

Abstract
To get insight into the limiting factors existing for the efficient production of fungal peroxidase in filamentous fungi, the expression of the Phanerochaete chrysosporium lignin peroxidase H8 (lipA) and manganese peroxidase (MnP) H4 (mnp1) genes in Aspergillus niger has been studied. For this purpose, a protease-deficient A. niger strain and different expression cassettes have been used. Northern blotting experiments indicated high steady-state mRNA levels for the recombinant genes. Manganese peroxidase was secreted into the culture medium as an active protein. The recombinant protein showed specific activity and a spectrum profile similar to those of the native enzyme, was correctly processed at its N terminus, and had a slightly lower mobility on sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Recombinant MnP production could be increased up to 100 mg/liter upon hemoglobin supplementation of the culture medium. Lignin peroxidase was also secreted into the extracellular medium, although the protein was not active, presumably due to incorrect processing of the secreted enzyme. Expression of the lipA and mnp1 genes fused to the A. niger glucoamylase gene did not result in improved production yields.
White rot fungi are unique in their ability to mineralize lignin using an array of extracellular enzymes. In the best-studied white rot fungus, the basidiomycete Phanerochaete chrysosporium, two heme peroxidases, lignin peroxidase (LiP) (EC 1.11.1.7) and manganese peroxidase (MnP) (EC 1.11.1.7), along with a H2O2 generating system, are the major components of the lignin degradation system (14, 16). During the last two decades these ligninolytic enzymes have been intensively studied in relation to potential biotechnical applications, such as biopulping, biobleaching, and soil bioremediation (4, 17, 21, 23).
To make such industrial applications feasible, an efficient production system for these enzymes is needed. In their natural host, both proteins are synthesized during secondary metabolism in response to nutrient limitation, and only limited amounts are produced (for a review, see reference 33). Although studies on P. chrysosporium culturing conditions have been carried out in an attempt to optimize LiP and MnP production (for an overview, see reference 11), they have not resulted in a suitable large-scale production system for these proteins.
Several groups have investigated the recombinant expression of fungal peroxidases in different hosts. Expression in Escherichia coli resulted in production of the inactive apoproteins in inclusion bodies (9, 49). Active LiP and MnP could be produced in insect cells by using the baculovirus expression system (19, 20, 30). However, this system suffers from low yields and high production costs and is not suitable for use on a large scale.
Gold and coworkers (26, 37) reported the development of a homologous expression system for MnP and LiP in P. chrysosporium. The constitutively expressed P. chrysosporium glyceraldehyde phosphate dehydrogenase (gpd) promoter was used to drive the expression of the recombinant genes, now using nutrient-rich media in which the endogenous genes are not expressed. However, and despite the use of the strong promoter, production levels of the recombinant proteins remained at the same low level as is normally produced by the endogenous genes under starvation conditions.
We have explored the possibility of producing these fungal peroxidases in another filamentous fungi. A number of filamentous fungal species are capable of secreting large amounts of proteins into the medium and are therefore exploited for the production of homologous and heterologous proteins. Expression of recombinant proteins of fungal origin is usually very efficient, and production levels of grams per liter are within reach (42). However, the reports presented so far on the expression of fungal peroxidases in filamentous fungi indicate that their (over)production is difficult to accomplish. The expression of the lignin peroxidase gene of Phlebia radiata in Trichoderma reesei, although resulting in detectable mRNA levels, failed to produce any extracellular LiP (34). Also, significant lip transcript but weak extracellular activity was found upon expression of a LiPH8 cDNA clone in a Tunisian Aspergillus niger strain (1). More success was obtained in the expression of the P. chrysosporium manganese peroxidase (mnp1) gene in Aspergillus oryzae: active protein was produced in the culture medium, but at levels similar to that of the parental host (38).
Several factors hampering the overproduction of recombinant proteins in filamentous fungi have been established in the last decade (for a review, see reference 3). Specifically, for heme-containing proteins, limited heme availability has been indicated as a limiting factor (2, 12).
The aim of this study is to gain more insight into the parameters affecting the overproduction of recombinant fungal heme-containing peroxidases in filamentous fungi. For this study, a protease-deficient A. niger strain and different expression constructs and culture conditions were used.
MATERIALS AND METHODS
Expression cassettes.
LiP isozyme H8 (lipA) and MnP isozyme H4 (mnp1) cDNAs were a gift from D. Cullen (Institute of Microbial & Biochemical Technology, Madison, Wis.). Three lipA and two mnp1 expression cassettes were constructed using a PCR cloning approach, and the cloned PCR products were checked by sequencing. Table 1 shows the oligonucleotides, vectors, and restriction sites used in the cloning strategy, and Table 2 lists the oligonucleotide sequences. Constructs pLipA.I and pMnp1.I contained LiP and MnP complete coding sequences, respectively. In pLipA.II, the 27-amino-acid (aa) LiP prepro sequence was replaced by the 24-aa glucoamylase (GLA) prepro sequence. In vectors pGLA::LipA and pGLA::Mnp1, the DNA fragment encoding the mature LiP or MnP protein, respectively, was fused to a DNA fragment encoding the N-terminal 514-aa sequence of the A. niger GLA. A DNA sequence encoding a recognition site for a fungal processing protease was placed between the GLA and the peroxidase sequences to allow in vivo splicing of the fused proteins (8). In all cases, the A. niger glaA promoter, the 5′ untranslated region of the glaA mRNA, and the Aspergillus nidulans trpC terminator were used to drive the expression of the LiP and MnP encoding sequences (Fig. 1).
TABLE 1.
Cloning strategya
| Expression vector | Template for PCR | Primers
|
Cloning vector | Cloning site restriction fragment | Cloning site vector | |
|---|---|---|---|---|---|---|
| Forward | Reverse | |||||
| pLipA.I | LipA cDNA | LipA5E/N | Lip3B/H | pAN52-10Notc | NcoIe-HindIII | NcoI-HindIII |
| pLipA.II | LipA cDNA | LipA5E/N | Lip3B/H | pAN52-6Notc | BssHII-HindIII | BssHII-HindIII |
| pGLA::LipAb | LipA cDNA | LipA5E/N | Lip3B/H | pAN56-2d | BssHII-BamHI | NarI-BglIIe |
| pMn1.I | Mnp1 cDNA | MNP15E/N | MNP13B/H | pAN52-10Not | NcoI-HindIII | NcoI-HindIII |
| pGLA::Mnp1b | Mnp1 cDNA | MNP1E/B | MNP13B/H | pAN56-2 | BbsI-BamHI | NarI-BglIIe |
For each expression vector are indicated primers used for amplification of the LipA or Mnp1 cDNA clone and addition of cloning sites, recipient Aspergillus expression vector, and restriction sites used in the final cloning procedure.
For pGLA::LipA and pGLA::Mnp1, the Kex2 site containing linker NarI-NVISKR-BssHII was used to join vector and insert molecules.
These vectors are derivatives from pAN52-4 (Punt, unpublished) with improved cloning sites.
GenBank accession number z32690.
Partial digestion.
TABLE 2.
Sequences of the oligonucleotides used for cloning
| Oligonucleotide | Sequence |
|---|---|
| EcoRINcoI | |
| LipA5E/Na | GGAATTCCATGGCCTTCAAGCAG |
| MAFKQ | |
| BamHIHindIII | |
| Lip3B/Ha | CGGGATCCAAGCTTAAGCACCCGGAGG |
| StopA G P P | |
| EcoRINcoI | |
| MNP15E/Na | GGAATTCCATGGCCTTCGGTTCT |
| M A F G S | |
| BamHIHindII | |
| MNP13B/Ha | CGGGATCCAAGCTTAGGCAGGGCCATC |
| Stop A P G D | |
| EcoRI BbsI | |
| MNP1E/Ba | GCGAATTCGAAGACCTCGCGCAGTCTGTCCAGA |
| A V C P D | |
| BssHII | |
| OLIGONARAb | CGCGCTTGGAAATCACATT |
| R K S I V N | |
| NarI | |
| OLIGONARBb | CGAATGTGATTTCCAAG |
| N V I S K |
Used as primer for PCR.
Used to construct the linker NarI-NVISKR-BssHII.
FIG. 1.

lipA and mnp1 expression cassettes. For an explanation, see Materials and Methods and Table 1.
Strains and transformation procedures.
E. coli DH5 was used for construction and propagation of vector molecules.
A. niger MGG029 (prtT gla::fleor pyrG) was used as the recipient strain in transformation experiments. MGG029 was obtained by parasexual recombination of A. niger AB1.13#7 (cspA1 fwnA trpA argB leuA nicA prtT), a derivative of AB1.13 (25) and A. niger AB6.4 (cspA1 fwnA pyrG gla::fleor), a fawn-colored mutant of AB6.1 (7). After the two parental strains were crossed, the progeny were screened for prototrophy and phleomycin resistance. From the resulting heterokaryotic progeny, a diploid strain (MGG016) was isolated (6). Strain MGG029 was obtained after benomyl-induced haploidization of this diploid, selecting for phleomycin resistance, reduced milk-halo formation, and the pyrG mutant (uridine-requiring) phenotype.
Fungal cotransformation was basically carried out as described (31) using each of the peroxidase expression vectors and pAB4-1 (44) containing the A. niger pyrG selection marker, in a 10:1 ratio. Transformants were selected for uridine prototrophy. Cotransformants containing expression cassettes were selected by colony PCR, as previously described (45).
Screening for peroxidase activity.
Colony PCR-positive cotransformants were assayed for peroxidase activity using a modified plate assay method of Mayfield et al. (26): cotransformants were inoculated onto petri dishes containing Aspergillus minimal growth medium (AMM) (5), 5% maltose, 0.03% o-anisidine (Fluka, Buchs, Switzerland) and 1.4% agar. The plates were incubated at 30°C for 3 days and then flooded with a solution of 50 mM Na-phosphate buffer (pH = 4.5) and 50 μM H2O2 for MnP transformants and 50 mM Na-tartrate (pH = 3), 2 mM veratryl alcohol (Sigma), and 50 μM H2O2 for LiP transformants. Peroxidase-producing transformants developed a purple halo upon incubation at 30°C.
Culture conditions.
A. niger strains were grown from conidial inocula in 300-ml shake flasks containing 50 ml of AMM (5) supplemented with 0.5% Casamino Acids and either 5% maltose (AMM-maltose) or 5% maltodextrin (AMM-maltodextrin). For studies on heme and Fe2+ availability, AMM was supplemented with either 500 mg of hemin/liter, 5 g of hemoglobin/liter, 5 g of apohemoglobin/liter or 0.02 g of FeSO4 · 7H2O/liter. Apohemoglobin was prepared according to Nakahara and Shoun (28). Cultures were incubated at 30°C and 300 rpm, and samples were taken at different time points after inoculation. The mycelium was separated from the culture medium by filtration through Miracloth and washed with physiological salt, and total protein extracts were prepared as described elsewhere (43). The filtered culture medium was dialyzed overnight with 50 mM sodium succinate buffer (pH = 4.5) for MnP-producing strains or 50 mM sodium tartrate (pH = 6) for LiP-producing strains.
Molecular methods.
Molecular methods were carried out essentially as previously described (36). Fungal DNA isolations were performed as described by Kolar et al. (24). Total fungal RNA was isolated using the RNAzol kit from CINNA/BIOTECX. Probes used for Northern analysis experiments were a 2-kb SfiI-BamHI fragment from pAN56-2 (14a) (GenBank accession number z32690) containing a 72-bp 5′ untranslated region of the glaA mRNA hybridizing to all peroxidase transcripts, and a 1.5-kb HindIII fragment from pAB5-2 (A. niger gpdA fragment) (47). For Southern analysis, chromosomal DNA was digested with MluI, which cuts at the glaA promoter and at the trpC terminator in each of the peroxidase expression cassettes. A 0.7-kb BamHI-XmnI fragment of the glaA promoter was used as a probe. For Western blotting experiments, polyclonal antibodies against LiPH8 (αLiP-Ma) and MnPH4 (kindly provided by D. Cullen), a second anti-LiPH8 (αLiP-Fr) polyclonal antibody (kindly provided by E. Record, INRA, Marseille, France), an anti-GLA monoclonal antibody (MAb 1.5; A. Mateo-Rosell, unpublished data) and an anti-GLA polyclonal antibody (32) were used. In immunodetection experiments employing the anti-LiPH8 or anti-MnPH4 polyclonal antibodies, 1% GLA was used as a blocking agent. In other cases, 1% bovine serum albumin was the blocking agent used. Quantification of both Northern and Western analysis band intensities was performed with the GeneTools (Syngene) software.
Enzyme assays.
MnP activity was measured by monitoring the oxidation of diammonium 2,2′-azinobis(3-ethylbenzthiazoline-6-sulfonate) (ABTS) in the presence of 20 μM Na-oxalate at 415 nm as previously described (13). LiP activity was measured by monitoring the oxidation of veratryl alcohol at 310 nm (39).
Purification of the recombinant proteins.
One of the MnP-producing transformants, MGG029(pMnp1.I)#25, was used to purify recombinant MnP (rMnP). Six liters of a 3-day culture in AMM-maltodextrin was filtered through Miracloth and centrifuged at 20,000 rpm in a Beckman C5-6R centrifuge to remove mycelial debris. The supernatant was concentrated to ∼800 ml at 4°C using a hollow-fiber filter system (5-kDa cutoff, 3,500 cm2; Omega Filtron) and dialyzed against 10 mM Na-acetate (pH = 6). The concentrate was applied to a 100-ml SourceQ column equilibrated with 10 mM Na-acetate (pH = 6) in a Biopilot system. The proteins were eluted with a linear gradient of 1 M to 10 mM Na-acetate (pH = 6). Elution was followed both at 280 nm (total protein) and at 405 nm (heme protein). Fractions containing ABTS oxidizing activity were pooled, desalted, and concentrated to 15 ml using an 8-ml SourceQ column eluted with a steep Na-acetate gradient. This concentrate was then applied to an 1,800-ml Superdex75 column, and proteins were eluted in 50 mM Na-succinate (pH = 4.5). Purity of the heme protein peak fractions was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)/silver staining using the Phast system (Pharmacia, Piscataway, N.J.) and by measuring their spectrum profile. Fractions containing the highest A407/A280 ratio were dialyzed against 10 mM Na-acetate (pH = 6) and concentrated.
Recombinant LiP (rLiP) was concentrated and partially purified from a 5-liter, 48-h AMM-maltose culture of a pLipA.I-containing transformant. The rLiP medium sample was filtrated, concentrated, dialyzed, and SourceQ fractionated as described for rMnP but using 20 mM Bis-Tris (pH = 6.5) as dialysis buffer and a 100 mM to 1 M Bis-Tris (pH = 6.5) elution gradient instead of the Na-acetate-based buffer. SourceQ fractions were analyzed by Western blotting and concentrated with a 5-kDa-cutoff FILTRON Omegacell at 4°C.
RESULTS
Transformation and screening.
In a cotransformation experiment, strain MGG029 was transformed with a mixture of plasmid pAB4-1 and each of the lipA or mnp1 expression vectors. Transformants were selected for their ability to grow in AMM plates without uridine. More than 300 uridine prototrophic transformants were obtained per plasmid pair.
Cotransformants containing the corresponding lipA or mnp1 expression vector were identified by colony PCR using specific primers (Table 1 and data not shown). Three colony PCR-positive transformants per construct were analyzed by Southern hybridization, and multicopy integration of the expression cassettes was found in all cases (3 to 6 copies; data not shown).
Transformants were analyzed for peroxidase production using an activity plate assay based on the oxidation of o-anisidine. Transformants containing the pMnp1.I expression cassette developed a purple halo, indicating extracellular peroxidase activity. However, none of the pGLA::Mnp1-, pLipA.I-, pLipA.II-, or pGLA::LipA-containing transformants showed significant halo formation.
Northern analysis.
Since the activity plate assay revealed differences in extracellular enzyme activity between different expression cassettes, it was important to determine whether the various transcript levels showed a similar difference. N402(pAB6-10)B1, a multicopy (20 copies) GLA2-producing strain (46), which produces up to 900 mg of extracellular GLA/liter, was taken as a reference. As shown in Fig. 2a, Northern blot analysis revealed mRNA of the expected size in all five transformants. The existence of a double transcript band in pLipA.I-, pLipA.II-, and pMnP1.I-containing transformants is due to the two polyadenylation sites used in the A. nidulans trpC terminator (27). Transcript signal intensities were measured and corrected for loading differences using the pgdA probe (Fig. 2b and c). Although specific normalized mRNA levels varied among transformants, significant hybridization signals were found in all cases and were comparable to the levels observed for the N402(pAB6-10)B1 strain. These results indicate that at the transcriptional level, no major bottlenecks exist for the production of MnP and LiP in A. niger MGG029.
FIG. 2.

Northern blotting analysis of total RNA isolated from one representative transformant per expression cassette after 48-h culturing on AMM-maltose. (a) pglaA probe. (b) gpd probe. (c) Ratio between each transformant corrected glaA signal and the N024(pAB6-10)B1 corrected glaA signal. The expected mRNA sizes are indicated.
Analysis of protein production.
Two transformants per construct showing the highest mRNA levels were chosen for a study of peroxidase protein production in shake-flask cultures. Transformants were incubated for 72 h in AMM-maltose, and samples were taken at 24-h intervals. Medium samples were assayed for peroxidase activity as described in Materials and Methods, and both culture medium samples and mycelium extracts were subjected to Western analysis using LiP, MnP, and GLA antibodies.
Maximum yields were obtained at the 48-h time point. In agreement with the results obtained with the plate assay, the culture medium of pMnp1.I-containing transformants showed ABTS oxidizing activity, and Western analysis revealed the presence of an anti-MnP cross-reactive protein band of approximately the size of the native MnP (Fig. 3a). In contrast, in the medium of transformants containing the fusion expression vector pGLA::Mnp1, no MnP cross-reactive material could be detected, and accordingly, no peroxidase activity was found. However, these transformants did secrete the GLA part of the fusion protein into the medium, which could be detected as a major 70-kDa protein band and some degradation products (Fig. 3b). Similar results were obtained when the mycelium extracts were analyzed. Intracellular MnP was detected in pMnp1.I-containing but not in pGLA::MnP1-containing transformants. In the latter, a weak 120-kDa protein band was observed which cross-reacted with both MnP and GLA antibodies, possibly corresponding to the uncleaved GLA::MnP protein (Fig. 3c and d).
FIG. 3.

Western blotting analysis of MnP transformants. Culture samples at 48 h of one representative transformant per construct are shown. (a and b) medium samples; (c and d) mycelium extracts. Blots were probed with either a polyclonal anti-MnP antibody (a and c) or a monoclonal anti-GLA antibody (b and d).
In the case of the LiP transformants, no veratryl alcohol oxidizing activity was measurable in the culture medium of transformants containing any of the three constructs. However, when these samples were analyzed by Western blotting, LiP cross-reactivity was observed (Fig. 4a). Most cross-reactive material was found as two protein bands of approximately 30 and 15 kDa, and only a small fraction of the protein produced migrated at the position of the native LiP control (∼42 kDa). These cross-reactive bands were also observed in the mycelium extracts (Fig. 4b). This result was found regardless of the construct used for expression of the lipA gene. As in the case of the pGLA::MnP transformants, an analysis of medium and mycelium extracts of the GLA::LiP fusion with the anti-GLA antibody showed synthesis and secretion of the GLA counterpart (data not shown).
FIG. 4.

Western blotting analysis of LiP transformants. Culture samples at 48 h of one representative transformant per construct are shown. (a) Medium samples; (b) mycelium extracts. Blots were probed with the anti-LiP-Ma polyclonal antibody.
To confirm the LipH8 identity of the anti-LiP cross-reactive protein bands, the culture medium of transformant MGG029(pLipA-I)#5 was concentrated and proteins were fractionated and analyzed as described in Materials and Methods. The anti-LiP-Ma cross-reacting 42-, 30-, and 15-kDa protein bands were submitted to N-terminal amino acid sequencing. Unfortunately, no interpretable sequence resulted from this analysis, probably due to N-terminal blockage of the polypeptides. However all three protein bands cross-reacted with a second polyclonal antibody raised against LiPH8 (Fig. 5 and results not shown), which supports the LiPH8 nature of these polypeptides. No MnP, LiP, or GLA was detected in the parent strain transformed only with a vector containing the selection marker, MGG029(pAB4-1).
FIG. 5.
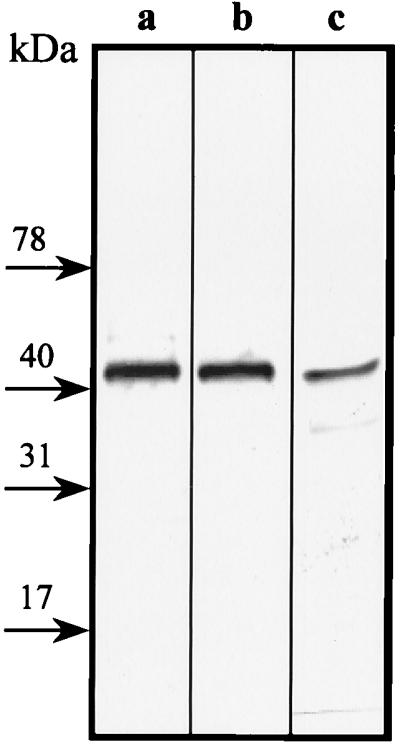
Immunodetection of rLiP. A purification fraction containing the anti-LiP cross-reactive 42-kDa protein band was detected either with anti-LiP-Ma polyclonal antibody (a) or with anti-LiP-Fr polyclonal antibody (b). (c) P. chrysosporium LiPH8 control probed with anti-LiP-Fr polyclonal antibody.
Heme and Fe2+ supplementation studies.
To study whether heme availability was a limiting factor in the production of fungal peroxidases in A. niger, the effects of several medium additives on the production of MnP and LiP were studied. Two pMnP1.I transformants and the wild-type control, MGG029(pAB4-1), were cultured in AMM-maltodextrin supplemented with either hemin, hemoglobin, apohemoglobin, or a fourfold excess of FeSO4 in comparison to AMM. Medium samples were taken at 12-h intervals up to 72 h, dialyzed, and analyzed for ABTS oxidizing activity. Although absolute activity values varied, the two pMnP.1 strains showed similar behavior. Maximum activity was reached after 36 h. At this point, hemin- and hemoglobin-supplemented media showed, respectively, a 7- and 10-fold increase in activity over that of the nonsupplemented medium. Medium supplemented with apohemoglobin showed a threefold increase in activity, whereas FeSO4 supplementation had no significant effect (Table 3). Transformants containing only the pAB4-1 vector showed no activity, regardless of the culture medium (data not shown). Western blot analysis revealed that the observed differences in activity corresponded to different amounts of rMnP in the culture medium (Fig. 6a and c). To verify that these differences in rMnP production were not merely the consequence of general metabolic effects, the secretion of a non-heme-related protein was analyzed in parallel by Western blotting. For this purpose we used a 70-kDa extracellular amylase, which can be detected with a polyclonal anti-GLA antiserum (P. J. Punt, unpublished data). Proteins bands were quantified, and signal intensities were related to the nonsupplemented medium. This experiment showed that extracellular amylase levels were similar in all five media (protein signal variations of less than 20% related to the unsupplemented medium), whereas anti-MnP signals varied accordingly with the measured activities (Fig. 6). Moreover, when equal amounts of ABTS oxidizing activity (in units/milliliter) of the five different medium samples were analyzed by Western blotting, similar amounts of rMnP were observed, indicating that the rMnP produced in the different media had similar specific activity.
TABLE 3.
Extracellular rMnP production of strain MGG029(pMn1.I)#25 after 36 h of growth in differently supplemented AMM-maltodextrin media
| Medium | Activity (ΔAbs/min/ml)a | Extracellular rMnP (mg/liter)c |
|---|---|---|
| AMM-maltodextrin | 6.0 ± 0.8b | 9.6 ± 1.2 |
| AMM-maltodextrin + 0.02 g/liter of FeSO4 | 6.7 ± 0.9 | 10.7 ± 1.4 |
| AMM-maltodextrin + 500 mg/liter of hemin | 41.8 ± 0.8 | 66.9 ± 1.2 |
| AMM-maltodextrin + 5 g/liter of apohemoglobin | 19.7 ± 0.6 | 31.5 ± 0.9 |
| AMM-maltodextrin + 5 g/liter of hemoglobin | 66.2 ± 12.1 | 105.9 ± 19.4 |
Activity was measured by ABTS oxidation (see Materials and Methods), and protein values were calculated based on MnP specific activity.
The values presented are the averages of at least two measurements in two different experiments, ± standard deviations.
Values are means ± standard deviations.
FIG. 6.

Western blotting analysis of the extracellular protein production of strain MGG029(pMnp1.I)#25 cultured in differently supplemented AMM-maltodextrin media. −, no supplementation; Fe, FeSO4; APO, apohemoglobin; HEM, hemin; HMG, hemoglobin. (a) Production of MnP: blot was probed with an anti-MnP polyclonal antibody. (b) Production of amylase (AMY): blot was probed with a cross-reacting anti-GLA polyclonal antibody. (c) Band intensities of the MnP signals (RMnP) and amylase signals (RAMY), relative to the unsupplemented medium.
A similar supplementation experiment was also done for glaA::mnp1- and lipA-expressing strains. In these transformants, heme supplementation did not result in any detectable MnP or LiP activity levels in the culture medium (data not shown). However, hemoglobin supplementation did increase the amount of the 30- and 15-kDa LiP cross-reactive bands found in the medium of the analyzed LiP transformants (data not shown).
Purification and characterization of rMnP.
rMnP was purified from the culture medium of strain MGG029(pMnp1.I)#25 by a two-step purification procedure. By using simultaneously 280-nm- and 405-nm-wavelength filters, the purification of rMnP could be easily monitored, showing that MnP was practically the only heme protein present in the culture medium. The purest rMnP fraction had an A407/A280 ratio of 5.1, comparable to that of the native enzyme, and its spectrum profile (Fig. 7) was very similar to reported data (38). The specific activity determined by ABTS oxidation was 0.44 ΔAbs/min/μg of enzyme. Under the same conditions, a specific activity of 0.63 ΔAbs/min/μg was measured for a commercially available native protein preparation. Upon N-terminal sequencing of the extracellular rMnP, the sequence AlaValXxxProAsp, where Xxx represents an unknown amino acid, was obtained, which matches the native MnP N terminus (AlaValCysProAsp). SDS-PAGE analysis of the purified recombinant protein confirmed the observation of slightly lower mobility than that of nMnP (Fig. 4a and results not shown).
FIG. 7.

Spectrum profile of rMnP. Numbers at left are absorbances expressed in arbitrary units.
DISCUSSION
To study the production of LipA and Mnp1 in A. niger, we constructed expression cassettes containing the cDNAs encoding these proteins to express them either with their own signal peptides or as GLA fusions. The use of a protein fusion strategy to improve the production of heterologous proteins has been proven to be successful (7, 8, 18, 22) and to date has not yet been described for the expression of fungal peroxidases in filamentous fungi.
The host of choice, A. niger MGG029, was constructed to combine in one strain various characteristics favorable for studies on the production of heterologous proteins. This strain is deficient in the expression of several protease genes due to a regulatory mutation (25; F. H. J. Schuren, unpublished data). Lacking the glaA gene, MGG029 produces no endogenous GLA. This facilitates analysis when GLA fusion constructs are employed, since detection of GLA secretion confirms successful expression of the fusion gene. Finally, the strain carries a nonfunctional pyrG, which enables the use of the pyrG selection marker.
Active MnP was secreted into the culture medium of transformants carrying the expression vector pMnp1.I, as was detected by both a colorimetric plate assay and Western analysis. Surprisingly, no MnP activity or anti-MnP cross-reacting material could be detected in the culture medium of transformants carrying the GLA::MnP fusion construct. This was puzzling, since the GLA part of the fusion was efficiently secreted. A possible explanation for this is that the presence of the GLA part of the fusion could interfere with essential maturation events for the MnP protein, leading to the degradation of rMnP. On the other hand, expression of lipA as a fusion protein did result in secretion of LiP protein, yet this was inactive. Further experiments to delve into these observations are in progress. This is, to our knowledge, the first reported case where a fusion approach failed. Interestingly, a similar observation has been made in our laboratory by J. G. M. Hessing and F. H. J. Schuren (unpublished data) in their studies on the expression of a laccase gene in Aspergillus spp. These results suggest that the secretion of carrier target fusion protein is more complex than previously thought and indicate that the success of the fusion approach is dependent not only on the availability of a potentially efficient carrier but also on the nature of the cargo protein. In contrast to the situation with mnp1, in the lipA-containing transformants no enzyme activity could be detected for any of the three constructs used. However, when culture medium of these transformants was analyzed by Western blotting, two dominant anti-LiP cross-reacting protein bands, of approximately 30 and 15 kDa, were observed (Fig. 4a). Little or no cross-reactive material of the size of the native LiP could be observed in the crude medium samples, although a clear 42-kDa protein could be detected with two different anti-LiP antibodies when the medium of a pLipA.I-containing transformant was concentrated. This suggests that although LiP is synthesized in MGG029, the protein is incorrectly processed into two protein fragments, whereby peroxidase activity is lost. These two cross-reacting bands were also observed in the mycelial extracts (Fig. 4b), indicating that the suggested processing is mycelium associated and not the result of extracellular degradation.
In other studies on the expression of lignin peroxidase in filamentous fungi (1, 35; D. Cullen, personal communication), the major production bottlenecks have been shown to occur at the protein level. In these previous works, little or no extracellular LiP protein could be detected, although sufficient mRNA was available. In our case, we do observe extracellular protein production but no measurable activity, probably due to incorrect processing of the LiP protein. We believe this processing is related to the LiP sequence and not to the requirement for a heme cofactor, since it was not observed for other heme peroxidases expressed in A. niger, such as MnP (this work) or the chloroperoxidase from Caldariomyces fumago (Conesa et al., unpublished data). Our results suggest that approaches to avoid this processing will be required to obtain efficient production of LiP in A. niger. Interestingly, analysis of the deduced amino acid sequence of lipA revealed a number of monobasic motifs (SerArg, SerLys) which could be potential protease processing sites. None of these sites is present at the corresponding position of the MnP sequence. Currently, we are investigating the use of site-directed mutagenesis to address this issue.
Analysis of purified rMnP showed similar spectral properties. Although the specific activity of rMnP was somewhat lower than those of the native enzyme, similar results were obtained by Stewart et al. (38) in their studies on the expression of mnp1 in A. oryzae. In our work we also show that correct processing of the MnP signal peptide occurs in A. niger. In contrast to Stewart's results, rMnP produced in A. niger showed a slightly lower mobility on SDS-PAGE than the nMnP. This difference could be the result of a higher degree of glycosylation of the recombinant enzyme and apparently has no major effect on the activity of rMnP. MnP is both N- and O-glycosylated, although Nie et al. (29) have showed that glycosylation is not essential for the enzyme activity of MnP. Overglycosylation has also been observed in the production of recombinant chloroperoxidase (Conesa et al., unpublished) and phytase (50) in Aspergillus.
The initial yields we obtained for rMnP in A. niger MGG029 were 5 to 10 mg/liter, which is low compared with those for most other fungal proteins expressed in filamentous fungi. This low yield range was not caused by unefficient transcription, since mnp1 mRNA levels were comparable to those of an efficiently secreted protein (Fig. 2). Similarly, efficient transcription was observed for the other mnp1 and lipA expression cassettes used in this study.
Low heme availability has been suggested as a limiting factor for the production of heme proteins in different expression systems (2, 12, 48). In our work, we have studied this limitation in more detail. Heme supplementation in the form of hemoglobin or hemin resulted in a significant increase of extracellular MnP activity due to a concomitant increase in protein production (Fig. 6a and c) and not to an increase in specific activity. This indicates that apoforms of rMnP may be unstable during or after secretion and that only the holoprotein accumulates in the extracellular medium. The observed production increase was not due to the additional Fe2+ supplied by hemin and hemoglobin, since Fe2+ supplementation alone had no effect on MnP activity. We observed that hemoglobin supplementation resulted in a higher rMnP production than the addition of hemin. The reason for this could be that hemoglobin may play a role not only in supplying heme but also in providing a protein excess in the culture medium. This protein excess may protect rMnP from proteolytic degradation, as is also suggested by the fact that addition of apohemoglobin or bovine serum albumin (data not shown) has a positive effect on the rMnP yields. The host strain we have used is considerably reduced in, but not totally devoid of, extracellular protease activity, and it is likely that some proteolytic degradation of rMnP is still taking place.
Our results in the heme supplementation experiments seem therefore to support the hypothesis regarding limited heme availability. However, in some cases, heme-containing proteins were successfully overexpressed without specific heme requirements being reported (15, 40, 41). However, heme limitation has not been assessed in most of these studies. Furthermore, since these examples concern intracellular proteins, other factors, such as the cellular localization of the heme proteins, heme incorporation, and/or other posttranslational modifications, may also be important. To date, very little is known about heme and heme protein biosynthesis in filamentous fungi. Elrod et al. (10) showed that overexpression of heme biosynthetic enzymes in A. oryzae led to increased yields of a heme-containing fungal peroxidase, although these strains still responded to heme supplementation. Further research is needed to unravel these processes.
In conclusion, we have reported the efficient production of LiP and MnP in A. niger. Although LiP was incorrectly processed and not active, our system provides a promising starting point for further research. Up to 100 mg of extracellular rMnP/liter could be produced in A. niger MGG029 under hemoglobin supplementation conditions. This is a considerable increase compared to previous reports and indicates that not only the composition of the culture medium but also the choice of the production strain is important for the efficient production of these difficult enzymes.
ACKNOWLEDGMENTS
We thank Ron van den Dool and Wim van Hartingsveldt for their collaboration in rMnP purification, Daan de Kloe for his assistance in the construction of A. niger MGG029, Dan Cullen for providing LiP and MnP clones and antisera, Eric Record for providing the anti-LiP antibody, and N. van Luijk for a critical reading of the manuscript.
REFERENCES
- 1.Aifa M S, Sayadi S, Gargouri A. Heterologous expression of lignin peroxidase of Phanerochaete chrysosporium in Aspergillus niger. Biotechnol Lett. 1999;21:849–853. [Google Scholar]
- 2.Andersen, H. D., E. B. Jensen, and K. G. Welinder. September 1992. A process for producing heme proteins. European Patent Application EP 0 505 311 A2.
- 3.Archer D B, Peberdy J F. The molecular biology of secreted enzyme production by fungi. Crit Rev Biotechnol. 1997;17:273–306. doi: 10.3109/07388559709146616. [DOI] [PubMed] [Google Scholar]
- 4.Aust S D. Degradation of environmental pollutants by Phanerochaete chrysosporium. Microb Ecol. 1990;20:197–209. doi: 10.1007/BF02543877. [DOI] [PubMed] [Google Scholar]
- 5.Bennett J W, Lasure L L. Growth media. In: Bennett J W, Lasure L L, editors. More gene manipulation in fungi. New York, N.Y: Academic Press, Inc.; 1991. pp. 441–457. [Google Scholar]
- 6.Bos C J. Ph.D. thesis. Wageningen, The Netherlands: Wageningen Agricultural University; 1986. Induced mutation and somatic recombination as tools for genetic analysis and breeding of imperfect fungi; p. 156. [Google Scholar]
- 7.Broekhuijsen M P, Mattern I E, Contreras R, Kinghorn J R, van den Hondel C A. Secretion of heterologous proteins by Aspergillus niger: production of active human interleukin-6 in a protease-deficient mutant by KEX2-like processing of a glucoamylase-hIL6 fusion protein. J Biotechnol. 1993;31:135–145. doi: 10.1016/0168-1656(93)90156-h. [DOI] [PubMed] [Google Scholar]
- 8.Contreras R, Carrez D, Kinghorn J R, van den Hondel C A, Fiers W. Efficient KEX2-like processing of a glucoamylase-interleukin-6 fusion protein by Aspergillus nidulans and secretion of mature interleukin-6. Biotechnology (New York) 1991;9:378–381. doi: 10.1038/nbt0491-378. [DOI] [PubMed] [Google Scholar]
- 9.Doyle W A, Smith A T. Expression of lignin peroxidase H8 in Escherichia coli: folding and activation of the recombinant enzyme with Ca2+ and haem. Biochem J. 1996;315:15–19. doi: 10.1042/bj3150015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elrod, S. L., J. R. Cherry, and A. Jones. December 1997. A method for increasing hemoprotein production in filamentous fungi. International Patent Application patent WO 97/47746.
- 11.Feijoo G, Dosoretz C, Lema J M. Production of lignin-peroxidase by Phanerochaete chrysosporium in a packed bed bioreactor operated in semicontinuous mode. J Biotechnol. 1995;42:247–253. [Google Scholar]
- 12.Fowler T, Rey M W, Vaha-Vahe P, Power S D, Berka R M. The catR gene encoding a catalase from Aspergillus niger: primary structure and elevated expression through increased gene copy number and use of a strong promoter. Mol Microbiol. 1993;9:989–998. doi: 10.1111/j.1365-2958.1993.tb01228.x. [DOI] [PubMed] [Google Scholar]
- 13.Glenn J K, Gold M H. Purification and characterization of an extracellular Mn(II)-dependent peroxidase from the lignin-degrading basidiomycete, Phanerochaete chrysosporium. Arch Biochem Biophys. 1985;242:329–341. doi: 10.1016/0003-9861(85)90217-6. [DOI] [PubMed] [Google Scholar]
- 14.Gold M H, Alic M. Molecular biology of the lignin-degrading basidiomycete Phanerochaete chrysosporium. Microbiol Rev. 1993;57:605–622. doi: 10.1128/mr.57.3.605-622.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14a.Gordon C L, Khalaj V, Ram A F, Archer D B, Brookman J L, Trinci A P, Jeenes D J, Doonan J H, Wells B, Punt P J, van den Hondel C A, Robson G D. Glucoamylase::green fluorescent protein fusions to monitor protein secretion in Aspergillus niger. Microbiology. 2000;146:415–426. doi: 10.1099/00221287-146-2-415. [DOI] [PubMed] [Google Scholar]
- 15.Gromada A, Fiedurek J. Optimization of catalase biosynthesis in submerged cultures of Aspergillus niger mutant. J Basic Microbiol. 1997;37:85–91. doi: 10.1002/jobm.3620370203. [DOI] [PubMed] [Google Scholar]
- 16.Hammel K E, Jensen K A, Jr, Mozuch M D, Landucci L L, Tien M, Pease E A. Ligninolysis by a purified lignin peroxidase. J Biol Chem. 1993;268:12274–12281. [PubMed] [Google Scholar]
- 17.Higson F K. Degradation of xenobiotics by white rot fungi. Rev Environ Contam Toxicol. 1991;122:111–152. doi: 10.1007/978-1-4612-3198-1_4. [DOI] [PubMed] [Google Scholar]
- 18.Jeenes D J, Marczinke B, MacKenzie D A, Archer D B. A truncated glucoamylase gene fusion for heterologous protein secretion from Aspergillus niger. FEMS Microbiol Lett. 1993;107:267–271. doi: 10.1111/j.1574-6968.1993.tb06041.x. [DOI] [PubMed] [Google Scholar]
- 19.Johnson T M, Li J K. Heterologous expression and characterization of an active lignin peroxidase from Phanerochaete chrysosporium using recombinant baculovirus. Arch Biochem Biophys. 1991;291:371–378. doi: 10.1016/0003-9861(91)90148-c. [DOI] [PubMed] [Google Scholar]
- 20.Johnson T M, Pease E A, Li J K, Tien M. Production and characterization of recombinant lignin peroxidase isozyme H2 from Phanerochaete chrysosporium using recombinant baculovirus. Arch Biochem Biophys. 1992;296:660–666. doi: 10.1016/0003-9861(92)90624-6. [DOI] [PubMed] [Google Scholar]
- 21.Karam J, Nicell J A. Potential applications of enzymes in waste treatment. J Chem Tech Biotechnol. 1997;69:141–153. [Google Scholar]
- 22.Keränen S, Penttilä M. Production of recombinant proteins in the filamentous fungus Trichoderma reesei. Curr Opin Biotechnol. 1995;6:534–537. doi: 10.1016/0958-1669(95)80088-3. [DOI] [PubMed] [Google Scholar]
- 23.Kirk T K, Farrell R L. Enzymatic “combustion”: the microbial degradation of lignin. Annu Rev Microbiol. 1987;41:465–505. doi: 10.1146/annurev.mi.41.100187.002341. [DOI] [PubMed] [Google Scholar]
- 24.Kolar M, Punt P J, van den Hondel C A, Schwab H. Transformation of Penicillium chrysogenum using dominant selection markers and expression of an Escherichia coli lacZ fusion gene. Gene. 1988;62:127–134. doi: 10.1016/0378-1119(88)90586-0. [DOI] [PubMed] [Google Scholar]
- 25.Mattern I E, van Noort J M, van den Berg P, Archer D B, Roberts I N, van den Hondel C A. Isolation and characterization of mutants of Aspergillus niger deficient in extracellular proteases. Mol Gen Genet. 1992;234:332–336. doi: 10.1007/BF00283855. [DOI] [PubMed] [Google Scholar]
- 26.Mayfield M B, Kishi K, Alic M, Gold M H. Homologous expression of recombinant manganese peroxidase in Phanerochaete chrysosporium. Appl Environ Microbiol. 1994;60:4303–4309. doi: 10.1128/aem.60.12.4303-4309.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mullaney E J, Hamer J E, Roberti K A, Yelton M M, Timberlake W E. Primary structure of the trpC gene from Aspergillus nidulans. Mol Gen Genet. 1985;199:37–45. doi: 10.1007/BF00327506. [DOI] [PubMed] [Google Scholar]
- 28.Nakahara K, Shoun H. N-terminal processing and amino acid sequence of two isoforms of nitric oxide reductase cytochrome P450nor from Fusarium oxysporum. J Biochem (Tokyo) 1996;120:1082–1087. doi: 10.1093/oxfordjournals.jbchem.a021525. [DOI] [PubMed] [Google Scholar]
- 29.Nie G, Reading N S, Aust S D. Relative stability of recombinant versus native peroxidases from Phanerochaete chrysosporium. Arch Biochem Biophys. 1999;365:328–334. doi: 10.1006/abbi.1999.1180. [DOI] [PubMed] [Google Scholar]
- 30.Pease E A, Aust S D, Tien M. Heterologous expression of active manganese peroxidase from Phanerochaete chrysosporium using the baculovirus expression system. Biochem Biophys Res Commun. 1991;179:897–903. doi: 10.1016/0006-291x(91)91903-p. [DOI] [PubMed] [Google Scholar]
- 31.Punt P J, van den Hondel C A. Transformation of filamentous fungi based on hygromycin B and phleomycin resistance markers. Methods Enzymol. 1992;216:447–457. doi: 10.1016/0076-6879(92)16041-h. [DOI] [PubMed] [Google Scholar]
- 32.Punt P J, Zegers N D, Busscher M, Pouwels P H, van den Hondel C A. Intracellular and extracellular production of proteins in Aspergillus under the control of expression signals of the highly expressed Aspergillus nidulans gpdA gene. J Biotechnol. 1991;17:19–33. doi: 10.1016/0168-1656(91)90024-p. [DOI] [PubMed] [Google Scholar]
- 33.Reddy C A, D'Souza T M. Physiology and molecular biology of the lignin peroxidases of Phanerochaete chrysosporium. FEMS Microbiol Rev. 1994;13:137–152. doi: 10.1111/j.1574-6976.1994.tb00040.x. [DOI] [PubMed] [Google Scholar]
- 34.Saloheimo M, Barajas V, Niku-Paavola M-L, Knowles J K C. A lignin peroxidase-encoding cDNA from the white fungus Phlebia radiata: characterization and expression in Trichoderma reesei. Gene. 1989;85:343–351. doi: 10.1016/0378-1119(89)90427-7. [DOI] [PubMed] [Google Scholar]
- 35.Saloheimo M, Niku-Paavola M-L. Heterologous production of a lignolytic enzyme: expression of the Phlebia radiata laccase gene in Trichoderma reesei. Biotechnology. 1991;9:987–990. [Google Scholar]
- 36.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 37.Sollewijn Gelpke M D, Mayfield-Gambill M, Lin Cereghino G P, Gold M H. Homologous expression of recombinant lignin peroxidase in Phanerochaete chrysosporium. Appl Environ Microbiol. 1999;65:1670–1674. doi: 10.1128/aem.65.4.1670-1674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stewart P, Whitwam R E, Kersten P J, Cullen D, Tien M. Efficient expression of a Phanerochaete chrysosporium manganese peroxidase gene in Aspergillus oryzae. Appl Microbiol Biotechnol. 1996;62:860–864. doi: 10.1128/aem.62.3.860-864.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tien M, Kirk T K. Lignin-degrading enzyme from Phanerochaete chrysosporium: purification, characterization, and catalytic properties of a unique H2O2-requiring oxygenase. Proc Natl Acad Sci USA. 1984;81:2280–2284. doi: 10.1073/pnas.81.8.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urban P, Cullin C, Pompon D. Maximizing the expression of mammalian cytochrome P-450 monooxygenase activities in yeast cells. Biochimie. 1990;72:463–472. doi: 10.1016/0300-9084(90)90070-w. [DOI] [PubMed] [Google Scholar]
- 41.van den Brink J M, van den Hondel C A, van Gorcom R F. Optimization of the benzoate-inducible benzoate p-hydroxylase cytochrome P450 enzyme system in Aspergillus niger. Appl Microbiol Biotechnol. 1996;46:360–364. doi: 10.1007/BF00166230. [DOI] [PubMed] [Google Scholar]
- 42.van den Hondel C A M J J, Punt P J, van Gorcom R F M. Heterologous gene expression in filamentous fungi. In: Bennett J W, Lasure L L, editors. More gene manipulations in fungi. New York, N.Y: Academic Press; 1991. pp. 396–428. [Google Scholar]
- 43.van Gorcom R F, Pouwels P H, Goosen T, Visser J, van den Broek H W, Hamer J E, Timberlake W E, van den Hondel C A. Expression of an Escherichia coli beta-galactosidase fusion gene in Aspergillus nidulans. Gene. 1985;40:99–106. doi: 10.1016/0378-1119(85)90028-9. [DOI] [PubMed] [Google Scholar]
- 44.van Hartingsveldt W, Mattern I E, van Zeijl C M, Pouwels P H, van den Hondel C A. Development of a homologous transformation system for Aspergillus niger based on the pyrG gene. Mol Gen Genet. 1987;206:71–75. doi: 10.1007/BF00326538. [DOI] [PubMed] [Google Scholar]
- 45.van Zeijl C M, van de Kamp E H, Punt P J, Selten G C, Hauer B, van Gorcom R F, van den Hondel C A. An improved colony-PCR method for filamentous fungi for amplification of PCR-fragments of several kilobases. J Biotechnol. 1997;59:221–224. doi: 10.1016/s0168-1656(97)00170-3. [DOI] [PubMed] [Google Scholar]
- 46.Verdoes J C, Punt P J, Schrickx J M, van Verseveld H W, Stouthamer A H, van den Hondel C A. Glucoamylase overexpression in Aspergillus niger: molecular genetic analysis of strains containing multiple copies of the glaA gene. Transgenic Res. 1993;2:84–92. doi: 10.1007/BF01969381. [DOI] [PubMed] [Google Scholar]
- 47.Verdoes J C, Punt P J, Stouthamer A H, van den Hondel C A. The effect of multiple copies of the upstream region on expression of the Aspergillus niger glucoamylase-encoding gene. Gene. 1994;145:179–187. doi: 10.1016/0378-1119(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 48.Weber J M, Ponti C G, Kappeli O, Reiser J. Factors affecting homologous overexpression of the Saccharomyces cerevisiae lanosterol 14 alpha-demethylase gene. Yeast. 1992;8:519–533. doi: 10.1002/yea.320080704. [DOI] [PubMed] [Google Scholar]
- 49.Whitwam R, Tien M. Heterologous expression and reconstitution of fungal Mn peroxidase. Arch Biochem Biophys. 1996;333:439–446. doi: 10.1006/abbi.1996.0413. [DOI] [PubMed] [Google Scholar]
- 50.Wyss M, Pasamontes L, Friedlein A, Remy R, Tessier M, Kronenberger A, Middendorf A, Lehmann M, Schnoebelen L, Rothlisberger U, Kusznir E, Wahl G, Muller F, Lahm H W, Vogel K, van Loon A P. Biophysical characterization of fungal phytases (myo-inositol hexakisphosphate phosphohydrolases): molecular size, glycosylation pattern, and engineering of proteolytic resistance. Appl Environ Microbiol. 1999;65:359–366. doi: 10.1128/aem.65.2.359-366.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]