

ABSTRACT
As a result of a high-throughput compound screening campaign using Mycobacterium tuberculosis-infected macrophages, a new drug candidate for the treatment of tuberculosis has been identified. GSK2556286 inhibits growth within human macrophages (50% inhibitory concentration [IC50] = 0.07 μM), is active against extracellular bacteria in cholesterol-containing culture medium, and exhibits no cross-resistance with known antitubercular drugs. In addition, it has shown efficacy in different mouse models of tuberculosis (TB) and has an adequate safety profile in two preclinical species. These features indicate a compound with a novel mode of action, although still not fully defined, that is effective against both multidrug-resistant (MDR) or extensively drug-resistant (XDR) and drug-sensitive (DS) M. tuberculosis with the potential to shorten the duration of treatment in novel combination drug regimens. (This study has been registered at ClinicalTrials.gov under identifier NCT04472897).
KEYWORDS: GSK2556286, mouse, Mycobacterium tuberculosis, pharmacology, relapse, tuberculosis
INTRODUCTION
According to World Health Organization (WHO) estimates for 2019, 10 million people were newly diagnosed with tuberculosis (TB), and 1.4 million died (1), making TB the single greatest cause of death globally by a single infectious agent prior to the coronavirus disease 2019 (COVID-19) pandemic. Multidrug-resistant TB (MDR-TB) threatens TB control in many countries, with approximately 363,000 new cases globally in 2019 (1). Furthermore, the incidence of extensively drug-resistant TB (XDR-TB), defined as MDR-TB plus resistance to at least one second-line injectable drug (e.g., amikacin, kanamycin, or capreomycin) and a fluoroquinolone, was over 12,000 in 2019. Cases of XDR-TB have now been reported in over 100 countries (1). Despite regulatory approvals for bedaquiline (B), delamanid (D), and pretomanid (Pa) in the past decade to treat MDR- or XDR-TB, there remains an unmet need for novel drugs with new mechanisms of action that are effective against drug-susceptible and drug-resistant forms of TB and shorten the duration of treatment required to prevent relapse.
To date, virtually all approved drugs used to treat TB were identified through phenotypic screens against actively replicating Mycobacterium tuberculosis in artificial nutrient-rich media, or they were repurposed from other infectious indications (2). The first-line TB drug pyrazinamide (Z) is the notable exception, having been identified by screening for activity in a murine TB model (3, 4). Few other pathogens rival M. tuberculosis in their ability to adapt to and persist within the infected host. Alternative screening methodologies that better represent the environmental conditions and stresses encountered by M. tuberculosis within the host have gained favor in recent years and may increase the efficiency with which new molecules with novel sterilizing activity are identified to complement existing TB drugs (5).
Over the last decade, we and others hypothesized that the macrophage, as a primary target of infection by M. tuberculosis and a niche in which the pathogen persists in established lesions, might represent an improved surrogate model to facilitate the discovery of novel TB drugs (6, 7). The cytochrome bc1:aa3 complex inhibitor telacebec is the first TB drug to reach clinical trials that was initially identified by a phenotypic high-content screening approach using a macrophage infection model (8, 9). Nonetheless, it is active against M. tuberculosis in standard nutrient-rich media as well as in macrophages. More recently, novel compounds with selective activity within macrophages were identified and shown to have cholesterol-dependent activity against extracellular M. tuberculosis in vitro (10). Previous observations suggest that cholesterol uptake and utilization are essential for pathogen survival in the host and indicate that these pathways are potential targets for novel TB drugs (10, 11). Despite these encouraging results, no molecule identified as having such macrophage-specific, cholesterol-dependent activity in vitro has progressed to clinical proof-of-concept studies. Here, we describe the discovery of GSK2556286, a novel inhibitor of M. tuberculosis extracellularly in the presence of cholesterol and within human macrophages, and provide evidence of favorable in vivo efficacy and safety profiles justifying further development as an attractive companion drug with the potential to shorten the duration of treatment in novel combination regimens for drug-susceptible and drug-resistant TB.
RESULTS
Microbiological profile.
To identify compounds that effectively inhibit the intracellular growth of M. tuberculosis, we screened a library of compounds against bacteria residing within human (THP-1) macrophage-like differentiated monocytes. The exploitation of this screening approach led to the identification of GSK2556286 (Fig. 1), a compound with potent activity (Table 1) against M. tuberculosis inside infected macrophages (50% inhibitory concentration [IC50] = 0.07 μM in THP-l cells) and the unusual phenotype of requiring the presence of cholesterol to demonstrate activity in axenic culture (IC50 = 0.71 to 2.12 μM). The maximal percent inhibition of growth achieved by GSK2556286 in these studies was 86% (range, 62 to 89.4%).
FIG 1.
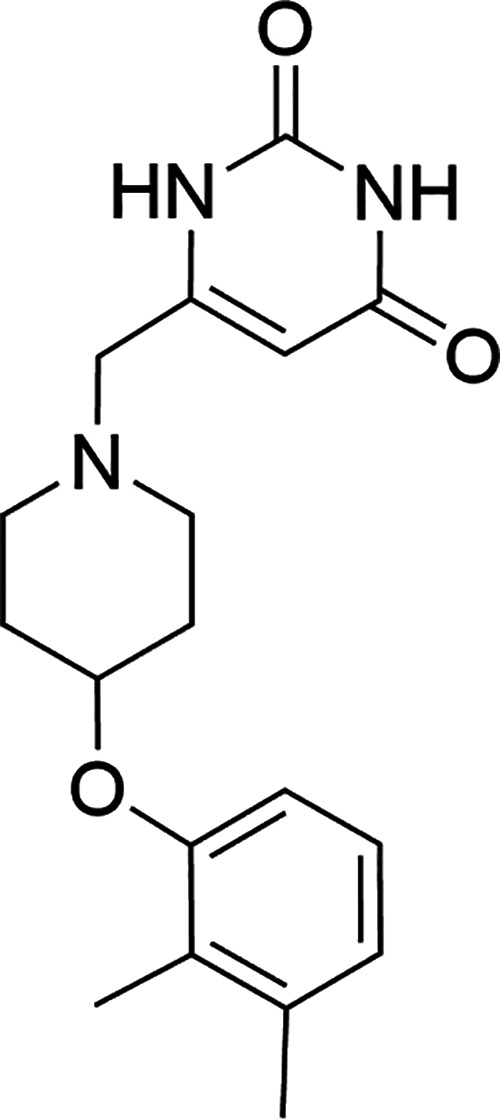
Chemical structure of GSK2556286.
TABLE 1.
In vitro activity of GSK2556286 under various conditions
| Compound | IC50 (μM) against M. tuberculosis strain H37Rv or Erdman |
||||
|---|---|---|---|---|---|
| Intracellular activity of H37Rv in THP-1 cells | Extracellular activity |
||||
| H37Rv |
Erdman |
||||
| Glucose medium | Cholesterol medium | Glucose medium | Cholesterol medium | ||
| GSK2556286 | 0.07 | >125 | 2.12 | >50 | 0.71 |
| Rifampicin | 0.0008 | 0.15 | 0.2 | 0.1 | 0.1 |
| Moxifloxacin | 0.16 | 0.11 | 0.5 | 0.28 | 0.4 |
To investigate the potential mode of action, we isolated spontaneous GSK2556286-resistant mutants by plating M. tuberculosis Erdman cultivated in vitro or in the lungs of infected C3HeB/FeJ mice on solid medium containing GSK2556286 at 96 μM (8× MIC in solid medium including cholesterol). In total, 29 colonies isolated from GSK2556286-containing plates in the in vitro and in vivo experiments were confirmed to have IC90 values in the presence of cholesterol that were 10-fold higher than those of the wild-type parent. Whole-genome sequencing and further analysis revealed that 14 out of 29 mutants had mutations mapping to the Rv1625c gene (cya) (see Table S1 in the supplemental material), which encodes a class IIIa membrane-anchored adenylyl cyclase that is nonessential for growth under routine in vitro conditions and has been implicated in resistance to other compounds with cholesterol-dependent activity (10, 12). The remainder of the isolated resistant mutants remain under analysis to identify mutations responsible for resistance.
None of the isolated resistant mutants with cya mutations had a complete deletion of the cya gene. Therefore, a cya knockout mutant created in the H37Rv strain background was evaluated to confirm the role of cya in GSK2556286 resistance. The IC50 value in cholesterol medium was >50 μM, which is 25-fold higher than the IC50 value of the wild-type strain. These results demonstrated that the cya gene has a role in resistance to GSK2556286 in M. tuberculosis.
Additional drug susceptibility testing of a selection of GSK2556286-resistant mutants (EM08, EM10, EM19, EM33, and EM63) showed susceptibility to a selection of commonly used antitubercular drugs (Table 2; Table S2) under axenic conditions and in infected macrophages (Table 3). Interestingly, moxifloxacin and the aminoglycosides amikacin and kanamycin tended to have lower MICs, and bedaquiline tended to have higher MICs, against the GSK2556286-resistant mutants. However, these observed MIC differences are small and fall within or near the outer limits of the expected normal variation around a single MIC result of 0.5 to 2 times the observed result (13). Therefore, they are of uncertain significance but worthy of further investigation.
TABLE 2.
Antitubercular drug activity against selected GSK2556286-resistant M. tuberculosis strains in liquid medium
| Drug | Activity against M. tuberculosis strainc |
|||||
|---|---|---|---|---|---|---|
| EM05b | EM08a | EM10a | EM19a | EM33b | EM63a | |
| Moxifloxacin | 0.4 | 0.4 | 0.3 | 0.4 | 0.5 | 0.3 |
| Linezolid | 0.7 | 0.7 | 0.8 | 0.6 | 1.6 | 1.2 |
| Pretomanid | 1.0 | 1.0 | 1.0 | 1.4 | 1.7 | 1.4 |
| Rifampicin | 0.9 | 0.9 | 0.9 | 0.9 | 1.4 | 1.4 |
| Bedaquiline | 2.2 | 2.2 | 1.7 | 1.0 | 2.2 | 2.2 |
| Ethionamide | 1.4 | 1.4 | 0.9 | 1.2 | 1.7 | 0.9 |
| Ethambutol | 1.6 | 1.6 | 1.4 | 1.9 | 1.7 | 1.1 |
| Kanamycin | 0.4 | 0.4 | 0.5 | 0.5 | 0.4 | 0.8 |
| Streptomycin | 0.6 | 0.5 | 0.6 | 0.8 | 0.8 | 0.6 |
| Amikacin | 0.3 | 0.3 | 0.4 | 0.3 | 0.3 | 0.4 |
| d-Cycloserine | 1.0 | 0.8 | 0.8 | 1.1 | 0.8 | 0.9 |
| PAS | 1.1 | 1.3 | 1.0 | 0.6 | 0.4 | 0.8 |
cya mutation confirmed.
The presence or absence of a cya mutation remains to be confirmed.
Activity is presented as the ratio of the IC90 of the mutant/IC90 of wild-type EM01.
TABLE 3.
Antitubercular drug activity against selected GSK2556286-resistant M. tuberculosis strains infecting THP-1 cells
| Compound | Activity against M. tuberculosis strainc |
|
|---|---|---|
| EM19a | EM33b | |
| Isoniazid | 0.57 | 0.54 |
| Rifampicin | 0.77 | 0.47 |
| Moxifloxacin | 0.32 | 0.30 |
cya mutation confirmed.
The presence or absence of a cya mutation remains to be confirmed.
Activity is presented as the ratio of the IC50 of the mutant/IC50 of wild-type EM01.
Calculation of the spontaneous frequency of resistance was attempted but, to date, has been challenging due primarily to the inability to completely inhibit the growth of apparently susceptible bacteria on cholesterol-containing solid culture media, which prevents the accurate calculation of the proportion of genotypically resistant CFU among the total number of CFU plated. Further efforts are ongoing to refine the methodology to enable accurate assessments of the spontaneous frequency of resistance to GSK2556286.
GSK2556286 displayed consistent in vitro activity in the presence of cholesterol against a panel of clinical isolates with various drug resistance phenotypes, including isolates from MDR- and XDR-TB cases (Table S3). The MIC of GSK2556286 that inhibited the growth of at least 90% of isolates (MIC90) was determined for 45 clinical isolates (from the National Institutes of Health [NIH]) plus 3 laboratory strains, with different resistance phenotypes, including drug-sensitive (DS), MDR, XDR, or other resistance phenotypes (Table S4), as well as two additional species belonging to the M. tuberculosis complex, Mycobacterium africanum and Mycobacterium bovis, in order to evaluate the activity of GSK2556286 against more genetically diverse species of the complex. The MIC90 was 1.2 μM (MIC range, 0.3 to 1.4 μM), similar to that determined for laboratory strains Erdman and H37Rv (0.71 and 2.12 μM, respectively), in cholesterol-containing medium.
To further investigate the in vitro activity of GSK2556286 against a more diverse panel of clinical isolates, IC50 values were determined against a reference set of 20 well-characterized clinical isolates representing the 7 known global lineages of the human-adapted M. tuberculosis complex (14). The results are shown in Table S5, along with any sequence differences in the putative operon containing cya (i.e., Rv1622c to Rv1625c) compared to the reference Erdman strain. Among the isolates representing the geographically widespread “modern” lineages (lineages 2 to 4), all 6 isolates from lineages 3 and 4 and 2 of 4 isolates from lineage 4 had IC50 values in the same range as those of the H37Rv and Erdman strains. Two other lineage 4 isolates, including one with an Ala157Thr change in cya, had IC50 values that were approximately 10-fold higher. Interestingly, all isolates from lineage 1 and 6 strains (which harbor cya or intergenic mutations) showed reduced sensitivity to GSK2556286, suggesting that phylogenetic differences in cya gene sequences may affect susceptibility to GSK2556286. However, further studies are necessary to establish causation.
Chemical and structural information and physicochemical properties.
GSK2556286A (Fig. 1) is a substituted 4-aryloxypiperidine with a low-to-moderate molecular weight (MW = 329.39).
The white-to-slightly colored solid is crystalline, with a high melting point (200°C) and a chromatographic logD and pharmaceutical formulation index (PFI) of 4.4 and 6.4, respectively. GSK2556286A has excellent stability in the solid and solution states with respect to temperature and light, giving confidence that a solid oral product with a suitable shelf life can be developed. GSK2556286 is practically insoluble in water, fasted-state simulated intestinal fluid (FaSSIF), fed-state simulated intestinal fluid (FeSSIF), and aqueous solution in a pH range of 5 to 9. It is very slightly soluble in simulated gastric fluid (SGF) and aqueous solution in a pH range of 2 to 4. The developability classification system (DCS) class (7) borders on class IIa/b at the predicted dose, suggesting potential issues with solubility and dissolution at high doses (Fig. S1).
GSK2556286 was obtained in an excellent yield at a multigram scale through the nucleophilic substitution of a 6-(chloromethyl)-uracil derivative (GSK1) with a 4-(2,3-dimethylphenoxy)piperidine hydrochloride salt (GSK2), both commercially available, catalyzed by triethylamine (Fig. 2).
FIG 2.
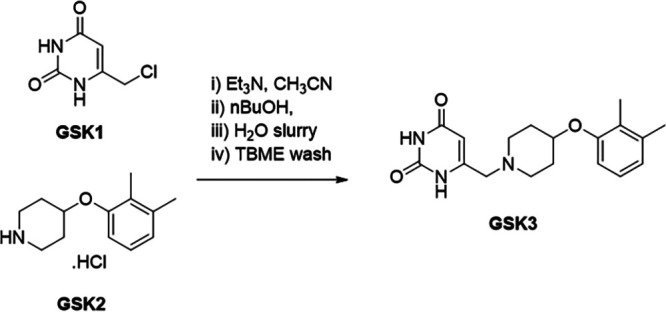
Synthetic route to GSK2556286 (GSK3). TBME, tert-butyl methyl ether.
In vitro absorption, distribution, metabolism, and elimination and pharmacokinetic (DMPK) profiles and potential for drug-drug interactions.
Physicochemical properties and in vitro absorption, distribution, metabolism, and elimination (ADME) and in vivo drug metabolism and pharmacokinetic (DMPK) profiles in preclinical species (mouse, rat, and dog) were evaluated to support the progression of GSK2556286 and dose prediction modeling in humans. GSK2556286 displayed notably higher solubility in SGF than that in other biologically relevant media. The compound exhibited high passive permeability in the hMDR1-MDCK-II cell line, and although it was shown to be an in vitro substrate for P-glycoprotein, based on its permeability and existing in vivo preclinical pharmacokinetic (PK) data, permeability is not expected to limit the oral absorption of GSK2556286 in humans. Low intrinsic clearance (CLint) was determined in both human microsomes and hepatocytes. Low plasma protein binding (PPB) and low-to-moderate blood-to-plasma partitioning ratios (B/P ratios) were observed in human and preclinical species (Table S6).
The pharmacokinetics of GSK2556286 after intravenous (i.v.) and oral administration at various doses were evaluated in rodents and dogs. The compound exhibited low-to-moderate blood clearance, as predicted by CLint in hepatocytes, and a moderate volume of distribution. Absorption was rapid, and oral bioavailability at pharmacologically relevant doses was high in mice and moderate in rats and dogs, in agreement with the expected first-pass effect (Table S7).
Human PK parameters were calculated for GSK2556286 using a physiologically based pharmacokinetic (PBPK) modeling approach (GastroPlus), based on physicochemical, preclinical (in vitro and in vivo), and in vitro human data. The PBPK models accurately predicted the i.v. and oral PK results from preclinical studies in mice, rats, and dogs, and the prediction estimates low human blood clearance (3.3 mL/min/kg of body weight), a moderate volume of distribution (3.5 L/kg), and high oral bioavailability (≥60% for predicted clinical doses). Taking as a reference the minimum area under the concentration-time curve (AUC) and the maximum concentration of the drug in serum (Cmax) at 10 mg/kg associated with a maximum effect as a single drug in the chronic BALB/c mouse infection model (see below), a human dose of between 150 and 300 mg/day was predicted (based on targeting AUC and Cmax, respectively).
To assess the risk of drug-drug interactions, direct inhibition of CYP isoforms was investigated by assessing the enzyme activities (CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) in an incubation mixture of microsomes with NADPH, in the presence and absence of GSK2556286. This preliminary evaluation showed that although it did not substantially inhibit CYP1A2, -2C9, -2C19, -2D6, and -3A4 (IC50 values of >25 μM), there is a moderate risk of CYP3A4-mediated perpetrator drug interactions assuming a CYP3A4 IC50 value of 25 μM and predicted human PK parameters for a 150-mg dose of GSK2556286 (Table S8).
Safety profile.
GSK2556286 was evaluated in single-dose oral toxicity studies in rats, dogs, and cynomolgus monkeys and in repeated-dose oral toxicity studies of up to 4 weeks in duration in Wistar Han rats and cynomolgus monkeys under good laboratory practice (GLP) conditions and performed according to International Council on Harmonisation (ICH) guidelines (15). In addition, GSK2556286 was evaluated in a battery of in vitro and in vivo safety pharmacology (respiratory, cardiovascular, and neurobehavioral tests) and genotoxicity studies.
In the definitive repeated-dose oral toxicity studies in rats and monkeys, adverse systemic effects were limited to rats in the high-dose group (1,000 mg/kg/day). No adverse effects were observed at exposures of up to an AUC0–t (AUC from 0 h to time t) of 65.2 μg · h/mL and a Cmax of 5.89 μg/mL in male rats, an AUC0–t of 129 μg · h/mL and a Cmax of 14.6 μg/mL in female rats, and an AUC0–t of 158 μg · h/mL and a mean Cmax of 9.96 μg/mL in cynomolgus monkeys (gender averaged). GSK2556286 did not produce acute cardiovascular effects in rats or monkeys, respiratory effects in monkeys, or adverse neurobehavioral effects in rats in single- or repeated-dose studies with up to 1,000 mg/kg/day, and the weight of evidence from in vitro and in vivo assessments indicates that GSK2556286 does not present a genotoxic hazard to humans. The preclinical safety profile supports continued progression to a first-time-in-humans (FTIH) trial (ClinicalTrials.gov identifier NCT04472897).
Efficacy in murine models of TB. (i) Dose-ranging activity as monotherapy.
Initial dose-ranging in vivo efficacy studies were conducted at GlaxoSmithKline (GSK) in a previously described acute TB infection model in which C57BL/6 mice were treated for 8 days, beginning 1 day after infection (16). The most dynamic portion of the fitted dose-response curve was between doses of 10 and 200 mg/kg, with the greatest effect (a CFU count 1.8 log10 units lower than that in untreated mice) being observed in mice receiving 200 mg/kg (Fig. 3A). An exposure-response relationship was observed at up to 50 mg/kg (the highest-dose group sampled to determine drug exposure) (Fig. 3B; Fig. S2). Based on these results, GSK2556286 was tested at doses ranging between 10 and 200 mg/kg in two different murine models of chronic TB infection in use at Johns Hopkins University (17, 18). In BALB/c mice, chronic M. tuberculosis infection promotes the development of inflammatory cellular lung lesions in which M. tuberculosis resides virtually entirely intracellularly, especially in macrophages, including foamy macrophages, where its survival depends on the utilization of cholesterol and other lipids as carbon sources. In addition to forming similar cellular lung lesions, C3HeB/FeJ mice form caseating granulomatous lung lesions, in which M. tuberculosis is found extracellularly in the acellular central caseum as well as inside neutrophils and foamy macrophages in the cellular cuff of the granuloma (19, 20).
FIG 3.

Lung CFU counts after 8 days of GSK2556286 treatment in an acute infection model in C57BL/6 mice. Data points represent individual mouse results. Open triangles represent mice from an initial experiment with a higher dose range. Solid circles represent mice from a second experiment with a lower, but overlapping, dose range. Solid lines represent fitted sigmoidal dose-response (A) and whole-blood exposure-response (B) curves, where AUClast is the AUC0–24. Horizontal dotted lines show thresholds for a 2-log10 reduction in CFU compared to untreated mice and the lower limit of CFU detection, as indicated.
GSK2556286 showed a statistically significant bactericidal effect when used as a single agent for 1 month (4 weeks) in chronic infection models in both mouse strains (Fig. 4; Table S9). In BALB/c mice, all GSK2556286 doses tested were superior to no treatment (P < 0.0001), and the magnitude of the bactericidal effect was similar to that of isoniazid. The maximal effect was achieved at a dose of ≤10 mg/kg, which corresponded to a Cmax of 1.38 μg/mL and an AUC from 0 h to infinity (AUC0–∞) of 6.61 μg · h/mL in uninfected BALB/c mice (Table S10), similar to the exposures achieved after a single dose in infected C57BL/6 mice. No increase in activity was observed with higher doses despite a linear increase in exposure to a Cmax of 6.76 μg/mL and an AUC0–∞ of 34.72 μg · h/mL after repeated doses of 50 mg/kg in BALB/c mice (Table S10), indicating that the maximal effect of GSK2556286 monotherapy in the chronic infection model in BALB/c mice is observed at doses near 10 mg/kg or lower. In C3HeB/FeJ mice, all doses above 10 mg/kg were significantly better than no treatment (P < 0.05) before adjustment for multiple comparisons, although only the 40-mg/kg dose was significantly different from no treatment after adjusting for multiple comparisons. Although the CFU counts after 1 month of GSK2556286 treatment in C3HeB/FeJ mice were not significantly lower than the day 0 CFU counts when analyzed as individual groups and adjusting for multiple comparisons, a post hoc analysis that combined all GSK2556286-treated groups to increase statistical power showed that GSK2556286 treatment significantly reduced the CFU counts compared to the day 0 CFU counts (P = 0.0385). Isoniazid was superior to each dose of GSK2556286 in C3HeB/FeJ mice (P < 0.01).
FIG 4.

Lung CFU counts after 4 weeks of GSK2556286 treatment in chronic infection models in BALB/c and C3HeB/FeJ mice. Data points represent individual mouse results. Horizontal bars represent the mean and median values for BALB/c and C3HeB/FeJ mice, respectively. D0, day 0; INH, isoniazid; GSK’286, GSK2556286.
(ii) Contribution to bactericidal activity in combination therapy.
Given the requirement for combination chemotherapy in the treatment of TB and the urgent need for novel regimens comprised of drugs that retain activity against MDR- and XDR-TB strains, the efficacy of GSK2556286 was evaluated in a subacute infection model in BALB/c mice that enables the evaluation of drug regimens against a higher bacterial burden (18). GSK2556286 (50 mg/kg) was coadministered with bedaquiline (B) and pretomanid (Pa), and the efficacy of this regimen was compared to that of BPa plus linezolid (L), which comprises a novel short-course regimen (21, 22) that was recently approved for the treatment of XDR-TB and refractory MDR-TB. The addition of GSK2556286 to the BPa combination significantly increased efficacy, compared to BPa alone, after 2 months of treatment (P < 0.001) (Table 4).
TABLE 4.
Efficacy of GSK2556286 combined with B and Pa in a BALB/c mouse model of TBa
| Regimen | Mean lung log10 CFU ± SD |
||
|---|---|---|---|
| Day 0 | Mo 1 | Mo 2 | |
| Untreated | 7.87 ± 0.12 | ||
| BPa | 5.54 ± 0.32 | 3.17 ± 0.20 | |
| BPaL | 4.73 ± 0.28 | 0.73 ± 0.52 | |
| BPa+GSK2556286 | 5.39 ± 0.18 | 1.74 ± 0.76 | |
For comparison, the three-drug combination BPaL is included.
(iii) Contribution to treatment-shortening activity in combination therapy.
Although the bactericidal activity of this novel 3-drug combination was not as great as that of BPaL (P < 0.01), the 6-log10 magnitude of the killing effect and the clear contribution of GSK2556286 to the combination led us to assess the potential of GSK2556286 to contribute sterilizing activity when incorporated into 3- and 4-drug regimens with B, Pa, and L in the subacute BALB/c mouse infection model, using the proportion of mice with relapse-free cure as the primary endpoint. The standard of care, RHZ (rifampicin plus isoniazid and pyrazinamide), was also included as a reference for bactericidal outcomes after 1 and 2 months of treatment and the relapse endpoint after 4 months based on previous data indicating that RHZ requires more than 3 months to observe significant reductions in the proportion of mice that relapse (22–24).
After 2 months of treatment, regimens combining GSK2556286 with BPa, BL, or BPaL resulted in significantly lower lung CFU counts than with the first-line RHZ control (P < 0.0001, P = 0.0417, and P < 0.0001, respectively) (Table 5). BPa plus GSK2556286 (BPa+GSK2556286) and BPaL+GSK2556286 were not significantly different from BPaL at this time point, but BL+GSK2556286 and PaL+GSK2556286 were significantly less active than BPaL (P < 0.0001). With respect to the relapse outcome, treatment with BPaL, BPa+GSK2556286, and BPaL+GSK2556286 for 2 months resulted in lower proportions of mice relapsing than with treatment with RHZ for 4 months. Although it remains to be determined clinically whether BPaL is effective when administered for less than 6 months, these results indicate the treatment-shortening potential of regimens combining GSK2556286 with BPa and BPaL, as well as BPaL itself, compared to RHZ. BL+GSK2556286 required 3 months of treatment to achieve a relapse rate lower than that of RHZ for 4 months.
TABLE 5.
Efficacy of GSK2556286 combined with various 2- and 3-drug combinations of B, Pa, and L in a BALB/c mouse model of TB
| Regimen | Mean lung log10 CFU ± SD |
Proportion of mice relapsing after treatment fora: |
||||
|---|---|---|---|---|---|---|
| Day 0 | Mo 1 | Mo 2 | 2 mo | 3 mo | 4 mo | |
| Untreated | 7.30 ± 0.10 | |||||
| RHZ | 5.12 ± 0.14 | 2.55 ± 0.07 | NT | NT | 7/15 | |
| BPaL | 3.10 ± 0.17 | 0.00 ± 0.00 | 4/15 | 1/15 | NT | |
| BPa+GSK2556286 | 3.77 ± 0.36 | 0.27 ± 0.54 | 5/15 | 1/15 | 0/15 | |
| BL+GSK2556286 | 4.16 ± 0.34 | 1.91 ± 0.33 | 14/15 | 2/14b | 1/14b | |
| PaL+GSK2556286 | 5.19 ± 0.19 | 2.87 ± 0.34 | 15/15 | 15/15 | 11/15 | |
| BPaL+GSK2556286 | 2.75 ± 0.36 | 0.00 ± 0.00 | 2/15 | 0/15 | NT | |
NT, not tested.
One of 15 mice died due to a gavage accident and could not be assessed for relapse.
The proportions of mice relapsing after 2 and 3 months of treatment with BPaL, BPa+GSK2556286, and BPaL+GSK2556286 did not significantly differ, indicating that GSK2556286 could replace L in the BPaL regimen without a loss of efficacy. On the other hand, PaL+GSK2556286 was associated with significantly more relapses (P < 0.0001) at each time point, and BL+GSK2556286 was associated with more relapses (P = 0.0005) after 2 months of treatment but was similar after 3 and 4 months.
(iv) Activity in combination therapy in the C3HeB/FeJ mouse model.
These 3- and 4-drug regimens in which GSK2556286 either was added to BPaL or replaced B, Pa, or L were also evaluated for CFU reductions in C3HeB/FeJ mice infected with M. tuberculosis H37Rv. After 1 month of treatment, none of these regimens was statistically significantly different from the RHZ or BPaL regimen (Fig. 5). After 2 months, only PaL+GSK2556286 was significantly worse than RHZ (P = 0.0006), suggesting that GSK2556286 could replace either Pa or L in the BPaL regimen without a loss of efficacy compared to BPaL or RHZ (group mean CFU counts are presented in Table S11).
FIG 5.

Efficacy of GSK2556286 (G) combined with the various 2- and 3-drug combinations of B (bedaquiline), Pa (pretomanid), and L (linezolid) for 1 month (A) or 2 months (B) in a C3HeB/FeJ mouse model of TB. For comparison, B, BG, and BPaL are included. Bars indicate median CFU counts. The median CFU on day 0, the start of treatment, is indicated by the dotted line.
DISCUSSION
GSK2556286 is a novel, small-molecule, antitubercular compound identified from high-throughput intramacrophage screening that is active against a variety of drug-sensitive and drug-resistant clinical isolates in axenic culture in the presence of cholesterol as a carbon source. Cholesterol uptake, catabolism, and broader utilization are important for the maintenance of the pathogen in the host, and other inhibitors of M. tuberculosis with activity revealed in the presence of cholesterol have been identified (10, 12).
To further understand the potential cholesterol-dependent mode of action, GSK2556286-resistant M. tuberculosis clones were isolated on medium containing cholesterol as the primary carbon source and analyzed by whole-genome sequencing. Approximately half of the resistant clones sequenced harbored mutations in the gene for the membrane-anchored adenylyl cyclase, cya, without altering the viability of the bacteria under laboratory conditions. The observed frameshift and premature stop codon mutations indicated that a loss of cya function results in resistance to GSK2556286, and this was confirmed with the cya knockout mutant. These findings are similar to those for other recently discovered cholesterol-dependent antitubercular leads that directly activate cya, induce bacterial cAMP production, and inhibit cholesterol production in wild-type M. tuberculosis in a cya-dependent fashion (9, 12). Increased intracellular levels of cAMP, one of the main secondary messengers in the cell, may negatively regulate cholesterol and propionate utilization by M. tuberculosis, reducing bacterial growth when it is dependent on utilizing these carbon sources (25). Therefore, GSK2556286 may likewise act by activating cya, inducing cAMP production, and negatively regulating cholesterol and propionate utilization. Ongoing studies to further evaluate the mode of action of GSK2556286, including its effects on cAMP levels and its impact in the presence of cholesterol, will be reported separately. Despite the need for further elucidation of the specific mechanism of action, GSK2556286-resistant mutants remained susceptible to a list of well-known antitubercular drugs, which suggests the novelty of this mechanism.
Given that the macrophage is a major cellular niche for M. tuberculosis, blocking the replication of the bacterium in this environment may enhance existing or future TB drug regimens (2, 6). However, human TB disease is also characterized by the development of caseation necrosis, leading to closed caseous foci and cavities in which M. tuberculosis is found extracellularly, in caseum. As caseum is also rich in cholesterol, those bacilli persisting extracellularly in the acellular zones of caseous foci could also be susceptible to GSK2556286, as they are in axenic culture in the presence of cholesterol (26). To demonstrate this therapeutic potential, GSK2556286 was evaluated, alone and in combination with other drugs, in two murine TB models, one of which (C3HeB/FeJ mice) is distinguished from other mouse models by its propensity to develop caseating lung lesions (17, 27, 28). When used alone, GSK2556286 exhibited bactericidal effects in chronic infection models in both BALB/c mice, where virtually all bacteria reside intracellularly, and C3HeB/FeJ mice, which form large caseating granulomas in which most bacteria are extracellular in caseum but many are also found in foamy macrophages in the cellular cuff surrounding caseous granulomas and in other cellular lesions. Interestingly, GSK2556286 was more potent in the chronic mouse infection models, achieving near-maximal effects at a lower dose, than in the acute infection model despite similar drug exposures in both BALB/c and C57BL/6 mice. We speculate that this difference may be based on a lower requirement for cholesterol utilization in the first 9 days of mouse infection.
When used in combination to identify novel efficacious drug regimens, GSK2556286 exhibited its potential to replace linezolid (L) in the BPaL regimen without significantly affecting efficacy. Whether the endpoint assessed was the bacterial burden after 2 months of treatment in either mouse strain or the proportion of BALB/c mice relapsing after 2 or 3 months of treatment, BPa+GSK2556286 and BPaL had similar efficacies. BPaL was recently approved as a novel short-course oral treatment for XDR-TB and treatment-refractory MDR-TB, but its implementation has been challenged by treatment-limiting linezolid toxicity (28). Should BPa+GSK2556286 prove safe and at least similarly efficacious in clinical trials, it may serve as a regimen or core component of a regimen capable of improving TB treatment.
Although the individual contribution of GSK2556286 to the regimen’s sterilizing activity was not shown directly in these studies, the BPaL and BPa+GSK2556286 regimens had similar sterilizing efficacies in the BALB/c mouse infection model. The contribution of linezolid to the sterilizing activity of the BPaL regimen has been repeatedly demonstrated in this model (22), and thus, these data strongly suggest that GSK2556286 is contributing to the overall efficacy of the BPa+GSK2556286 regimen.
Furthermore, although there were more relapses after 2 and 3 months of treatment when GSK2556286 was substituted for Pa in BPaL (14 versus 4, and 2 versus 1, respectively), BL+GSK2556286 treatment for 3 and 4 months resulted in fewer relapses (2 and 1, respectively) than RHZ treatment for 4 months (7 relapses) in BALB/c mice, and BL+GSK2556286 had bactericidal activity that could not be distinguished from that of RHZ or BPaL in C3HeB/FeJ mice.
In summary, GSK2556286 acts via a novel mode of action to achieve significant in vivo activity in murine models displaying both cellular and extracellular lesion compartments. This result combined with the compound’s low clearance values across a number of species, low propensity for drug-drug interaction liabilities, and adequate preliminary toxicology profile (genotoxicity, safety pharmacology, and general toxicology) present evidence supporting its progression as a new clinical candidate for the treatment of both MDR and drug-susceptible TB that has the potential to contribute to the shortening of TB chemotherapy. Accordingly, a first-time-in-humans (FTIH) trial is now under way (ClinicalTrials.gov identifier NCT04472897).
MATERIALS AND METHODS
Microbiological assays.
The MIC of GSK2556286 against extracellular M. tuberculosis laboratory strains was determined in standard medium with glucose as a carbon source and also in medium supplemented with cholesterol. For MIC determination in the absence of cholesterol, approximately 1 × 105 CFU/mL of M. tuberculosis H37Rv (ATCC 25618) or Erdman (TMCC 107) was added to 96-well flat-bottom plates containing 10 2-fold drug dilutions of GSK2556286 in Middlebrook 7H9 medium (Difco) supplemented with 2% glucose, 0.025% Tween 80, 0.05% tyloxapol, and 10% albumin-dextrose-catalase (ADC). Plates were placed in a sealed box to prevent drying and incubated at 37°C for 6 days. Twenty-five microliters of a resazurin solution (38.6 μM) (resazurin tablets for milk testing [reference number 330884Y; VWR International Ltd.] in 30 mL of phosphate-buffered saline [PBS]) was added to each well. Fluorescence was measured after 48 h at 37°C using a SpectraMax M5 microplate reader (Molecular Devices). Nonlinear regression analysis was used to fit the normalized fluorescence results into dose-response curves, and IC50 and IC90 values were determined using the Excel add-in XLFit. Reported data are the averages from at least two experiments.
The MIC of GSK2556286 in medium containing cholesterol as the carbon source was determined against the M. tuberculosis H37Rv and Erdman strains with a final inoculum of approximately 1.4 × 106 CFU/mL. Bacteria grown in 7H9 medium supplemented with 2% glucose and 0.025% tyloxapol to an optical density (OD) at 600 nm of around 0.5 were pelleted by centrifugation and washed twice with cholesterol medium (Middlebrook 7H9 medium supplemented with 1 g/L KH2PO4, 2.5 g/L Na2HPO4, 0.5 g/L asparagine, 50 mg/L ferric ammonium citrate, 10 mg/L MgSO4·7H2O, 0.5 mg/L CaCl2, and 0.1 mg/L ZnSO4 with 0.01% cholesterol as the sole carbon source). Pellets were resuspended in cholesterol medium and incubated for at least 3 days at 37°C. Plates containing GSK2556286 were inoculated and incubated at 37°C for an additional 7 days, a resazurin solution (38.6 μM) was added, and the plates were incubated for 48 h before fluorescence was measured. Rifampicin (catalog number R3501; Sigma) was used as a positive control at up to 0.36 μM. IC50 and IC90 values were determined as described above. Reported data are the averages from at least two experiments.
The intracellular antibacterial activity of GSK2556286 was determined using human THP-1 cells maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS), 1 mM pyruvate, and 2 mM l-glutamine, and the cells were incubated at 37°C with 5% CO2. An M. tuberculosis H37Rv reporter strain carrying the firefly luciferase gene (under the control of the hsp60 promoter) was grown in Middlebrook 7H9 broth supplemented with 10% ADC, 0.4% glycerol, and 0.05% Tween 80 until the mid-log phase. THP-1 cells were infected at a multiplicity of infection (MOI) of 1:1 in antibiotic-free RPMI 1640 medium containing 10% FBS, 1 mM pyruvate, 2 mM l-glutamine, and 20 nM phorbol 12-myristate 13-acetate (PMA) for 4 h at 37°C with 5% CO2. Following a 4-h incubation period, infected cells were harvested and plated onto 96-well plates containing either GSK2556286 (up to 25 μM), rifampicin (up to 0.73 μM as a positive control), or dimethyl sulfoxide (DMSO) (<0.5% final concentration). After 5 days of incubation, cell luminescence was measured using the Promega Bright-Glo kit and the SpectraMax M5 plate reader. The percentages of inhibition were calculated relative to the DMSO control well. For each compound, the average value from duplicate samples was calculated, and a sigmoidal dose-response (variable-slope) curve was fit by nonlinear regression (GraphPad) to enable the estimation of the IC50.
MIC determination against a panel of clinical isolates on cholesterol-based medium.
The MIC of GSK2556286 was determined against 45 clinical isolates of M. tuberculosis with different resistance phenotypes maintained by the National Institutes of Health. M. tuberculosis strains HN878, CDC1551, Erdman, and H37Rv were included as controls. Individual M. africanum and M. bovis isolates were included to represent other members of the M. tuberculosis complex. Briefly, bacteria were grown to an OD of 0.2 to 0.6 in 7H9 medium supplemented with bovine serum albumin, tyloxapol, and cholesterol as a sole carbon source and added (2 × 104 bacteria per well) to 96-well plates containing GSK2556286 (at concentrations ranging up to 50 μM). para-Aminosalicylic acid (PAS), isoniazid, and bedaquiline were used as positive controls, and DMSO was used as a negative control. Plates were incubated for up to 3 weeks at 37°C. At various time points, plates were read with an inverted enlarging mirror plate reader and graded as either growth or no growth to determine the MIC. The time point was dependent on the growth rate of the strain in the drug-free control medium (generally between 1 and 2 weeks). After 2 weeks of incubation, resazurin was added to the plates. Following incubation at 37°C for 24 h, results were read visually with an inverted enlarging mirror plate reader (blue indicates growth inhibition, and pink indicates growth). The lowest concentration to inhibit growth was defined as the MIC.
IC50 determination against a reference panel of clinical isolates representing global lineages of the human-adapted M. tuberculosis complex.
A panel of 20 clinical isolates of the M. tuberculosis complex representing each of the seven human-adapted lineages (14) was obtained from the Belgian Coordinated Collections of Microorganisms, Institute of Tropical Medicine, Antwerp, Belgium. Bacteria grown in 7H9 medium with 10% ADC and tyloxapol were adapted to medium supplemented with cholesterol (0.01%). Wells containing GSK2556286 concentrations in doubling dilutions ranging up to 100 μM and control wells containing rifampicin (1.22 μM) or DMSO diluent alone (at the highest [<2%] final concentration) were inoculated with 1.4 × 106 CFU/mL of each isolate and incubated for 17 days. Tetrazolium salt [2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt (XTT)] was added at 300 μg/mL, and the absorbance was measured after an additional 24-h incubation. The IC50 of GSK2556286 was defined as the concentration producing an absorbance value that was 50% of that observed in the DMSO-only control.
Selection of GSK2556286-resistant mutants in vitro.
The MIC of GSK2556286 in cholesterol-containing agar medium was used to establish the concentration of GSK2556286 for the selection of resistant mutants. Stocks of M. tuberculosis Erdman (2 × 109 CFU/mL) were thawed and diluted 1:5 in PBS, and 0.1-mL aliquots were plated onto 7H11 agar medium containing cholesterol or cholesterol plus dextrose as the carbon sources, with or without GSK2556286 at 96 μM (8× MIC). The bacterial colonies were counted after 4 or 6 weeks of incubation.
For genetic characterization, genomic DNA (gDNA) from single colonies was extracted with the MasterPure DNA purification kit from Epicentre (catalog number MCD85201) according to the manufacturer’s instructions. DNA libraries were generated according to the Nextera XT Illumina protocol (Nextera XT library prep kit, catalog number FC-131-1024). A total of 0.2 ng/μL purified gDNA was used to initiate the protocol. The multiplexing step was performed using a Nextera XT index kit (catalog number FC-131-1096). The libraries were sequenced using a 2× 150-bp paired-end run with a NextSeq high-output reagent kit on a NextSeq sequencer according to the manufacturer’s instructions (Illumina). Quality assessment was performed using the prinseq-lite program (29), applying the following parameters: Min_length of 50, Trim_qual_right of 20, Trim_qual_type of mean, and Trim_qual_window of 20. R1 and R2 from Illumina sequencing were joined using fastq-join from the ea-tools suite (30).
Creation of an Rv1625c knockout mutant.
A knockout mutant was created in the H37Rv strain by replacing Rv1625c with a hygromycin resistance cassette using the recombineering approach developed by Murphy et al. (31) Gene replacement was confirmed by PCR.
Selection of GSK2556286-resistant mutants in vivo.
C3HeB/FeJ mice received a low-dose aerosol infection with 31 CFU/lung of M. tuberculosis Erdman. Starting at 8 weeks postinfection, mice were given oral doses of GSK2556286 at 100 mg/kg, 5 days a week for 6 weeks. Lungs were harvested, and homogenates were prepared and plated in serial 10-fold dilutions on plates with and without GSK2556286 at 100 μM.
Mouse efficacy studies.
All housing and procedures involving mice were approved by the Institutional Animal Care and Use Committee at Johns Hopkins University School of Medicine or underwent ethical review at GSK, and all studies were carried out in accordance with the Animals (Scientific Procedures) Act 1986 and the GSK policy on the care, welfare, and treatment of animals.
Mice.
Female C57BL/6 mice aged 8 to 12 weeks were purchased from Harlan. Female specific-pathogen-free BALB/c mice and C3HeB/FeJ mice, each aged 5 to 6 weeks, were purchased from Charles River (Wilmington, MA) and Jackson Laboratories (Bar Harbor, ME), respectively. Mice were housed in a biosafety level 3 animal facility.
Mycobacterial strain.
M. tuberculosis H37Rv was mouse passaged, frozen in aliquots, and subcultured in Middlebrook 7H9 broth with 10% oleic acid-albumin-dextrose-catalase (OADC) (Fisher, Pittsburgh, PA) and 0.05% Tween 80 prior to infection.
Infection.
Mice were infected with M. tuberculosis H37Rv. For the acute infection model, C57BL/6 mice were infected intratracheally with approximately 105 CFU (29) and initiated on treatment the day after infection. For the chronic infection models in both mouse strains, aerosol infection was performed using an inhalation exposure system (Glas-col, Terre Haute, IN) and a thawed aliquot of the bacterial culture that was diluted 1:50 with sterile PBS for infecting BALB/c mice and 1:100 for infecting C3HeB/FeJ mice, with the goal of implanting approximately 100 and 50 to 75 CFU, respectively, in the lungs. Mice were held for 6 weeks before beginning treatment. The subacute infection model in BALB/c mice was initiated with a late-log-phase culture in 7H9 broth (optical density at 600 nm of 0.8 to 1), with the goal of implanting 3.5 to 4 log10 CFU. Mice were held for 2 weeks before beginning treatment.
Drug treatments.
All drugs were administered orally via gavage. For dose-ranging monotherapy studies, GSK2556286 was formulated in 1% methylcellulose, and isoniazid at 10 mg/kg was prepared in distilled water. Both drugs were administered orally. In the acute infection model, GSK2556286 doses ranging from 0.3 to 500 mg/kg of body weight were administered for 8 consecutive days. In the chronic infection models, GSK2556286 doses of 10, 40, 100, and 200 mg/kg of body weight were administered once daily, 5 days per week, for 4 weeks. Positive and negative controls received isoniazid and no treatment, respectively.
For experiments evaluating the activity of GSK2556286 in combinations with bedaquiline, pretomanid, and linezolid, GSK2556286 and isoniazid were formulated as described above. The following other drugs were formulated and administered at the indicated doses as previously described (22, 23): rifampicin (10 mg/kg) and pyrazinamide (150 mg/kg) in distilled water, bedaquiline (25 mg/kg) in an acidified 10% (2-hydroxypropyl)-β-cyclodextrin (HPCD) solution, pretomanid (100 mg/kg) in a 20% HPCD–lecithin (CM-2) formulation, and linezolid (100 mg/kg) in 0.5% methylcellulose.
Assessment of GSK2556286 pharmacokinetics.
Whole blood was sampled at 0.5, 1, and 6 h postdose (and 24 h after 18- to 50-mg/kg doses) from C57BL/6 mice (n = 2 samples per time point) in the second acute infection model experiment. Whole blood was also sampled from uninfected BALB/c mice (n = 3 samples per time point) 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, and 24 h after a single dose of 10 mg/kg or the last of 4 daily doses of 50 mg/kg under the same dosing conditions as the ones used in the efficacy studies. Whole-blood samples were lysed with distilled water and stored frozen prior to quantification. GSK2556286 concentrations were determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Pharmacokinetic analysis was performed by noncompartmental methods using WinNonlin Phoenix version 6.3 (Pharsight Corporation).
Assessment of efficacy.
Two microbiological outcomes were assessed: lung CFU counts during treatment and the proportion of mice relapsing after the completion of treatment. Lungs were collected and homogenized in glass grinders at prespecified time points during and after drug treatment. The homogenates were serially diluted in PBS and plated onto Middlebrook 7H11 agar plates supplemented with 10% (vol/vol) OADC (Gibco), cycloheximide (10 mg/mL), carbenicillin (50 mg/mL), polymyxin B (25 mg/mL), and trimethoprim (20 mg/mL), except that the medium used in the acute infection model studies did not contain selective antibiotics. Homogenates from mice in the acute infection model studies and mice receiving drug combinations were plated onto the same agar media but with the addition of activated charcoal powder (0.4%, wt/vol) to prevent drug carryover. Colonies were counted after 4 and 6 weeks of incubation at 37°C to ensure that all cultivable bacteria would be detected. Relapse after 2, 3, and 4 months of treatment with drug combinations was assessed by holding cohorts of 15 mice per group for an additional 3 months without treatment before sacrificing the mice and plating the entire lung homogenate, as described above. Relapse was defined as the detection of ≥1 CFU.
Statistical analysis.
CFU counts (x) were log transformed (as x + 1) before analysis. A sigmoidal inhibitory dose-response curve was fit to the log10 CFU data obtained from the acute mouse infection model, as previously described (15). Group means from BALB/c mice were compared by one-way analysis of variance with Dunnett’s posttest to control for multiple comparisons. Group relapse proportions were compared using Fisher’s exact test, adjusting for multiple comparisons. The Kruskal-Wallis test and Dunn’s nonparametric posttest to adjust for multiple comparisons were used to test for significance on nonnormally distributed CFU data from C3HeB/FeJ mice. GraphPad Prism version 6 (GraphPad, San Diego, CA) was used for all analyses. The use of 15 mice per group for relapse assessment provides approximately 80% power to detect 40 percentage-point differences in the relapse rate, after setting the alpha value at 0.01 to adjust for up to 5 simultaneous two-sided comparisons. Smaller differences may not be meaningful in terms of shortening the duration of treatment.
ACKNOWLEDGMENTS
We thank Kevin Pethe, Jichan Jang, and Min Chung for their collaboration with the screening at Institute Pasteur Korea. We thank Ken Duncan and Peter Warner from the Bill & Melinda Gates Foundation, Steve Berthel from the New Venture Fund, and GSK technical and administrative support staff. We thank Dirk Schnappinger and Curtis Engelhart for providing the Rv1625c knockout strain and Anne Lenaerts for assistance with the in vitro and in vivo selection of resistant mutants.
This work was funded, in part, by the Tres Cantos Open Lab Foundation (grant number TC049), by the European Union’s 7th framework program (FP7-2007-2013) under an Orchid grant agreement (number 261378), by the Division of Intramural Research of the NIAID/NIH, and by the Bill & Melinda Gates Foundation (OPP1178195).
Footnotes
Supplemental material is available online only.
Contributor Information
Eric L. Nuermberger, Email: enuermb@jhmi.edu.
Elena Jiménez, Email: elena.n.jimenez@gsk.com.
REFERENCES
- 1.World Health Organization. 2020. Global tuberculosis report 2020. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Cole ST. 2016. Inhibiting Mycobacterium tuberculosis within and without. Philos Trans R Soc Lond B Biol Sci 371:20150506. 10.1098/rstb.2015.0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kushner S, Dalalian D, Sanjurjo JL, Bach FL, Jr, Safir SR, Smith VK, Jr, Williams JH. 1952. Experimental chemotherapy of tuberculosis. II. The synthesis of pyrazinamides and related compounds. J Am Chem Soc 74:3617–3621. 10.1021/ja01134a045. [DOI] [Google Scholar]
- 4.Malone L, Schurr A, Lindh H, McKenzie D, Kiser JS, Williams JH. 1952. The effect of pyrazinamide (aldinamide) on experimental tuberculosis in mice. Am Rev Tuberc 65:511–518. [PubMed] [Google Scholar]
- 5.Parish T. 2020. In vitro drug discovery models for Mycobacterium tuberculosis relevant for host infection. Expert Opin Drug Discov 15:349–358. 10.1080/17460441.2020.1707801. [DOI] [PubMed] [Google Scholar]
- 6.Christophe T, Jackson M, Jeon HK, Fenistein D, Contreras-Dominguez M, Kim J, Genovesio A, Carralot JP, Ewann F, Kim EH, Lee SY, Kang S, Seo MJ, Park EJ, Skovierova H, Pham H, Riccardi G, Nam JY, Marsollier L, Kempf M, Joly-Guillou ML, Oh T, Shin WK, No Z, Nehrbass U, Brosch R, Cole ST, Brodin P. 2009. High content screening identifies decaprenyl-phosphoribose 2′ epimerase as a target for intracellular antimycobacterial inhibitors. PLoS Pathog 5:e1000645. 10.1371/journal.ppat.1000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberger J, Butler J, Dressman J. 2018. A refined developability classification system. J Pharm Sci 107:2020–2032. 10.1016/j.xphs.2018.03.030. [DOI] [PubMed] [Google Scholar]
- 8.de Jager VR, Dawson R, van Niekerk C, Hutchings J, Kim J, Vanker N, van der Merwe L, Choi J, Nam K, Diacon AH. 2020. Telacebec (Q203), a new antituberculosis agent. N Engl J Med 382:1280–1281. 10.1056/NEJMc1913327. [DOI] [PubMed] [Google Scholar]
- 9.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, Christophe T, Lee H, Kempf M, Jackson M, Lenaerts AJ, Pham H, Jones V, Seo MJ, Kim YM, Seo M, Seo JJ, Park D, Ko Y, Choi I, Kim R, Kim SY, Lim S, Yim S-A, Nam J, Kang H, Kwon H, Oh C-T, Cho Y, Jang Y, Kim J, Chua A, Tan BH, Nanjundappa MB, Rao SPS, Barnes WS, Wintjens R, Walker JR, Alonso S, Lee S, Kim J, Oh S, Oh T, Nehrbass U, Han S-J, No Z, et al. 2013. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med 19:1157–1160. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 10.VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB, Memmott C, Crowe AM, Eltis LD, Perola E, Deininger DD, Wang T, Locher CP, Russell DG. 2015. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog 11:e1004679. 10.1371/journal.ppat.1004679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pandey AK, Sassetti CM. 2008. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci USA 105:4376–4380. 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilburn KM, Montague CR, Qin B, Woods AK, Love MS, McNamara CW, Schultz PG, Southard TL, Huang L, Petrassi HM, VanderVen BC. 2022. Pharmacological and genetic activation of cAMP synthesis disrupts cholesterol utilization in Mycobacterium tuberculosis. PLoS Pathog 18:e1009862. 10.1371/journal.ppat.1009862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Odds FC. 2003. Synergy, antagonism, and what the chequerboard puts between them. J Antimicrob Chemother 52:1. 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]
- 14.Borrell S, Trauner A, Brites D, Rigouts L, Loiseau C, Coscolla M, Niemann S, De Jong B, Yeboah-Manu D, Kato-Maeda M, Feldmann J, Reinhard M, Beisel C, Gagneux S. 2019. Reference set of Mycobacterium tuberculosis clinical strains: a tool for research and product development. PLoS One 14:e0214088. 10.1371/journal.pone.0214088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.International Council on Harmonisation. 2013. M3 (R2) Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals. https://www.ema.europa.eu/en/ich-m3-r2-non-clinical-safety-studies-conduct-human-clinical-trials-pharmaceuticals. [PubMed]
- 16.Rullas J, García JI, Beltrán M, Cardona PJ, Cáceres N, García-Bustos JF, Angulo-Barturen I. 2010. Fast standardized therapeutic-efficacy assay for drug discovery against tuberculosis. Antimicrob Agents Chemother 54:2262–2264. 10.1128/AAC.01423-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lanoix JP, Lenaerts AJ, Nuermberger EL. 2015. Heterogeneous disease progression and treatment response in a C3HeB/FeJ mouse model of tuberculosis. Dis Model Mech 8:603–610. 10.1242/dmm.019513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nuermberger EL. 2017. Preclinical efficacy testing of new drug candidates. Microbiol Spectr 5:TBTB2-0034-2017. 10.1128/microbiolspec.TBTB2-0034-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Driver ER, Ryan GJ, Hoff DR, Irwin SM, Basaraba RJ, Kramnik I, Lenaerts AJ. 2012. Evaluation of a mouse model of necrotic granuloma formation using C3HeB/FeJ mice for testing of drugs against Mycobacterium tuberculosis. Antimicrob Agents Chemother 56:3181–3195. 10.1128/AAC.00217-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenthal IM, Tasneen R, Peloquin CA, Zhang M, Almeida D, Mdluli KE, Karakousis PC, Grosset JH, Nuermberger EL. 2012. Dose-ranging comparison of rifampin and rifapentine in two pathologically distinct murine models of tuberculosis. Antimicrob Agents Chemother 56:4331–4340. 10.1128/AAC.00912-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conradie F, Diacon AH, Ngubane N, Howell P, Everitt D, Crook AM, Mendel CM, Egizi E, Moreira J, Timm J, McHugh TD, Wills GH, Bateson A, Hunt R, Van Niekerk C, Li M, Olugbosi M, Spigelman M, Nix-TB Trial Team. 2020. Treatment of highly drug-resistant pulmonary tuberculosis. N Engl J Med 382:893–902. 10.1056/NEJMoa1901814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tasneen R, Betoudji F, Tyagi S, Li SY, Williams K, Converse PJ, Dartois V, Yang T, Mendel CM, Mdluli KE, Nuermberger EL. 2016. Contribution of oxazolidinones to the efficacy of novel regimens containing bedaquiline and pretomanid in a mouse model of tuberculosis. Antimicrob Agents Chemother 60:270–277. 10.1128/AAC.01691-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li SY, Tasneen R, Tyagi S, Soni H, Converse PJ, Mdluli K, Nuermberger EL. 2017. Bactericidal and sterilizing activity of a novel regimen with bedaquiline, pretomanid, moxifloxacin, and pyrazinamide in a murine model of tuberculosis. Antimicrob Agents Chemother 61:e00913-17. 10.1128/AAC.00913-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams K, Minkowski A, Amoabeng O, Peloquin CA, Taylor D, Andries K, Wallis RS, Mdluli KE, Nuermberger EL. 2012. Sterilizing activities of novel combinations lacking first- and second-line drugs in a murine model of tuberculosis. Antimicrob Agents Chemother 56:3114–3120. 10.1128/AAC.00384-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson RM, Bai G, DeMott CM, Banavali NK, Montague CR, Moon C, Shekhtman A, VanderVen B, McDonough KA. 2017. Chemical activation of adenylyl cyclase Rv1625c inhibits growth of Mycobacterium tuberculosis on cholesterol and modulates intramacrophage signaling. Mol Microbiol 105:294–308. 10.1111/mmi.13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarathy JP, Dartois V. 2020. Caseum: a niche for Mycobacterium tuberculosis drug-tolerant persisters. Clin Microbiol Rev 33:e00159-19. 10.1128/CMR.00159-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Irwin SM, Driver E, Lyon E, Schrupp C, Ryan G, Gonzalez-Juarrero M, Basaraba RJ, Nuermberger EL, Lenaerts AJ. 2015. Presence of multiple lesion types with vastly different microenvironments in C3HeB/FeJ mice following aerosol infection with Mycobacterium tuberculosis. Dis Model Mech 8:591–602. 10.1242/dmm.019570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan H, Yan BS, Rojas M, Shebzukhov YV, Zhou H, Kobzik L, Higgins DE, Daly MJ, Bloom BR, Kramnik I. 2005. Ipr1 gene mediates innate immunity to tuberculosis. Nature 434:767–772. 10.1038/nature03419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sparrowe J, Jimenez M, Rullas J, Martínez AE, Ferrer S. 2015. Refined intratracheal intubation technique in the mouse, complete protocol description for lower airways models. Glob J Anim Sci Res 3:363–369. [Google Scholar]
- 30.Aronesty E. 2011. ea-utils: command-line tools for processing biological sequencing data. https://github.com/ExpressionAnalysis/ea-utils.
- 31.Murphy KC, Campellone KG. 2003. Lambda red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol 4:11. 10.1186/1471-2199-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1 to S11 and Fig. S1 and S2. Download aac.00132-22-s0001.pdf, PDF file, 0.3 MB (265KB, pdf)