

Abstract
Large DNA constructs (>10 kb) are invaluable tools for genetic engineering and the development of therapeutics. However, the manufacture of these constructs is laborious, often involving multiple hierarchical rounds of preparation. To address this problem, we sought to test whether Golden Gate assembly (GGA), an in vitro DNA assembly methodology, can be utilized to construct a large DNA target from many tractable pieces in a single reaction. While GGA is routinely used to generate constructs from 5 to 10 DNA parts in one step, we found that optimization permitted the assembly of >50 DNA fragments in a single round. We applied these insights to genome construction, successfully assembling the 40 kb T7 bacteriophage genome from up to 52 parts and recovering infectious phage particles after cellular transformation. The assembly protocols and design principles described here can be applied to rapidly engineer a wide variety of large and complex assembly targets.
Keywords: Golden Gate assembly, DNA assembly, T7 phage, synthetic biology
Introduction
Large DNA constructs are widely used in synthetic biology to develop genetically engineered organisms for therapeutic uses and chemical production. Typically, these constructs are much too long to be directly synthesized and are consequently assembled from multiple shorter DNA fragments.1 For example, in vivo recombination methods permit assembly of >200 kb from as many as 50 fragments in a single transfection.2−5 Nevertheless, in vitro DNA assembly methods, such as Golden Gate assembly (GGA), remain popular to generate constructs due to their ease of use despite typical standards being limited to 5–10 DNA fragments per reaction.6,7 Thus, hierarchical assembly schemes involving multiple rounds of molecular cloning, construct purification, and sequence verification are employed for the in vitro assembly of large constructs. Recent work suggests that GGA can routinely accommodate many additional fragments per reaction, potentially reducing the number of, or even entirely avoiding, multiple assembly rounds.8−11
GGA permits one-tube assembly of multiple DNA fragments in a predetermined order using a type IIS restriction enzyme to generate fragments with complementary single-stranded DNA overhangs joined by DNA ligase.12,13 Type IIS restriction enzymes cleave outside of their recognition sequences, meaning the recognition sequence is eliminated from the final construct sequence, allowing for the seamless one-tube assembly of DNA fragments.14 When fusion sites are outside of coding sequences, the connections used may be chosen arbitrarily; this feature has given rise to more than a dozen “modular” cloning systems with standardized fusion sites and prefabricated reusable parts.8,15 These systems are attractive engineering platforms, as they have empirically vetted reaction conditions and allow for parts to be easily shared between groups following the same standards.
While GGA permits scarless assembly even within coding regions, the desired assembly sequence cannot contain the recognition sequence of the type IIS restriction enzyme used in the assembly, as these sites would be cleaved in the final assembly. Typically, this limitation is overcome by introducing silent mutations to remove internal sites within coding regions; mutagenesis outside coding regions can require guesswork and careful control to remove internal sites without significant perturbation to the system. Recent work using methylation to protect recognition sequences from digestion offers an additional solution to this problem.16 Another limitation of GGA is that the method relies on the accurate and efficient ligation of assembly pieces by a DNA ligase to avoid improperly ordered fragments and low assembly yield. T4 DNA ligase is the most frequently utilized DNA ligase in GGA, as it joins assembly fragments more efficiently and with less bias than other commercially available DNA ligases; however, T4 DNA ligase is prone to ligation of some mismatched sequences, which can result in constructs with mis-ordered, duplicated, or missing assembly pieces.9 To avoid erroneous assembly products caused by ligation errors, GGA is typically limited to sets of overhang sequences that contain multiple mismatches between all non-cognate pairs or pre-vetted sets of fusion sites. This strategy has worked well for small DNA targets but generally limits users to <10 fragments.
Recent work by our laboratory has significantly expanded the capacity of GGA by using ligase fidelity data to select fusion sites, a process termed data-optimized assembly design (DAD), with the successful assembly of 35 fragments into a 5 kb cassette in a single reaction using this strategy.11 Here, we sought to test the limits of the fragment number and final construct size when applying DAD to GGA in two practical applications. In the first, we assemble a 4.9 kb lac operon cassette into a destination vector from 52 constituent fragments. This one-pot assembly is enabled using DAD junction selection tools and an optimized reaction protocol. To demonstrate a more stringent screening condition, we construct the 40 kb T7 bacteriophage genome in a single assembly round. First, we show efficient construction of the phage genome and reconstitution of infectious phage particles from 10 amplicons of ∼4 kb each. Then, we demonstrate the successful assembly of the phage genome from 52 amplicon fragments of ∼800 bp each. We find that the genome assembly and phage reconstitution can be carried out in less than 1 day despite the large size of the target and complexity of the assembly. This work suggests that GGA can be used to rapidly generate large DNA targets in a single assembly round from many small, easily manipulated pieces.
Methods
52-Fragment Lactose Operon Assembly Fragment Design and Purification
Assembly junction sequences were selected using the optimization described in our previous work, and the corresponding suite of webtools is available at https://goldengate.neb.com (Table S1).11 Assembly fragments were amplified from pre-cloned templates purchased from GenScript (Table S2). Assembly fragments were generated by PCR (Q5 Hot-Start High-Fidelity 2× Master Mix) with oligonucleotide primers (IDT) (Table S3) and purified using the Monarch PCR & DNA Cleanup Kit. Fragment quality was evaluated using the Agilent Bioanalyzer 2100, and each assembly part was quantified using the Qubit assay (Thermo Fisher). GGA reactions (5 μL final volume) were carried out with 3 nM of each DNA fragment and 0.5 μL of the appropriate NEB GGA Mix in 1× T4 DNA ligase buffer; the BsaI-HFv2 mix was used to reconstitute the lac operon cassette. Reactions to reconstitute the lac operon cassette were incubated for 48 h at 37 °C and then subjected to a final heat-soak step at 60 °C for 5 min before being incubated at 4 °C prior to transformation (Table S4).
All assembly products were transformed into T7 Express chemically competent Escherichia coli cells, and the assembly fidelity was scored as described previously.9,11 Briefly, transformations were performed using 2 μL of each assembly reaction added to 50 μL of competent T7 Express cells. Transformation reactions were incubated on ice for 30 min and then incubated at 42 °C for 10 s, with a final 5 min recovery period on ice. SOC outgrowth medium (950 μL) was added, and the cells were incubated for 1 h at 37 °C with vigorous rotation. The outgrowth was spread onto prewarmed agar plates [Luria–Bertani (LB) broth supplemented with 1 mg/mL of dextrose, 1 mg/mL of MgCl2, 30 μg/mL of chloramphenicol, 200 μM of IPTG, and 80 μg/mL of X-gal]. Plates were inverted and placed at 37 °C overnight and then stored at 4 °C for 8 h before scoring a colony color phenotype.
T7 Phage Genome Assembly Fragment Design and Production
Silent mutations to permit assembly of the T7 phage genome (GenBank: V01146.1 and Supporting Information) by SapI or BsmBI were designed using the DNASTAR SeqBuilder Pro software package. The desired phage genomic DNA sequence was split into approximately equally sized assembly pieces using the New England Biolabs (NEB) SplitSet tool (https://ligasefidelity.neb.com/) to select high-fidelity junction points between assembly fragments. Most of the assembly junctions were selected using 200 base pair search windows to ensure maximum assembly fidelity. The junctions near sites of intended silent mutations were limited to five base pair search windows to ensure that the same PCR primers could simultaneously introduce the desired mutation(s) and append the necessary type IIS restriction sites. PCR primers to generate assembly pieces were designed using the DNASTAR SeqBuilder Pro software, and the optimum annealing temperatures were calculated for each primer pair using the NEB Tm calculator tool (https://tmcalculator.neb.com). Of note, recent upgrades to the NEB assembly tool (https://goldengate.neb.com) now support automated primer designs along with junction selection facilitated by DAD, thus eliminating the need to use multiple programs for primer design. Additionally, the optimizer code used in the SplitSet tool is available under a non-commercial use license on request at https://www.neb.com/forms/overhang-optimizer-code.
PCR reactions (50 μL final volume each) to generate amplicon assembly pieces were carried out with 100 pg of template T7 phage genomic DNA, amplification primers, and Q5 Hot-Start PCR Master Mix (NEB) using the manufacturer’s recommended protocol. For assembly pieces that adjoin the terminal ends of the phage genome, the template wt T7 bacteriophage genomic DNA was circularized prior to amplification using the Blunt/TA Ligase Master Mix (NEB). Amplification primer sequences are listed in Table S3. The resulting amplicons were purified using the Monarch DNA Cleanup Kit (NEB). Importantly, each assembly piece was stringently quality checked by electrophoresis using an Agilent Bioanalyzer 2100 to ensure that every amplicon was free of non-specific amplification products and primer dimers. Lastly, assembly pieces were quantified using the Qubit assay (Thermo Fisher). Of note, together the purification, quality check, and quantification steps described here produce high-quality assembly pieces; we recommend that similarly rigorous protocols be used to prepare parts for amplicon assembly reactions, as impure or inaccurately quantified assembly pieces are likely to yield suboptimal results.
GGA Reactions and Plaque Forming Assay
Genome assembly reactions (5 μL final volume) to construct the circular or linear T7 phage genome from 10 assembly pieces were carried out by combining 3 nM of each assembly fragment (final concentration, 80 ng/uL of total DNA) with SapI (0.75 units) and T4 DNA ligase (125 units) in 1× T4 DNA ligase buffer (50 mM Tris-HCl, 10 mM MgCl2, 10 mM Dithiothreitol 1 mM ATP, pH 7.5, T 25 °C). Reactions were cycled between 37 and 16 °C for 5 min at each temperature for 90 cycles, followed by a 60 °C heat-soak step for 5 min. The assembly reactions were then chilled to 4 °C prior to transformation. Reactions to assemble a circular T7 phage genome from 52 fragments (5 μL final volume) were carried out by combining 3 nM of each assembly piece (final concentration, 80 ng/uL total DNA) with the NEB Golden Gate enzyme mix containing BsmBIv2 and T4 DNA ligase (0.5 μL) in 1× T4 DNA ligase buffer. Reactions were cycled between 42 and 16 °C for 5 min at each temperature for either 30 or 90 cycles, followed by a 60 °C heat-soak step for 5 min and then a final temperature held at 4 °C prior to transformation.
Reconstitution of infectious phage particles was carried out by transforming 1 μL of the completed assembly reaction mixture into NEB 10-beta electrocompetent cells using the manufacturer’s recommended protocol with 1 mM electroporation cuvettes and a Gene Pulser Xcell electroporator (BioRad). Transfected cell mixtures were recovered using 975 μL of NEB 10-beta/stable outgrowth media, then combined with 3 mL of 50 °C molten top-agar (LB medium containing 0.9% agar), and plated on LB agar plates. The resulting plates were cooled on the benchtop for 20 min to allow the top-agar to solidify before inversion and incubation at 37 °C until phage plaques were visible, approximately 4 h. Plates containing plaques were stored at 4 °C; individual plaques were collected and stored in 20 μL of phage dilution buffer (50 mM Tris-HCl, 10 mM MgCl2, 75 mM NaCl, pH 7.5 at 25 °C) at 4 °C.
Phage Plaque PCR Reactions and Restriction Digest
Plaque PCR reactions (50 μL final volume each) were carried out by combining 1 μL of the diluted plaque stock with amplification primers and LongAmp Taq PCR Master Mix using the manufacturer’s recommended protocol. Amplification primer sequences can be found in Table S3. As a control, PCR reactions with 500 pg of template DNA isolated from the wild-type T7 bacteriophage were carried out in parallel. Amplicons from the PCR reactions were purified using the Monarch DNA cleanup kit (NEB). Amplicon digests (10 μL final volume) were carried out using NdeI (20 units) or SapI (10 units) with approximately 200 ng of the DNA substrate in a final concentration of 1× NEB rCutSmart buffer (50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 100 μg/mL of BSA, pH 7.9 at 25 °C) overnight at 37 °C. As a control, mock digests lacking restriction enzymes were carried out in parallel. Digestion reactions were resolved by electrophoresis using an Agilent Bioanalyzer 2100 with the DNA 12000 kit.
Phage gDNA Preparation and Long-read Sequencing
Phage genomic DNA was extracted and prepared as described previously17 Briefly, phage plaques were used to infect/lyse cultures of exponentially growing NEB 5-alpha cells (grown in LB medium). Lysates were cleared by centrifugation, and bacteriophage precipitation was carried out by adding 10% polyethylene glycol 8000 and 1 M NaCl (final concentrations) to the cleared cell lysate with an overnight incubation at 4 °C. Precipitated phages were harvested by centrifugation and resuspended in phage dilution buffer supplemented with DNase (10 μg/mL) and RNase (5 μg/mL) to degrade host nucleic acids. Phage samples were purified by CsCl density ultracentrifugation. Purified bacteriophage samples were lysed by treatment with 1% sodium dodecyl sulfate, 25 mM EDTA, and 200 μg/mL of proteinase K (NEB) for 30 min at 55 °C. Genomic DNA from lysed phages was purified by phenol-chloroform precipitation, gently spooled onto a glass rod, and purified/concentrated using ethanol precipitation.
DNA sequencing libraries of phage genomic DNA were prepared using the Ligation Sequencing Kit (SQK-LSK109; Oxford Nanopore Technologies) and the NEBNext DNA repair, End repair, and Ligation reagents with 200 ng of input DNA as per the manufacturers’ instructions. Samples were barcoded using the Rapid Barcoding Kit (SQK-RBK004) and sequenced using the MinION platform with the R9.4.1 flow cell (Oxford Nanopore Technologies). Data acquisition and base-calling were done using the MinKNOW v3.4.5 software.
Results and Discussion
We initially sought to carry out a complex assembly reaction and clone a 4.9 kb cassette of the lac operon into an E. coli destination vector from 52 constituent parts in a single assembly round (Figure 1). Importantly, the lac operon cassette system used here mimics a traditional cloning reaction wherein, upon transformation of the assembly reaction into E. coli cells, we can observe colonies harboring correctly or incorrectly assembled constructs. This test system was engineered to provide a colorimetric readout to differentiate between transformants harboring correctly and incorrectly assembled products.9
Figure 1.

One-pot GGA of 52 fragments into a destination vector. (A) Schematic of the 52-fragment lac operon cassette assembly. Assembly inserts were generated by PCR amplification and assembled into a destination vector containing an antibiotic resistance marker. (B) Example outgrowth plate used for colorimetric scoring by a reverse blue-white screen. Correctly assembled 52 insert constructs form blue colonies upon cellular transformation, and incorrectly assembled constructs produce white colonies.
DNA fragments comprising the lac operon cassette were generated by PCR and assembled in a reaction containing BsaI-HFv2 and T4 DNA ligase at 37 °C for 48 h (Tables S1–S4). Assembly reactions were then transformed into chemically competent E. coli cells, and the resulting transformants were scored as having correctly or incorrectly assembled insert sequences. Notably, under these optimized reaction conditions, we found that 49% of the observed transformants harbored correctly assembled constructs (Figure 1 and Table S4). The reaction generated >500 transformants with correctly assembled constructs per 100 μL of E. coli outgrowth plated, a surprisingly high yield given the complexity of the assembly reaction. To confirm successful assembly of all 52 inserts, constructs were purified from a subset of colonies and analyzed by PCR and Sanger sequencing. All constructs from colonies scored as having correct assemblies were found to have inserts of the anticipated size and sequence, and constructs from colonies scored as having incorrect sequences contained truncated inserts (Figure S1). Taken together, these data show that >50 fragments can be assembled in one reaction using a high-temperature reaction protocol and rational junction selection by DAD. However, we note challenges in assembling >35-fragments in one reaction without a stringent selection system, as we observed significant lot-to-lot variation in the yield and fidelity of this assembly reaction, likely owing to difficulties in preparing and normalizing 52 individual amplicon assembly pieces (Table S4).
In order to test complex assemblies in a system with a more stringent selection, we sought to construct the 40 kb T7 bacteriophage genome in a single reaction. We initially sought to compare the assembly of the 40 kb T7 bacteriophage genome in its native linear conformation to a circular assembly, reasoning that the latter would be more resistant to cellular exonucleases and provide more efficient transfection from naked DNA. To compare, we designed assemblies of 10 ∼4 kb fragments, using the seven-base recognition site SapI as the restriction enzyme, since only four silent mutations located within annotated coding sequences were required to eliminate native sites from the T7 phage genome (Table S5, Figure S2, and SI file). The SplitSet tool was used to select 10 fusion sites, equally spaced in the genome, predicted to be an overall high-fidelity set of 10 three-base overhang fusion sites (Table S6).11 To ensure that the proper silent mutations could be introduced, the search was limited to within five bases of the four desired mutations, with the other six sites permitting a 200 nt search window to find an overall maximum fidelity set. Attempts to sub-clone these fragments into E. coli propagation vectors were unsuccessful, likely because many phage genes are toxic in a bacterial host. Thus, we used fragments generated by PCR amplification directly in the assembly, appending the SapI recognition sequences onto each assembly fragment, and simultaneously removing pre-existing SapI recognition sites from the native T7 phage genome sequence (Tables S3, S5, and Figure S3).
Assembly reactions and transformations were carried out as described in methods (Scheme 1). When assembling the genome from 10 fragments, we observed an average of >50 phage plaques per microliter of assembly reaction (>635 pfu/ug input DNA) transformed into E. coli cells (Figure 2A and Table 1). To ensure the observed phage plaques resulted from the in vitro assembly and not the assembly of the DNA fragments within the E. coli cells by DNA repair mechanisms or carryover contamination from the wild-type genome, we carried out control reactions lacking T4 DNA ligase. We did not observe phage plaques upon the transformation of these mock assembly reactions (Table 1). Additionally, we selected six phage plaques for screening by plaque PCR and restriction enzyme digestion to verify that the assembled genomes included every assembly amplicon in the proper order and harbored the desired silent mutations (Figures 2B,C and S4). All the plaques selected for additional screening had the expected genome arrangement and desired silent mutations, indicating successful one-pot assembly of the linear phage genome and reconstitution of the infectious phage.
Scheme 1. T7 Phage Genome Assembly and Infectious Phage Reconstitution.
Figure 2.
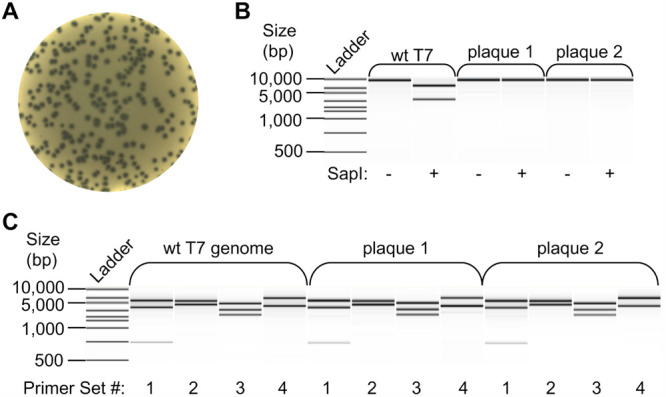
Amplification and digestion of wt T7 and assembled phage genomes. Amplicon digestion reactions to compare the genome arrangements of phages reconstituted from wt T7 bacteriophage genomic DNA (wt T7 genome) or in vitro GGA reactions to create linear (plaque 1) or circular (plaque 2) genomes were resolved using a Bioanalyzer 2100 instrument. (A) Representative plate showing phage plaques reconstituted from assembled genomes on an E. coli lawn. (B) Digestions of a 10 kb amplicon of the wt T7 phage genome or plaques from assembled phage genomes with SapI (+) or mock-treated (−). The assembled genomes (plaques 1 and 2) are inert to cleavage by SapI, whereas the parental T7 bacteriophage genomic DNA is sensitive to SapI digestion, indicating successful mutagenesis of the assembled T7 bacteriophage genomes. (C) NdeI restriction digests of four amplicons1−4 spanning the 40 kb T7 phage genome for each sample are shown. Comparison of amplicon digests between samples shows an identical digestion pattern, indicating the same genome arrangement. See Figure S2 for a schematic of the T7 phage genome showing the locations of the expected restriction sites and primer annealing regions.
Table 1. Phage Plaque Yield from the 10-fragment T7 Phage Genome Assembly Reactionsa.
| genome topologyb | assembly enzymesc | replicate 1 | replicate 2 | replicate 3 | average |
|---|---|---|---|---|---|
| linear | SapI alone | 0 | 0 | 0 | 0 |
| linear | SapI + T4 ligase | 81 | 50 | 45 | 59 |
| circular | SapI Alone | 0 | 0 | 0 | 0 |
| circular | SapI + T4 ligase | 33,600 | 36,400 | 38,200 | 36,100 |
The table shows the number of phage plaques observed per microliter of assembly reaction transformed into NEB 10-beta cells for three experimental replicates and their average. One microliter of assembly reaction contains approximately 35 ng of DNA.
Assembly reactions were designed to produce two variants of the phage genome with identical sequences that have either blunt termini (linear) or a circular configuration (circular).
Genome assembly reactions were carried out with SapI and T4 DNA ligase. As a control, mock assembly reactions with SapI alone were carried out in parallel.
A Schematic of the T7 phage genome assembly and infectious phage reconstitution assay is depicted. DNA fragments are assembled into a complete copy of the 40 kb phage genome in one reaction using GGA. Completed assembly reactions are transformed into NEB 10-beta electrocompetent cells to reconstitute infectious phage particles.
While the construction of the phage genome in the native linear topology was successful, the circular genome assembly reactions produced >30,000 plaques per microliter (>375,000 pfu/ug input DNA) transfected into E. coli cells; a >500-fold increase in the number of plaque-forming units compared to the linear conformation (Table 1 and Figure 2B,C) is likely due to the protection of the assembled genome from cellular exonucleases. Circularization of genomes by GGA may be beneficial for many assembly targets; however, it is not clear that all targets with a linear packaged genome topological conformation will replicate properly in cells when starting from a circular arrangement.
Next, we sought to test the limits of single-reaction fragment capacity in the assembly of a large DNA target by GGA. Based on our previous work, we predicted that as many as 50 fragments could be assembled in one round using four-base fusion sites, although even with junctions optimized for high fidelity, we expected many misassembled constructs with this number of fusion sites. However, given the high density of coding sequences in its genome, we reasoned that an improperly assembled T7 phage would be unlikely to produce a viable phage upon cellular transfection. Therefore, we designed a 52-fragment assembly reaction to construct the phage genome with fragment fusion sites selected by DAD. Of the commonly used four-base type IIS cutters, BsmBI was chosen due to the presence of fewer native sites than BsaI or BbsI (Table S7 and Figure S2). These sites were further all in CDS annotated in GenBank. Nevertheless, 16 mutations were required to remove all BsmBI sites from the genome (Table S8and SI file). To ensure that we used a set of 16 high-fidelity fusion sites compatible with a larger set with maximal fidelity, SplitSet was again used, requiring a fusion site within 5 nt of each of these required mutations and searching for an additional 36 sites within 200 nt windows approximately equally distributed across the genome (Figure S5 and Table S6). After identification of a high-fidelity fusion site set, assembly fragments were generated by PCR amplification of the native phage gDNA with primers that appended flanking BsmBI recognition sequences on each fragment and introduced silent mutations to remove pre-existing BsmBI recognition sites from the phage genome and to generate a final circular construct (Tables S3, S6, S8 and Figure S5).
Genome assembly reactions were carried out using BsmBI-v2 and T4 DNA ligase (Methods). Successful phage reconstitution was assessed by the plaque-forming assay described above (Scheme 1). Assembly reactions carried out for 5 h generated an average of 14 phage plaques per microliter of assembly reaction (175 pfu/ug input DNA) transfected into E. coli cells (Table 2). We observed a moderate increase in plaque yield for assembly reactions incubated for 15 h, with the assembly averaging 41 plaques per microliter of assembly reaction (512 pfu/ug input DNA). Thus, while the assembly reaction and phage reconstitution protocol can be carried out in a single day even at a 52-fragment complexity, extended duration GGA reactions significantly improved the reaction yield. To verify the correct assembly of the phage genome, we carried out plaque PCR and restriction digest of five phage plaques and found that all the genomes contained every assembly fragment in the proper order and harbored the intended silent mutations (Figure S6). Additionally, we prepared phage genomic DNA from 10 phage plaques and five plaques from each time point and sequenced these genomes using nanopore long-read sequencing. All 10 genomes were correctly assembled copies with the intended silent mutations (Tables 3, S9 and S10). We found that three genomes from the assembled phage plaques contained novel single nucleotide polymorphisms (SNPs); however, none of these mutations were observed within 40 base pairs of a fusion site. Thus, these mutations likely resulted from PCR errors and not from inaccurate digestion or ligation of assembly fragments.
Table 2. Phage Plaque Yield from the 52-fragment T7 Phage Genome Assembly Reactionsa.
| reaction time (h)b | assembly enzymesc | replicate 1 | replicate 2 | replicate 3 | average |
|---|---|---|---|---|---|
| 5 | BsmBI alone | 0 | 0 | 0 | 0 |
| 5 | BsmBI + T4 ligase | 13 | 10 | 18 | 14 |
| 15 | BsmBI alone | 0 | 0 | 0 | 0 |
| 15 | BsmBI + T4 ligase | 38 | 40 | 45 | 41 |
The table shows the number of phage plaques observed per microliter of assembly reaction transformed into NEB 10-beta cells for three experimental replicates and their average.
Assembly reactions to construct the phage genome were cycled between 42 and 16 °C for 5 min at each temperature for 30 cycles (5 h) or 90 cycles (15 h) prior to electroporation.
Genome assembly reactions were carried out with BsmBI and T4 DNA ligase or BsmBI alone as a control.
Table 3. Genome Sequencing Results from 52-fragment Phage Plaquesa.
| plaque # | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| position | reference | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| mutations to remove pre-existing BsmBI sitesb | |||||||||||
| 2137 | G | A | A | A | A | A | A | A | A | A | A |
| 2641 | C | T | T | T | T | T | T | T | T | T | T |
| 10867 | C | T | T | T | T | T | T | T | T | T | T |
| 12767 | G | A | A | A | A | A | A | A | A | A | A |
| 15366 | T | A | A | A | A | A | A | A | A | A | A |
| 18262 | G | A | A | A | A | A | A | A | A | A | A |
| 18983 | G | A | A | A | A | A | A | A | A | A | A |
| 20752 | C | T | T | T | T | T | T | T | T | T | T |
| 22582 | G | A | A | A | A | A | A | A | A | A | A |
| 24596 | T | A | A | A | A | A | A | A | A | A | A |
| 24734 | T | A | A | A | A | A | A | A | A | A | A |
| 31347 | T | A | A | A | A | A | A | A | A | A | A |
| 32694 | C | G | G | G | G | G | G | G | G | G | G |
| 33108 | C | T | T | T | T | T | T | T | T | T | T |
| 33396 | T | A | A | A | A | A | A | A | A | A | A |
| 35247 | C | T | T | T | T | T | T | T | T | T | T |
| novel SNPsc | |||||||||||
| 7609 | T | G | |||||||||
| 14378 | A | G | |||||||||
| 26773 | G | T | |||||||||
Phage genomic DNA was isolated from the phage plaques of the 52-fragment assembly reactions (plaques 1–5 from 5 h reactions and plaques 6–10 from 15 h reactions) and sequenced using nanopore sequencing. Mutations differentiating the genomic sequences of the reconstituted phages compared to the reference T7 strain (GenBank: V01146.1) are shown.
Assembly reactions were designed to create phage genomes with 16 silent mutations to permit assembly with the BsmBI-type IIS restriction enzyme. All assembled genomes contained these mutations.
SNPs appeared in several of the assembled genomes; however, these mutations were not within 40 base pairs of assembly junction sites. More information on these mutations can be found in Tables S8, S9, and S10.
In comparing the T7 phage assembly test systems described above, we note that the 52-fragment assembly reaction produced ∼800-fold fewer plaques than the equivalent 10-fragment assembly reactions of the same target, although the stringent selection imposed by the need for the complete genome to allow viability still permitted multiple successful assemblies to be easily identified. Using less stringent selections, such as the previously described Lac cassette system dependent only on antibiotic resistance genes in the plasmid, we found that the 52-fragment assembly failed under standard cycled conditions and produced only a few colonies with overall low and variable fidelity when assembled using a long incubation at 37 °C. This observed drop in efficiency across the systems suggests that ∼50 fragments are approaching the practical limits of assembly capacity in our hands and require a highly stringent selection or extensive screening to permit isolation of correct assemblies. Our previous work has shown that the 35-fragment assembly can be carried out with very high fidelity even without stringent selection when selecting junctions using DAD, with assemblies of 50+ fragments well within the limits of GGA when a stringent selection can be applied.11
Furthermore, we are optimistic that our assembly design principles could be applied to even larger assembly sequences. Notably, previous work has shown that assembly of targets >200 kb in vitro by GGA is feasible, albeit from only four large fragments.16 Still, further investigation will be needed to test whether targets of this size can be assembled from 10 or more pieces as described here. Larger assembly targets are more challenging to handle, introduce, and maintain in cells; however, cellular transformation may be avoided in some applications altogether by using in vitro transcription/translation and/or cell-free DNA replication systems. For example, pioneering work by the Noireaux laboratory has demonstrated successful cell-free production of several model bacteriophages from genomic DNA samples using a cellular-extract system, including the bacteriophage T4 from its 170 kb genome.18 Indeed, combining high-capacity GGA with other cell-free workflows offers an attractive option for groups interested in expanding throughput or wishing to increase their control over production workflows.
Overall, our results show that GGA can be used to assemble large DNA targets in one reaction from many assembly pieces, demonstrating that large constructs with many fusion sites can be assembled in a single round. A similar strategy could be applied to engineering other bacteriophages or similarly sized targets, allowing efficient metabolic engineering and genomic studies. Moreover, the large fragment capacity allows for the assembly of toxic and/or large DNA constructs via subdivision into small parts that are easily manipulated and propagated using standard molecular biology techniques. When designing GGA assemblies of new systems, care must be taken to choose a restriction enzyme that permits the number of needed fragments yet minimizes the number of mutations that must be introduced to remove native sites. Importantly, mutations outside of coding regions require careful control experiments on domesticated systems to ensure that no changes were made that affected gene expression or other functions, and mutations within known regulatory elements such as promoters should be avoided. With this caveat in mind, this work has the potential to facilitate rapid engineering of large DNA targets, as GGA is amenable to automated workflows and supports the standardized modular assembly of construct variants with minimal ad-hoc design. In summary, our work demonstrates an efficient and cost-effective means to create and engineer variants of large/complex DNA constructs through improved design of assembly reactions.
Acknowledgments
We thank Tasha José (New England Biolabs) for providing illustrations as well as Rebecca Kucera and Eric Cantor (New England Biolabs) for providing reagents.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.1c00525.
Author Contributions
J.M.P. performed experiments and wrote the original draft of the paper. V.P. performed data analysis and provided bioinformatic support. K.B. helped write and revise the paper. N.P. performed experiments. G.J.S.L. supervised the research and helped write and revise the paper.
The authors declare the following competing financial interest(s): When performing this research and drafting this manuscript, all authors were employees of New England Biolabs, a manufacturer and vendor of molecular biology reagents including DNA ligases and Type IIS restriction enzymes. New England Biolabs funded the work and paid the salaries of all authors. This does not alter our adherence to journal policies on sharing data and materials.
Supplementary Material
References
- Zhang W.; Mitchell L. A.; Bader J. S.; Boeke J. D. Synthetic Genomes. Annu. Rev. Biochem. 2020, 89, 77–101. 10.1146/annurev-biochem-013118-110704. [DOI] [PubMed] [Google Scholar]
- Gibson D. G.; Benders G. A.; Andrews-Pfannkoch C.; Denisova E. A.; Baden-Tillson H.; Zaveri J.; Stockwell T. B.; Brownley A.; Thomas D. W.; Algire M. A.; Merryman C.; Young L.; Noskov V. N.; Glass J. I.; Venter J. C.; Hutchison C. A. 3rd; Smith H. O. Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 2008, 319, 1215–1220. 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
- Mitchell L. A.; Wang A.; Stracquadanio G.; Kuang Z.; Wang X.; Yang K.; Richardson S.; Martin J. A.; Zhao Y.; Walker R.; Luo Y.; Dai H.; Dong K.; Tang Z.; Yang Y.; Cai Y.; Heguy A.; Ueberheide B.; Fenyö D.; Dai J.; Bader J. S.; Boeke J. D. Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science 2017, 355, eaaf4831 10.1126/science.aaf4831. [DOI] [PubMed] [Google Scholar]
- Tsuge K.; Sato Y.; Kobayashi Y.; Gondo M.; Hasebe M.; Togashi T.; Tomita M.; Itaya M. Method of preparing an equimolar DNA mixture for one-step DNA assembly of over 50 fragments. Sci. Rep. 2015, 5, 10655. 10.1038/srep10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma E. D.; Dashko S.; van Breemen L.; Taylor Parkins S. K.; van den Broek M.; Daran J.-M.; Daran-Lapujade P. A supernumerary designer chromosome for modular in vivo pathway assembly in Saccharomyces cerevisiae. Nucleic Acids Res. 2021, 49, 1769–1783. 10.1093/nar/gkaa1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casini A.; Storch M.; Baldwin G. S.; Ellis T. Bricks and blueprints: methods and standards for DNA assembly. Nat. Rev. Mol. Cell Biol. 2015, 16, 568–576. 10.1038/nrm4014. [DOI] [PubMed] [Google Scholar]
- Eisenstein M. How to build a genome. Nature 2020, 578, 633–635. 10.1038/d41586-020-00511-9. [DOI] [PubMed] [Google Scholar]
- Martella A.; Matjusaitis M.; Auxillos J.; Pollard S. M.; Cai Y. EMMA: An Extensible Mammalian Modular Assembly Toolkit for the Rapid Design and Production of Diverse Expression Vectors. ACS Synth. Biol. 2017, 6, 1380–1392. 10.1021/acssynbio.7b00016. [DOI] [PubMed] [Google Scholar]
- Potapov V.; Ong J. L.; Kucera R. B.; Langhorst B. W.; Bilotti K.; Pryor J. M.; Cantor E. J.; Canton B.; Knight T. F.; Evans T. C. Jr.; Lohman G. J. S. Comprehensive Profiling of Four Base Overhang Ligation Fidelity by T4 DNA Ligase and Application to DNA Assembly. ACS Synth. Biol. 2018, 7, 2665–2674. 10.1021/acssynbio.8b00333. [DOI] [PubMed] [Google Scholar]
- HamediRad M.; Weisberg S.; Chao R.; Lian J.; Zhao H. Highly Efficient Single-Pot Scarless Golden Gate Assembly. ACS Synth. Biol. 2019, 8, 1047–1054. 10.1021/acssynbio.8b00480. [DOI] [PubMed] [Google Scholar]
- Pryor J. M.; Potapov V.; Kucera R. B.; Bilotti K.; Cantor E. J.; Lohman G. J. S. Enabling one-pot Golden Gate assemblies of unprecedented complexity using data-optimized assembly design. PLoS One 2020, 15, e0238592 10.1371/journal.pone.0238592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C.; Kandzia R.; Marillonnet S. A one pot, one step, precision cloning method with high throughput capability. PLoS One 2008, 3, e3647 10.1371/journal.pone.0003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marillonnet S.; Grützner R. Synthetic DNA Assembly Using Golden Gate Cloning and the Hierarchical Modular Cloning Pipeline. Curr. Protoc. Mol. Biol.Curr. Protoc. 2020, 130, e115 10.1002/cpmb.115. [DOI] [PubMed] [Google Scholar]
- Szybalski W.; Kim S. C.; Hasan N.; Podhajska A. J. Class-IIS restriction enzymes - a review. Gene 1991, 100, 13–26. 10.1016/0378-1119(91)90345-c. [DOI] [PubMed] [Google Scholar]
- Weber E.; Engler C.; Gruetzner R.; Werner S.; Marillonnet S. A modular cloning system for standardized assembly of multigene constructs. PLoS One 2011, 6, e16765 10.1371/journal.pone.0016765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D.; O’Callaghan C. A. MetClo: methylase-assisted hierarchical DNA assembly using a single type IIS restriction enzyme. Nucleic Acids Res. 2018, 46, e113 10.1093/nar/gky596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.-J.; Weigele P. R.. Detection of Modified Bases in Bacteriophage Genomic DNA. DNA Modifications; Methods in Molecular Biology; Springer: Clifton, N.J., 2021; Vol. 2198, pp 53–66. [DOI] [PubMed] [Google Scholar]
- Rustad M.; Eastlund A.; Jardine P.; Noireaux V. Cell-free TXTL synthesis of infectious bacteriophage T4 in a single test tube reaction. Synth. Biol. 2018, 3, ysy002. 10.1093/synbio/ysy002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.