

Abstract
Substance use disorders (SUDs) are conditions in which the use of legal or illegal substances, such as nicotine, alcohol or opioids, results in clinical and functional impairment. SUDs and more generally, substance use, are genetically complex traits that are enormously costly on an individual and societal basis. The past few years have seen remarkable progress in our understanding of the genetics, and therefore the biology, of substance use and abuse. Various studies — including of well-defined phenotypes in deeply phenotyped samples, as well as broadly defined phenotypes in meta-analysis and biobank samples — have revealed multiple risk loci for these common traits. A key emerging insight from this work establishes a biological and genetic distinction between quantity and/or frequency measures of substance use (which may involve low levels of use without dependence), versus symptoms related to physical dependence.
Table of contents blurb:
In this Review, Gelernter and Polimanti discuss how recent large-scale studies have provided insights into the genetics and biology of substance use and abuse. By considering a range of addictive substances (both legal and illegal), they describe the genetic commonalities and distinctions among use and dependency phenotypes for these substances.
Introduction
It is well known and often said that use of addictive substances [G] is highly destructive to individuals and to society as a whole. Substance use [G], legal and illegal, is widespread in most populations. Individuals who use substances frequently or heavily can lose control of their substance use behaviours, resulting in substance use disorders [G] (SUDs) — substance use that is no longer under the person’s full control, often because of physiological substance dependence [G]. Over the years, there has been considerable debate over the extent to which SUDs should be considered habits or behaviours on the one hand, or medical illnesses on the other. A rather dim view of these behaviours has been taken by some under the assumption that they can be controlled with sufficient effort; this view has influenced the stigma our society attaches to SUDs. The biological data, including the genetic data, tell a different story though: what begins as habit or volitional behaviour becomes a genetically influenced brain disease where substance abuse [G] creates a continuing need for the substance in the affected individual.
Substance use is notoriously difficult to treat. Treatment strategies have tended to focus on replacement (as for nicotine and opioid replacement) and abstinence, sometimes together with self-help groups or psychotherapy1–3. To provide better treatments, we require better understanding of the underlying biology. SUDs have long been known to be moderately heritable. In particular, genetic influences on SUD traits were shown decades ago via genetic epidemiology methods: twin and adoption studies4. This research was critical in establishing a basis for gene mapping in SUDs, but the twin-study framework was well established a decade ago and has advanced little relative to molecular gene mapping successes. Genetic studies have been considered a key tool for identifying targets to develop more effective treatments. However, we are just starting to comprehend and dissect the highly polygenic architecture of these complex traits. The main steps forward in this field have occurred recently and over just a few years. This is attributable to the advent of large biobanks and consortia that are allowing study of sample sizes that until recently were unimaginable.
Large-scale genome-wide studies of complex traits illustrate several key propositions. First, bigger is better. Large genome-wide association studies (GWAS) can uncover novel biology. Second, similarly to other complex traits5, genetic findings from European populations (EUR; e.g., European-Americans) greatly exceed those from other populations (e.g., African-Americans (AAs)). Third, candidate genes [G] selected based on known biology, which have a dismal record in the field as a whole6, 7, have been successfully replicated by large-scale SUD GWAS in a few instances.
In this Review, we focus on the largest and the most consequential studies based on genome-wide designs (Figure 1; Table 1). We discuss legal and illegal substances separately. Why so? This is not simply an administrative, or legal, issue. The environmental effects of exposure to various substances play a critical role in determining how an individual’s genetic risk to become addicted [G] plays out; and legality plays a large role in exposure. Also, societies have tended to legalize those substances that are considered to be less harmful (at least at the time they are legalized) or to which exposure is deemed inevitable and impossible to control. The unequivocally illegal substances, for example cocaine and opioids, are considered very harmful, and their use has comparatively low prevalence; the unequivocally legal ones, especially alcohol and tobacco, are freely available to almost everyone and are used more widely. Cannabis straddles the two categories. We describe emerging biological insights, including from large-effect loci, and genetic overlap among SUDs and related traits. We also discuss how genomics studies can be complemented by other omics approaches such as epigenomics and transcriptomics for a more complete biological understanding. Studies based on animal models and pedigree analyses are not included in detail due to space limitations, but we address the ongoing debate about optimal use of animal models for SUD genetics.
Figure 1 |. From epidemiology and gene discovery to biology of substance use disorders.
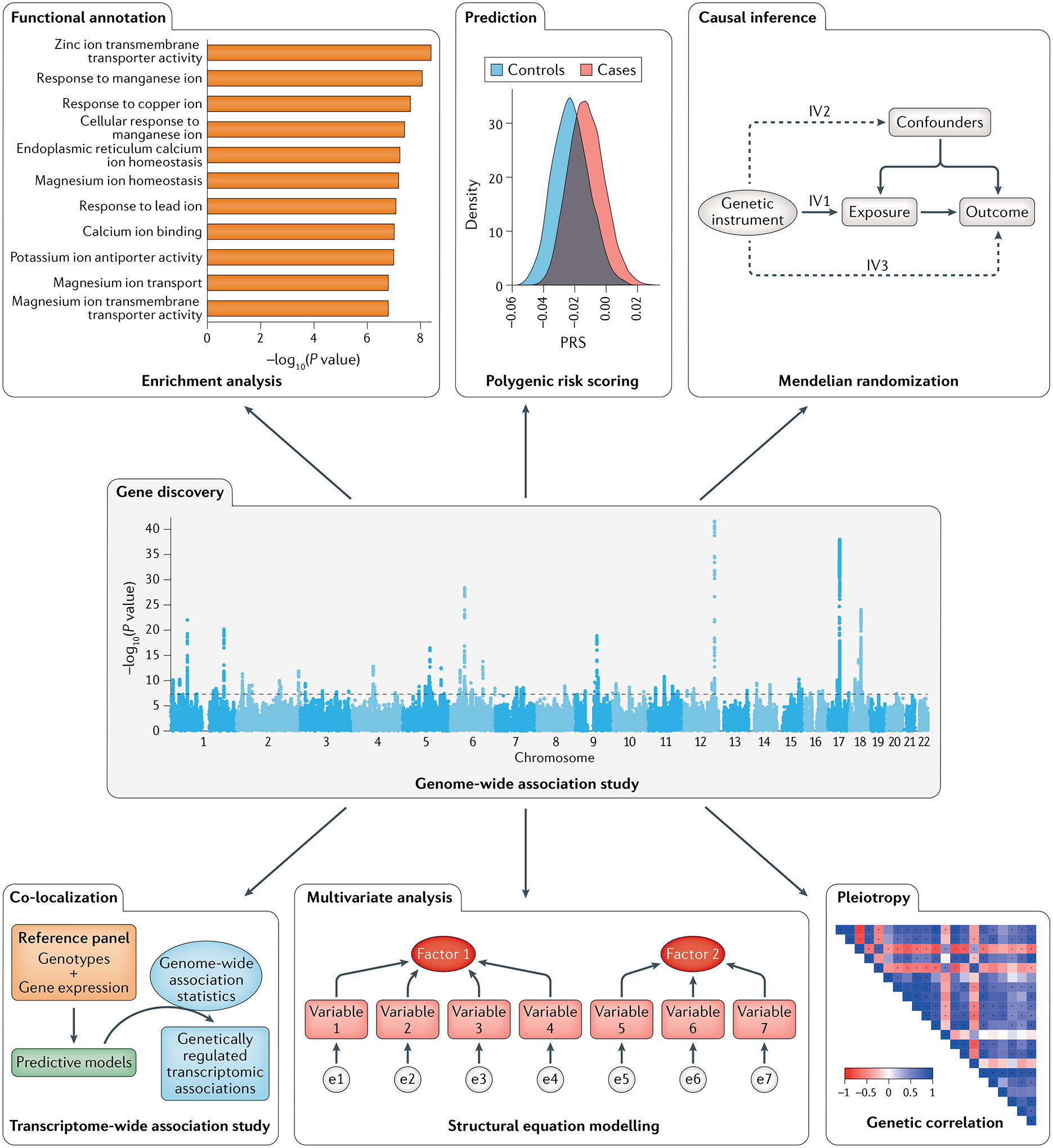
Genome-wide association datasets can be used as the basis for multiple analytical approaches to disentangle the biology of the traits investigated (e.g., cellular processes and molecular functions) and to understand the mechanisms underlying the associations observed in epidemiological studies.
Table 1 |.
Main phenotypes related to SUDs investigated in the largest genome-wide association studies available
| SUD outcome | Description | GWS loci | SNP-based heritability | Sample size | Refs |
|---|---|---|---|---|---|
| Alcohol | |||||
| Alcohol dependence | Alcohol dependence (AD) diagnosis based on DSM-IV or DSM-III-R criteria | 1 | 0.090±0.019 | 14,904/37,944 | 8 |
| Alcohol use disorder | AUD diagnosis based on ICD-9 and ICD-10 codes | 10 | 0.056±0.004 | 55,584 /218,807 | 14 |
| Problematic alcohol use | Meta-analysis of cohorts assessed using different phenotypic definitions including AD, AUD, and AUDIT-P | 42 | 0.068±0.004 | 300,789* | 13 |
| Alcohol use disorder identification test:consumption | AUDIT-C scores were derived from electronic health records | 13 | 0.068±0.005 | 272,842 | 14 |
| Drinks per week | Average number of standard drinks a subject reports consuming each week | 99 | 0.042±0.002 | 941,280 | 15 |
| Tobacco smoking | |||||
| Nicotine dependence | Nicotine dependence category (mild, moderate, severe) defined on the basis of the Fagerström Test for Nicotine Dependence (FTND) scores | 5 | 0.086±0.012 | 58,000 | 185 |
| Age of initiation of regular smoking | The age at which an individual first became a regular smoker | 10 | 0.047±0.003 | 341,427 | 15 |
| Cigarettes per day | Average number of cigarettes smoked per day binned in categories | 55 | 0.08±0.008 | 337,334 | 15 |
| Smoking initiation | Binary phenotype with ever regular smokers coded as cases and never regular smokers coded as controls | 378 | 0.078±0.002 | 557,337/ 674,754 | 15 |
| Smoking cessation | Binary phenotype with current smokers coded as cases and former smokers coded as controls | 24 | 0.046±0.002 | 139,453/407,766 | 15 |
| Smoking trajectories | Longitudinal trajectory groups for smoking status based on electronic health records | 20 | 0.187±0.01 0.058±0.005 |
286,118 | 28 |
| Opioids | |||||
| Opioid dependence | Opioid dependence (OD) diagnosis based on DSM-IV criteria. Opioid exposure defined as being exposed to opioids at least once | 0/1 | NS 0.28±0.1 |
4,503/4,173/32,500 | 41 |
| Opioid use disorder | OUD diagnosis and opioid exposure based on ICD-9 and ICD-10 codes | 1 | 0.113±0.018 | 15,756/99,039 | 44 |
| Opioid exposure | Opioid exposure defined as being exposed to opioids at least once | 1 | NS | 4,173/32,500 | 41 |
| Cannabis | |||||
| Cannabis use | Self-reported information on whether the participants had ever used cannabis during their lifetimes | 8 | 0.11±0.01 | 180,934* | 34 |
| Cannabis use disorder | CUD diagnosis based on DSM-IV or DSM-III-R criteria or ICD-10 codes | 2 | 0.067±0.006 | 20,916/363,116 | 37 |
| Age at first cannabis use | Age at first cannabis use assessed from questionnaires or clinical interviews | 5 | NS | 24,953 | 35 |
| Cocaine | |||||
| Cocaine dependence | DSM-IV symptom count for cocaine dependence DSM-IV cocaine dependence diagnosis |
1/0 | NA 0.30±0.06 |
9,760 2,085/4,293 |
46,47 |
For each trait, we report a brief description, the number of genome-wide significant (GWS) loci, the SNP-based heritability, and the sample size. For binary traits, we reported the number of cases and controls (i.e., cases/controls). For traits meta-analysing different types of phenotypic definitions, we reported the effective sample size (*). For smoking trajectories, we report the SNP-based heritability calculated for contrast I (current versus never) and contrast II (current versus mixed). With respect to opioid dependence, the case–control analysis was conducted considering opioid-exposed controls and opioid-unexposed controls separately. For this phenotype, we reported information (i.e., GWS loci, SNP-based heritability, and sample size) for both analyses (cases/opioid-exposed controls/opioid-unexposed controls). With respect to cocaine dependence, we reported results from two studies. AUD, alcohol use disorder; AUDIT, Alcohol Use Disorders Identification Test for consumption (AUDIT-C) or problems (AUDIT-P); CUD, cannabis use disorder; DSM, Diagnostic and Statistical Manual of Mental Disorders; ICD, International Classification of Diseases; NA, not available in the referenced publication; NS, not significant (p>0.05); SUD, substance use disorder.
Legal substances
Alcohol.
Alcohol use disorder (AUD) and related traits provide one of the very few examples of the survival into the current ‘Big Data’ era of large-effect loci initially identified by candidate gene studies8. We discuss these loci — the alcohol metabolism genes alcohol dehydrogenase 1B (class I), beta polypeptide (ADH1B) and aldehyde dehydrogenase 2 family member (ALDH2) — in the predisposition to alcohol use, abuse, and dependence, in the section “From large-effect risk loci to disease biology”. However, these large-effect alleles explain very little of the overall phenotypic variance, which is accounted for by the contribution of thousands of alleles with small effects (i.e., polygenicity). Large-scale GWAS are beginning to reveal the polygenic architecture of several alcohol-related traits, while identifying genetic differences between them that were not appreciated in the days of small-scale studies. Many traits have been considered in GWAS of alcohol use (Box 1). Alcohol use disorder (AUD by the Diagnostic and Statistical Manual of Mental Disorders fifth edition (DSM-5); alcohol dependence (AD) by DSM-IV and earlier) seeks to capture physiological dependence, which is an inability to discontinue use as opposed to use per se (as for all SUD diagnoses). The Alcohol Use Disorders Identification Test (AUDIT) is in clinical use and is collected by some biobank projects (Box 2). The AUDIT can be separated into two sections, the AUDIT-C, focused on consumption, and the AUDIT-P, focused on problems caused by alcohol use9. The AUDIT was designed to be able to detect less-severely affected drinkers. As such it identifies alcohol users who do not meet diagnostic criteria [G] for AD (as per DSM-IV10) or AUD (as per DSM-511). A GWAS including data from the UK Biobank sample and 23andMe (N = 141,958 participants) investigated AUDIT scores, but also AUDIT-C and AUDIT-P separately, and found several novel AUDIT associations (e.g., junctional cadherin 5 associated, (JCAD) and solute carrier family 39 member 13 (SLC39A13)); an additional key finding was a different pattern of genetic correlations for the two AUDIT subscales12. AUDIT-P, but not AUDIT-C, was correlated with a range of psychiatric traits. This very important observation has been confirmed and amplified upon in numerous other studies13, 14 and a similar pattern has been observed for some other substances: namely that dependence traits tend to be correlated more strongly with other psychiatric phenotypes than quantity/frequency-of-use traits.
Box 1 |. Major instruments used to define SUD traits in large-scale genome-wide association studies.
Different phenotyping strategies have been used by genetic studies to derive information regarding substance use disorder (SUD) traits from several thousand participants. Below we describe the main instruments used in large-scale genome-wide association studies.
DSM
The Diagnostic and Statistical Manual of Mental Disorders (DSM) is the evolving handbook published by the American Psychiatric Association used as the authoritative guide to the diagnosis of mental disorders. For addictive substances, DSM diagnostic criteria permit diagnoses of substance abuse, dependence, and use disorders, depending on the DSM version used; the diagnostic definitions are updated during the subsequent DSM revisions. In the latest DSM version, DSM-5189, substance use disorder (SUD) diagnosis combines the DSM-IV categories of substance abuse and substance dependence into a single disorder measured on a continuum from mild to severe based on criterion count.
ICD
The International Classification of Disease (ICD)190 is a standardized set of criteria maintained by the World Health Organization that is used globally to identify health trends and statistics globally and to report diseases and health conditions for clinical and research purposes. SUDs can be defined on the basis of algorithms that consider the occurrence of ICD codes related to substance abuse and dependence in the electronic health records of the participants involved. Similar to DSM, the ICD code set is revised over the years. Recent SUD genetic studies were mostly based on ICD-9 and ICD-10 codes.
AUDIT
The Alcohol Use Disorders Identification Test (AUDIT)9 is a 10-item screening instrument developed by the World Health Organization to evaluate alcohol consumption, drinking behaviours, and alcohol-related problems. The first three AUDIT items comprise the AUDIT-C which assesses quantity and frequency of drinking and heavy or binge drinking. The last seven AUDIT items contain the AUDIT-P which covers problems: symptoms related to alcohol dependence and problems resulting from drinking.
FTND
The Fagerström Test for Nicotine Dependence (FTND)24 is a standard instrument for assessing the severity of nicotine dependence. It includes six items that evaluate the quantity of cigarette consumption, the compulsion to use, and dependence, yielding a score ranging from 0 (no dependence) to 10 (highest dependence level).
Box 2 |. The major biobanks and collaborative projects that have provided insights into SUD genetics.
The high polygenicity seen in substance use disorders (SUDs) and related traits requires the analysis of large cohorts that may include hundreds of thousands of participants. To generate these large sample sizes, collaborative efforts are needed to integrate the work of many investigators and the support of adequate funding institutions.
Biobanks
23andMe191 is a biotechnology company that offers direct-to-consumer genetic testing. To date, 80% of the >12,000,000 23andMe consumers have opted-in to participate in research projects, completing online surveys regarding health-related outcomes. In collaborations with academic groups and pharmaceutical companies, these data are being used to investigate the genetics of common diseases and traits. 23andMe summary-level data can be accessed through the publication dataset access program via a data transfer agreement.
BioBank Japan (BBJ)192 is a registry of patients suffering from lifestyle-related diseases and various cancers. DNA was collected from all participants at baseline. Serum samples and clinical information were collected annually until 2013 for 200,000 participants. From 2013 to 2017, BBJ collected DNA and clinical information from an additional 67,000 patients with common diseases.
China Kadoorie Biobank193 recruited >510,000 adult participants from 10 geographically defined regions of China. Data were collected by questionnaires and physical measurements. Blood samples collected for each participant are stored long term for future studies. Additional clinical information is collected through linkage with established registries and health insurance databases.
deCODE Genetics194 is a biopharmaceutical company based in Reykjavík, Iceland. To date, deCODE gathered genotypic and medical data from >160,000 volunteer participants, comprising half of Iceland’s adult population. In addition to their Icelandic datasets, deCODE investigators analyse genetic and medical data from around the globe using proprietary statistical algorithms and informatics tools.
FinnGen195 is a personalized medicine project supported by public and private funding that is bringing together Finnish universities, hospitals, biobanks, and international pharmaceutical companies to improve human health through genetic research and identify new therapeutic targets and diagnostics. The project started in 2017 and it is expected to continue for ten years, recruiting 500,000 participants. FinnGen summary-level data are shared openly with the scientific community.
iPSYCH196 is a Danish national project with the goal of identifying the genetic causes and creating the basis for better treatment and prevention of psychiatric disorders. This cohort includes more than 130,000 Danes with genetic and environmental information relevant to the study of mental health.
The Million Veteran Program (MVP)197 is a national research program funded by the US Veterans Administration to learn how genes, lifestyle, and military exposures affect health and illness. Participants are US veterans. Since launching in 2011, >825,000 Veteran partners have been recruited in the MVP cohort. The MVP sample is remarkable for its population diversity, as it includes not only European-ancestry subjects but also substantial numbers of African-ancestry and Latinx subjects, and smaller numbers of other US populations. Reflecting historical participation in the US military, the MVP sample is mostly male. MVP limits access to its primary data to US Department of Veteran Affairs (VA) investigators at present, but makes summary statistics available freely at the time of publication of results.
The UK Biobank198 is an open-access resource available to investigate a wide range of serious and life-threatening illnesses. The cohort includes >500,000 participants with genome-wide data and detailed information regarding their diet, cognitive function, work history, health status, and other relevant phenotypes. Individual-level data are made available to investigators worldwide after an application process.
Collaborative efforts
GWAS and Sequencing Consortium of Alcohol and Nicotine (GSCAN)15 is an international genetic association meta-analysis consortium, aiming to aggregate genetic association findings of alcohol-drinking and tobacco-smoking traits across studies including millions of individuals.
International Cannabis Consortium (ICC)34 is a worldwide collaborative effort that aims to identify genetic variants underlying individual differences in cannabis use phenotypes, including lifetime cannabis use, age at first cannabis use, and quantity/frequency of cannabis use.
Psychiatric Genomics Consortium (PGC)199 is a collaborative effort including >800 investigators from >150 institutions in >40 countries with the goal to advance genetic discovery of biologically, clinically, and therapeutically meaningful insights. The PGC-SUD workgroup focuses on the study of use and misuse of alcohol, cannabis, cocaine, opioids, tobacco, and other illicit substances.
The easier the trait ascertainment, the larger the available samples; for the alcohol-related trait “drinks per week”, data have been collected in numerous studies. The current largest meta-analysis conducted by the GWAS & Sequencing Consortium of Alcohol and Nicotine use (GSCAN) for an alcohol-related trait concerns drinks per week, included a sample size of 941,280, with 99 risk loci identified (e.g., phosphodiesterase 4B (PDE4B) and cullin 3 (CUL3))15. This quantity/frequency measure tended not to show genetic correlation with other psychiatric traits, similar to AUDIT-C. As for AUDIT-C GWAS results, these quantity/frequency results are of great interest, but of less clear relevance to pathological AUD; much of the information would have derived from behaviour in the normal range.
While many databases collect quantity/frequency information for alcohol consumption, information regarding AUD per se — the more severe trait — is sparser. For more severe traits, the Million Veteran Program (MVP) sample has been particularly useful (Box 2). There have been three major GWAS of alcohol-related traits based on the MVP. The first examined maximum habitual alcohol use (MaxAlc)16, and included 126,936 EUR and 17,029 AA subjects. Consistent with previous reports, ADH1B was the lead locus for both populations. Also as in previous reports, different lead single nucleotide polymorphisms (SNPs) were seen in the two populations: rs1229984 for EUR; and rs2066702 for AAs. Three other genome-wide significant (GWS) MaxAlc loci were identified in EUR, including corticotropin-releasing hormone receptor 1 (CRHR1); the protein product of this gene is involved in stress and immune responses17.
Another MVP GWAS considered both AUD and AUDIT-C14 in a multi-ancestry sample (N = 274,424) using electronic health record (EHR) data. ADH1B was again the lead locus (for both traits), and the lead SNPs were the same as those observed in the MaxAlc study, but this larger study identified 18 GWS loci — five associated with both traits (e.g., ADH1B), eight associated with AUDIT-C only (e.g., VRK serine/threonine kinase 2 (VRK2) and klotho beta (KLB)), and five associated with AUD diagnosis only (e.g., SIX homeobox 3 (SIX3) and dopamine receptor D2 (DRD2)). AUD and AUDIT-C had a genetic correlation of only 0.52; unsurprisingly, considering the moderate correlation between these two traits, they showed differing correlations with other traits. AUD tended to be positively correlated with other psychiatric traits (such as schizophrenia) whereas AUDIT-C was not correlated with psychiatric diagnoses, but was correlated with some healthy traits and behaviours (e.g., educational attainment). This might be because AUDIT-C indexes alcohol consumption more in the normal ‘social drinking’ range, whereas AUD is more sensitive to problematic drinking, i.e. in the pathological range.
The largest GWAS to date with direct relevance to AUD is a meta-analysis effort, combining AUD per se (from MVP and the Psychiatric Genomics Consortium) and the alcohol use component of the AUDIT related to medical problems, i.e. the AUDIT-P (from the UK Biobank); the meta-analyzed trait was considered to be “problematic alcohol use (PAU)”. This study included 435,563 European-ancestry individuals and identified 29 independent risk variants (e.g., thrombospondin type 1 domain containing 7B (THSD7B) and cell adhesion molecule 2 (CADM2))13. In addition to the gene discovery, the large sample size permitted a much more powerful investigation of PAU polygenicity. For example, 138 significant genetic correlations with other traits were observed. Positive genetic correlations included major depression, depressive symptoms, attention-deficit/hyperactivity disorder (ADHD), schizophrenia, and bipolar disorder, among psychiatric traits, as well as insomnia; negative correlations included subjective well-being and age of smoking initiation. With respect to physical health, phenome-wide association analysis with a polygenic risk score (PRS) for PAU implicated several disorders associated with alcohol and tobacco abuse, including alcoholic liver disease and chronic airway obstruction and bronchitis.
Although this study roughly tripled the number of PAU risk loci, only EUR subjects were included. This illustrates a well-known and critical issue in human genetics5: there are comparatively few studies in other populations. Studies in small Asian samples have alighted on ADH1B and/or ALDH218–20 but have not yet moved beyond the large-effect alleles mapping in these alcohol metabolizing enzyme-encoding genes.
Nicotine.
Tobacco use genetics is one of the best studied among the addictive disorders. The medical importance to the research community is obvious owing to the well-known increases in risk for cancer, cardiac disease, and many other systemic illnesses associated with nicotine, and it is so important medically that tobacco use information is collected in most investigations of medical traits. Furthermore, tobacco use is highly prevalent, resulting in much useful information that can be obtained even from population-based studies. There have been numerous well-powered studies related to tobacco use; we focus here on the largest and most powerful such studies.
One of the strongest findings in SUD genetics pertains to nicotine use — the relationship between markers mapped to the gene cluster encoding neuronal acetylcholine receptor (CHRNA3–CHRNA5–CHRNB4) and smoking heaviness or dependence21–23. These genes encode cholinergic nicotinic receptor alpha 3 and 5 and beta 4 subunits and their protein products interact directly with nicotine. We review the functional consequences of genetic variation in the CHRNA3–CHRNA5–CHRNB4 gene cluster in the section “From large-effect risk loci to disease biology”. There have been comparatively many large-scale GWAS studies of behaviours related to cigarette smoking; the largest to date is, as for alcohol use, GSCAN15, which considered up to 1.2 million subjects for “smoking initiation” and hundreds of thousands for several other smoking phenotypes including cigarettes per day (where the lead variant mapped to CHRNA5 with P = 1.2×10−278) and smoking cessation. These phenotypes, however, relate mostly to quantity and frequency of use, rather than nicotine dependence; cessation and initiation of use, while very important clinically, are also different from dependence. This study was well-powered and identified remarkably many significant risk loci, e.g. 378 independent loci for smoking initiation.
Considering what was seen with AUD and quantity/frequency measures of alcohol use, it would seem important to evaluate the analogous dependence trait for smoking behaviours. Several definitions of nicotine dependence are in common use, based on the Fagerström Test for Nicotine Dependence (FTND)24 or on DSM-IV or DSM-5 (Box 1). In addition to the CHRNA3–CHRNA5–CHRNB4 locus, DNA (cytosine-5-)-methyltransferase 3 beta (DNMT3B) was identified as associated with nicotine dependence (FTND) in a meta-analysis study that included 38,602 smokers25, with both EUR and AAs contributing to the finding. Several studies have reported significant association with related phenotypes, including “time to first cigarette” e.g. REF26. An FTND GWAS including 58,000 smokers observed an 8.6% SNP-based heritability and identified five GWS loci (e.g., teneurin transmembrane protein 2 (TENM2) and dopamine beta-hydroxylase (DBH)) that were enriched for transcriptomic regulatory mechanisms in the cerebellum27.
A GWAS on smoking traits from the MVP also included meta-analysis for smoking initiation and cessation from GSCAN28. The MVP EHR data also permitted analysis of nicotine use trajectories as a phenotype; 18 risk loci (e.g., neuronal growth regulator 1 (NEGR1) and cyclin and CBS domain divalent metal cation transport mediator 2 (CNNM2)) for this trait were identified in EUR. In addition to participants of European descent, this study has the merit of including comparatively large numbers of AAs and Latinx subjects, although only a few discoveries in those samples, still much smaller than the EUR sample, were reported.
Illegal substances
Cannabis.
Cannabis is the most commonly used illegal substance throughout most of the world29; in the United States, its status straddles the ‘legal’ and ‘illegal’ categories in many states, where use was decriminalized and then legalized for medical and/or recreational use, despite continuing illegality on the federal level. This has corresponded to gradually increasing societal acceptance of cannabis use, the natural outcome of which has been increased use and childhood exposure30. In this context, working out the genetic risk for cannabis use disorder (CUD) has taken on increased importance. The first two cannabis-relevant GWAS that yielded significant results mapped risk loci for CUD in AAs in one study31 and in EUR in the other32. The next several efforts, which put together considerably larger samples via meta-analysis, did not address CUD per se, but related traits, including lifetime cannabis use33, 34 and age of first cannabis use35. The relationship these latter two traits bear to CUD is not entirely clear: these traits would appear to bear strong relationships to environmental exposures, drug availability and personality traits such as sensation seeking, as opposed to the transition from use to dependence. Larger studies directly relevant to CUD have shown association implicating CHRNA2 (2387 cases, 48,985 controls)36 and to several loci (e.g., forkhead box P2 (FOXP2)) including a confirmation of CHRNA2 effects in a recent Psychiatric Genomics Consortium (PGC; Box 2) meta-analysis37. The latter study, which included 20,916 cases and >300,000 controls, identified two loci, and showed partial genetic overlap between CUD and cannabis use (rg=0.50), generally consistent with the differences observed between quantity/frequency versus dependence measures of alcohol use12–14. Thus, the situation with CUD is illustrative of illegal SUDs in general: lower prevalence than alcohol and nicotine use, fewer available samples to study, and reduced gene discovery that is seen mostly in populations of European descent.
Opioids.
In the last three years, five opioid use disorder (OUD) GWAS have yielded genome-wide significant loci (e.g., repulsive guidance molecule BMP co-receptor a (RGMA), cornichon family AMPA receptor auxiliary protein 3 (CNIH3) and potassium voltage-gated channel subfamily C member 1 (KCNC1))38–41 and identified potential biological pathways involved in OUD pathogenesis. However, the limited sample size of these studies did not permit investigation of the polygenic architecture of OUD. The PGC-SUD workgroup combined these GWAS datasets together with additional cohorts, comparing 4,503 opioid-dependence (OD) cases, 4,173 opioid-exposed (OE) controls, and 32,500 opioid-unexposed controls41. There were GWS loci (e.g., the SDCCAG8 SHH signalling and ciliogenesis regulator gene), and a different genetic-overlap pattern observed across the traits investigated. A PRS based on a GWAS of risk-tolerance42 was positively associated with OD (OD cases versus unexposed or exposed controls) and OE (exposed controls versus unexposed controls). A PRS based on a GWAS of neuroticism43 was positively associated with OD (OD cases versus unexposed controls and OD cases versus exposed controls) but not with opioid exposure (exposed versus unexposed controls). These analyses highlight the difference between dependence and exposure and the importance of considering the definition of controls (exposed versus unexposed) in opioid studies: only subjects who are exposed to a substance have the opportunity to become dependent; thus unexposed controls can also be considered “diagnosis-unknown”. The importance of this distinction depends largely on the risk of eventually becoming dependent upon esposure.
The largest study to date44 incorporated MVP and Yale–Penn data, and included over 114,000 informative EUR and AA subjects in total. This study identified significant association of OUD with a well-known functional OPRM1 variant, A118G (Asn40Asp) — even in the largest study to date there was only one clear GWS finding. OPRM1 encodes the μ opioid receptor gene, considered the main biological target of opioid drugs. Opioids such as methadone and morphine are full agonists at the μ-opioid receptor. OPRM1 has been the subject of intense interest, particularly this same common missense A118G SNP. Opioid dosing in therapeutic contexts is important clinically, and another GWAS considered the trait of methadone dose45. In AAs only, this study identified a significant association of methadone dose to a variant upstream of OPRM1, thus it could possibly have an effect through the same gene as that identified in the largest-to-date GWAS. The sample size for this latter study was very small and the results could be a false positive, but the proximity of this signal to the OUD GWAS signal is intriguing.
Cocaine.
Very little work of genome-wide scope has been accomplished for cocaine dependence or for other stimulants; all cocaine-related work has relied primarily on the Yale–Penn cohort. The first46 of the two available GWAS identified several risk variants (e.g., family with sequence similarity 53, member B (FAM53B)) that despite the years since the completion of the study, still await not only replication, but any suitably powered replication attempt. On the other hand, the FAM53B locus has seen replication of sorts in an animal model47. The second study48, which incorporated data used in the first together with additional cohorts, did not identify GWS risk loci but showed consistent genetic overlap of cocaine dependence with other psychiatric disorders and behavioural traits. Another analysis of the Yale–Penn data showed that phenotypic heterogeneity and gene–environment interplay affect gene discovery (e.g., transmembrane protein 51 (TMEM51)) in cocaine use disorder49. Despite the morbidity and mortality associated with these traits, they are not common enough to be approachable through biobank data so far, and there is regional variation (cocaine use is more predominant in some parts of the US, whereas methamphetamine use is more common in others) tending to increase heterogeneity.
From large-effect risk loci to disease biology
Alcohol-drinking and tobacco-smoking behaviours are among the few complex phenotypes that have common risk alleles with relatively large effect sizes (Box 3). Although these loci explain only a small proportion of the genetic heritability, the evidence generated by their investigation represents a proof-of-concept regarding how to translate genetic associations into disease biology.
Box 3 |. Biological mechanisms of genes associated with large effect on SUD traits.
Substance use disorders (SUDs) are highly polygenic traits where their predisposition is due to the additive effect of thousands of loci mostly with small effect sizes. However, traits related to alcohol drinking and tobacco smoking present several common variants with relatively large effect size, which is unusual for a complex psychiatric trait. These large effects are attributable to the fact that these molecular changes affect key biological processes involved in the metabolism or the response of the human body to alcohol and nicotine, i.e., they act pharmacogenomically. Further details are provided in the main text. The large-effect loci include the following genes:
ADH1B (alcohol dehydrogenase 1B) encodes a subunit of the alcohol dehydrogenase enzyme which oxides alcohol to acetaldehyde (refs. 53 and 54). Alleles increasing alcohol oxidation (A/G variant, rs1229984*A, previously denoted ADH1B*2; C/T variant, rs2066702*T, previously denoted ADH1B*3) lead to accumulation of acetaldehyde and are associated with increased aversive alcohol-related intoxication effects, and decreased risk of not only alcohol dependence but also an extensive range of alcohol-related traits. rs1229984 is observed in many populations but especially in Asians and to a lesser extent Europeans. rs2066702 is virtually exclusive to African populations (ref. 54). For both, the minor allele increases enzyme activity in the encoded protein product, and is protective with respect to alcohol use, dependence, and indeed an extensive range of alcohol-related traits (ref. 53).
ALDH2 (aldehyde dehydrogenase 2 family member) encodes an aldehyde dehydrogenase enzyme that oxidizes acetaldehyde to acetate; the key functional SNP is rs671 G/A. rs671*A (previously denoted ALDH2*2) is associated with reduced acetaldehyde oxidation activity which causes increased acute alcohol sensitivity (and decreased risk of alcohol dependence) (ref. 53). The protective very-low-activity variant is observed only in East Asian populations.
CHRNA5–CHRNA3–CHRNB4 (cholinergic receptor nicotinic alpha 5, alpha 3, and beta 4 subunits) is a cluster of genes located on chromosome 15. These loci encode subunits of the nicotinic acetylcholine receptor (nAChR), a ligand-gated ion channel involved in synaptic signal transmission. Coding changes in these nAChR subunits (e.g., CHRNA5Asp398Asn, rs16969968 G/A) affect the nAChR activity, reducing the aversive effects of nicotine (ref. 56). The locus includes several functional risk alleles.
CYP2A6 (cytochrome P450 family 2 subfamily A member 6) encodes monooxygenase that is involved in nicotine and cotinine oxidation, accounting for up to 80% of nicotine clearance. Genetic variants associated with reduced CYP2A6 activity and consequently reduced nicotine metabolism appear to affect the brain concentration of nicotine influencing circuits involved in reward and impulsivity processes (refs. 67, 68, 72).
AHRR (aryl-hydrocarbon receptor repressor) encodes a protein involved in the aryl-hydrocarbon receptor signalling cascade. It is known to mediate dioxin toxicity but is likely to have additional important biological functions. Tobacco smoking is associated with a very strong decrease in DNA methylation (i.e., hypomethylation) of the CpG sites located in the AHRR gene (ref. 131).
Alcohol-metabolism genes: ADH1B and ALDH2.
Genes encoding alcohol-metabolism enzymes represented obvious candidates in the study of alcohol use disorder and other alcohol-drinking behaviours and were among the first-studied candidate loci. Alcohol metabolism includes two key steps: alcohol oxidation to acetaldehyde by alcohol dehydrogenases (ADHs), followed by acetaldehyde oxidation to acetate by aldehyde dehydrogenases (ALDHs). Acetaldehyde can cause a wide range of aversive reactions and consequences such as unpleasant or dangerous physical reactions including facial flushing and gastroinstestinal tract cancers50. In ADH1B, the Arg48His amino acid substitution (rs1229984) increases alcohol oxidation activity with respect to the the more common variant in worldwide populations. Analogously, the Glu504Lys amino acid substitution (rs671) in ALDH2 drastically reduces acetaldehyde oxidation activity of the encoded enzyme. Both ADH1BArg48His and ALDH2Glu504Lys cause increases in acetaldehyde levels with respect to a given dose of ethanol and consequently, increases its adverse effects. Due to the high frequency of ADH1BArg48His and ALDH2Glu504Lys variation in some East Asian populations and their pharmacokinetic properties, initial studies explored the association of these alleles with drinking behaviours and alcohol use disorder in Chinese and Japanese individuals51. ADH1BArg48His and ALDH2Glu504Lys showed robust associations with facial flushing, increased skin temperature, increase in pulse rate, and ventilation52. This is the case because after drinking alcohol, carriers of ADH1BHis48 and ALDH2Lys504 have increased acetaldehyde levels and consequently more acetaldehyde-induced toxic effects, which induce a protective effect with respect to alcohol intake53, 54. The same mechanism is observed in populations of African descent for ADH1BArg370Cys (rs2066702; ADH1B*3) which causes an increase in the alcohol oxidation activity. ADH1B and ALDH2 alleles were also identified as risk loci of cancers localized in tissues directly exposed to alcohol ingestion55. With greater power and larger sample sizes, ADH1B variation has now been shown to be the most important genetic influence on alcohol intake and dependence in EUR populations as well.
The CHRNA5–CHRNA3–CHRNB4 nicotinic receptor gene cluster and the CYP2A6 nicotine-metabolising gene.
Nicotine is the primary addictive compound among the complex mixture in tobacco smoke. The addictive effect of nicotine is considered to be due to its binding of the nicotinic acetylcholine receptors (nAChRs)56. Ligand-gated ion channels (including these) are widely distributed in the nervous system where they modulate the release of several neurotransmitters, including dopamine, gamma-aminobutyric acid, and glutamate57. Variants mapped to genes encoding nAChR subunits (specifically, the CHRNA5–CHRNA3–CHRNB4 gene cluster) were identified as associated with nicotine dependence in a candidate-gene study58. The initial findings were replicated and expanded by numerous subsequent analyses, also including large-scale GWAS15. Multiple variants causing coding changes in the protein products of CHRNA5, CHRNA3, and CHRNB4 were confirmed as associated with a wide range of traits related to tobacco smoking, such as nicotine dependence, smoking severity and heaviness, smoking cessation, and nicotine aversive effects59. Beyond the association with smoking behaviours, CHRNA5–CHRNA3–CHRNB4 variants showed associations with harmful downstream consequences of tobacco smoking, including lung cancer, chronic obstructive pulmonary disease, and reduced pulmonary function60, 61.
The functional role of CHRNA5Asp398Asn (rs16969968) has been investigated via different approaches. The CHRNA5Asn398 allele encodes an α5 subunit that forms nACh receptors with lower activity, increased short-term desensitization, and lower calcium relative permeability62. In mice, nicotine negatively affected cognitive performance attention of wild-type mice but not α5 (Chrna5) knockouts, in line with the reduced nACh activity63. Viral-mediated expression of the α5 subunit in the medial habenula in brain restores the aversive effect of α5 knockouts to the levels observed in the wild-type animals. When their α5 expression is inhibited, wild-type animals showed nicotine aversion at the level of α5 knockouts64. In humans (overnight-abstinent smokers) who received intravenous administration of nicotine, CHRNA5Asn398 carriers showed an attenuated aversive response65. The mechanism proposed to explain these concordant results is that nAChRs containing α5 subunits limit nicotine effects due to the stimulation of the projections of the medial habenula into the interpeduncular nucleus that causes the aversive reaction. This illustrates an application of studies in model animals to further our understanding of the biologiocal effects of a genetic risk variant in humans.
Nicotine biological effects are also modulated by metabolism, with multiple steps and several enzymatic pathways63. Cytochrome P450 family 2 subfamily A member 6 (CYP2A6) accounts for up to 80% of nicotine clearance. In particular, CYP2A6 is involved in two key reactions, nicotine oxidation to nicotine iminium ion, and cotinine oxidation to hydroxycotinine. The hydroxycotinine/cotinine ratio (known as nicotine metabolic ratio (NMR)) is a biomarker of CYP2A6 activity and nicotine clearance66. NMR heritability is about 60–80%, and CYP2A6 variants account for up to 30% of the phenotypic variantion67. In line with this functional effect, CYP2A6 variation showed consistent associations with nicotine dependence, smoking, smoking cessation, lung cancer, and other related traits68–71. A brain imaging study showed that in smokers the CYP2A6 effect on brain concentration of nicotine influences circuits involved in reward and impulsivity processes72.
These biological mechanisms are inherently pharmacogenomic [G] effects. SUD genetics is, in a very real sense, a pharmacogenomic application: it reflects the interaction of the body with exogenous substances. The principles are essentially the same as when dealing with a therapeutic endogenous substances; indeed, some abusable substances, like opioids used for pain, are themselves sometimes therapeutic, depending on context.
Genetic overlap of addictions and related traits
The genetic architecture of complex traits — including SUDs — is mainly characterized by two phenomena: polygenicity (cumulative contribution of thousands of risk alleles with very small individual effects)73 and pleiotropy (risk alleles are shared across human diseases and traits)74. Below, we review how different methods have advanced our understanding of the polygenicity and pleiotropy of SUDs.
Genetic correlation.
The high comorbidity observed within SUDs (i.e., SUDs with other SUDs) and with respect to other psychiatric disorders reflects that there are shared genetic and non-genetic risk factors linking them (Figure 2).
Figure 2 |. Genetic correlation among SUD traits and other phenotypes.
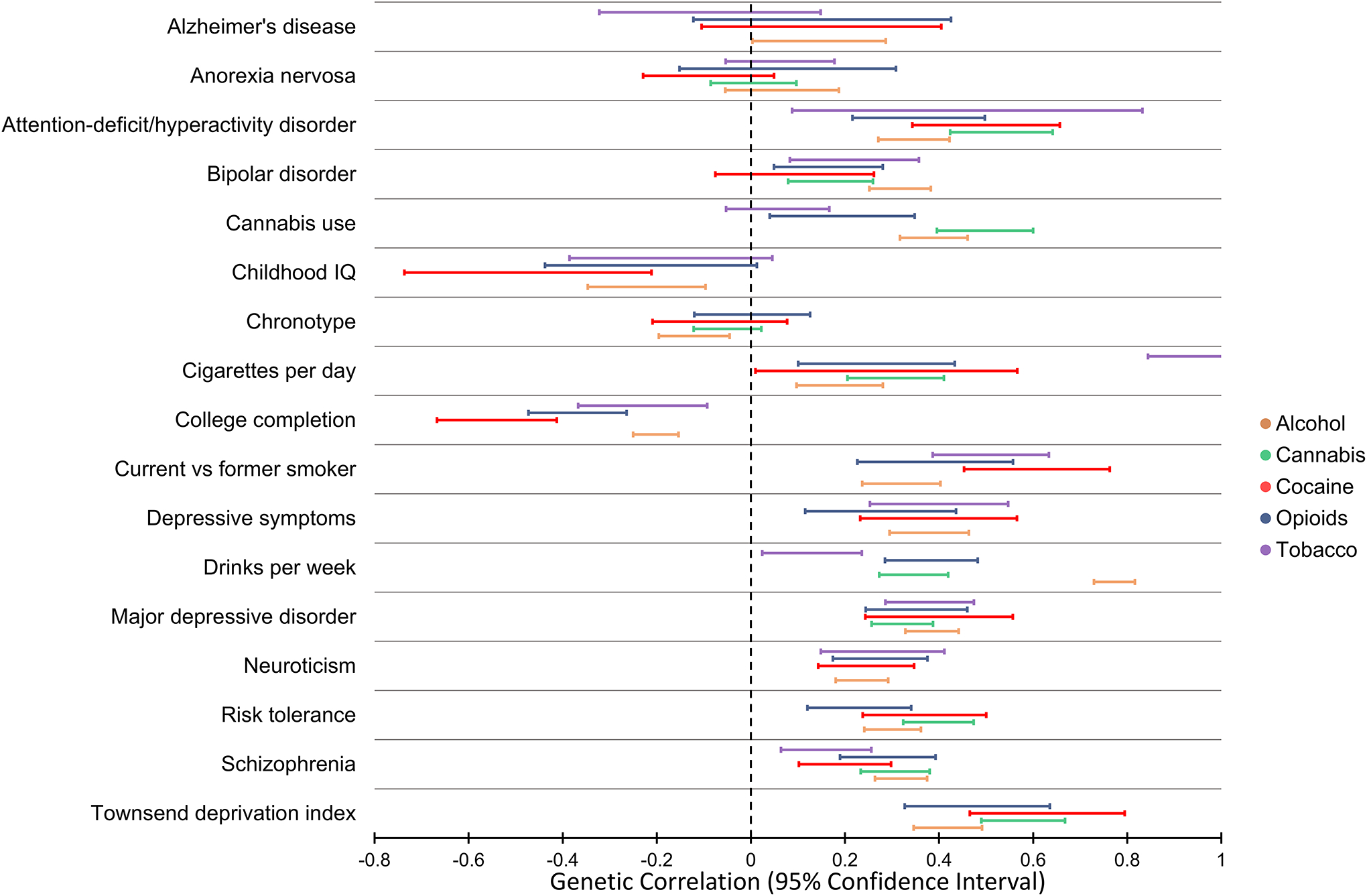
Genetic correlation of problematic alcohol use, cannabis use disorder, cocaine dependence, opioid use disorder, and nicotine dependence with psychiatric disorders, behavioural traits, and other complex phenotypes. The 95% confidence interval of the genetic correlation estimates were obtained from previous studies13, 37, 44, 48, 185 that applied the linkage disequilibrium score regression method. The traits included are those tested with respect to at least four out of the five addictions considered. There are some patterns of genetic correlation that are consistent across the five addictions presented. The major differences among them are related to the substance-specific genetic correlations, i.e. nicotine addiction vs. cigaretters per day, alcohol addiction vs. drinks per week, and cannabis addiction vs. cannabis use.
As genome-wide SNP data became widely available, novel approaches were developed to estimate pairwise genetic correlations using GWAS results75; e.g. the development of computational tools using genome-wide association statistics (estimated effect sizes and standard errors for each variant analyzed in a GWAS) instead of individual-level data (genotypes and trait information for each participant tested in a GWAS)76. Methods based on genome-wide association summary statistics no longer contain data from individual subjects and greatly reduce privacy concerns and other logistic issues related to individual-level genetic data, permitting wide data sharing that has allowed a growing number of investigators to explore the genetic correlation between SUDs and other complex traits.
Estimating genetic correlation across multiple psychiatric disorders using GWAS data has become a standard analysis after the primary GWAS. Although SNP-based heritability is, based on current data, low (Table 1), these analyses have confirmed that SUDs are genetically correlated with other psychiatric disorders and behavioural traits, with the strongest overlap (besides other SUDs) observed for depression, anxiety, posttraumatic stress disorder, neuroticism, and risk-taking behaviours42, 77–83 (Figure 2). However, as detailed above, differences have been observed between SUD traits defined based on dependence criteria and phenotypes related to substance consumption/use. The dependence-based traits tend to have significantly higher genetic correlation with psychopathology-related traits than the use-based traits12, 14, 41, 82. This pattern — dependence for a given substance is genetically different from abuse — is seen across the small number of substances where there is sufficient data to make a comparison (e.g. alcohol and cannabis), but for many other SUDs insufficient data exist (e.g. for opioids and cocaine) for this pattern to be potentially confirmed as universal. An exception to the difference between frequency/quantity versus dependence is nicotine, where a high genetic correlation is observed (rg=0.95)27; this may be explicable on the basis that nicotine is so addictive and available that regular smoking very readily engenders physiological dependence84.
Polygenic risk scores.
The availability of large GWAS permits us to weight and to model the cumulative effect of hundreds or even thousands of small-effect variants into polygenic risk scores (PRS)85. There are a wide range of methods available to model the polygenicity of complex traits determining the genetic risk of an individual86 (or that part of the risk affected by available common variant information). Similarly, the strength of prediction between PRS and outcome can be measured with several goodness-of-fit metrics86, such as the effect size estimate, phenotypic variance explained, the area under the receiver–operator curve (AUC), and the P-value corresponding to a null hypothesis of no association. For SUDs, recent studies are applying these approaches mainly based on large-scale GWAS of traits related to alcohol, cannabis, and tobacco use and dependence. SUD PRS show limited predictive power on an individual basis — meaning they are not (yet) good at predicting disease risk in individuals — but are useful to understand genetic overlap of SUDs with psychiatric and behavioural traits. Due to the wide range of methods applied and SUD-related traits tested, it is hard to compare studies using different metrics, such as testing PRS on other datasets when the PRS were generated from datasets based on different sample sizes and phenotype definitions. With respect to PRS derived from traits related to alcohol consumption and dependence (e.g., AUDIT-C and AUDIT-P), several associations were observed across traits related to cocaine, amphetamine, and MDMA (3,4-Methylenedioxymethamphetamine; also known as ‘ecstasy’)87 in addition to alcohol use phenotypes assssed in independent cohorts (i.e., alcohol dependence, AUD symptom count, maximum drinks, increased likelihood of AD onset, and ICD-based alcohol-related disorders88,89). In a cross-ancestry analysis, AD PRS derived from Thai and European-American GWAS were associated with AD in Han Chinese, although the effect was mostly due to the contribution of the ALDH2 and ADH1B loci19. Similarly to alcohol-related studies, PRS derived from smoking traits showed associations across traits related to multiple substances (e.g., cocaine, amphetamine, hallucinogens, ecstasy, and cannabis initiation, as well as DSM-5 AUD)87,90,91. For cannabis, PRS derived from use (versus dependence) phenotypes showed association with depression, self-harm behaviours, and cannabis use assessed in an independent cohort92. In addition to testing SUD PRS prediction other traits, some studies have investigated how PRS derived from other psychiatric traits predict (are associated with) SUD phenotypes. For example, cocaine dependence was significantly associated with multiple PRS related to schizophrenia, ADHD, major depressive disorder, children’s aggressive behaviour, and antisocial behaviour48. With respect to dual diagnoses93, schizophrenia PRS was associated with having any SUD diagnosis94 and, in an independent study, the same PRS was associated with lifetime tobacco smoking in women but not in men, with significant interactions of the PRS with sex and birth decade95. Several investigations have been conducted to understand the interplay between SUD genetic risk and environmental factors. In a recent review article about this topic96, the most reported frameworks were differential susceptibility and diathesis-stress, with substantial heterogeneity across environmental exposures, genetic factors, and outcomes tested.
PRS that embody genome-wide risk for a trait can, depending on the GWAS data available on which the PRS is based, be used to predict risk in any array-genotyped individual and the statistical significance of this prediction can be measured. Current PRS studies identified several significant associations between SUD genetic liability and a wide range of psychiatric and behavioral traits. However, there is still a wide gulf between a statistically significant prediction of risk and one that is clinically useful on an individual level. At this point, we have not yet approached clinical utility for PRS in risk prediction, and although some commercial tests have been marketed that purport to do exactly that, we view them skeptically, and where evaluated rigorously, they have failed to hold up. For example one test that purports to predict OUD risk, actually predicts not the subject’s OUD risk but his or her ancestral population97.
Mendelian randomization.
The consistent genetic correlation among SUDs and other psychiatric disorders could be due to shared genetic effects or causal effects between the traits98. Mendelian randomization (MR) methods leverage instrumental variables based on genetic information to distinguish simple association from causation99. Alcohol drinking and tobacco smoking behaviours have been investigated using known risk alleles in the ALDH2, ADH1B and CHRNA5–CHRNA3–CHRNB4 loci to test causal relationships with respect to mental and physical health100–108. MR approaches based on polygenic instrumental variables are more powerful, and allow for exploration of a wider range of hypotheses, including testing the difference between phenotypes based on diagnostic criteria and those based on consumption and frequency use82. Two-sample MR approaches, which are based exclusively on genome-wide association statistics, reduced the limitations due to the use of individual-level data, allowing for a more extensive range of studies109. Polygenic instruments were investigated to test the causal links between traits related to tobacco smoking with psychiatric disorders and behavioural traits. Smoking initiation is affected by educational attainment but not by cognitive ability, suggesting a contribution to health inequality in less-educated people110. Among personality traits, genetic liabilities to neuroticism and extraversion have a causal effect with smoking severity and initiation, respectively111. Studies investigating causality between smoking behaviours and schizophrenia showed conflicting results112, 113, whereas depression and bipolar disorder appear to have a bidirectional relationship with lifetime smoking and smoking initiation112, 114. For physical health, several novel causal effects were observed such as smoking initiation on stroke risk115 and fracture risk116 or body mass index on being a smoker117. Evidence of bidirectional associations was observed between cannabis use and schizophrenia34, 118, 119. Conversely, ADHD showed a causal effect on cannabis initiation together with smoking initiation and severity77, 120. With respect to alcohol-drinking behaviours, both major depression and ADHD have causal effects on AUD risk but not on alcohol consumption82, 120. Although these studies provided novel insights useful to understand the mechanisms underlying comorbidities of SUDs, MR analyses present several limitations that should be considered carefully when interpreting their findings in the context of SUDs and other complex traits. Each MR design is based on specific assumptions121. Although several kinds of sensitivity analyses can be employed to evaluate possible biases122, it is hard to determine whether other unaccounted confounders are affecting the results. Additionally, the low SNP-based heritability, the high polygenicity, and the complex pleiotropy of SUD-related traits strongly affect the statistical power and realiability of MR methods. Other analytical approaches therefore need to be used to extend and validate MR results. Longitudinal studies can be a viable option although they also present certain limitations123. The gold standard for causal inference remain randomized controlled trials. However, there are several issues (e.g., ethical quandaries) that limit their application to SUD research.
Integrating omics into addiction studies
High-throughput technologies generating large amounts of individual data besides gene variant information have expanded, permitting investigators to analyze human variation across different omics data types (such as epigenomics and transcriptomics)124. Different tissues and cell types are expected to be more or less informative depending on the specific SUD traits studied. Brain is often the organ of greatest interest (but not necessarily, because peripheral metabolism is, as detailed above, also important). However, study of brain tissue is possible only postmortem, and this entails substantial compromises. As discussed above, analyses of human GWAS results have demonstrated that the exact phenotype studied — substance use, initiation, quantity, physiological dependence, and so on — is critically important. But for deceased subjects, phenotypic information is often limited. Sometimes we find ourselves in the position of needing, for example, to interpret epigenetic findings for subjects who have died of opioid overdose. But were they opioid dependent? Did they overdose accidentally, or did they die of suicide? If the latter, were they opioid-exposed prior to the terminal event? When we study peripheral tissues from living subjects, we can have more extensive information about phenotype. There is an additional key consideration if omics results are to have clinical utility: utilization in the clinic depends on measurements being made on tissue that can be derived from living subjects. In the sections below, we focus on two main omics domains investigated with respect to SUD traits. As for GWAS, the general pattern that there are much more data for legal than for illegal SUDs pertains.
Epigenomics.
Epigenetic changes are modifications that regulate gene function in response to endogenous and exogenous processes125. There has been some investigation of candidate loci126–128, but epigenome-wide association studies (EWAS), mostly done in peripheral tissues, provide a better understanding of the link between addictive substances and epigenetic regulation.
Large studies in this research field are, however, mostly lacking; some of the largest concern smoking traits. DNA methylation alteration extensively associates with smoking and is a plausible link between smoking and adverse health129. A large-scale EWAS of cigarette smoking in >15,500 participants identified thousands of methylation sites associated with smoking status, with different patterns between current and formers smokers130. A striking effect of tobacco smoking was observed in reducing the methylation of CpG sites located in the AHRR locus, which encodes the aryl hydrocarbon receptor repressor131. This association is one of the strongest and most consistently replicated epigenetic relationships for psychiatric traits so far; in fact, several algorithms have been developed to predict smoking status on the basis of epigenetic variation132. Different aspects of how tobacco smoking affects methylation changes have been explored, including the role of nicotine metabolism, the effect of prenatal exposure, and the risk of lung cancer in smokers133–135.
There is nothing else comparable to the remarkable AHRR–smoking relationship in the rest of the SUD epigenetics literature, but alcohol consumption was also associated with epigenetic changes across hundreds of methylation sites in the human genome136, 137. Longitudinal investigations showed that the majority of these methylation changes are reversible in the context of long-term variation in alcohol consumption138. Chronic alcohol consumption appears to be related to methylation changes leading to neuroadaptations that may underlie some of the mechanisms of dependence risk and persistence139. Across multiple CpG sites associated with AUD, there is a consistent correlation of methylation profiles in buccal cells with putamen brain tissues140, highlighting the possibility to investigate brain-related epigenetic changes in peripheral tissues. In a cross-tissue and cross-phenotypic analysis of AUD-induced genome-wide methylomic variation, epigenetic changes in PCSK9 (proprotein convertase subtilisin/kexin type 9) were a possible contributor of the effect of alcohol on lipid metabolism and cardiovascular risk141. A methylome-wide analysis of 1,287 adolescents showed that methylation of the SPDEF (E-twenty-six transcription factor) gene moderated the association of psychosocial stress with alcohol and tobacco abuse142. Epigenetic dysregulation appears to be involved in reprogramming the medial prefrontal cortex after prolonged exposure to cycles of alcohol intoxication and withdrawal, which promotes the escalation of voluntary alcohol intake and aversion-resistant alcohol seeking143 (i.e., AUD). Epigenome-wide analysis in patients during acute alcohol withdrawal and after 2 weeks of recovery as well as in age-matched controls showed that, although acute alcohol withdrawal in severely dependent patients was associated with extensive epigenetic changes, the differences between patients and controls diminished after recovery, suggesting partial reversibility of alcohol- and withdrawal-related methylation144. Leveraging epigenetic clock algorithms [G]145, epigenetic ageing in AD appears to be tissue-specific with positive age acceleration in blood and liver, but no significant effect was observed likely due to the small sample size (N<50)146.
Very limited information is available regarding the epigenetic changes associated with other addiction disorders. A study of OD in 220 EA women identified differentially methylated CpG sites in genes involved in chromatin remodelling, DNA binding, cell survival, and cell projection147. A genome-wide DNA methylation analysis of 48 heavy cannabis users confirmed the association of AHRR and F2RL3 genes with tobacco smoking in cannabis users and, for users of cannabis only, identified nominally significant methylation changes enriched for neuronal signalling (glutamatergic synapse and long-term potentiation) and cardiomyopathy148. But results from small studies such as these warrant cautious interpretiation.
Transcriptomics.
Understanding transcriptomics supports understanding of disease pathophysiology. Investigation of different tissue and cell types can provide different information regarding transcriptomic changes. Although studies of pathological states are limited by sample availability and phenotype ascertainment, postmortem analyses provide an important approach towards understanding the normal brain, and the relationship between SUDs and brain transcriptomic regulation.
A candidate-locus postmortem brain analysis showed that prodynorphin is downregulated in the dorsolateral prefrontal cortex of individuals with a diagnosis of AD or alcohol abuse when compared to controls149. The investigators hypothesized that prodynorphin down-regulation could lead to neurotransmission disinhibition, which could contribute to the formation of alcohol-related behaviour149. The cerebral cortex transcriptome-wide profile of AUD showed a lack of overlap with the gene expression changes observed in other psychiatric disorders (i.e., autism, schizophrenia, bipolar disorder, and depression)150. In nucleus accumbens, subjects with AD showed transcriptomic downregulation of gene modules enriched for neuronal-specific marker genes and upregulation of gene modules enriched for astrocyte and microglial specific marker genes151. The neuronal-specific modules were related to genes involved in oxidative phosphorylation, mitochondrial dysfunction, and MAPK signalling. The glial-specific modules were related to genes involved in the immune function151.
Another approach to investigate transcriptomic changes is based on cultured cell line models. In induced pluripotent stem cell (iPSC)-derived neural cell cultures obtained from healthy individuals and those with AD, significant changes in the expression of the candidate loci GABRA1, GABRG2, and GABRD were observed following 21-day alcohol exposure152. A model based on forebrain neural cells showed a module of 58 co-expressed genes that were uniformly decreased following alcohol exposure153. These alcohol-responsive genes are related to biological functions related to cell cycle, Notch signalling, and cholesterol biosynthesis pathways. An early neural differentiation model showed a wide range of ethanol-mediated transcriptional alterations, including a strong association among modulators involved in protein modification, protein synthesis, and gene expression154. A dopaminergic neuronal model based on SH-SY5Y-differentiated cells showed that cocaine exposure is associated with transcriptomic changes in genes involved in transcription regulation, cell cycle, adhesion, cell projection, mitogen-activated protein kinase, cAMP response element-binding protein, and neurotrophin and neuregulin signalling155. In the same model, cocaine exposure was associated with the down-regulation of several microRNAs, which are involved in post-transcriptional regulation of gene expression in the brain156. RNA transcriptomic analyses in iPSC-derived human neural cells revealed that tetrahydrocannabinol (THC) administration, either by acute or chronic exposure, dampened neuronal transcriptional response following potassium chloride-induced neuronal depolarization with significant alterations to synaptic, mitochondrial and glutamate signalling157.
Although post-mortem brain samples and models based on cultured human brain cells provide a reliable approach to investigate the human transcriptome, investigation of peripheral tissues can permit studies of larger samples. In a blood-based transcriptome-wide study, 132 of 18,238 genes tested were differentially expressed between current smokers and never smokers and the loci identified were involved in the immune system, blood coagulation, natural killer cell, and cancer pathways158. By comparing former smokers with current and never-smokers, it was possible to distinguish different status: reversible for 94 genes, slowly reversible for 31 genes, and irreversible for 6 genes158. In the adipose tissue of 542 healthy female twins, DNA methylation and gene expression changes were observed in five genes (AHRR, CYP1A1, CYP1B1, CYTL1, and F2RL3) in response to tobacco smoking159. Based on neonatal umbilical cord blood, prenatal smoking was associated with the transcriptomic downregulation of fetal brain regulatory genes (BDNF, PLP1, and MBP) in active-smoker mothers but not among passive smokers160. In prospectively collected saliva samples, prenatal opioid exposure was associated with sex-dependent effects on hypothalamic feeding regulatory genes (DRD2 and NPY2R) with correlations with neonatal opioid withdrawal syndrome including hyperphagia and severity of withdrawal state161.
Genome-wide gene expression in whole blood of 90 heavy cannabis users and 100 cannabis-naïve participants showed that expression of PPFIA2 (protein tyrosine phosphatase receptor type F polypeptide-interacting-protein alpha-2) is increased in cannabis users and is negatively correlated with neuropsychological function in both groups162. Peripheral genome-wide gene expression in individuals with cocaine use disorder identified that the expression of genes involved in inflammation and immune functions (IRF1 and GBP5) were negatively correlated with anhedonia scores163.
The availability of transcriptome-informative datasets for a wide range of human tissues and cells has led to the development of computational methods to integrate transcriptomic data with information related to genetic variation. These approaches can be used to calculate the overrepresentation of genes expressed in certain tissues and cell types164 and also to predict the component of the transcriptomic changes regulated by genetic variation and test its association with the phenotype of interest165, 166. Applying these methods, investigators were able to derive additional information regarding SUD pathophysiology from genome-wide association data34, 167. For instance, the integration of genetic and transcriptomic information can permit calculation of the genetically regulated component of tissue-specific transcriptomic changes associated with substance-use traits168 or to estimate the colocalization of risk alleles associated with alcohol consumption with expression quantitative trait loci169.
In summary, omics studies are improving our understanding of the biological mechanisms between genetic variation and phenotypes, and environmental effects on biology. Although a focus on brain studies has immediate intuitive appeal, the limitations of such studies (such as sample size and phenotype understanding) must also be recognized. And as for studies of genetic sequence variation, the field is generally limited by insufficient sample size, a problem that is more acute for illegal than for legal substance dependencies.
Human and animal research in SUD genetics
Much ongoing SUD genetic research is based on animal models170. There is disagreement regarding the translational value of animal models as a starting point in SUD genetic research and their predictive power with respect to clinical scenarios171. Investigators supporting the relevance to humans of animal models of addiction put forward examples of medications developed based on molecular targets and circuits facilitated by animal studies: naloxone and acamprosate for alcoholism, buprenorphine-naloxone for opioid addiction, and varenicline for nicotine addiction172. However, in 2019, the US National Institute on Mental Health (NIMH) released a new notice regarding the use of animals in mental health research173. Based on the main conclusion that there is not a true animal model of a psychiatric disorder, it was decided that NIMH-supported studies should not establish particular animal models to understand a human mental illness, but instead, investigate areas of biology of relevance to mental illnesses174. Conversely, the US National Institute on Drug Abuse (NIDA) appears to prioritize the use of non-human animal models to understand the genomic architecture of SUDs and addictive behaviours172, 175–177.
A clear difficulty is the identification of phenotypes for study in animal models, and establishing how those phenotypes related to human phenotypes. As we have emphasized throughout this Review, it was only recently demonstrated that substance use and dependence may have important genetic differences; this is best established with respect to alcohol. Since this is a recent discovery, and based on the evidence available up to a few years ago, it would have been easy to argue that these traits were qualitatively similar and only quantitatively different. Since we barely know how to distinguish different traits that relate to use of the same substance in humans, what can we really say about what traits in animals might be analogous to human traits? Which animal traits might relate to quantity or frequency of use, which to dependence, and which to neither? We do not have a full understanding of how SUD biology in model organisms relates to biology in humans. If we were to base genetic discovery for SUDs on non-human models, the utility of this approach would depend on the actual correspondence between relevant animal and human traits. But as for other psychiatric traits, this is very hard to establish. We can have animals that are more or less susceptible to developing substance self-administration, but we do not have the means to determine why this is so, or how the animal’s motivations map onto human systems.
Animal studies provide very important contributions to understanding the molecular basis of human diseases and traits. However, similar to what is recognized by the National Advisory Mental Health Council Workgroup on Genomics for other psychiatric disorders178, we argue that there is no “probably-true” animal model mapping directly onto human SUDs. Psychiatric and behavioural traits appear to have a genetic architecture even more polygenic than other complex traits due to the action of two main forces: background selection and diagnostic heterogeneity179, 180. Both of these mechanisms are human-specific and we believe it is problematic to model them in animals. These limitations should be recognized when using animal models to investigate SUD pathogenesis. The utility of animal models for evaluating the biological properties and effects of genes and specific variants that were first identified in human studies is widely accepted, and an important method in understanding genetic phenomena identified in human subjects. On balance we believe that the utility of animal studies for complex behavioural traits like SUD risk in humans, is mostly limited to evaluation and testing of findings from humans, rather than in enhancing gene discovery for human traits (that are not necessarily congruent to animal traits).
Conclusions and perspectives
There has been enormous progress in SUD genetics research towards the goal of understanding the molecular risk factors for SUDs, mostly in the past few years. For those traits that are well-represented in biobanks, such as alcohol and tobacco use traits, the prospects are very good for continued progress, discovery of more risk loci, and improvement in our knowledge of their biology. The prospects are not quite so good for illegal SUDs that tend to be less-well-represented in biobanks. For these traits, we will need directed recruitment as well as biobank data. Biobanks have both strengths and weaknesses for discovery. For less-prevelent or stigmatized traits, even large biobank samples may not provide sufficient information to investigate SUD polygenic architecture adequately. A further limitation of EHR data and some biobank assessments is that they meaure state rather than trait, whereas we are generally more interested in lifetime diagnoses than the research participant’s characteristics at a specific point in time. As recently shown181, alcohol consumption, tobacco smoking, and phenotyping of other traits are subject to misreports and longitudinal changes, causing biases in gene discovery and follow-up analyses. Appropiate phenotyping strategies are needed to avoid this possible confounder, especially with respect to self-reported frequency and quantity of substance use.
Until quite recently the only risk genes that were well established for SUDs acted pharmacogenomically — metabolizing enzymes and receptor variants. Now, for some traits, we have many more significant risk variants, and have moved into brain biology. The single greatest advance to emerge from more powerful GWAS, in our view, has been the characterization of the genetic differences between quantity/frequency traits and dependence traits across multiple substances. Despite these remarkable advances, we can still account for only a small proportion of genetic risk based on currently identified variants. Accordingly, we are very far from widespread clinical application of these data. Recent studies based on whole-genome sequencing (WGS) data showed that the ‘missing heritability’182 of complex traits (i.e., the difference between twin-based heritability and GWAS-based heritability; Figure 3) appears to be due, at elast for some traits, to uncommon variants located in regions with low linkage disequilibrium183, 184 that cannot be ascertained by genotyping array. Accordingly, the WGS being generted by large-scale efforts (e.g., AllOfUs, the Million Veteran Program (MVP), and the Trans-Omics for Precision Medicine (TOPMed) program) are likely to contribute to reduce SUD missing heritabilities. However, as these resources involve a population-based design and are not specifically assessed for SUD research, they may have only limited impact for SUDs with low prevalence in the general population. In the next few years, we expect that our understanding of SUD genetics will grow rapidly, comensurate with the increasing availability of large-scale datasets for each trait and the advanced computational methods that continue to be developed to investigate them.
Figure 3 |. Twin-based versus SNP-based heritabilities of alcohol, cannabis, cocaine, opioid, and tobacco addictions.
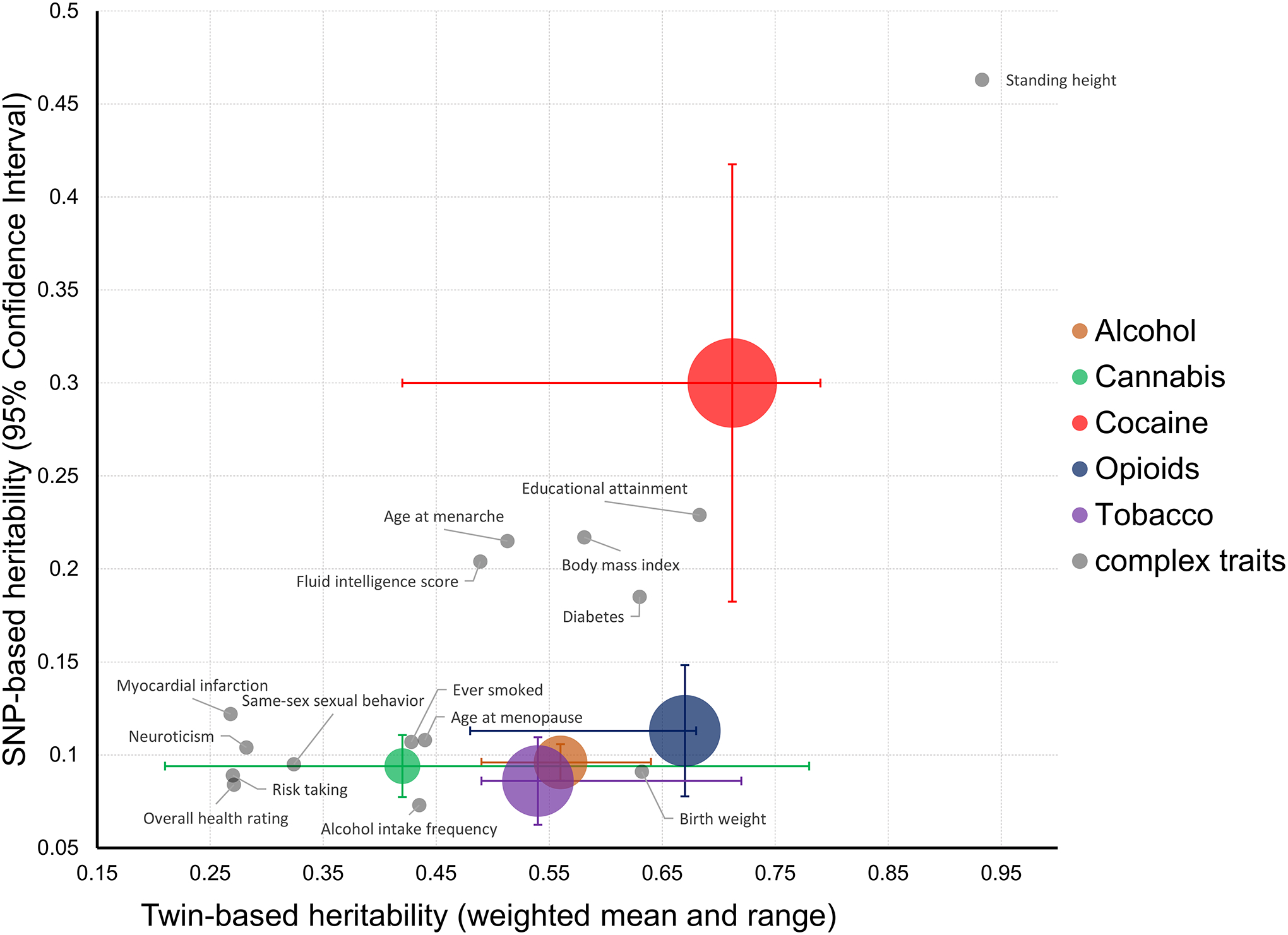
Twin-based heritabilities (weighted means and ranges) were previously estimated in US surveys of addictive agents in adult twin pairs186. SNP-based heritabilities were previously estimated by genome-wide association studies using linkage disequilibrium score regression13, 37, 44, 48, 185. Bubble size represents the relative risk of addiction for each substance187. Vertical bars represent 95% confidence intervals for the SNP-based estimate, and horizontal bars represent the twin-based range. Family-based and SNP-based heritabilities of complex traits previously calculated using UK Biobank data188 are also plotted. The differences between twin-based and SNP-based heritabilities (i.e., the ‘missing heritability’) for four of the addictions shown is in line with those observed among other human traits and diseases. The only exception appears to be cocaine addiction that presents a large 96% confidence interval due to the small sample size of the largest genome-wide association study available for this trait to date.
Acknowledgements
The authors are supported by grants from the National Institutes of Health (R01DA012690, R01AA026364, U01MH109532, P50AA012870, R01DA037974, R21DA047527, and R21DC018098) and the Department of Veterans Affairs (1I01CX001849). The authors thank Dan Levey, Yaira Nunez, Chelsea Tyrrell, and Frank Wendt for their helpful comments.
Glossary
- Addictive substances
Psychoactive substances affecting mental processes and causing brain changes associated with the development of physiological dependence
- Substance use
The use of drugs or alcohol, including extrinsic substances such as cigarettes, cannabis, illegal drugs, prescription drugs, inhalants, and solvents
- Substance use disorders
(SUDs). According to the Diagnostic and Statistical Manual of Mental Disorders fifth edition (DSM-5) definition, this diagnostic category combines substance abuse and substance dependence into a single disorder measured on a continuum from mild to severe
- Substance dependence
Mental illness characterized by behavioural, cognitive, and physiological symptoms developed after repeated substance use that make it difficult to discontinue use, often despite harmful effects. These symptoms, which extend beyond purely psychological effects, are commonly known as physiological dependence or physical dependence
- Substance abuse
The harmful or hazardous use of psychoactive substances, including alcohol and licit and illicit drugs
- Addicted
Addiction is a psychiatric condition manifested by compulsive substance use despite its harmful consequences
- Diagnostic criteria
Criteria reflecting signs, symptoms, and tests that are useful to guide the care of patients and understand prognosis
- Pharmacogenomics
The study of how genomic variation affects individual responses to drugs and drug metabolism
- Candidate Genes
Loci hypothesized to be associated with a complex traits on the basis of prevailing theories and positional mapping from linkage studies and/or cytogenetic studies
- Epigenetic Clock Algorithms
Age predictors based on DNA methylation
Footnotes
Competing interests
J.G. and R.P. are paid for their editorial work for ‘Complex Psychiatry’ journal. J.G. is named as an inventor on PCT patent application #15/878,640 entitled: “Genotype-guided dosing of opioid agonists,” filed January 24, 2018.
References
- 1.Volkow ND, Jones EB, Einstein EB & Wargo EM Prevention and Treatment of Opioid Misuse and Addiction: A Review. JAMA Psychiatry 76, 208–216 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Rehm J et al. Alcohol Use Disorders in Primary Health Care: What Do We Know and Where Do We Go? Alcohol Alcohol 51, 422–7 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Kiluk BD, Fitzmaurice GM, Strain EC & Weiss RD What defines a clinically meaningful outcome in the treatment of substance use disorders: reductions in direct consequences of drug use or improvement in overall functioning? Addiction 114, 9–15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prescott CA, Khoddam R & Arpawong TE in eLS 1–11. [Google Scholar]
- 5.Sirugo G, Williams SM & Tishkoff SA The Missing Diversity in Human Genetic Studies. Cell 177, 26–31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Border R et al. No Support for Historical Candidate Gene or Candidate Gene-by-Interaction Hypotheses for Major Depression Across Multiple Large Samples. Am J Psychiatry 176, 376–387 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson EC et al. No Evidence That Schizophrenia Candidate Genes Are More Associated With Schizophrenia Than Noncandidate Genes. Biol Psychiatry 82, 702–708 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walters RK et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat Neurosci 21, 1656–1669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saunders JB, Aasland OG, Babor TF, de la Fuente JR & Grant M Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO Collaborative Project on Early Detection of Persons with Harmful Alcohol Consumption--II. Addiction 88, 791–804 (1993). [DOI] [PubMed] [Google Scholar]
- 10.APA. Diagnostic and Statistical Manual of Mental Disorders IV (American Psychiatric Association, Washington DC, 1994). [Google Scholar]
- 11.Association, A.P. Diagnostic and Statistical Manual of Mental Disorders the Fifth Edition (DSM-5) (American Psychiatric Association; 2013). [Google Scholar]
- 12.Sanchez-Roige S et al. Genome-wide association study meta-analysis of the Alcohol Use Disorder Identification Test (AUDIT) in two population-based cohorts (N=141,958). Am J Psychiatry 176, 107–118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou H et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat Neurosci 23, 809–818 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kranzler HR et al. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat Commun 10, 1499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu M et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 51, 237–244 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gelernter J et al. Genome-wide Association Study of Maximum Habitual Alcohol Intake in >140,000 U.S. European and African American Veterans Yields Novel Risk Loci. Biol Psychiatry 86, 365–376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Inda C et al. Different cAMP sources are critically involved in G protein-coupled receptor CRHR1 signaling. J Cell Biol 214, 181–95 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quillen EE et al. ALDH2 is associated to alcohol dependence and is the major genetic determinant of “daily maximum drinks” in a GWAS study of an isolated rural Chinese sample. Am J Med Genet B Neuropsychiatr Genet 165B, 103–10 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun Y et al. Genome-wide association study of alcohol dependence in male Han Chinese and cross-ethnic polygenic risk score comparison. Transl Psychiatry 9, 249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gelernter J et al. Genomewide Association Study of Alcohol Dependence and Related Traits in a Thai Population. Alcohol Clin Exp Res 42, 861–868 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sherva R et al. Variation in nicotinic acetylcholine receptor genes is associated with multiple substance dependence phenotypes. Neuropsychopharmacology 35, 1921–31 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tobacco & Genetics, C. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet 42, 441–7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saccone NL et al. Multiple independent loci at chromosome 15q25.1 affect smoking quantity: a meta-analysis and comparison with lung cancer and COPD. PLoS Genet 6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heatherton TF, Kozlowski LT, Frecker RC & Fagerstrom KO The Fagerstrom Test for Nicotine Dependence: a revision of the Fagerstrom Tolerance Questionnaire. Br J Addict 86, 1119–27 (1991). [DOI] [PubMed] [Google Scholar]
- 25.Hancock DB et al. Genome-wide association study across European and African American ancestries identifies a SNP in DNMT3B contributing to nicotine dependence. Mol Psychiatry 23, 1911–1919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J et al. Genome-Wide Meta-Analyses of FTND and TTFC Phenotypes. Nicotine Tob Res 22, 900–909 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quach BC et al. Expanding the genetic architecture of nicotine dependence and its shared genetics with multiple traits. Nat Commun 11, 5562 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu K et al. Genome-wide Association Study of a Longitudinal Phenotype of Smoking and Meta-analysis of Smoking Status in up to 842,000 Individuals. Nat Commun (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasin DS et al. US Adult Illicit Cannabis Use, Cannabis Use Disorder, and Medical Marijuana Laws: 1991–1992 to 2012–2013. JAMA Psychiatry 74, 579–588 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hasin DS US Epidemiology of Cannabis Use and Associated Problems. Neuropsychopharmacology 43, 195–212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sherva R et al. Genome-wide Association Study of Cannabis Dependence Severity, Novel Risk Variants, and Shared Genetic Risks. JAMA Psychiatry 73, 472–80 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agrawal A et al. Genome-wide association study identifies a novel locus for cannabis dependence. Mol Psychiatry 23, 1293–1302 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stringer S et al. Genome-wide association study of lifetime cannabis use based on a large meta-analytic sample of 32 330 subjects from the International Cannabis Consortium. Transl Psychiatry 6, e769 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasman JA et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nat Neurosci 21, 1161–1170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minica CC et al. Genome-wide association meta-analysis of age at first cannabis use. Addiction 113, 2073–2086 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Demontis D et al. Genome-wide association study implicates CHRNA2 in cannabis use disorder. Nat Neurosci 22, 1066–1074 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson EC et al. A large-scale genome-wide association study meta-analysis of cannabis use disorder. Lancet Psychiatry (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gelernter J et al. Genome-wide association study of opioid dependence: multiple associations mapped to calcium and potassium pathways. Biol Psychiatry 76, 66–74 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nelson EC et al. Evidence of CNIH3 involvement in opioid dependence. Mol Psychiatry 21, 608–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cheng Z et al. Genome-wide Association Study Identifies a Regulatory Variant of RGMA Associated With Opioid Dependence in European Americans. Biol Psychiatry 84, 762–770 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Polimanti R et al. Leveraging genome-wide data to investigate differences between opioid use vs. opioid dependence in 41,176 individuals from the Psychiatric Genomics Consortium. Mol Psychiatry (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karlsson Linner R et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet 51, 245–257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nagel M et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat Genet 50, 920–927 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Zhou H et al. Association of OPRM1 Functional Coding Variant With Opioid Use Disorder: A Genome-Wide Association Study. JAMA Psychiatry (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith AH et al. Genome-wide association study of therapeutic opioid dosing identifies a novel locus upstream of OPRM1. Mol Psychiatry 22, 346–352 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gelernter J et al. Genome-wide association study of cocaine dependence and related traits: FAM53B identified as a risk gene. Mol Psychiatry 19, 717–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dickson PE et al. Systems genetics of intravenous cocaine self-administration in the BXD recombinant inbred mouse panel. Psychopharmacology (Berl) 233, 701–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cabana-Dominguez J, Shivalikanjli A, Fernandez-Castillo N & Cormand B Genome-wide association meta-analysis of cocaine dependence: Shared genetics with comorbid conditions. Prog Neuropsychopharmacol Biol Psychiatry 94, 109667 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Sun J, Kranzler HR, Gelernter J & Bi J A genome-wide association study of cocaine use disorder accounting for phenotypic heterogeneity and gene-environment interaction. J Psychiatry Neurosci 45, 34–44 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Edenberg HJ The genetics of alcohol metabolism: role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res Health 30, 5–13 (2007). [PMC free article] [PubMed] [Google Scholar]
- 51.Eng MY, Luczak SE & Wall TL ALDH2, ADH1B, and ADH1C genotypes in Asians: a literature review. Alcohol Res Health 30, 22–7 (2007). [PMC free article] [PubMed] [Google Scholar]
- 52.Luczak SE et al. ALDH2 and ADH1B interactions in retrospective reports of low-dose reactions and initial sensitivity to alcohol in Asian American college students. Alcohol Clin Exp Res 35, 1238–45 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edenberg HJ & McClintick JN Alcohol Dehydrogenases, Aldehyde Dehydrogenases, and Alcohol Use Disorders: A Critical Review. Alcohol Clin Exp Res 42, 2281–2297 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Polimanti R & Gelernter J ADH1B: From alcoholism, natural selection, and cancer to the human phenome. Am J Med Genet B Neuropsychiatr Genet 177, 113–125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Matejcic M, Gunter MJ & Ferrari P Alcohol metabolism and oesophageal cancer: a systematic review of the evidence. Carcinogenesis 38, 859–872 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Brunzell DH, Stafford AM & Dixon CI Nicotinic receptor contributions to smoking: insights from human studies and animal models. Curr Addict Rep 2, 33–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Albuquerque EX, Pereira EF, Alkondon M & Rogers SW Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev 89, 73–120 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saccone SF et al. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet 16, 36–49 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lassi G et al. The CHRNA5-A3-B4 Gene Cluster and Smoking: From Discovery to Therapeutics. Trends Neurosci 39, 851–861 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bosse Y & Amos CI A Decade of GWAS Results in Lung Cancer. Cancer Epidemiol Biomarkers Prev 27, 363–379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wain LV et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir Med 3, 769–81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dani JA Neuronal Nicotinic Acetylcholine Receptor Structure and Function and Response to Nicotine. Int Rev Neurobiol 124, 3–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Valentine G & Sofuoglu M Cognitive Effects of Nicotine: Recent Progress. Curr Neuropharmacol 16, 403–414 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Antolin-Fontes B, Ables JL, Gorlich A & Ibanez-Tallon I The habenulo-interpeduncular pathway in nicotine aversion and withdrawal. Neuropharmacology 96, 213–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jensen KP et al. A CHRNA5 Smoking Risk Variant Decreases the Aversive Effects of Nicotine in Humans. Neuropsychopharmacology 40, 2813–21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tanner JA et al. Nicotine metabolite ratio (3-hydroxycotinine/cotinine) in plasma and urine by different analytical methods and laboratories: implications for clinical implementation. Cancer Epidemiol Biomarkers Prev 24, 1239–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loukola A et al. A Genome-Wide Association Study of a Biomarker of Nicotine Metabolism. PLoS Genet 11, e1005498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tanner JA et al. Variation in CYP2A6 and nicotine metabolism among two American Indian tribal groups differing in smoking patterns and risk for tobacco-related cancer. Pharmacogenet Genomics 27, 169–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park SL et al. Association of CYP2A6 activity with lung cancer incidence in smokers: The multiethnic cohort study. PLoS One 12, e0178435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tanner JA & Tyndale RF Variation in CYP2A6 Activity and Personalized Medicine. J Pers Med 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Polimanti R, Jensen KP & Gelernter J Phenome-wide association study for CYP2A6 alleles: rs113288603 is associated with hearing loss symptoms in elderly smokers. Sci Rep 7, 1034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li S, Yang Y, Hoffmann E, Tyndale RF & Stein EA CYP2A6 Genetic Variation Alters Striatal-Cingulate Circuits, Network Hubs, and Executive Processing in Smokers. Biol Psychiatry 81, 554–563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Boyle EA, Li YI & Pritchard JK An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watanabe K et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet 51, 1339–1348 (2019). [DOI] [PubMed] [Google Scholar]
- 75.Weissbrod O, Flint J & Rosset S Estimating SNP-Based Heritability and Genetic Correlation in Case-Control Studies Directly and with Summary Statistics. Am J Hum Genet 103, 89–99 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pasaniuc B & Price AL Dissecting the genetics of complex traits using summary association statistics. Nat Rev Genet 18, 117–127 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Soler Artigas M et al. Attention-deficit/hyperactivity disorder and lifetime cannabis use: genetic overlap and causality. Mol Psychiatry (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Few LR et al. Genetic variation in personality traits explains genetic overlap between borderline personality features and substance use disorders. Addiction 109, 2118–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lo MT et al. Genome-wide analyses for personality traits identify six genomic loci and show correlations with psychiatric disorders. Nat Genet 49, 152–156 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nievergelt CM et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat Commun 10, 4558 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gelernter J et al. Genome-wide association study of post-traumatic stress disorder reexperiencing symptoms in >165,000 US veterans. Nat Neurosci 22, 1394–1401 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Polimanti R et al. Evidence of causal effect of major depression on alcohol dependence: findings from the psychiatric genomics consortium. Psychol Med 49, 1218–1226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Levey DF et al. Reproducible Genetic Risk Loci for Anxiety: Results From approximately 200,000 Participants in the Million Veteran Program. Am J Psychiatry 177, 223–232 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Benowitz NL Nicotine addiction. The New England journal of medicine 362, 2295–2303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Torkamani A, Wineinger NE & Topol EJ The personal and clinical utility of polygenic risk scores. Nat Rev Genet 19, 581–590 (2018). [DOI] [PubMed] [Google Scholar]
- 86.Pain O et al. Evaluation of Polygenic Prediction Methodology within a Reference-Standardized Framework. bioRxiv, 2020.07.28.224782 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chang LH et al. Association between polygenic risk for tobacco or alcohol consumption and liability to licit and illicit substance use in young Australian adults. Drug Alcohol Depend 197, 271–279 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Johnson EC et al. Polygenic contributions to alcohol use and alcohol use disorders across population-based and clinically ascertained samples. Psychol Med, 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnson EC et al. The Genetic Relationship Between Alcohol Consumption and Aspects of Problem Drinking in an Ascertained Sample. Alcohol Clin Exp Res 43, 1113–1125 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang LH et al. Associations between polygenic risk for tobacco and alcohol use and liability to tobacco and alcohol use, and psychiatric disorders in an independent sample of 13,999 Australian adults. Drug Alcohol Depend 205, 107704 (2019). [DOI] [PubMed] [Google Scholar]
- 91.Vink JM et al. Polygenic risk scores for smoking: predictors for alcohol and cannabis use? Addiction 109, 1141–51 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Johnson EC et al. Exploring the relationship between polygenic risk for cannabis use, peer cannabis use and the longitudinal course of cannabis involvement. Addiction 114, 687–697 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Polimanti R, Agrawal A & Gelernter J Schizophrenia and substance use comorbidity: a genome-wide perspective. Genome Med 9, 25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hartz SM et al. Association Between Substance Use Disorder and Polygenic Liability to Schizophrenia. Biol Psychiatry 82, 709–715 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang SH et al. Association between polygenic liability for schizophrenia and substance involvement: A nationwide population-based study in Taiwan. Genes Brain Behav 19, e12639 (2020). [DOI] [PubMed] [Google Scholar]
- 96.Pasman JA, Verweij KJH & Vink JM Systematic Review of Polygenic Gene-Environment Interaction in Tobacco, Alcohol, and Cannabis Use. Behav Genet 49, 349–365 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hatoum AS et al. Genetic Data Can Lead to Medical Discrimination: Cautionary tale of Opioid Use Disorder. medRxiv, 2020.09.12.20193342 (2020). [Google Scholar]
- 98.Hemani G, Bowden J & Davey Smith G Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum Mol Genet 27, R195–R208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dimou NL & Tsilidis KK A Primer in Mendelian Randomization Methodology with a Focus on Utilizing Published Summary Association Data. Methods Mol Biol 1793, 211–230 (2018). [DOI] [PubMed] [Google Scholar]
- 100.Brand JS et al. Associations of maternal quitting, reducing, and continuing smoking during pregnancy with longitudinal fetal growth: Findings from Mendelian randomization and parental negative control studies. PLoS Med 16, e1002972 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ostergaard SD et al. Associations between Potentially Modifiable Risk Factors and Alzheimer Disease: A Mendelian Randomization Study. PLoS Med 12, e1001841; discussion e1001841 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pedersen KM et al. Smoking and Increased White and Red Blood Cells. Arterioscler Thromb Vasc Biol 39, 965–977 (2019). [DOI] [PubMed] [Google Scholar]
- 103.Bjorngaard JH et al. Heavier smoking increases coffee consumption: findings from a Mendelian randomization analysis. Int J Epidemiol 46, 1958–1967 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Johnsen MB et al. The causal role of smoking on the risk of headache. A Mendelian randomization analysis in the HUNT study. Eur J Neurol 25, 1148–e102 (2018). [DOI] [PubMed] [Google Scholar]
- 105.Caramaschi D et al. Maternal smoking during pregnancy and autism: using causal inference methods in a birth cohort study. Transl Psychiatry 8, 262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Taylor M et al. Is smoking heaviness causally associated with alcohol use? A Mendelian randomization study in four European cohorts. Int J Epidemiol 47, 1098–1105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Taylor AE et al. Investigating the possible causal association of smoking with depression and anxiety using Mendelian randomisation meta-analysis: the CARTA consortium. BMJ Open 4, e006141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Guo R, Wu L & Fu Q Is There Causal Relationship of Smoking and Alcohol Consumption with Bone Mineral Density? A Mendelian Randomization Study. Calcif Tissue Int 103, 546–553 (2018). [DOI] [PubMed] [Google Scholar]
- 109.Hemani G et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sanderson E, Davey Smith G, Bowden J & Munafo MR Mendelian randomisation analysis of the effect of educational attainment and cognitive ability on smoking behaviour. Nat Commun 10, 2949 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sallis HM, Davey Smith G & Munafo MR Cigarette smoking and personality: interrogating causality using Mendelian randomisation. Psychol Med 49, 2197–2205 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wootton RE et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: a Mendelian randomisation study. Psychol Med, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gage SH et al. Investigating causality in associations between smoking initiation and schizophrenia using Mendelian randomization. Sci Rep 7, 40653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vermeulen JM et al. Smoking and the risk for bipolar disorder: evidence from a bidirectional Mendelian randomisation study. Br J Psychiatry, 1–7 (2019). [DOI] [PubMed] [Google Scholar]
- 115.Larsson SC, Burgess S & Michaelsson K Smoking and stroke: A mendelian randomization study. Ann Neurol 86, 468–471 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yuan S, Michaelsson K, Wan Z & Larsson SC Associations of Smoking and Alcohol and Coffee Intake with Fracture and Bone Mineral Density: A Mendelian Randomization Study. Calcif Tissue Int 105, 582–588 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Carreras-Torres R et al. Role of obesity in smoking behaviour: Mendelian randomisation study in UK Biobank. BMJ 361, k1767 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gage SH et al. Assessing causality in associations between cannabis use and schizophrenia risk: a two-sample Mendelian randomization study. Psychol Med 47, 971–980 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vaucher J et al. Cannabis use and risk of schizophrenia: a Mendelian randomization study. Mol Psychiatry 23, 1287–1292 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Treur JL et al. Investigating causality between liability to ADHD and substance use, and liability to substance use and ADHD risk, using Mendelian randomization. Addict Biol, e12849 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Davies NM, Holmes MV & Davey Smith G Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 362, k601 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Burgess S, Bowden J, Fall T, Ingelsson E & Thompson SG Sensitivity Analyses for Robust Causal Inference from Mendelian Randomization Analyses with Multiple Genetic Variants. Epidemiology 28, 30–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Farabee D, Schulte M, Gonzales R & Grella CE Technological aids for improving longitudinal research on substance use disorders. BMC Health Serv Res 16, 370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hasin Y, Seldin M & Lusis A Multi-omics approaches to disease. Genome Biol 18, 83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kanherkar RR, Bhatia-Dey N & Csoka AB Epigenetics across the human lifespan. Front Cell Dev Biol 2, 49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ruggeri B et al. Methylation of OPRL1 mediates the effect of psychosocial stress on binge drinking in adolescents. J Child Psychol Psychiatry 59, 650–658 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Muschler M et al. Epigenetic alterations of the POMC promoter in tobacco dependence. Eur Neuropsychopharmacol 28, 875–879 (2018). [DOI] [PubMed] [Google Scholar]
- 128.Ebrahimi G et al. Elevated levels of DNA methylation at the OPRM1 promoter region in men with opioid use disorder. Am J Drug Alcohol Abuse 44, 193–199 (2018). [DOI] [PubMed] [Google Scholar]
- 129.Kaur G, Begum R, Thota S & Batra S A systematic review of smoking-related epigenetic alterations. Arch Toxicol 93, 2715–2740 (2019). [DOI] [PubMed] [Google Scholar]
- 130.Joehanes R et al. Epigenetic Signatures of Cigarette Smoking. Circ Cardiovasc Genet 9, 436–447 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bojesen SE, Timpson N, Relton C, Davey Smith G & Nordestgaard BG AHRR (cg05575921) hypomethylation marks smoking behaviour, morbidity and mortality. Thorax 72, 646–653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bollepalli S, Korhonen T, Kaprio J, Anders S & Ollikainen M EpiSmokEr: a robust classifier to determine smoking status from DNA methylation data. Epigenomics 11, 1469–1486 (2019). [DOI] [PubMed] [Google Scholar]
- 133.Gupta R et al. Epigenome-wide association study of serum cotinine in current smokers reveals novel genetically driven loci. Clin Epigenetics 11, 1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fragou D, Pakkidi E, Aschner M, Samanidou V & Kovatsi L Smoking and DNA methylation: Correlation of methylation with smoking behavior and association with diseases and fetus development following prenatal exposure. Food Chem Toxicol 129, 312–327 (2019). [DOI] [PubMed] [Google Scholar]
- 135.Baglietto L et al. DNA methylation changes measured in pre-diagnostic peripheral blood samples are associated with smoking and lung cancer risk. Int J Cancer 140, 50–61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Xu K et al. Epigenome-Wide DNA Methylation Association Analysis Identified Novel Loci in Peripheral Cells for Alcohol Consumption Among European American Male Veterans. Alcohol Clin Exp Res 43, 2111–2121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu C et al. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry 23, 422–433 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dugue PA et al. Alcohol consumption is associated with widespread changes in blood DNA methylation: Analysis of cross-sectional and longitudinal data. Addict Biol 26, e12855 (2021). [DOI] [PubMed] [Google Scholar]
- 139.Zhang H & Gelernter J Review: DNA methylation and alcohol use disorders: Progress and challenges. Am J Addict 26, 502–515 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hagerty SL, Bidwell LC, Harlaar N & Hutchison KE An Exploratory Association Study of Alcohol Use Disorder and DNA Methylation. Alcohol Clin Exp Res 40, 1633–40 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lohoff FW et al. Methylomic profiling and replication implicates deregulation of PCSK9 in alcohol use disorder. Mol Psychiatry 23, 1900–1910 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tay N et al. Allele-Specific Methylation of SPDEF: A Novel Moderator of Psychosocial Stress and Substance Abuse. Am J Psychiatry 176, 146–155 (2019). [DOI] [PubMed] [Google Scholar]
- 143.Heilig M et al. Reprogramming of mPFC transcriptome and function in alcohol dependence. Genes Brain Behav 16, 86–100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Witt SH et al. Acute alcohol withdrawal and recovery in men lead to profound changes in DNA methylation profiles: a longitudinal clinical study. Addiction 115, 2034–2044 (2020). [DOI] [PubMed] [Google Scholar]
- 145.Fransquet PD, Wrigglesworth J, Woods RL, Ernst ME & Ryan J The epigenetic clock as a predictor of disease and mortality risk: a systematic review and meta-analysis. Clin Epigenetics 11, 62 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rosen AD et al. DNA methylation age is accelerated in alcohol dependence. Transl Psychiatry 8, 182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Montalvo-Ortiz JL, Cheng Z, Kranzler HR, Zhang H & Gelernter J Genomewide Study of Epigenetic Biomarkers of Opioid Dependence in European- American Women. Sci Rep 9, 4660 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Osborne AJ et al. Genome-wide DNA methylation analysis of heavy cannabis exposure in a New Zealand longitudinal cohort. Transl Psychiatry 10, 114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bazov I et al. Downregulation of the neuronal opioid gene expression concomitantly with neuronal decline in dorsolateral prefrontal cortex of human alcoholics. Transl Psychiatry 8, 122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gandal MJ et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 359, 693–697 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mamdani M et al. Integrating mRNA and miRNA Weighted Gene Co-Expression Networks with eQTLs in the Nucleus Accumbens of Subjects with Alcohol Dependence. PLoS One 10, e0137671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lieberman R, Kranzler HR, Levine ES & Covault J Examining the effects of alcohol on GABAA receptor mRNA expression and function in neural cultures generated from control and alcohol dependent donor induced pluripotent stem cells. Alcohol 66, 45–53 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jensen KP et al. Alcohol-responsive genes identified in human iPSC-derived neural cultures. Transl Psychiatry 9, 96 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mandal C et al. Gene expression signatures after ethanol exposure in differentiating embryoid bodies. Toxicol In Vitro 46, 66–76 (2018). [DOI] [PubMed] [Google Scholar]
- 155.Fernandez-Castillo N et al. Transcriptomic and genetic studies identify NFAT5 as a candidate gene for cocaine dependence. Transl Psychiatry 5, e667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cabana-Dominguez J, Arenas C, Cormand B & Fernandez-Castillo N MiR-9, miR-153 and miR-124 are down-regulated by acute exposure to cocaine in a dopaminergic cell model and may contribute to cocaine dependence. Transl Psychiatry 8, 173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Guennewig B et al. THC exposure of human iPSC neurons impacts genes associated with neuropsychiatric disorders. Transl Psychiatry 8, 89 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Vink JM et al. Differential gene expression patterns between smokers and non-smokers: cause or consequence? Addict Biol 22, 550–560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Tsai PC et al. Smoking induces coordinated DNA methylation and gene expression changes in adipose tissue with consequences for metabolic health. Clin Epigenetics 10, 126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Salihu HM et al. Evidence of altered brain regulatory gene expression in tobacco-exposed fetuses. J Perinat Med 45, 1045–1053 (2017). [DOI] [PubMed] [Google Scholar]
- 161.Yen E et al. Sex-Dependent Gene Expression in Infants with Neonatal Opioid Withdrawal Syndrome. J Pediatr 214, 60–65 e2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.He Y et al. Liprin alfa 2 gene expression is increased by cannabis use and associated with neuropsychological function. Eur Neuropsychopharmacol 29, 643–652 (2019). [DOI] [PubMed] [Google Scholar]
- 163.Fries GR et al. Anhedonia in cocaine use disorder is associated with inflammatory gene expression. PLoS One 13, e0207231 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gamazon ER et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet 47, 1091–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gusev A et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 48, 245–52 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Salvatore JE et al. Beyond genome-wide significance: integrative approaches to the interpretation and extension of GWAS findings for alcohol use disorder. Addict Biol 24, 275–289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Marees AT et al. Post-GWAS analysis of six substance use traits improves the identification and functional interpretation of genetic risk loci. Drug Alcohol Depend 206, 107703 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Thompson A et al. Functional validity, role, and implications of heavy alcohol consumption genetic loci. Sci Adv 6, eaay5034 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cates HM et al. National Institute on Drug Abuse genomics consortium white paper: Coordinating efforts between human and animal addiction studies. Genes Brain Behav 18, e12577 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Spanagel R Animal models of addiction. Dialogues Clin Neurosci 19, 247–258 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Volkow ND & Koob G Brain disease model of addiction: why is it so controversial? Lancet Psychiatry 2, 677–679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.National Institute of Mental Health. 2019. NOT-MH-19–053: Notice of NIMH’s Considerations Regarding the Use of Animal Neurobehavioral Approaches in Basic and Pre-clinical Studies. https://grants.nih.gov/grants/guide/notice-files/NOT-MH-19-053.html.
- 174.Gordon JA 2019. A Hypothesis-Based Approach: The Use of Animals in Mental Health Research. https://www.nimh.nih.gov/about/director/messages/2019/a-hypothesis-based-approach-the-use-of-animals-in-mental-health-research.shtml.
- 175.Volkow ND, Koob GF & McLellan AT Neurobiologic Advances from the Brain Disease Model of Addiction. N Engl J Med 374, 363–71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Volkow ND & Boyle M Neuroscience of Addiction: Relevance to Prevention and Treatment. Am J Psychiatry 175, 729–740 (2018). [DOI] [PubMed] [Google Scholar]
- 177.Uhl GR, Koob GF & Cable J The neurobiology of addiction. Ann N Y Acad Sci 1451, 5–28 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.National Advisory Mental Health Council Workgroup on Genomics. 2019. Report of the National Advisory Mental Health Council Workgroup on Genomics: Opportunities and Challenges of Psychiatric Genetics. https://www.nimh.nih.gov/about/advisory-boards-and-groups/namhc/reports/report-of-the-national-advisory-mental-health-council-workgroup-on-genomics.shtml#recommendations.
- 179.Wendt FR et al. Natural selection influenced the genetic architecture of brain structure, behavioral and neuropsychiatric traits. bioRxiv, 2020.02.26.966531 (2020). [Google Scholar]
- 180.O’Connor LJ et al. Extreme Polygenicity of Complex Traits Is Explained by Negative Selection. Am J Hum Genet 105, 456–476 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Xue A et al. Genome-wide analyses of behavioural traits are subject to bias by misreports and longitudinal changes. Nat Commun 12, 20211 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Young AI Solving the missing heritability problem. PLoS Genet 15, e1008222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wainschtein P et al. Recovery of trait heritability from whole genome sequence data. bioRxiv, 588020 (2019). [Google Scholar]
- 184.Wessel J et al. Rare Non-coding Variation Identified by Large Scale Whole Genome Sequencing Reveals Unexplained Heritability of Type 2 Diabetes. medRxiv, 2020.11.13.20221812 (2020). [Google Scholar]
- 185.Quach BC et al. Expanding the Genetic Architecture of Nicotine Dependence and its Shared Genetics with Multiple Traits: Findings from the Nicotine Dependence GenOmics (iNDiGO) Consortium. bioRxiv, 2020.01.15.898858 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Goldman D, Oroszi G & Ducci F The genetics of addictions: uncovering the genes. Nat Rev Genet 6, 521–32 (2005). [DOI] [PubMed] [Google Scholar]
- 187.Goldstein A & Kalant H Drug policy: striking the right balance. Science 249, 1513–21 (1990). [DOI] [PubMed] [Google Scholar]
- 188.Ganna A et al. Large-scale GWAS reveals insights into the genetic architecture of same-sex sexual behavior. Science 365 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (Washington, DC, 2013). [Google Scholar]
- 190.World Health, O (World Health Organization, Geneva, 2005). [Google Scholar]
- 191.Check Hayden E The rise and fall and rise again of 23andMe. Nature 550, 174–177 (2017). [DOI] [PubMed] [Google Scholar]
- 192.Hirata M et al. Overview of BioBank Japan follow-up data in 32 diseases. J Epidemiol 27, S22–S28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chen Z et al. China Kadoorie Biobank of 0.5 million people: survey methods, baseline characteristics and long-term follow-up. Int J Epidemiol 40, 1652–66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hjorleifsson S & Schei E Scientific rationality, uncertainty and the governance of human genetics: an interview study with researchers at deCODE genetics. Eur J Hum Genet 14, 802–8 (2006). [DOI] [PubMed] [Google Scholar]
- 195.Locke AE et al. Exome sequencing of Finnish isolates enhances rare-variant association power. Nature 572, 323–328 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Pedersen CB et al. The iPSYCH2012 case-cohort sample: new directions for unravelling genetic and environmental architectures of severe mental disorders. Mol Psychiatry 23, 6–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gaziano JM et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J Clin Epidemiol 70, 214–23 (2016). [DOI] [PubMed] [Google Scholar]
- 198.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Sullivan PF et al. Psychiatric Genomics: An Update and an Agenda. Am J Psychiatry 175, 15–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]