

Abstract
Serotonin 2A receptors (5-HT2ARs) mediate the hallucinogenic effects of psychedelic drugs and are a key target of the leading class of medications used to treat psychotic disorders. These findings suggest that dysfunction of 5-HT2ARs may contribute to the symptoms of schizophrenia, a mental illness characterized by perceptual and cognitive disturbances. Indeed, numerous studies have found that 5-HT2ARs are reduced in the brains of individuals with schizophrenia. However, the mechanisms that regulate 5-HT2AR expression remain poorly understood. Here we show that a physiologic environmental stimulus, sleep deprivation, significantly upregulates 5-HT2ARs levels in the mouse frontal cortex in as little as 6–8 hours (for mRNA and protein, respectively). This induction requires the activity-dependent immediate early gene transcription factor early growth response 3 (Egr3) as it does not occur in Egr3 deficient (−/−) mice. Using chromatin immunoprecipitation, we show that EGR3 protein binds to the promoter of Htr2a, the gene that encodes the 5-HT2ARs, in the frontal cortex in vivo, and drives expression of in vitro reporter constructs via two EGR3 binding sites in the Htr2a promoter. These results suggest that EGR3 directly regulates Htr2a expression, and 5-HT2ARs levels, in the frontal cortex in response to physiologic stimuli. Analysis of publicly available post-mortem gene expression data revealed that both EGR3 and HTR2A mRNA are reduced in the prefrontal cortex of schizophrenia patients compared to controls. Together these findings suggest a mechanism by which environmental stimuli alter levels of a brain receptor that may mediate the symptoms, and treatment, of mental illness.
Introduction
The serotonin 2A receptor (5-HT2AR) is extensively expressed in the cerebral cortex where it is believed to play a critical role in perception, cognition, and psychosis. Much of the evidence for this comes from studies on the effects of drugs that bind to the 5-HT2AR 1–3. This includes agonists, such as lysergic acid (LSD), psilocybin, and mescaline, which cause hallucinations 4–6, as well as antagonists and inverse agonists, including second-generation antipsychotics, which reverse the perceptual disturbances of psychiatric illnesses such as schizophrenia 6–8. Additional findings supporting a role for the 5-HT2AR in the symptoms of psychosis include a long history of studies showing abnormal levels of 5-HT2ARs in the brains of patients diagnosed with schizophrenia. While some studies did not detect differences 9–11, and one group identified increased levels 12, 13, the vast majority of these studies, including a meta-analysis 14, have revealed reduced 5-HT2AR levels in schizophrenia subjects. These include post-mortem studies measuring 5-HT2AR ligand binding and mRNA expression, and as well as in vivo Positron Emission Tomography (PET) scan investigations 14–24. Notably, studies in antipsychotic-naïve patients indicate that the reduction in 5-HT2AR levels is not simply a consequence of medication treatment 16, 18. Despite the longstanding recognition of the importance of 5-HT2ARs in the response to psychedelic drugs, and the symptoms and treatment of psychotic disorders, the processes that regulate expression of this critical receptor remain unknown.
Our prior work identified that mice lacking the immediate early gene (IEG) Egr3 (Egr3−/− mice) have reduced levels of 5-HT2ARs in the frontal cortex 25. This suggested that Egr3 is required for expression of Htr2a, the gene that encodes the 5-HT2AR. If so, we reasoned that stimuli that activate expression of Egr3 may also induce Htr2a expression. Sleep deprivation (SD) is a physiological stimulus that upregulates Egr3 in the mouse cerebral cortex 26. Using this approach, we found that just six hours of SD increases Htr2a mRNA levels in the mouse cortex and, moreover, this required Egr3 27.
This discovery that Htr2a expression can be rapidly induced in the rodent brain in response to a physiologic stimulus is supported by a prior study in humans. In 2012 Elmenhorst and colleagues reported that preventing healthy subjects from sleeping for a period of 24 hours resulted in a significant increase in 5-HT2AR binding in the neocortex, detected on PET scan 28.
These findings raised the question of whether EGR3, an activity dependent transcription factor, directly regulates the Htr2a gene, and thereby mediates the environmental induction of Htr2a mRNA in the cortex. In the current study we have addressed the questions of where in the cerebral cortex SD is inducing Htr2a expression, does this also result in increased 5-HT2AR protein, and is EGR3 directly regulating the Htr2a gene to effect this environmentally-induced receptor expression. We show that six hours of SD significantly upregulates Htr2a in the prefrontal cortex and adjacent sensorimotor cortex, and that 8 hours of SD upregulates 5-HT2AR levels in the same regions. In both cases this upregulation requires Egr3. Further, we show that the EGR3 protein binds to the Htr2a promoter in vivo and drives expression of an in vitro reporter construct, suggesting that the activity dependent IEG transcription factor EGR3 directly regulates expression of Htr2a in response to the acute environmental stimulus of SD.
Materials and Methods
See online supplementary methods for details and primer sequences.
Animals
Previously generated Egr3−/− mice 29, backcrossed to a C57BL/6 background for >30 generations, were housed on a 14/10 h light/dark schedule with ad libitum access to food and water. Matched pairs of littermate Egr3−/− and Wildtype (WT) mice were designated at weaning and randomly assigned to experimental groups. Male mice ages 3–5 months were used for qRT-PCR studies. Male and female mice we used for in situ hybridization (4 months old) and autoradiography (7–9 weeks old) experiments. WT male C57BL/6NTac mice (2.5–4 months old) were obtained from Taconic Biosciences for Western blot and chromatin immunoprecipitation (ChIP) experiments. Mice were acclimated to the facility for two weeks before experiments. Animal studies were performed in accordance with the University of Arizona Institutional Animal Care and Use Committee (IACUC) guidelines under an approved IACUC protocol.
Sleep deprivation (SD)
SD was initiated at the onset of “lights on” (8:00 a.m.) and conducted for 6h (for mRNA studies) or 8h (for protein), using the “Gentle handling” method as previously described 27. Investigators were blinded to genotype during SD experiments. The number and type of stimulation required to keep each animal awake during the SD did not differ between WT and Egr3−/− mice (Supplementary Figure 1).
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Tissue was dissected on ice, transferred to RNAlater (Thermo Fisher Scientific), frozen on dry ice and stored at −80°C. Tissue homogenization and RNA isolation was performed in TRI reagent (Life Technologies) using ceramic beads and a MagMAX express processor (Applied Biosystems). mRNA was reverse transcribed using M-MLV reverse transcriptase kit (Life Technologies) and qRT-PCR performed using FastStart SYBR green master mix (Roche). Each sample was amplified in triplicate for the gene of interest and the housekeeping gene phosphoglycerate kinase 1 (Pgk1). Fold changes in gene expression were calculated and data were plotted using the 2-ΔΔCt method 30. Relative gene expression of all samples was first normalized to Pgk1 (ΔCt). Then, average ΔCt data were normalized to WT SD controls for data points representing WT animals that underwent SD, or Egr3−/− SD controls for Egr3−/− animals that underwent SD.
In situ hybridization
In situ hybridization was performed on 12μm sections using the RNAscope Multiplex Fluorescent Reagent Kit V2 (Catalogue # 323100). The Htr2a probe was purchased from Advanced Cell Diagnostics, Inc (Probe-Mm-Htr2a-C3, 401291-C3) and developed with the fluorescent Opal dye 570 (Akoya Biosciences, Cat. # FP1488001KT) at a concentration of 1:750. Sections were counterstained with DAPI (Vector Laboratories). Qualitative analyses performed via thorough review of all tissue sections and in reference to anatomical regions described in The Allen Mouse Brain Atlas (Reference Atlas, Interactive P56, Coronal) 31, 32.
Radioligand Binding Analysis – Autoradiography
Six 12μm sections of anterior frontal cortex (Bregma 2.8 – Bregma 2.34) and posterior frontal cortex (Bregma 1.70 – Bregma 0.86), and two sections of mid-cortex (Bregma 0.02 – 0.82), and posterior cortex (Bregma −1.34 – 2.30) were thaw mounted on the same slide. A total of ten slides/brain were serially collected and stored at −80°C. Tissue was treated with selective 5HT2AR antagonist 3-H-MDL100907 [R(+)-α-(2,3-dimethoxyphenyl)-1-[2-(4-fluorphenyl)-ethyl]-4-piperidin-methanol] (specific activity; 59.22 Ci/mmol) and detected by autoradiography. Non-drug exposed control tissue was used to assess non-specific binding.
Western blot
Six hours after SD, the frontal cortex was dissected. Right and left hemispheres were used for Western blot and ChIP experiments respectively. Tissue was Dounce homogenized in RIPA buffer and centrifuged at 12,000 rpm, 4°C x 20 min. Supernatant protein was denatured at 95 °C x 5 min and 20ug of protein was loaded per lane on an Any kD TGX polyacrylamide gel (Bio-Rad Laboratories). Samples were transferred to nitrocellulose membrane and blocked overnight at 4°C in 5% non-fat dry milk in Tris-buffered saline with 0.05% Tween 20 (TBS-T). Blocked membranes were incubated for 2h at room temperature (RT) with rabbit anti-EGR3 antibody (1:500, kindly donated by Dr. Mary Kay Lobo, University of Maryland School of Medicine, original source: Baraban Lab, Johns Hopkins University School of Medicine 33) and mouse anti-β actin antibody (1:5000, Sigma-Aldrich, Cat. # A5441) diluted in 1% non-fat dry milk/TBS-T. Following washes, membranes were incubated with secondary antibodies IRDye800CW (1:10,000 for EGR3, Li-COR Biosciences, Cat. # 926–32211) and IRDye680RD (1:10,000 for β-actin, Li-COR Biosciences, Cat. # 926–68072) for 1h at RT. Blots were washed in TBS with 0.1% Tween 20, and imaged. Protein levels were determined as ratios of EGR3 to β-actin internal control and reported as percentage of the control group. For Western blot experiments, recombinant EGR3 protein was synthesized using the TnT Coupled Reticulocyte Lysate System (Promega, Cat. # L4611) using template pcDNA 3.1(−) mouse Egr3, a gift from Peter Johnson (Addgene plasmid # 107998; http://n2t.net/addgene:107998; RRID:Addgene_107998) 34.
Promoter Analysis
The genomic region, including 4 kb upstream of the Htr2a transcription start site (NM_172812, chr14:74636840–74640839), was obtained from the UCSC genome browser (https://genome.ucsc.edu/). Consensus binding sites for EGR3 were identified using the software ‘Find Individual Motif Occurrences’ (FIMO, http://meme-uite.org/tools/fimo) 35. The motif occurrences with a p value less than 0.0001 were selected.
Chromatin immunoprecipitation
Following 6h of SD, frontal cortex was dissected, minced, incubated in 1% formaldehyde to crosslink DNA-protein, and quenched with 125 mM glycine. Right and left hemispheres were used for Western blot and ChIP experiments respectively. Samples were homogenized, and chromatin sheared to 200 bp-1000 bp at 4°C for 25 cycles. Fragment size was verified via agarose gel electrophoresis. Samples were incubated with 70μl of anti-EGR3 antibody 33 complexed to magnetic beads (Invitrogen) x 16h at 4°C. Control IgG IP was performed in parallel. Beads were sequentially washed with low salt, high salt, and LiCl wash buffers. Samples were reverse crosslinked at 65°C overnight and treated with 2μl RNase (Roche) at 37°C x 1h and 2μl proteinase K (Invitrogen) at 55°C x 2h. DNA was isolated via QIAGEN kit and qPCR was performed. Relative occupancy (aka. fold enrichment) of the EGR3 proteins at each Htr2a promoter site was estimated using the following equation 2^(ΔCt MOCK- ΔCt SPECIFIC), where ΔCT MOCK and ΔCT SPECIFIC are mean normalized threshold cycles of PCR performed in triplicate on DNA samples from MOCK (anti-IgG antibody) and transcription factor EGR3 immunoprecipitations to the input IPs.
Promoter reporter vector design
The Htr2a promoter luciferase reporters were generated by Genecopeia Inc. using the dual-reporter system. The “Htr2a proximal promoter” reporter includes genomic DNA from 1061 bp upstream to 200 bp downstream of the Htr2a gene transcription start site (TSS) and contains the putative EGR3 binding sequence GCGCGGGGGAGGGG. The “Htr2a distal promoter” reporter includes genomic DNA from −2727 bp to −2841 bp upstream of the Htr2a TSS and contains the putative EGR3 binding site AGGAGGGGGAGTCT. The control Arc promoter luciferase reporter includes genomic DNA ranging from 1049 bp upstream to 200 bp downstream of the TSS of the Arc gene, which contains a previously validated EGR3 binding site 36. The “non-promoter” luciferase reporter, containing non-promoter sequence (TGCAGATATCCTCGCCC), was used as a negative control.
Cell culture and transfection
Neuro2a cells (ATCC) were seeded into 6-well plates and grown to 70–90% confluence. 3h prior to transfection, culture medium was replaced with 3ml of fresh medium. For each promoter construct, 1.5μg of CMV-Egr3 vector (or CMV vector control) was co-transfected with 1μg of promoter reporter, using 3.75μl of Lipofectamine 3000 reagent, 5μl of P3000 reagent and 250μl of Opti-MEM per well. Cells were incubated at 37°C in 5% CO2: 95% air. Transfections were conducted in triplicate and replicated twice.
Luciferase signal measurement
Twenty-four hours after transfection, 0.2ml of medium from each cell culture was collected and luciferase activities were measured using the secrete-pair dual luminescence assay kit (Genecopoeia). Each sample was run in duplicate. For all measurements, the GLuc value was first normalized to the internal control SEAP luciferase value (GLuc/SEAP ratio) and then to the non-promoter luciferase reporter. Following collection of medium, cells were processed for protein isolation and Western blot analysis to determine EGR3 protein levels.
NCBI GEO Data Analysis
Published data were obtained from NCBI GEO, Gene Expression Omnibus 37 dataset GSE53987 38. The microarray platform file GPL570 was used to ascertain gene-specific probe IDs for EGR3 and HTR2A, which were used to download gene expression data from prefrontal cortex (Brodmann Area 46).
Statistical Analyses
All statistical analyses were carried out using GraphPad prism with significance set at p < 0.05. For statistical analyses, unpaired, two-tailed, Student’s t-tests, two-way analysis of variance (ANOVA) with Bonferroni post-hoc tests, or two-tailed Mann-Whitney U tests were performed to determine significance for conditions in which there were more than two groups or two factors. Outliers were identified and excluded using the ROUT method 39 in GraphPad Prism with a False Discovery Rate (FDR) of 1%. Data were examined graphically within each group, and no strong deviation from normality was observed. Power analyses were performed based on previous data using G*Power (Heinrich-Heine Universität Düsseldorf). Data were plotted as means ± standard error of the mean (SEM).
Results
We had previously reported that SD rapidly upregulates Htr2a expression in the mouse cortex 27. However, since this study was performed in cortical homogenates, it was unclear in what cortical regions this upregulation was taking place. This is important since levels of both Htr2a mRNA and 5-HT2AR protein display a distinctive anterior-posterior gradient in the rodent cortex 40–42. This raised the question of whether SD was increasing Htr2a expression in regions where the receptor is already expressed or inducing it de novo in regions where it is normally not expressed. We therefore examined the cortical region in which SD induces Htr2a expression.
We first tested whether we could replicate published in situ hybridization findings showing that SD upregulates Egr3 26 using quantitative reverse transcription (qRT) PCR. Figure 1A shows the SD protocol and coordinates for regional brain dissection. In WT mice, we found that 6h of SD did not increase Egr3 expression in the most anterior region of frontal cortex (AFC) but did significantly upregulate Egr3 mRNA in the posterior part of the frontal cortex (PFC), as well as in more posterior regions of cortex (labeled “mid-posterior cortex (MPC)) (Fig.s 1B – 1D).
Figure 1. Sleep deprivation upregulates Egr3 in a region-dependent manner in the frontal cortex.
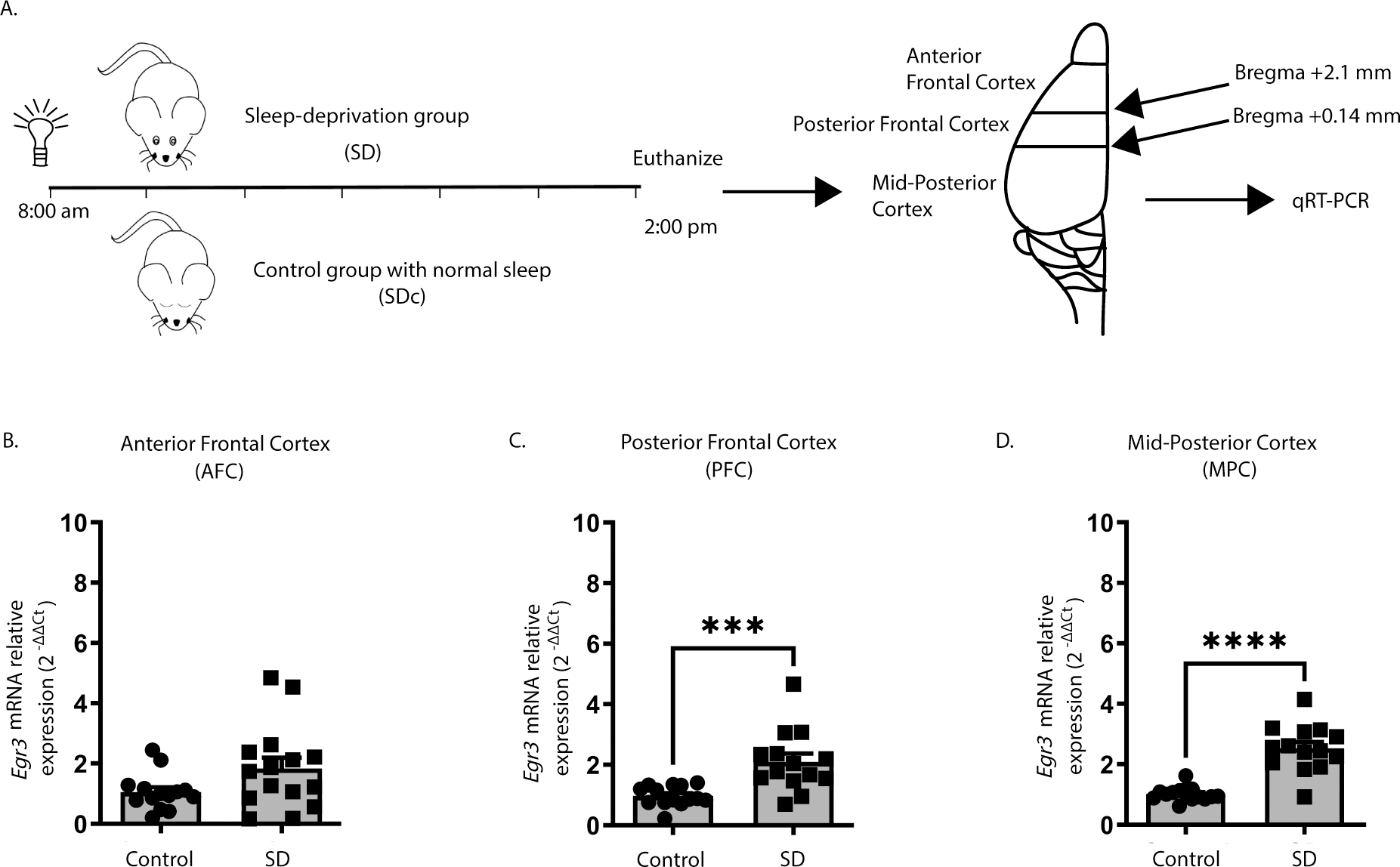
(A) SD protocol. In WT mice quantitative RT-PCR shows that 6h of SD (B) does not increase Egr3 expression in AFC regions (t27 = 1.956; p = 0.0609; SDc, n = 14; SD, n = 15 ), but significantly upregulates Egr3 mRNA in (C) PFC (t26 = 3.979, p = 0.0005; SDc, n = 14; SD, n = 14) and (D) MPC (t27 = 7.307; p < 0.0001; SDc, n = 15; SD, n = 14) regions, compared to SDc. Unpaired student’s t-test, *** p < 0.001, **** p < 0.0001. Values represent means ± SEM. (Abbreviations: AFC- anterior frontal cortex; PFC- posterior frontal cortex; MPC- mid to posterior cortex; SD- sleep deprivation; SDc- SD control; WT- wildtype; h: hours).
We next examined whether 6h of SD can upregulate Htr2a expression in the same cortical regions, and whether this requires Egr3 (Fig.s 2A – 2C). In the AFC, SD increased Htr2a expression when both genotypes were analyzed together (two-way ANOVA), but not in either WT or Egr3−/− groups independently (post-hoc analyses were not significant) (Fig. 2A). However, in the PFC of WT mice, SD significantly increased Htr2a mRNA compared to SDc, a result that was absent in Egr3−/− mice (Fig. 2B). In the MPC, SD also increased Htr2a mRNA in WT mice, though the relative amount of the increase was less than in the PFC. Again, this effect did not occur in Egr3−/− mice (Fig. 2C).
Figure 2. Sleep deprivation upregulates Htr2a in an Egr3-dependent, and region-specific, manner.
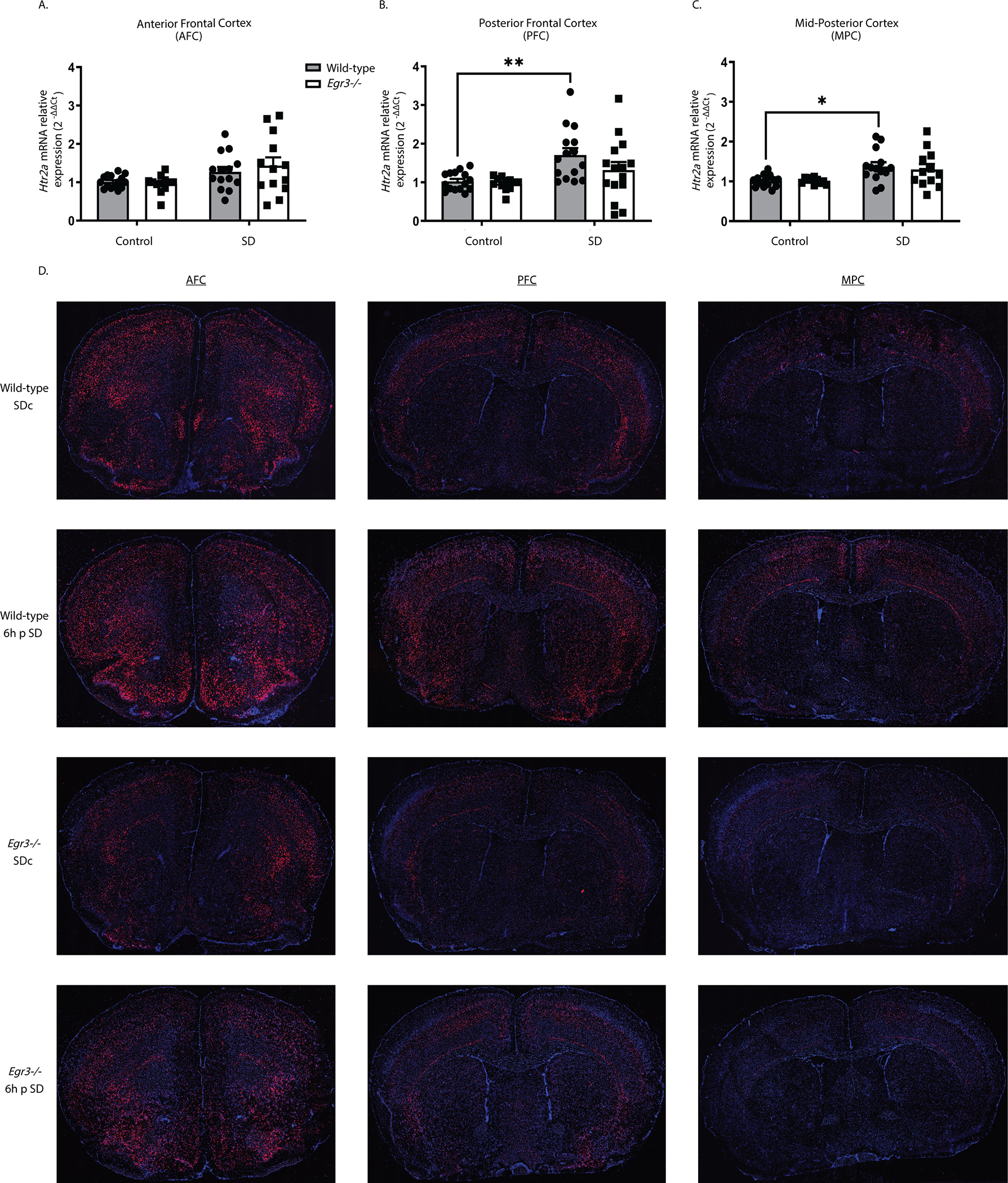
(A – C) In WT and Egr3−/− mice qRT-PCR shows that 6h of SD (A) increases Htr2a overall in the AFC when both genotypes were analyzed (two-way ANOVA, sig. main effect of SD (F1, 50 = 8.279, p = 0.0059; WT: SDc, n = 15; SD, n = 14; Egr3−/−: SDc, n = 12; SD, n = 13)) but not in either genotype alone (post-hoc analyses showed no sig. differences between genotypes or SD conditions). (B) However, SD significantly upregulates Htr2a expression in the PFC of WT, but not Egr3−/−, mice, compared to SDc (two-way ANOVA, sig. main effect of SD (F 1, 53 = 12.08, p = 0.0010; WT: SDc, n = 15; SD, n = 15; Egr3−/−: SDc, n = 12; SD, n = 15); post-hoc analyses showed a sig. increase of Htr2a mRNA after SD (vs. SDc) in WT mice (p = 0.0088), but not in the Egr3 −/− mice (p = 0.6777)). (C) In the MPC, SD increased Htr2a in WT, but not Egr3−/−, mice, compared to SDc (two-way ANOVA, sig. main effect of SD (F 1,47 = 13.14, p = 0.0007; WT: SDc, n = 15; SD, n = 14; Egr3−/−: SDc, n = 10; SD, n = 12)); post-hoc analyses showed sig. increase of Htr2a mRNA after SD (vs. SDc) in WT mice (p = 0.0205) but not in the Egr3 −/− mice (p = 0.2343). (D) Representative images from n = 3 per group of RNAscope in situ hybridization demonstrating Htr2a expression in SDc and SD WT and Egr3−/− mice. Bonferroni-corrected comparisons: * p < 0.05, ** p < 0.01. Values represent means ± SEM. (Abbreviations: p - post).
To further characterize the regional distribution of Htr2a expression in response to SD, and in the absence of Egr3, we conducted RNAscope in situ hybridization. Figure 2D shows that 6h of SD increases Htr2a expression in WT mice in a region-specific manner. Compared to WT mice, Egr3−/− mice have lower levels of Htr2a mRNA throughout most of the cortex, sparing selected brain regions. SD may produce small increases in some regions of Egr3−/− mice, though this is less consistent than in WT mice.
In WT mice that did not undergo SD (SDc), RNAscope in situ hybridization showed a distribution pattern of Htr2a mRNA similar to prior studies in rats using radioactive in situ hybridization 40, 41. This includes high levels of expression in the anterior frontal cortex, particularly in a thin layer (L) of cells at the L1/L2 border and in the deep L2/3 – L5 (L4 not present in anterior frontal cortex), with relatively lower signal in L6a. Also consistent with prior studies, Htr2a mRNA levels decrease with progression toward more posterior cortical regions, with notable reductions in L2/3, less drastic reductions in expression in L5, overall minimal L6a expression, and strong expression in L6b bordering the corpus callosum. In the posterior frontal cortex (Bregma +2.1 – +0.14) expression of Htr2a is present in L2/3 in the somatomotor areas (MOs, MOp) and medial somatosensory cortex (SSp), but notably absent from the lateral SSp, and SSs, with posterior progression. This absence of Htr2a in L2/3 in the SSp/s is evident in prior studies in rat as well 40, 41.
In areas outside of the cortex, Htr2a expression in WT SDc animals is seen in the piriform cortex (PIR), olfactory tubercle (OT), taenia tecta dorsal and ventral (TTd/TTv), anterior olfactory nucleus (AON), and substantia innomata (SI) with strong expression in the claustrum (CL) and endopiriform cortex (dorsal) (EPd). Low level Htr2a expression is also seen in patches in the caudate/putamen (CP) as well as in the lateral and medial septum (LS, MS). Low level expression is also seen in the hypothalamus.
Following sleep deprivation in WT mice, the overall level of Htr2a in the anterior frontal cortex (anterior to Bregma +2.1) does not appear to be increased compared to SDc, however the laminar distribution that is apparent in SDc animals becomes less clearly delineated, suggesting a potential increase in expression in some regions and decrease in expression in others. In contrast to the cortex, in this anterior part of the brain SD produces a marked increase in expression in the olfactory regions, including the AON, OT, as well as the PIR (particularly the pyramidal layer, PIR2), EPd, and TTd & TTv, compared to WT SDc mice.
In the PFC (Bregma +2.1 – +0.14) SD increases overall Htr2a expression in WT mice, while also resulting in a more diffuse pattern, with loss of clear laminar signal, compared to SDc. Cortical areas in which SD produces the greatest increases in Htr2a levels include the anterior cingulate (ACAv, ACAd), & somatomotor areas (MOs, MOp), as well as more lateral regions, including the agranular insular area (AI). Notably, Htr2a expression in L6b is unchanged or only slightly increased by SD in WT mice. SD induces diffuse Htr2a expression in the CP, and potentially small increases in the MS and hypothalamus (this was not consistent across different animals in the same group).
Compared with WT mice, in Egr3−/− mice that did not undergo SD (SDc) Htr2a expression is reduced throughout the cortex, an effect that becomes more pronounced with progression in the posterior direction. The exceptions to this Egr3-depencence of Htr2a expression are the cells in L6b, as well as in the CL and EP and PIR, in which expression is only mildly reduced in Egr3−/− mice. Despite the reduced expression, a clear laminar distribution remains in the SDc mice, with most of the residual cortical expression in L5 & L4, and almost no expression evident in L2/3.
Despite the reduced expression of Htr2a in Egr3−/− mice, SD does appear to increase Htr2a mRNA in some brain regions of these animals. This induction is not consistently seen in the anterior frontal cortex but is clear in more posterior cortical regions (most evident in the ACA and MO, the same regions where SD increases expression in WT mice). L6b expression is only mildly increased, if at all, by SD in Egr3−/− mice. In regions outside of the cortex, SD increases Htr2a expression in the EPd, PIR, TT, AON, and OT, in Egr3−/− mice, though not to the levels seen in WT mice. Expression in the CL is not obviously affected by SD in Egr3−/− mice.
To determine if SD also increases 5-HT2AR protein levels, we performed receptor autoradiography with the selective 5-HT2AR antagonist 3H-M100907 on brain sections from WT and Egr3−/− mice at baseline and following 8h SD (to allow time for translation of mRNA) (Fig. 3A). We found that, in the AFC, SD results in an overall increase in 5-HT2AR binding that differs between genotypes, but this increase is not significant in either WT or Egr3−/− genotypes alone (Fig. 3B). However, SD increases WT expression sufficiently to produce a significant difference in 5-HT2AR levels between WT and Egr3−/− mice that is not present in SDc animals (Fig. 3B, comparison between WT and Egr3−/− in the SD groups). In the PFC, 8h of SD significantly increases 5-HT2AR levels in WT mice but not in Egr3−/− mice (Fig. 3C). In addition, 5-HT2AR levels are significantly greater in WT than Egr3−/− mice in this region both at baseline (replicating our prior radioligand binding assay findings 25), and following SD. In the MPC, where endogenous Htr2a expression is lower than in more anterior cortical regions, SD does not increase 5-HT2AR levels in WT or Egr3−/− mice (Fig. 3D), and levels of 5-HT2AR are lower in Egr3−/− mice than WT mice under at baseline and following SD. Figures 3E–G show representative autoradiographic images from WT and Egr3−/− mice under SDc and SD conditions.
Figure 3. SD increases 5-HT2AR levels in the PFC of WT mice in an Egr3-dependent manner.
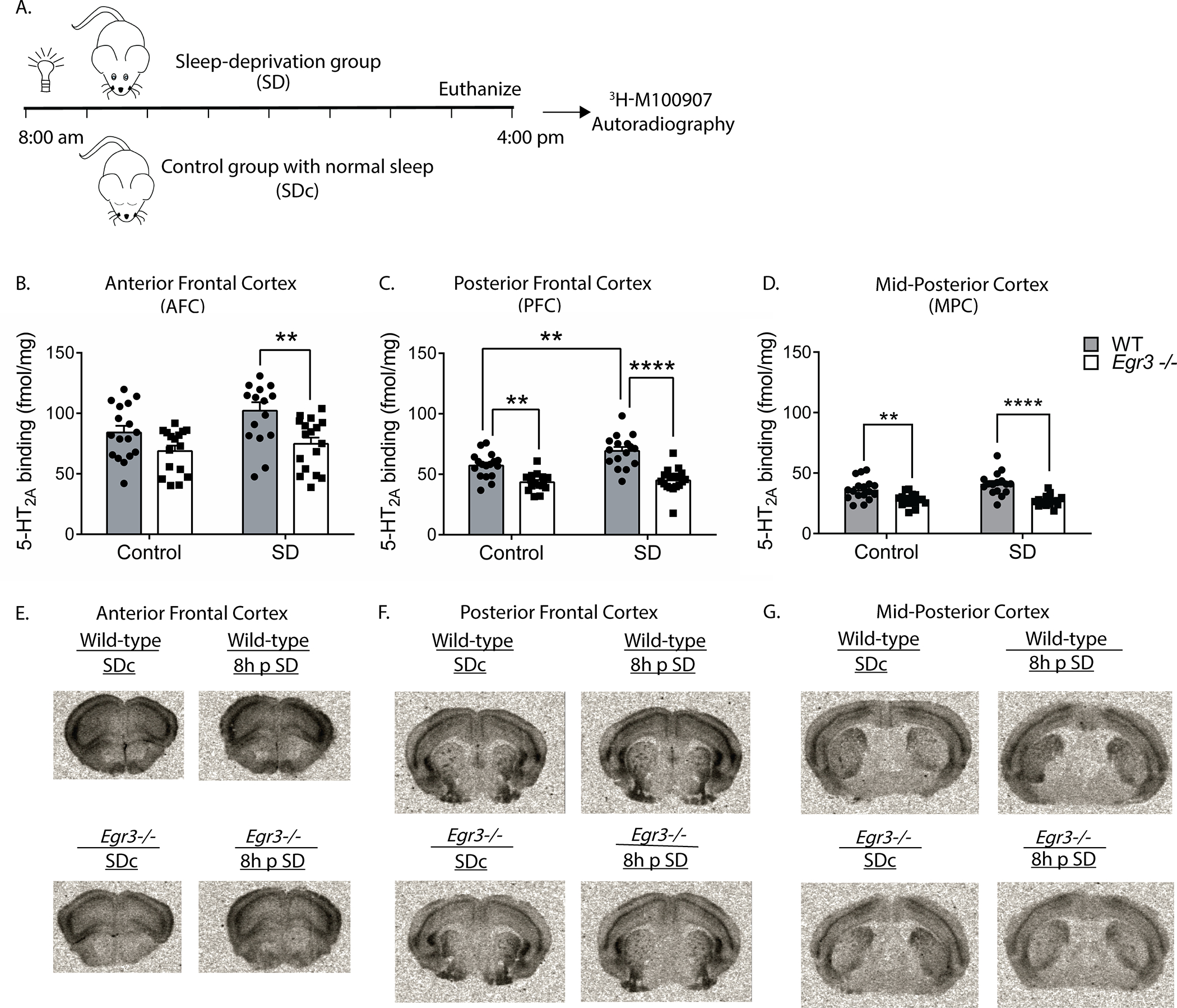
(A) 8h SD protocol. Quantification of 3H-M100907 binding autoradiography shows that SD, compared with SDc: (B) in AFC results in significantly greater 5-HT2AR levels in WT mice than Egr3−/− mice after SD (two-way ANOVA, sig. main effects of SD (F 1, 62 = 4.61, p = 0.0358) and genotype (F 1, 62 = 14.78, p = 0.0003); (C) in the PFC SD significantly upregulates 5-HT2AR levels in WT, but not Egr3−/−, mice (two-way ANOVA, sig. interaction between SD and genotype (F 1, 62 = 4.18, p = 0.0451)). (D) In the MPC, SD did not significantly increase 5-HT2AR levels; notably, 5-HT2ARs were lower in Egr3−/− mice than WT under both basal (SDc) and SD conditions (two-way ANOVA, sig. main effect of genotype (F 1, 62 = 38.79, p < 0.0001)) but not of SD (F 1, 62 = 1.371, p = 0.2461). Representative 3H-M100907 autoradiography images of brain tissue sections from (E) AFC, (F) PFC, and (G) MPC. For experiments in (B-D) WT: SDc, n = 17; SD, n = 16; Egr3−/−: SDc, n = 16; SD, n = 17. Bonferroni-corrected comparisons: * p < 0.05, ** p < 0.01, *** p < 0.001. Values represent means ± SEM.
These data reveal the novel finding that 5-HT2ARs can be upregulated in the PFC in a matter of hours in response to an environmental stimulus, and that this requires Egr3. The results suggest that EGR3, an activity dependent IEG transcription factor, may directly regulate expression of Htr2a in response to environmental events. For this to be happening, EGR3 would have to be expressed in the same cells as Htr2a and EGR3 consensus binding sequences would have to be present in the Htr2a promoter. Our prior study demonstrated that the former is true 27. To determine the other requirement, we used the FIMO program 35 which revealed two high probability EGR3 binding sites in the Htr2a promoter, a distal binding site at −2777 bp (site A, AGGAGGGGGAGTCT) and a proximal site at −61 bp (site B, GCGCGGGGGAGGGG) upstream of the start ATG (Fig. 4A).
Figure 4. EGR3 binds to the Htr2a promoter in frontal cortex.
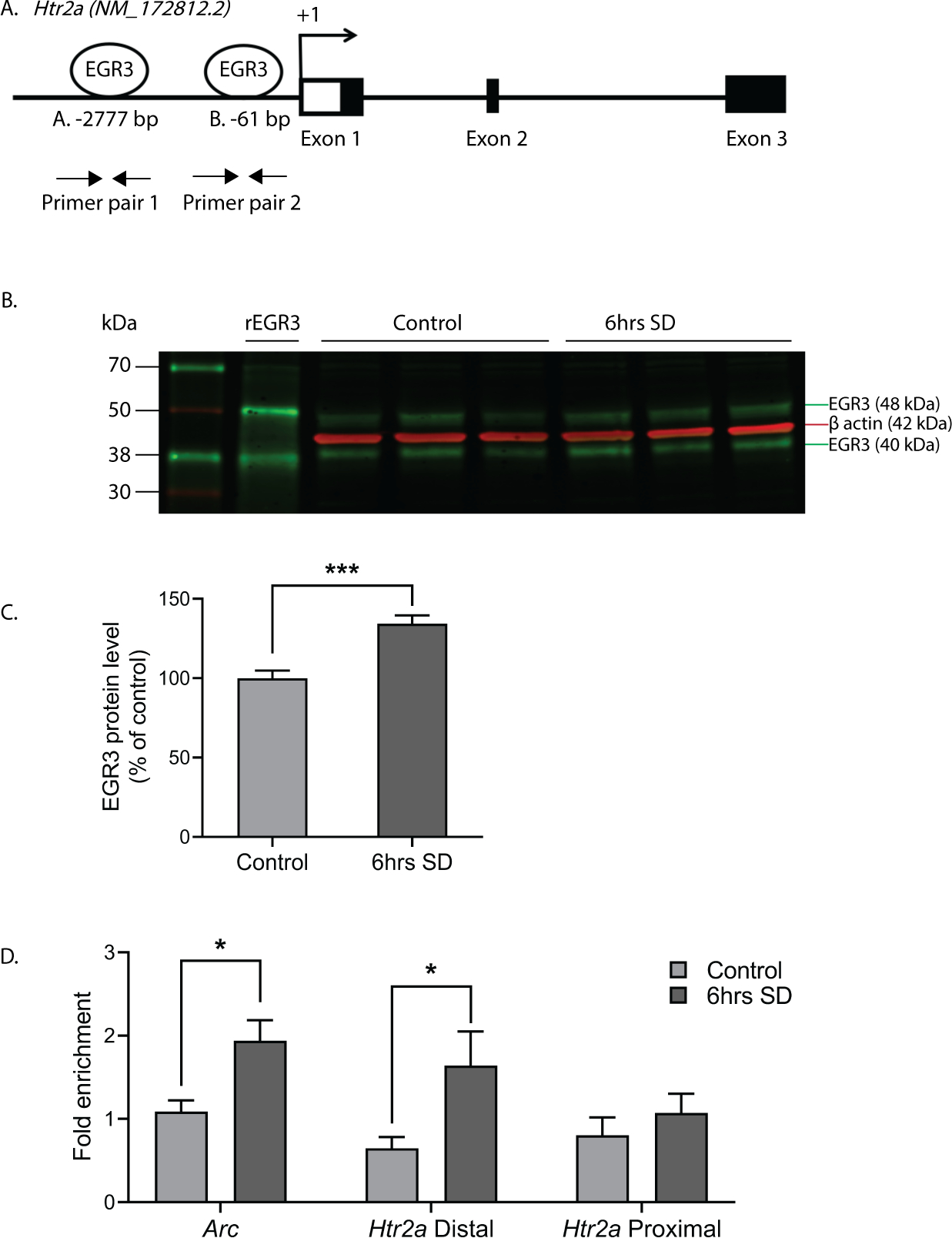
(A) Schematic showing high probability EGR3 consensus binding sites in the Htr2a promoter. (B) Representative Western blot image (n = 3 shown of n = 11 per group, Chameleon Duo Pre-Stained Protein Ladder (Li-COR Biosciences) and (C) Western blot average protein levels, show significant upregulation of activity dependent EGR3 protein following 6h of SD in WT frontal cortex (t20 = 4.778; p = 0.0001; n = 11 per group). (D) ChIP-qPCR shows SD increases binding of EGR3 to Arc promotor (positive control; SDc, n = 8; SD, n = 10) and Htr2a distal promoter (SDc, n = 9; SD, n = 9), but not Htr2a proximal promoter (SDc, n = 9; SD, n = 11), in frontal cortex tissue (Respectively, t16 = 2.802; p = 0.0128, t16 = 2.297; p = 0.0354, and t18 = 0.8463; p = 0.4085). Unpaired student’s t-test, * p < 0.05, *** p < 0.001. Values represent means ± SEM. (Abbreviations: rEGR3 - recombinant EGR3 protein).
To determine whether EGR3 protein binds to these sites in mouse cortex, we conducted chromatin immunoprecipitation (ChIP). As an IEG, Egr3 is expressed in a stimulus dependent manner. Under basal, unstimulated conditions, levels of EGR3 protein in the brain are low, and little binding to the promoters of target genes would be expected. SD, which increases Egr3 mRNA (26, 27 and Fig. 1), should increase EGR3 protein levels and, we expected, binding to the Htr2a promoter. Figures 4B – 4C show that 6h of SD significantly increases EGR3 protein levels in the frontal cortex of WT mice, measured by Western blot. Next, we conducted ChIP on samples of chromatin from WT mice that underwent 6h of SD compared to SDc mice. We used the promoter region of activity-regulated cytoskeleton associated protein (Arc) that includes a validated EGR3 binding domain as a positive control 36. Figure 4D shows that SD significantly increased EGR3 binding to the Arc promoter region, as well as to the distal Htr2a promoter, compared to SDc conditions. EGR3 binding to the proximal promoter was not significantly changed following SD.
To confirm that the binding of EGR3 to the Htr2a promoter results in a change in gene expression, we conducted in vitro luciferase-reporter assays. We co-transfected neuro2a cells with luciferase/SEAP constructs driven by either the positive control Arc promoter 36, the distal Htr2a promoter, or the proximal Htr2a promoter, with either a CMV vector overexpressing EGR3, or a control empty CMV vector (Fig. 5A–C). Figure 5D shows Western blot results validating EGR3 overexpression in the cell culture assays. We found that both regions of the Htr2a promoter containing high-probability EGR3 binding sites (Fig. 4A) drive expression of luciferase in response to EGR3 expression. EGR3 expression induces a 4.9-fold increase in positive control Arc promoter-driven luciferase (Fig. 5A) and 3.9-fold increase in the Htr2a distal promoter-driven luciferase (Fig. 5B). In addition, although the proximal Htr2a promoter did not show a statistically significant increase in EGR3 binding in the in vivo ChIP assay (Fig. 4D), in vitro expression of EGR3 induced a significant 4.2-fold increase in Htr2a proximal promoter-driven luciferase signal, compared to CMV vector alone (Fig. 5C). These results suggest that the physiologic stimulus of SD upregulates EGR3, which directly binds to the Htr2a promoter and activates Htr2a expression, results in increased levels of cortical 5-HT2ARs in the mouse brain.
Figure 5. EGR3 drives gene expression via binding sites in the Htr2a promoter and EGR3 and HTR2A expression is reduced in schizophrenia brains.
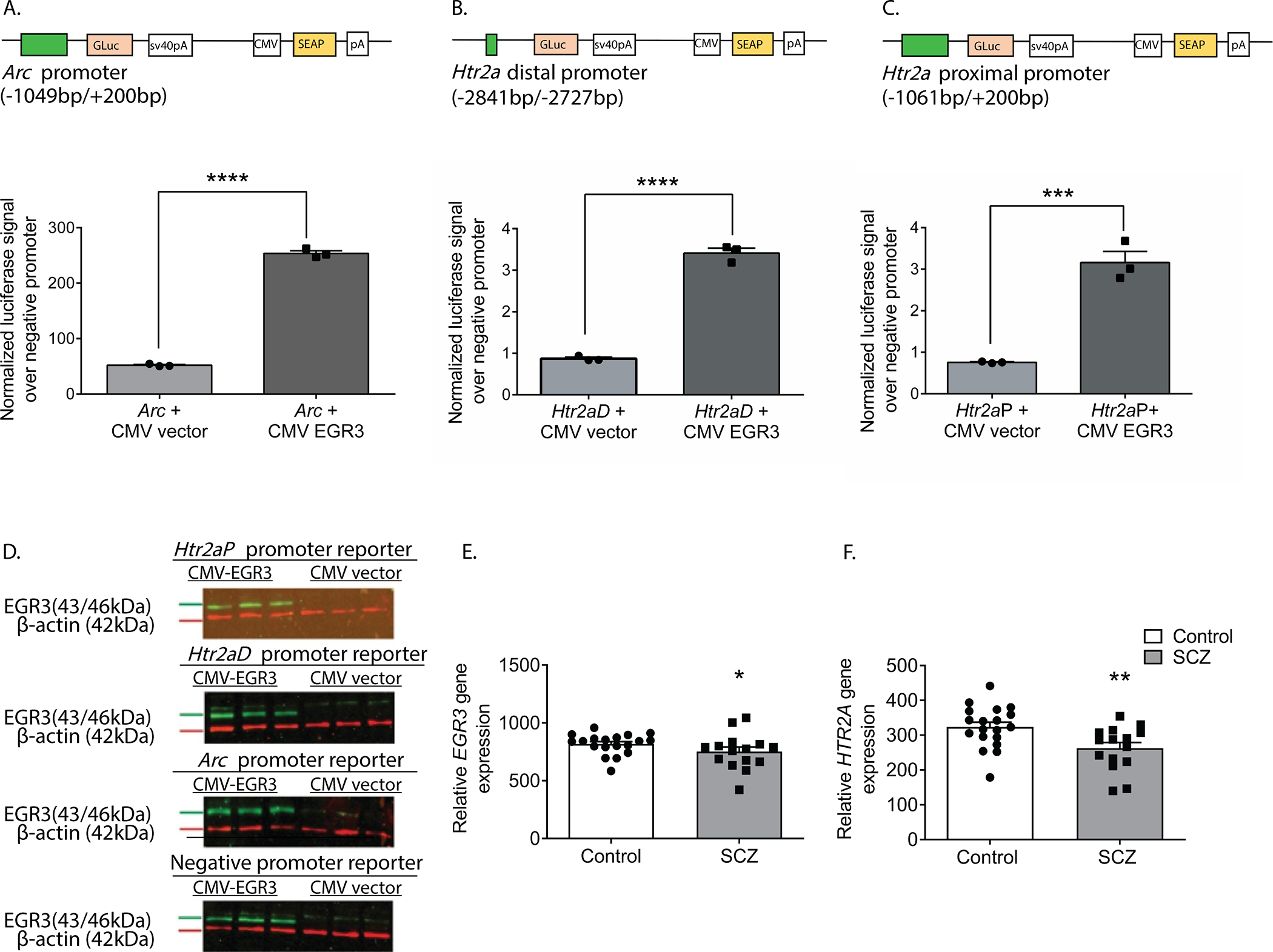
(A-C) Schematics of dual luciferase/SEAP reporter constructs containing EGR consensus binding sites and results of in vitro assays in neuro2a cells. CMV-driven EGR3 overexpression significantly upregulates expression of luciferase reporters driven by (A) Arc promoter (t4 = 42.28; p < 0.0001), (B) Htr2aD distal promoter (t4 = 21.17; p < 0.0001), and (C) Htr2aP proximal promoter (t4 = 8.977, p = 0.0009) regions. (D) Western blot validation of EGR3 expression following transfection with CMV-EGR3 versus CVM empty vector, from cultures expressing reporter constructs driven by promoters from Arc, Htr2aD, Htr2aP, or negative promoter control vector. Unpaired student’s t-test, (A-D) n = 3 per group. (E, F) EGR3 and HTR2A mRNA levels are significantly decreased in human brain tissue samples from the prefrontal cortex of schizophrenia patients compared to controls. Microarray (Robust Multi-Array Average) gene expression data derived from NCBI Geo database GSE53987 showing significant decrease in (E) EGR3 (U = 81, p = 0.0331) expression and (F) HTR2A (U = 63, p = 0.0050) expression, in control (n = 19) vs. schizophrenia (n = 15) patients. Mann-Whitney U test, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Values represent means ± SEM. (Abbreviations: CMV - cytomegalovirus, GLuc - Gaussia luciferase, SEAP - secreted alkaline phosphatase).
As discussed above, numerous studies have reported deficient levels of 5-HT2ARs in schizophrenia patients, as well as decreased levels of HTR2A in postmortem patient brain tissue. If our findings showing that EGR3 regulates Htr2a in mice are also true in humans, then the deficiency in HTR2A expression in the brains of schizophrenia patients could be a consequence of reduced expression of EGR3, a gene that has also been found to be reduced in schizophrenia patient brains. To further explore this possibility, we analyzed the results of a published gene expression dataset from postmortem prefrontal cortex tissue samples from schizophrenia patient and control brains in the NCBI GEO Database 38. Figure 5E and 5F show that both EGR3 and HTR2A expression are reduced in the schizophrenia subject samples compared to controls.
Discussion
The results we report here reveal several novel mechanisms of how 5-HT2AR levels in the brain are regulated. Our findings demonstrate that a physiological stimulus, SD, rapidly upregulates expression of both Htr2a expression and levels of membrane bound 5-HT2AR in the mouse brain in a matter of several hours. They also demonstrate that this environmentally induced upregulation requires the activity dependent IEG transcription factor EGR3, which binds to the Htr2a promoter in the frontal cortex in vivo and is able to activate expression of Htr2a promoter-driven reporter constructs in vitro. Thus, these results identify a direct transcriptional regulator of the 5-HT2AR in the frontal cortex and reveal a previously unrecognized characteristic of 5-HT2AR regulation; that it is environmental-stimulus responsive.
Our finding that 5-HT2ARs can be rapidly upregulated in the mouse frontal cortex in response to SD is supported by a study in humans that employed a similar stimulus. Elmenhorst and colleagues conducted a PET study using the 5-HT2AR selective radioligand [18F]altanserin to determine the effects on 24h of SD on 5-HT2AR levels in the frontal cortex of healthy subjects. Comparing PET scans conducted following a night of normal sleep to scans conducted the following day, after 24h of total sleep deprivation, they found a 9.6% increase of [18F]altanserin binding in neocortical regions, including the medial inferior frontal gyrus, insula, and anterior cingulate, parietal, sensomotoric, and ventrolateral prefrontal cortices 28. Their study was the first investigation of the effects of SD on cerebral cortex 5-HT2AR levels in either humans or animals 28. In discussing the possible mechanisms that may underlie their finding, the authors point to the fact that SD increases brain levels of serotonin (5-HT) in rodents 43. However, the effects of serotonin, and other agonists, on 5-HT2AR levels are complicated, with reports showing both upregulation and downregulation in different systems and in a manner that may be influenced by concentration or dosage 44. Our findings suggest, instead, that this increase in 5-HT2AR levels may result from direct upregulation of Htr2a by the IEG EGR3, which has been shown to be activated in the cortex of mice by 6h of SD by the Allen Institute for Brain Research group 26 and replicated by our laboratory here and in our prior study 27.
We chose to use SD as a stimulus in the current study because it is an environmental intervention that we and others have shown increases Egr3 expression 26, 27 and thus allowed us to test our hypothesis that induction of EGR3 should upregulate expression of Htr2a. As a class, IEGs are activated in the brain in response to a wide range of stimuli, including many types of stress 45. However, the expression of Egr3 in response to specific stressors has been less well investigated than that of many other IEGs. So, regardless of whether SD causes mice to experience “stress”, it is an effective intervention that demonstrates how rapidly an environmental stimulus can upregulate 5-HT2AR mRNA and protein levels in the brain; in just 6–8 hours.
Roles of 5-HT2ARs and Egr3 in stress response and memory
A significant body of research has suggested that stress influences 5-HT2AR expression. Numerous types of stress, including physical stressors of immobilization, chronic forced swimming and toe pinch, social stress, including maternal separation, as well as in utero exposure to lipopolysaccharide (to mimic bacterial infection), have been found to elevate levels of 5-HT2AR mRNA and protein, as well as to increase the frequency of 5-HT2AR-mediated behaviors (head-twitch response to 5-HT2AR agonists) (reviewed in Table 1 of 46). Though not all types of stress produce this effect 46. These findings suggest that increased expression of 5-HT2ARs may play a role in the brain’s response to environmental events, particularly stressful ones.
One of the critical responses of the brain to environmental events is the formation of memories. In fact, numerous studies suggest a role for 5-HT2ARs in memory formation. 5-HT2ARs facilitate associative learning (reviewed in Table 2 of 46), play a role in the retrieval of recognition memory 47, and modulate reconsolidation of contextual recognition memory 48. If EGR3 is regulating the increase in 5-HT2ARs expression in response to environmental stimuli, this suggests that the Egr3 gene should also be required for memory formation. In fact, our prior studies showed that Egr3 deficient (−/−) mice fail to habituate to a startling stimulus (demonstrating deficits in associative memory), display social interaction responses consistent with an inability to remember familiar mice (including persistent, non-habituating social investigation and increased aggressive behavior), and show deficits in Y-maze navigation indicative of defects in spatial memory formation 49.
We also found that Egr3−/− mice show a heightened response to stress 49. While at first this may appear inconsistent with the literature indicating that 5-HT2ARs are upregulated in response to stress, the findings are actually aligned. Without Egr3, mice fail to remember having experienced events, such as being handled by an investigator or having encountered mice they have lived with their whole lives, and they respond with the same high level of reactivity, and cortisol release, as mice being exposed for the first time 49. Thus, the findings that increased 5-HT2AR levels are important for associative and contextual learning, and the fact that Egr3−/− mice have deficient levels of 5-HT2AR that fail to normally upregulate in response to environmental stimuli (25, 27 and current results), provide a potential explanation for at least part of the heightened stress response of Egr3−/− mice 49. More research will be necessary to determine if Egr3 mediates the change in 5-HT2AR levels in response to other types of stress.
Regulation of Htr2a expression
Studies examining the mechanisms by which 5-HT2AR levels are regulated have largely focused on effects of agonist and antagonist actions. These studies demonstrate the complex nature of 5-HT2AR regulation. The endogenous ligand 5-HT, as well as both agonists and antagonists, can trigger rapid internalization of 5-HT2ARs 44, 50, 51. Though some studies have found that specific agonists can induce processes such as receptor desensitization without altering surface density 52, and other studies have shown a paradoxical increase in receptor density following antagonist exposure in specific cell types 53.
Yet, little is known about transcriptional regulation of Htr2a, or that environmental stimuli rapidly alter 5-HT2AR levels. One study employed bioinformatic resources and structural equation modeling to analyze the putative effect of a polymorphism in the human HTR2A promoter on transcription factor binding in subjects with chronic fatigue syndrome 54. Their findings suggested a diagnosis-specific interaction between methylation of an E47 transcription factor binding site and HTR2A expression levels in peripheral blood mononuclear cells in a manner influenced by cortisol level. However, the functionality of these transcription factor binding sites was not examined in that study 54. Thus, our findings are novel in identifying direct regulation of Htr2a by an activity dependent IEG.
5-HT2ARs in schizophrenia
5-HT2ARs are abundantly expressed in the neocortex and play important roles in cognition, mood and sleep, processes that are disrupted across numerous psychiatric disorders. Investigations of drugs that act as agonists at 5-HT2ARs have recently experienced a resurgence in the search for treatments for severe psychiatric symptoms ranging from depression 55 to anxiety disorders such as PTSD 56–58. Yet decades of clinical practice and research have demonstrated a particular importance of 5-HT2ARs in schizophrenia.
Schizophrenia is characterized by abnormalities in perception, thinking and memory (exemplified by hallucinations, delusions and cognitive deficits). The fact that 5-HT2ARs mediate the hallucinogenic effects of numerous drugs including LSD, psilocybin, and mescaline 5, 6 suggest the possibility that this receptor may also influence the hallucinations and perceptual disturbances of schizophrenia. One of the most important discoveries that demonstrated a critical role for 5-HT2ARs in schizophrenia symptomatology was the discovery that clozapine, one of the most effective antipsychotic medications to date, binds with high affinity to this receptor 59. In fact, 5-HT2AR binding is an essential feature of second-generation antipsychotics, which were modeled after clozapine and are the first-line of treatment for psychotic disorders 8, 59. Furthermore, the 5-HT2AR inverse agonist pimavanserin is effective for treatment of psychosis 7. Finally, as far back as 1976 numerous post-mortem and in vivo studies have revealed that 5-HT2AR (or 5-HT2R) levels are reduced in schizophrenia patients’ brains, and specifically in the prefrontal cortex 14, 15, 19, 22, 60, 61.
The prefrontal cortex has long been hypothesized to be a central brain region involved in schizophrenia pathogenesis 62, 63. Activity in this region is essential for spatial working memory, dysfunction in which may contribute to the cognitive deficits that characterize schizophrenia (reviewed in 64). Our results show that the environmental stimulus of SD increases levels of Egr3 and 5-HT2AR mRNA and protein in the antero-posterior domains of mouse cortex corresponding to the human prefrontal cortex, particularly the anterior cingulate cortex (ACAv, ACAd) 65. These are also regions where we found that SD-induced 5-HT2AR expression is Egr3-dependent.
The fact that this process is mediated by an activity-dependent IEG transcription factor suggests the intriguing possibility that the reduced 5-HT2AR levels reported in schizophrenia patients may be a consequence of disrupted neural activity, resulting in insufficient activation of IEGs including EGR3. Such a hypothesis is intriguing in the context of recent findings that neural circuit connectivity, and the synchrony of neural oscillations, between the prefrontal cortex and other brain regions is disrupted in schizophrenia patients 66–68. It is also supported by findings of abnormal IEG expression in patients’ brains (69 and Fig. 5E).
In further support of this hypothesis, numerous EGR family genes are associated with risk for schizophrenia 70, 71. Although genome wide association studies (GWAS) have recently identified numerous loci believed to increase genetic risk for illnesses like schizophrenia, the mechanism by which environment may interact with these regions remains elusive. It is notable that, EGR1, EGR4, and NAB2 (a transcriptional co-regulator that alters gene expression via binding to the EGRs) each map to one of the 145 GWAS loci for schizophrenia 71. Although EGR3 itself is not within a GWAS locus, it interacts in co-regulatory feedback loops with the EGRs and NAB2 70, 72, and EGR3 expression is reduced in the brains of schizophrenia patients 73, 74, including in our analyses of data from the NCBI GEO database, which shows reduced levels of both EGR3 and HTR2A mRNA in the prefrontal cortex of schizophrenia patients compared with controls (Fig. 5E and 5F) 38.
Another feature of schizophrenia, although less commonly discussed, is sleep disruption, which includes fragmentation and overall decreased duration of sleep 75, 76. Our finding that SD increases expression of Egr3 and 5-HT2AR mRNA and protein may initially appear inconsistent with the numerous reports that expression of these genes is reduced in schizophrenia patients, since sleep disruption and loss is a common characteristic of this mental illness 75, 76. There could be several explanations for this. First, as an IEG, expression of Egr3 is rapidly increased in response to a stimulus and returns to baseline within hours of the stimulus 77. So, the timing of the study, or time of death (in the case of post-mortem studies), may influence whether the gene expression consequences of sleep disruption will still be detectable. However, an explanation we feel is more likely is that the altered neural activity in the prefrontal cortex of schizophrenia patients, which may be related to documented deficiencies in synaptic density 78 and abnormal circuit function 64, 66–68, could result in deficient induction of EGR3 in response to environmental stimuli, including SD. Our data suggest this would consequently result in deficient upregulation of 5-HT2ARs. Finally, genetic variations that affect expression of EGR family genes may also reduce the stimulus-dependent upregulation of 5-HT2ARs. The literature suggesting that this upregulation plays an important role in contextual and associative memory formation 46 suggests that such insufficient 5-HT2AR expression might contribute to the cognitive deficits of schizophrenia.
Together, these findings suggest that dysfunction in activity dependent EGR family IEGs, which include, and result in, decreased activity of EGR3, may contribute to the reported deficits in 5-HT2AR expression in schizophrenia patient brains. These findings thereby shed light on a potential mechanism whereby environment interacts with genetic variations to influence neurobiology that may contribute to the symptoms, and treatment, of neuropsychiatric illness.
Supplementary Material
Acknowledgments
We are grateful to L Muppana, and D Elizalde for animal colony maintenance and technical assistance, to A. Aden, A. Barkatullah, M. Charbel, and E. Offenberg for assistance with SD studies, to Javier González-Maeso, PhD and Allan Gulledge PhD for expert advice, and to the laboratory of Stanley Watson, MD, PhD, for providing instruction and reagents for situ hybridization. Funding: This work was supported by National Institutes of Health R01 MH097803 (ALG) and R21 MH113154–01A1 (ALG).
Footnotes
Conflicts of Interest
The authors have no conflicts of interest to report.
Supplementary information is available at MP’s website.
References
- 1.Raote I, Bhattacharya A, Panicker MM. Serotonin 2A (5-HT2A) Receptor Function: Ligand-Dependent Mechanisms and Pathways. In: Chattopadhyay A (ed). Serotonin Receptors in Neurobiology: Boca Raton (FL), 2007. [PubMed] [Google Scholar]
- 2.Geyer MA, Vollenweider FX. Serotonin research: contributions to understanding psychoses. Trends Pharmacol Sci 2008; 29(9): 445–453. [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Gimenez JF, Gonzalez-Maeso J. Hallucinogens and Serotonin 5-HT2A Receptor-Mediated Signaling Pathways. Curr Top Behav Neurosci 2018; 36: 45–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aghajanian GK, Marek GJ. Serotonin and hallucinogens. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 1999; 21(2 Suppl): 16S–23S. [DOI] [PubMed] [Google Scholar]
- 5.Nichols DE. Hallucinogens. Pharmacol Ther 2004; 101(2): 131–181. [DOI] [PubMed] [Google Scholar]
- 6.Halberstadt AL, Geyer MA. Multiple receptors contribute to the behavioral effects of indoleamine hallucinogens. Neuropharmacology 2011; 61(3): 364–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meltzer HY, Mills R, Revell S, Williams H, Johnson A, Bahr D et al. Pimavanserin, a serotonin(2A) receptor inverse agonist, for the treatment of parkinson’s disease psychosis. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 2010; 35(4): 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meltzer HY, Huang M. In vivo actions of atypical antipsychotic drug on serotonergic and dopaminergic systems. Progress in brain research 2008; 172: 177–197. [DOI] [PubMed] [Google Scholar]
- 9.Laruelle M, Abi-Dargham A, Casanova MF, Toti R, Weinberger DR, Kleinman JE. Selective abnormalities of prefrontal serotonergic receptors in schizophrenia. A postmortem study. Archives of general psychiatry 1993; 50(10): 810–818. [DOI] [PubMed] [Google Scholar]
- 10.Reynolds GP, Rossor MN, Iversen LL. Preliminary studies of human cortical 5-HT2 receptors and their involvement in schizophrenia and neuroleptic drug action. J Neural Transm Suppl 1983; 18: 273–277. [PubMed] [Google Scholar]
- 11.Eastwood SL, Burnet PW, Gittins R, Baker K, Harrison PJ. Expression of serotonin 5-HT(2A) receptors in the human cerebellum and alterations in schizophrenia. Synapse 2001; 42(2): 104–114. [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez-Maeso J, Ang RL, Yuen T, Chan P, Weisstaub NV, Lopez-Gimenez JF et al. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 2008; 452(7183): 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muguruza C, Moreno JL, Umali A, Callado LF, Meana JJ, Gonzalez-Maeso J. Dysregulated 5-HT(2A) receptor binding in postmortem frontal cortex of schizophrenic subjects. Eur Neuropsychopharmacol 2013; 23(8): 852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selvaraj S, Arnone D, Cappai A, Howes O. Alterations in the serotonin system in schizophrenia: a systematic review and meta-analysis of postmortem and molecular imaging studies. Neuroscience and biobehavioral reviews 2014; 45: 233–245. [DOI] [PubMed] [Google Scholar]
- 15.Rasmussen H, Frokjaer VG, Hilker RW, Madsen J, Anhoj S, Oranje B et al. Low frontal serotonin 2A receptor binding is a state marker for schizophrenia? Eur Neuropsychopharmacol 2016; 26(7): 1248–1250. [DOI] [PubMed] [Google Scholar]
- 16.Hurlemann R, Matusch A, Kuhn KU, Berning J, Elmenhorst D, Winz O et al. 5-HT2A receptor density is decreased in the at-risk mental state. Psychopharmacology (Berl) 2008; 195(4): 579–590. [DOI] [PubMed] [Google Scholar]
- 17.Dean B, Hayes W. Decreased frontal cortical serotonin2A receptors in schizophrenia. Schizophrenia research 1996; 21(3): 133–139. [DOI] [PubMed] [Google Scholar]
- 18.Ngan ET, Yatham LN, Ruth TJ, Liddle PF. Decreased serotonin 2A receptor densities in neuroleptic-naive patients with schizophrenia: A PET study using [(18)F]setoperone. The American journal of psychiatry 2000; 157(6): 1016–1018. [DOI] [PubMed] [Google Scholar]
- 19.Lopez-Figueroa AL, Norton CS, Lopez-Figueroa MO, Armellini-Dodel D, Burke S, Akil H et al. Serotonin 5-HT1A, 5-HT1B, and 5-HT2A receptor mRNA expression in subjects with major depression, bipolar disorder, and schizophrenia. Biological psychiatry 2004; 55(3): 225–233. [DOI] [PubMed] [Google Scholar]
- 20.Matsumoto I, Inoue Y, Iwazaki T, Pavey G, Dean B. 5-HT2A and muscarinic receptors in schizophrenia: a postmortem study. Neurosci Lett 2005; 379(3): 164–168. [DOI] [PubMed] [Google Scholar]
- 21.Serretti A, Drago A, De Ronchi D. HTR2A gene variants and psychiatric disorders: a review of current literature and selection of SNPs for future studies. Curr Med Chem 2007; 14(19): 2053–2069. [DOI] [PubMed] [Google Scholar]
- 22.Garbett K, Gal-Chis R, Gaszner G, Lewis DA, Mirnics K. Transcriptome alterations in the prefrontal cortex of subjects with schizophrenia who committed suicide. Neuropsychopharmacol Hung 2008; 10(1): 9–14. [PubMed] [Google Scholar]
- 23.Kang K, Huang XF, Wang Q, Deng C. Decreased density of serotonin 2A receptors in the superior temporal gyrus in schizophrenia--a postmortem study. Progress in neuro-psychopharmacology & biological psychiatry 2009; 33(5): 867–871. [DOI] [PubMed] [Google Scholar]
- 24.Burnet PW, Eastwood SL, Harrison PJ. 5-HT1A and 5-HT2A receptor mRNAs and binding site densities are differentially altered in schizophrenia. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 1996; 15(5): 442–455. [DOI] [PubMed] [Google Scholar]
- 25.Williams AA, Ingram WM, Levine S, Resnik J, Kamel CM, Lish JR et al. Reduced levels of serotonin 2A receptors underlie resistance of Egr3-deficient mice to locomotor suppression by clozapine. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 2012; 37(10): 2285–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thompson CL, Wisor JP, Lee CK, Pathak SD, Gerashchenko D, Smith KA et al. Molecular and anatomical signatures of sleep deprivation in the mouse brain. Front Neurosci 2010; 4: 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maple AM, Zhao X, Elizalde DI, McBride AK, Gallitano AL. Htr2a Expression Responds Rapidly to Environmental Stimuli in an Egr3-Dependent Manner. ACS Chem Neurosci 2015; 6(7): 1137–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elmenhorst D, Kroll T, Matusch A, Bauer A. Sleep deprivation increases cerebral serotonin 2A receptor binding in humans. Sleep 2012; 35(12): 1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tourtellotte WG, Milbrandt J. Sensory ataxia and muscle spindle agenesis in mice lacking the transcription factor Egr3. Nature genetics 1998; 20(1): 87–91. [DOI] [PubMed] [Google Scholar]
- 30.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25(4): 402–408. [DOI] [PubMed] [Google Scholar]
- 31.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007; 445(7124): 168–176. [DOI] [PubMed] [Google Scholar]
- 32.Allen Mouse Brain Atlas. https://mouse.brain-map.org/static/atlas, 2004, Accessed Date Accessed 2004 Accessed.
- 33.O’Donovan KJ, Baraban JM. Major Egr3 isoforms are generated via alternate translation start sites and differ in their abilities to activate transcription. Molecular and cellular biology 1999; 19(7): 4711–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salotti J, Sakchaisri K, Tourtellotte WG, Johnson PF. An Arf-Egr-C/EBPbeta pathway linked to ras-induced senescence and cancer. Molecular and cellular biology 2015; 35(5): 866–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics 2011; 27(7): 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Carter J, Gao X, Whitehead J, Tourtellotte WG. The neuroplasticity-associated arc gene is a direct transcriptional target of early growth response (Egr) transcription factors. Molecular and cellular biology 2005; 25(23): 10286–10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30(1): 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lanz TA, Joshi JJ, Reinhart V, Johnson K, Grantham LE 2nd, Volfson D. STEP levels are unchanged in pre-frontal cortex and associative striatum in post-mortem human brain samples from subjects with schizophrenia, bipolar disorder and major depressive disorder. PloS one 2015; 10(3): e0121744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Motulsky HJ, Brown RE. Detecting outliers when fitting data with nonlinear regression - a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 2006; 7: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mengod G, Pompeiano M, Martinez-Mir MI, Palacios JM. Localization of the mRNA for the 5-HT2 receptor by in situ hybridization histochemistry. Correlation with the distribution of receptor sites. Brain research 1990; 524(1): 139–143. [DOI] [PubMed] [Google Scholar]
- 41.Pompeiano M, Palacios JM, Mengod G. Distribution of the serotonin 5-HT2 receptor family mRNAs: comparison between 5-HT2A and 5-HT2C receptors. Brain research Molecular brain research 1994; 23(1–2): 163–178. [DOI] [PubMed] [Google Scholar]
- 42.Weber ET, Andrade R. Htr2a Gene and 5-HT(2A) Receptor Expression in the Cerebral Cortex Studied Using Genetically Modified Mice. Front Neurosci 2010; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez-Rodriguez F, Wilson CL, Maidment NT, Poland RE, Engel J. Total sleep deprivation increases extracellular serotonin in the rat hippocampus. Neuroscience 2003; 121(2): 523–530. [DOI] [PubMed] [Google Scholar]
- 44.Van Oekelen D, Luyten WH, Leysen JE. 5-HT2A and 5-HT2C receptors and their atypical regulation properties. Life Sci 2003; 72(22): 2429–2449. [DOI] [PubMed] [Google Scholar]
- 45.Senba E, Ueyama T. Stress-induced expression of immediate early genes in the brain and peripheral organs of the rat. Neurosci Res 1997; 29(3): 183–207. [DOI] [PubMed] [Google Scholar]
- 46.Murnane KS. Serotonin 2A receptors are a stress response system: implications for post-traumatic stress disorder. Behav Pharmacol 2019; 30(2 and 3-Spec Issue): 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bekinschtein P, Renner MC, Gonzalez MC, Weisstaub N. Role of medial prefrontal cortex serotonin 2A receptors in the control of retrieval of recognition memory in rats. The Journal of neuroscience : the official journal of the Society for Neuroscience 2013; 33(40): 15716–15725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morici JF, Miranda M, Gallo FT, Zanoni B, Bekinschtein P, Weisstaub NV. 5-HT2a receptor in mPFC influences context-guided reconsolidation of object memory in perirhinal cortex. Elife 2018; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gallitano-Mendel A, Izumi Y, Tokuda K, Zorumski CF, Howell MP, Muglia LJ et al. The immediate early gene early growth response gene 3 mediates adaptation to stress and novelty. Neuroscience 2007; 148(3): 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhattacharyya S, Puri S, Miledi R, Panicker MM. Internalization and recycling of 5-HT2A receptors activated by serotonin and protein kinase C-mediated mechanisms. Proceedings of the National Academy of Sciences of the United States of America 2002; 99(22): 14470–14475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Willins DL, Berry SA, Alsayegh L, Backstrom JR, Sanders-Bush E, Friedman L et al. Clozapine and other 5-hydroxytryptamine-2A receptor antagonists alter the subcellular distribution of 5-hydroxytryptamine-2A receptors in vitro and in vivo. Neuroscience 1999; 91(2): 599–606. [DOI] [PubMed] [Google Scholar]
- 52.Roth BL, Palvimaki EP, Berry S, Khan N, Sachs N, Uluer A et al. 5-Hydroxytryptamine2A (5-HT2A) receptor desensitization can occur without downregulation. The Journal of pharmacology and experimental therapeutics 1995; 275(3): 1638–1646. [PubMed] [Google Scholar]
- 53.Akiyoshi J, Hough C, Chuang DM. Paradoxical increase of 5-hydroxytryptamine2 receptors and 5-hydroxytryptamine2 receptor mRNA in cerebellar granule cells after persistent 5-hydroxytryptamine2 receptor stimulation. Mol Pharmacol 1993; 43(3): 349–355. [PubMed] [Google Scholar]
- 54.Falkenberg VR, Gurbaxani BM, Unger ER, Rajeevan MS. Functional genomics of serotonin receptor 2A (HTR2A): interaction of polymorphism, methylation, expression and disease association. Neuromolecular Med 2011; 13(1): 66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carhart-Harris RL, Bolstridge M, Rucker J, Day CM, Erritzoe D, Kaelen M et al. Psilocybin with psychological support for treatment-resistant depression: an open-label feasibility study. Lancet Psychiatry 2016; 3(7): 619–627. [DOI] [PubMed] [Google Scholar]
- 56.Grob CS, Danforth AL, Chopra GS, Hagerty M, McKay CR, Halberstadt AL et al. Pilot study of psilocybin treatment for anxiety in patients with advanced-stage cancer. Archives of general psychiatry 2011; 68(1): 71–78. [DOI] [PubMed] [Google Scholar]
- 57.Griffiths RR, Johnson MW, Carducci MA, Umbricht A, Richards WA, Richards BD et al. Psilocybin produces substantial and sustained decreases in depression and anxiety in patients with life-threatening cancer: A randomized double-blind trial. Journal of psychopharmacology 2016; 30(12): 1181–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nichols DE, Johnson MW, Nichols CD. Psychedelics as Medicines: An Emerging New Paradigm. Clin Pharmacol Ther 2017; 101(2): 209–219. [DOI] [PubMed] [Google Scholar]
- 59.Meltzer HY. The role of serotonin in antipsychotic drug action. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 1999; 21(2 Suppl): 106S–115S. [DOI] [PubMed] [Google Scholar]
- 60.Bennett JP Jr., Enna SJ, Bylund DB, Gillin JC, Wyatt RJ, Snyder SH. Neurotransmitter receptors in frontal cortex of schizophrenics. Archives of general psychiatry 1979; 36(9): 927–934. [DOI] [PubMed] [Google Scholar]
- 61.Arora RC, Meltzer HY. Serotonin2 (5-HT2) receptor binding in the frontal cortex of schizophrenic patients. J Neural Transm Gen Sect 1991; 85(1): 19–29. [DOI] [PubMed] [Google Scholar]
- 62.Goldman-Rakic PS. Prefrontal cortical dysfunction in schizophrenia : the relevance of working memory. Psychopathology and the Brain 1991: 1–23. [Google Scholar]
- 63.Berman KF, Weinberger DR. The prefrontal cortex in schizophrenia and other neuropsychiatric diseases: in vivo physiological correlates of cognitive deficits. Progress in brain research 1990; 85: 521–536; discussion 536–527. [DOI] [PubMed] [Google Scholar]
- 64.Kupferschmidt DA, Gordon JA. The dynamics of disordered dialogue: Prefrontal, hippocampal and thalamic miscommunication underlying working memory deficits in schizophrenia. Brain Neurosci Adv 2018; 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Laubach M, Amarante LM, Swanson K, White SR. What, If Anything, Is Rodent Prefrontal Cortex? eNeuro 2018; 5(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sheffield JM, Barch DM. Cognition and resting-state functional connectivity in schizophrenia. Neuroscience and biobehavioral reviews 2016; 61: 108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bahner F, Meyer-Lindenberg A. Hippocampal-prefrontal connectivity as a translational phenotype for schizophrenia. Eur Neuropsychopharmacol 2017; 27(2): 93–106. [DOI] [PubMed] [Google Scholar]
- 68.Hu ML, Zong XF, Mann JJ, Zheng JJ, Liao YH, Li ZC et al. A Review of the Functional and Anatomical Default Mode Network in Schizophrenia. Neurosci Bull 2017; 33(1): 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018; 362(6420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Marballi KK, Gallitano AL. Immediate Early Genes Anchor a Biological Pathway of Proteins Required for Memory Formation, Long-Term Depression and Risk for Schizophrenia. Frontiers in behavioral neuroscience 2018; 12: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pardinas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nature genetics 2018; 50(3): 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kumbrink J, Kirsch KH, Johnson JP. EGR1, EGR2, and EGR3 activate the expression of their coregulator NAB2 establishing a negative feedback loop in cells of neuroectodermal and epithelial origin. Journal of cellular biochemistry 2010; 111(1): 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamada K, Gerber DJ, Iwayama Y, Ohnishi T, Ohba H, Toyota T et al. Genetic analysis of the calcineurin pathway identifies members of the EGR gene family, specifically EGR3, as potential susceptibility candidates in schizophrenia. Proceedings of the National Academy of Sciences of the United States of America 2007; 104(8): 2815–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mexal S, Frank M, Berger R, Adams CE, Ross RG, Freedman R et al. Differential modulation of gene expression in the NMDA postsynaptic density of schizophrenic and control smokers. Brain research Molecular brain research 2005; 139(2): 317–332. [DOI] [PubMed] [Google Scholar]
- 75.Kaskie RE, Graziano B, Ferrarelli F. Schizophrenia and sleep disorders: links, risks, and management challenges. Nat Sci Sleep 2017; 9: 227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferrarelli F Sleep Abnormalities in Schizophrenia: State of the Art and Next Steps. The American journal of psychiatry 2021; 178(10): 903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamagata K, Kaufmann WE, Lanahan A, Papapavlou M, Barnes CA, Andreasson KI et al. Egr3/Pilot, a zinc finger transcription factor, is rapidly regulated by activity in brain neurons and colocalizes with Egr1/zif268. Learning & memory 1994; 1(2): 140–152. [PubMed] [Google Scholar]
- 78.Glausier JR, Lewis DA. Dendritic spine pathology in schizophrenia. Neuroscience 2013; 251: 90–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.