

Abstract
Background and Objectives
Muscle-specific kinase (MuSK) antibody–positive myasthenia gravis (MuSK + MG) is a form of MG with bulbar-predominant symptoms often resistant to conventional treatments. Patients with MuSK + MG may have an electrodiagnostic (EDX) profile distinct from other MG. This study compares EDX features of MuSK + MG with acetylcholine receptor (AChR) antibody–positive MG (AChR + MG) to discern whether any unique EDX pattern exists that can aid in clinical diagnosis.
Methods
From January 1, 2010, through December 31, 2020, all patients with MuSK + MG at our institution were identified and randomly matched to an AChR + MG cohort in a 1:2 ratio based on sex, age at onset, and subsequently Myasthenia Gravis Foundation of America (MGFA) clinical severity for a case-control study. Each patient's clinical profile, treatment, and EDX testing were summarized and analyzed.
Results
Twenty-two patients with MuSK + MG (18 female) and 44 patients with AChR + MG were studied. The average symptom duration at presentation was shorter in the MuSK + MG group (4.7 years) compared with AChR + MG (10.9 years). Myotonic discharges were rare in both groups but more frequently observed in patients with MuSK + MG (10%) identified in 5 muscles in 2 patients compared with AChR + MG (2%) noted in only 1 muscle in 1 patient. Patients with MuSK + MG more often had myopathic appearing motor unit potentials (MUPs) (41% vs 30%) compared with AChR + MG. Myopathic appearing MUPs were found in milder cases of MuSK + MG (MGFA class I–IIB) compared with AChR + MG (MGFA Class IIB–V).
Discussion
Patients with MuSK + MG may have a recognizable EDX profile from AchR + MG that includes (1) myotonic discharges, (2) greater occurrence of myopathic appearing MUPs in clinically mild disease, and (3) symptoms leading to earlier testing.
Myasthenia gravis (MG) is an autoimmune disease of the neuromuscular junction characterized by fatigable weakness that improves with rest.1 Although MG can involve any muscle, weakness most often affects specific muscle groups including ocular and bulbar segments with some patients having respiratory or more generalized (limb) disease.2 The role of autoantibodies in the pathogenesis and diagnosis of MG is well established. The acetylcholine receptor (AChR) antibody was the first discovered and is the most common antibody present in up to 80% of patients with generalized MG.3 Muscle-specific tyrosine kinase (MuSK) antibody has been more recently identified and is present in 20%–40% of AChR-seronegative patients with MG.4
MuSK + MG differs clinically from AChR + MG with bulbar-predominant symptoms that are resistant to anticholinesterase inhibitors and some disease-modifying therapies.5 Diagnosing patients with MuSK + MG can be challenging.6,7 MuSK + MG's predilection for bulbar and respiratory muscles in addition to occasionally observed muscle atrophy may mimic other neuromuscular disorders, e.g., bulbar-predominant amyotrophic lateral sclerosis,8 causing some providers to not consider it as a diagnostic possibility. Also, although serologic testing is available for MuSK + MG, serum antibody studies may not be readily available and take several days to return possibly delaying diagnosis. For these reasons, a rapid and reproducible tool to evaluate for MuSK + MG such as electrodiagnostic (EDX) testing9 should be further explored.
The ability to distinguish MuSK + MG from other disorders with EDX testing is supported by recent literature.10-13 Electrical myotonic discharges are an unusual EDX feature in neuromuscular junction disorders but have been reported in case studies of patients with MuSK + MG.10,11 Another study suggested that myopathic motor unit potentials (MUPs) were detected more frequently in MuSK + MG compared with AChR + MG in EDX testing.12 This finding was confirmed by a separate study that also found myopathic MUPs in a greater number of proximal muscles in patients with MuSK + MG compared with patients with AChR + MG.13 Collectively, these data indicate that MuSK + MG may have a suggestive EDX profile that, if further defined, may help facilitate a timely diagnosis. The aim of this study was to assess for distinctive EDX characteristics that distinguish MuSK + MG from the more common AChR + MG.
Methods
All patients with MuSK antibodies at levels >0.02 nmol/L tested at Mayo Clinic Laboratories between January 1, 2010, and December 31, 2020, were identified via searching through the clinical antibody database and patient data explorer at our institution. Patients were included if they carried the clinical diagnosis of MuSK antibody–positive MG. The following information was manually reviewed and collected from each patient's medical record: sex, age at symptom onset, age at diagnosis, time from symptom onset to EDX testing, symptom distribution, severity of symptoms, MuSK and AChR antibody titers, needle electromyography (EMG) and nerve conduction study (NCS) data including the degree of decrement on repetitive nerve stimulation (RNS), and medications used for treatment. Needle EMG data included the presence of fibrillation potentials, fasciculation potentials, myotonic discharges, voluntary MUP parameters (including recruitment, amplitude, duration, and percent polyphasics), and single-fiber EMG (SFEMG) results including the presence of abnormal jitter. MUPs were graded in a semiquantitative fashion at the time of acquisition by the physician performing the EMG and were compared with reference MUP parameter values. MUPs that were identified as having rapid (early) recruitment, low amplitude, and/or short duration, similar to those seen in primary muscle diseases, were considered to be myopathic appearing. Neurogenic MUPs were defined as MUPs that were graded as having reduced recruitment, high amplitude, long duration, and/or increased complexity. At the time of performing the EMG, the examining physician commented if myotonic discharges or MUP instability were present for each muscle tested.
Patients with AChR + MG with serology also performed at Mayo Clinic Laboratories were identified during the same time period via our clinical database search tool and matched to the MuSK + MG cases based on sex, age at symptom onset, and severity of symptoms using the Myasthenia Gravis Foundation of America (MGFA) severity class scale14 to achieve a 1:2 MuSK + MG to AChR + MG comparison. The same data as listed above were then collected for each patient with AChR + MG. Descriptive data are presented as median and range for continuous variables and frequency and percent for categorical variables.
Standard Protocol Approvals, Registrations, and Patient Consents
This case-control study was reviewed and approved by the Mayo Clinic Institutional Review Board.
Results
A total of 23 patients with MuSK + MG (18 female) were identified. One patient did not carry the diagnosis of MG and was excluded from the study. The remaining 22 patients were matched to 44 patients with AChR + MG as detailed in the Methods section of this article. Demographic characteristics between groups were similar (Table 1).
Table 1.
Patient Demographics

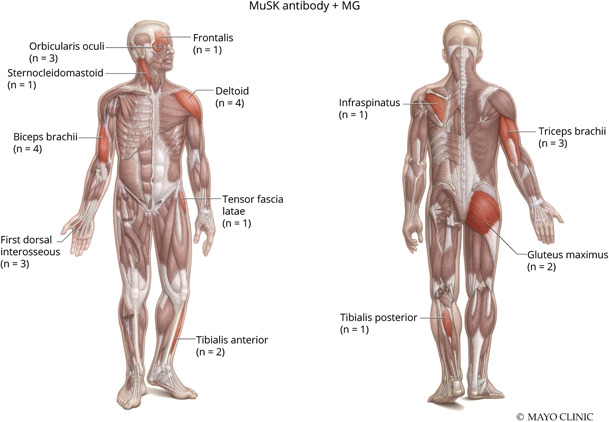
NCS/EMG data for both groups are presented in Table 2. Twenty of the 22 patients with MuSK + MG had NCS/EMG testing available for review. Patients with MuSK + MG (75%) and AChR + MG (64%) had similar frequencies of abnormal RNS, defined by a >10% decrement in amplitude or area between the first and last response in a train of 4 stimuli at slow (2 Hz) stimulation. Patients with MuSK + MG demonstrated myotonic discharges in 5 muscles and were observed in 2 of 20 (10%) patients. In comparison, 1 of 44 (2%) patients with AChR + MG had a single myotonic discharge in 1 muscle orbicularis oculi. Patients with MuSK + MG had a greater frequency of myopathic-appearing MUPs per patient (45%, 9/20) compared with patients with AChR + MG (30%, 13/44). Figures 1 and 2 depict the total number and specific muscles with myopathic-appearing MUPs recorded in patients with MuSK + MG and AChR + MG, respectively. Patients with MuSK + MG had a greater number of facial muscles tested (15%, 4/26) on needle EMG, whereas patients with AChR + MG had a greater number of axial muscles tested (14%, 4/28). Of interest, MUP variability was more often recorded in AChR + MG (45%, 20/44) than MuSK + MG (35%, 7/20).
Table 2.
Nerve Conduction and EMG Data

Figure 1. Myopathic Muscles Noted in Patients With MuSK + MG.

Used with permission of the Mayo Foundation for Medical Education and Research, all rights reserved. MuSK = muscle specific kinase; MG = myasthenia gravis.
Figure 2. Myopathic Muscles Noted in Patients With AChR + MG.

Used with permission of the Mayo Foundation for Medical Education and Research, all rights reserved. AChR = acetylcholine receptor; MG = myasthenia gravis.
Single-fiber EMG data from Table 2 showed that patients with MuSK + MG had a greater than average mean consecutive difference (100.0 μs) compared with AChR + MG (15.9 μs) supporting the idea of greater muscle membrane instability. The single-fiber studies were performed on patients with MuSK + MG with otherwise relatively normal needle EMGs. Most of the patients (89%, 8/9) of the MuSK MG group who had myopathic appearing MUPs did not have single fiber performed, and the patients with myotonic discharges also did not have single-fiber studies performed.
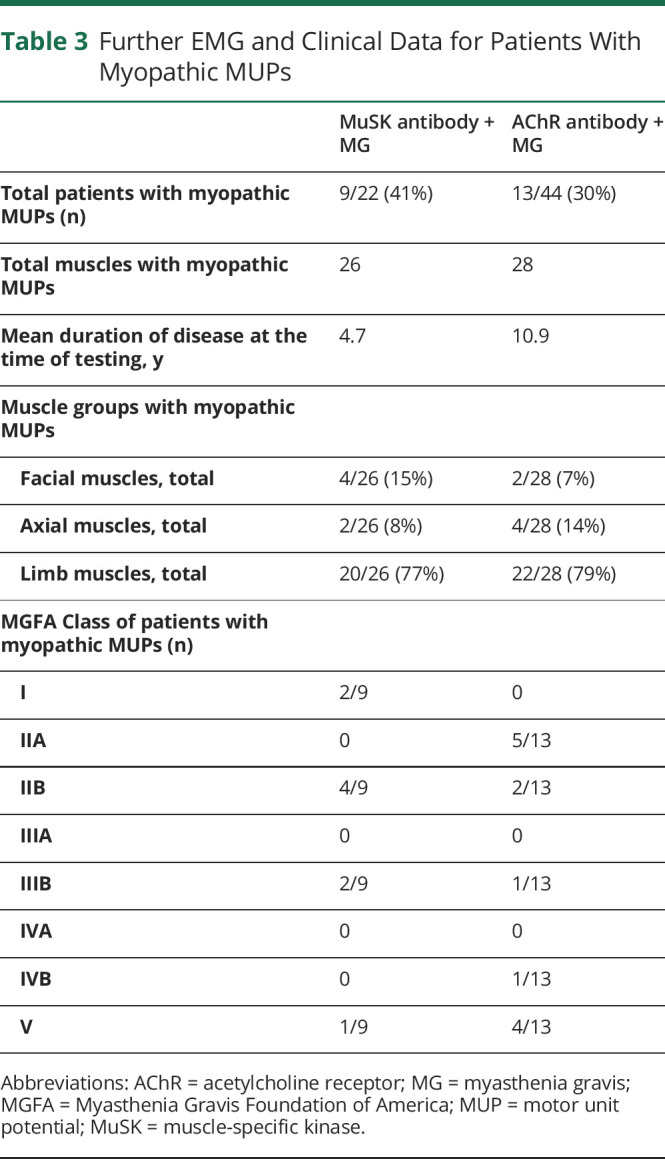
The characteristics of patients who had myopathic-appearing EDX findings are further detailed in Table 3. The mean symptom duration at the time of testing was shorter in the MuSK + MG group (4.7 years) compared with AChR + MG (10.9 years). The majority of patients with myopathic-appearing MUPs (66%, 6/9) were seen in patients with MuSK + MG with lower disease severity (MGFA Class I–IIB) compared with the AChR + MG group where most patients (62%, 8/13) had a greater clinical disease severity (MGFA Class IIB through V). Neurogenic MUPs from superimposed conditions were infrequently noted (approximately 10% in both cohorts) and included C5 radiculopathy, L5 radiculopathy, lumbosacral radiculopathy, facial neuropathy, peripheral neuropathy, and West Nile associated motor neuron disease. Notably, myotonic discharges were not recorded in muscles with neurogenic MUPs.
Table 3.
Further EMG and Clinical Data for Patients With Myopathic MUPs
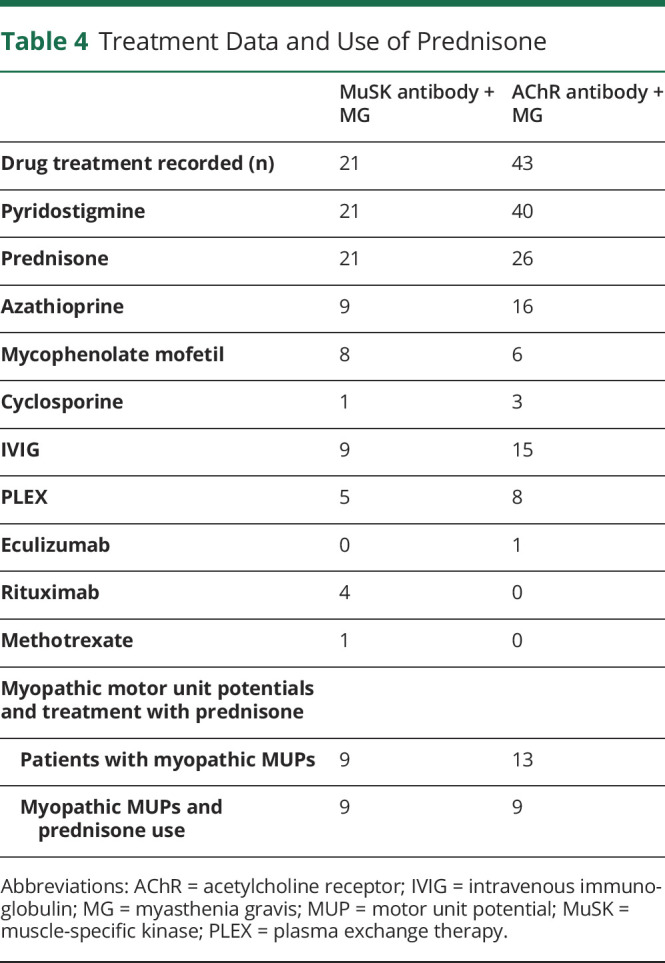
Treatment data are summarized in Table 4 with a variety of medications used for each group. All 9 patients with MuSK + MG with myopathic-appearing EDX findings were treated with prednisone. Eight of these patients were receiving corticosteroid treatment at the time of EDX while 1 patient received steroids only after EDX testing. In comparison, 26 patients with AChR + MG were treated with prednisone before or at the time of EDX testing, and the majority of these patients who were treated with prednisone (65%, 17/26) did not have myopathic-appearing MUPs. Nine of 13 patients with AChR + MG with myopathic-appearing MUPs had been previously treated with prednisone.
Table 4.
Treatment Data and Use of Prednisone
Discussion
The current study presents both clinical and EDX differences that may help distinguish MuSK + MG from AChR + MG. Clinically, MuSK + MG had a higher percentage of bulbar muscle weakness, which has been previously established.5 On EDX evaluation, we found that patients with MuSK + MG had greater numbers and higher percentages of myotonic discharges and myopathic-appearing MUPs compared with patients with AChR + MG. These data also suggest that patients with MuSK + MG may have myopathic-appearing MUPs earlier in their disease course and in lower MGFA classes compared with AChR + MG who are otherwise similar in age at symptom onset and sex. Although previous case reports support these EDX findings,10-13 this study uniquely combines this information in a large series that is matched in a case-control format including MGFA clinical severity score.
Despite the differences noted above, several clinical and EDX similarities were also seen between our MuSK + MG and AChR + MG groups. Clinically, both groups showed similar age at symptom onset, age at diagnosis, and proportion of ocular and generalized weakness. The 2 groups also shared some EDX features including similar degree of RNS decrement and frequency of abnormal needle EMG and SFEMG. As both groups of patients are variants of the larger disease MG, some common characteristics are expected.15
We postulate that these novel differences in EDX testing between MuSK + MG and AChR + MG are attributable to antibody-mediated pathophysiologic mechanisms. MuSK is a postsynaptic transmembrane protein at the neuromuscular junction that triggers and maintains clustering of AChRs, which is crucial for generating postsynaptic action potentials facilitating muscle contraction through acetylcholine transmission and sodium conductance.16 Interruption of AChR clustering via MuSK antibodies may theoretically lead to muscle membrane instability due to compromise of synaptic architecture and preserved, but asynchronous, sodium conductance of fewer AChR channels. Ultimately, this membrane instability may result in myotonic discharges and earlier myopathic-appearing MUPs in milder disease. The elevated mean consecutive difference measuring jitter seen in our MuSK + MG cohort's single-fiber EMGs also supports increased muscle membrane instability. A similar mechanism of muscle membrane instability via altered sodium conductance is also proposed for postexercise myotonia seen in paramyotonia congenita and hyperkalemic periodic paralysis, and myopathic MUP remodeling has been reported in congenital myasthenia related to alterations of MuSK genotype.17,18
This mechanism differs from AChR antibodies that block the binding of acetylcholine vesicles to AChR-associated sodium channels preventing depolarization altogether.19 The muscle membrane itself may not be as directly affected and thus requires a longer time frame or a more severe disease phenotype before myotonic discharges or myopathic-appearing MUPs would be observed on EDX.
Confounding variables such as age, sex, and highest achieved MGFA clinical severity were controlled for via matching. One might argue that these differences could be influenced by patient treatment, especially prednisone. Of interest, all patients with MuSK + MG with myopathic-appearing MUPs were treated with prednisone at some point during their disease course, and most patients (8 of 9 patients) had prednisone before or at the time of EDX testing. Although there are reported cases of corticosteroid-induced myopathic MUPs, steroid therapy is typically not associated with detectable EDX abnormalities in most patients.20,21 Moreover, the majority of our patients with AChR + MG treated with prednisone (17/26, 65%) did not have myopathic-appearing MUPs on needle EMG. Although medications can influence testing results, the current data argue against a direct association between prednisone and the observed needle EMG changes.
Defining EDX characteristics suggestive of MuSK + MG has important clinical relevance as this information may help expedite diagnosis and guide physicians to order appropriate laboratory testing. Also, a suggestive MuSK + MG EDX profile may provide enough diagnostic evidence to accelerate prescribing an appropriate treatment plan while serologic antibody levels are pending. As MuSK + MG can be difficult to treat with conventional methods, early evidence of MuSK + MG may help provide appropriate expectations of response to typical myasthenic treatments and may also guide physicians to select a specific immunosuppressant agent such as rituximab, which is uniquely beneficial to MuSK + MG.22
There are several limitations to this study. First, the small sample size makes statistical comparison between groups difficult to interpret, which is a problem inherent to a small observational study. Second, given that this study was a retrospective review of EMG reports, identification of MUP instability and myotonic discharges relied on the examining physician to manually document these findings, which may not have always occurred. Also, MUPs were graded by the electromyographers in a semiquantitative fashion, and potential interexaminer variation in grading MUPs cannot be excluded, especially in facial muscles where abnormally short duration or early recruitment of MUPs can be difficult to distinguish from normally small duration motor units. Future research would ideally include a larger MuSK + MG and AChR + MG cohort that is statistically powered to detect specific EDX differences and perhaps a prospective study carefully documenting EDX differences using quantitative measurements.
This study highlights potentially distinguishing EDX features that should prompt consideration of MuSK + MG. Identification of myotonic discharges and myopathic-appearing MUPs on EDX evaluation of a patient with a clinical suspicion of an acquired neuromuscular junction disorder may accelerate diagnosis, focus testing, and expedite proper treatment.
Appendix. Authors
Study Funding
No targeted funding reported.
Disclosure
The authors report no disclosures relevant to the manuscript. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
References
- 1.Jayam Trouth A, Dabi A, Solieman N, Kurukumbi M, Kalyanam J. Myasthenia gravis: a review. Autoimmune Dis. 2012;2012:874680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grob D, Arsura EL, Brunner NG, Namba T. The course of myasthenia gravis and therapies affecting outcome. Ann N Y Acad Sci. 1987;505:472-499. [DOI] [PubMed] [Google Scholar]
- 3.Peeler CE, De Lott LB, Nagia L, Lemos J, Eggenberger ER, Cornblath WT. Clinical utility of acetylcholine receptor antibody testing in ocular myasthenia gravis. JAMA Neurol. 2015;72(10):1170-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vincent A, Leite MI. Neuromuscular junction autoimmune disease: muscle specific kinase antibodies and treatments for myasthenia gravis. Curr Opin Neurol. 2005;18(5):519-525. [DOI] [PubMed] [Google Scholar]
- 5.Hatanaka Y, Hemmi S, Morgan MB, et al. Nonresponsiveness to anticholinesterase agents in patients with MuSK-antibody-positive MG. Neurology. 2005;65(9):1508-1509. [DOI] [PubMed] [Google Scholar]
- 6.Ciafaloni E. Myasthenia gravis and congenital myasthenic syndromes. Continuum (Minneap Minn). 2019;25(6):1767-1784. [DOI] [PubMed] [Google Scholar]
- 7.Rodolico C, Bonanno C, Toscano A, Vita G. MuSK-associated myasthenia gravis: clinical features and management. Front Neurol. 2020;11:660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Farrugia ME, Robson MD, Clover L, et al. MRI and clinical studies of facial and bulbar muscle involvement in MuSK antibody-associated myasthenia gravis. Brain. 2006;129(6):1481-1492. [DOI] [PubMed] [Google Scholar]
- 9.Pasnoor M, Dimachkie MM, Farmakidis C, Barohn RJ. Diagnosis of myasthenia gravis. Neurol Clin. 2018;36(2):261-274. [DOI] [PubMed] [Google Scholar]
- 10.Joundi RA, Israelian G, Ghavanini A, Kassardjian CD. Myotonic discharges in anti-MuSK myasthenia. Can J Neurol Sci. 2018;45(6):707-708. [DOI] [PubMed] [Google Scholar]
- 11.Magnussen M, Karakis I, Harrison TB. The myotonic plot thickens: electrical myotonia in antimuscle-specific kinase myasthenia gravis. Case Rep Neurol Med. 2015;2015:242691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nikolic A, Basta I, Stojanovic VR, Stevic Z, Peric S, Lavrnic D. Myopathic changes detected by quantitative electromyography in patients with MuSK and AChR positive myasthenia gravis. J Clin Neurosci. 2016;27:126-129. [DOI] [PubMed] [Google Scholar]
- 13.Rostedt Punga A, Ahlqvist K, Bartoccioni E, et al. Neurophysiological and mitochondrial abnormalities in MuSK antibody seropositive myasthenia gravis compared to other immunological subtypes. Clin Neurophysiol. 2006;117(7):1434-1443. [DOI] [PubMed] [Google Scholar]
- 14.Jaretzki A III, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task force of the medical scientific advisory board of the myasthenia gravis foundation of America. Neurology. 2000;55(1):16-23. [DOI] [PubMed] [Google Scholar]
- 15.Deymeer F, Gungor-Tuncer O, Yilmaz V, et al. Clinical comparison of anti-MuSK- vs anti-AChR-positive and seronegative myasthenia gravis. Neurology. 2007;68(8):609-611. [DOI] [PubMed] [Google Scholar]
- 16.Hughes BW, Kusner LL, Kaminski HJ. Molecular architecture of the neuromuscular junction. Muscle Nerve. 2006;33(4):445-461. [DOI] [PubMed] [Google Scholar]
- 17.Kimura J. Electrodiagnosis in Diseases of Nerve and Muscle: Principles and Practice. Oxford University Press; 2013. Accessed April 20, 2021. oxfordmedicine.com/view/10.1093/med/9780199738687.001.0001/med-9780199738687. [Google Scholar]
- 18.Pinto MV, Saw JL, Milone M. Congenital vocal cord paralysis and late-onset limb-girdle weakness in MuSK-congenital myasthenic syndrome. Front Neurol. 2019;10:1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koneczny I, Herbst R. Myasthenia Gravis: pathogenic effects of autoantibodies on neuromuscular architecture. Cells. 2019;8(7):671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khaleeli AA, Edwards RH, Gohil K, et al. Corticosteroid myopathy: a clinical and pathological study. Clin Endocrinol (Oxf). 1983;18(2):155-166. [DOI] [PubMed] [Google Scholar]
- 21.Surmachevska N, Tiwari V. Corticosteroid induced myopathy. In: StatPearls [Internet]: StatPearls Publishing; 2021. [PubMed] [Google Scholar]
- 22.El-Salem K, Yassin A, Al-Hayk K, Yahya J, Al-Shorafat D, Dahbour S. Treatment of MuSK-associated myasthenia gravis. Curr Treat Options Neurol. 2014;16(4):283. [DOI] [PubMed] [Google Scholar]