients with early‐stage liver cirrhosis in terms of death, progression to advanced cirrhosis, and development of HCC. 24 , 41 Poor prognosis genes, associated with a four‐fold higher rate of tumor incidence, 41 were highly enriched in genes activated by chronic HFD/SW (NES = 1.81, p < .0001; Figure 6B). In contrast, good prognosis genes were enriched in genes downregulated in livers of mice fed HFD/SW (NES = −2.53, p < .001; Figure 6C). To understand which type(s) of tumors could potentially develop in the livers of mice fed HFD/SW, we compared our expression data with human signatures of HCC subclasses from early‐stage tumors. 25 Genes activated in HFD/SW livers were strongly associated with the development of human S1 subclass tumors (NES = 1.98, p < 1e‐7; Figure 6C), which, in addition to the S2 subclass, is also associated with poor prognosis. 25 , 42 Molecular pathways for human S1 subclass genes activated by HFD/SW include locomotion/cell migration, cytokine production, extracellular matrix degradation, and blood vessel development (Figure 6D). These processes support a relationship with the S1 subclass that is suggested to be related to a more invasive/disseminative phenotype and associated with a greater risk of early recurrence. 25 Overall, these findings demonstrate that in mice, chronic HFD/SW promotes NASH‐driven HCC and activation of molecular pathways related to poor prognosis and the development of HCC in humans.
FIGURE 6.
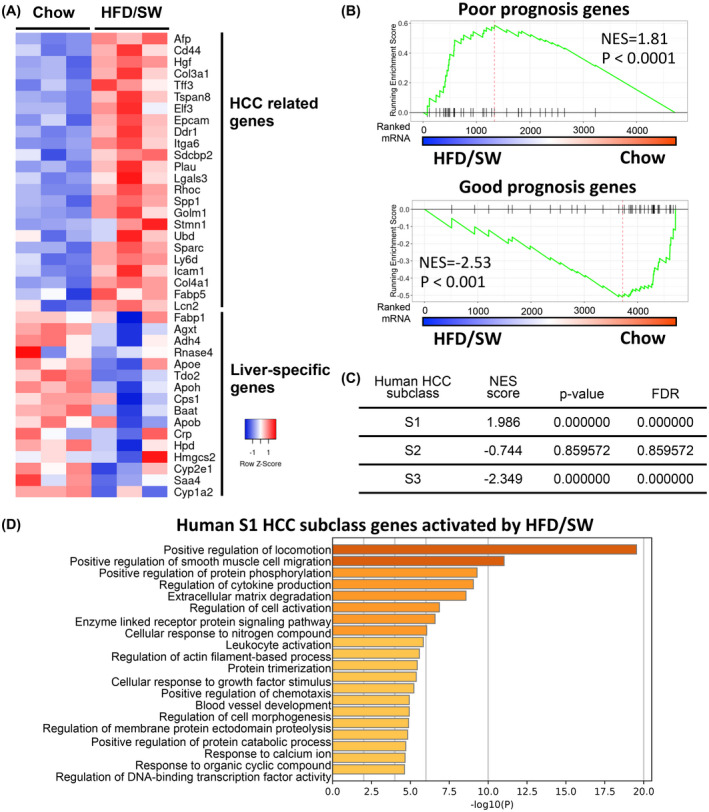
Chronic HFD/SW induces signaling pathways associated with HCC. (A) Heatmap of expression of HCC‐related and liver‐specific genes in livers from mice fed chow and HFD/SW for 54 weeks. (B) GSEA of gene signatures associated with poor prognosis (73 genes) or good prognosis (113 genes) in human HCC patients compared to those in livers of mice on chow and HFD/SW. (C) GSEA of gene signatures associated with human HCC subclasses S1, S2, and S3 defined by Hoshida et al 25 compared to HFD/SW‐induced HCC at 54 weeks. NES, normalized enrichment score; FDR, false discovery rate. (D) Molecular pathways activated by HFD/SW related to human S1 subclass genes. GSEA, Gene Set Enrichment Analysis; HCC, hepatocellular carcinoma; SW, sugar water.
4. DISCUSSION
The progression from hepatosteatosis towards NASH significantly increases the risk of developing HCC, the third leading cause of cancer‐related deaths worldwide. 2 , 4 , 43 The epidemic of obesity and its related NAFLD/NASH complications in humans has been associated with excessive consumption of calories derived from high fat, cholesterol, and sugar diets particularly enriched in fructose. 44 , 45 , 46 Nevertheless, the mechanisms of NASH‐driven HCC are still unclear, and efforts to develop animal models that mimic the gradual progression of fatty liver to NASH and HCC as observed in humans are important for understanding the etiology of this complex disease. 11
Previous diet‐induced mouse models found that HFD alone while promoting obesity, insulin resistance, and hepatic steatosis, was unable to progress to NASH 9 (Table 1). Methionine‐ and choline‐deficient diets promote steatosis and fibrosis; however, this model causes rapid weight loss, does not result in HCC, and does not mimic the etiology and metabolic features of human NASH. 47 Accumulating evidence has shown that progression toward hepatocellular ballooning, a hallmark of NASH, and fibrosis with HFD requires additional dietary manipulations, such as a choline‐deficient diet or HFD combined with high cholesterol and sugar diet (HF/Chol/SD). 48 , 49 Nevertheless, while choline‐deficient HFD causes a NASH phenotype as well as HCC, the penetrance of HCC induction is very low. 48 Similarly, a model based on a high trans‐fat and sugar diet led to obesity, steatosis, and HCC, but did not cause hepatocellular ballooning, and it remains to be determined whether the metabolic and immune mechanisms reflect the human disease. 50 Moreover, mouse models of NASH‐driven HCC thus far have relied on specific transgenic or isogenic strains that are less amenable to genetic manipulations. 9 , 15 To address these deficiencies, we developed a new preclinical model of Western diet‐induced NASH and HCC in C57BL/6NJ mice, a genetic background readily amenable to gene‐targeted approaches. Based on a previous Western diet model 15 and to mimic diet‐induced NASH in humans, we demonstrate reliable progression to NASH and HCC in C57BL/6NJ mice fed HFD, containing 0.2% cholesterol, plus high fructose/glucose SW. Importantly, these mice exhibited key clinical histopathological and molecular characteristics of the human disease, including p62 accumulation that has been implicated in HCC initiation in humans, 10 as well as human gene signatures of NASH, poor prognosis, and an environment susceptible to the development of HCC. In contrast, most previous models did not examine these factors important for human HCC (Table 1). While similar to our findings, a recent study also found consistent NASH and HCC development with HF/Chol/SD in C57BL/6J mice, 49 whereas p62 accumulation and association with poor prognosis of HCC in humans were not examined. Furthermore, the 2% high cholesterol diet used has been considered to cause “toxic steatohepatitis” and is less physiologically relevant. 51 Similarly, other models using various toxins, such as diethylnitrosamine and carbon tetrachloride, a hepatotoxin that induces genotoxicity and oxidative DNA damage, that increase the onset of HCC are not physiological as the majority of NASH in humans does not involve exposure to these toxins.
TABLE 1.
Comparison of diet‐induced NASH and HCC mouse models
| Model | Obesity | Insulin resistance | Steatosis | Inflammation/ ER stress | Ballooning | Fibrosis | p62 accumulation | HCC | Human NASH gene signature | Human prognosis gene signature | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HFD | ++ | ++ | ++ | + | — | — | — | — | — | nd | [9] |
| MCD | — | — | + | + | — | ++ | nd | — | nd | nd | [47] |
| CD | — | — | + | + | — | ++ | nd | — | nd | nd | [61] |
| MCD HFD | ++ | ++ | ++ | ++ | — | ++ | nd | — | — | nd | [62] |
| CD HFD | ++ | ++ | ++ | ++ | ++ | + | nd | + | nd | nd | [48] |
| Trans‐fat/SD | ++ | ++ | ++ | ++ | — | ++ | nd | ++ | nd | nd | [50] |
|
C57BL/6J HF/Chol/SD a |
++ | ++ | ++ | ++ | ++ | ++ | nd | ++ | ++ | nd | [49] |
|
MUP‐uPA Tg HFD |
++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | nd | [9] |
|
DIAMOND HF/Chol/SD b + SW |
++ | ++ | ++ | ++ | ++ | ++ | nd | ++ | ++ | ++ | [15] |
|
C57BL/6NJ HF/Chol/SD b + SW |
++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | Current study |
Abbreviations: —, not present or negative; +, moderately present; ++, highly present; CD, choline‐deficient diet; DIAMOND, isogenic BL6J/129 strain; ER, endoplasmic reticulum; HCC, hepatocellular carcinoma; HF/Chol/SD, high fat + high cholesterol + high sugar diet; HFD, high‐fat diet; MCD, methionine‐ and choline‐deficient diet; MUP‐uPA Tg, mice with transient overexpression of urokinase plasminogen activator in hepatocytes; NASH, non‐alcoholic steatohepatitis; nd, not determined; SW, sugar water; Trans‐fat/SD, trans‐fat + high sugar diet.
2% cholesterol.
0.2% cholesterol.
Our model of chronic HFD/SW feeding led to similar metabolic dysfunctions as observed in obesity‐related NAFLD/NASH in humans. These HFD/SW‐fed mice developed obesity, increased serum cholesterol and NEFAs, liver steatosis, and marked glucose intolerance. In addition, HFD/SW caused significant increases in hepatocellular ballooning, inflammatory cell infiltration, and fibrosis, the latter correlating with elevated stellate cell activation, recapitulating clinical histopathological characteristics of NASH in humans. 11 , 29 , 52 Although initially elevated, parameters including liver steatosis, triglycerides, and hepatocellular ballooning showed a trend for reduction after 54 weeks, whereas fibrosis and liver damage, indicated by increased serum ALT and AST levels, remained elevated. In human NASH patients, the gradual progression of fibrosis toward cirrhosis has been associated with diminished liver architecture and depletion in liver lipids, which can lead to complete fat loss or “burn‐out” NASH. 53 Therefore, these findings indicate chronic HFD/SW might result in the transition to a more advanced fibrosis phenotype while reducing liver lipids, similar to the progression in humans.
Mechanistic studies suggest NASH is the result of multiple “hits” to the liver, where in addition to steatosis other events such as hepatocellular injury, inflammation, ER stress, and oxidative stress contribute to its development. 54 Indeed, at the molecular level, chronic HFD/SW also induced de novo lipogenesis as shown by increased FASN, the rate‐limiting reaction in de novo fatty acid biosynthesis, 55 and enhanced ER stress markers including CHOP and JNK. This is consistent with the role of high fructose diets driving liver lipogenic gene expression and triglyceride accumulation and induction of liver ER stress. 15 , 44 FASN expression in NAFLD livers correlated with the degree of hepatic steatosis, 56 whereas ER stress has been implicated in the pathogenesis of insulin resistance, hepatosteatosis, and NASH, conditions that increase the risk of HCC. 9 , 57
In addition, our transcriptome analyses clearly indicated that HFD/SW‐induced activation of genes related to pro‐inflammatory responses and fibrosis, in agreement with histopathological findings in these mice and known key characteristics of NASH. 11 , 29 , 38 Importantly, in contrast to several widely used NASH mouse models with little to no overlap in gene expression with human NASH livers, 23 we found genes activated in human NASH were highly enriched in genes upregulated in HFD/SW fed mice, suggesting that they closely recapitulate the transcription profiles of human NASH. Therefore, our findings indicate that molecular pathways associated with NASH in humans are also activated in C57BL/6NJ mice by the chronic HFD/SW regimen.
These mice also spontaneously developed HCC without the need for feeding toxic diets or carcinogens that induce liver damage. We found that levels of p62, a ubiquitin‐binding autophagy receptor and a hallmark of NASH‐driven HCC, 10 , 37 were markedly elevated in non‐tumor liver tissue of mice fed HFD/SW. p62 is the major component of Mallory‐Denk bodies, accumulates in premalignant hepatocytes, and has been detected in many human HCC specimens. 58 A previous study elegantly indicated that p62 accumulation is required for progression from premalignancy to malignancy, likely by maintaining the survival of stressed HCC‐initiating cells, enabling these cells to accumulate multiple oncogenic mutations. 10 Non‐tumor liver tissue from patients with early‐stage HCC displayed elevated p62 expression that correlated with increased cancer recurrence and reduced disease‐free survival. 10
NASH‐driven HCC is commonly associated with chronic liver damage, hepatocyte death, and inflammation that lead to a compensatory proliferative response and the onset of liver cancer. 59 In agreement, chronic HFD/SW feeding caused significant hepatocyte damage and apoptosis that was accompanied by hepatocellular proliferation. Importantly, this compensatory response to chronic HFD/SW correlated with the development of HCC, and many genes are known to be activated in HCC progenitors as well as in well‐differentiated HCC were also upregulated. In addition, analysis of human gene signatures that predict the prognosis of HCC patients 41 revealed that HFD/SW feeding of C57BL/6NJ mice induced the expression of genes associated with poor prognosis. These findings indicate that HFD/SW promotes a NASH environment markedly more prone to liver cancer. Among several previous NASH‐driven HCC murine models, only HFD/SW feeding of a unique isogenic strain 15 had a significant correlation in gene expression with human NASH‐associated HCC samples. 60 Furthermore, similar to our findings, transcriptome comparison with human HCC subclass gene signatures suggests that tumors derived from these isogenic mice were significantly more related to proliferative or invasive human HCC subclasses. 15
In summary, the preclinical model described here reproduces many metabolic, histopathological, and molecular hallmarks present in the obesity‐related progression toward NASH and HCC as observed in humans. The clinical translatability of these findings emphasizes the significance of chronic high fat/sugar diets in driving this disease, as their consumption is associated with human NAFLD/NASH complications. 45 Importantly, as the C57BL/6NJ background is commonly used for gene‐targeted knockout and transgenic mice, this model will be useful for future mechanistic studies of how NASH progresses to HCC and the development of new targets for the treatment of these devastating diseases.
AUTHOR CONTRIBUTIONS
Sarah Spiegel and Christopher D. Green conceived the study; Sarah Spiegel and Arun J. Sanyal designed the model; Christopher D. Green performed experiments, analyzed data, designed RNA‐seq analyses, and assembled the figures; Cynthia Weigel and Ryan D. R. Brown performed staining for cytokeratin‐18 and Oil Red O; Pierre Bedossa conducted blinded histology scoring; Mikhail Dozmorov analyzed RNA‐seq data; Christopher D. Green wrote the paper and Sarah Spiegel reviewed and edited the manuscript.
DISCLOSURES
Dr. Sanyal has stock options in Genfit, Galmed, Exhalenz, Durect, Tiziana, Algernon, and Indalo. He has served as a consultant to Intercept, Gilead, Bristol Myers Squibb, Novartis, Pfizer, Lilly, Novo Nordisk, Astra Zeneca, Medimmune, Merck, Allergan, Albireo, Boehringer Ingelheim, Celgene, NGM, Glympse, Conatus, Genentech, Tern, Takeda, Hemoshear, Immuron, Surrozen, Poxel, Path AI, Second Genome, Zydus, Chiasma, Surrozen, Poxel, Blade, Pliant, Liposcience, Cymabay, Salix, Ferring, and Teva. His institution has received grants from Intercept, Gilead, Novartis, Merck, Astra Zeneca, Mallinckrodt, Pfizer, Lilly, Salix, and Bristol Myers Squibb. VCU has a patent application for the DIAMOND mouse model. The other authors declare that they have no competing financial interests.
ACKNOWLEDGMENTS
SS was supported by the National Institutes of Health Grant R01GM043880. We thank the University of Cincinnati Mouse Metabolic Phenotyping Center for the analysis of serum lipids, Dr. William Korzun for ALT/AST measurements, and the Virginia Commonwealth University Cancer Mouse Models Core Laboratory, supported, in part, with funding from the Massey Cancer Center from NIH‐NCI Cancer Center Support Grant P30CA016059.
Green CD, Weigel C, Brown RDR, et al. A new preclinical model of western diet‐induced progression of non‐alcoholic steatohepatitis to hepatocellular carcinoma. FASEB J. 2022;36:e22372. doi: 10.1096/fj.202200346R
Contributor Information
Christopher D. Green, Email: christopher.d.green@vcuhealth.org.
Sarah Spiegel, Email: sarah.spiegel@vcuhealth.org.
DATA AVAILABILITY STATEMENT
The RNA‐seq data generated for this study have been deposited in the NCBI Gene Expression Omnibus under GSE197884. All relevant data are within the paper and are available from the corresponding authors upon reasonable request.
REFERENCES
- 1. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol. 2013;10:686‐690. [DOI] [PubMed] [Google Scholar]
- 2. Diehl AM, Day C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med. 2017;377:2063‐2072. [DOI] [PubMed] [Google Scholar]
- 3. Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99‐S112. [DOI] [PubMed] [Google Scholar]
- 4. Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol. 2012;56:704‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6. [DOI] [PubMed] [Google Scholar]
- 6. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16:411‐428. [DOI] [PubMed] [Google Scholar]
- 7. Loomba R, Abraham M, Unalp A, et al. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology. 2012;56:943‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nakagawa H, Umemura A, Taniguchi K, et al. ER stress cooperates with hypernutrition to trigger TNF‐dependent spontaneous HCC development. Cancer Cell. 2014;26:331‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Umemura A, He F, Taniguchi K, et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC‐initiating cells. Cancer Cell. 2016;29:935‐948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, Karin M. Preclinical models for studying NASH‐driven HCC: how useful are they? Cell Metab. 2019;29:18‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clapper JR, Hendricks MD, Gu G, et al. Diet‐induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol. 2013;305:G483‐G495. [DOI] [PubMed] [Google Scholar]
- 13. Lim JS, Mietus‐Snyder M, Valente A, Schwarz J‐M, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251‐264. [DOI] [PubMed] [Google Scholar]
- 14. Walenbergh SM, Shiri‐Sverdlov R. Cholesterol is a significant risk factor for non‐alcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2015;9:1343‐1346. [DOI] [PubMed] [Google Scholar]
- 15. Asgharpour A, Cazanave SC, Pacana T, et al. A diet‐induced animal model of non‐alcoholic fatty liver disease and hepatocellular cancer. J Hepatol. 2016;65:579‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lattouf R, Younes R, Lutomski D, et al. Picrosirius red staining: a useful tool to appraise collagen networks in normal and pathological tissues. J Histochem Cytochem. 2014;62:751‐758. [DOI] [PubMed] [Google Scholar]
- 17. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 18. Rohrbach TD, Asgharpour A, Maczis MA, et al. FTY720/fingolimod decreases hepatic steatosis and expression of fatty acid synthase in diet‐induced nonalcoholic fatty liver disease in mice. J Lipid Res. 2019;60:1311‐1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mehlem A, Hagberg CE, Muhl L, Eriksson U, Falkevall A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat Protoc. 2013;8:1149‐1154. [DOI] [PubMed] [Google Scholar]
- 20. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu G, Wang LG, Han Y, He Q‐Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Teufel A, Itzel T, Erhart W, et al. Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology. 2016;151(513‐525):e510. [DOI] [PubMed] [Google Scholar]
- 24. Hoshida Y, Villanueva A, Kobayashi M, et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N Engl J Med. 2008;359:1995‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hoshida Y, Nijman SM, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69:7385‐7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Babicki S, Arndt D, Marcu A, et al. Heatmapper: web‐enabled heat mapping for all. Nucleic Acids Res. 2016;44:W147‐W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hansen HH, Ægidius HM, Oró D, et al. Human translatability of the GAN diet‐induced obese mouse model of non‐alcoholic steatohepatitis. BMC Gastroenterol. 2020;20:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cariou B, Byrne CD, Loomba R, Sanyal AJ. Nonalcoholic fatty liver disease as a metabolic disease in humans: a literature review. Diabetes Obes Metab. 2021;23:1069‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Younossi ZM. Non‐alcoholic fatty liver disease—a global public health perspective. J Hepatol. 2019;70:531‐544. [DOI] [PubMed] [Google Scholar]
- 30. Ratziu V, Charlotte F, Heurtier A, et al. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology. 2005;128:1898‐1906. [DOI] [PubMed] [Google Scholar]
- 31. Caldwell S, Ikura Y, Dias D, et al. Hepatocellular ballooning in NASH. J Hepatol. 2010;53:719‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guy CD, Suzuki A, Burchette JL, et al. Costaining for keratins 8/18 plus ubiquitin improves detection of hepatocyte injury in nonalcoholic fatty liver disease. Hum Pathol. 2012;43:790‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine‐driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121:977‐990. [DOI] [PubMed] [Google Scholar]
- 34. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 35. Li Y, Xu A, Jia S, Huang J Recent advances in the molecular mechanism of sex disparity in hepatocellular carcinoma. Oncol Lett. 2019;17:4222‐4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sato Y, Nakata K, Kato Y, et al. Early recognition of hepatocellular carcinoma based on altered profiles of alpha‐fetoprotein. N Engl J Med. 1993;328:1802‐1806. [DOI] [PubMed] [Google Scholar]
- 37. Inami Y, Waguri S, Sakamoto A, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xiong X, Kuang H, Ansari S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single‐cell secretome gene analysis. Mol Cell. 2019;75(644‐660):e645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He G, Dhar D, Nakagawa H, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL‐6 signaling. Cell. 2013;155:384‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shalapour S, Lin XJ, Bastian IN, et al. Inflammation‐induced IgA+ cells dismantle anti‐liver cancer immunity. Nature. 2017;551:340‐345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hoshida Y, Villanueva A, Sangiovanni A, et al. Prognostic gene expression signature for patients with hepatitis C‐related early‐stage cirrhosis. Gastroenterology. 2013;144:1024‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ahn KS, O'Brien DR, Kim YH, et al. Associations of serum tumor biomarkers with integrated genomic and clinical characteristics of hepatocellular carcinoma. Liver Cancer. 2021;10:593‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Satapathy SK, Sanyal AJ. Epidemiology and natural history of nonalcoholic fatty liver disease. Semin Liver Dis. 2015;35:221‐235. [DOI] [PubMed] [Google Scholar]
- 44. Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci. 2016;61:1282‐1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jensen T, Abdelmalek MF, Sullivan S, et al. Fructose and sugar: a major mediator of non‐alcoholic fatty liver disease. J Hepatol. 2018;68:1063‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ioannou GN. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol Metab. 2016;27:84‐95. [DOI] [PubMed] [Google Scholar]
- 47. Machado MV, Michelotti GA, Xie G, et al. Mouse models of diet‐induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One. 2015;10:e0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wolf MJ, Adili A, Piotrowitz K, et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross‐talk with hepatocytes. Cancer Cell. 2014;26:549‐564. [DOI] [PubMed] [Google Scholar]
- 49. Mollerhoj MB, Veidal SS, Thrane KT, et al. Hepatoprotective effects of semaglutide, lanifibranor and dietary intervention in the GAN diet‐induced obese and biopsy‐confirmed mouse model of NASH. Clin Transl Sci. 2022;15(5):1167‐1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dowman JK, Hopkins LJ, Reynolds GM, et al. Development of hepatocellular carcinoma in a murine model of nonalcoholic steatohepatitis induced by use of a high‐fat/fructose diet and sedentary lifestyle. Am J Pathol. 2014;184:1550‐1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Farrell GC, van Rooyen D. Liver cholesterol: is it playing possum in NASH? Am J Physiol Gastrointest Liver Physiol. 2012;303:G9‐G11. [DOI] [PubMed] [Google Scholar]
- 52. Lackner C. Hepatocellular ballooning in nonalcoholic steatohepatitis: the pathologist's perspective. Expert Rev Gastroenterol Hepatol. 2011;5:223‐231. [DOI] [PubMed] [Google Scholar]
- 53. Yeh MM, Brunt EM. Pathology of nonalcoholic fatty liver disease. Am J Clin Pathol. 2007;128:837‐847. [DOI] [PubMed] [Google Scholar]
- 54. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple‐hit pathogenesis of non‐alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65:1038‐1048. [DOI] [PubMed] [Google Scholar]
- 55. Chirala SS, Wakil SJ. Structure and function of animal fatty acid synthase. Lipids. 2004;39:1045‐1053. [DOI] [PubMed] [Google Scholar]
- 56. Dorn C, Riener MO, Kirovski G, et al. Expression of fatty acid synthase in nonalcoholic fatty liver disease. Int J Clin Exp Pathol. 2010;3:505‐514. [PMC free article] [PubMed] [Google Scholar]
- 57. Reibe S, Febbraio MA. Relieving ER stress to target NASH‐driven hepatocellular carcinoma. Nat Rev Endocrinol. 2019;15:73‐74. [DOI] [PubMed] [Google Scholar]
- 58. Denk H, Stumptner C, Fuchsbichler A, et al. Are the Mallory bodies and intracellular hyaline bodies in neoplastic and non‐neoplastic hepatocytes related? J Pathol. 2006;208:653‐661. [DOI] [PubMed] [Google Scholar]
- 59. Hou J, Zhang H, Sun B, Karin M. The immunobiology of hepatocellular carcinoma in humans and mice: basic concepts and therapeutic implications. J Hepatol. 2020;72:167‐182. [DOI] [PubMed] [Google Scholar]
- 60. Pinyol R, Torrecilla S, Wang H, et al. Molecular characterisation of hepatocellular carcinoma in patients with non‐alcoholic steatohepatitis. J Hepatol. 2021;75:865‐878. [DOI] [PubMed] [Google Scholar]
- 61. Kodama Y, Kisseleva T, Iwaisako K, et al. c‐Jun N‐terminal kinase‐1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. 2009;137(1467‐1477):e1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kita Y, Takamura T, Misu H, et al. Metformin prevents and reverses inflammation in a non‐diabetic mouse model of nonalcoholic steatohepatitis. PLoS One. 2012;7:e43056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The RNA‐seq data generated for this study have been deposited in the NCBI Gene Expression Omnibus under GSE197884. All relevant data are within the paper and are available from the corresponding authors upon reasonable request.