

Abstract
Microsporidia are poorly understood, ubiquitous eukaryotic parasites that are completely dependent on their hosts for replication. With the discovery of microsporidia species naturally infecting the genetically tractable transparent nematode C. elegans, this host has been used to explore multiple areas of microsporidia biology. Here we review results about microsporidia infections in C. elegans, which began with the discovery of the intestinal-infecting species Nematocida parisii. Recent findings include new species identification in the Nematocida genus, with more intestinal-infecting species, and also a species with broader tissue tropism, the epidermal and muscle-infecting species Nematocida displodere. This species has a longer polar tube infection apparatus, which may enable its wider tissue range. After invasion, multiple Nematocida species appear to fuse host cells, which likely promotes their dissemination within host organs. Localized proteomics identified Nematocida proteins that have direct contact with the C. elegans intestinal cytosol and nucleus, and many of these host-exposed proteins belong to expanded, species-specific gene families. On the host side, forward genetic screens have identified regulators of the Intracellular Pathogen Response (IPR), which is a transcriptional response induced by both microsporidia and the Orsay virus, which is also a natural, obligate intracellular pathogen of the C. elegans intestine. The IPR constitutes a novel immune/stress response that promotes resistance against microsporidia, virus, and heat shock. Overall, the Nematocida/C. elegans system has provided insights about strategies for microsporidia pathogenesis, as well as innate defense pathways against these parasites.
Keywords: C. elegans, Microsporidia, Nematocida, Tissue tropism, Syncytium, Intracellular Pathogen Response, Host-exposed proteins
5.1. Introduction
Study of the genetically tractable nematode Caenorhabditis elegans has resulted in seminal discoveries ranging from RNA interference (RNAi) to apoptotic signaling pathways (Singh 2021). C. elegans has several advantages for research in the lab including reproduction by selfing of hermaphrodites, which allows for propagation without crossing, a 3-day reproductive life cycle, and a 1-mm-long transparent body plan that facilitates imaging. Over 20 years ago, C. elegans was established as a powerful system to study innate immune responses in a whole animal model, using bacterial and fungal pathogens that cause lethal infections in this host (Darby et al. 1999; Mahajan-Miklos et al. 1999; Pujol et al. 2001). The majority of infection studies in C. elegans have been performed with clinically relevant bacterial pathogens, but these studies have now broadened to include natural pathogens, including microsporidia infections as described below.
The Microsporidia phylum contains over 1400 species of obligate intracellular pathogens related to fungi, and they can infect a wide range of hosts, from single-celled protists to humans (Han and Weiss 2017). Historical observations of microsporidia species that infect non-C. elegans nematodes date back to the early 1900s, but it was not until 2008 that a microsporidia species was identified that infects C. elegans. This pathogen was found in a wild C. elegans from a compost pit outside Paris, France, and causes a lethal intestinal infection. Based on ribosomal sequence, it was given a new genus and species name Nematocida parisii (in Latin Nematocida means nematode killer and parisii means from Paris) in the Microsporidia phylum (Troemel et al. 2008). Since that time, the Nematocida genus has grown to include eight species, which have been identified from wild Caenorhabditis and related nematodes across the globe (Zhang et al. 2016). Based on these studies, microsporidia are a common cause of infection for C. elegans in the wild, and N. parisii infection is the most common microsporidia species to infect C. elegans. This chapter will describe the isolation and characterization of these microsporidia species, their impact on host physiology, and the C. elegans innate immune response to these naturally occurring intracellular infections.
5.2. Identification of Microsporidia That Infect C. elegans
Species in the Caenorhabditis genus are free-living, bacterivorous nematodes that belong to the family Rhabditidae. Among the fifty-plus species in the Caenorhabditis genus, C. elegans and C. briggsae are the best studied (Stevens et al. 2019). The standard laboratory strains of each of these species are “domesticated” in that they have been grown for decades in a controlled laboratory environment. Driven by the interest to describe the natural ecology and biology of these species, researchers increased efforts to sample wild worms from across the world in the early 2000s, and there are now hundreds of wild-caught isolates of these two species, as well as several other Caenorhabditis species (Kiontke et al. 2011; Lee et al. 2021; Schulenburg and Felix 2017). In the course of isolating wild-caught nematodes from their natural habitats including rotting fruits, decaying plant stems, and compost heaps, co-occurring natural microbes have also been isolated, including pathogens like microsporidia.
Of the eight Nematocida species isolated from wild Caenorhabditis and related nematodes, six have so far been shown to infect C. elegans. These are N. parisii, Nematocida ironsii, Nematocida ausubeli, Nematocida displodere, Nematocida major, and Nematocida homosporus (Table 5.1) (Zhang et al. 2016). In addition, one non-Nematocida species called Pancytospora epiphaga can infect C. elegans. N. parisii has been isolated from wild C. elegans and wild C. briggsae hosts from various regions in France. Notably, the ERTm5 strain originally isolated from C. briggsae in Kauai (Hawaii, USA) was named N. parisii based on large subunit ribosomal sequence (Balla et al. 2015) but was later re-named N. ironsii based on whole genome sequencing and molecular divergence (Reinke et al. (2017) and see below). This finding indicates that other strains assigned as N. parisii based on large subunit ribosomal sequence may be reassigned once their genomes are fully sequenced. After N. ironsii, the species with closest sequence identity to N. parisii is N. ausubeli. This species was originally called Nematocida sp. 1 and is described with this name in several publications (Bakowski et al. 2014b; Balla et al. 2016; Reinke et al. 2017; Troemel et al. 2008). It subsequently was named N. ausubeli for Fred Ausubel, a pioneer in the field of C. elegans host/pathogen interactions (Zhang et al. 2016). N. ausubeli was originally isolated from C. briggsae in Kerala, India, and has subsequently been isolated in wild-caught C. elegans from France and from Portugal; in wild-caught C. briggsae from Cape Verde; and in wild-caught C. remanei from Germany. N. major has been isolated from C. briggsae in Guadalupe and Thailand and from C. tropicalis in Guadalupe. This species has not yet been isolated from C. elegans in the wild, but it is capable of infecting C. elegans in the lab. Similarly, N. homosporus and P. epiphaga have both been isolated from non-C. elegans nematode species in the wild, but can infect C. elegans in the lab. The fourth species so far shown to infect C. elegans in the wild is N. displodere, which was isolated from a wild C. elegans in France. This species is distinct from other Nematocida species in that it can infect multiple tissues, in addition to the intestine, providing an opportunity to learn more about what governs tissue tropism ((Luallen et al. 2016) and see below).
Table 5.1.
List of microsporidia species that can infect C. elegans in the wild or the lab
| Microsporidia species | Isolated from | Infects C. elegans | Number of published strains | Tissues infected | Genomes sequenced | Other names | References |
|---|---|---|---|---|---|---|---|
| Nematocida parisii | Caenorhabditis elegans, Caenorhabditis briggsae (France) | In wild, in lab | 17 | Intestine | Strains ERTm1, ERTm3 | Troemel et al., PLOS Biology (2008); Cuomo et al., Genome Research (2012); Zhang et al., PLOS Pathogens (2016) | |
| Nematocida ironsii | Caenorhabditis briggsae (Hawaii, USA) | In wild, in lab | 1 | Intestine | Strains ERTm5 | N. parisii | Balla et al., PLOS Pathogens (2015); Zhang et al., PLOS Pathogens (2016); Reinke et al., Nature Communications (2017) |
| Nematocida ausubeli | Caenorhabditis elegans (France, Portugal); Caenorhabditis briggsae (India, Cape Verde, Germany); Caenorhabditis remanei (Germany) | In wild, in lab | 10 | Intestine | Strains ERTm2, ERTm6 | N. sp. 1 | Troemel et al., PLOS Biology (2008); Cuomo et al., Genome Research (2012); Bakowski et al., Genome Announcement (2014b); Zhang et al., PLOS Pathogens (2016) |
| Nematocida displodere | Caenorhabditis elegans (France) | In wild, in lab | 1 | Intestine, coelomocytes, neurons, epidermis, muscle | JUm2807 | Luallen et al., PLOS Pathogens (2016); Zhang et al., PLOS Pathogens (2016) | |
| Nematocida major | Caenorhabditis briggsae (Thailand, Guadeloupe); Caenorhabditis tropicalis (Guadeloupe) | In lab | 3 | Intestine | Zhang et al., PLOS Pathogens (2016) | ||
| Nematocida homosporus | Oscheius tipulae (France); Rhabditella typhae (Portugal) | In lab | 2 | Intestine | Zhang et al., PLOS Pathogens (2016) | ||
| Pancytospora epiphaga | Caenorhabditis brenneri (Colombia) | In lab | 1 | Epidermis | Zhang et al., PLOS Pathogens (2016) | ||
| Nematocida minor | Oscheius tipulae (Czech Republic, Armenia) | No | 2 | Intestine | Zhang et al., PLOS Pathogens (2016) | ||
| Nematocida ciargi | Procephalobus sp. (Spain) | No | 1 | Intestine | Zhang et al., PLOS Pathogens (2016) |
While infection by all Nematocida species appear capable of killing C. elegans hosts, different species have varying levels of virulence. Side-by-side comparisons between N. ausubeli and N. parisii demonstrate that N. ausubeli is more virulent, as assessed by N. ausubeli causing more severe reduction in body size and a greater reduction in the number of eggs laid by infected hosts (Balla et al. 2016). Furthermore, N. ausubeli replicates more quickly inside C. elegans and forms spores earlier than N. parisii. Side-by-side comparisons of N. displodere and N. parisii virulence have not been described, but C. elegans begins shedding N. parisii spores earlier than N. displodere spores, likely because N. parisii spores are actively shed via host exocytosis from the intestine, whereas N. displodere spores gradually build up inside the animal until it bursts (Luallen et al. 2016; Szumowski et al. 2014). Further distinctions among the different Nematocida species genomes, life cycles, and interactions with hosts are described below.
5.3. Nematocida Genomes, Transcriptomes, and Proteomes
Next-generation sequencing has provided whole genome assembly for the four microsporidia species found to infect C. elegans in the wild (N. parisii, N. ausubeli, N. ironsii, and N. displodere). Microsporidia have the smallest known genomes among eukaryotes, with the human-infecting species Encephalitozoon intestinalis as one of the smallest at only 2.3 Mb (Corradi et al. 2010). The first Nematocida genome sequences were obtained as part of the Microsporidian Genome consortium and included two strains of N. parisii (ERTm1 from Paris, France, and ERTm3 from Santeuil, France) (Cuomo et al. 2012) and two strains of N. ausubeli (ERTm2 from Kerala, India, and ERTm6 from the Cape Verde Islands) (Bakowski et al. 2014b; Cuomo et al. 2012). N. parisii ERTm1 and ERTm3 each have genomes of about 4.1 Mb, with 2661 predicted genes for ERTm1 and 2770 for ERTm3. Transcriptome analysis aided annotation of the N. parisii genome and indicated that 2546 of the ERTm1 genes were expressed during at least one of five stages of the N. parisii intracellular replicative life cycle and/or during the spore stage (Cuomo et al. 2012). Genome size was more divergent between the two N. ausubeli sequenced strains, at 4.7 Mb with 2770 predicted genes for ERTm2 and 4.3 Mb with 2433 genes for ERTm6. Both N. parisii and N. ausubeli appear to be diploid, with the highest degree of heterozygosity seen in N. ausubeli ERTm2 at 1 SNP every 82 bases (Cuomo et al. 2012). Large regions in N. parisii and N. ausubeli genomes have lost heterozygosity, which is suggestive of a recent or rare recombination event as part of a sexual or parasexual cycle. Morphological evidence of sexual cycles had previously been described for microsporidia species infecting other hosts (Becnel et al. 2005).
These genome sequencing studies were part of a larger phylogenomics effort to compare diverse microsporidia genomes (Cuomo et al. 2012). This effort identified gene gains and losses specific to the Microsporidia phylum and elucidated possible strategies used for the unique life cycle of these parasites. For example, compared to other eukaryotes, microsporidia have lost the cell cycle inhibitor retinoblastoma, which appears to be one of the earliest steps in the evolution of the Microsporidia phylum and may have facilitated their rapid replication inside host cells (Cuomo et al. 2012; Haag et al. 2014). Furthermore, these studies and others at the same time (Capella-Gutierrez et al. 2012) firmly established microsporidia as being most closely related to fungi, which had previously been a topic of controversy (Keeling and McFadden 1998).
Following the efforts of the Microsporidian Genomes Consortium, the genomes of one strain each of N. displodere and N. ironsii have also been described. As mentioned earlier, N. ironsii ERTm5 has the same large subunit ribosomal sequence as N. parisii, but whole genome sequencing revealed only 92.3% identity between ERTm5 and ERTm1 DNA, thus leading to its definition as a distinct species (Reinke et al. 2017). For comparison, N. parisii strains ERTm1 and ERTm3 share 99.8% genome sequence identity (Cuomo et al. 2012). The N. ironsii ERTm5 genome spans 4.4 Mb with 2709 predicted genes and is the closest to that of N. parisii. At 3 Mb, the N. displodere strain JUm2807 has a smaller genome than either N. parisii, N. ironsii, or N. ausubeli (Luallen et al. 2016). This reduction is due in part to a decrease in intergenic regions: 85.8% of N. displodere’s genome is protein coding, while 69.2% and 63.7% are protein coding in N. parisii and N. ausubeli, respectively. In addition, the N. displodere genome has only 2278 predicted genes, which is fewer than N. parisii, N. ironsii, or N. ausubeli. N. displodere proteins share only 48% amino acid sequence similarity with N. parisii or N. ausubeli proteins, supporting the assignment of this species as an outgroup in the Nematocida genus compared to N. parisii and N. ausubeli and also compared to other species. Of note, genome sequencing indicated that all Nematocida genomes contain expanded species-specific gene families, with the largest being a family with 235 members found in N. displodere (Luallen et al. 2016; Reinke et al. 2017). Thus, this family comprises about 10% of the protein-coding potential in the N. displodere genome. This gene family is characterized by the presence of a RING domain, which is a protein-protein interaction domain found in most E3 ubiquitin ligases.
To determine which microsporidia proteins might enable its obligate intracellular life cycle, localized proteomics was performed to identify Nematocida proteins that are “host-exposed” to the C. elegans host cytosol or nucleus (Reinke et al. 2017). Specifically, spatially restricted enzymatic tagging was performed by expressing the promiscuous biotin ligase APX in either the C. elegans intestinal cytosol or intestinal nucleus, which allows APX to attach a biotin tag to lysine resides on any nearby proteins in these cellular compartments. APX-expressing transgenic animals were then infected with either N. parisii or N. ausubeli. To identify Nematocida proteins located in either the intestinal cytosol or intestinal nuclei, biotin-tagged proteins were isolated by streptavidin affinity pull-down, followed by liquid chromatography and tandem mass spectrometry. Overall, this analysis identified 82 Nematocida proteins (72 from N. parisii and 10 from N. ausubeli) with high confidence of interacting with intestinal cell tissue, including several proteins found in the nucleus. These host-exposed proteins were enriched for those that contained transmembrane domains and/or signal sequences. In addition, most of these host-exposed proteins were found to be rapidly evolving and species-specific. In fact, only 12% of N. parisii host-exposed proteins have orthologues outside of the “clade” of N. parisii, N. ironsii, and N. ausubeli, whereas 63% of proteins as a whole in the N. parisii genome have orthologs outside of this clade. These host-exposed proteins appear to not just be species-specific, but also belong to large gene families. Such families appear to be a general feature of microsporidia genomes. The size and divergence of these gene families may reflect the outcome of host/pathogen arms races that drive both sides to expand and diversify the sequence of proteins directly involved in the battle for survival during intracellular replication (Lazetic and Troemel 2020).
5.4. Nematocida Life Cycles and Their Impact on C. elegans Cell Biology
The life cycles of Nematocida species share many similarities with other microsporidia species, including the spore stage being the transmissible form, and all parasite replication and differentiation occurring inside of host cells, before spores exit to infect new hosts (Fig. 5.1). As N. parisii strain ERTm1 is the best characterized of the microsporidia species infecting C. elegans, we will first describe its life cycle and then compare it to other species below.
Fig. 5.1.
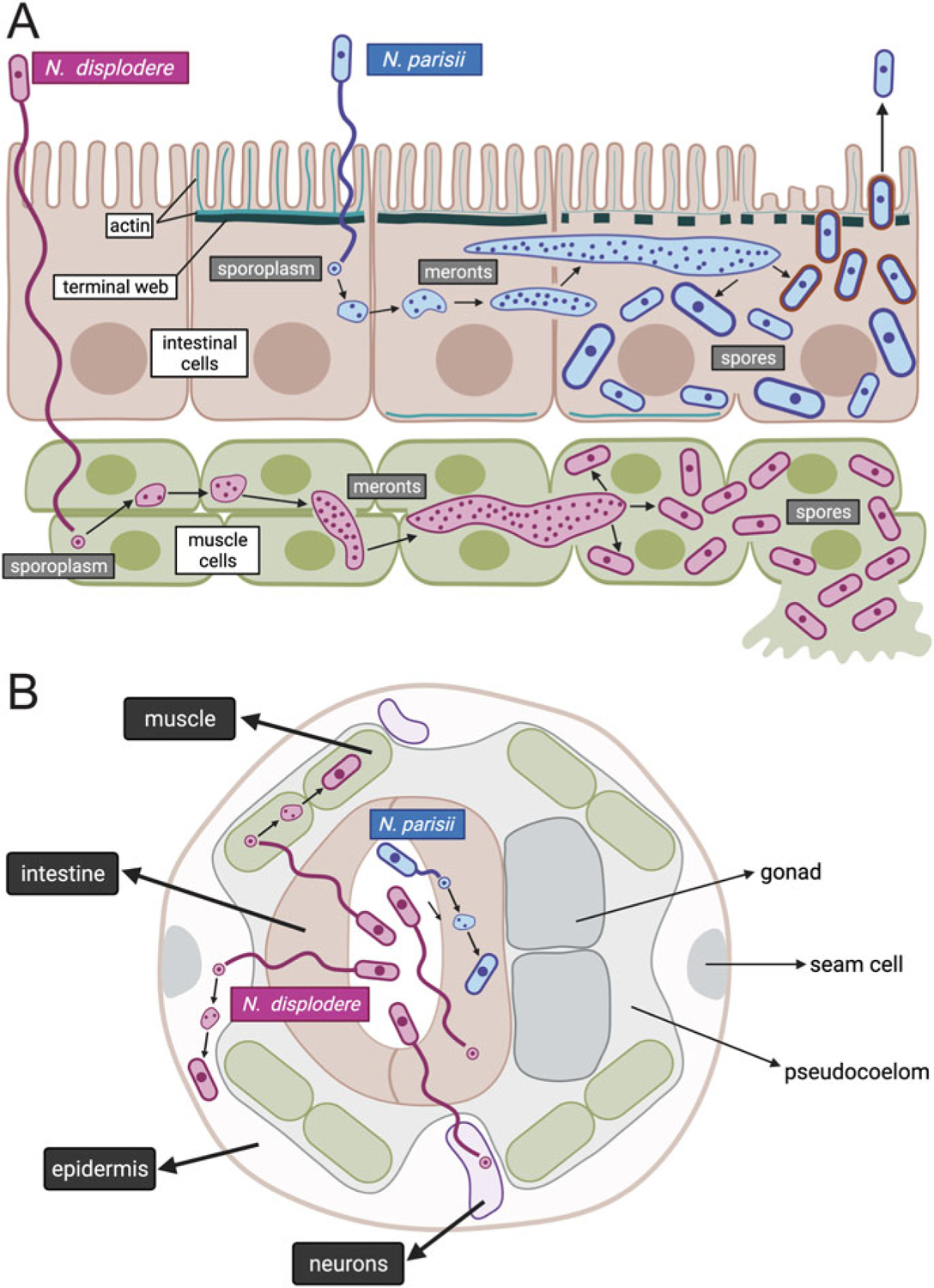
Life cycle of N. parisii and N. displodere inside the C. elegans host. Longitudinal (a) and cross-sectional (b) diagrams of Nematocida infection of the C. elegans intestine, muscle, epidermis, and neurons. N. parisii spore in blue is shown in the intestinal lumen, firing a polar tube that delivers a sporoplasm (a membrane-bound parasite cell) into the cytoplasm of C. elegans intestinal cells. This sporoplasm develops in direct contact with the host cytoplasm, replicating its nuclei without undergoing cell division, to develop into a multinucleate meront. This meront can spread across intestinal cells, causing the intestinal organ to become a syncytial structure with shared cytoplasmic contents. As N. parisii meronts develop into spores, gaps appear in the terminal web (made of actin and intermediate filaments), which is thought to remove a barrier to exit. Once N. parisii meronts develop into spores, they are found in separate membrane-bound compartments of unknown origin. These spores then become coated with the small GTPase RAB-11 (in red) and fuse with the apical membrane, to be released non-lytically back into the lumen. N. displodere spore in purple is also shown in the intestinal lumen, firing a polar tube that is longer than the polar tube of N. parisii. The N. displodere polar tube is hypothesized to reach all the way from the lumen into muscle cells, epidermal cells, and neurons to deliver sporoplasms inside of these cells. N. displodere sporoplasms develop into meronts, which can lead to fusion of muscle cells to form a syncytium (the epidermis is already a syncytium). In contrast to the non-lytic exit of N. parisii, N. displodere spores appear to burst out of cells
N. parisii is transmitted fecal orally, with infection starting after C. elegans ingests infectious spores that enter the lumen of the intestine (Fig. 5.1) (Troemel et al. 2008). The C. elegans intestine is composed of 20 polarized epithelial cells with structural and functional similarity to intestinal epithelial cells in mammalian hosts, except that these intestinal cells are non-renewable (Dimov and Maduro 2019). While in the intestinal lumen, N. parisii spores fire an infection apparatus common to all microsporidia species called a polar tube, which is coiled inside of the spore before receiving a cue to fire. As the polar tube fires, a small parasite cell called a sporoplasm is injected through the tube and delivered into a C. elegans intestinal cell. The exact cues and mechanisms used by N. parisii to fire the polar tube and invade host cells are poorly understood, but a recent report describes an intestinally secreted host factor that promotes microsporidia invasion, perhaps through enabling more efficient polar tube firing (El Jarkass et al. 2022). Notably, the process of intestinal cell invasion is fast, as sporoplasms inside intestinal cells can be observed within minutes after feeding C. elegans infectious N. parisii spores (Balla et al. 2019). Like other microsporidia species, N. parisii sporoplasms develop into multinucleated, replicating cells called meronts, which then differentiate back into spores that exit into the lumen. From there, spores can be defecated out of the animal to infect new hosts. The intestine is the only tissue where N. parisii replicates, and spore exit appears to be directional. Specifically, all spores exit apically from intestinal cells into the lumen, and have not been seen to exit basolaterally into the pseudocoloemic space of the animal (Estes et al. 2011).
N. parisii sporoplasms and meronts appear to replicate in direct contact with host intestinal cytosol. In particular, they have their own cellular plasma membrane, but do not appear to have a separate host membrane surrounding them at this stage (Szumowski et al. 2014). Further, N. parisii causes several restructuring events in the host while going through its life cycle. Here we will only briefly summarize such events related to spore exit, as they have been previously reviewed (El Jarkass and Reinke 2020; Troemel 2016). One restructuring event is a partial loss of polarity, with apically restricted cytoskeletal component actin appearing on the basolateral side of intestinal cells (Estes et al. 2011). This relocalization may trigger another restructuring event, which is the appearance of gaps in the terminal web. The terminal web is a conserved actin and intermediate filament-rich structure found in the apical region of most polarized epithelial cells, including epithelial cells of the C. elegans intestine. These gaps in the terminal web appear precisely when N. parisii meronts differentiate into spores, and thus formation of gaps may remove a barrier to enable spores to exit into the lumen (Estes et al. 2011). As meronts differentiate into spores, they enter a separate membrane-bound compartment that feeds into the host recycling endocytosis pathway. Near the apical membrane, these compartments get coated with the C. elegans small GTPase RAB-11, which is a host trafficking factor that facilitates fusion of these spore-containing compartments with the apical membrane to direct non-lytic exit of spores back into the intestinal lumen (Szumowski et al. 2014; Szumowski et al. 2016). Overall, these restructuring events appear to enable a large number of N. parsii spores to be shed from worms while minimizing damage to the host intestine.
In contrast to the intestinal-specific tropism described for N. parisii and other Nematocida species, the more recently described species N. displodere is able to replicate in several tissues in C. elegans. N. displodere replicates most robustly in muscle and epidermis cells, with rare replication seen in coelomocytes (phagocytic cells with no known role in immunity), neurons, and intestinal cells (Fig. 5.1) (Luallen et al. 2016). N. displodere bursts out of worms (displodere in Latin means to cause to explode), which may occur because it is not possible to non-lytically exit out of the non-intestinal tissues where N. displodere replicates and differentiates most efficiently. After N. displodere replicates in the epidermis and muscle, it differentiates into spores that exit from the animal by bursting through the C. elegans vulva, or egg-laying structure. N. displodere infection begins through feeding, and thus the pathogen appears to access all of these diverse tissues by starting from the intestinal lumen. Interestingly, the N. displodere polar tube is longer than that of N. parisii (12.6 μm vs. 4.0 μm). These observations, together with the observation that N. displodere sporoplasms are found inside non-intestinal tissues less than 2 minutes after feeding spores to worms, suggest that N. displodere spores fire their polar tubes from the intestinal lumen all the way through both the apical and basal side of intestinal cells to invade non-intestinal tissue like the epidermis and muscle, although this event has not been directly visualized (Fig. 5.1). Another notable characteristic of N. displodere is having only one spore size, while N. parisii has two distinct spore sizes. The functional significance of these spore differences is unknown. It should be noted that the morphological and life cycle differences between N. displodere and N. parisii are accompanied by differences in genome content as described above, including the strikingly expanded gene family that has 235 members in N. displodere. This family has been named NemLGF2 and has only 1–3 members each in N. parisii, N. ironsii, and N. ausubeli genomes (Reinke et al. 2017). Perhaps this family is responsible for the distinct lifestyle and tissue tropism of N. displodere, which has the broadest tissue tropism described so far for any C. elegans microbial pathogen.
One pathogen-driven restructuring event seen in common with infections by N. displodere, N. parisii, and N. ausubeli is the fusion of host cells (Balla et al. 2016). This fusion occurs while meronts are replicating, with N. parisii able to spread across several intestinal cells after first invading only a single intestinal cell. Imaging experiments with photoconvertible GFP demonstrated that N. parisii growth across intestinal cells causes sharing of cytoplasmic contents, indicating that what once was 20 separate intestinal cells becomes a large syncytial organ during infection. Similar results were found with N. ausubeli, which also only grows in the intestine. In contrast, N. displodere did not spread across intestinal cells in the rare instances where it replicated there, but instead spread across muscle cells after invading a single cell. These results indicate that the ability to fuse host cells is conserved across Nematocida species, with each species triggering syncytial formation in the host organ where it grows best. This cell-to-cell spread by Nematocida meronts likely enables faster, more efficient dissemination than if they were to differentiate back into spores, exit the infected cell, and then invade uninfected host cells using the polar tube invasion strategy. These findings provide a new perspective on how microsporidia can invade host cells, using a strategy independent of spores and polar tubes.
What regulates the differentiation of microsporidia meronts into spores? Experiments using differing doses of N. parisii spores and differing sizes of C. elegans host animals indicated that sporulation is triggered when meronts reach a critical density inside the intestine (Balla et al. 2016). An RNAi screen identified predicted C. elegans transcription factors as host factors that regulate sporulation of N. parisii inside intestinal cells. These host factors include members of the Myc family of transcription factors (Botts et al. 2016). N. parisii replication appears to proceed normally in C. elegans animals defective for these factors, but N. parisii differentiation is compromised. The exact role for C. elegans Myc transcription factors in regulating N. parisii sporulation is unclear, but one possibility is that they regulate expression of C. elegans metabolites or other cues that are sensed by N. parisii upon high meront density within the intestine to enter the sporulation program.
5.5. C. elegans Natural Variation in Resistance to Nematocida Infections
The original N. parisii isolate ERTm1 came from a wild-caught C. elegans near Paris, France, but ERTm1 is able to infect the N2 laboratory strain and other strains of C. elegans as well (Troemel et al. 2008). A comparison of wild-caught C. elegans strains found that the C. elegans Hawaiian isolate CB4856 displays higher resistance to infection with a Hawaiian isolate of Nematocida called N. ironsii ERTm5 (Balla et al. 2015). There is a lower initial colonization of N. ironsii inside CB4856 intestinal cells compared to N2 intestinal cells, and CB4856 intestinal cells can also clear N. ironsii infection over time, unlike N2, which always succumbs to infection by N. ironsii, as well as by other microsporidia species (Balla et al. 2015; Balla et al. 2019). Competition experiments showed that this increased resistance of CB4856 provided a fitness advantage over N2 in the presence of N. ironsii. Interestingly, the increased resistance and clearance (the reduction in parasite load over time) ability of CB4856 is restricted to the first larval (L1) stage of development, with no difference between N2 and CB4856 at later larval stages. Of note, the L1 stage is the only stage where infection reduces progeny production, with a greater reduction of progeny production caused by infection of N2 compared to infection of CB4856. While infection by N. ironsii shortens life span of N2 and CB4856 at any stage of development, this pathogen somewhat surprisingly does not reduce progeny production in older animals, despite establishing a similarly robust infection. Therefore, L1 may be the only stage where increased resistance against N. ironsii can promote the evolutionary success of C. elegans.
Quantitative genetics using recombinant-inbred lines identified four regions in the CB4856 genome associated with resistance (Balla et al. 2015). Introgressing two of these regions from the CB4856 genome into the N2 genome conferred resistance, and introgressing them from the N2 genome into the CB4856 genome conferred susceptibility, thus confirming the role of these two regions in resistance to Nematocida infection, acting additively. While the causative alleles in these genomic regions have not been identified, they are enriched for genes encoding components of cullin-ring E3 ubiquitin ligases. Ubiquitin ligases are enzymes that add ubiquitin tags to proteins and other targets to alter their fates, which can include autophagy-mediated degradation of intracellular pathogens in a process called xenophagy (Kuo et al. 2018). For this reason, the role of ubiquitin and the downstream process of autophagy was further analyzed in the ability of the CB4856 strain to clear N. ironsii (Balla et al. 2019). Ubiquitin and LGG-1/ATG8, a common marker for autophagy, were previously shown to target N. parisii sporoplasms in N2 animals. The level of targeting was relatively low, and ubiquitin-mediated autophagy played a modest but significant functional role in resistance against infection, although clearance was not examined in this study (N2 is not able to clear infection) (Bakowski et al. 2014a). In support of the possibility that more efficient xenophagy could explain the increased resistance of CB4856 hosts, this strain was found to have much higher levels of ubiquitin targeting sporoplasms compared to N2. For example, 84% of parasite cells were targeted at 15 hours post-inoculation (hpi) in CB4856 animals, and only 11% targeted at 15 hpi in N2 animals (Balla et al. 2019). Ubiquitin targeting correlated with parasite clearance in terms of parasite species, as analysis in CB4856 hosts showed there was high levels of targeting to N. ironsii cells, which are cleared, and little targeting to N. parisii and N. ausubeli parasite cells, which are not cleared. Furthermore, increased ubiquitin targeting was seen in infections of CB4856 L1 but not L4 animals, again showing a correlation between ubiquitin targeting to parasite cells and their subsequent clearance.
Despite this strong correlation between host targeting of ubiquitin to N. ironsii parasite cells and their subsequent clearance, a functional role for ubiquitin and the downstream process of autophagy has not yet been shown. Ubiquitin is an essential gene, and C. elegans has an enormous number of ubiquitin ligases, making it difficult to test their roles. Ubiquitin ligase-mediated autophagy converges on core components however, including the ATG8 homolog LGG-1 mentioned above, which is commonly used as a marker for autophagy in C. elegans, as well as the other C. elegans ATG8 homolog LGG-2, which is most similar to LC3 in mammals. Interestingly, it was seen that LGG-2 targeting parasite cells correlated with clearance, with higher levels of LGG-2 targeting N. ironsii cells in CB4856 compared to N. ironsii cells in N2 hosts. There was no difference in LGG-1 targeting N. ironsii cells in CB4856 compared to N2. However, clearance still occurred normally in lgg-2 mutants in CB4856, indicating this gene is not required for clearance. One possibility is that when LGG-2 is absent, LGG-1 can take its place. Of note, LGG-1 has redundant functions with LGG-2 for other processes in the N2 background. However, this model is difficult to test in CB4856 animals, because lgg-1 is an essential gene in N2 (and thus likely also in CB4856), and RNAi knockdown is relatively ineffective in CB4856 animals.
While the host factors that cause clearance of N. ironsii over time are yet undefined, lgg-2 was found to be required for the 50% decrease in N. ironsii initial colonization inside CB4856 intestinal cells compared to N2 intestinal cells (Balla et al. 2019). Deletion of lgg-2 had no effect in N2 animals, but in CB4856 animals, it led to an increase in intracellular colonization by N. ironsii to the same levels as N2 animals. This result indicates a specific role for lgg-2 in the increased resistance to intracellular colonization by N. ironsii in CB4856 animals. Thus, the role of different defense components can vary depending on the host strain background. Given the numerous roles for autophagy components in host defense outside of xenophagic clearance of intracellular pathogens, it will be interesting to define how lgg-2 controls intracellular colonization of microsporidia infection.
5.6. C. elegans Host Transcriptional Response to Microsporidia Infection: The Intracellular Pathogen Response
To determine the C. elegans transcriptional response to microsporidia infection, RNA-seq transcriptomic profiling was performed on N. parisii-infected N2 animals isolated at five different points of infection representing N. parisii invasion, growth, and sporulation (Bakowski et al. 2014a). At each infection time point, there was a significant enrichment of differentially expressed genes associated with the intestine, as compared to uninfected controls. Through comparisons with other gene sets, the genes regulated by N. parisii infection showed little similarity to genes regulated by bacterial extracellular pathogens or known immunity signaling pathways. However, there was a significant similarity between genes upregulated by N. parisii and genes upregulated by Orsay virus infection. Orsay virus is a three-gene, positive-sense, single-stranded RNA virus isolated from wild C. elegans in France (Felix et al. 2011). Despite their vast molecular differences, the Orsay virus and N. parisii are both obligate intracellular pathogens of the C. elegans intestine, suggesting they trigger a previously undefined immune response. The common gene set induced by both pathogens was termed the Intracellular Pathogen Response, or IPR (Fig. 5.2) (Bakowski et al. 2014a; Reddy et al. 2017). Analysis of a subset of IPR genes by qRT-PCR indicated that N. ausubeli is a weaker IPR activator than N. parisii in C. elegans, despite having a higher negative impact on host fitness than N. parisii (Balla et al. 2016; Zhang et al. 2016). One possible explanation is that N. ausubeli actively inhibits the IPR to enable a more virulent infection compared to N. parisii, or perhaps N. ausubeli is not as easily recognized by the host to induce the IPR.
Fig. 5.2.
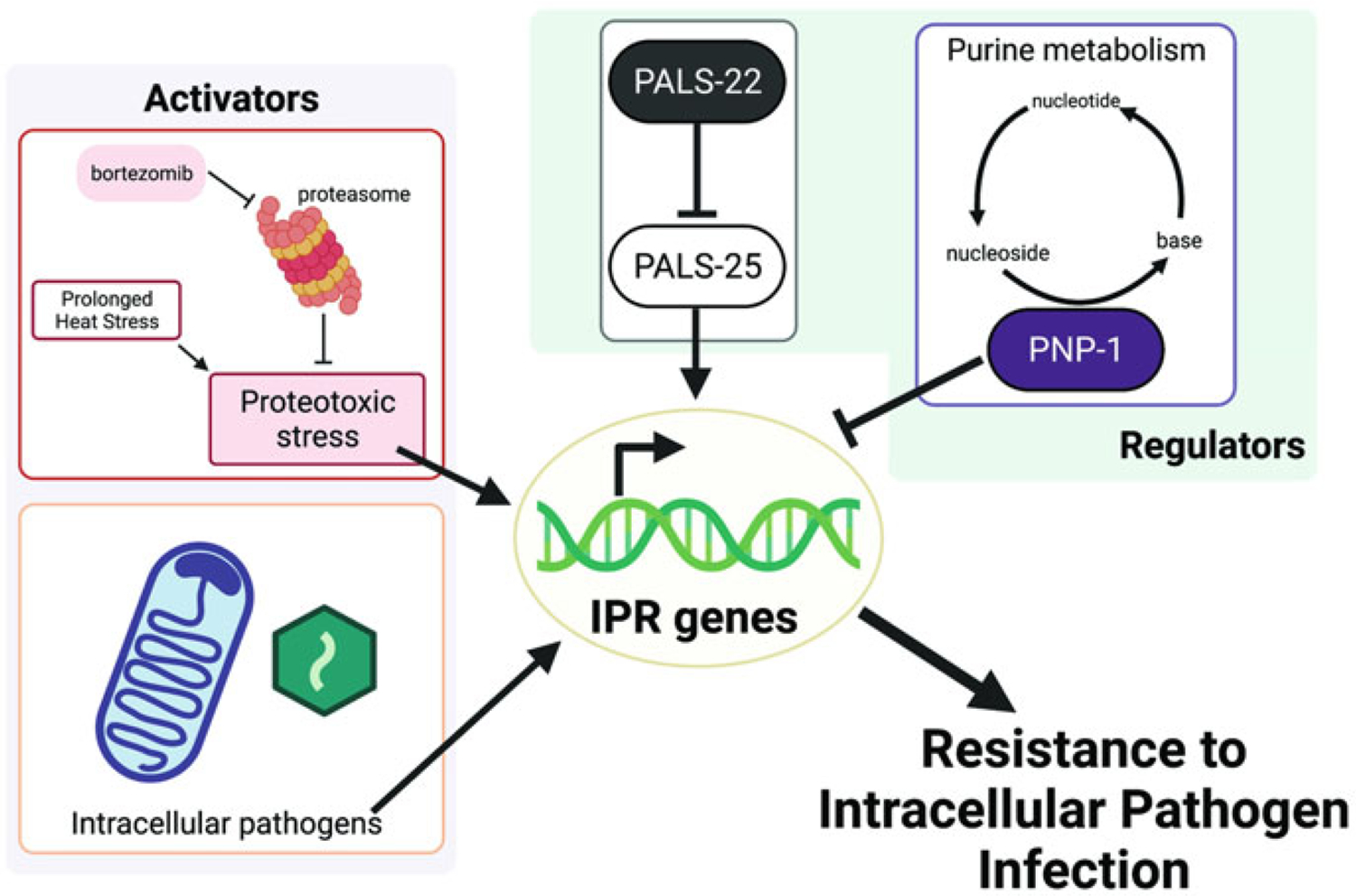
Model of the Intracellular Pathogen Response. Intestinal intracellular pathogens, like N. parisii and the Orsay virus, upregulate mRNA expression of a common set of C. elegans genes called the Intracellular Pathogen Response, or IPR genes. IPR genes can also be induced by proteotoxic stress, as well as by perturbation to purine metabolism. Independently of these triggers, they are also regulated by a pair of antagonistic paralogs called PALS-22 and PALS-25. Upregulation of IPR genes leads to increased resistance to pathogen infection
What is the function of IPR genes? IPR genes are enriched for those that encode components of multi-subunit cullin-ring ubiquitin ligases (Bakowski et al. 2014a). Ubiquitin localizes to about 4–7% of N. parisii sporoplasms in the N2 host, and this localization was shown to be reduced in half by RNAi knockdown of the cullin cul 6, a core component of cullin-ring ubiquitin ligases. Almost no ubiquitin localizes to parasite cells at later stages of N. parisii development, such as multinucleate meronts and spores. However, large clusters of conjugated ubiquitin appear throughout C. elegans intestinal cells, while N. parisii meronts are replicating and differentiating into spores, including sites distant from the parasite. Such ubiquitin clusters are a hallmark of perturbations in protein homeostasis (proteostasis), indicating that intracellular growth of microsporidia impairs proteostasis. Interestingly, IPR genes can also be induced by perturbations to proteostasis, such as genetic or pharmacological inhibition of the proteasome. RNA-seq analysis demonstrated that proteasome blockade induces almost all IPR genes (Reddy et al. 2019). Importantly, IPR genes are distinct from genes regulated by other stress response pathways, such as the HSF-1/heat shock response and the SKN-1/proteotoxic stress response, indicating the IPR constitutes a novel response pathway (Reddy et al. 2017, 2019).
In addition to ubiquitin ligase components, IPR genes include the pals genes, which are defined by a loosely conserved pals protein signature (Leyva-Diaz et al. 2017). The biochemical functions of pals genes are unknown, but pals stands for protein containing ALS2CR12 signature, which is found in the single pals gene in humans called ALS2CR12, of unknown function. There is also a single pals gene in mouse and none found in Drosophila melanogaster. The pals gene family expanded in the C. elegans genome to over 39 members, and these genes are found in clusters in the genome (Leyva-Diaz et al. 2017). 26 pals genes are IPR genes in that they are upregulated by both N. parisii and viral infection, and phylogenetic analysis indicates sequence similarity among upregulated genes (Fig. 5.3). One of these genes is pals-5, which is highly induced in the intestine upon infection and is used as a robust readout for the IPR through the use of a pals-5p::GFP reporter (Bakowski et al. 2014a).
Fig. 5.3.
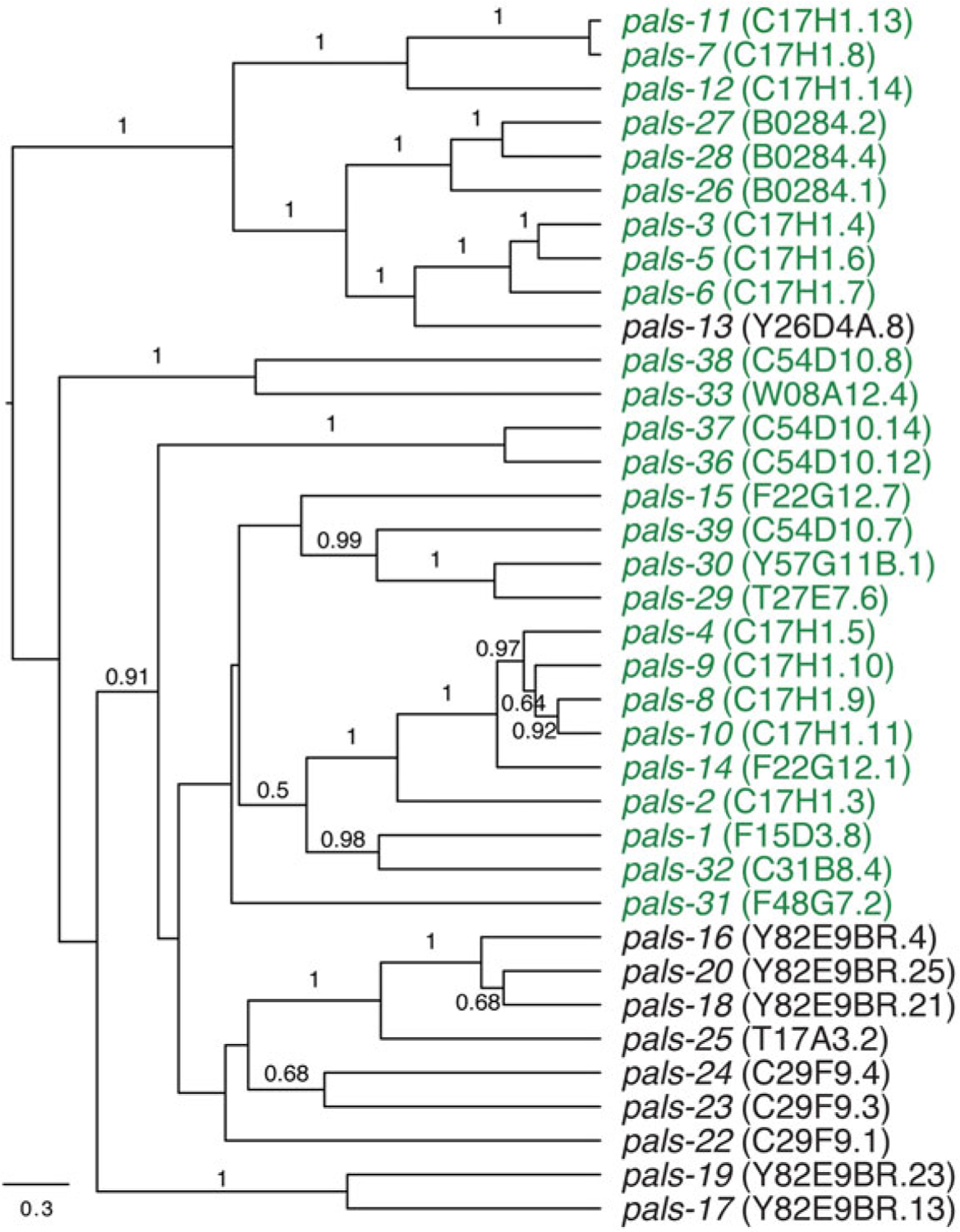
Summary of pals genes. Phylogenetic tree of sequence relationships of pals genes. Green text indicates genes that are upregulated by both N. parisii and Orsay virus infection. Adapted with permission from Reddy et al. 2017
Several forward genetic screens monitoring pals-5p::GFP expression have identified C. elegans regulators of the IPR and have provided insight into the physiological function of this pathogen response. The first regulator of IPR gene expression identified in C. elegans is called pals-22, which is a negative regulator found through a genetic screen for mutants with constitutively high pals-5p::GFP expression (Reddy et al. 2017). Expression of pals-22 is not regulated by infection; rather it acts as a negative regulator of induced pals and all other IPR genes. pals-22 mutants have several phenotypes, including (1) increased resistance to natural pathogens of the intestine such as N. parisii and the Orsay virus; (2) increased resistance to the natural oomycete pathogen Myzocytiopsis humicola, a eukaryotic pathogen that infects the epidermis; (3) increased transgene silencing; (4) increased susceptibility to Pseudomonas aeruginosa bacterial infection; (5) shortened life span; and (6) increased tolerance of proteotoxic stress (Fasseas et al. 2021; Leyva-Diaz et al. 2017; Reddy et al. 2017; Reddy et al. 2019). It is this last phenotype that has been characterized in the most detail, using the phenotype of thermotolerance as a readout. Here, a combination of genetics and biochemistry demonstrated that the increased thermotolerance in pals-22 mutants is dependent on cul-6, acting together with other components in a multi-subunit ubiquitin ligase complex (Panek et al. 2020; Reddy et al. 2017). The target(s) of this ubiquitin ligase remain to be defined, but their identification may provide novel insights into maintenance of proteostasis, especially during intracellular infection.
To better understand how pals-22 regulates the IPR and defense against natural pathogens, suppressor screens were performed in a pals-22 mutant background to find mutants with loss of constitutive pals-5p::GFP expression. These screens identified eight alleles in the gene pals-25, which is immediately downstream of pals-22 in the genome, and is transcribed together with pals-22 as part of an operon (Reddy et al. 2019). Like pals-22, pals-25 expression is not induced by intracellular infection. All pals-22 mutant phenotypes, including increased IPR gene expression, increased thermotolerance, and altered immunity, as well as reduced fitness in other contexts, are reversed to wild-type levels by mutations in pals-25. Because pals-22 and pals-25 direct opposite phenotypes and are in the same gene family, they are termed “antagonistic paralogs,” with pals-22 acting as a negative regulator of pals-25, which is a downstream activator of the IPR. As PALS-22 and PALS-25 proteins are not enriched in the nucleus, they do not appear to be transcription factors, and it is not known how they regulate IPR gene transcription. By determining the overlap between genes upregulated by N. parisii infection, those upregulated in pals-22 mutants, and also reversed back to wild-type levels in pals-22 pals-25 double mutants, the IPR has been refined to include approximately 80 genes, including induced pals genes. Overall, these results show that pals-25 is a global activator of pals-5 and other IPR gene expression, but it is still unclear which induced PALS protein or other IPR proteins function to restrict microsporidia growth downstream of pals-22/25. Answering this question could provide better insight into how C. elegans controls intracellular pathogens in the intestine.
Genome analysis of 330 wild C. elegans strains suggests that there may be balancing selection acting at the pals-22 and pals-25 locus, with distinct alleles being maintained in C. elegans populations in the wild. Interestingly, the CB4856 Hawaiian C. elegans strain has predicted loss-of-function mutations in both pals-22 and pals-25 (Thompson et al. 2015). There are no obvious orthologs of pals-22 and pals-25 in the 27 other nematode genomes examined, including C. inopinata, the recently described sister species of C. elegans (Leyva-Diaz et al. 2017; Reddy et al. 2019). The pals gene family is smaller in other Caenorhabditis species compared to C. elegans, with eight pals genes in C. briggsae, eight in C. brenneri, and 18 in C. remanei (Leyva-Diaz et al. 2017). It is difficult to confidently assess orthology among pals genes in different Caenorhabditis species, because of the high level of sequence divergence within this gene family. For example, C. elegans PALS-22 and PALS-25 proteins only have 19.4% identity on the amino acid level. However, the induction of pals genes by intracellular infection appears to be conserved between C. elegans and C. briggsae (Chen et al. 2017). While C. briggsae cannot be infected by the Orsay virus, it can be infected by the related Santeuil virus, found naturally infecting C. briggsae (Felix et al. 2011). Comparison of the genes expressed upon Orsay virus infection in C. elegans and those expressed upon Santeuil virus infection in C. briggsae identified 58 homologous genes differentially regulated by both viruses. These genes included all 8 C. briggsae pals genes, with one of them only modestly induced. Taken together, these results suggest that the IPR may be an evolutionarily conserved immune response to intracellular pathogens in Caenorhabditis nematodes.
While pals-22 and pals-25 control IPR gene expression and rewire C. elegans physiology, they are dispensable, as their deletion from the genome results in animals with wild-type phenotypes. For example, pals-22 pals-25 double mutants still upregulate IPR gene expression upon exposure to all known triggers of the IPR, including N. parisii infection, virus infection, proteotoxic stress, and chronic heat stress, among other triggers (Reddy et al. 2019). Thus, pals-22 and pals-25 appear to have been inserted in the C. elegans genome as an ON/OFF switch for the IPR, but act in parallel to other factors that mediate induction (Fig. 5.2). Induction of the IPR is best understood for viral infection, which is sensed by DRH-1, a C. elegans homolog of mammalian RIG-I-like receptors that sense double-stranded viral RNA and other viral replication products. DRH-1 is required for induction of IPR genes upon viral infection, but not upon other triggers (Sowa et al. 2020). How microsporidia infection and other triggers induce the IPR is unknown.
While there appear to be many independent upstream triggers of the IPR, there does appear to be a common downstream regulator through which these triggers induce gene expression. The predicted bZIP transcription factor ZIP-1 was identified in two independent RNAi-based screens as being required for inducing pals-5p:: GFP expression, and it appears to be important for induction of this reporter upon all known IPR triggers (Lažetić et al. 2022). Interestingly, RNA-seq analysis demonstrated that ZIP-1 is required for induction of less than half of the IPR genes. This finding indicates there are other transcription factors required for regulating these zip-1-independent IPR genes. Despite only regulating some IPR genes, zip-1 is required to promote resistance to Orsay virus and N. parisii infection and is particularly important in a mutant background where IPR genes are constitutively expressed. These results indicate that ZIP-1 controls IPR genes that protect against intracellular infection.
Recently, forward genetic screens identified pnp-1 as another negative regulator of the IPR, functioning in parallel to pals-22 and upstream of zip-1 (Tecle et al. 2021). pnp-1 encodes a purine nucleoside phosphorylase (PNP), which is an enzyme that acts in the purine salvage pathway to recycle guanine and adenine nucleotides. RNA-seq profiling of pnp-1 mutants demonstrated that they have constitutive upregulation of almost all IPR genes, including the induced pals genes. In keeping with this constitutive upregulation of IPR genes, pnp-1 mutants are resistant to Orsay virus and N. parisii infection, and zip-1 is important for this resistance. Restoring expression of pnp-1 in C. elegans intestinal epithelial cells rescues pnp-1 mutant phenotypes, including IPR gene expression and resistance against N. parisii, indicating that purine metabolism in the intestine regulates the IPR. It would be interesting to investigate if loss of pnp-1 in C. briggsae results in resistance to Nematocida and/or Santeuil virus infection in this host.
The phenotypes of pnp-1 highlight the relevance of purine metabolism to obligate intracellular infections like those caused by microsporidia. Purine metabolism pathways are conserved across all domains of life and consist of the energy-costly de novo pathway and the far less energy-costly salvage pathway. PNPs function only in the purine salvage pathway and are evolutionarily conserved from bacteria to man. In contrast to the lack of pals-22 sequence conservation, pnp-1 sequences are highly conserved, for example, 95% homology between C. elegans and C. briggsae. PNPs convert the purine nucleoside inosine into the purine base hypoxanthine, which is then submitted to the action of various enzymes to generate purine nucleotides. Metabolomics analysis confirmed that pnp-1 mutants have increased levels of inosine and decreased levels of hypoxanthine, as expected if pnp-1 functions as a PNP in C. elegans. Considering that microsporidia are completely dependent on the host for nucleotides, it seems plausible that perturbations in purine metabolism in the host could impair microsporidia growth. Moreover, buildup of purine metabolite precursors (such as inosine) or the absence of others (such as hypoxanthine) due to infection could be sensed by the host which then deploys defense mechanisms against infection, such as the IPR. Interestingly, human mutations in purine salvage enzymes like PNP and adenosine deaminase result in immunodeficiency due to T cell dysfunction, but little is known about the role of PNP and other purine salvage enzymes in epithelial cells, which are the common sites of virus and microsporidia infection, and where pnp-1 functions in C. elegans. Perhaps purine metabolism enzyme mutations in humans provide an evolutionary advantage in the context of viral or microsporidia infection in epithelial cells, which are often the first cells to be infected.
The work with pals-22 and pnp-1 demonstrates that IPR induction promotes resistance within a generation. Recent work indicates that N. parisii infection and IPR induction can provide an immunity benefit to the progeny of infected animals (Willis et al. 2021). Thus, immunity against microsporidia can be maternally inherited in C. elegans. N. parisii infection itself is not required for inheritance of resistance, only IPR activation in the parental generation. Mothers with IPR activation, such as pals-22 mutants, have progeny without IPR gene activation, but nonetheless these progeny are resistant to N. parisii infection. Several other IPR activators confer an immune benefit against N. parisii to the next generation, including viral infection, and mutations in another negative regulator of IPR gene expression, lin-35, which is a homolog of the cell cycle regulator retinoblastoma. This inherited immunity does come at a fitness cost however; progeny from infected parents are smaller, less fecund and display increased susceptibility to other types of stresses. Inherited resistance to N. parisii infection is observed in C. briggsae, indicating that this is an evolutionary conserved trait (Burton et al. 2021). It would be interesting to determine whether inherited immunity can be triggered by infection with other Nematocida species or by loss of pnp-1, and if zip-1 is required for this process. It is not well understood how IPR activation leads to increased resistance either within a generation or across a generation, but as more is uncovered, it will be interesting to compare whether similar or different mechanisms are at play.
5.7. Perspectives and Future Directions
The power of the C. elegans host system has facilitated progress on several fronts of microsporidia research. For example, the ease of imaging the transparent body plan of C. elegans has enabled visualization of Nematocida species fusing host cells together during replication to create syncytial structures that facilitate spread of this pathogen. These studies were notable because they described a polar tube-independent invasion mechanism of microsporidia. The ease of isolating wild-caught C. elegans and related nematodes has provided a growing collection of natural microsporidian pathogens of nematodes, which offer new models for studying important questions about microsporidia pathogenesis, such as what determines their host and tissue tropism. Localized proteomics in C. elegans intestinal cells highlighted how species-specific gene families from microsporidia encode proteins that directly interface with host tissue during the host/pathogen battle, which may drive the expansion and diversification of these families. Similarly, forward genetics has highlighted the expansion and diversification of species-specific gene families involved in host defense against these pathogens (Lazetic and Troemel 2020). Genetic and transcriptomic studies have identified the IPR, a shared immune/stress response pathway induced by natural microsporidian and viral infections. The IPR is regulated by metabolism of purine nucleotides, which are crucial factors stolen by both microsporidia and viruses to enable their replication. The short life cycle of C. elegans facilitated the discovery that activation of the IPR can confer an immune benefit to the next generation. However, many questions remain. For example, it is unknown how C. elegans senses microsporidia to trigger the IPR, how the IPR confers resistance to infection, and whether there is an analogous system in mammals, which lack obvious sequence orthologs of IPR regulators pals-22/25 and many other IPR genes. In addition, the mechanisms by which Nematocida species fuse C. elegans organs into syncytia are unknown. Further elucidation of these findings and investigation of their potential roles in mammalian hosts will be exciting areas for future inquiry.
Despite such progress, research of microsporidia infections in C. elegans is hampered by the same problem that all microsporidia researchers face: manipulation of the microsporidia genome (Reinke and Troemel 2015). The lack of genetics or DNA transformation techniques makes it challenging to determine which Nematocida factors trigger the various restructuring events described above, cause disease, or activate the IPR. Heterologous expression of pathogen proteins provides one avenue to explore these questions. Here, the host-exposed Nematocida proteins identified through localized proteomics, including proteins from the species-specific expanded gene families, provide attractive candidates to explore (Reinke et al. 2017). Without genetic manipulation of microsporidia, we are like detectives who meticulously investigate a crime scene but never manage to specifically identify the perpetrators. Although genetics and transformation techniques are currently not available in microsporidia, the development of highly sensitive reporter systems such as nanoluciferase will provide useful tools for developing these techniques in the future (Sfarcic et al. 2019). Together with CRISPR-Cas9-mediated gene editing, novel genetic tools will help define the microsporidia factors that enable its elaborate intracellular life and the host response to these ubiquitous, fascinating parasites.
Acknowledgments
Thanks to Michalis Barkoulas, Lakshmi Batachari, Crystal Chhan, Marie-Anne Felix, Vladimir Lazetic, and David Wang for helpful comments on the manuscript.
Funding
This work was supported by NIH under R01 AG052622, GM114139 to ERT, and NIGMS/NIH award K12GM068524 to ET.
Footnotes
Conflict of Interest The authors declare that there is no conflict of interest.
Ethical Approval The chapter is a review of previously published accounts; as such, no animal or human studies were performed.
References
- Bakowski MA et al. (2014a) Ubiquitin-mediated response to microsporidia and virus infection in C. elegans. PLoS Pathog 10:e1004200. 10.1371/journal.ppat.1004200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakowski MA, Priest M, Young S, Cuomo CA, Troemel ER (2014b) Genome sequence of the microsporidian species Nematocida sp1 strain ERTm6 (ATCC PRA-372). Genome Announc 2: e00905–14. 10.1128/genomeA.00905-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla KM, Andersen EC, Kruglyak L, Troemel ER (2015) A wild C. elegans strain has enhanced epithelial immunity to a natural microsporidian parasite. PLoS Pathog 11:e1004583. 10.1371/journal.ppat.1004583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla KM, Luallen RJ, Bakowski MA, Troemel ER (2016) Cell-to-cell spread of microsporidia causes Caenorhabditis elegans organs to form syncytia. Nat Microbiol 1:16144. 10.1038/nmicrobiol.2016.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla KM, Lazetic V, Troemel ER (2019) Natural variation in the roles of C. elegans autophagy components during microsporidia infection. PLoS One 14:e0216011. 10.1371/journal.pone.0216011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becnel JJ, White SE, Shapiro AM (2005) Review of microsporidia-mosquito relationships: from the simple to the complex. Folia Parasitol (Praha) 52:41–50 [PubMed] [Google Scholar]
- Botts MR, Cohen LB, Probert CS, Wu F, Troemel ER (2016) Microsporidia intracellular development relies on myc interaction network transcription factors in the host. G3 (Bethesda) 6:2707–2716. 10.1534/g3.116.029983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton NO et al. (2021) Intergenerational adaptations to stress are evolutionarily conserved, stress-specific, and have deleterious trade-offs. bioRxiv. 10.1101/2021.05.07.443118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Marcet-Houben M, Gabaldon T (2012) Phylogenomics supports microsporidia as the earliest diverging clade of sequenced fungi. BMC Biol 10:47. 10.1186/1741-7007-10-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Franz CJ, Jiang H, Jiang Y, Wang D (2017) An evolutionarily conserved transcriptional response to viral infection in Caenorhabditis nematodes. BMC Genomics 18:303. 10.1186/s12864-017-3689-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi N, Pombert JF, Farinelli L, Didier ES, Keeling PJ (2010) The complete sequence of the smallest known nuclear genome from the microsporidian Encephalitozoon intestinalis. Nat Commun 1:77. 10.1038/ncomms1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuomo CA et al. (2012) Microsporidian genome analysis reveals evolutionary strategies for obligate intracellular growth. Genome Res 22:2478–2488. 10.1101/gr.142802.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby C, Cosma CL, Thomas JH, Manoil C (1999) Lethal paralysis of Caenorhabditis elegans by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 96:15202–15207. 10.1073/pnas.96.26.15202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimov I, Maduro MF (2019) The C. elegans intestine: organogenesis, digestion, and physiology cell tissue res, p 377:383–396. 10.1007/s00441-019-03036-4 [DOI] [PubMed] [Google Scholar]
- El Jarkass HT, Reinke AW (2020) The ins and outs of host-microsporidia interactions during invasion, proliferation and exit. Cell Microbiol 22:e13247. 10.1111/cmi.13247 [DOI] [PubMed] [Google Scholar]
- El Jarkass HT, Mok C, Schertzberg MR, Fraser AG, Troemel ER, Reinke AW (2022, Jan 7) An intestinally secreted host factor promotes microsporidia invasion of C. elegans. Elife 11:e72458. 10.7554/eLife.72458. Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes KA, Szumowski SC, Troemel ER (2011) Non-lytic, actin-based exit of intracellular parasites from C. elegans intestinal cells. PLoS Pathog 7:e1002227. 10.1371/journal.ppat.1002227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasseas MK, Grover M, Drury F, Essmann CL, Kaulich E, Schafer WR, Barkoulas M (2021) Chemosensory Neurons Modulate the Response to Oomycete Recognition in Caenorhabditis elegans Cell Rep 34:108604. 10.1016/j.celrep.2020.108604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix MA et al. (2011) Natural and experimental infection of Caenorhabditis nematodes by novel viruses related to nodaviruses. PLoS Biol 9:e1000586. 10.1371/journal.pbio.1000586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag KL, James TY, Pombert JF, Larsson R, Schaer TM, Refardt D, Ebert D (2014) Evolution of a morphological novelty occurred before genome compaction in a lineage of extreme parasites. Proc Natl Acad Sci U S A 111:15480–15485. 10.1073/pnas.1410442111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Weiss LM (2017) Microsporidia: obligate intracellular pathogens within the fungal Kingdom. Microbiol Spectr 5:97–113. 10.1128/microbiolspec.FUNK-0018-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, McFadden GI (1998) Origins of microsporidia. Trends Microbiol 6:19–23. 10.1016/S0966-842X(97)01185-2 [DOI] [PubMed] [Google Scholar]
- Kiontke KC, Felix MA, Ailion M, Rockman MV, Braendle C, Penigault JB, Fitch DH (2011) A phylogeny and molecular barcodes for Caenorhabditis, with numerous new species from rotting fruits. BMC Evol Biol 11:339. 10.1186/1471-2148-11-339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CJ, Hansen M, Troemel E (2018) Autophagy and innate immunity: insights from invertebrate model organisms. Autophagy 14:233–242. 10.1080/15548627.2017.1389824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazetic V, Troemel ER (2020) Conservation lost: host-pathogen battles drive diversification and expansion of gene families. FEBS J. 10.1111/febs.15627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lažetić V, Wu F, Cohen LB et al. (2022) The transcription factor ZIP-1 promotes resistance to intracellular infection in Caenorhabditis elegans. Nat Commun 13:17. 10.1038/s41467-021-27621-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D et al. (2021) Balancing selection maintains hyper-divergent haplotypes in Caenorhabditis elegans Nat Ecol Evol 5:794–807. 10.1038/s41559-021-01435-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyva-Diaz E, Stefanakis N, Carrera I, Glenwinkel L, Wang G, Driscoll M, Hobert O (2017) Silencing of repetitive DNA is controlled by a member of an unusual Caenorhabditis elegans gene family. Genetics 207:529–545. 10.1534/genetics.117.300134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luallen RJ, Reinke AW, Tong L, Botts MR, Felix MA, Troemel ER (2016) Discovery of a natural microsporidian pathogen with a broad tissue tropism in Caenorhabditis elegans. PLoS Pathog 12:e1005724. 10.1371/journal.ppat.1005724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan-Miklos S, Tan MW, Rahme LG, Ausubel FM (1999) Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell 96:47–56. 10.1016/s0092-8674(00)80958-7 [DOI] [PubMed] [Google Scholar]
- Panek J, Gang SS, Reddy KC, Luallen RJ, Fulzele A, Bennett EJ, Troemel ER (2020) A cullin-RING ubiquitin ligase promotes thermotolerance as part of the intracellular pathogen response in Caenorhabditis elegans. Proc Natl Acad Sci U S A 117:7950–7960. 10.1073/pnas.1918417117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol N et al. (2001) A reverse genetic analysis of components of the toll signaling pathway in Caenorhabditis elegans. Curr Biol 11:809–821. 10.1016/s0960-9822(01)00241-x [DOI] [PubMed] [Google Scholar]
- Reddy KC et al. (2017) An intracellular pathogen response pathway promotes Proteostasis in C. elegans. Curr Biol 27:3544–3553. 10.1016/j.cub.2017.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy KC et al. (2019) Antagonistic paralogs control a switch between growth and pathogen resistance in C. elegans. PLoS Pathog 15:e1007528. 10.1371/journal.ppat.1007528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke AW, Troemel ER (2015) The development of genetic modification techniques in intracellular parasites and potential applications to microsporidia. PLoS Pathog 11:e1005283. 10.1371/journal.ppat.1005283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke AW, Balla KM, Bennett EJ, Troemel ER (2017) Identification of microsporidia host-exposed proteins reveals a repertoire of rapidly evolving proteins. Nat Commun 8:14023. 10.1038/ncomms14023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulenburg H, Felix MA (2017) The natural biotic environment of Caenorhabditis elegans. Genetics 206:55–86. 10.1534/genetics.116.195511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfarcic I, Bui T, Daniels EC, Troemel ER (2019) Nanoluciferase-based method for detecting gene expression in Caenorhabditis elegans. Genetics 213:1197–1207. 10.1534/genetics.119.302655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J (2021) Harnessing the power of genetics: fast forward genetics in Caenorhabditis elegans. Mol Gen Genomics 296:1–20. 10.1007/s00438-020-01721-6 [DOI] [PubMed] [Google Scholar]
- Sowa JN, Jiang H, Somasundaram L, Tecle E, Xu G, Wang D, Troemel ER (2020) The Caenorhabditis elegans RIG-I homolog DRH-1 mediates the intracellular pathogen response upon viral infection. J Virol 94. 10.1128/JVI.01173-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens L et al. (2019) Comparative genomics of 10 new Caenorhabditis species. Evol Lett 3:217–236. 10.1002/evl3.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szumowski SC, Botts MR, Popovich JJ, Smelkinson MG, Troemel ER (2014) The small GTPase RAB-11 directs polarized exocytosis of the intracellular pathogen N. parisii for fecal-oral transmission from C. elegans. Proc Natl Acad Sci U S A 111:8215–8220. 10.1073/pnas.1400696111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szumowski SC, Estes KA, Popovich JJ, Botts MR, Sek G, Troemel ER (2016) Small GTPases promote actin coat formation on microsporidian pathogens traversing the apical membrane of Caenorhabditis elegans intestinal cells. Cell Microbiol 18:30–45. 10.1111/cmi.12481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecle E, Chhan CB, Franklin L, Underwood RS, Hanna-Rose W, Troemel ER (2021) The purine nucleoside phosphorylase pnp-1 regulates epithelial cell resistance to infection in C. elegans. PLoS Pathog 17:e1009350. 10.1371/journal.ppat.1009350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson OA et al. (2015) Remarkably divergent regions punctuate the genome assembly of the Caenorhabditis elegans Hawaiian strain CB4856 genetics, p 200:975–989. 10.1534/genetics.115.175950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel ER (2016) Host-microsporidia interactions in caenorhabditis elegans, a model nematode host. Microbiol Spectr 4(5). 10.1128/microbiolspec.FUNK-0003-2016 [DOI] [PubMed] [Google Scholar]
- Troemel ER, Felix MA, Whiteman NK, Barriere A, Ausubel FM (2008) Microsporidia are natural intracellular parasites of the nematode Caenorhabditis elegans. PLoS Biol 6:2736–2752. 10.1371/journal.pbio.0060309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis AR, Zhao W, Sukhdeo R, Wadi L, El Jarkass HT, Claycomb JM, Reinke AW (2021) A parental transcriptional response to microsporidia infection induces inherited immunity in offspring. Sci Adv 7(19):eabf3114. 10.1126/sciadv.abf3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Sachse M, Prevost MC, Luallen RJ, Troemel ER, Felix MA (2016) A large collection of novel nematode-infecting microsporidia and their diverse interactions with Caenorhabditis elegans and other related nematodes. PLoS Pathog 12:e1006093. 10.1371/journal.ppat.1006093 [DOI] [PMC free article] [PubMed] [Google Scholar]