

Abstract
Accumulating evidence indicates that epilepsy has a higher risk of inducing memory impairment and dementia. However, the underlying onset mechanism remains unclear. Here, we found that mice with spontaneous epilepsy induced by endothelial CDK5 deficiency exhibited hippocampal‐dependent memory impairment at 6 months of age, but not at 2 months of age. Moreover, the persistent epileptic seizures induce aberrant changes in phosphorylation of CaMKII protein in the hippocampus of spontaneous epileptic mice. Using genome‐wide RNA sequencing and intergenic interaction analysis of STRING, we found that in addition to epilepsy‐related genes, there are changes in synaptic organization pathway node genes, such as Bdnf and Grin1. The synapse‐related proteins by Western blot analysis, such as NMDA receptors (NR1 and NR2B), PSD95, and the phosphorylation of synapsin1, are progressively decreased during epileptic seizures in Cdh5‐CreERT2;CDK5f/f mice. Notably, we found that valproate (VPA) and phenytoin (PHT) augment mRNA expression and protein levels of synapse‐related genes and ameliorate memory impairment in Cdh5‐CreERT2;CDK5f/f mice. Our study elucidates a potential mechanism of memory deficits in epilepsy, and pharmacological reversal of synaptic pathology targeting might provide a new therapeutic intervention for epileptic memory deficits.
Keywords: endothelial CDK5, mechanism, memory impairment, spontaneous epilepsy, synapse‐related proteins
The mechanism of cognitive deficits in epilepsy remains unclear. Here, we found that persistent epileptic seizures induce cognitive deficits by cognitional behavior analysis. Accordingly, changes of synapse‐related gene (Phka1, Btaf1, Magt1, and Grin1) and aberrant alternation of NMDA receptors, PSD95, phosphorylation of CaMKII were found in the hippocampus of spontaneous epilepsy mice. More importantly, antiepileptic drugs treatment increased synaptic protein expression and partly ameliorated cognitive dysfunction.
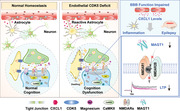
1. INTRODUCTION
Epilepsy is one of the most common and disabling neurological disorders. 1 , 2 Decreased memory has long been recognized as an epileptic comorbidity, which can occur in children with epilepsy and is more common in clinically refractory epilepsy. 3 , 4 Elderly individuals with new‐onset epilepsy are also at a higher risk of developing dementia, especially those with cerebrovascular diseases. 5 , 6 It has been reported that cognitive impairment in epilepsy is usually related to potential etiology, onset age, location and frequency, as well as antiepileptic drugs. Nevertheless, less is known about the underlying mechanism of cognitive impairment during epilepsy.
As is widely acknowledged, the hippocampus stands as an essential region responsible for the formation and consolidation of memory, which is an important cognitive process. 7 , 8 , 9 The decreasing activation of mossy cells, which are glutamatergic cell populations in the hilum of the hippocampal dentate gyrus, is sufficient to hamper spatial contextual coding and induce memory deficits in chronically epileptic mice. 10 Normal synaptic transmission plays a key role in long/short‐term memory formation, 11 , 12 and synaptic alterations are reported in epileptic patients and mouse model. 13 , 14 However, whether these changes are a cause or a consequence of epilepsy remains a mystery. Moreover, levetiracetam, as an antiepileptic drug, was reported to have a beneficial effect on cognitive networks in drug‐resistant temporal lobe epileptic patients by impeding abnormal network activation. 15 However, the mechanism by which epilepsy contributes to hippocampal memory dysfunction remains unclear.
Recently, our previous studies have shown that brain endothelial CDK5 deletion induces progressive reactive astrogliosis and results in the development of spontaneous epilepsy in Cdh5‐CreERT2;CDK5f/f mice. 16 Spontaneous epileptic mice show decreased astrocytic GLT1‐mediated currents through endothelial chemokine (C‐X‐C motif), ligand 1 (CXCL1), and its receptor chemokine receptor 2 (CXCR2) in astrocytes. In the present study, LoxP/Cre strategy was used to generate endothelial‐specific CDK5 knockout mice, and our data indicated that the mice exhibited spontaneous epilepsy accompanied by memory impairment at 6 months old. Our transcriptomic analysis indicated that synapse‐related genes and synapse‐related proteins (PSD95, NR1, and NR2B subunits of NMDA receptors) were decreased in spontaneously epileptic mice. Remarkably, the decreased expression of the NR1 and NR2B subunits of NMDA receptors was reversed by VPA and PHT co‐administration, which was associated with coordinated alleviation of memory impairment in spontaneous epileptic mice. In summary, potent pharmacological interference and synaptic function improvement may help prevent memory impairment during vasogenic epilepsy, potentially providing an important strategy for the treatment of this devastating disease.
2. RESULTS
2.1. The risk of cognitive dysfunction or dementia in patients with epilepsy
To clarify the correlation of epilepsy and memory dysfunction or dementia, a summary of all the meta‐analyses reports from epilepsy is provided in (Figure 1A). For the correlation of epilepsy and memory dysfunction or dementia, the meta‐analysis of the five case–control studies 17 , 18 , 19 , 20 , 21 showed that the relative risk (RR) of developing memory dysfunction or dementia was 3.99 times higher (95% CI: 3.36–4.74) in epileptic cohort compared with reference cohort without epilepsy (Figure 1B). The above analysis shows that there is a considerable intersection between epilepsy and cognitive dysfunction, adding that a better understanding of the common mechanisms in these disorders may contribute to the improvement of epileptogenesis and dementia.
FIGURE 1.
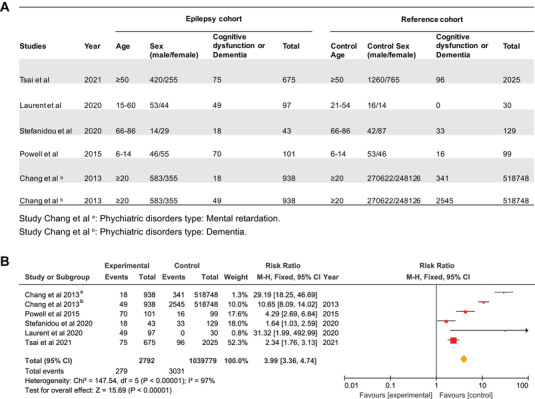
The risk of cognitive dysfunction or dementia in patients with epilepsy. (A) Characteristics of the analytic sample. (B) Results from meta‐analyses of observational and trials about the impact of associations between epilepsy and memory dysfunction or dementia analyzed through RevMan 5 software
2.2. Endothelial CDK5 conditional deletion induces spontaneous epilepsy
To eliminate the effect of gene deficit on development, Cdh5‐CreERT2;CDK5f/f mice were injected intraperitoneally (IP) with tamoxifen at P30 to induce Cre activity in endothelial cells (ECs) to generate an endothelial CDK5‐deficient strain (Figure 2A). EEG recordings for local field potential suggested that epileptic waves were observed in the hippocampal CA1 region of Cdh5‐CreERT2;CDK5f/f mice at 6 months old but not in the cortex of both CKO and control mice (Figure 2B,C). The amplitude, power density, and percentage of total power (%) were significantly higher at the theta band (4–8 Hz) in Cdh5‐CreERT2;CDK5f/f mice than in CDK5f/f mice (Figure 2D–F). Consistent with previous results, these data suggested that endothelial CDK5‐deficit strain induced a spontaneous epileptic phenotype in Cdh5‐CreERT2;CDK5f/f mice.
FIGURE 2.
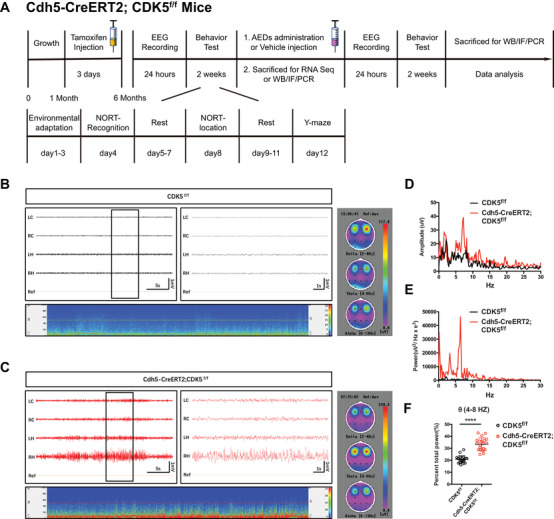
Endothelial CDK5 deficits caused seizure in mice. (A) The schematic diagram of experimental process. (B and C) Representative EEG recording spectrogram of CDK5f/f (B) and Cdh5‐CreERT2;CDK5f/f (C) (LC: left cortex, RC: right cortex, LH: left hippocampus, RH: right hippocampus), oscillation subtypes were distinguished and analyzed, respectively, according to frequency: δ (0–4 Hz), θ (4–8 Hz), and α (8–13 Hz). (D and E) Amplitude (μV) and power (μV2/Hz × e3) curve were analyzed using Labchart 8 software. (F) Quantitative analysis of single subtype: θ (4–8 Hz)/total power ratio. ****p < 0.0001, Student's t‐test. Both error bars indicate the mean value ± SEM
2.3. Progression of epilepsy leads to impaired hippocampal‐dependent memory
To verify the potential concomitant effects of endothelial spontaneous epilepsy, several behavioral tests on memory were performed. We previously reported that Cdh5‐CreERT2;CDK5f/f mice showed an age‐dependent increase in the prevalence and frequency of seizures. Here, we checked memory behavior at 4 weeks (2‐month‐old mice) and 20 weeks (6‐month‐old mice) after tamoxifen treatment. First, in novel object recognition analysis, the decrease in the discrimination index suggested that the ability of mice to recognize either novel objects or novel locations was also impaired at 6 months old but not at 2 months old in Cdh5‐CreERT2;CDK5f/f mice (Figure 3A,B). Second, the CDK5‐deficient mice also exhibited a significant decrease in discrimination index of Y‐maze test at 6 months old, not at 2 months old (Figure 3C). Finally, the levels of phosphorylated CaMKII (Thr286), a critical signaling molecule in hippocampal memory formation, were also reduced in Cdh5‐CreERT2;CDK5f/f mice (Figure 3D,E). Calcineurin (CaN), a Ca2+‐calmodulin‐dependent serine/threonine protein phosphatase, was not changed in Cdh5‐CreERT2;CDK5f/f mice (Figure 3F,G). Collectively, these data suggest that endothelial CDK5 knockout impairs hippocampal‐dependent memory.
FIGURE 3.
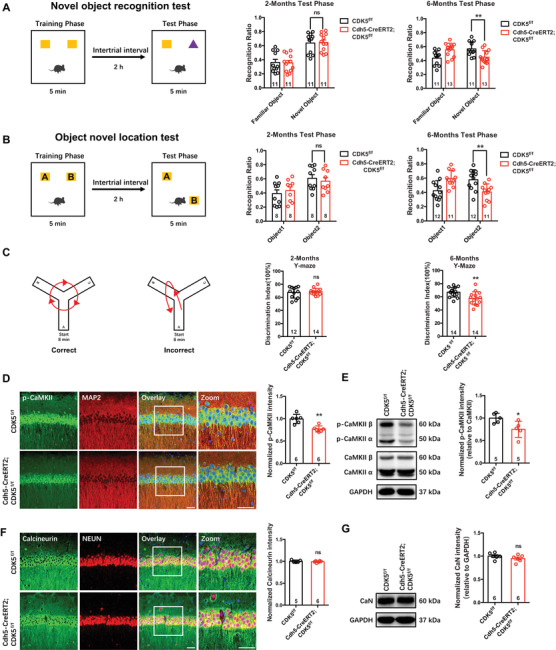
Persistent seizures lead to hippocampus‐dependent memory impairment. (A–C) Hippocampus‐dependent memory behaviors were performed in CDK5f/f and Cdh5‐CreERT2;CDK5f/f mice. The schematic diagram of novel object recognition test and the discrimination index value (A), the schematic diagram of object new location memory and the discrimination index value (B), the schematic diagram of Y‐maze and the discrimination index value (C). (D and E) Representative phospho‐CaMKII (p‐CaMKII, Thr286) (green) staining in CA1 of mice (D, left, counterstained with microtubule‐associated protein 2 [MAP2] [red] and nuclear marker DAPI [blue]) and quantification of normalized p‐CaMKII intensity (D, right, **p < 0.01, Student's t‐test). Representative immunoblotting bands of p‐CaMKII (Thr286), CaMKII, and GAPDH (E, left), and quantitative analyses by densitometry for p‐CaMKII, *p < 0.05 (E, right). (F and G) Representative calcineurin (CaN) (green) staining in CA1 of mice (F, left, counterstained with neuronal marker [NEUN] [red] and nuclear marker DAPI [blue]) and quantification of normalized CaN intensity (F, right, ns: not significant, Student's t‐test). Representative immunoblotting bands of CaN and GAPDH (G, left), and quantitative analyses by densitometry for CaN (G, right). Scale bar: 20 μm, magnified images: 50 μm
2.4. Endothelial CDK5 deficiency causes aberrant changes in synapse‐related genes in the hippocampus
To inspect the molecular mechanism that contributes to memory impairment, we performed digital gene expression by whole‐genome RNA sequencing and genomics analysis on hippocampal tissue samples obtained from Cdh5‐CreERT2;CDK5f/f and CDK5f/f mice. Using gene expression microarrays combined with network analysis, we found 55,573 robustly expressed genes in the transcripts. A total of 1222 differentially expressed genes (DEGs; 577 upregulated genes and 645 downregulated genes) in mutant mice compared to control mice were screened (Figure 4A,B).
FIGURE 4.
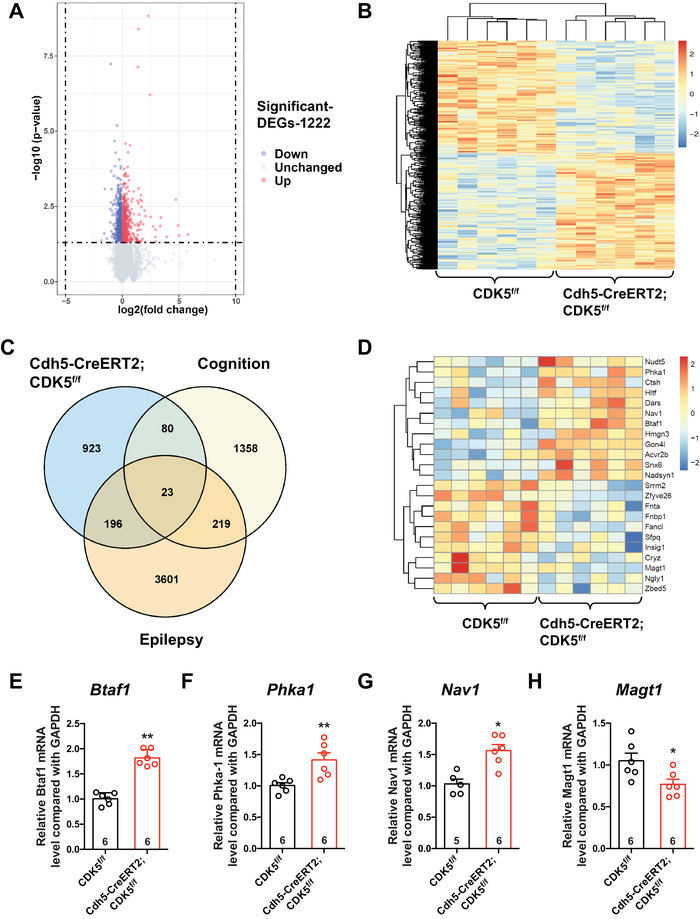
Changes of gene expression in the hippocampus of endothelial CDK5‐deficit mice. (A) Volcano plot of genes expression profiler. (B) Heatmap of 1222 differentially expressed genes. (C) Venn plot representing the number of intersecting genes. (D) Heatmap of 23 intersecting genes. (E–H) The relative mRNA level of indicated genes (Btaf1, Phka1, Nav1, Magt1) in hippocampus of CDK5f/f and Cdh5‐CreERT2;CDK5f/f mice
To explore the correlation between epilepsy and memory dysfunction caused by CDK5 deletion in ECs, transcripts were cross‐analyzed with epilepsy (GSE60772) and memory dysfunction (GSE80312) transcripts. Eventually, we sorted 23 target genes (12 upregulated genes and 11 downregulated genes) from the cross‐analysis data (Figure 4C,D). Next, we used quantitative real‐time PCR (RT‐PCR) to confirm the mRNA levels of 23 DEGs filtered from the three transcripts above. The RT‐PCR results suggested that the mRNA levels of Btaf1, Phka1, and Nav1, which possess a strong correlation with epilepsy, were upregulated significantly (Figure 4E–G). Accordingly, the magnesium ion transporter‐encoding gene Magt1 was observed to have decreased mRNA levels (Figure 4H). Together, the mRNA levels of Btaf1, Phka1, Nav1, and Magt1 were related to epilepsy.
In addition, through the Gene Ontology (GO) database, the differentially expressed genes were classified by cellular component (CC), molecular function (MF), and biological process (BP), and we found that the screened differentially expressed genes were involved in the regulation of neuron‐to‐neuron synapses, postsynaptic specialization, and postsynaptic density (Figure 5A–C). After further enrichment analysis of the synaptic organization pathway by KEGG signaling pathway, 38 differential genes were obtained. Genes involved in the synaptic organization pathway were screened, among which 13 genes were decreased and 25 genes were increased (Figure 5D). Through gene‐to‐gene interaction analysis by STRING, we found that Bdnf 22 , 23 and Grin1 24 , 25 were two important node genes in the synaptic organization pathway (Figure 5E). Grin1 mRNA was downregulated in Cdh5‐CreERT2;CDK5f/f mice, and Bdnf mRNA was upregulated (Figure 5F,G). Therefore, out data suggest that aberrant changes in synapse‐related genes in the hippocampus might contribute to the memory impairment in endothelial CDK5‐deficiency mice.
FIGURE 5.
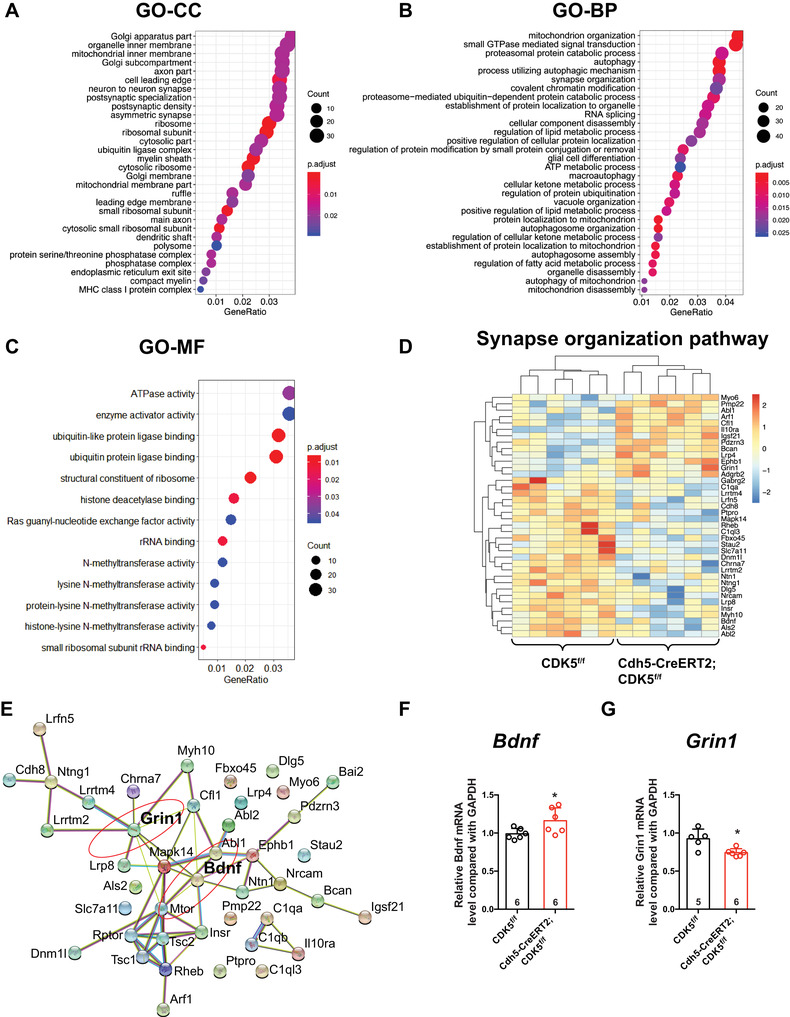
Endothelial CDK5 deficiency causes changes of synapse‐related gene in hippocampus. (A) Cellular component analysis. (B) Biological process analysis. (C) Molecular function analysis. (D) Heatmap of synapse organization pathway genes. (E) Protein–protein interaction (PPI) network of synapse organization pathway genes. (F and G) The relative mRNA level of Grin1 and Bdnf genes of hippocampus in both strains. *p < 0.05, **p < 0.01, Student's t‐test. All error bars indicate the mean value ± SEM
2.5. Expression of synapse‐related proteins decreased in the hippocampus of Cdh5‐CreERT2;CDK5f/f mice
To examine whether structural proteins of neuronal synapses were changed in the hippocampus of Cdh5‐CreERT2;CDK5f/f mice, we analyzed the protein levels of Synapsin1 (a presynaptic marker) and PSD‐95 (a postsynaptic marker in excitatory neurons). The immunofluorescence data showed that the phosphorylation of Synapsin1 and the protein level of PSD95 were significantly decreased (Figure 6A,B). Similar to the immunofluorescence data, the Western blot data showed that the protein level of Synapsin1 had no change, but p‐Synapsin1 significantly decreased in Cdh5‐CreERT2;CDK5f/f mice at 6 months of age (Figure 6D), which suggested that phosphorylation‐dependent regulation of synaptic vesicle clusters was decreased. Moreover, PSD95 and synaptic receptors (NR1, NR2B) were dampened in Cdh5‐CreERT2;CDK5f/f mice (Figure 6D,F,G), and GluR1 and p‐GluR1 showed no changes (Figure 6E). However, immunofluorescence staining data show that the number of hippocampal neurons was not changed in the CA1, DG, or CA3 regions (Figure S1A,B). Synaptic activation is a key link in a chain of molecular and biochemical events involved in memory formation. 11 , 12 Together, the progression of epilepsy‐impeded setup of synaptic proteins is close correlated with memory impairment in endothelial CDK5‐deficiency mice.
FIGURE 6.
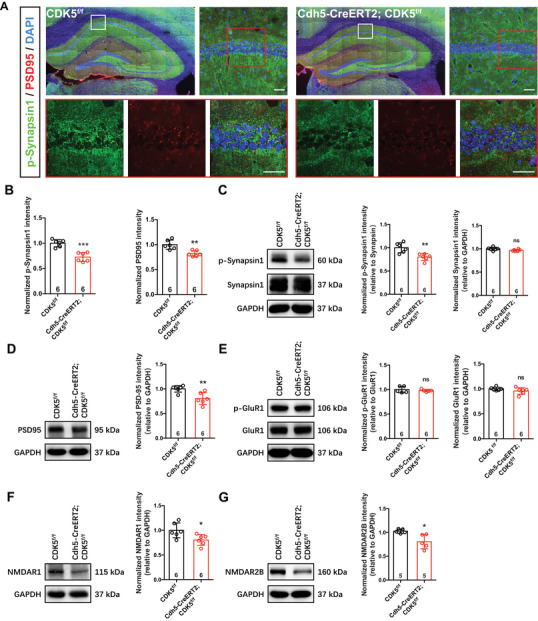
Synapse‐related proteins decreased in hippocampus of Cdh5‐CreERT2;CDK5f/f mice. (A and B) Representative p‐Synapsin1 (green) and PSD95 (red) staining in hippocampus of mice (A, counterstained with DAPI [blue], top right, scale bar: 20 μm; bottom, scale bar: 50 μm) and quantification of normalized p‐Synapsin1 and PSD95 intensity (B) (**p < 0.01, ***p < 0.001, Student's t‐test). (C and D) Representative immunoblots of synapse structure‐related proteins p‐Synapisn1/Synapsin1 (C) and PSD95 (D), and quantitative analyses by densitometry (**p < 0.01, ns: not significant, Student's t‐test). (E–G) Representative immunoblots of synapse receptors NMDARs (NR1 and NR2B) (F and G) and GluR1/phospho‐GluR1 (p‐GluR1) (E), and quantitative analyses by densitometry (*p < 0.05, **p < 0.01, ns: not significant, Student's t‐test, NR2B, n = 5; NR1, GluR1/p‐GluR1). All error bars indicate the mean value ± SEM
2.6. Antiepilepsy drugs alleviated seizures, improved memory, and aligned with normalized synaptic protein levels
VPA is one of the most widely used anticonvulsant and mood‐stabilizing agents for treating epilepsy. 26 PHT is the recommended second‐line intravenous anticonvulsant for the treatment of pediatric convulsive status epilepticus. 27 To explore whether VPA and PHT alleviate spontaneous epileptic seizures, EEG recordings were performed after intraperitoneal injection of VPA for 7 consecutive days. The data showed that VPA or VPA+PHT treatment attenuated epileptic seizures in Cdh5‐CreERT2;CDK5f/f mice, and the power of the theta band (4–8 Hz) was also decreased significantly after VPA or VPA+PHT administration (Figure 7A–D). Although the number of spontaneous recurrent seizures (SRSs) at 24 h and the total time of SRS at 24 h did not change (Figure 7E,F), the average time of SRS changed significantly after VPA or VPA+PHT administration compared to CKO+vehicle, and there was no difference between VPA and VPA+PHT treatment (Figure 7G).
FIGURE 7.
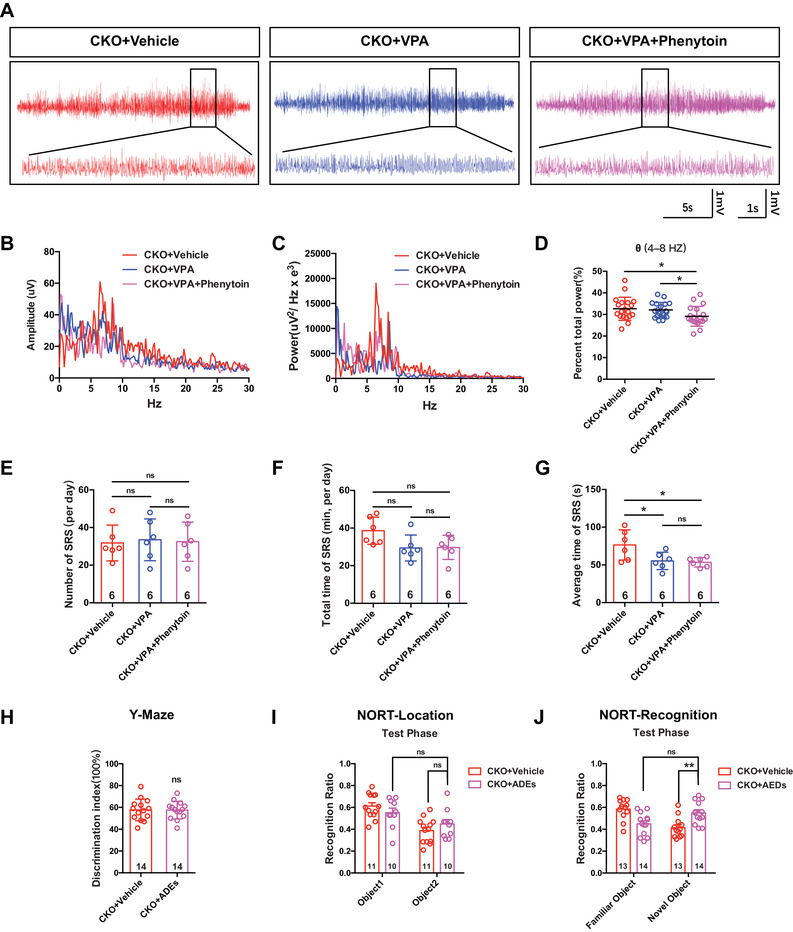
Antiepilepsy drugs alleviated seizure and ameliorate memory. (A) Representative EEG recording spectrogram for 24 h in Cdh5‐CreERT2;CDK5f/f mice administered with valproic acid (VPA, 300 mg/kg/day) or with VPA (300 mg/kg/day) combined phenytoin (PHT, 30 mg/kg/day) for 7 days consecutively. (B and C) Amplitude (B) and power curve (C) were analyzed using Labchart 8 software. (D) Quantitative analysis of single subtype: θ (4–8 Hz)/total power ratio (*p < 0.05, two‐way ANOVA). (E–G) The spontaneous recurrent seizure (SRS) was analyzed using Labchart 8 from aspects of average duration (E), total times per day (F), and total duration per day (G) (E–G, *p < 0.05, ns: not significant, two‐way ANOVA). (H–J) Memory behavioral tests were performed. Y‐maze (H), novel object recognition (I), and object new location recognition (J) (*p < 0.05, **p < 0.01, ns: not significant, (H) Student's t‐test, (I and J) two‐way ANOVA). All error bars indicate the mean value ± SEM
To verify whether antiepileptic drug administration ameliorates memory deficits, we conducted Y‐maze and novel object recognition tests (NORT) after antiepileptic drugs (AEDs, VPA+PHT) treatment. The data showed that AEDs administration improved the NORT object recognition ability (Figure 7J), but had no effects on the Y‐maze or NORT location behaviors (Figure 7H,I). Moreover, the phosphorylation of CaMKII also increased after AEDs treatments, as shown by Western blot and immunofluorescence staining analyses (Figure 8A,B).
FIGURE 8.
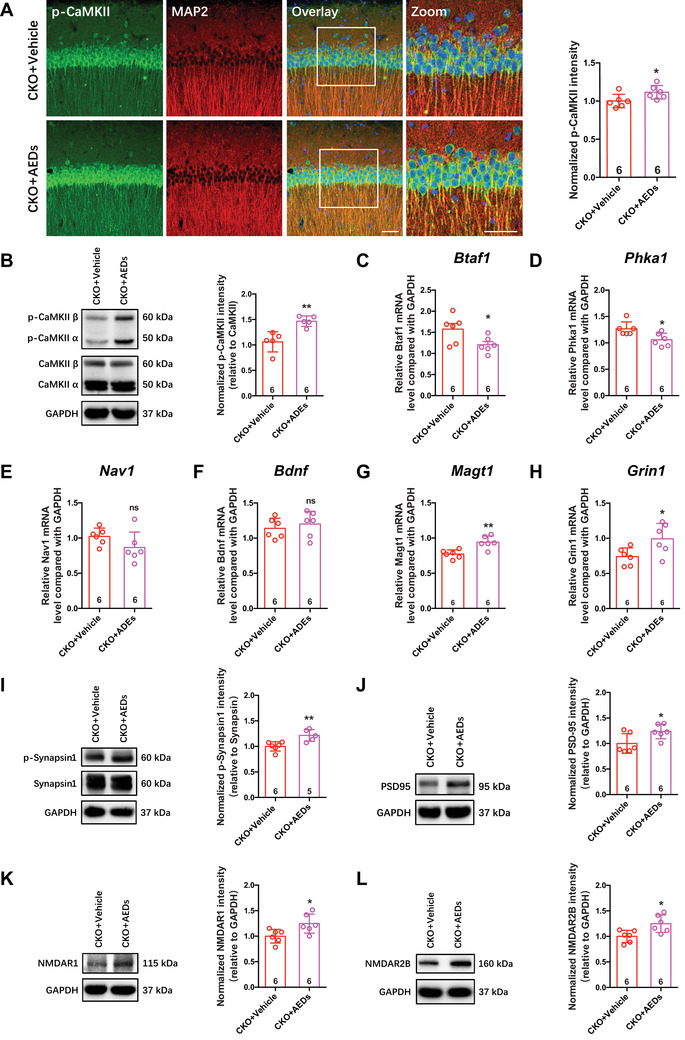
Antiepilepsy drugs increased synaptic proteins level. (A and B) Representative p‐CaMKII (Thr286) (green) staining in CA1 of mice (A, left, counterstained with MAP2 [red] and DAPI [blue], scale bar: 20 μm, magnified images: 50 μm) and quantification of normalized p‐CaMKII intensity (B, right, *p < 0.05, Student's t‐test). Representative immunoblotting bands of p‐CaMKII (Thr286), CaMKII and GAPDH, and quantitative analyses by densitometry for p‐CaMKII (B). (C–G) The relative mRNA level of indicated genes in hippocampus. Btaf1 (C), Phka1 (D), Nav1 (E), Bdnf (F), Magt1 (G), and Grin1 (H) (*p < 0.05, **p < 0.01, ns: not significant, Student's t‐test). (I–L) Representative immunoblotting bands of synapse‐related proteins in hippocampus and quantitative analyses by densitometry (*p < 0.05, **p < 0.01, Student's t‐test). All error bars indicate the mean value ± SEM
Moreover, to clarify the effects of AEDs on the mRNA levels of epilepsy‐related and synapse‐related proteins in Cdh5‐CreERT2;CDK5f/f mice, the mRNA levels of Btaf1, Phka1, Nav1, Bdnf, Magt1, and Grin1 were confirmed by qRT‐PCR. Antiepileptic drug administration decreased the expression of Phka1 and Btaf1 and elevated the expression of Grin1 and Magt1 (Figure 8C–H). Finally, we tested the protein concerning synaptic structure and its receptors. Western blot data indicated that AEDs restored the protein levels of the synaptic structure‐related proteins p‐Synapsin1/Synapsin1 and PSD95 and the synaptic receptors NR1 and NR2B (Figure 8I–L).
Hence, AEDs exerted a therapeutic effect on vasogenic epilepsy derived from endothelial CDK5 loss, which results in downregulating epilepsy‐related Phka1 and Btaf1 and upregulating synaptic function‐related Grin1 and Magt1. We speculate that improvement of memory and improved phosphorylation of CaMKII in the hippocampus may be associated with restoration of synaptic structural components p‐Synapsin1 and PSD95, as well as synaptic receptors NR1 and NR2B function.
3. DISCUSSION
To date, the mechanism of memory deficits in epilepsy remains unclear. An imbalance of synaptic excitation and inhibition is considered to be the basis of epilepsy; however, whether the imbalance leads to memory impairment is also unclear. Here, clinical meta‐analysis of epilepsy patients showed that the risk of memory impairment or dementia was higher in epilepsy patients than in controls. We utilized the spontaneous epilepsy model to discover that persistent epileptic seizures induce memory deficits by memory behavior analysis. Accordingly, synapse‐related gene and protein changes were found in the hippocampus of Cdh5‐CreERT2;CDK5f/f mice. More importantly, AEDs treatment increased synaptic protein levels and partly ameliorated memory dysfunction.
In adult hippocampus, CDK5 modulates synaptic plasticity, memory formation, and long‐term behavioral changes. 28 , 29 Neuronal hyperactivation of CDK5 has been reported to be involved in the pathological process of many neurodegenerative diseases, including AD. 30 , 31 Here, we found that endothelial CDK5‐deficit mice exhibited memory impairment and decreased phosphorylation of CaMKII after tamoxifen treatment for 20 weeks and not for 4 weeks. Long‐term potentiation (LTP) has an important role in hippocampus‐dependent learning and memory and plays a basic role in memory processes. 32 , 33 CaMKII is a key protein kinase in hippocampal neural plasticity and memory. 34 , 35 The enzyme CaMKII can be autophosphorylated at T286 on the alpha subunits and at 287 on the beta subunits, 36 which makes CaMKII activity persist even after the calcium concentration drops to baseline levels. 37 The persistent activation of CaMKII is sufficient to trigger LTP, and CaMKII activation is necessary for LTP. In epileptic Cdh5‐CreERT2;CDK5f/f mice, although the total protein level of CaMKII had no change and no neurons were lost, autophosphorylated CaMKII decreased, which may play an important role in memory deficits caused by epilepsy.
NMDARs, as glutamate receptors, are blocked by Mg2 + ions, which are only removed when neurons depolarize. MagT1 is located in the cell membrane and facilitates Mg2+ influx, which maintains intracellular Ca2+ levels and protects neurons from hyperexcitatory Ca2+ overload. 38 Here, we showed that the decreased mRNA level of MagT1 may contribute to seizures in CDK5‐deficient mice. On the other hand, LTP maintenance requires different downstream molecular events, such as CaMKII activation, PSD, NR2B, NR1, and AMPAR. 39 Activated CaMKII diffuses to the synapse and specifically binds to NR2B, which plays a key role in LTP induction and learning. 40 , 41 Moreover, PSD95, which is present in excitatory synapses, stabilizes the surface expression of NMDAR and is necessary for CaMKII, 42 NMDAR, and CaMKII–NMDAR complex binding in the postsynaptic membrane. 43 In the present study, transcriptomics analysis revealed synaptic gene changes, which were also verified by decreased protein levels of PSD95, NR1, and NR2B in tamoxifen‐treated Cdh5‐CreERT2;CDK5f/f mice. Decreased protein levels of PSD95, NR1, and NR2B affect the formation and maintenance of hippocampal neural plasticity, resulting in memory impairment. The ATP release/CD73/A2AR pathway preceded and related with synaptic dysfunction and neuronal damage. 44 Therefore, it is of interest to further address the role of metabolic changes in the context of vasogenic epilepsy in the future.
PHT and VPA are clinical first‐line antiepileptic drugs. PHT is used to control seizures by modulating voltage‐gated sodium channels, 45 and VPA is effective in most seizure types. 46 According to our study, VPA treatment, or combination with PHT, could decrease epileptic gene mRNA expression and alleviate seizures. VPA is approved for the treatment of mania associated with manic depression, epileptic seizures, and migraine headaches. 26 However, the mechanism of VPA remains unknown. Interestingly, we found that VPA treatment increased the phosphorylation of CaMKII and the protein levels of NR2B, NR1, and PSD95, which may contribute to improve memory behaviors. Nevertheless, the mechanism of VPA on synapses needs to be explored in the future.
In summary, our data indicated that the mice exhibited memory impairments in the process of spontaneous epilepsy. To explore the cellular and molecular characteristics underlying spontaneous epileptic memory impairments, we generated a transcriptomic atlas of the hippocampus in CDK5f/f mice and Cdh5‐CreERT2;CDK5f/f mice. We identified the transcriptional alterations underlying the dysregulation of epilepsy‐ and synapse‐related gene changes in the hippocampus and unveiled synapse‐related protein aberrations associated with epileptic memory impairments. Moreover, VPA or PHT co‐administration rescued impaired hippocampal synaptic plasticity, resulting in memory improvement. Overall, our study describes a mechanism of memory deficits in epilepsy and identifies new effects of VPA on synapses, which provides new insights into preventive or therapeutic interventions for epileptic memory deficits. In the future, the study of the neuronal circuit mechanism of memory dysfunction in vasogenic epilepsy and the pharmacological regulation of targeting this neural circuit will promote the treatment of this disease.
4. MATERIALS AND METHODS
4.1. Experimental animals
Mice were housed under the condition of a 12‐h light/dark schedule with a controlled atmospheric environment (22°C ± 1°C, 40%–60% humidity) and had free access to standard diet and water. Certain experimental operations were applied to the animals following at least 1 week of acclimation to their habitat. All animal studies and experimental procedures were approved by the Animal Experimentation Committee at Nanjing Medical University in China (1804007).
The generation of endothelial‐specific CDK5 knockout mice applied LoxP/Cre strategy. To construct Cdh5‐CreERT2;CDK5f/f mice, endothelial tamoxifen‐inducible driver Cdh5‐CreERT2 mice (acquired from Prof. Ralf Adams, Max Planck Institute, G¨ottingen, Germany) were crossed with mice with a loxP‐flanked CDK5 gene (stock no. 014156; The Jackson Laboratory). Cre activity would be induced at 4 weeks by tamoxifen injections (Sigma‐Aldrich, 0.1 mg/g bodyweight, intraperitoneal), as described previously. 16
4.2. Behavioral analysis
Mice at 4 weeks (2‐month‐old mice) and 20 weeks (6‐month‐old mice) after tamoxifen treatment were used for the experimental procedures. Homologous CDK5f/f mice were treated as the control group. Y‐maze, novel object location test, and novel object recognition test were performed.
4.2.1. Y‐maze test
Spontaneous alternation behavior of Cdh5‐CreERT2;CDK5f/f mice relative to CDK5f/f mice was recorded for spatial memory evaluation in the Y‐maze task, as described previously. 47 Briefly, mice were placed at the end of one arm of the apparatus with three identical black plexiglass arms (length: 50 cm, width: 16 cm, height: 32 cm) to allow them to freely explore the three arms of the maze during an 8‐min session. An alternation was defined by every three different choices through recording the sequence of arm entries visually. The calculation of discrimination index (%) was (actual alternations/maximum alternations) × 100.
4.2.2. Novel object recognition test
An object recognition task on the basis of the mice tending to discriminate between familiar and novel objects was carried out according to a previously validated method to evaluate recognition memory. 48 Two same objects (A and B) were placed in symmetric positions in the chamber (50 × 50 × 60 cm) center. During the acquisition phase training, mice were put into the chamber to explore the objects (A and B) for 5 min. Two hours later, exploratory behavior was assessed for 5 min after a novel object C replacing object B. The discrimination index (%) was calculated as Times of exploring novel (C)/Total times of exploring (A+C).
4.2.3. Object novel location test
To test the spatial memory of mice, two same objects (A and B) were placed in symmetric positions in the chamber (50 × 50 × 60 cm) center. During the acquisition phase training, mice were made to explore in the chamber for 5 min. The position of object B was changed to the diagonal area of object A 2 h after the acquisition phase training, then the mice were put into the chamber to explore the objects for 5 min. The discrimination index (%) was calculated as Times of exploring novel (B)/Total times of exploring (A+B).
4.3. EEG recording
EEG recording could be used to monitor spontaneous seizure activity as reported previously. 16 In brief, bipolar twisted stainless steel electrodes (0.2 mm in diameter; Plastics One) were placed bilaterally in hippocampus CA1 region (−2.0 mm rostral to bregma, ±1.5 mm to midline, and 1.5 mm ventral to bregma) of the mice. Then, the electrodes were connected into a hole through a plastic pedestal (6 Channel, Plastics one), followed by being fixed to the skull with dental cement. After a week of recovery, EEG recording was conducted using a Vanguard system (Lamont) continuously in freely moving mice at a sampling rate of one utilizing a high‐frequency filter of 70 Hz for 24 h. The definition of epileptic seizures was field potentials two‐fold greater than the basal potential following durations longer than 10 s. Raw recording data were analyzed by Labchart 8 software.
4.4. Drugs administration
VPA (valproic acid sodium salt, Sigma P4543) and PHT (phenytoin sodium, Sigma D4505) were procured from Sigma‐Aldrich, Co., St. Louis, MO, USA. After EEG recording, the epileptic mice were screened for intraperitoneal injection of VPA (300 mg/kg/day) individually or VPA (300 mg/kg/day) + PHT (30 mg/kg/day) combined administration for 7 days consecutively. Then, EEG and memory‐related behavioral tests were performed.
4.5. Immunohistochemistry
Immunohistochemistry was performed as previously described. 47 After fixation in 4% PFA at 4°C, overnight, the brain was fully preserved in 30% sucrose PBS solution for cryoprotection. Then, 40‐μm slices from the brains were cut in a frozen microtome equipment later. The slices were permeabilized with 0.1% Triton X‐100 for 15 min, then blocked in 5% bovine serum albumin (BSA) for another hour at room temperature. For immunolabeling, the slices were incubated with indicated primary antibodies: anti‐CaMKII (1:300, Fukunaga et al., 1988), anti‐phospho‐Thr286‐CaMKII (1:300, Fukunaga et al., 1988), anti‐calcineurin (1:300, Fukunaga et al., 1988), anti‐MAP2 (1:1000, Millipore, Cat# 05‐346), anti‐NEUN (1:500, Millipore, Cat# ABN78), anti‐phospho‐Ser603‐Synapsin1 (1:300, Millipore, AB5583), and anti‐PSD95 (1:300, Thermo, MA1‐045). After incubation for two nights at 4°C, slices were incubated with Alexa Fluor 488‐conjugated anti‐mouse IgG (1:300, A21202, Life Technologies) and Alexa Fluor 594‐conjugated anti‐rabbit IgG (1:300, A21207, Life Technologies) for 1 h at RT. Using Zeiss LSM 800 confocal microscope for images and fluorescence values and co‐localization were later analyzed with ImageJ software.
4.6. Western blot analysis
Western blot analysis was performed as previously described. 49 It has been reported that memory spatiotemporal alterations manifest differently in circuits in different subregions of the hippocampus, but are fully integrated in the CA1 neuronal network as independent, multiplexed representations. 50 Therefore, we extracted total extracts of the whole tissue of the hippocampal CA1 region for the detection of related protein indicators. In brief, samples containing equivalent amounts of protein were separated by SDS‐PAGE (10%–12%) and transferred onto PVDF membranes (Millipore). The primary antibodies were listed as follows: phospho‐Thr286‐CaMKII (1:3000, Fukunaga et al., 1988), CaMKII (1:3000, Fukunaga et al., 1988), anti‐phospho‐Ser603‐Synapsin1 (1:1000, Millipore, AB5583), anti‐Synapsin1 (1:1000, Fukunaga et al., 1988), anti‐PSD95 (1:5000, Thermo, MA1‐045), anti‐NMDAR1 (1:1000, Abcam, ab109182), anti‐NMDAR2B (1:1000, Alomone, AGC‐003), anti‐phospho‐Ser831‐GluR1 (1:1000, Millipore, 04‐823), anti‐GluR1 (1:1000, Santa Cruz, SC‐7608), and GAPDH (1:5000, Cell Signaling Technology, Cat# 2118). EZ‐ECL Chemiluminescence Detection Kit was used to visualize immunoreactive proteins (Biological Industries). Quantification of the bands was performed using ImageJ software (US National Institutes of Health). GAPDH was used as an internal control.
4.7. RNA‐seq data analysis
According to false discovery rate (FDR), differentially expressed genes (DEGs) were selected (value <0.05). Heatmaps were produced using the R package heatmap. FPKM values were z‐scaled and used as input data for heatmap. We used R package Venn diagram to fetch the intersection of CDK5 CKO sequencing data, GSE80312 and GSE128300. 51 , 52 , 53 Gene Ontology‐biological process (GO‐BP) and Gene Ontology‐cellular component (GO‐CC) terms were enriched and visualized using the R package cluster Profiler. 54 p‐Value <0.05 was set as the cutoff value for the enrichment analysis. STRING database (www.string‐db.org) was used to predict protein–protein interaction (PPI) network of DEGS of synapse organization term. 55
4.8. Real‐time quantitative PCR
In order to validate the mRNA levels of differentially expressed genes obtained by RNA‐seq analysis, brain hippocampus tissues were dissected. The total RNA of the samples was extracted using RNAiso Plus (Takara, Shiga, Japan), as described previously. 56 First strand cDNA was synthesized by reverse transcription with 500 ng total RNA using the Prime Script RT reagent Kit Perfect Real Time (Takara) driven by indicated primer pairs in a 20‐μl reaction system according to the manufacturer's instructions. qRT‐PCR was performed with a Mastercycler ep realplex (Eppendorf, Hamburger, Germany) in 96‐well plates using SYBR Premix Ex Taq (Takara) to detect the mRNA expression level of 23 DEGs. The primer sequences are provided in Table S1. PCR condition is 95°C for 2 min, 40 cycles of each 95°C for 15 s, 55°C for 15 s, and 68°C for 25 s. Melting curve analysis was performed with the default settings on the instrument from 50°C to 85°C. Commercial software was used to calculate the RNA expression C t values automatically. β‐actin or GAPDH was used as an internal control. Data were expressed relative to a calibrator using the 2‐(DDC t) ± s formula.
4.9. Statistical analysis
Statistical analysis was performed using GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA). An unpaired two‐tailed Student's t‐test was used for datasets, including two mere independent groups. For multigroup comparisons, one‐way ANOVA was performed by Tukey's multiple comparison test. Two‐way ANOVA followed by Bonferroni's multiple comparison test was performed. Data results were presented as means ± SEM and p < 0.05 was considered to be statistically significant.
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
ZM.L., XX.L. contributed equally to this work, conducted experiments, analyzed data and write articles, C.L., ZC.W., Y.S. and HY.S. conducted behavioral analysis and data collation, X.C., Y.Z., JW.L., RF.Z. analyzed of transcriptomic data, BH.H., WF.Y., D.H., GJ.J and T.S. assisted with the experiments and reviewed paper. L.Z., F.H., YM.L. designed and coordinated the experiments, supervised the work, reviewed and edited paper.
ETHICS APPROVAL
All animal studies and experimental procedures were approved by the Animal Experimentation Committee at Nanjing Medical University in China (1804007).
Supporting information
SUPPORTING INFORMATION
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundations of China (81973300 to Ying‐Mei Lu; 82104162 to Xiu‐Xiu Liu; 82073946 to Wei‐Feng Ye); Natural Science Foundation of Jiangsu Province (BK20210539 to Xiu‐Xiu Liu); Natural Science Foundation of Zhejiang Province (LY18H310009 to Guo‐Jun Jiang).
Li Z‐M, Liu X‐X, Li C, et al. Decreased synapse‐associated proteins are associated with the onset of epileptic memory impairment in endothelial CDK5‐deficient mice. MedComm. 2022;3:e128. 10.1002/mco2.128
Zheng‐Mao Li and Xiu‐Xiu Liu contributed equally to this study.
Contributor Information
Feng Han, Email: fenghan169@njmu.edu.cn.
Ying‐Mei Lu, Email: lufx@njmu.edu.cn.
DATA AVAILABILITY STATEMENT
The RNA‐seq data generated in this study are available at NCBI’s Gene Expression Omnibus GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi; accession # GSE198170). The other datasets are available from the corresponding authors upon reasonable request.
REFERENCES
- 1. Devinsky O, Vezzani A, O'Brien TJ, et al. Epilepsy. Nat Rev Dis Primers. 2018;4:18024. [DOI] [PubMed] [Google Scholar]
- 2. Vezzani A, Fujinami RS, White HS, et al. Infections, inflammation and epilepsy. Acta Neuropathol. 2016;131(2):211‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Centers for Disease Control and Prevention . Influenza vaccination practices of physicians and caregivers of children with neurologic and neurodevelopmental conditions ‐ United States, 2011–12 influenza season. MMWR Morb Mortal Wkly Rep. 2013;62(36):744‐746. [PMC free article] [PubMed] [Google Scholar]
- 4. Iyengar PJ, Wamala J, Ratto J, et al. Prevalence of nodding syndrome–Uganda, 2012–2013. MMWR Morb Mortal Wkly Rep. 2014;63(28):603‐606. [PMC free article] [PubMed] [Google Scholar]
- 5. Sen A, Jette N, Husain M, Sander JW. Epilepsy in older people. Lancet. 2020;395(10225):735‐748. [DOI] [PubMed] [Google Scholar]
- 6. Vu LC, Piccenna L, Kwan P, O'Brien TJ. New‐onset epilepsy in the elderly. Br J Clin Pharmacol. 2018;84(10):2208‐2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jeffery KJ. The hippocampus: from memory, to map, to memory map. Trends Neurosci. 2018;41(2):64‐66. [DOI] [PubMed] [Google Scholar]
- 8. Lisman J, Buzsaki G, Eichenbaum H, Nadel L, Ranganath C, Redish AD. Viewpoints: how the hippocampus contributes to memory, navigation and cognition. Nat Neurosci. 2017;20(11):1434‐1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zeidman P, Maguire EA. Anterior hippocampus: the anatomy of perception, imagination and episodic memory. Nat Rev Neurosci. 2016;17(3):173‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bui AD, Nguyen TM, Limouse C, et al. Dentate gyrus mossy cells control spontaneous convulsive seizures and spatial memory. Science. 2018;359(6377):787‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castellucci VF, Carew TJ, Kandel ER. Cellular analysis of long‐term habituation of the gill‐withdrawal reflex of Aplysia californica . Science. 1978;202(4374):1306‐1308. [DOI] [PubMed] [Google Scholar]
- 12. Carew T, Castellucci VF, Kandel ER. Sensitization in Aplysia: restoration of transmission in synapses inactivated by long‐term habituation. Science. 1979;205(4404):417‐419. [DOI] [PubMed] [Google Scholar]
- 13. Rossini L, De Santis D, Mauceri RR, et al. Dendritic pathology, spine loss and synaptic reorganization in human cortex from epilepsy patients. Brain. 2021;144(1):251‐265. [DOI] [PubMed] [Google Scholar]
- 14. Lybrand ZR, Goswami S, Zhu J, et al. A critical period of neuronal activity results in aberrant neurogenesis rewiring hippocampal circuitry in a mouse model of epilepsy. Nat Commun. 2021;12(1):1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wandschneider B, Stretton J, Sidhu M, et al. Levetiracetam reduces abnormal network activations in temporal lobe epilepsy. Neurology. 2014;83(17):1508‐1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu XX, Yang L, Shao LX, et al. Endothelial Cdk5 deficit leads to the development of spontaneous epilepsy through CXCL1/CXCR2‐mediated reactive astrogliosis. J Exp Med. 2020;217(1):e20180992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chang HJ, Liao CC, Hu CJ, Shen WW, Chen TL. Psychiatric disorders after epilepsy diagnosis: a population‐based retrospective cohort study. PLoS One. 2013;8(4):e59999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Laurent A, Artiges E, Mellerio C, et al. Metabolic correlates of cognitive impairment in mesial temporal lobe epilepsy. Epilepsy Behav. 2020;105:106948. [DOI] [PubMed] [Google Scholar]
- 19. Powell K, Walker RW, Rogathe J, et al. Cognition and behavior in a prevalent cohort of children with epilepsy in rural northern Tanzania: a three‐year follow‐up study. Epilepsy Behav. 2015;51:117‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stefanidou M, Beiser AS, Himali JJ, et al. Bi‐directional association between epilepsy and dementia: the Framingham Heart Study. Neurology. 2020;95(24):e3241‐e3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tsai ZR, Zhang HW, Tseng CH, et al. Late‐onset epilepsy and subsequent increased risk of dementia. Aging (Albany NY). 2021;13(3):3573‐3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hashimotodani Y, Nasrallah K, Jensen KR, Chavez AE, Carrera D, Castillo PE. LTP at hilar mossy cell‐dentate granule cell synapses modulates dentate gyrus output by increasing excitation/inhibition balance. Neuron. 2017;95(4):928‐943.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Garriga‐Canut M, Schoenike B, Qazi R, et al. 2‐Deoxy‐D‐glucose reduces epilepsy progression by NRSF‐CtBP‐dependent metabolic regulation of chromatin structure. Nat Neurosci. 2006;9(11):1382‐1387. [DOI] [PubMed] [Google Scholar]
- 24. Epi4K Consortium , Epilepsy Phenome/Genome Project , Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501(7466):217‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mielnik CA, Binko MA, Chen Y, et al. Consequences of NMDA receptor deficiency can be rescued in the adult brain. Mol Psychiatry. 2021;26(7):2929‐2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sgobio C, Ghiglieri V, Costa C, et al. Hippocampal synaptic plasticity, memory, and epilepsy: effects of long‐term valproic acid treatment. Biol Psychiatry. 2010;67(6):567‐574. [DOI] [PubMed] [Google Scholar]
- 27. Lyttle MD, Rainford NEA, Gamble C, et al. Levetiracetam versus phenytoin for second‐line treatment of Paediatric Convulsive Status Epilepticus (EcLiPSE): a multicentre, open‐label, randomised trial. Lancet. 2019;393(10186):2125‐2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bibb JA, Chen J, Taylor JR, et al. Effects of chronic exposure to cocaine are regulated by the neuronal protein Cdk5. Nature. 2001;410(6826):376‐380. [DOI] [PubMed] [Google Scholar]
- 29. Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Cyclin‐dependent kinase 5 is required for associative learning. J Neurosci. 2002;22(9):3700‐3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu SL, Wang C, Jiang T, Tan L, Xing A, Yu JT. The role of Cdk5 in Alzheimer's disease. Mol Neurobiol. 2016;53(7):4328‐4342. [DOI] [PubMed] [Google Scholar]
- 31. Ogara MF, Belluscio LM, de la Fuente V, et al. CDK5‐mediated phosphorylation of p19INK4d avoids DNA damage‐induced neurodegeneration in mouse hippocampus and prevents loss of cognitive functions. Biochim Biophys Acta. 2014;1843(7):1309‐1324. [DOI] [PubMed] [Google Scholar]
- 32. Nicoll RA. A brief history of long‐term potentiation. Neuron. 2017;93(2):281‐290. [DOI] [PubMed] [Google Scholar]
- 33. Davenport CM, Rajappa R, Katchan L, et al. Relocation of an extrasynaptic GABAA receptor to inhibitory synapses freezes excitatory synaptic strength and preserves memory. Neuron. 2021;109(1):123‐134.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3(3):175‐190. [DOI] [PubMed] [Google Scholar]
- 35. Hojjati MR, van Woerden GM, Tyler WJ, et al. Kinase activity is not required for alphaCaMKII‐dependent presynaptic plasticity at CA3‐CA1 synapses. Nat Neurosci. 2007;10(9):1125‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chao LH, Stratton MM, Lee IH, et al. A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin‐ dependent kinase II holoenzyme. Cell. 2011;146(5):732‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pi HJ, Otmakhov N, El Gaamouch F, Lemelin D, De Koninck P, Lisman J. CaMKII control of spine size and synaptic strength: role of phosphorylation states and nonenzymatic action. Proc Natl Acad Sci U S A. 2010;107(32):14437‐14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Levitsky DO, Takahashi M. Interplay of Ca(2+) and Mg(2+) in sodium‐calcium exchanger and in other Ca(2+)‐binding proteins: magnesium, watchdog that blocks each turn if able. Adv Exp Med Biol. 2013;961:65‐78. [DOI] [PubMed] [Google Scholar]
- 39. Murakoshi H, Wang H, Yasuda R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nature. 2011;472(7341):100‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411(6839):801‐805. [DOI] [PubMed] [Google Scholar]
- 41. Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48(2):289‐301. [DOI] [PubMed] [Google Scholar]
- 42. Won S, Incontro S, Nicoll RA, Roche KW. PSD‐95 stabilizes NMDA receptors by inducing the degradation of STEP61. Proc Natl Acad Sci U S A. 2016;113(32):E4736‐E4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen X, Vinade L, Leapman RD, et al. Mass of the postsynaptic density and enumeration of three key molecules. Proc Natl Acad Sci U S A. 2005;102(32):11551‐11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Augusto E, Goncalves FQ, Real JE, et al. Increased ATP release and CD73‐mediated adenosine A2A receptor activation mediate convulsion‐associated neuronal damage and hippocampal dysfunction. Neurobiol Dis. 2021;157:105441. [DOI] [PubMed] [Google Scholar]
- 45. Farber NB, Jiang XP, Heinkel C, Nemmers B. Antiepileptic drugs and agents that inhibit voltage‐gated sodium channels prevent NMDA antagonist neurotoxicity. Mol Psychiatry. 2002;7(7):726‐733. [DOI] [PubMed] [Google Scholar]
- 46. Tomson T, Battino D, Perucca E. Valproic acid after five decades of use in epilepsy: time to reconsider the indications of a time‐honoured drug. Lancet Neurol. 2016;15(2):210‐218. [DOI] [PubMed] [Google Scholar]
- 47. Tan C, Lu NN, Wang CK, et al. Endothelium‐derived semaphorin 3G regulates hippocampal synaptic structure and plasticity via neuropilin‐2/PlexinA4. Neuron. 2019;101(5):920‐937.e13. [DOI] [PubMed] [Google Scholar]
- 48. Tian Y, Yabuki Y, Moriguchi S, et al. Melatonin reverses the decreases in hippocampal protein serine/threonine kinases observed in an animal model of autism. J Pineal Res. 2014;56(1):1–11. 10.1111/jpi.12081 [DOI] [PubMed] [Google Scholar]
- 49. Sasaki T, Han F, Shioda N, et al. Lithium‐induced activation of Akt and CaM kinase II contributes to its neuroprotective action in a rat microsphere embolism model. Brain Res. 2006;1108(1):98‐106. [DOI] [PubMed] [Google Scholar]
- 50. Eichenbaum H. On the integration of space, time, and memory. Neuron. 2017;95(5):1007‐1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen H, Boutros PC. VennDiagram: a package for the generation of highly‐customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kobow K, Ziemann M, Kaipananickal H, et al. Genomic DNA methylation distinguishes subtypes of human focal cortical dysplasia. Epilepsia. 2019;60(6):1091‐1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ung DC, Iacono G, Méziane H, et al. , Ptchd1 deficiency induces excitatory synaptic and cognitive dysfunctions in mouse. Molecular Psychiatry. 2018;23(5):1356–1367. 10.1038/mp.2017.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality‐controlled protein‐protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362‐D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang H, Hong LJ, Huang JY, et al. P2RX7 sensitizes Mac‐1/ICAM‐1‐dependent leukocyte‐endothelial adhesion and promotes neurovascular injury during septic encephalopathy. Cell Res. 2015;25(6):674‐690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION
Data Availability Statement
The RNA‐seq data generated in this study are available at NCBI’s Gene Expression Omnibus GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi; accession # GSE198170). The other datasets are available from the corresponding authors upon reasonable request.