

ABSTRACT
Mycobacterium tuberculosis encounters numerous stress conditions within the host, but how it is able to mount a coupled stress response remains unknown. Growing evidence suggests that under acidic pH, M. tuberculosis modulates redox homeostasis. In an attempt to dissect the mechanistic details of responses to multiple stress conditions, here we studied the significance of connectivity of extracytoplasmic sigma factors with PhoP. We show that PhoP impacts the mycothiol redox state, and the H37Rv ΔphoP deletion mutant strain displays a significantly higher susceptibility to redox stress than the wild-type bacilli. To probe how the two regulators PhoP and redox-active sigma factor SigH contribute to redox homeostasis, we show that SigH controls expression of redox-active thioredoxin genes, a major mycobacterial antioxidant system, and under redox stress, SigH, but not PhoP, is recruited at the target promoters. Consistent with these results, interaction between PhoP and SigH fails to impact redox-dependent gene expression. This is in striking contrast to our previous results showing PhoP-dependent SigE recruitment within acid-inducible mycobacterial promoters to maintain pH homeostasis. Our subsequent results demonstrate reduced PhoP-SigH interaction in the presence of diamide and enhanced PhoP-SigE interaction under low pH. These contrasting results uncover the underlying mechanism of the mycobacterial adaptive program, coupling low pH with maintenance of redox homeostasis.
IMPORTANCE M. tuberculosis encounters reductive stress under acidic pH. To investigate the mechanism of coupled stress response, we show that PhoP plays a major role in mycobacterial redox stress response. We observed a strong correlation of phoP-dependent redox-active expression of thioredoxin genes, a major mycobacterial antioxidant system. Further probing of functioning of regulators revealed that while PhoP controls pH homeostasis via its interaction with SigE, direct recruitment of SigH, but not PhoP-SigH interaction, controls expression of thioredoxin genes. These strikingly contrasting results showing enhanced PhoP-SigE interaction under acidic pH and reduced PhoP-SigH interaction under redox conditions uncover the underlying novel mechanism of the mycobacterial adaptive program, coupling low pH with maintenance of redox homeostasis.
KEYWORDS: low-pH stress, Mycobacterium tuberculosis, PhoP, oxidation reduction, redox, oxidative stress, Sigma factor, thioredoxin, transcription coregulation, protein-protein interactions
INTRODUCTION
Survival and persistence of mycobacteria in response to various environmental cues rely on the coupling between signal sensing and induction of the appropriate adaptive program. Therefore, understanding molecular mechanisms of adaptation in response to environmental changes represents a major aspect of Mycobacterium tuberculosis biology. Although recent years have seen substantial progress in the molecular characterization of the bacillus, much work is still needed to understand how M. tuberculosis copes with the various environments it encounters in the course of an infection. Adaptations to such conditions must require a complex regulatory mechanism of gene expression.
Mycobacterial gene expression in response to acidic pH significantly overlaps with the PhoP regulon (1). In keeping with this, a large subset of low-pH-inducible genes, which are also regulated by PhoPR system, are induced immediately following M. tuberculosis phagocytosis and remain induced during macrophage infection (2–5). These include genes involved in biosynthesis of cell envelope lipids, which contribute to arresting phagosomal maturation (6), indicating a major shift in anabolic metabolism under acidic pH. PhoPR, in addition, controls major virulence factors central to tuberculosis (TB) pathogenesis, including ESX-1-dependent secretion of ESAT-6 (7–9), which interferes with phagosomal maturation, neutralizes the phagosome, and modulates carbon source availability in the mycobacterial cytoplasm from the fatty acids and cholesterol of the phagosome (10). Thus, during onset of macrophage infection, PhoPR activation is linked to acidic pH and the available carbon source, suggesting a physiological link between pH, carbon source, and macrophage pathogenesis.
Under acidic pH, M. tuberculosis overexpresses redox-active proteins, including thioredoxins, alkyl hydroxy-peroxidase reductase, and the regulatory protein WhiB3. PhoPR-dependent WhiB3 regulation (11) is known to control synthesis of lipids, such as sulfolipids, poly- and di-acyl trehaloses, and phthiocerol dimycoserosates (PDIM) (12). Thus, WhiB3-controlled lipid synthesis functions as a reductive sink to maintain redox homeostasis during hypoxia (12) and macrophage infection (13). These results suggest that under acidic pH, M. tuberculosis encounters reductive stress, a phenomenon in which cells are unable to replenish oxidized cofactors like NAD+/NADP+, resulting in accumulation of NADH and NADPH (14). In keeping with this, mycobacteria display a reduced cytoplasmic potential under acidic pH (10). Thus, acidic pH appears to promote reductive stress, and a network of responses controlled by major regulators like PhoPR and WhiB3 function to mitigate reductive stress (for a review, see reference 15). In a paradoxical situation, cells encountering reductive stress might face elevated oxidative stress as metabolic adaptations needed to oxidize NADH/NADPH often result in the generation of reactive oxygen species (ROS). In fact, elevated ROS level and altered sensitivity to thiol oxidative stress have been reported in mycobacteria at acidic pH relative to neutral conditions (16).
Previous work from our laboratory has shown that out of a total of 13 mycobacterial sigma factors, PhoP interacts with two extracytoplasmic sigma factors, SigE and SigH (17), implicated in numerous stress responses. At the transcriptional level, SigH activity is regulated by autoregulation of the sigH promoter. However, at the posttranslational level, SigH activity is regulated via its interaction with the cognate anti-sigma factor RshA (18). Under oxidizing conditions, RshA-SigH interaction is disrupted, enabling SigH to bind to the core RNA polymerase (RNAP) and activate transcription. It has been shown that phosphorylation of RshA by an essential serine-threonine protein kinase, PknB, inhibits SigH-RshA interaction, leading to decreased inhibition of SigH function (19). However, when cells encounter SDS-induced surface stress, heat stress, or prolonged nutrient starvation or upon infection of macrophages, the mRNA level of sigE is elevated (20–22). Thus, the SigE regulon comprises genes involved in mycobacterial stress induced by SDS and genes coding for different transcription regulators and enzymes involved in fatty acid degradation, as well as expression of heat shock proteins (23). Similar to the SigH-RshA interaction, SigE activity is controlled by the RseA-SigE interaction (24), and PknB-dependent phosphorylation and subsequent degradation of RseA regulate SigE activity (25). Together, these results elegantly elaborate functioning of SigE and SigH in mycobacterial stress response.
We have previously shown that PhoP-SigE interaction plays a critical role in mycobacterial pH homeostasis. However, the physiological significance of the PhoP-SigH interaction remained unknown. Having shown that both PhoP and SigH function as regulators of mycobacterial redox response (10, 26, 27), we sought to investigate whether PhoP-SigH interaction controls integration of pH stress and reductive stress to govern metabolic plasticity of mycobacteria (10, 13, 15). We found that a mutant M. tuberculosis H37Rv strain lacking phoP is significantly more susceptible to redox stress than the wild-type (WT) bacilli. Using a mycothiol-specific reporter sensitive to a redox environment, we provide biochemical correlation of the above results suggesting that PhoP impacts mycothiol redox stress under both low-pH conditions and under redox stress. Consistent with these results, we found a significant correlation of phoP-dependent and redox-specific expression of mycobacterial thioredoxin genes. On probing the functioning of regulators, we suggest a model that provides new biological insights into the metabolic events necessary to maintain mycobacterial thiol redox homeostasis. Although PhoP controls pH homeostasis via its interaction with SigE (17), here we demonstrate that direct recruitment of SigH alone, but not PhoP-SigH interaction, regulates expression of mycobacterial thioredoxin genes. Together, these results uncover the underlying mechanism of an adaptive program that couples mycobacterial thiol redox homeostasis with low-pH conditions.
RESULTS
Mycobacterial response to oxidative stress is controlled by PhoP.
To carry out a systematic investigation on the role of PhoP in redox stress and in mounting stress response, we first investigated the impact of PhoP deletion on susceptibility of M. tuberculosis to redox stress. To this end, WT H37Rv, the phoP::Kanr phoP deletion mutant strain, and the phoP::Kanr mutant complemented strain were grown in the presence of increasing concentrations of diamide, a thiol-specific oxidant. Mycobacterial metabolic activity was monitored by microplate-based assays using alamarBlue, an oxidation-reduction indicator (see Fig. S1A in the supplemental material). In this assay, change of a nonfluorescent blue to a fluorescent pink is directly linked to bacterial metabolic activity. Although Fig. S1A suggested a MIC value of WT H37Rv as ~10 mM, whereas the MIC of the mutant was ~5 mM, indicating an approximately 2-fold difference in diamide sensitivity between the two strains, our assessment of difference in sensitivity, however, is based on the Fig. S1A-derived quantification of 5 mM diamide data (shown in Fig. S1B). In this piece of data, we considered the fluorescence intensity of WT H37Rv (incubated in the presence of 5 mM diamide) as 100%, and the mutant, under identical conditions, displayed approximately ~10% of the intensity of WT H37Rv. Based on these values, we suggested an ~10-fold difference in diamide sensitivity between the two strains. For the phoP::Kanr strain, a significantly lower metabolic activity (<10%) relative to WT bacilli in the presence of diamide suggests a role of PhoP as a regulator of the mycobacterial redox stress response (Fig. S1B). Importantly, the metabolic activity of the mutant in the presence of diamide could be restored by stable expression of a copy of the phoP gene (as described in Materials and Methods) (Fig. S1A). We also evaluated survival of the WT and phoP::Kanr strains when subjected to 5 mM diamide for 48 h; survival was determined by enumerating CFU values (Fig. 1A). In the presence of diamide, the phoP::Kanr strain displayed a significantly stronger growth inhibition (4.5 ± 2-fold) relative to WT H37Rv. More importantly, stable PhoP expression could significantly restore diamide-dependent growth inhibition of the phoP::Kanr strain, suggesting that PhoP plays a major role in detoxifying redox stress. We also observed a significantly enhanced sensitivity of the phoP::Kanr strain to 50 μM cumene hydrogen peroxide (CHP) relative to WT H37Rv, indicating that the sensitivity of the mutant was not to any specific oxidant per se (Fig. 1B). Here, again, complementation of the phoP::Kanr mutant completely rescued sensitivity of the mutant bacilli to CHP. Together, these results conclusively demonstrate a specific role of PhoP in detoxifying thiol-oxidizing stress.
FIG 1.

phoP plays a major role in mycobacterial redox stress response. (A and B) The WT H37Rv and phoP::Kanr strains were grown in the presence of either 5 mM diamide for 48 h or 50 μM cumene hydroperoxide (CHP) for 24 h, and CFU values were enumerated. For each strain, the percentage of bacterial survival was determined relative to growth of the corresponding strain in the presence of carryover DMSO. The growth experiments were performed in biological duplicates, each with two technical repeats. (C and D) To examine whether phoP is linked to mycobacterial redox potential, we compared the intramycobacterial mycothiol redox states of the WT and phoP::Kanr strains grown either (C) in the presence of 5 mM diamide or (D) under low-pH conditions. In this experiment, we used plasmid Mrx1-roGPF2, where mycoredoxin is fused to redox-sensitive GFP, enabling real-time measurement of the mycothiol redox state by analysis of fluorescence emission at 510 nm after excitation of the samples at 405 and 488 nm (see Materials and Methods). The results show average values from biological triplicates, each with two technical repeats (*, P ≤ 0.05; **, P ≤ 0.01).
We next grew the WT H37Rv and phoP::Kanr strains in the presence of 5 mM diamide to investigate redox-dependent change in mycothiol redox state (Fig. 1C). Most eukaryotes and a large number of prokaryotes utilize glutathione to maintain their redox balance (28, 29). However, the glutathione system is lacking in M. tuberculosis (30), and mycothiol (MSH) is considered the substitute for glutathione. The nonprotein thiol MSH, which is produced in millimolar concentrations in cells (31), is capable of reducing oxidized cysteine residues of proteins by mycoredoxin (31–34). In keeping with this, mshB and mshD mutants of M. tuberculosis are more susceptible to cumene hydroperoxide (CHP) and H2O2, respectively (35, 36), than WT bacilli. Importantly, development of a green fluorescence-based sensor (with roGFP2 as the fluorescent probe) in which mycoredoxin is fused to redox-sensitive green fluorescent protein (roGFP) has enabled real-time measurement of the mycothiol redox state in mycobacteria (37). Singh and coworkers have utilized this probe to demonstrate that acidic pH inside phagosomes induces reductive stress in M. tuberculosis cells residing within macrophages (13). This probe was chosen because of its largest dynamic range, pH insensitivity, additional brightness, and resistance to photoswitching (37, 38). Also, it is noteworthy that by determining the ratio of fluorescence intensities as a measure of the redox state of the cell or compartment where it is expressed, the errors due to variation of roGFP2 under different growth phases are significantly reduced. Using this probe, we observed that mycothiol redox state of WT H37Rv remains significantly higher (or more oxidizing) than that of the phoP::Kanr strain even when grown under normal conditions. This suggests that PhoP contributes to mycobacterial thiol redox homeostasis even under the unstressed condition (Fig. 1C). When subjected to oxidative stress by growth in 5 mM diamide, the difference in the redox states was further enhanced, with WT H37Rv showing a significant increase in mycothiol redox state relative to the unstressed condition (1.7 ± 0.02-fold) (Fig. 1C). This was in contrast to the phoP::Kanr mutant, which showed a smaller change (1.3 ± 0.03-fold) in the mycothiol redox state in cells grown under oxidative stress relative to normal conditions. In this experiment, we used plasmid Mrx1-roGPF2, in which mycoredoxin is fused to roGFP, allowing real-time measurement of the mycothiol redox state by analysis of fluorescence emission at 510 nm after excitation of the samples at 405 and 488 nm. This difference is highly significant since even a minor change in ratios of fluorescence intensities reflects a significant change in the intracellular redox state of mycobacteria (13, 39–41). Thus, because the phoP::Kanr strain displayed a significantly lower intramycobacterial mycothiol redox state than WT H37Rv under normal conditions as well as under redox stress, we conclude that PhoP also plays a role in mycobacterial thiol redox homeostasis.
We also measured the intramycobacterial mycothiol redox state of the WT and phoP::Kanr strains grown under low-pH conditions (Fig. 1D), where cells have been reported to experience a reductive stress. For the WT bacilli, we observed a significantly (1.3 ± 0.1-fold) lower mycothiol redox state under low-pH conditions than the bacilli grown under normal conditions (pH 7.0), as was expected (13, 16). In contrast, the phoP::Kanr strain under identical conditions showed a 1.5 ± 0.04-fold-lower mycothiol redox state than the mutant grown under normal pH (pH 7.0). These results demonstrate the importance of PhoP on maintaining mycobacterial redox homeostasis not only under normal conditions, but also under both oxidative stress conditions as well as low pH or reductive stress conditions. Together, these findings underline the critical role played by PhoP in redox modulation in mycobacteria.
Deletion of sigH impacts mycobacterial growth under redox stress.
Previous studies have established that SigH plays a role in mycobacterial oxidative stress regulation (27, 42). However, it was important for us to examine a possible role of other mycobacterial sigma factors because sigma factors are central to much of the success of M. tuberculosis at adapting to various host environments through complex transcriptional programs. To carry out this comprehensive analysis, we performed a detailed study using global gene expression analysis of cells exposed to 5 mM diamide. Our results identified 25 significantly upregulated (>3.5-fold; P < 0.01) genes (Fig. 2A; Table S1 [Excel spreadsheet]). Most of these belong to either heat stress response (hsp, Rv2466c, and Rv3054c) or oxidative stress response (trxB1, trxB2, sigE, and sigH). When we compared global expression profiles of sigma factors for bacterial cells grown with or without 5 mM diamide (Fig. 2B), the two sigma factors SigE and SigH showed an approximately ~30- to 50-fold redox-inducible activation of expression. Under identical conditions, we could establish that no other sigma factor-encoding genes displayed such a significant difference in their expression (26, 42).
FIG 2.

Redox stress-inducible mycobacterial genes. (A) RNA-seq-derived heat map of WT H37Rv treated with 5 mM diamide versus WT H37Rv exposed to carryover DMSO. The data show a list of significantly induced genes (≥3.5-fold; P < 0.01), most of which belong to the SigH regulon. (B) To investigate redox-active sigma factors, expression of 13 mycobacterial sigma factor-encoding genes was compared by RT-qPCR using WT H37Rv grown in absence or presence of 5 mM diamide (compare empty and filled columns) as described in Materials and Methods. The results show average values from biological duplicates, each with two technical repeats (**, P ≤ 0.01). Nonsignificant difference is not indicated. (C) To compare the metabolic activity of the sigH::Kanr mutant with WT H37Rv, cells were grown in the presence of increasing concentrations of diamide, and alamarBlue assays were carried out as described in Materials and Methods. Bacterial metabolic activity under normal conditions (in the presence of carryover DMSO concentrations) and in the presence of 2.5 mM diamide was assessed by comparing the data from the mutant relative to those from the corresponding WT H37Rv cultures (considered 100%). (D and E) To examine if SigH contributes to mycothiol redox potential, we compared the intramycobacterial mycothiol redox states of the WT H37Rv and sigH::Kanr strains grown (D) under normal conditions versus in the presence of 5 mM diamide and (E) at pH 7.0 versus acidic pH (pH 4.5). Mycothiol redox state was determined as described in the legends to Fig. 1C and D.
We next investigated the impact of deletion of SigH on susceptibility to redox stress. Thus, the WT H37Rv and sigH::Kanr mutant strains were grown in the presence of increasing concentrations of diamide, and metabolic activity was monitored by using the microplate-based alamarBlue assay (Fig. S2) as described above. To determine the fold difference in susceptibility of the WT and the mutant bacilli to diamide, we quantified and plotted the fluorescence data (Fig. 2C). We observed that while sigH::Kanr and WT bacilli grew comparably well under normal conditions of growth, sigH::Kanr bacilli showed a significant reduction in metabolic activity (>10-fold) compared to the WT bacilli in the presence of 2.5 mM diamide. This difference in growth was solely due to the absence of SigH since stable expression of SigH in the mutant bacteria could rescue the redox-dependent growth defect. As SigH was important for redox stress response, we also measured the impact of loss of SigH on intramycobacterial mycothiol redox level (Fig. 2D). We observed that the sigH deletion mutant showed a significantly lower mycothiol redox state than WT bacilli both under normal conditions and in the presence of 5 mM diamide. When this was examined under conditions of low-pH-induced reductive stress, we observed that even under low pH, the sigH::Kanr strain displayed a lower mycothiol redox state (Fig. 2E). These observations are in agreement with decades of elegant research establishing the critical importance of SigH for mycobacterial survival under oxidative stress (18, 26, 27).
Repression of genes associated with thiol redox homeostasis depends on the phoP locus.
M. tuberculosis PhoP has been implicated in numerous functions, including complex lipid biosynthesis and virulence regulation of mycobacteria (3, 5, 6, 8, 9). More recently, it has been shown that under low-pH conditions, mycobacteria encounter reductive stress, and maintenance of redox homeostasis involves the phoPR regulatory system (10, 15). Considering a higher susceptibility of the phoP::Kanr mutant to 5 mM diamide (relative to the WT bacilli) (Fig. 1), we were interested to see if PhoP also had a role in expression of redox-relevant genes. We carried out global transcriptional profiling by RNA sequencing using WT H37Rv and phoP::Kanr mutant cells grown in the presence of 5 mM diamide (Fig. 3). Importantly, we observed that several redox-active genes that include genes coding for thioredoxins (trxB1 and trxB2), lipid synthesis (papA4), cysteine synthesis (cysM), and several predicted oxidoreductases (Rv2454C and Rv3463) belong to the top 15 genes (>2-fold; P < 0.05) that show the highest level of expression in response to redox stress (Fig. 3A; Table S2 [Excel spreadsheet]). This piece of data strongly suggests that PhoP has a role in regulating genes involved in thiol homeostasis and has a major role in the thiol-associated stress response. What was surprising, however, was that while the phoP::Kanr mutation led to the induction of expression of genes such as those coding for thioredoxins that tackle the oxidative stress response, the phoP::Kanr mutant was actually more sensitive to oxidative stress rather than resistant.
FIG 3.
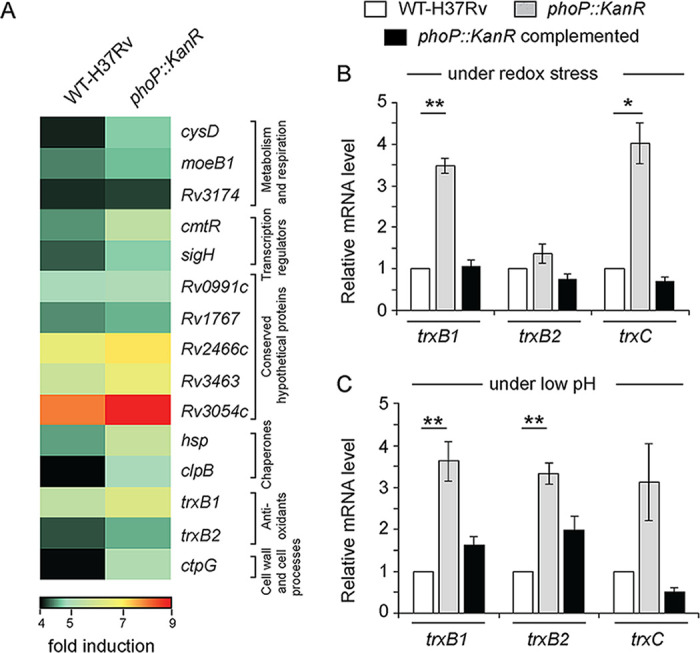
PhoP regulates expression of redox-inducible genes associated with the SigH regulon. (A) RNA-seq-derived heat-map of upregulated (≥2.0-fold; P < 0.05) genes in the WT H37Rv and phoP::Kanr mutant strains incubated in the presence of 5 mM diamide relative to their expression levels under normal conditions of growth. (Only the top 15 genes are shown.) The data show a list of significantly induced genes, most of which belong to the SigH regulon. Notably, thioredoxin (trx) genes representing one of the major mycobacterial antioxidant systems are found to be part of the redox-active PhoP regulon. (B and C) Expression of redox-active thioredoxin genes in the WT, phoP::Kanr, and complemented phoP::Kanr strains, grown (B) in the presence of 5 mM diamide or (C) under acidic pH conditions, were investigated by RT-qPCR measurements, and fold differences in expression levels with standard deviations from replicate experiments were determined from at least three independent RNA preparations (*, P < 0.05; **, P < 0.01).
We thus decided to investigate expression of thioredoxin genes, which belong to the PhoP regulon, more carefully (Fig. 3A). The mycobacterial thioredoxin system, is one of the antioxidant systems dedicated to ensuring growth and survival of the bacilli within the host (30, 43). Although bacterial thioredoxin reductases have been shown as drug targets (44, 45), the mechanism of regulation of thioredoxin gene expression remains largely unknown. In quantitative real-time PCR (RT-qPCR) experiments, we observed that the phoP::Kanr mutant showed a significantly higher expression of thioredoxin genes, particularly trxB1 and trxC, in the presence of 5 mM diamide (Fig. 3B). Although we observed an ≈4-fold change in expression level of trxC in RT-qPCR measurements, the differential expression was insignificant in RNA-seq data (Table S2 [Excel spreadsheet]). Furthermore, stable phoP expression in the complemented mutant could effectively decrease expression of these genes to WT levels. Interestingly, under normal conditions of growth, the thioredoxin genes in the mutant displayed a largely comparable expression to that of the WT bacilli (Fig. S3A). These results suggest that PhoP functions as a stress-specific repressor of trx genes, which control many important cellular processes. Since PhoPR is activated under acidic pH, a condition that causes reductive stress in mycobacteria (10, 13, 15), we also examined the expression profile of thioredoxin genes in the WT H37Rv and phoP::Kanr mutant strains grown under low-pH conditions (Fig. 3C). Here, again, under acidic pH or reductive stress, thioredoxin promoters showed significantly higher expression in phoP::Kanr bacilli than in WT bacilli. Thus, both oxidative and reductive stress conditions were leading to the induction of thioredoxin genes in the phoP::Kanr strain. From these results, we conclude that PhoP is a redox stress-specific repressor of mycobacterial thioredoxin genes.
SigH and SigE were both very strongly induced under conditions of redox stress (Fig. 2A and B). To evaluate their role in the regulation of the redox-active genes, we examined regulation of thioredoxin genes in strains with either the SigH or SigE gene deleted. In the case of SigH, which has previously been shown to regulate redox-active transcriptome of mycobacteria (26, 27, 42), we observed that thioredoxin genes showed a significantly reduced expression in the sigH::Kanr mutant relative to the WT bacilli under redox stress, and stable expression of SigH in the sigH::Kanr mutant could partly restore gene expression (Fig. S3B). Likewise, thioredoxin genes showed a reduced level of expression in the sigH::Kanr strain even under low pH (Fig. S3C), but not under normal conditions of growth (Fig. S3D). These studies, in agreement with previous reports, conclusively demonstrate the critical role of SigH in mycobacterial redox stress.
Because PhoP was also shown to be critical for the regulation of redox regulation, we examined the overlap between the genes that were being regulated by PhoP, as seen under conditions of oxidative stress (diamide treatment), with those regulated by SigH. Figure S3E highlights the overlap of redox-inducible genes belonging to the PhoP and SigH regulons, respectively (Table S3 [Excel spreadsheet]). While the thioredoxin promoters under redox stress showed a significantly lower level in the sigH::Kanr mutant (Fig. S3B), these genes showed a noticeable induction in the phoP::Kanr mutant (Fig. 3B). In agreement with the redox-dependent activation by SigH, under low-pH conditions, we also found a reproducible activation of thioredoxin genes by SigH (Fig. S3C). In the case of SigE, we observed that expression of thioredoxin genes in the WT H37Rv and sigE::Hygr strains grown under normal conditions (Fig. S3F) as well as under stress (Fig. S3G and H) showed no significant difference, thus eliminating any role for SigE in the regulation of the trx genes.
Redox stress-specific SigH recruitment accounts for activation of thioredoxin promoters.
To investigate SigH recruitment within target promoters, we next expressed FLAG-tagged SigH in WT-H37Rv, and performed chromatin immunoprecipitation (ChIP) followed by qPCR measurements (Fig. 4A). Our results demonstrate that under redox stress, SigH is effectively recruited within thioredoxin promoters. However, no significant SigH recruitment was obtained within the identical promoters under normal conditions of growth (compare empty and filled columns in Fig. 4A). From these results, we conclude that redox stress-specific SigH recruitment within thioredoxin promoters maintains mycobacterial thiol redox homeostasis. In contrast, under an identical experimental setup, we were unable to detect PhoP recruitment within the target promoters under either condition of mycobacterial growth (Fig. 4B). However, as a positive control (9), the upstream regulatory region of espA (espAup) showed a considerable PhoP recruitment under normal conditions. Together, these results suggest that induction of redox-active thioredoxin promoter activity is attributable to direct recruitment of SigH within these promoters, and the role of PhoP appears indirect in redox-dependent promoter activation. It should be noted that ChIP data showing no significant recruitment of PhoP or SigH within these promoters under normal conditions are consistent with a lack of regulatory expression of thioredoxin genes by these regulators under normal conditions of growth (Fig. S3A and D, respectively).
FIG 4.

Redox stress-specific recruitment of SigH within thioredoxin promoters. In vivo recruitment of (A) SigH and (B) PhoP within thioredoxin promoters was examined by ChIP-qPCR using WT H37Rv grown under normal conditions and in the presence of 5 mM diamide. FLAG-tagged SigH or PhoP was expressed in WT-H37Rv, and immunoprecipitation was carried out with anti-FLAG antibody to determine fold PCR enrichment due to binding of regulators, as described in Materials and Methods. “trxC ORF” refers to a part of the trxB2 gene that remains adjacent to trxC, and “espAup” refers to the upstream regulatory region (positions −957 to −580 relative to the transcription start site of espA [Rv3616c]) comprising PhoP binding site (9). Each piece of data collected in duplicate qPCR measurements (technical repeats) using biological duplicates was normalized relative to ChIP samples from cells grown under normal conditions (*, P ≤ 0.05; **, P ≤ 0.01). To examine PhoP recruitment within espAup (as a positive control), IP samples from cells grown under redox stress were normalized to 1.
While expression of these genes under redox stress was noticeably activated in the phoP::Kanr strain (Fig. 3B), under identical conditions, these genes showed a significant lack of activation in the sigH::Kanr strain (Fig. S3B). To understand a possible molecular connectivity between PhoP and SigH, we sought to investigate whether either of the two regulators controls expression of the other. Thus, we compared sigH expression in the phoP::Kanr strain and phoP expression in the sigH::Kanr strain (Fig. S4A and C, respectively). In RT-qPCR experiments, under normal conditions SigH expression remains insignificantly higher in the phoP::Kanr strain than in WT H37Rv (Fig. S4A). In contrast, phoP expression is significantly induced in the sigH::Kanr mutant relative to WT H37Rv, both under normal conditions and under redox stress (Fig. S4C; Tables S9 and S10 [Excel spreadsheet]). More importantly, stable SigH expression in the sigH::Kanr strain complemented PhoP expression, suggesting that SigH appears to function as a repressor of PhoP expression. We also compared phoP expression in WT H37Rv under normal conditions of growth and redox stress (Fig. S4E). Our results showed that phoP expression was significantly reduced under redox stress relative to that in mycobacterial cells grown under normal conditions. However, the fold difference in expression remained insignificant. Together, a significant activation of sigH expression coupled with a decrease in (or insignificant alteration) of phoP expression likely accounts for a strongly reduced PhoP-SigH interaction under redox stress.
The C-terminal domain of PhoP interacts with SigH and the N-terminal domain with SigE.
We previously showed that PhoP interacts with both SigE and SigH (17), the redox-active sigma factors. While PhoP-SigE interactions have been implicated in pH homeostasis (17), the physiological consequence of PhoP-SigH interaction remains unknown. The ability of PhoP to repress several redox-active genes and the demonstration that SigH can activate the same subset of genes suggest cross talk between the two proteins, but these findings do not shed significant light on the reasons why the two proteins might also be physically interacting. Towards a better understanding, we wanted to investigate whether PhoP utilizes the same interaction interface with both the regulators. Thus, we expressed different domains of PhoP in M. smegmatis along with SigE and SigH independently, and interactions between the mycobacterial regulators were examined by M-PFC (mycobacterial protein fragment complementation assay) (Fig. 5) as described previously (46). In this assay, the interacting proteins upon expression in M. smegmatis reconstitute functional dihydrofolate reductase (DHFR), and therefore, bacteria coexpressing interacting proteins can now grow on trimethoprim (Trim) plates. Our M-PFC results show that while the C-terminal domain of PhoP (PhoPC), interacts with SigH (Fig. 5A), it is the N-terminal domain of PhoP (PhoPN) that interacts with SigE (Fig. 5B). To investigate the role of phosphorylation of PhoP in PhoP-SigE/SigH interactions, we designed M-PFC experiments using a mutant, PhoPD71N, which is unable to be phosphorylated at Asp71 (47). (Fig. 5C and D). Our results demonstrate that unlike PhoP, under identical conditions, M. smegmatis cells coexpressing PhoPD71N/SigH (Fig. 5C) and PhoPD71N/SigE (Fig. 5D) pairs fail to grow in plates containing Trim, suggesting that phosphorylation of PhoP is necessary for PhoP-SigE/SigH interactions.
FIG 5.
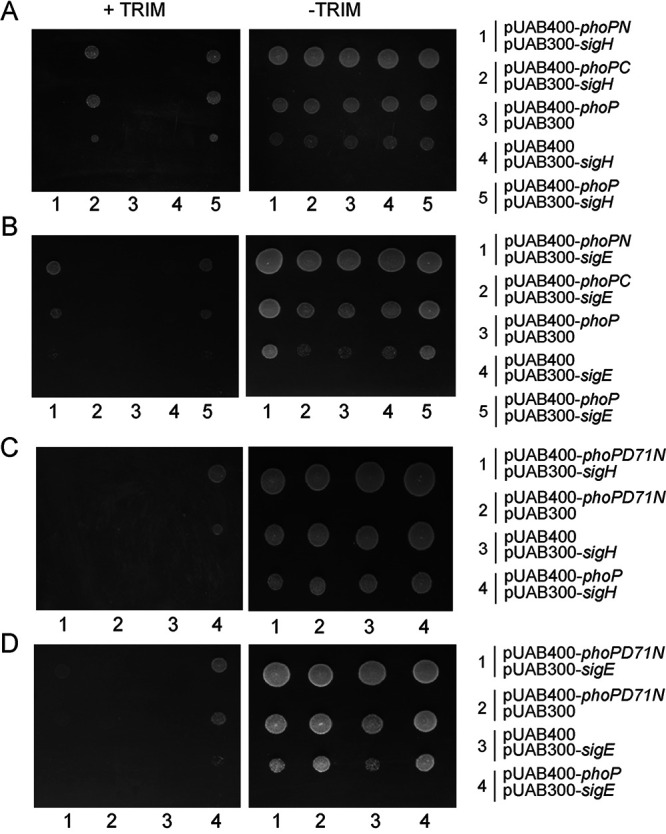
Probing the SigE/SigH-interacting domains of PhoP. (A and B) In M-PFC experiments, M. smegmatis cells coexpressing M. tuberculosis PhoP or its indicated domains and SigH (A) and SigE (B) were grown on 7H10/Hyg/Kan plates in the presence or absence of Trim. Likewise, in panels C and D, M. smegmatis cells coexpressing M. tuberculosis PhoPD71N and SigH (C) or SigE (D) were grown on 7H10/Hyg/Kan plates in the presence or absence of Trim. Coexpression fusion plasmids pUAB400-phoP/pUAB300 and pUAB400/pUAB300-sigH or pUAB400-phoP/pUAB300 and pUAB400/pUAB300-sigE (expressing WT and mutant phoP genes, as indicated on the figure) were used as the respective empty vector controls. As positive controls, coexpression plasmids pUAB400-phoP/pUAB300-sigH or pUAB400-phoP/pUAB300-sigE encoding PhoP/SigH or PhoP/SigE pairs, respectively, showed M. smegmatis growth in the presence of Trim. All of the strains grew well in the absence of Trim.
To further validate the results, we performed in vitro pulldown assays (Fig. S5A and B). In this experiment, we immobilized glutathione S-transferase (GST)-tagged PhoP domains (PhoPN or PhoPC) on glutathione-Sepharose, and incubated it with purified His-tagged SigH (Fig. S5A) or SigE (Fig. S5B) proteins. GST tag alone and glutathione Sepharose beads were used as controls. Importantly, the results from pulldown assays are in agreement with M-PFC data showing that N- and C-terminal domains of PhoP interact with SigE and SigH, respectively. These results suggest that the same molecule of PhoP has the ability to interact with both SigE and SigH simultaneously. Next, we assessed interactions between PhoPD71N and SigE/SigH proteins using in vitro pulldown assays (Fig. S5C and D). Consistent with the M-PFC data, our results demonstrate that PhoPD71N, but not PhoP, fails to coelute with SigH (Fig. S5C) and SigE (Fig. S5D). Although we do not know the extent of phosphorylation of M. tuberculosis PhoP when expressed in Mycobacterium smegmatis, from the above results, we conclude that phosphorylation of PhoP appears to play a major role in PhoP-SigE/SigH interactions. The fact that PhoPD71N fails to interact with SigE and SigH under the conditions where PhoP interacts with both the sigma factors effectively further suggests that the observed phosphorylation-dependent interactions are unlikely to be due to overexpression of the regulator and, therefore, remain physiologically relevant. Notably, previous studies from our laboratory had shown effective DNA binding by PhoPD71N comparable to that of unphosphorylated PhoP (48), despite its inability to function as a transcriptional activator (6).
Contrasting mode of stress-dependent PhoP-sigma factor interactions.
We have shown above that SigH is recruited within thioredoxin promoters in a redox-dependent manner (Fig. 4A). However, under identical conditions, recruitment of PhoP was undetectable (Fig. 4B). Mechanistically, this is in striking contrast to our previous results showing SigE recruitment within low-pH-inducible promoters in a PhoP-dependent manner (17), where we showed recruitment of both PhoP and SigE within acid-inducible promoters during low-pH-dependent mycobacterial adaptation. Therefore, it was of interest to investigate the PhoP-SigH interaction in vivo under stress conditions. In a phoP::Kanr background, we expressed a His-tagged PhoP under the control of the 19-kDa antigen promoter in mycobacterial expression vector p19Kpro (49). These cells were grown under normal and stress conditions (under low pH and in the presence of 5 mM diamide, respectively). The whole-cell lysates with a comparable amount of total protein were incubated with Ni-nitrilotriacetic acid (NTA), and His-tagged PhoP was eluted to examine for the presence of an association with either SigH or SigE (Fig. 6A and B). For cells grown under normal conditions, the eluent revealed the clear presence of SigH, suggesting in vivo interaction between PhoP and SigH (lane 1, top panel, Fig. 6A). In striking contrast, we noted an almost undetectable signal of SigH in the eluent using cell lysates of the same strain, grown in the presence of 5 mM diamide (lane 2, top panel, Fig. 6A). This was despite the fact that SigH was present in mycobacterial cell lysates, grown under normal conditions and in the presence of diamide (lanes 3 and 4, top panel, Fig. 6A). However, SigE was found to be associated with PhoP under both normal conditions as well as in the presence of 5 mM diamide (compare lanes 1 and 2, middle panel, Fig. 6A). These studies indicate that while PhoP interacts with SigE both under normal conditions and under redox stress, in the case of SigH, this interaction occurs only under normal conditions and is significantly lower in cells grown in the presence of diamide.
FIG 6.
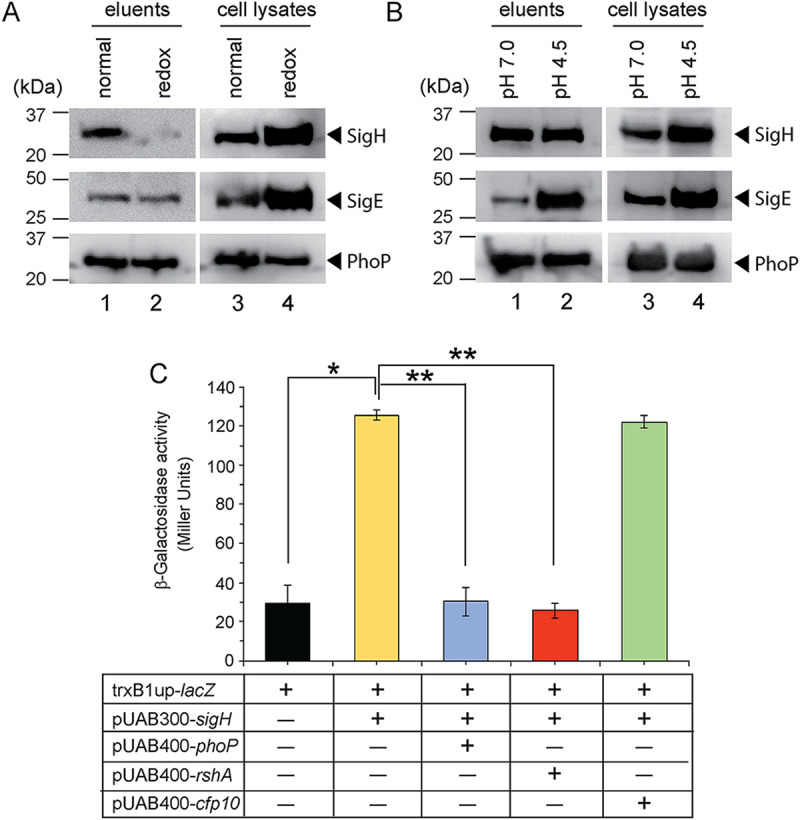
In vivo interactions of PhoP and extracytoplasmic sigma factors. To investigate stress-specific interactions of PhoP and sigma factors (SigE and SigH), the phoP::Kanr mutant, expressing a His-tagged PhoP under the control of 19-kDa mycobacterial antigen promoter in p19Kpro (49), was grown (A) under normal conditions and in the presence of 5 mM diamide or (B) at pH 7.0 and acidic pH (pH 4.5), respectively. The cell lysates with comparable amounts of total protein were incubated with Ni-NTA, and bound proteins were eluted. Panels A and B display immunoblots of eluents (lanes 1 and 2) and the corresponding cell lysates (lanes 3 and 4) detecting SigH (top panel), SigE (middle panel), and PhoP (bottom panel) in Western blots using antibodies directed against the corresponding purified proteins. (C) To examine the effect of PhoP on SigH-dependent activation of the trxB1 promoter, M. smegmatis strains harboring transcriptional fusion of the trxB1 regulatory region upstream of lacZ and the indicated constructs expressing SigH or other regulators were grown under normal conditions, and β-galactosidase activity was measured as described in Materials and Methods. In each case, average promoter activity was determined from two biological repeats, each with two technical repeats (*, P < 0.05; **, P < 0.01). Note that in the reporter assays described above, PhoP and SigH were expressed from pUAB400-phoP and pUAB300-sigH, respectively (as described in the legend to Fig. 5).
Since mycobacteria under low pH encounter reductive stress, we next compared PhoP-SigH interactions in mycobacterial cells grown under pH 7.0 and acidic pH. Interestingly, under normal growth conditions and conditions of low pH or reductive stress, PhoP continued to interact with SigH (compare lane 1 and lane 2, top panel, Fig. 6B). This indicates that PhoP-SigH interactions are disrupted only under oxidative stress and not under normal or even reductive (low-pH) conditions. However, in the case of SigE, we observed a striking stimulation of PhoP-SigE interaction under acidic pH relative to normal conditions of growth (compare lane 1 and lane 2, middle panel, Fig. 6B). In all cases, the levels of PhoP were comparable in the eluents, as well as in the cell lysates, for cells grown under normal conditions, redox stress, and low-pH conditions, respectively. These results provide two novel and key insights into the PhoP-dependent regulation of target promoters: (i) PhoP-independent redox-specific SigH recruitment within thioredoxin promoters and (ii) PhoP-dependent low-pH-inducible SigE recruitment within acid-inducible mycobacterial promoters of the PhoP regulon. Furthermore, the fact that PhoP has different domains for its interaction with SigH and SigE suggests that the physical interactions between PhoP and the two sigma factors would enable very rapid responses to redox stress and allow the titration of different proteins to maintain a delicate balance during the stress response. This also brings in an additional layer of regulation upon redox stress. Thus, the stress-specific recruitment of extracytoplasmic sigma factors (SigE or SigH), controlled by PhoP-SigE/SigH interactions or lack thereof, mitigates stress response, regulates mycobacterial physiology under various conditions of the host environment, and enables intracellular survival of the bacilli.
To investigate the physiological significance of PhoP-SigH interactions under normal conditions of mycobacterial growth and to compare relative efficiencies of PhoP and RshA, we studied SigH-dependent activation of trxB1, a representative promoter (referred to as trxB1up). Note that in the reporter assays described above, PhoP and SigH were expressed from pUAB400-phoP and pUAB300-sigH, respectively (as described in the legend to Fig. 5). As expected, expression of SigH in reporter assays using M. smegmatis showed a significant activation of trxB1 relative to empty vector control (Fig. 6C). However, upon simultaneous expression of PhoP, we observed a striking inhibition of SigH-dependent activation of the promoter, suggesting that PhoP significantly inhibits SigH-dependent activation of trxB1 expression. Notably, RshA, which is known to function as an anti-sigma factor of SigH (18, 50), under identical conditions displayed a strong inhibition of SigH-dependent trxB1 promoter activation. However, as a negative control, under identical conditions, CFP-10 was unable to inhibit SigH-dependent activation of trxB1 expression. These results confirm that (i) PhoP under normal conditions binds to SigH (Fig. 5; Fig. S5) and inhibits SigH-dependent activation of thioredoxin genes, and (ii) PhoP appears to be as effective as RshA at inhibiting SigH-dependent activation of trxB1 promoter activity. The fact that the levels of efficiency of inhibition of SigH-dependent promoter activity by both PhoP and RshA remain largely comparable further suggests that the functional effect of PhoP-mediated inhibition is quite robust and, therefore, unlikely to be of little or no physiological significance. A possible mechanism would be that under normal conditions, perhaps there is interaction between PhoP, SigH, and RshA that effectively maintains the basal level of redox-active gene expression via blocking of SigH-dependent activation. However, under redox stress, dissociation of RhsA destabilizes the PhoP-SigH interaction, leading to release of SigH to activate redox-inducible gene expression. Clearly, more experiments are required to assess the relative affinities of RshA and PhoP for SigH, especially in the presence of other relevant regulators at physiological concentrations.
DISCUSSION
Endogenous oxidative stress represents a significant challenge to survival and growth of microbes adapted to an aerobic lifestyle (51). However, intracellular pathogens like M. tuberculosis also encounter exogenous oxidative stress generated by the host (52). Expectedly, M. tuberculosis is equipped with a number of dedicated antioxidant systems to ensure survival and growth within the host macrophages (53–58). Among these, the thioredoxin system controls many important cellular processes, such as antioxidant pathways, DNA and protein repair enzymes, and activation of redox-sensitive transcription factors (30, 43). Furthermore, depletion of thioredoxin reductase (TrxB2) causes lytic death of mycobacteria and is essential for growth and survival in vitro and in mice (44, 59–61). Since mycobacteria under acidic pH accumulate NADH/NADPH and generate redox stress, as part of an adaptive response, reactive oxygen species (ROS) are generated as a metabolic requirement to oxidize these cofactors. Along this line, M. tuberculosis under acidic pH displays increased ROS synthesis, higher sensitivity to thiol stress, and a lower cytoplasmic redox potential than at normal pH (13, 16). Abramovitch and coworkers showed that a reductive thiol environment was generated upon microbial growth using reduced carbon sources under low pH with a greater reduction in cytoplasm for a phoP mutant than for WT bacilli (10). Further supporting an adaptive shift of redox poise under acidic pH, WT H37Rv displayed a lower mycothiol redox state under acid stress (13). However, the mechanism that couples pH stress with modulation of redox homeostasis remains obscure.
During its life cycle within the human host, M. tuberculosis encounters numerous stress conditions, such as hypoxia in animal models like mice, rabbit, guinea pigs, and nonhuman primates (62–64), and increased levels of glutathione (GSH) inside lung granulomas of guinea pigs (65). Because of inhibition of respiration by NO or hypoxia (52), the NADH/NAD+ ratio goes up within the bacilli and M. tuberculosis encounters reductive stress (66). Previous studies have established correlation of inducible NO synthase expression with infection with M. tuberculosis in surgically resected lungs of human TB patients (63) and in a murine model of TB (64). Likewise, there are reports of elevated expression of HO-1 (which degrades heme molecules to release CO) in (i) lungs of M. tuberculosis-infected mice (relative to uninfected animals) (67) and (ii) plasma of untreated HIV-1 coinfected TB patients that undergoes substantial reduction with anti-TB treatment (68). These results suggest that despite tight regulation of host metabolic pathways, intracellular M. tuberculosis modulates its metabolic plasticity for survival and persistence (69). In this study, we explored the possibility of whether PhoP-SigH interaction contributes to mycobacterial redox homeostasis for the following reasons. First, coupling of pH stress and reductive stress has been shown to involve the phoPR regulatory system (10, 15). Second, SigH has been implicated in regulating expression of thioredoxin genes, a major mycobacterial antioxidant system (42). Third, global expression profiling of mycobacterial sigma factors under redox stress reveals a striking induction of SigH expression among all 13 sigma factors (Fig. 2).
Although the sigE::Hygr mutant displayed significantly lower expression of pH-inducible genes under low pH with a direct involvement of SigE in mycobacterial pH homeostasis (17), here we show that the sigH::Kanr strain is significantly more susceptible to diamide (oxidative stress) than WT H37Rv (Fig. 2C). Consistent with its role as a redox-active sigma factor activating mycobacterial thioredoxin genes (see Fig. S3B and C in the supplemental material), SigH is recruited within the thioredoxin promoters in a redox stress-dependent manner (Fig. 4A), just as for low-pH-driven recruitment of SigE within acid-inducible M. tuberculosis promoters (17). However, the most novel insight is derived from the findings that mycobacterial responses to acidic pH stress and redox stress, which remain somewhat connected (10, 13, 15), are controlled by the same regulator, PhoP, interacting with two specific extracytoplasmic sigma factors, SigE and SigH. Interestingly, PhoP shares the C-terminal domain to interact with SigH and the N-terminal domain to interact with SigE (Fig. 5A and B; Fig. 5SA and B). However, while the acidic condition of bacterial growth significantly promotes PhoP-SigE interactions, the in vivo PhoP-SigH interaction is strikingly reduced under redox stress relative to normal conditions of growth (Fig. 6), thereby facilitating SigH-dependent redox-active transcription of target genes. In a nutshell, our results showing stress-specific activation of low-pH and redox stress-inducible genes, which rely on strikingly elevated PhoP-SigE interactions and significantly reduced PhoP-SigH interactions (sharing two different interaction interfaces of PhoP), respectively, facilitate an integrated view of our results and most likely remain central to the mechanism of the coupled stress response in mycobacteria.
Also, our finding that SigH appears to function as a repressor of PhoP expression (Fig. S4C; Tables S9 and S10 [Excel spreadsheet]) accounts for both SigH-dependent activation and PhoP-dependent repression of thioredoxin genes. Coupled with a lower expression of phoP under redox stress (relative to normal conditions of growth) (Fig. S4E), it is conceivable that under redox stress with a striking overexpression of sigH, a significantly reduced level of PhoP is available to interact with SigH, and as a result, most of the cellular SigH pool will remain available to transcribe thioredoxin genes. However, it is speculative that this is one of the physiologically relevant reasons because the effect is relatively weaker (and also less significant), and there is no direct evidence that links lowering the PhoP concentration to activation of thioredoxin genes.
The above results are now summarized in a schematic model (Fig. 7). According to this model, under normal conditions, the PhoP-SigH interaction might be limiting the availability of SigH, with PhoP functioning like an anti-sigma factor. Therefore, possibly due to lower availability of free SigH, a functional SigH-bound RNAP is not formed, thereby maintaining the basal expression levels of redox-responsive genes. Thus, under normal conditions, there is no detectable SigH recruitment within redox-inducible thioredoxin promoters. In contrast, under redox stress, SigH expression is strongly induced, and PhoP-SigH interaction is significantly interrupted. As a result, SigH-loaded RNAP transcribes thioredoxin genes, and stress-specific SigH recruitment strongly induces expression of these genes to effectively mitigate oxidative stress. On the other hand, during low pH, mycobacteria encounter reductive stress, thereby allowing SigE-loaded RNAP to transcribe PhoP-dependent acid-inducible genes. However, interaction between PhoP and SigH involves only a fraction of a free cellular pool of SigH, while a considerable part of free SigH remains available to mitigate reductive stress. Although we cannot rule out the possibility that downregulation of thioredoxin genes by PhoP and upregulation by SigH are two independent and unrelated phenomena, at present, we consider this an unnecessary complexity and thus favor a more integrated view of the results suggesting that PhoP-SigH interactions, or lack thereof, contribute to the context-dependent regulation of thioredoxin gene expression.
FIG 7.

Schematic model depicting low-pH- and redox-inducible mycobacterial gene expression. While PhoP restricts redox-inducible expression of mycobacterial thioredoxin genes under normal conditions, SigH is an activator of these genes under redox stress. Thus, under redox stress, the PhoP-SigH interaction is of no physiological consequence. Under normal conditions, thioredoxin genes are not influenced by either of the two regulators (PhoP or SigH). We propose that under normal conditions, perhaps there is interaction between PhoP, SigH, and RshA that effectively maintains the basal level of redox-active gene expression via blocking of SigH-dependent activation. However, under redox stress, dissociation of RhsA destabilizes the PhoP-SigH interaction, leading to release of SigH to activate redox-inducible gene expression. However, under acidic pH conditions, due to enhanced PhoP-SigE interaction, both PhoP and SigE are recruited within low-pH-inducible promoters of mycobacteria, and the transcription initiation promotes activation of acid-inducible genes. In summary, the PhoP-SigE interaction under acid stress contributes to pH homeostasis, whereas the reduced PhoP-SigH interaction under redox stress contributes to mycobacterial thiol redox homeostasis.
Because PhoP shares different interaction interfaces with SigE and SigH (Fig. 5; Fig. S5), our model further accommodates functioning of both of the sigma factors under normal conditions of mycobacterial growth. Given the fact that PhoP plays an indirect role in regulating expression of the major antioxidant system, we are unable to explain how a mutant M. tuberculosis H37Rv strain lacking phoP is significantly sensitive to redox stress. We believe that many other PhoPR-regulated redox-active genes contribute to mycobacterial redox stress response and account for the mutant’s susceptibility to redox stress. As a large number of redox-active overlapping genes (regulated by both PhoP and SigH) include essential genes involved in metabolic pathways, respiration, and expression of transcription regulators (Fig. S3E), most likely a strongly reduced PhoP-SigH interaction is physiologically relevant when mycobacteria encounter redox stress. However, under normal conditions, interaction between the two regulators prevents unnecessary activation of the SigH regulon, consisting of ~39 genes (27), which would otherwise be energy demanding, and therefore, a second molecular control mechanism (in addition to RshA-SigH) would be in place to precisely limit SigH activation to only under the most appropriate stress conditions. These results are consistent with a significant activation of mycobacterial heat shock-inducible genes (Rv2466c and Rv3054c) in the phoP::Kanr mutant under normal conditions (26, 70). We consider the possibility that under normal conditions, perhaps there is interaction between PhoP, SigH, and RshA, which effectively maintains a basal level of redox-active gene expression via blocking of SigH-dependent activation. However, under redox stress, dissociation of RhsA destabilizes the PhoP-SigH interaction, leading to release of SigH to activate redox-inducible gene expression.
We argue that a strong induction of SigH (Fig. 2) coupled with a striking decline in the PhoP-SigH in vivo interaction (Fig. 6A) under redox stress would be necessary for the induction of redox proteins for the following reasons. First, a very significant reduction (>10-fold, based on limits of detection in these assays) in PhoP-SigH interaction during redox stress compared to normal conditions of growth strongly suggests that the effect appears to be robust. It should be noted that the fold difference in reduction of PhoP-SigH in vivo interaction (Fig. 6A) is somewhat comparable to a low-pH-induced in vivo activation of PhoP-SigE interaction (Fig. 6B). Second, the other convincing evidence is derived from regulation of gene expression data as determined by RT-qPCR measurements (Fig. 3B and C; Fig. S3B-C). Note that neither PhoP (Fig. S3A) nor SigH (Fig. S3D) shows an impact on expression of thioredoxin genes under normal conditions, when they interact in vivo (Fig. 6). In contrast, both of these regulators significantly impact expression of thioredoxin genes under redox stress (Fig. 3B; Fig. S3B) when they fail to interact in vivo (Fig. 6A). Third, we provide evidence to show that PhoP functions as a specific inhibitor of SigH-dependent activation of thioredoxin gene expression (Fig. 6C), further suggesting the physiological significance of PhoP-SigH interactions under normal conditions.
Thus, the results reported here argue for a novel and unexpected mode of regulation on top of an already complex regulatory mechanism, which distinguishes regulators like PhoP from other members of the family. It would be worth investigating whether such sigma factor-dependent control of transcriptional regulators contributing to mycobacterial physiology remains a general feature involving several other sigma factors and regulators. Given the nature of the complex lifestyle of pathogenic mycobacteria within the continuously changing complex physiology of the host, perhaps these subtle features are related to the need for multiple regulatory functions during establishment and maintenance of infection.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Escherichia coli DH5α was used for routine subcloning, and the E. coli BL21 λ(DE3) strain was utilized for expression of recombinant proteins. M. tuberculosis H37Rv strains and M. smegmatis mc2155, an electroporation-efficient mutant strain of mc26 (71), were grown at 37°C in Middlebrook 7H9 broth (Difco) containing 0.2% glycerol, 0.05% Tween 80, and 10% Middlebrook ADC (albumin-dextrose-catalase) or on 7H10 agar medium (Difco) containing 0.5% glycerol and 10% OADC (oleic acid-albumin-dextrose-catalase) enrichment, unless mentioned otherwise, and transformed by electroporation as described previously (9).
Construction of the sigE::Hygr (sigE deletion mutant) and sigH::Kanr (sigH deletion mutant) strains and complementation of the mutants were described previously (23, 27). Deletion mutants were constructed by disrupting sigE and sigH via insertion of a Hygr cassette into a unique BglII site and a Kanr cassette into a unique KpnI site of sigE and sigH, respectively. Likewise, the phoP deletion mutant was generated by disrupting phoP via insertion of a Kanr cassette into a unique EcoRV site within the gene (3). The disrupted DNA fragments were then cloned into a suicide vector, pSM270 (23, 72). Next, these constructs were transformed into M. tuberculosis H37Rv (3). After 4 weeks of incubation, Kanr/Hygr, and sucrose-resistant colonies were selected. Southern blot analyses were performed using chromosomal DNA of the mycobacterial strains to confirm gene disruption in the corresponding mutants. To complement M. tuberculosis mutants, PCR fragments comprising open reading frames (ORFs) encompassing 2.04-kb sigE, 2.02-kb sigH, and 3.6-kb phoP-phoR were cloned in the multicloning site of integrative plasmids pSM316 (Kanr), pYUB413 (Hygr), and pMV306 (Hygr), respectively.
For acid stress, cells at an optical density at 600 nm (OD600) of 0.4 to 0.6 were pelleted, washed twice with 7H9 medium buffered with 100 mM morpholinepropanesulfonic acid (MOPS) or 1 N HCl at pH 7.0 or pH 4.5, respectively, finally resuspended in medium of the indicated pH, and further grown for the indicated times at 37°C. For redox stress, mycobacterial strains were grown to mid-log phase (OD600 of ~0.4 to 0.6) and exposed to 5 mM diamide for the indicated times at 37°C. To evaluate susceptibility of M. tuberculosis to the indicated stress conditions, mycobacterial strains were grown to mid-log phase (OD600 of 0.4) in 7H9-ADS (albumin-dextrose-sodium chloride), and cells were collected by centrifugation and washed twice with phosphate-buffered saline (PBS) containing 0.05% Tween 80 (PBST). For diamide stress, cells were inoculated into fresh 7H9-ADS medium containing 5 mM diamide (Sigma) or carryover dimethyl sulfoxide (DMSO) at an OD600 of ~0.05 and further grown for 48 h at 37°C. For CHP treatment, freshly inoculated cells at an OD600 of ~0.05 were grown to an OD600 of ~0.2, CHP (Sigma) was added to a final concentration of 50 μM, and cells were further grown for 24 h at 37°C. Finally, CFU were enumerated following 21 days of growth on 7H11-OADS (oleic acid-albumin-dextrose-sodium chloride) agar plates. The following antibiotics were used as appropriate: ampicillin (Amp), 100 μg/mL; hygromycin (Hyg), 150 μg/mL for E. coli or 50 μg/mL for mycobacterial strains; kanamycin (Kan), 50 μg/mL for E. coli or 20 μg/mL for mycobacterial strains; streptomycin (Strep), 100 μg/mL for E. coli or 20 μg/mL for mycobacterial strains.
Cloning, expression, and purification.
The oligonucleotide primer sequences used in cloning and the recombinant plasmids used in this study are listed in Table S4 in the supplemental material. M. tuberculosis constructs overexpressing PhoP, SigE, and SigH were described earlier (17, 73). To express GST-tagged phoPN (carrying 423 bp of the N-terminal phoP ORF) and phoPC (carrying 321 bp of the C-terminal phoP ORF), PCR products were amplified and cloned in pGEX-4T1 as described for pGEX-phoP (73). To express FLAG-tagged phoP and sigH in WT H37Rv, the corresponding ORFs were cloned and expressed from the promoter of the 19-kDa antigen of mycobacterial expression vector p19Kpro (49). M. tuberculosis RshA was cloned in pUAB400 between MunI and HindIII sites and expressed in M. smegmatis. Transcriptional fusion to lacZ was constructed by cloning the PCR-amplified upstream regulatory fragment of the M. tuberculosis trxB1 (trxB1up) into the ScaI site of pSM128 (a promoterless integrative lacZ reporter vector [74] with a streptomycin resistance gene) using primer pairs FPtrxB1up/RPtrxB1up, respectively (Table S5). This promoter fragment comprised upstream of the coding region, including the first 141 coding bases of the trxB1 gene, respectively. In all cases, nucleotide sequences of the constructs were verified by automated DNA sequencing.
Proteins.
M. tuberculosis PhoP and its domain constructs, SigE (Rv1221) and SigH (Rv3223c), were purified as recombinant proteins either by being immobilized on glutathione-Sepharose (GE Healthcare) or by immobilized metal-affinity chromatography (Ni-NTA) (Qiagen) as described previously (17, 48, 73). Purity of the proteins was verified by SDS-PAGE, and protein contents were determined by the bicinchoninic acid (BCA) reagent with bovine serum albumin (BSA) as the standard and expressed in equivalents of protein monomers. For immunoblotting, cell lysates or purified proteins were resolved by 12% SDS-PAGE, electroblotted onto polyvinyl difluoride (PVDF) membranes (Millipore), and visualized by Western blotting using anti-His (Invitrogen), or anti-GST (GE Healthcare) antibodies. Anti-mouse (Thermo Scientific) and anti-goat (Invitrogen) antibodies conjugated to horseradish peroxidase were used as secondary antibodies, and blots were developed with Luminata Forte chemiluminescence reagent (Millipore). The SigE- and SigH-specific antibodies were raised and purified by Alpha Omega Sciences (India). To produce SigE- and SigH-specific polyclonal antibodies in rabbits, 0.3 mg of purified recombinant protein emulsified in Freund’s complete adjuvant was administered as the primary dose subcutaneously, followed by two booster immunizations in Freund's incomplete adjuvant after 14 and 30 days into a New Zealand White rabbit (~3.8-kg body weight). The titers were determined by the endpoint method, blood samples were collected, and serum was separated and stored at –20°C. Total IgG was purified from the serum by affinity chromatography using protein A resin.
Microplate-based alamarBlue assay.
The indicated mycobacterial strains were cultured in 7H9 medium (Difco) with 10% ADS (albumin-dextrose-NaCl) as described previously (75). In this assay, change of a nonfluorescent blue to a fluorescent pink is directly linked to bacterial growth. Increasing concentrations of diamide were added to the wells of a 96-well plate containing 0.05 mL 7H9 medium and 0.05 mL of mycobacterial cells (OD600 of 0.02), freshly diluted from the log-phase culture at an OD600 of ~0.4. The assay included appropriate negative and positive controls, and the plate was incubated at 37°C for 7 days. Finally, 0.02 mL of 0.02% resazurin (sodium salt; MP Biomedicals), prepared in sterile 7H9 medium, was added to each of the wells of the plate, and the color change was monitored after incubation for 16 h at 37°C. Excitation was at 530 nm, and fluorescence emission was recorded at 590 nm. Efficiency of inhibition was evaluated with respect to control wells lacking diamide. To assess bacterial sensitivity to increasing diamide concentrations, the following controls were included in alamarBlue assays. These included a “no-bacterium” control comprising growth medium only, an “only-bacterium” control without any diamide, a “DMSO control” with bacterial cells grown in the presence of carryover DMSO, and a “rifampin control,” ensuring mycobacterial growth inhibition to confirm validity of the assay.
In vitro measurements of mycothiol redox level.
To determine the intramycobacterial mycothiol redox level, the indicated strains expressing Mrx1-roGFP2 were grown in 7H9 medium (supplemented with 10% ADS) to log phase, and cultures were divided into two parts. The first part was grown in two halves: under normal conditions of pH 7.0 and acidic conditions of pH 4.5 for 24 h. Similarly, the second part was grown in two halves in the presence of 5 mM diamide or carryover DMSO for 20 min. Following growth, in all cases cells were treated with 10 mM N-ethylmaleimide for 5 min and fixed with 4% paraformaldehyde for 15 min at room temperature. Next, cultures were washed three times with 1× phosphate-buffered saline (PBS), and the ratios of emission intensities (510 nm) were measured after excitation of the samples at 405 and 488 nm in an Infinite MPlex microplate reader (Tecan).
RNA isolation.
RNA was extracted from exponentially growing mycobacterial cells in Middlebrook 7H9 medium. Briefly, 25 mL of bacterial culture was grown to mid-log phase (OD600 of ~0.4 to 0.6) and combined with 40 mL of 5 M guanidinium thiocyanate solution containing 1% β-mercaptoethanol and 0.5% Tween 80. Cells were pelleted by centrifugation and lysed by being resuspended in 1 mL TRIzol (Ambion) in the presence of Lysing matrix B (100-μm silica beads; MP Bio) using a FastPrep-24 bead beater (MP Bio) at a speed setting of 6.0 for 30 s. The procedure was repeated for 2 to 3 cycles with incubation on ice between pulses. Next, cell lysates were centrifuged at 13,000 rpm for 10 min; supernatant was collected and processed for RNA isolation using a Direct-zol RNA isolation kit (Zymo Research) as per the manufacturer’s recommendation. Following extraction, RNA was treated with DNase I (Promega) to degrade contaminating DNA, and integrity was assessed using a NanoDrop (ND-1000) spectrophotometer. RNA samples were further checked for intactness of 23S and 16S rRNA using formaldehyde-agarose gel electrophoresis and a Qubit fluorimeter (Invitrogen).
RNA sequencing and data analysis.
Total RNA was isolated using Direct-zol RNA MiniPrep kit (Zymo Research Corporation), the purity of isolated RNA was determined using the NanoDrop (Thermo Scientific) and quantified using Qubit (Invitrogen), and RNA integrity was checked using the Agilent 2200 Tape Station system (Agilent Technologies). Library construction, RNA sequencing, and data analysis were carried out by AgriGenome Labs Private, Ltd. (Cochin), India. Briefly, rRNA was removed using si-tools Pan-prokaryote ribopool probes, and further library preparation was carried out using the TruSeq stranded RNA library prep kit. Prepared libraries were sequenced using the Illumina HiSeq ×10 platform, and 150-bp paired-end reads were generated. Threads that passed quality control were mapped onto the reference genome of M. tuberculosis H37Rv using Hisat2 (v2.0.5). The abundance of transcripts in individual samples was estimated using Cufflinks (v2.2.1) (76). Differential gene expression analysis of treated samples with respect to control was performed using the Cuffdiff (v2.2.1) program, and differentially expressed genes (DEGs) with an adjusted P value of <0.05 and log2 fold change value of ≥2 or ≤−2 were used for further analysis. Common and unique DEGs were visualized by Venn diagram using venny software. Heat maps were generated using log2 fold change values of ≥2 or ≤−2 of treated versus control samples using GENE-E software.
Quantitative real-time PCR.
cDNA synthesis and PCRs were carried out using total RNA extracted from each bacterial culture grown with or without specific stress and the Superscript III platinum-SYBR green one-step quantitative real-time PCR (RT-qPCR) kit (Invitrogen) with appropriate primer pairs (2 μM) using a Realplex PCR detection system (Eppendorf). The oligonucleotide primer sequences used in RT-qPCR experiments are listed in Table S5. Global expression of mycobacterial sigma factors was studied by RT-qPCR as described previously (17). The endogenously expressed M. tuberculosis gapdh gene (Rv1436) was used as an internal control, and the fold difference in gene expression was calculated using the threshold cycle (ΔΔCT) method (77). Control reactions with Platinum Taq DNA polymerase (Invitrogen) confirmed the absence of genomic DNA in all our RNA preparations. To validate RNA-seq data, RT-qPCR was utilized by determining specific mRNA levels relative to a control set displaying no differential expression. To determine enrichment due to PhoP and/or SigH binding targets in the immunoprecipitated (IP) DNA samples, 1 μL of IP or mock IP (no-antibody control) DNA was used with SYBR green (Invitrogen) along with target promoter-specific primers.
ChIP-qPCR.
Chromatin immunoprecipitation (ChIP) experiments were carried out with some modifications to the protocol described previously (17). PhoP and SigH were expressed in WT H37Rv as FLAG-tagged protein, and in vivo recruitment of the regulators was examined by ChIP-qPCR using anti-FLAG antibody (Thermo) to detect promoter regions of interest. Mycobacterial cells were grown to the mid exponential phase (OD600 of 0.5), and formaldehyde was added to a final concentration of 1%. After incubation for 20 min, glycine was added to a final concentration of 0.5 M to quench the reaction and incubated for further 10 min. Cross-linked cells were harvested by centrifugation and washed twice with ice-cold immunoprecipitation (IP) buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 5% glycerol). Cell pellets were resuspended in 1.5 mL IP buffer containing protease inhibitor cocktail tablet (Roche). Cells were lysed, and insoluble matter was removed by centrifugation at 13,000 rpm for 10 min at 4°C. Then, supernatant containing DNA was sheared to an average size of ~500 bp using a Bioruptor (Sonics, VibraCell) with settings of 20 s on and 40 s off for 3 to 5 min and split into two aliquots. Ten microliters of sample was analyzed on agarose gel to check the size of the DNA fragments. Each 0.7-mL aliquot was incubated with 20 μL protein A/G UltraLink resin (Pierce) on a rotary shaker for 20 min at 4°C to eliminate complexes bound to the resin nonspecifically. The supernatant was removed and incubated with either no antibody (mock-IP) or specific anti-FLAG antibody and 50 μL protein A/G UltraLink resin, preincubated with 50 μg bovine serum albumin in 500 μL of IP buffer on a rotary shaker at 4°C overnight. The beads were then washed to remove nonspecific interactions with 500 μL of IP buffer twice, once each with low-salt buffer (0.1% SDS, 0.1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl [pH 8.0] and 150 mM NaCl), high-salt buffer (0.1% SDS, 0.1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl [pH 8.0], and 500 mM NaCl), and lithium chloride (LiCl) wash buffer (250 mM LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 20 mM Tris-HCl [pH 8.0]), and twice with Tris-EDTA (TE) buffer (10 mM Tris-HCl [pH 8.0] and 1 mM EDTA [pH 8.0]). Finally, the elution was carried out using buffer containing 10 mM Tris-HCl (pH 8.0), 1 mM EDTA (pH 8.0), 0.1% SDS, and 0.1 M NaHCO3. The eluate was reverse cross-linked at 65°C overnight; proteinase K (Thermo) treatment was performed for 1 h at 58°C. Next, the DNA was extracted with phenol-chloroform treatment followed by precipitation using ethanol.
Gene-specific qPCR was carried out using appropriate dilutions of IP DNA in a reaction buffer containing SYBR green mix (Invitrogen), 2 μM PAGE-purified oligonucleotide primers (Table S5) and 1 U of Platinum Taq DNA polymerase (Invitrogen). Typically, 40 cycles of amplification were carried out using a real-time PCR detection system (Eppendorf/Life technologies). To determine fold enrichment, PCR signal from anti-FLAG IP was compared with PCR signal from the “no-antibody” control (mock sample) to assess region-specific recruitment of the regulators, and specificity was examined by ChIP-qPCR of the same IP samples using gapdh/16S rRNA gene-specific primers. In all cases, melting curve analysis confirmed amplification of a single product.
Mycobaterial protein fragment complementation assays.
M-PFC experiments were performed as described previously (46, 75). To express domains of M. tuberculosis PhoP in M. smegmatis, domain-spanning regions were cloned in the integrative vector pUAB400 (Kanr) between MfeI and HindIII sites to generate pUAB400-phoPN and pUAB400-phoPC, respectively, as described previously (78). Likewise, SigE- and SigH-expressing episomal constructs pUAB300-sigE and pUAB300-sigH, respectively, were described earlier (17). The oligonucleotide primer sequences and plasmids used to generate constructs used in M-PFC experiments are listed in Table S4, and each construct was verified by DNA sequencing. Cotransformed cells were selected on 7H10/Kan/Hyg plates in both the absence and presence of 10 to 12.5 μg/mL trimethoprim (Trim). As positive controls, PhoP/SigE- or PhoP/SigH-expressing pairs were used.
Coimmunoprecipitation.
The phoP::Kanr mutant expressing His-tagged PhoP was grown to an OD600 of ~0.4 to 0.6 and exposed to specific stress conditions as described below. For acid stress, cells at an OD600 of 0.4 to 0.6 were pelleted, washed twice with 7H9 buffered with 100 mM MOPS or 1 N HCl to pH 7.0 or pH 4.5, respectively, resuspended in medium of the indicated pH, and grown further for 10 to 12 h at 37°C. For redox stress, mycobacterial strains were grown to mid-log phase (OD600 of ≈0.4 to 0.6), directly exposed to 5 mM diamide or carryover DMSO (normal conditions of pH 6.4) without changing any media, and grown for 3 h at 37°C. Cultures were fixed using 1% formaldehyde for 15 min, followed by the addition of 250 mM glycine to neutralize cross-linking. Cell pellets were resuspended in lysis buffer (20 mM Tris [pH 8.0], 150 mM NaCl, 1 mM MgCl2, 1 mM Na3PO4, 1 mM NaF, 0.50% NP-40, 1× protease inhibitor, 1 mM PMSF) and lysed in the presence of silica beads (100-μm diameter, Lysing matrix B; MP Biomedicals) in a FastPrep at a speed setting of 5.5 10 to 12 times for 30 s. The supernatant was collected by centrifugation and filtered through a low-binding Millex 0.22-μm-pore membrane filter (Millipore). Whole-cell lysates containing 2 to 3 mg of total protein were incubated with Ni-nitrilotriacetic acid (Ni-NTA) beads at 4°C for 2 to 3 h, followed by 4 to 5 washes with wash buffer (20 mM Tris [pH 8.0], 150 mM NaCl, 1 mM MgCl2, 1 mM Na3PO4, 1 mM NaF, 0.10% NP-40, 1 mM PMSF), and finally proteins were eluted using 250 mM imidazole. Samples were analyzed by SDS-PAGE and visualized by Western blotting using protein-specific antibodies. In all cases, aliquots of cell lysates and eluents were used to determine the protein concentration using BCA protein estimation kit (Pierce).
Regulation of mycobacterial promoter activity in M. smegmatis.
To express M. tuberculosis PhoP, RshA, or CFP-10 in M. smegmatis, electro-competent cells were transformed with pUAB400-phoP, pUAB400-rshA, or pUAB400-cfp10 (Table S4), expressing wild-type PhoP, RshA, or CFP-10, respectively, as described earlier (17). To express M. tuberculosis SigH, M. smegmatis was transformed with pUAB300-sigH (Table S4). M. smegmatis harboring the trxB1up-lacZ fusion was cotransformed with SigH and the PhoP/RshA/CFP-10 expression construct and grown to an OD600 of ≈1.0 as described earlier (6). Cultures were inoculated in fresh medium (1:100) and allowed to grow until they reached an OD600 of ≈0.8 to 1.0. Cells were centrifuged, and cell pellets were washed with phosphate-buffered saline (PBS). Next, cells were resuspended in 0.5 mL phosphate buffer (pH 7.2) containing 75 mM NaCl, cell suspensions were sonicated, and the β-galactosidase activity of cell extracts was determined by using the chromogenic substrate 2-nitrophenyl-β-d-galactopyranoside (ONPG) at a final concentration of 1 mg/mL. The reaction mixtures were incubated at 37°C for 10 min, the reactions were terminated by adding 0.2 M Na2CO3, β-galactosidase activity was measured by recording absorbance at 420 nm, and enzyme activity was calculated in Miller units.
Statistical analysis.
Data are presented as arithmetic means of the results obtained from multiple replicate experiments ± standard deviations. Statistical significance was determined by Student’s paired t test using Microsoft Excel or Graph Pad Prism. Statistical significance was accepted at P values of <0.05.
Supporting material.
This article contains supporting information in the supplemental material. The data include five supporting figures, two supporting tables (Table S4 and Table S5), and one supporting Excel spreadsheet, which consists of data for the heat map and Venn diagram showing genes that are differentially regulated (>2-fold; P < 0.05) either in WT H37Rv or in the indicated mutants under normal conditions of growth or specific stress conditions (Tables S1 to S3 and Tables S6 to S10). Comparisons involve genes annotated in the H37Rv genome, and the data provided represent average values from two biological repeats.
Data availability.
All RNA sequencing data have been deposited in the GEO database under accession no. GSE171775. All other relevant data are part of the article and the supplemental material.
ACKNOWLEDGMENTS
We thank G. Marcela Rodriguez and Issar Smith (The Public Health Research Institute, UMDNJ) for the phoP::Kanr, complemented phoP::Kanr, sigE::Hygr, and complemented sigE::Hygr strains, Riccardo Manganelli (University of Padova) for the sigH::Kanr and complemented sigH::Kanr strains, Amit Singh (Indian Institute of Science) for the Mrx1-roGFP2 plasmid, Adrie Steyn (University of Alabama) for pUAB300/pUAB400 plasmids, Prabhat Ranjan Singh for help with ChIP experiments, and Vijjamarri Anil Kumar for preliminary results. We are grateful to Anand Bachhawat and Sanjeev Khosla for critically reviewing the manuscript. Library construction, RNA sequencing, and data analysis were carried out by AgriGenome Labs Private, Ltd. (Cochin), India.
This work was supported by intramural funding from CSIR-IMTECH (OLP-0170), CSIR (MLP-0049), and a research grant (to D.S.) from SERB (EMR/2016/004904), Department of Science and Technology (DST). H.G., P.P., and H.K. were supported by CSIR predoctoral fellowships. The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Dibyendu Sarkar, Email: dibyendu@imtech.res.in.
Patricia A. Champion, University of Notre Dame
REFERENCES
- 1.Rohde KH, Abramovitch RB, Russell DG. 2007. Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe 2:352–364. 10.1016/j.chom.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Perez E, Samper S, Bordas Y, Guilhot C, Gicquel B, Martin C. 2001. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol Microbiol 41:179–187. 10.1046/j.1365-2958.2001.02500.x. [DOI] [PubMed] [Google Scholar]
- 3.Walters SB, Dubnau E, Kolesnikova I, Laval F, Daffe M, Smith I. 2006. The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol Microbiol 60:312–330. 10.1111/j.1365-2958.2006.05102.x. [DOI] [PubMed] [Google Scholar]
- 4.Martin C, Williams A, Hernandez-Pando R, Cardona PJ, Gormley E, Bordat Y, Soto CY, Clark SO, Hatch GJ, Aguilar D, Ausina V, Gicquel B. 2006. The live Mycobacterium tuberculosis phoP mutant strain is more attenuated than BCG and confers protective immunity against tuberculosis in mice and guinea pigs. Vaccine 24:3408–3419. 10.1016/j.vaccine.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalo Asensio J, Maia C, Ferrer NL, Barilone N, Laval F, Soto CY, Winter N, Daffe M, Gicquel B, Martin C, Jackson M. 2006. The virulence-associated two-component PhoP-PhoR system controls the biosynthesis of polyketide-derived lipids in Mycobacterium tuberculosis. J Biol Chem 281:1313–1316. 10.1074/jbc.C500388200. [DOI] [PubMed] [Google Scholar]
- 6.Goyal R, Das AK, Singh R, Singh PK, Korpole S, Sarkar D. 2011. Phosphorylation of PhoP protein plays direct regulatory role in lipid biosynthesis of Mycobacterium tuberculosis. J Biol Chem 286:45197–45208. 10.1074/jbc.M111.307447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson BK, Colvin CJ, Needle DB, Mba Medie F, Champion PA, Abramovitch RB. 2015. The carbonic anhydrase inhibitor ethoxzolamide inhibits the Mycobacterium tuberculosis PhoPR regulon and Esx-1 secretion and attenuates virulence. Antimicrob Agents Chemother 59:4436–4445. 10.1128/AAC.00719-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frigui W, Bottai D, Majlessi L, Monot M, Josselin E, Brodin P, Garnier T, Gicquel B, Martin C, Leclerc C, Cole ST, Brosch R. 2008. Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS Pathog 4:e33. 10.1371/journal.ppat.0040033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anil Kumar V, Goyal R, Bansal R, Singh N, Sevalkar RR, Kumar A, Sarkar D. 2016. EspR-dependent ESAT-6 protein secretion of Mycobacterium tuberculosis requires the presence of virulence regulator PhoP. J Biol Chem 291:19018–19030. 10.1074/jbc.M116.746289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker JJ, Johnson BK, Abramovitch RB. 2014. Slow growth of Mycobacterium tuberculosis at acidic pH is regulated by phoPR and host-associated carbon sources. Mol Microbiol 94:56–69. 10.1111/mmi.12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng L, Chen S, Hu Y. 2018. PhoPR positively regulates whiB3 expression in response to low pH in pathogenic mycobacteria. J Bacteriol 200:e00766-17. 10.1128/JB.00766-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh A, Crossman DK, Mai D, Guidry L, Voskuil MI, Renfrow MB, Steyn AJ. 2009. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog 5:e1000545. 10.1371/journal.ppat.1000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mehta M, Rajmani RS, Singh A. 2016. Mycobacterium tuberculosis WhiB3 responds to vacuolar pH-induced changes in mycothiol redox potential to modulate phagosomal maturation and virulence. J Biol Chem 291:2888–2903. 10.1074/jbc.M115.684597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farhana A, Guidry L, Srivastava A, Singh A, Hondalus MK, Steyn AJ. 2010. Reductive stress in microbes: implications for understanding Mycobacterium tuberculosis disease and persistence. Adv Microb Physiol 57:43–117. 10.1016/B978-0-12-381045-8.00002-3. [DOI] [PubMed] [Google Scholar]
- 15.Baker JJ, Dechow SJ, Abramovitch RB. 2019. Acid fasting: modulation of Mycobacterium tuberculosis metabolism at acidic pH. Trends Microbiol 27:942–953. 10.1016/j.tim.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coulson GB, Johnson BK, Zheng H, Colvin CJ, Fillinger RJ, Haiderer ER, Hammer ND, Abramovitch RB. 2017. Targeting Mycobacterium tuberculosis sensitivity to thiol stress at acidic pH kills the bacterium and potentiates antibiotics. Cell Chem Biol 24:993–1004.e4. 10.1016/j.chembiol.2017.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bansal R, Anil Kumar V, Sevalkar RR, Singh PR, Sarkar D. 2017. Mycobacterium tuberculosis virulence-regulator PhoP interacts with alternative sigma factor SigE during acid-stress response. Mol Microbiol 104:400–411. 10.1111/mmi.13635. [DOI] [PubMed] [Google Scholar]
- 18.Song T, Dove SL, Lee KH, Husson RN. 2003. RshA, an anti-sigma factor that regulates the activity of the mycobacterial stress response sigma factor SigH. Mol Microbiol 50:949–959. 10.1046/j.1365-2958.2003.03739.x. [DOI] [PubMed] [Google Scholar]
- 19.Park ST, Kang CM, Husson RN. 2008. Regulation of the SigH stress response regulon by an essential protein kinase in Mycobacterium tuberculosis. Proc Natl Acad Sci USA 105:13105–13110. 10.1073/pnas.0801143105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manganelli R, Dubnau E, Tyagi S, Kramer FR, Smith I. 1999. Differential expression of 10 sigma factor genes in Mycobacterium tuberculosis. Mol Microbiol 31:715–724. 10.1046/j.1365-2958.1999.01212.x. [DOI] [PubMed] [Google Scholar]
- 21.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. 2002. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol 43:717–731. 10.1046/j.1365-2958.2002.02779.x. [DOI] [PubMed] [Google Scholar]
- 22.Graham JE, Clark-Curtiss JE. 1999. Identification of Mycobacterium tuberculosis RNAs synthesized in response to phagocytosis by human macrophages by selective capture of transcribed sequences (SCOTS). Proc Natl Acad Sci USA 96:11554–11559. 10.1073/pnas.96.20.11554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manganelli R, Voskuil MI, Schoolnik GK, Smith I. 2001. The Mycobacterium tuberculosis ECF sigma factor sigmaE: role in global gene expression and survival in macrophages. Mol Microbiol 41:423–437. 10.1046/j.1365-2958.2001.02525.x. [DOI] [PubMed] [Google Scholar]
- 24.Dona V, Rodrigue S, Dainese E, Palu G, Gaudreau L, Manganelli R, Provvedi R. 2008. Evidence of complex transcriptional, translational, and posttranslational regulation of the extracytoplasmic function sigma factor sigma(E) in Mycobacterium tuberculosis. J Bacteriol 190:5963–5971. 10.1128/JB.00622-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barik S, Sureka K, Mukherjee P, Basu J, Kundu M. 2010. RseA, the SigE specific anti-sigma factor of Mycobacterium tuberculosis, is inactivated by phosphorylation-dependent ClpC1P2 proteolysis. Mol Microbiol 75:592–606. 10.1111/j.1365-2958.2009.07008.x. [DOI] [PubMed] [Google Scholar]
- 26.Raman S, Song T, Puyang X, Bardarov S, Jacobs WR, Jr, Husson RN. 2001. The alternative sigma factor SigH regulates major components of oxidative and heat stress responses in Mycobacterium tuberculosis. J Bacteriol 183:6119–6125. 10.1128/JB.183.20.6119-6125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manganelli R, Voskuil MI, Schoolnik GK, Dubnau E, Gomez M, Smith I. 2002. Role of the extracytoplasmic-function sigma factor sigma(H) in Mycobacterium tuberculosis global gene expression. Mol Microbiol 45:365–374. 10.1046/j.1365-2958.2002.03005.x. [DOI] [PubMed] [Google Scholar]
- 28.Toone EJ. 2011. Advances in enzymology and related areas of molecular biology. Preface. Adv Enzymol Relat Areas Mol Biol 78:ix–xi. [PubMed] [Google Scholar]
- 29.Penninckx MJ, Elskens MT. 1993. Metabolism and functions of glutathione in micro-organisms. Adv Microb Physiol 34:239–301. 10.1016/S0065-2911(08)60031-4. [DOI] [PubMed] [Google Scholar]
- 30.Lu J, Holmgren A. 2014. The thioredoxin antioxidant system. Free Radic Biol Med 66:75–87. 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 31.Newton GL, Unson MD, Anderberg SJ, Aguilera JA, Oh NN, delCardayre SB, Av-Gay Y, Fahey RC. 1999. Characterization of Mycobacterium smegmatis mutants defective in 1-d-myo-inosityl-2-amino-2-deoxy-alpha-d-glucopyranoside and mycothiol biosynthesis. Biochem Biophys Res Commun 255:239–244. 10.1006/bbrc.1999.0156. [DOI] [PubMed] [Google Scholar]
- 32.Van Laer K, Buts L, Foloppe N, Vertommen D, Van Belle K, Wahni K, Roos G, Nilsson L, Mateos LM, Rawat M, van Nuland NA, Messens J. 2012. Mycoredoxin-1 is one of the missing links in the oxidative stress defence mechanism of mycobacteria. Mol Microbiol 86:787–804. 10.1111/mmi.12030. [DOI] [PubMed] [Google Scholar]
- 33.Rawat M, Johnson C, Cadiz V, Av-Gay Y. 2007. Comparative analysis of mutants in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Biochem Biophys Res Commun 363:71–76. 10.1016/j.bbrc.2007.08.142. [DOI] [PubMed] [Google Scholar]
- 34.Rawat M, Newton GL, Ko M, Martinez GJ, Fahey RC, Av-Gay Y. 2002. Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob Agents Chemother 46:3348–3355. 10.1128/AAC.46.11.3348-3355.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buchmeier NA, Newton GL, Fahey RC. 2006. A mycothiol synthase mutant of Mycobacterium tuberculosis has an altered thiol-disulfide content and limited tolerance to stress. J Bacteriol 188:6245–6252. 10.1128/JB.00393-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchmeier NA, Newton GL, Koledin T, Fahey RC. 2003. Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol Microbiol 47:1723–1732. 10.1046/j.1365-2958.2003.03416.x. [DOI] [PubMed] [Google Scholar]
- 37.Bhaskar A, Chawla M, Mehta M, Parikh P, Chandra P, Bhave D, Kumar D, Carroll KS, Singh A. 2014. Reengineering redox sensitive GFP to measure mycothiol redox potential of Mycobacterium tuberculosis during infection. PLoS Pathog 10:e1003902. 10.1371/journal.ppat.1003902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanson GT, Aggeler R, Oglesbee D, Cannon M, Capaldi RA, Tsien RY, Remington SJ. 2004. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem 279:13044–13053. 10.1074/jbc.M312846200. [DOI] [PubMed] [Google Scholar]
- 39.Mehta M, Singh A. 2019. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis and survival in response to reactive oxygen and nitrogen species. Free Radic Biol Med 131:50–58. 10.1016/j.freeradbiomed.2018.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tyagi P, Dharmaraja AT, Bhaskar A, Chakrapani H, Singh A. 2015. Mycobacterium tuberculosis has diminished capacity to counteract redox stress induced by elevated levels of endogenous superoxide. Free Radic Biol Med 84:344–354. 10.1016/j.freeradbiomed.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan MZ, Bhaskar A, Upadhyay S, Kumari P, Rajmani RS, Jain P, Singh A, Kumar D, Bhavesh NS, Nandicoori VK. 2017. Protein kinase G confers survival advantage to Mycobacterium tuberculosis during latency-like conditions. J Biol Chem 292:16093–16108. 10.1074/jbc.M117.797563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharp JD, Singh AK, Park ST, Lyubetskaya A, Peterson MW, Gomes AL, Potluri LP, Raman S, Galagan JE, Husson RN. 2016. Comprehensive definition of the SigH regulon of Mycobacterium tuberculosis reveals transcriptional control of diverse stress responses. PLoS One 11:e0152145. 10.1371/journal.pone.0152145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carmel-Harel O, Storz G. 2000. Roles of the glutathione- and thioredoxin-dependent reduction systems in the Escherichia coli and Saccharomyces cerevisiae responses to oxidative stress. Annu Rev Microbiol 54:439–461. 10.1146/annurev.micro.54.1.439. [DOI] [PubMed] [Google Scholar]
- 44.Harbut MB, Vilcheze C, Luo X, Hensler ME, Guo H, Yang B, Chatterjee AK, Nizet V, Jacobs WR, Jr, Schultz PG, Wang F. 2015. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc Natl Acad Sci USA 112:4453–4458. 10.1073/pnas.1504022112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu J, Vlamis-Gardikas A, Kandasamy K, Zhao R, Gustafsson TN, Engstrand L, Hoffner S, Engman L, Holmgren A. 2013. Inhibition of bacterial thioredoxin reductase: an antibiotic mechanism targeting bacteria lacking glutathione. FASEB J 27:1394–1403. 10.1096/fj.12-223305. [DOI] [PubMed] [Google Scholar]
- 46.Singh A, Mai D, Kumar A, Steyn AJ. 2006. Dissecting virulence pathways of Mycobacterium tuberculosis through protein-protein association. Proc Natl Acad Sci USA 103:11346–11351. 10.1073/pnas.0602817103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gupta S, Sinha A, Sarkar D. 2006. Transcriptional autoregulation by Mycobacterium tuberculosis PhoP involves recognition of novel direct repeat sequences in the regulatory region of the promoter. FEBS Lett 580:5328–5338. 10.1016/j.febslet.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 48.Pathak A, Goyal R, Sinha A, Sarkar D. 2010. Domain structure of virulence-associated response regulator PhoP of Mycobacterium tuberculosis: role of the linker region in regulator-promoter interaction(s). J Biol Chem 285:34309–34318. 10.1074/jbc.M110.135822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Smet KA, Kempsell KE, Gallagher A, Duncan K, Young DB. 1999. Alteration of a single amino acid residue reverses fosfomycin resistance of recombinant MurA from Mycobacterium tuberculosis. Microbiology 145:3177–3184. 10.1099/00221287-145-11-3177. [DOI] [PubMed] [Google Scholar]
- 50.Kumar S, Badireddy S, Pal K, Sharma S, Arora C, Garg SK, Alam MS, Agrawal P, Anand GS, Swaminathan K. 2012. Interaction of Mycobacterium tuberculosis RshA and SigH is mediated by salt bridges. PLoS One 7:e43676. 10.1371/journal.pone.0043676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Imlay JA. 2013. The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11:443–454. 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar A, Farhana A, Guidry L, Saini V, Hondalus M, Steyn AJ. 2011. Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev Mol Med 13:e39. 10.1017/S1462399411002079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ng VH, Cox JS, Sousa AO, MacMicking JD, McKinney JD. 2004. Role of KatG catalase-peroxidase in mycobacterial pathogenesis: countering the phagocyte oxidative burst. Mol Microbiol 52:1291–1302. 10.1111/j.1365-2958.2004.04078.x. [DOI] [PubMed] [Google Scholar]
- 54.Bryk R, Lima CD, Erdjument-Bromage H, Tempst P, Nathan C. 2002. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science 295:1073–1077. 10.1126/science.1067798. [DOI] [PubMed] [Google Scholar]
- 55.Piddington DL, Fang FC, Laessig T, Cooper AM, Orme IM, Buchmeier NA. 2001. Cu,Zn superoxide dismutase of Mycobacterium tuberculosis contributes to survival in activated macrophages that are generating an oxidative burst. Infect Immun 69:4980–4987. 10.1128/IAI.69.8.4980-4987.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saini V, Cumming BM, Guidry L, Lamprecht DA, Adamson JH, Reddy VP, Chinta KC, Mazorodze JH, Glasgow JN, Richard-Greenblatt M, Gomez-Velasco A, Bach H, Av-Gay Y, Eoh H, Rhee K, Steyn AJC. 2016. Ergothioneine maintains redox and bioenergetic homeostasis essential for drug susceptibility and virulence of Mycobacterium tuberculosis. Cell Rep 14:572–585. 10.1016/j.celrep.2015.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jaeger T, Budde H, Flohe L, Menge U, Singh M, Trujillo M, Radi R. 2004. Multiple thioredoxin-mediated routes to detoxify hydroperoxides in Mycobacterium tuberculosis. Arch Biochem Biophys 423:182–191. 10.1016/j.abb.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 58.Nambi S, Long JE, Mishra BB, Baker R, Murphy KC, Olive AJ, Nguyen HP, Shaffer SA, Sassetti CM. 2015. The oxidative stress network of Mycobacterium tuberculosis reveals coordination between radical detoxification systems. Cell Host Microbe 17:829–837. 10.1016/j.chom.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang YJ, Ioerger TR, Huttenhower C, Long JE, Sassetti CM, Sacchettini JC, Rubin EJ. 2012. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog 8:e1002946. 10.1371/journal.ppat.1002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin K, O'Brien KM, Trujillo C, Wang R, Wallach JB, Schnappinger D, Ehrt S. 2016. Mycobacterium tuberculosis thioredoxin reductase is essential for thiol redox homeostasis but plays a minor role in antioxidant defense. PLoS Pathog 12:e1005675. 10.1371/journal.ppat.1005675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kuo HP, Wang CH, Huang KS, Lin HC, Yu CT, Liu CY, Lu LC. 2000. Nitric oxide modulates interleukin-1beta and tumor necrosis factor-alpha synthesis by alveolar macrophages in pulmonary tuberculosis. Am J Respir Crit Care Med 161:192–199. 10.1164/ajrccm.161.1.9902113. [DOI] [PubMed] [Google Scholar]
- 63.Choi HS, Rai PR, Chu HW, Cool C, Chan ED. 2002. Analysis of nitric oxide synthase and nitrotyrosine expression in human pulmonary tuberculosis. Am J Respir Crit Care Med 166:178–186. 10.1164/rccm.2201023. [DOI] [PubMed] [Google Scholar]
- 64.Voskuil MI, Schnappinger D, Visconti KC, Harrell MI, Dolganov GM, Sherman DR, Schoolnik GK. 2003. Inhibition of respiration by nitric oxide induces a Mycobacterium tuberculosis dormancy program. J Exp Med 198:705–713. 10.1084/jem.20030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Somashekar BS, Amin AG, Rithner CD, Troudt J, Basaraba R, Izzo A, Crick DC, Chatterjee D. 2011. Metabolic profiling of lung granuloma in Mycobacterium tuberculosis infected guinea pigs: ex vivo 1H magic angle spinning NMR studies. J Proteome Res 10:4186–4195. 10.1021/pr2003352. [DOI] [PubMed] [Google Scholar]
- 66.Boshoff HI, Xu X, Tahlan K, Dowd CS, Pethe K, Camacho LR, Park TH, Yun CS, Schnappinger D, Ehrt S, Williams KJ, Barry CE, III.. 2008. Biosynthesis and recycling of nicotinamide cofactors in mycobacterium tuberculosis. An essential role for NAD in nonreplicating bacilli. J Biol Chem 283:19329–19341. 10.1074/jbc.M800694200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kumar A, Deshane JS, Crossman DK, Bolisetty S, Yan BS, Kramnik I, Agarwal A, Steyn AJ. 2008. Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. J Biol Chem 283:18032–18039. 10.1074/jbc.M802274200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rockwood N, Costa DL, Amaral EP, Du Bruyn E, Kubler A, Gil-Santana L, Fukutani KF, Scanga CA, Flynn JL, Jackson SH, Wilkinson KA, Bishai WR, Sher A, Wilkinson RJ, Andrade BB. 2017. Mycobacterium tuberculosis induction of heme oxygenase-1 expression is dependent on oxidative stress and reflects treatment outcomes. Front Immunol 8:542. 10.3389/fimmu.2017.00542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trivedi A, Singh N, Bhat SA, Gupta P, Kumar A. 2012. Redox biology of tuberculosis pathogenesis. Adv Microb Physiol 60:263–324. 10.1016/B978-0-12-398264-3.00004-8. [DOI] [PubMed] [Google Scholar]
- 70.Sevalkar RR, Arora D, Singh PR, Singh R, Nandicoori VK, Karthikeyan S, Sarkar D. 2019. Functioning of mycobacterial heat shock repressors requires the master virulence regulator PhoP. J Bacteriol 201:e00013-19. 10.1128/JB.00013-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR, Jr.. 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol 4:1911–1919. 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- 72.Rodriguez GM, Voskuil MI, Gold B, Schoolnik GK, Smith I. 2002. ideR, an essential gene in Mycobacterium tuberculosis: role of IdeR in iron-dependent gene expression, iron metabolism, and oxidative stress response. Infect Immun 70:3371–3381. 10.1128/IAI.70.7.3371-3381.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gupta S, Pathak A, Sinha A, Sarkar D. 2009. Mycobacterium tuberculosis PhoP recognizes two adjacent direct-repeat sequences to form head-to-head dimers. J Bacteriol 191:7466–7476. 10.1128/JB.00669-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dussurget O, Timm J, Gomez M, Gold B, Yu S, Sabol SZ, Holmes RK, Jacobs WR, Jr, Smith I. 1999. Transcriptional control of the iron-responsive fxbA gene by the mycobacterial regulator IdeR. J Bacteriol 181:3402–3408. 10.1128/JB.181.11.3402-3408.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Singh R, Anil Kumar V, Das AK, Bansal R, Sarkar D. 2014. A transcriptional co-repressor regulatory circuit controlling the heat-shock response of Mycobacterium tuberculosis. Mol Microbiol 94:450–465. 10.1111/mmi.12778. [DOI] [PubMed] [Google Scholar]
- 76.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7:562–578. 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3:1101–1108. 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 78.Singh PR, Vijjamarri AK, Sarkar D. 2020. Metabolic switching of Mycobacterium tuberculosis during hypoxia is controlled by the virulence regulator PhoP. J Bacteriol 202:e00705-19. 10.1128/JB.00705-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1 to S3 and Tables S6 to S10. Download jb.00110-22-s0001.xlsx, XLSX file, 3.6 MB (3.7MB, xlsx)
Tables S4 and S5; Fig. S1 to S5. Download jb.00110-22-s0002.pdf, PDF file, 0.7 MB (767.5KB, pdf)
Data Availability Statement
All RNA sequencing data have been deposited in the GEO database under accession no. GSE171775. All other relevant data are part of the article and the supplemental material.