

ABSTRACT
β-Lactam antibiotics are the first choice for the treatment of most bacterial infections. However, the increased prevalence of β-lactamases, in particular extended-spectrum β-lactamases, in pathogenic bacteria has severely limited the possibility of using β-lactam treatments. Combining β-lactam antibiotics with β-lactamase inhibitors can restore treatment efficacy by negating the effect of the β-lactamase and has become increasingly important against infections caused by β-lactamase-producing strains. Not surprisingly, bacteria with resistance to even these combinations have been found in patients. Studies on the development of bacterial resistance to β-lactam/β-lactamase inhibitor combinations have focused mainly on the effects of single, chromosomal or plasmid-borne, β-lactamases. However, clinical isolates often carry more than one β-lactamase in addition to multiple other resistance genes. Here, we investigate how the evolutionary trajectories of the development of resistance to three commonly used β-lactam/β-lactamase inhibitor combinations, ampicillin-sulbactam, piperacillin-tazobactam, and ceftazidime-avibactam, were affected by the presence of three common β-lactamases, TEM-1, CTX-M-15, and OXA-1. First-step resistance was due mainly to extensive gene amplifications of one or several of the β-lactamase genes where the amplification pattern directly depended on the respective drug combination. Amplifications also served as a stepping-stone for high-level resistance in combination with additional mutations that reduced drug influx or mutations in the β-lactamase gene blaCTX-M-15. This illustrates that the evolutionary trajectories of resistance to β-lactam/β-lactamase inhibitor combinations are strongly influenced by the frequent and transient nature of gene amplifications and how the presence of multiple β-lactamases shapes the evolution to higher-level resistance.
KEYWORDS: β-lactam/β-lactamase inhibitor, antibiotic resistance, evolution, gene amplification
INTRODUCTION
β-Lactams are the most important class of antibiotics used for the treatment of Gram-negative bacterial infections (1). However, their extensive use has led to resistance development, and today, the prevalence of β-lactam resistance among bacterial pathogens has severely reduced their usability for treatment in many parts of the world. Mechanisms of resistance against β-lactams include decreased influx as well as increased efflux of the drug, target alteration, and drug inactivation (2). While drug inactivation by β-lactamase enzymes that hydrolyze the β-lactam ring is the most important clinical resistance mechanism among Gram-negative bacteria, combinations of several resistance mechanisms are common among clinical isolates and contribute to the level of resistance (2). To date, many hundreds of unique β-lactamase genes, either plasmid borne or chromosomally located, have been identified in various contexts (3). Some of the ones most commonly found in clinical isolates belong to the groups TEM, OXA, SHV, and CTX-M (3, 4). Early members of the TEM and OXA enzyme groups mainly catalyze the degradation of penicillins and early-generation cephalosporins, but through the accumulation of point mutations, they have developed extended-spectrum activity (named extended-spectrum β-lactamases [ESBLs]) toward later generations of cephalosporins and, in some cases, even against carbapenems (5–7).
With the increased prevalence of ESBL-producing bacteria, combinations of β-lactam antibiotics with β-lactamase inhibitors have become of growing clinical importance (1, 8, 9). The inactivation of the β-lactamase activity prevents the enzyme from acting on the incoming antibiotic, which restores the susceptibility of the cell (10, 11). This approach has attracted much interest from the pharmaceutical industry, and there is an increasing number of new potential β-lactamase inhibitors in the pharmaceutical pipeline (12–15). Three commonly used inhibitors are sulbactam, tazobactam, and avibactam, mainly effective against Ambler class A, C, and D β-lactamases (1).
Following the introduction of β-lactamase inhibitors into clinical practice, resistance against these compounds has been reported (16–21). The main resistance mechanism has been the overexpression of β-lactamases, which enables the bacterium to cope with higher doses of the drugs (16, 17, 19, 20, 22, 23), but single-amino-acid substitutions rendering the β-lactamase less sensitive to the combination treatment have also been found (4, 18, 21, 24). Most studies on inhibitor resistance have demonstrated how individual β-lactamases circumvent the inhibition of combination treatment. However, clinical isolates often encode multiple β-lactamases, often with overlapping catalytic spectra, which may lead to different forms of allogenous selection (25). It is therefore conceivable that the evolution of resistance to β-lactam/β-lactamase inhibitor combinations in a strain with several β-lactamases differs from that of a strain carrying only one enzyme variant due to the expanded selective landscape and differences in catalytic activity and inhibitor binding between the enzymes. Hence, here, we explored the evolutionary trajectories toward resistance to different combinations of β-lactam/β-lactamase inhibitors for a strain carrying a plasmid that encodes three different β-lactamases. The pUUH239.2 plasmid was originally isolated from an ESBL-producing Klebsiella pneumoniae clone that caused an outbreak at Uppsala University Hospital in Sweden (26). It carries 13 resistance-associated genes, including three β-lactamases: the two narrow-spectrum β-lactamases TEM-1 (class A) and OXA-1 (class D) and the ESBL CTX-M-15 (class A) (26, 27). The resistance cassette of the plasmid is highly similar to those of plasmids associated with Escherichia coli sequence type 131 (ST131), and the plasmid backbone is highly similar to that of the Klebsiella plasmid pKPN3 originally isolated from a K. pneumoniae ST258 strain (27). To investigate the evolutionary trajectories of resistance to β-lactamase/β-lactamase inhibitors, we chose three clinically used combinations, (i) ampicillin-sulbactam (SAM), (ii) piperacillin-tazobactam (TZP), and (iii) ceftazidime-avibactam (CZA) (28–30). We conducted one-step selections as well as short-term in vitro evolution experiments at increasing concentrations of the antibiotic-inhibitor. Isolated mutants and endpoint populations were characterized by their maximum growth rate, MIC, cross-resistance to other combinations, and whole-genome sequence to elucidate the genetic basis of resistance.
RESULTS
To study the effect of multiple β-lactamases on the development of resistance to β-lactam/β-lactamase inhibitor combinations, we used the multiresistance plasmid pUUH239.2 (carrying blaTEM-1, blaOXA-1, and blaCTX-M-15) in the well-studied E. coli MG1655 strain as a model system (27). The three combinations ampicillin-sulbactam, piperacillin-tazobactam, and ceftazidime-avibactam were chosen due to their use in the treatment of infections caused by E. coli (28, 29, 31, 32). Susceptibility to the combinations was determined in two ways throughout the study: (i) according to the standard EUCAST methodology measuring the MIC, where a fixed concentration of 4 mg/L of the inhibitor is used, and (ii) according to the concentration ratio that the drug and the respective inhibitor are administered to treat an infection (called MICratio here). The similar pharmacokinetic properties of the paired antibiotic-inhibitor combinations result in a maintained ratio during treatment, but the absolute concentrations will vary depending on the dosing interval and location in the body (33, 34). The MICratio therefore relates directly to the selective potential of the clinically used drug ratio at different concentrations, and we therefore used the clinical treatment ratios of the drug and the inhibitor for the selection of resistant strains. The only combination where there was a significant discrepancy between the MIC and the MICratio was SAM, where the plasmid-containing strain had an MIC of >256 mg/L, while the MICratio was only 32 mg/L (Table 1). This is explained by the inability of sulbactam to completely inactivate the β-lactamases at the fixed concentration of 4 mg/L used for standard susceptibility testing (Fig. 1A).
TABLE 1.
Susceptibility profiles of the parental strains and one-step selection mutants for SAM, TZP, and CZAa
| Strain | Combination for selection | Mean growth rate ± SD | SAM MICratio (mg/L) | SAM MIC (mg/L) | TZP MICratio (mg/L) | TZP MIC (mg/L) | TZ MIC (mg/L) | CZA MICratio (mg/L) | CZA MIC (mg/L) |
|---|---|---|---|---|---|---|---|---|---|
| WT | None | 1 ± 0.02 | 4 | 4 | 2 | 2 | 0.5 | 0.25 | 0.25 |
| WT/pUUH | None | 0.97 ± 0.01 | 32 | >256 | 16 | 8 | 64 | 0.25 | 0.25 |
| DA61263 | SAM | 0.84 ± 0.05 | 256 | >256 | 256 | 256 | 0.5 | 0.25 | 0.25 |
| DA61264 | 0.75 ± 0.01 | 256 | >256 | 256 | 256 | 0.5 | 0.25 | 0.25 | |
| DA61265 | 0.81 ± 0.01 | 256 | >256 | 64 | 256 | 64 | 1 | 0.5 | |
| DA61266 | 0.87 ± 0.02 | 128 | >256 | 32 | 128 | 0.5 | 0.5 | 0.25 | |
| DA61267 | 0.99 ± 0.04 | 256 | >256 | 64 | 256 | 64 | 0.5 | 0.25 | |
| DA61268 | TZP | 1.00 ± 0.02 | 64 | >256 | 128 | >256 | 64 | 1 | 0.25 |
| DA61269 | 0.98 ± 0.02 | 64 | >256 | 64 | 256 | 64 | 1 | 0.25 | |
| DA61270 | 1.05 ± 0.02 | 64 | >256 | 128 | 256 | 64 | 1 | 0.5 | |
| DA61271 | 0.97 ± 0.03 | 64 | >256 | 128 | 256 | 64 | 0.5 | 0.25 | |
| DA61272 | 0.97 ± 0.04 | 64 | >256 | 128 | 256 | 128 | 1 | 0.25 | |
| DA61259 | CZA | 0.85 ± 0.04 | 128 | >256 | 64 | 32 | 128 | 2 | 0.25 |
| DA61260 | 0.83 ± 0.05 | 64 | >256 | 32 | 128 | 64 | 4 | 0.25 | |
| DA61261 | 0.91 ± 0.04 | 32 | 128 | 8 | 4 | 8 | 4 | 0.5 | |
| DA61262 | 0.56 ± 0.16 | 128 | >256 | 64 | 128 | >256 | 2 | 0.25 | |
| DA62411 | 0.73 ± 0.01 | 128 | >256 | 64 | >256 | >256 | 4 | 0.25 | |
MICs for ampicillin and piperacillin were >256 mg/L for the parental strain and all mutants. Values in boldface type exceed the clinical breakpoints set by EUCAST. Clinical breakpoints are as follows: >8 mg/L for ampicillin-sulbactam (SAM), >8 mg/L for piperacillin-tazobactam (TZP), >4 mg/L for ceftazidime (TZ), and >8 mg/L for ceftazidime-avibactam (CZA). WT, wild type.
FIG 1.

Relationship between β-lactam MICs and inhibitor concentrations. (A) Ampicillin-sulbactam; (B) piperacillin-tazobactam; (C) ceftazidime-avibactam. Filled boxes represent E. coli MG1655 (DA5438), and clear circles represent E. coli MG1655 containing the pUUH239.2 plasmid (DA63522). Note that for SAM, the ampicillin MIC was >256 mg/L at sulbactam concentrations of <16 mg/L (gray circles), and no growth was detected at sulbactam concentrations of >16 mg/L for DA5438 and >32 mg/L for DA63522. Values are medians from 3 to 4 biological replicates.
Effect of inhibition of β-lactamases.
To investigate the relationship between inhibitor concentrations and antibiotic efficacy in response to the three β-lactamases, we expanded the standard susceptibility test by performing a checkerboard analysis with a wider range of inhibitor concentrations. As shown in Fig. 1, the susceptibility for the plasmid-free strain (DA5438) (filled symbols) was largely unchanged for all three antibiotics at the range of inhibitor concentrations tested, apart from at the highest concentrations of sulbactam and avibactam, in accordance with previous reports of the killing effects of these inhibitors at high concentrations (35, 36). For the plasmid-containing strain (DA63522) (open symbols), there was a clear dose dependence of the inhibitor on susceptibility to the antibiotic. Complete inhibition of the β-lactamases (susceptibility equal to that of the plasmid-free strain) required concentrations of 32 mg/L of sulbactam, 64 mg/L of tazobactam, and 1 mg/L of avibactam. There were also clear plateaus in the susceptibility curves, indicating that there is likely a different dose response for the different β-lactamases. Inversely, the highest tested concentrations of inhibitors that gave no detectable effect on the β-lactamases (MIC the same as that without the inhibitor) were 8 mg/L for sulbactam and <1 mg/L for tazobactam, while we never reached a value close to the MIC of ceftazidime (32 mg/L), even with the lowest tested concentration of avibactam (0.016 mg/L). This clearly illustrates the high potency of avibactam to inhibit all three tested β-lactamases even at very low concentrations.
First-step resistance selection.
To compare the frequencies and resistance mechanisms for the different drug combinations, we first performed one-step mutant selection on plates containing 1, 2, and 4 times the respective MICratios (Table 1). Mutants were obtained for all of the combinations on the plates at the respective MICratio but not above that. The mutant frequencies for SAM and TZP were 4 × 10−9 and 1.7 × 10−9 per cell/division, respectively, whereas for CZA, the frequency was 2 × 10−10 per cell/division. Five clones from each selection were subjected to susceptibility testing, growth rate determination, and whole-genome resequencing to determine genetic changes.
Interestingly, although no mutants could be obtained at concentrations above the MICratio, all isolated mutants had increased their MICratio 4- to 16-fold, indicating a discrepancy between the resistance levels of the mutants and the mutant-selecting concentrations (Table 1). The MICs for TZP-selected mutants reached the maximum tested concentrations of 256 mg/L, equaling a 32-fold increase. The CZA-selected mutants had MICs that did not differ from those of the parental strain, although the MICratio increased 8- to 16-fold. This can be explained by the inhibitor concentration still being much lower than 4 mg/L at these concentrations. Although the parental strain with pUUH239.2 already had a SAM MIC of >256 mg/L, the MICratio increased 4- to 8-fold. There was also strong cross-resistance for SAM-selected mutants against TZP (up to a 16-fold increase in the MICratio) but less so for TZP-selected mutants against SAM (2-fold increase in the MICratio). Some SAM- and TZP-selected mutants had small increases in the MIC or MICratio for CZA (2- to 4-fold). Most CZA-selected mutants had increased MICs against SAM and TZP but with substantial variation between mutants. Interestingly, for some mutants, collateral sensitivity was observed, as reported in previous studies (37). For example, three SAM-selected mutants lost their resistance to ceftazidime, and one CZA-selected mutant (DA61261), although with an 8-fold increased MICratio for CZA, had an increased susceptibility to ceftazidime alone and generally low MICs for the other combinations. Some selected mutants showed growth defects of up to 40%, but several mutants had no discernible reduction in growth, especially the mutants selected on TZP (Table 1).
Extensive gene amplification of β-lactamase genes causes high-level resistance.
We sequenced the first-step mutants to determine the genetic basis for the decreased susceptibility. In accordance with previous studies on resistance to β-lactam/β-lactamase inhibitor combinations, the majority of isolated mutants had extensive gene amplifications of the plasmid-borne β-lactamase genes that likely result in the overexpression of β-lactamase enzymes (Fig. 2; see also Table S3 in the supplemental material) (20). The three β-lactamase genes are located within a 41-kb resistance region of pUUH239.2 together with other resistance genes and six interspersed copies of IS26 (Fig. 2). Selection for each drug combination resulted in a specific pattern of high-level gene amplifications of the different β-lactamases. SAM mutants had highly increased copy numbers of blaTEM-1 (28 to 71 copies), most with amplification recombination points in original or newly inserted IS26 elements (Fig. 2). In addition, three of the SAM mutants had deletions of the regions containing the other two β-lactamases, blaCTX-M-15 and blaOXA-1. The mutants selected on TZP showed a different amplification pattern, with a >30-fold gene copy number of blaOXA-1 in three mutants. Two mutants, DA61268 and DA61270, instead had complex rearrangements with plasmid sequences inserted on the chromosome in individual positions in rRNA genes through transposition via IS26 elements. DA61268 had a deletion of the repAB and repA4 genes, involved in plasmid replication control, and a deletion of the macrolide resistance region, but apart from that, the whole plasmid appears to have been incorporated on the chromosome. DA61270 had the region between IS26 (2) - IS26 (5), containing both blaCTX-M-15 and blaOXA-1, inserted into an rRNA operon on the chromosome, while the rest of the plasmid sequences were lost.
FIG 2.
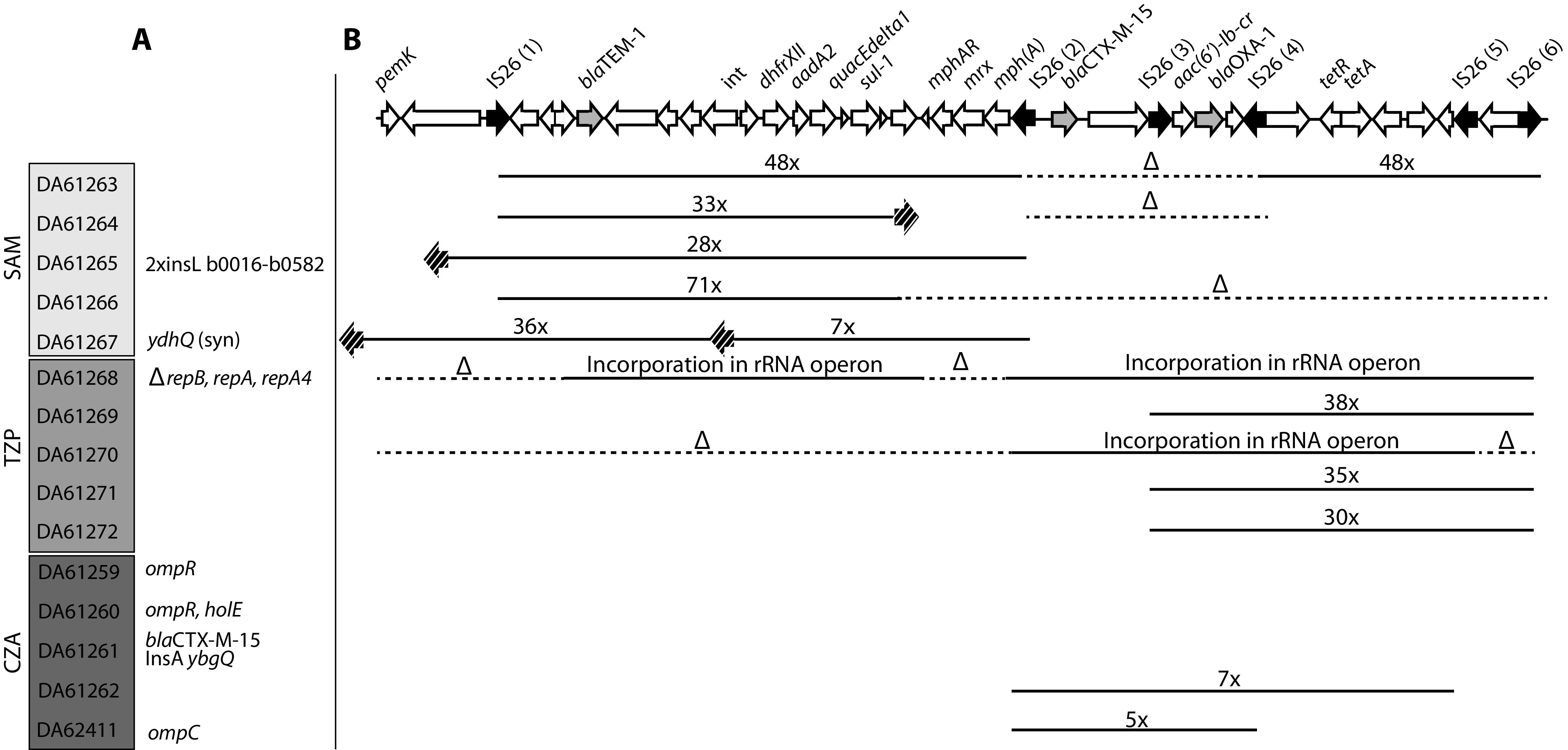
Genetic changes for one-step mutants. (A) Point mutations and other changes. (B) Deletions and amplifications in the resistance cassette. Black arrows indicate IS26 elements, and gray arrows display the three β-lactamases. Striped arrows indicate additional IS26 insertions. Solid lines display the amplification of a certain region of the cassette, whereas dotted lines with Δ indicate deletions. Numbers indicate the copy numbers of the amplified regions. The exact locations of all genetic changes can be found in Table S3 in the supplemental material.
The CZA-selected mutants showed a mix of β-lactamase gene amplifications and other mutations. Two mutants had increased copy numbers of blaCTX-M-15 and blaOXA-1 (5- and 7-fold) (Fig. 2). In addition, three of the CZA mutants contained mutations in the outer membrane porin OmpC or its regulator OmpR, which has previously been linked to decreased susceptibility to β-lactams (38). Interestingly, DA61261, which showed increased susceptibility to several β-lactams apart from CZA, carried a single-amino-acid change in blaCTX-M-15 (G238D, according to the Ambler classification [39]).
Individual overexpression of the three β-lactamases results in increased MICratio against different drug combinations.
To verify that the identified gene amplifications result in increased gene expression of the β-lactamases, we measured the relative transcription levels of a set of the isolated mutants (Fig. 3). DA61263 had a 48-fold gene copy number of blaTEM-1, which was mirrored by the 50-fold increased transcription of blaTEM-1. DA61269 had a 38-fold copy number of blaOXA-1 and a corresponding transcriptional increase of more than 20-fold. The two mutants that had inserted plasmid sequences in rRNA operons (DA61268 and DA61270) also showed increased expression of β-lactamases (up to 20-fold), especially blaOXA-1 (Fig. 3).
FIG 3.

Expression of β-lactamases determined using RT-qPCR in order to verify increased transcript levels. The gene expression of the mutants is relative to that of the parental strain DA63522. Standard deviations from three biological replicates are shown.
Since there appears to be a pattern where increased expression was seen for blaTEM-1 in SAM-selected mutants, blaOXA-1 for TZP-selected mutants, and blaCTX-M-15 for CZA-selected mutants, we analyzed the individual effects of the three β-lactamases using the l-arabinose-inducible pBAD18 expression system. As expected, in the absence of the inhibitor, the overexpression of all three β-lactamases gave very high increases in the MICs for ampicillin and piperacillin and, in the case of CTX-M-15, also for ceftazidime (Table 2). When overexpressed, all three β-lactamases also gave high MIC and MICratio values for SAM, but only TEM-1 and OXA-1 were able to achieve MICs equivalent to clinical resistance to TZP (8 mg/L of the antibiotic with 4 mg/L of the inhibitor), in line with the good impact that tazobactam is known to have on CTX-M enzymes (40). However, none of the β-lactamases were able to achieve high-level resistance to the CZA combination when overexpressed, although a decrease in susceptibility could be observed with the expression of TEM-1 and CTX-M-15. These patterns are coherent with the pattern of amplifications from the mutants selected on the different drug combinations.
TABLE 2.
Effects of the overexpression of the three β-lactamases TEM-1, OXA-1, and CTX-M-15 on different β-lactam/β-lactamase inhibitor combinationsa
| Plasmid | AMP MIC (mg/L) | SAM MICratio (mg/L) | SAM MIC (mg/L) | PIP MIC (mg/L) | TZP MICratio (mg/L) | TZP MIC (mg/L) | TZ MIC (mg/L) | CZA MICratio (mg/L) | CZA MIC (mg/L) |
|---|---|---|---|---|---|---|---|---|---|
| Empty pBAD18 | 4 | 4 | 2 | 2 | 2 | 2 | 0.25 | 0.25 | 0.125 |
| pBAD-blaTEM-1 | >256 | 256 | >256 | >256 | 64 | 8 | 2 | 1 | 0.25 |
| pBAD-blaCTX-M-15 | >256 | 32 | 64 | >256 | 16 | 4 | 256 | 0.5 | 0.25 |
| pBAD-blaOXA-1 | >256 | 64 | 128 | >256 | 128 | 32 | 0.25 | 0.25 | 0.125 |
AMP, ampicillin; SAM, ampicillin-sulbactam; PIP, piperacillin; TZP, piperacillin-tazobactam; TZ, ceftazidime; CZA, ceftazidime-avibactam. Values in boldface type exceed the clinical breakpoints set by EUCAST.
Short-term evolution enables clinical resistance levels for all β-lactam/β-lactamase inhibitor combinations.
While single-step selection on plates allows for rapid identification of initial genetic changes causing decreased susceptibility, it has the limitation that resistance mechanisms requiring combinations of multiple mutations cannot be selected easily. To investigate resistance development in a competitive environment that also allows the selection of multiple mutations, we conducted short-term experimental-evolution experiments in liquid cultures of E. coli MG1655 with pUUH239.2 at increasing concentrations of each of the three inhibitor combinations. For SAM and TZP, all lineages were able to achieve high-level resistance, already reaching our upper MICratio limit of 256 mg/L in 5 to 8 and 7 to 11 passages, respectively (Fig. 4A and B). The populations passaged in CZA reached a maximum MICratio of 8 to 64 mg/L of the antibiotic but reached the clinical resistance breakpoint within 8 to 11 passages (Fig. 4C). All SAM- and TZP-selected lineages and 6/8 CZA-selected lineages had MICs of 256 mg/L, and there was strong cross-resistance to SAM and TZP for all selected lineages (Table 3).
FIG 4.
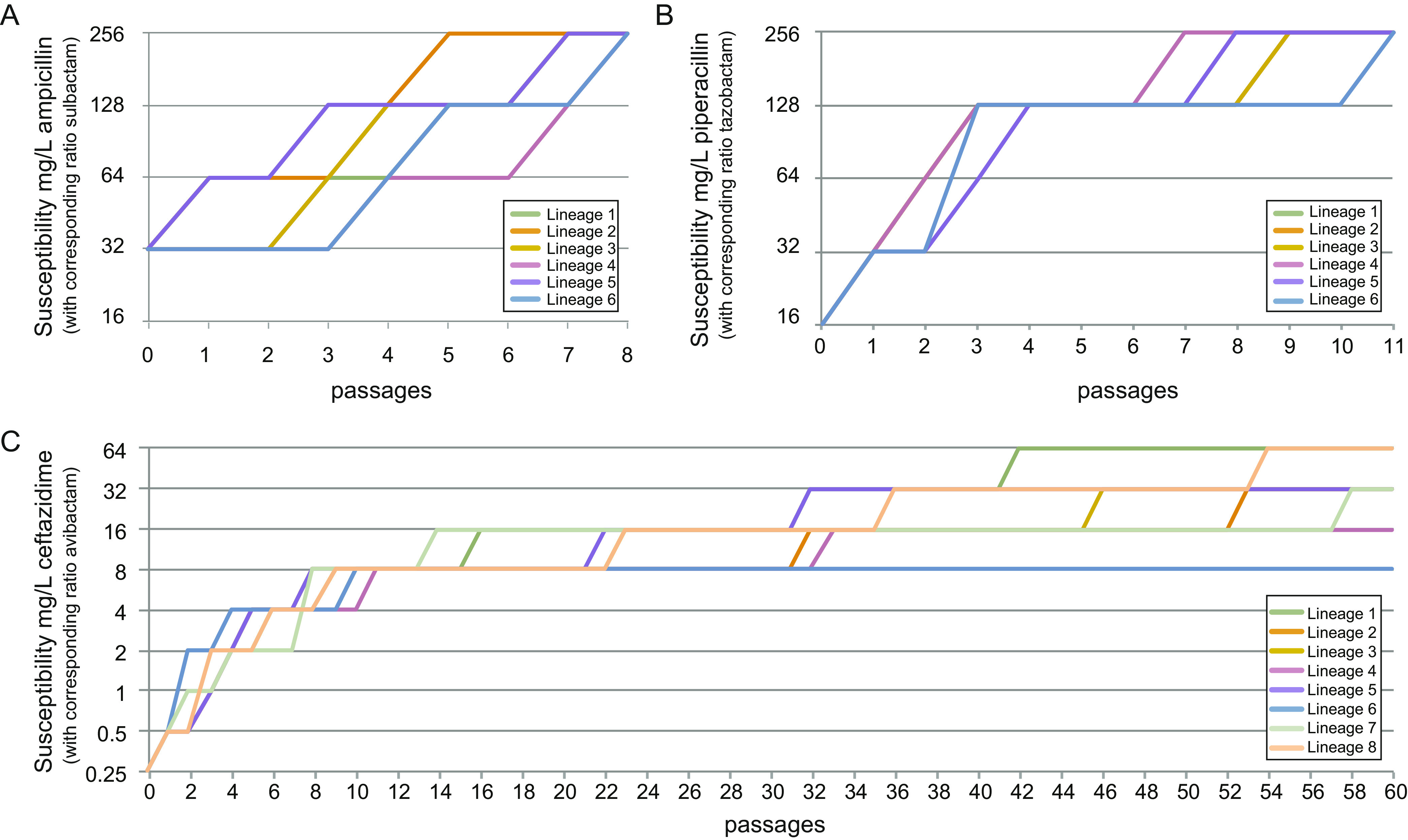
Continuous in vitro evolution at increasing concentrations of the three β-lactam/β-lactamase inhibitor combinations. Curves show the decreases in the susceptibility (MICratio) of a strain carrying pUUH239.2 to SAM (A), TZP (B), and CZA (C).
TABLE 3.
Susceptibility and growth rates of endpoint clones from evolved lineagesa
| Strain | Combination used for selection | Evolution endpoint concn (β-lactam/inhibitor) (mg/L) | Mean growth rate ± SD | MIC (mg/L) |
||
|---|---|---|---|---|---|---|
| SAM | TZP | CZA | ||||
| DA65027 | SAM | 256/128 | 0.46 ± 0.03 | >256 | >256 | 1 |
| DA65028 | 256/128 | 0.52 ± 0.01 | >256 | >256 | 1 | |
| DA65029 | 256/128 | 0.50 ± 0.01 | >256 | >256 | 2 | |
| DA65030 | 256/128 | 0.52 ± 0.04 | >256 | >256 | 2 | |
| DA65031 | 256/128 | 0.62 ± 0.02 | >256 | >256 | 1 | |
| DA65032 | 256/128 | 0.55 ± 0.01 | >256 | >256 | 2 | |
| DA65043 | TZP | 256/32 | 0.59 ± 0.02 | >256 | >256 | 0.25 |
| DA65044 | 256/32 | 0.40 ± 0.01 | >256 | >256 | 0.5 | |
| DA65045 | 256/32 | 0.40 ± 0.01 | >256 | >256 | 0.25 | |
| DA65046 | 256/32 | 0.60 ± 0.04 | >256 | >256 | 0.5 | |
| DA65047 | 256/32 | 0.47 ± 0.05 | >256 | >256 | 0.25 | |
| DA65048 | 256/32 | 0.44 ± 0.01 | >256 | >256 | 4 | |
| DA65159 | CZA | 64/16 | 0.32 ± 0.01 | >256 | 256 | 256 |
| DA65160 | 32/8 | 0.36 ± 0.01 | >256 | >256 | 256 | |
| DA65161 | 32/8 | 0.40 ± 0.02 | >256 | 256 | >256 | |
| DA65162 | 16/4 | 0.41 ± 0.01 | >256 | >256 | 32 | |
| DA65163 | 32/8 | 0.26 ± 0.03 | >256 | >256 | >256 | |
| DA65164 | 8/2 | 0.66 ± 0.01 | >256 | 64 | 16 | |
| DA65165 | 32/8 | 0.41 ± 0.01 | >256 | >256 | >256 | |
| DA65166 | 64/16 | 0.31 ± 0.01 | >256 | 256 | >256 | |
SAM, ampicillin-sulbactam; TZP, piperacillin-tazobactam; CZA, ceftazidime-avibactam. Values in boldface type exceed the clinical breakpoints set by EUCAST. For all combinations, resistance (R) breakpoints were >8 mg/L of the antibiotic with 4 mg/L of the inhibitor.
The endpoint populations from all evolved lineages were whole-genome sequenced, and for a subset, we also sequenced populations at earlier time points, when there were shifts in susceptibility, to determine the order in which mutations occurred. The absolute majority of the identified mutations were either nonsynonymous, disruptive (insertions/deletion), or predicted to affect promoters, indicating strong positive selection (Table S3). Due to the strong antibiotic selection pressure, most mutations are likely linked to resistance, but some mutations were likely due to medium adaptations, such as mutations in malT, as reported previously (41).
For the populations that evolved in the presence of SAM, the main driver of resistance was again β-lactamase gene amplifications. Especially blaCTX-M-15 and blaOXA-1 were amplified, which is in contrast to the one-step selection where exclusively blaTEM-1 was amplified and where the other two β-lactamases were lost from the plasmid in several cases. We also found subsequent mutations in ompR, ompC, and lipopolysaccharide (LPS)-associated genes (rfaC, rfaG, rclR, and galU) (Fig. 5A and Table S3). The TZP-evolved populations also showed extensive amplifications (up to 52-fold) but involving only blaOXA-1. These amplifications were found already in the first cycle, and the copy number increased further during passaging. For TZP, common mutational targets were also genes involved in the respiratory chain, especially cydA and cydB, as well as LPS-associated genes (rfaE, rfaG, and gmhB) (Fig. 5B and Table S3). In addition, two of the mutants displayed duplications of different regions on the chromosome spanning approximately 230 and 600 kb, respectively. Both regions have different genes important for cell envelope integrity, such as penicillin binding proteins (PBPs), and the stress response (for exact positions of the duplications, see Table S3).
FIG 5.
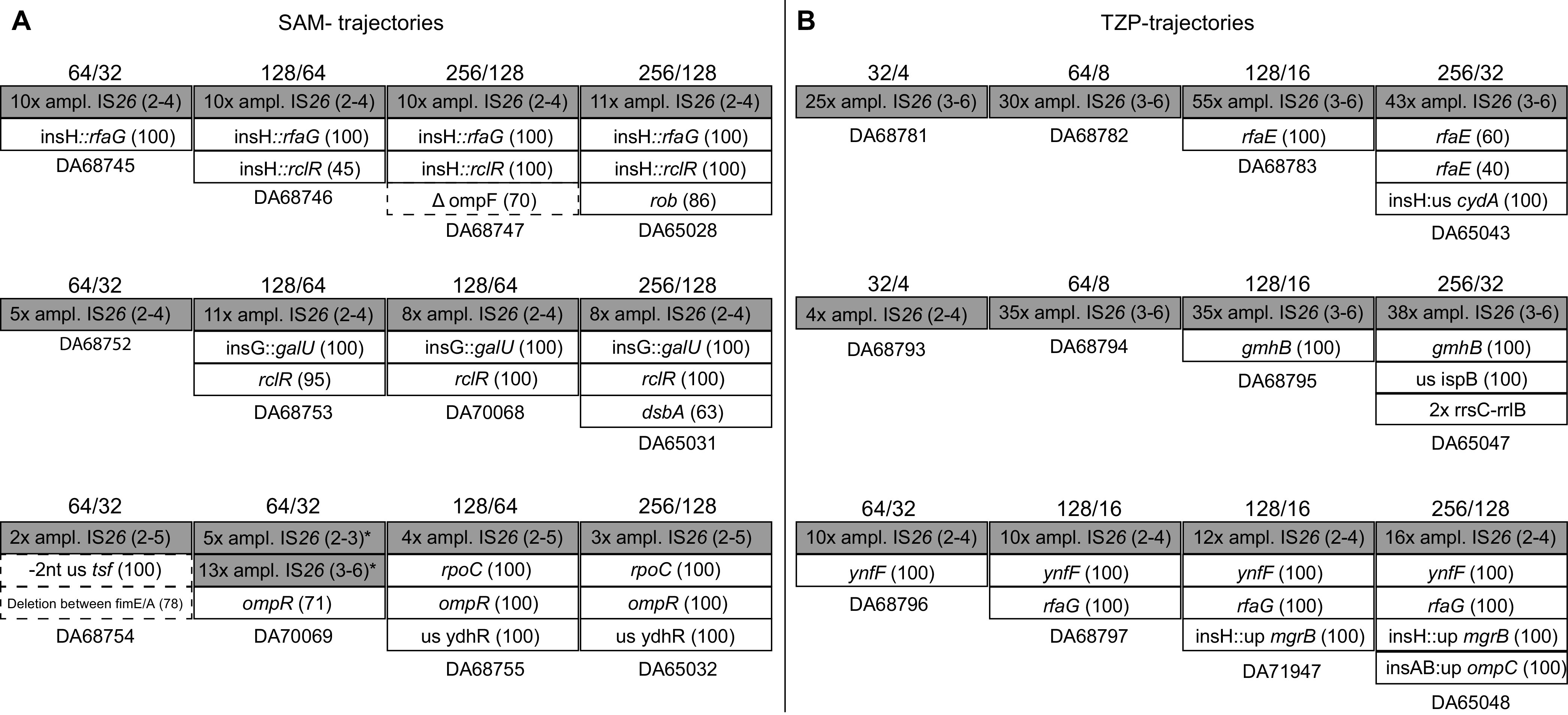
Trajectories of the development of resistance to ampicillin-sulbactam (A), piperacillin-tazobactam (B), and ceftazidime-avibactam (C). The accumulation of mutations over time is plotted from left to right. Numbers above the boxes indicate the concentrations of the antibiotic-inhibitor at which the respective population was selected.
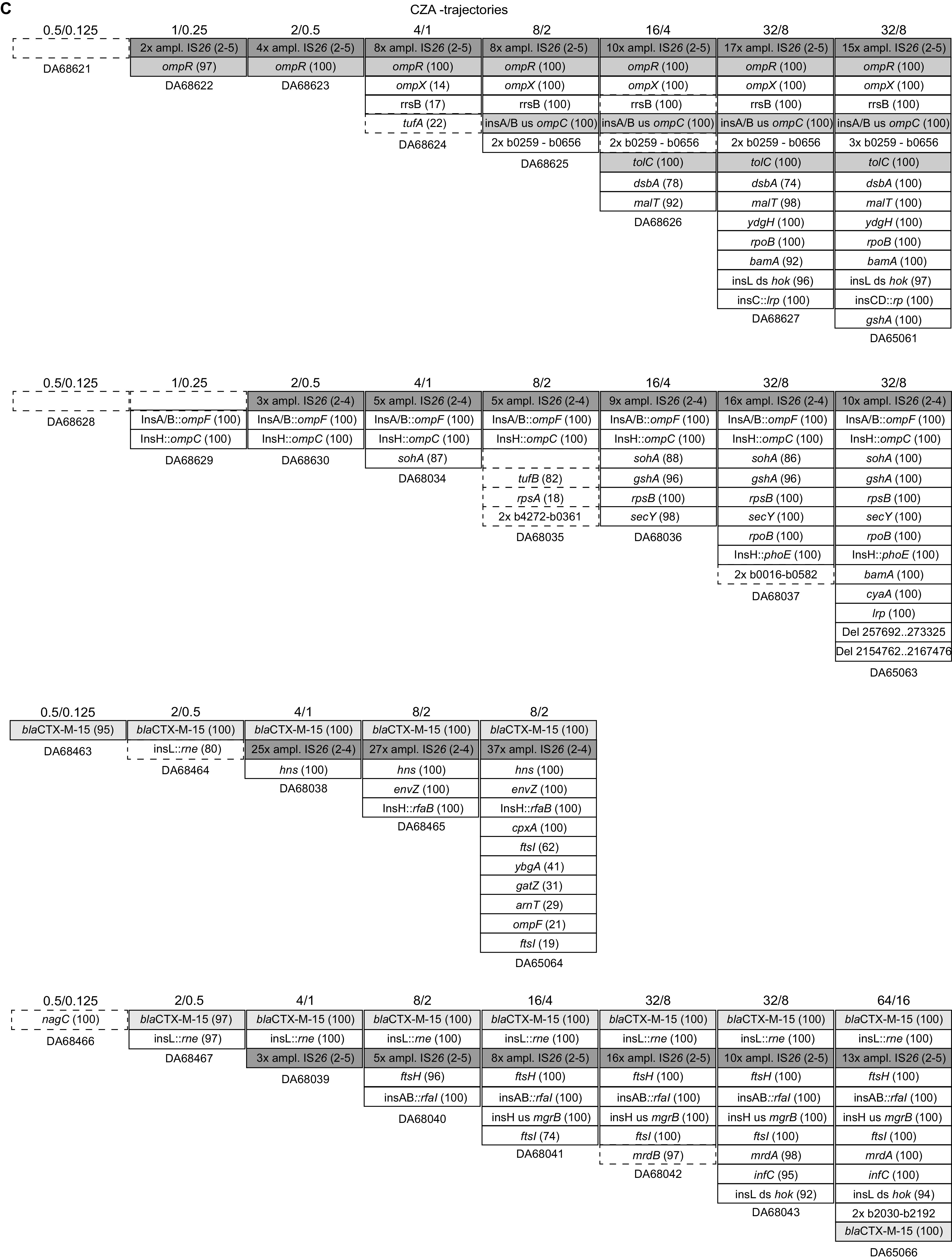
Evolution in the presence of CZA yielded a more complex picture, where high-level resistance depended on the occurrence of multiple mutations (on average, 15 changes per lineage in the end population, including mutations, deletions, and amplifications) (Fig. 5C and Table S3). This is in accordance with the observation that single-step mutants did not yield high-level CZA resistance. The CZA populations showed early amplifications of blaCTX-M-15 and blaOXA-1 and mutations in outer membrane porins (ompF and ompC) and/or their regulators (envZ and ompR). In five of the lineages, the essential gene bamA and/or the nonessential complex partner bamC, responsible for the assembly of outer membrane proteins, acquired mutations, one of which leads to a truncation of the protein. Four of the CZA lineages showed chromosomal amplification of regions similar to those of the TZP lineages. In three lineages, there were also mutations in the penicillin binding protein PBP3 (ftsI), PBP2 (mrdA), or PBP1a (mrcA) at later stages of evolution. Interestingly, as in the one-step selections, mutations in CTX-M-15 arose during evolution in three out of eight lineages. In two of these lineages, the amino acid change P167T occurred early (Table S3). This particular substitution has previously been observed to give enhanced ceftazidime hydrolysis (42). A second amino acid substitution, N132K, a conserved residue for group A β-lactamases (43), occurred in lineage 8 at the last cycle, together with P167T. In one of the lineages, the same mutation arose as that in the one-step selection mutant G238D, this time together with I142V. This displays the potential of altered CTX-M-15 in the evolution toward decreased susceptibility to CZA. The general evolutionary trajectories for CZA resistance seem to first be either amplification of blaCTX-M-15 and blaOXA-1 together with reduced porin expression or mutation of blaCTX-M-15 followed by amplification of the new allele (Fig. 5C). Subsequently, lineages with amplifications/porin loss accumulated a wide variety of different additional mutations, while the amplified CTX-M-15 mutants all yielded mutations in penicillin binding proteins (Fig. 6).
FIG 6.
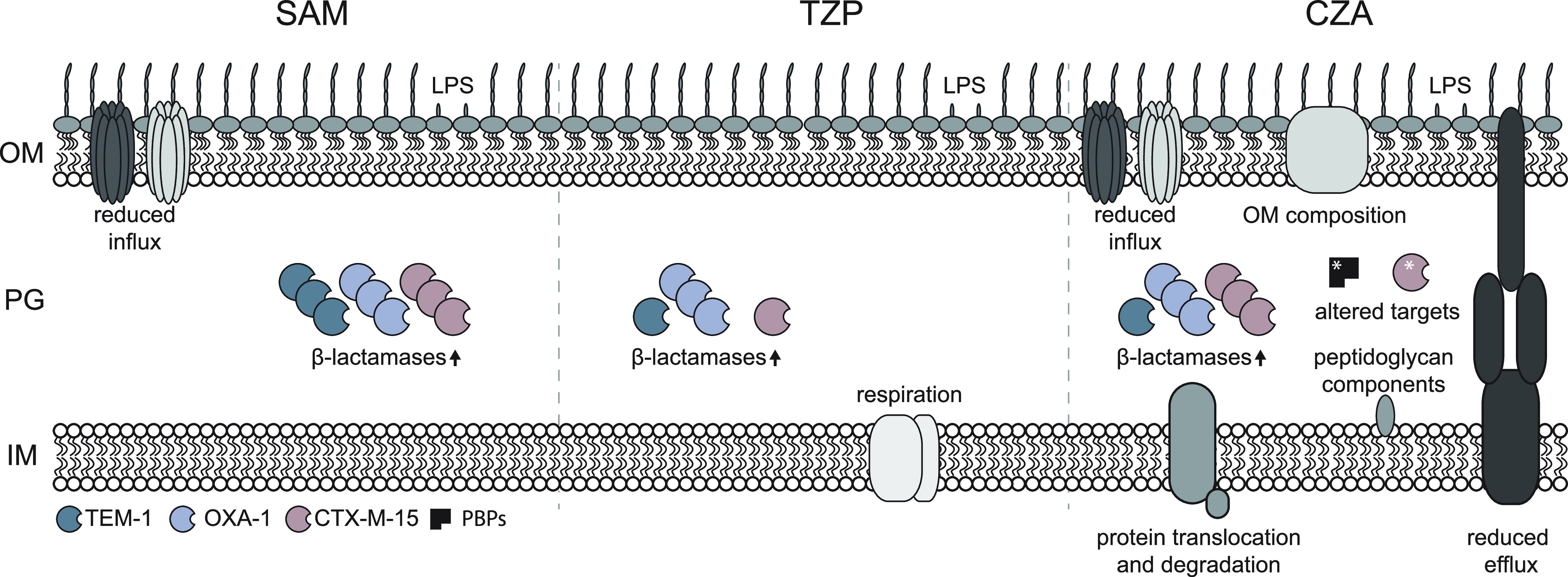
Overview of processes affected in achieving reduced susceptibility to β-lactams and β-lactamase inhibitors. The dominant mechanisms are illustrated for each combination. OM, outer membrane; PG, peptidoglycan; IM, inner membrane.
DISCUSSION
Combination therapy with β-lactam antibiotics and β-lactamase inhibitors has the potential to bring antibiotics back to life where resistance had previously limited the treatment options (44–46). In particular, the newer combination of ceftazidime and the non-β-lactam β-lactamase inhibitor avibactam has attracted great clinical interest. The difference between avibactam and other inhibitors, such as tazobactam and sulbactam, which are so-called suicide inhibitors, is its reversible binding to the β-lactamase (47). The use of these three combinations differs globally due to differences in resistance prevalence, but SAM may be given when traditional β-lactam antibiotics are ineffective but the infecting strain expresses non-extended-spectrum β-lactamases. TZP is instead used for infections caused by ESBL-encoding strains, and CZA is used in treatment regimens where ESBL- or carbapenemase-producing strains are causing the disease (30). However, it is not uncommon that clinical isolates encode multiple different β-lactamases and thereby have a broad spectrum of resistance to this important group of antibiotics. While the effect of, and development of resistance to, β-lactam/β-lactamase inhibitor combinations has been studied for individual β-lactamases, little is known about the effect of multiple enzymes in the same cell. Resistance to SAM has been investigated extensively, where the hyperproduction of, or point mutations in, TEM-1 has been shown to lead to high-level resistance (22, 48, 49). Even though the strain carrying pUUH239.2 is clinically resistant to SAM, by one-step selection, we could see that the amplification of the blaTEM-1 gene increased the resistance level further and at the same time gave cross-resistance to TZP. However, at the end of the short-term evolution experiments with SAM, the populations showed amplifications of blaCTX-M-15 and blaOXA-1, rather than the blaTEM-1 gene, indicating that this path is more successful for the bacterium when competing with other possible mutants. The mutation frequencies in vitro toward all combinations were relatively low, in the range of 10−10 to 10−9, indicative of a rare event or combinations of events. This is surprising given that gene duplications, especially between repetitive sequences such as insertion sequence (IS) elements, are several orders of magnitude more frequent than that (50). The likely explanation is that a duplication of a β-lactamase gene helps the cell to survive initially but is not enough to sustain growth at increased concentrations of the antibiotic-inhibitor unless the cell also manages to further increase the copy number significantly, which is a much rarer event. Gene amplification levels of up to 70-fold are very unlikely to spontaneously exist in the population and instead are most likely the result of additional amplification from initial duplications during the growth of cells on the selection plates. This would also explain why we were not able to select mutants at concentrations higher than 1-fold the MICratio, even though the isolated mutants tolerated much higher concentrations. This means that increased expression of β-lactamases is most likely to evolve at low levels of the drug but can substantially decrease the susceptibility once they have occurred. Previous studies have found the mechanisms of resistance to TZP to be similar to the ones for SAM, where amplifications or promoter mutations of blaTEM-1 or mutations in CTX-M enzymes increase resistance (17, 20, 37). We found strong cross-resistance to TZP by SAM-selected amplifications of blaTEM-1 but no such amplifications during TZP selections. Instead, clinical resistance was reached mainly via amplifications of blaOXA-1 both for the one-step mutants and for populations in the evolution experiments. These mutants also gave cross-resistance to SAM but not CZA. Resistance against CZA previously observed in clinical isolates has been due mainly to point mutations in the carbapenemase genes blaKPC-2 and blaOXA-48 (18, 21, 51), while mutations in blaCTX-M-14 and blaCTX-M-15 have been found to decrease susceptibility but not enough to reach the clinical breakpoint (18, 52). Here, we found two separate but complementary evolutionary pathways to CZA resistance. Initial changes were either a combination of blaCTX-M-15 and blaOXA-1 gene amplifications with loss-of-function mutations in porins or their regulators or initial mutation of blaCTX-M-15 followed by amplification of the new allele, reduced expression of porins, and mutations in penicillin binding proteins. In addition, we found a large number of mutations in genes involved in cell envelope synthesis and integrity at later stages of evolution. Antagonistic pleiotropic effects of the coproduction of different β-lactamases or combinations with other resistance mechanisms have, in view of the frequent occurrence of collateral sensitivity by in vitro-selected mutant CTX-M enzymes, been speculated to limit the in vivo selection of mutant enzymes resistant to inhibitors (37, 53). For combination therapy, β-lactamase enzyme mutations can be predicted to affect either the β-lactam or the inhibitor. The G238D substitution most likely interferes with the binding of avibactam since this mutation led to more susceptibility to ceftazidime alone. The P167T substitution has instead been observed to decrease susceptibility to ceftazidime and therefore also increases resistance to the CZA combination (52). Interestingly the second-residue exchange of one of the CZA mutants, N132K, is at an important site for both β-lactam and avibactam binding, creating a strong hydrogen bond to the substrate (54). This mutation could be a trade-off between being able to hydrolyze the β-lactam and avoiding strong binding of the β-lactamase inhibitor avibactam. Additional studies are needed to more exactly decipher how these mutations affect the catalytic potential of the enzyme.
A general trend for the evolved populations was a decrease in growth compared to the parental strain. This would potentially lead to lower competitive fitness under nonselective conditions. Amplifications of blaCTX-M-15 and blaOXA-1 could be directly associated with an increased cost of gene expression since one particular mutant, DA61262, had a decreased growth rate of about 40%, without additional background mutations. We previously found that amplifications of this region of the resistance cassette confer a fitness cost to the cell and that this cost is coupled to the increased expression of β-lactamases (55, 56). However, amplifications could play a beneficial role as stepping-stones for the bacterium during selective circumstances due to their higher frequency of occurrence than point mutations. In addition, their inherent instability could rapidly revert the cost and thereby also the resistance phenotype and make the strain more competitive again when the selective pressure has been removed. This could result in the phenomenon called heteroresistance, which can be a complicating factor when testing susceptibility in clinical laboratories (57).
To conclude, we find that the combination of different β-lactamases and the amplification of one or several β-lactamase genes are very important features for increased resistance to β-lactam/β-lactamase inhibitor combinations. The relatively high abundances of gene duplications in bacterial populations followed by additional amplifications let them act as stepping-stones toward higher-level, but rarer, resistance mutations. In addition, mutations in blaCTX-M-15 alone could make the bacterium less susceptible to CZA, and this effect was strengthened by the amplification of the mutated allele in combination with reduced drug influx and mutations in other central cell functions. These findings corroborate previous findings of the development of resistance of single β-lactamases to β-lactam/β-lactamase inhibitor combinations and further outline the trajectories of the evolution of resistance against this important group of drugs.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All constructed strains were derived from Escherichia coli MG1655 (see Table S1 in the supplemental material) and grown on Mueller-Hinton agar (MHA; Becton, Dickinson and Company) or in Mueller-Hinton broth (MHB; Becton, Dickinson and Company) with selection by trimethoprim (10 mg/L; Sigma-Aldrich) for the maintenance of the pUUH239.2 plasmid. The β-lactam antibiotics and β-lactamase inhibitors (Sigma-Aldrich) were used at clinical ratios of 2:1 for ampicillin-sulbactam, 8:1 for piperacillin-tazobactam, and 4:1 for ceftazidime-avibactam. Kanamycin (50 mg/L; Sigma-Aldrich) was used for the maintenance of pBAD18 vectors.
Susceptibility testing.
Susceptibility testing was performed by broth microdilution according to EUCAST guidelines (58). For β-lactam/β-lactamase inhibitor combinations, two different MICs were determined, the standard MIC according to EUCAST, with a variable concentration of the antibiotic and a fixed concentration of 4 mg/L of the inhibitor, and an MIC where the concentrations of both the antibiotic and inhibitor were changing but with fixed clinical ratios of 2:1 for SAM, 8:1 for TZP, and 4:1 for CZA, denoted MICratio here. Since arabinose induction is negatively affected by glucose (59), MICs for strains with β-lactamases cloned on pBAD18 expression vectors were measured in tryptone broth at 37°C. Induction from pBAD18 was done by the addition of 0.05% l-arabinose, and 50 mg/L kanamycin was included to select for the plasmid.
Checkerboard susceptibility assay.
Susceptibility to the β-lactam antibiotics at a wider range of β-lactamase inhibitor concentrations was evaluated with a checkerboard assay in 96-well plates. Checkerboard panels were prepared with 2-fold dilution series in MHB with final concentrations of 0.5 to 256 mg/L for ampicillin and piperacillin, 0.062 to 4 mg/L for ceftazidime, 2 to 128 mg/L for sulbactam, 1 to 64 mg/L for tazobactam, and 0.016 to 8 mg/L for avibactam. Cultures of DA5438 (E. coli MG1655) and DA63522 (E. coli MG1655/pUUH239.2) grown overnight were diluted in MHB and adjusted to a final inoculum of 5 × 105 CFU/mL. Plates were incubated at 37°C for 20 to 24 h. The experiments were performed with at least three biological replicates. E. coli ATCC 25922, E. coli ATCC 35218, and K. pneumoniae ATCC 700603 were used as quality control strains according to EUCAST guidelines (58).
In vitro selection of mutants with reduced susceptibility to combinations of β-lactam antibiotics and β-lactamase inhibitors.
Single-step mutants with decreased susceptibility to the antibiotic-inhibitor combinations were selected in fluctuation assays by plating 1 × 107 and 1 × 108 CFU/mL onto plates containing 1×, 2×, and 4× the MICratio of the respective drug combination and incubated overnight at 37°C. The mutant frequency was calculated as r/Nt, where r is the number of mutants (CFU per milliliter) from the selective plate and Nt is the total number of viable cells (CFU per milliliter) from nonselective plates.
Continuous-evolution experiments were performed with eight independent lineages by passaging bacterial cultures at increasing concentrations of the antibiotic-inhibitor with a daily bottleneck of about 107 CFU (1 μL of the culture grown overnight to 1 mL of fresh medium) for SAM and CZA and 106 CFU for TZP. The different bottleneck for TZP was due to observed inoculum effects at 107 CFU. Inocula were transferred to 1×, 2×, and 4× MICratio of the drug combination and incubated overnight. The culture with the highest concentration showing dense growth (visible by the naked eye as for broth microdilution susceptibility testing) was used for the next transfer at additional 1×, 2×, and 4× increases of the concentration in which it was previously grown. Since the culture in the evolution experiment with CZA did not grow immediately at or above the initial MICratio, these lineages were first passaged once at 1/2 MICratio before cycling at increasing concentrations.
Growth rate assay.
Growth rates were measured with four biological replicates at 37°C in MHB using a BioscreenC MBR (Oy Growth Curves Ad Ltd.), taking measurements at an optical density at 600 nm (OD600) every 4 min for 20 h. The maximum exponential growth rate was based on OD600 values of between 0.02 and 0.08. The growth rates of the mutants were calculated relative to that of the parental strain.
Determination of β-lactamase transcription levels.
The relative transcription levels of the β-lactamase genes blaTEM-1, blaOXA-1, and blaCTX-M-15 were measured for five mutants using real-time quantitative PCR (RT-qPCR). Cultures of three biological replicates grown overnight were diluted 1:500 in MHB and grown with antibiotic-inhibitor selection to an OD600 of 0.15. Cultures were added at a 1:2 ratio to RNAprotect reagent (Qiagen). RNA extraction was performed using the RNeasy minikit (Qiagen) according to the manufacturer’s instructions. DNA was removed from samples using the Turbo DNA-free kit (Ambion). A total of 500 ng RNA was used as the template for cDNA synthesis using a high-capacity reverse transcription kit (Applied Biosystems) according to the manufacturer’s instructions. One hundred fifty microliters of water was added to the cDNA, and the sample was diluted (1:10, 1:100, and 1:1,000). The diluted cDNA was used as the template for RT-qPCR using the MiniOpticon real-time PCR system (Bio-Rad, Hercules, CA). The chromosomal housekeeping genes hcaT and cysG were used as references. Primers used for the β-lactamase and housekeeping genes are listed in Table S2. The transcription levels were calculated as 2ΔC, where ΔC = CTref − CTtarget gene. The transcription levels of the mutants were calculated relative to those of the parental strain DA63522.
Whole-genome resequencing.
Genomic DNA was obtained with the Epicentre MasterPure DNA purification kit (Illumina Inc.). Samples were prepared with 2× 300-bp paired-end read lengths using the Nextera XT DNA library preparation kit (Illumina Inc.) according to the manufacturer’s instructions. The DNA fragment size distribution was confirmed using the Agilent high-sensitivity D1000 ScreenTape system (Agilent Technologies). A MiSeq desktop sequencer was used according to the manufacturer’s instructions (Illumina Inc.) to perform sequencing. The data were analyzed using the CLC Genomics Workbench version 21.1 (CLCbio, Qiagen). An in-house reference sequence of the parental strain, including the pUUH239.2 plasmid (GenBank accession number NC_016966), was used for the reference assembly of sequencing reads from the isolated mutants. Single nucleotide polymorphisms (SNPs), structural variations, and indels were determined using the respective tools in CLC Genomics Workbench. All genetic changes were also manually inspected, and for rearrangements (deletions, duplications, and inversions), de novo assembly and reference coverage analyses were used to identify the respective changes and measure coverage for amplified regions. For population sequence analysis, a cutoff of 20% was set as the lowest detection level for genetic variations.
Data availability.
Raw data from sequencing have been deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA798964.
ACKNOWLEDGMENTS
This study was supported by grants from the Swedish Research Council of Medicine and Health (2012-1511), the Carl Tryggers Stiftelse (CTS16:395), and the Åke Wibergs Stiftelse to L.S.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Bush K, Bradford PA. 2019. Interplay between β-lactamases and new β-lactamase inhibitors. Nat Rev Microbiol 17:295–306. 10.1038/s41579-019-0159-8. [DOI] [PubMed] [Google Scholar]
- 2.Jacoby GA, Munoz-Price LS. 2005. The new β-lactamases. N Engl J Med 352:380–391. 10.1056/NEJMra041359. [DOI] [PubMed] [Google Scholar]
- 3.Castanheira M, Simner PJ, Bradford PA. 2021. Extended-spectrum β-lactamases: an update on their characteristics, epidemiology and detection. JAC Antimicrob Resist 3:dlab092. 10.1093/jacamr/dlab092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bush K, Bradford PA. 2020. Epidemiology of β-lactamase-producing pathogens. Clin Microbiol Rev 33:e00047-19. 10.1128/CMR.00047-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gniadkowski M. 2008. Evolution of extended-spectrum β-lactamases by mutation. Clin Microbiol Infect 14:11–32. 10.1111/j.1469-0691.2007.01854.x. [DOI] [PubMed] [Google Scholar]
- 6.Palzkill T. 2018. Structural and mechanistic basis for extended-spectrum drug-resistance mutations in altering the specificity of TEM, CTX-M, and KPC β-lactamases. Front Mol Biosci 5:16. 10.3389/fmolb.2018.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans BA, Amyes SGB. 2014. OXA β-lactamases. Clin Microbiol Rev 27:241–263. 10.1128/CMR.00117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carcione D, Siracusa C, Sulejmani A, Leoni V, Intra J. 2021. Old and new beta-lactamase inhibitors: molecular structure, mechanism of action, and clinical use. Antibiotics (Basel) 10:995. 10.3390/antibiotics10080995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drawz SM, Bonomo RA. 2010. Three decades of β-lactamase inhibitors. Clin Microbiol Rev 23:160–201. 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bush K. 1988. Beta-lactamase inhibitors from laboratory to clinic. Clin Microbiol Rev 1:109–123. 10.1128/CMR.1.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bush K. 2010. Alarming β-lactamase-mediated resistance in multidrug-resistant Enterobacteriaceae. Curr Opin Microbiol 13:558–564. 10.1016/j.mib.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Butler MS, Paterson DL. 2020. Antibiotics in the clinical pipeline in October 2019. J Antibiot (Tokyo) 73:329–364. 10.1038/s41429-020-0291-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papp-Wallace KM. 2019. The latest advances in β-lactam/β-lactamase inhibitor combinations for the treatment of Gram-negative bacterial infections. Expert Opin Pharmacother 20:2169–2184. 10.1080/14656566.2019.1660772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.World Health Organization. 2021. 2020 antibacterial agents in clinical and preclinical development: an overview and analysis. World Health Organization, Geneva, Switzerland. https://www.who.int/publications-detail-redirect/9789240021303. Accessed 26 October 2021. [Google Scholar]
- 15.Brem J, Panduwawala T, Hansen JU, Hewitt J, Liepins E, Donets P, Espina L, Farley AJM, Shubin K, Campillos GG, Kiuru P, Shishodia S, Krahn D, Leśniak RK, Schmidt Adrian J, Calvopiña K, Turrientes M-C, Kavanagh ME, Lubriks D, Hinchliffe P, Langley GW, Aboklaish AF, Eneroth A, Backlund M, Baran AG, Nielsen EI, Speake M, Kuka J, Robinson J, Grinberga S, Robinson L, McDonough MA, Rydzik AM, Leissing TM, Jimenez-Castellanos JC, Avison MB, Da Silva Pinto S, Pannifer AD, Martjuga M, Widlake E, Priede M, Hopkins Navratilova I, Gniadkowski M, Belfrage AK, Brandt P, Yli-Kauhaluoma J, Bacque E, Page MGP, Björkling F, Tyrrell JM, Spencer J, Lang PA, et al. 2022. Imitation of β-lactam binding enables broad-spectrum metallo-β-lactamase inhibitors. Nat Chem 14:15–24. 10.1038/s41557-021-00831-x. [DOI] [PubMed] [Google Scholar]
- 16.Hubbard ATM, Mason J, Roberts P, Parry CM, Corless C, van Aartsen J, Howard A, Bulgasim I, Fraser AJ, Adams ER, Roberts AP, Edwards T. 2020. Piperacillin/tazobactam resistance in a clinical isolate of Escherichia coli due to IS26-mediated amplification of blaTEM-1B. Nat Commun 11:4915. 10.1038/s41467-020-18668-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou K, Tao Y, Han L, Ni Y, Sun J. 2019. Piperacillin-tazobactam (TZP) resistance in Escherichia coli due to hyperproduction of TEM-1 β-lactamase mediated by the promoter Pa/Pb. Front Microbiol 10:833. 10.3389/fmicb.2019.00833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Both A, Büttner H, Huang J, Perbandt M, Belmar Campos C, Christner M, Maurer FP, Kluge S, König C, Aepfelbacher M, Wichmann D, Rohde H. 2017. Emergence of ceftazidime/avibactam non-susceptibility in an MDR Klebsiella pneumoniae isolate. J Antimicrob Chemother 72:2483–2488. 10.1093/jac/dkx179. [DOI] [PubMed] [Google Scholar]
- 19.Bret L, Chaibi EB, Chanal-Claris C, Sirot D, Labia R, Sirot J. 1997. Inhibitor-resistant TEM (IRT) beta-lactamases with different substitutions at position 244. Antimicrob Agents Chemother 41:2547–2549. 10.1128/AAC.41.11.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schechter LM, Creely DP, Garner CD, Shortridge D, Nguyen H, Chen L, Hanson BM, Sodergren E, Weinstock GM, Dunne WM, van Belkum A, Leopold SR. 2018. Extensive gene amplification as a mechanism for piperacillin-tazobactam resistance in Escherichia coli. mBio 9:e00583-18. 10.1128/mBio.00583-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shields RK, Chen L, Cheng S, Chavda KD, Press EG, Snyder A, Pandey R, Doi Y, Kreiswirth BN, Nguyen MH, Clancy CJ. 2017. Emergence of ceftazidime-avibactam resistance due to plasmid-borne blaKPC-3 mutations during treatment of carbapenem-resistant Klebsiella pneumoniae infections. Antimicrob Agents Chemother 61:e02097-16. 10.1128/AAC.02097-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez JL, Cercenado E, Rodriguez-Creixems M, Vincente-Perez MF, Delgado-Iribarren A, Baquero F. 1987. Resistance to beta-lactam/clavulanate. Lancet 330:1473. 10.1016/S0140-6736(87)91180-9. [DOI] [PubMed] [Google Scholar]
- 23.Reguera JA, Baquero F, Pérez-Díaz JC, Martínez JL. 1991. Factors determining resistance to β-lactam combined with β-lactamase inhibitors in Escherichia coli. J Antimicrob Chemother 27:569–575. 10.1093/jac/27.5.569. [DOI] [PubMed] [Google Scholar]
- 24.Blazquez J, Baquero MR, Canton R, Alos I, Baquero F. 1993. Characterization of a new TEM-type beta-lactamase resistant to clavulanate, sulbactam, and tazobactam in a clinical isolate of Escherichia coli. Antimicrob Agents Chemother 37:2059–2063. 10.1128/AAC.37.10.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baquero F, Martínez JL, Novais Â, Rodríguez-Beltrán J, Martínez-García L, Coque TM, Galán JC. 2021. Allogenous selection of mutational collateral resistance: old drugs select for new resistance within antibiotic families. Front Microbiol 12:757833. 10.3389/fmicb.2021.757833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lytsy B, Sandegren L, Tano E, Torell E, Andersson DI, Melhus Å. 2008. The first major extended-spectrum β-lactamase outbreak in Scandinavia was caused by clonal spread of a multiresistant Klebsiella pneumoniae producing CTX-M-15. APMIS 116:302–308. 10.1111/j.1600-0463.2008.00922.x. [DOI] [PubMed] [Google Scholar]
- 27.Sandegren L, Linkevicius M, Lytsy B, Melhus Å, Andersson DI. 2012. Transfer of an Escherichia coli ST131 multiresistance cassette has created a Klebsiella pneumoniae-specific plasmid associated with a major nosocomial outbreak. J Antimicrob Chemother 67:74–83. 10.1093/jac/dkr405. [DOI] [PubMed] [Google Scholar]
- 28.Tamma PD, Aitken SL, Bonomo RA, Mathers AJ, van Duin D, Clancy CJ. 2020. Infectious Diseases Society of America guidance on the treatment of antimicrobial resistant Gram-negative infections. Infectious Diseases Society of America, Arlington, VA. https://www.idsociety.org/practice-guideline/amr-guidance/. Accessed 26 October 2021. [Google Scholar]
- 29.Tamma PD, Rodriguez-Baňo J. 2017. The use of noncarbapenem β-lactams for the treatment of extended-spectrum β-lactamase infections. Clin Infect Dis 64:972–980. 10.1093/cid/cix034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paul M, Carrara E, Retamar P, Tängdén T, Bitterman R, Bonomo RA, de Waele J, Daikos GL, Akova M, Harbarth S, Pulcini C, Garnacho-Montero J, Seme K, Tumbarello M, Lindemann PC, Gandra S, Yu Y, Bassetti M, Mouton JW, Tacconelli E, Rodríguez-Baño J. 2022. European Society of Clinical Microbiology and Infectious Diseases (ESCMID) guidelines for the treatment of infections caused by multidrug-resistant Gram-negative bacilli (endorsed by European Society of Intensive Care Medicine). Clin Microbiol Infect 28:521–547. 10.1016/j.cmi.2021.11.025. [DOI] [PubMed] [Google Scholar]
- 31.Harris PNA, Tambyah PA, Paterson DL. 2015. β-Lactam and β-lactamase inhibitor combinations in the treatment of extended-spectrum β-lactamase producing Enterobacteriaceae: time for a reappraisal in the era of few antibiotic options? Lancet Infect Dis 15:475–485. 10.1016/S1473-3099(14)70950-8. [DOI] [PubMed] [Google Scholar]
- 32.Rodríguez-Baño J, Navarro MD, Retamar P, Picón E, Pascual Á, Extended-Spectrum Beta-Lactamases-Red Española de Investigación en Patología Infecciosa/Grupo de Estudio de Infección Hospitalaria Group . 2012. β-Lactam/β-lactam inhibitor combinations for the treatment of bacteremia due to extended-spectrum β-lactamase-producing Escherichia coli: a post hoc analysis of prospective cohorts. Clin Infect Dis 54:167–174. 10.1093/cid/cir790. [DOI] [PubMed] [Google Scholar]
- 33.Ripa S, Ferrante L, Prenna M. 1990. Pharmacokinetics of sulbactam/ampicillin in humans after intravenous and intramuscular injection. Chemotherapy 36:185–192. 10.1159/000238765. [DOI] [PubMed] [Google Scholar]
- 34.Das S, Li J, Armstrong J, Learoyd M, Edeki T. 2015. Randomized pharmacokinetic and drug-drug interaction studies of ceftazidime, avibactam, and metronidazole in healthy subjects. Pharmacol Res Perspect 3:e00172. 10.1002/prp2.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berkhout J, Melchers MJ, van Mil AC, Nichols WW, Mouton JW. 2015. In vitro activity of ceftazidime-avibactam combination in in vitro checkerboard assays. Antimicrob Agents Chemother 59:1138–1144. 10.1128/AAC.04146-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shapiro AB. 2017. Kinetics of sulbactam hydrolysis by β-lactamases, and kinetics of β-lactamase inhibition by sulbactam. Antimicrob Agents Chemother 61:e01612-17. 10.1128/AAC.01612-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ripoll A, Baquero F, Novais Â, Rodríguez-Domínguez MJ, Turrientes M-C, Cantón R, Galán J-C. 2011. In vitro selection of variants resistant to β-lactams plus β-lactamase inhibitors in CTX-M β-lactamases: predicting the in vivo scenario? Antimicrob Agents Chemother 55:4530–4536. 10.1128/AAC.00178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Adler M, Anjum M, Andersson DI, Sandegren L. 2016. Combinations of mutations in envZ, ftsI, mrdA, acrB and acrR can cause high-level carbapenem resistance in Escherichia coli. J Antimicrob Chemother 71:1188–1198. 10.1093/jac/dkv475. [DOI] [PubMed] [Google Scholar]
- 39.Ambler RP, Coulson AF, Frère JM, Ghuysen JM, Joris B, Forsman M, Levesque RC, Tiraby G, Waley SG. 1991. A standard numbering scheme for the class A beta-lactamases. Biochem J 276:269–270. 10.1042/bj2760269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faheem M, Rehman MT, Danishuddin M, Khan AU. 2013. Biochemical characterization of CTX-M-15 from Enterobacter cloacae and designing a novel non-β-lactam-β-lactamase inhibitor. PLoS One 8:e56926. 10.1371/journal.pone.0056926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knöppel A, Knopp M, Albrecht LM, Lundin E, Lustig U, Näsvall J, Andersson DI. 2018. Genetic adaptation to growth under laboratory conditions in Escherichia coli and Salmonella enterica. Front Microbiol 9:756. 10.3389/fmicb.2018.00756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stepanova MN, Pimkin M, Nikulin AA, Kozyreva VK, Agapova ED, Edelstein MV. 2008. Convergent in vivo and in vitro selection of ceftazidime resistance mutations at position 167 of CTX-M-3 β-lactamase in hypermutable Escherichia coli strains. Antimicrob Agents Chemother 52:1297–1301. 10.1128/AAC.01060-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Philippon A, Slama P, Dény P, Labia R. 2016. A structure-based classification of class a β-lactamases, a broadly diverse family of enzymes. Clin Microbiol Rev 29:29–57. 10.1128/CMR.00019-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKinnon PS, Paladino JA, Grayson ML, Gibbons GW, Karchmer AW. 1997. Cost-effectiveness of ampicillin/sulbactam versus imipenem/cilastatin in the treatment of limb-threatening foot infections in diabetic patients. Clin Infect Dis 24:57–63. 10.1093/clinids/24.1.57. [DOI] [PubMed] [Google Scholar]
- 45.Polk HC, Jr, Fink MP, Laverdiere M, Wilson SE, Garber GE, Barie PS, Hebert JC, Cheadle WG. 1993. Prospective randomized study of piperacillin/tazobactam therapy of surgically treated intra-abdominal infection. The Piperacillin/Tazobactam Intra-Abdominal Infection Study Group. Am Surg 59:598–605. [PubMed] [Google Scholar]
- 46.Niinikoski J, Havia T, Alhava E, Pääkkönen M, Miettinen P, Kivilaakso E, Haapiainen R, Matikainen M, Laitinen S. 1993. Piperacillin/tazobactam versus imipenem/cilastatin in the treatment of intra-abdominal infections. Surg Gynecol Obstet 176:255–261. [PubMed] [Google Scholar]
- 47.Shirley M. 2018. Ceftazidime-avibactam: a review in the treatment of serious Gram-negative bacterial infections. Drugs 78:675–692. 10.1007/s40265-018-0902-x. [DOI] [PubMed] [Google Scholar]
- 48.Waltner-Toews RI, Paterson DL, Qureshi ZA, Sidjabat HE, Adams-Haduch JM, Shutt KA, Jones M, Tian G-B, Pasculle AW, Doi Y. 2011. Clinical characteristics of bloodstream infections due to ampicillin-sulbactam-resistant, non-extended-spectrum-β-lactamase-producing Escherichia coli and the role of TEM-1 hyperproduction. Antimicrob Agents Chemother 55:495–501. 10.1128/AAC.00797-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou XY, Bordon F, Sirot D, Kitzis MD, Gutmann L. 1994. Emergence of clinical isolates of Escherichia coli producing TEM-1 derivatives or an OXA-1 beta-lactamase conferring resistance to beta-lactamase inhibitors. Antimicrob Agents Chemother 38:1085–1089. 10.1128/AAC.38.5.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sandegren L, Andersson DI. 2009. Bacterial gene amplification: implications for the evolution of antibiotic resistance. Nat Rev Microbiol 7:578–588. 10.1038/nrmicro2174. [DOI] [PubMed] [Google Scholar]
- 51.Fröhlich C, Sørum V, Thomassen AM, Johnsen PJ, Leiros H-KS, Samuelsen Ø. 2019. OXA-48-mediated ceftazidime-avibactam resistance is associated with evolutionary trade-offs. mSphere 4:e00024-19. 10.1128/mSphere.00024-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Compain F, Dorchène D, Arthur M. 2018. Combination of amino acid substitutions leading to CTX-M-15-mediated resistance to the ceftazidime-avibactam combination. Antimicrob Agents Chemother 62:e00357-18. 10.1128/AAC.00357-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ripoll A, Galán J-C, Rodríguez C, Tormo N, Gimeno C, Baquero F, Martínez-Martínez L, Cantón R, SEIMC Quality Control Study Group . 2014. Detection of resistance to beta-lactamase inhibitors in strains with CTX-M beta-lactamases: a multicenter external proficiency study using a well-defined collection of Escherichia coli strains. J Clin Microbiol 52:122–129. 10.1128/JCM.02340-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lahiri SD, Bradford PA, Nichols WW, Alm RA. 2016. Structural and sequence analysis of class A β-lactamases with respect to avibactam inhibition: impact of Ω-loop variations. J Antimicrob Chemother 71:2848–2855. 10.1093/jac/dkw248. [DOI] [PubMed] [Google Scholar]
- 55.Adler M, Anjum M, Berg OG, Andersson DI, Sandegren L. 2014. High fitness costs and instability of gene duplications reduce rates of evolution of new genes by duplication-divergence mechanisms. Mol Biol Evol 31:1526–1535. 10.1093/molbev/msu111. [DOI] [PubMed] [Google Scholar]
- 56.Rajer F, Sandegren L. 2022. The role of antibiotic resistance genes in the fitness cost of multiresistance plasmids. mBio 13:e03552-21. 10.1128/mbio.03552-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nicoloff H, Hjort K, Levin BR, Andersson DI. 2019. The high prevalence of antibiotic heteroresistance in pathogenic bacteria is mainly caused by gene amplification. Nat Microbiol 4:504–514. 10.1038/s41564-018-0342-0. [DOI] [PubMed] [Google Scholar]
- 58.European Committee on Antimicrobial Susceptibility Testing. 2021. Breakpoint tables for interpretation of MICs and zone diameters, version 11.0. https://eucast.org/clinical_breakpoints/. Accessed 26 October 2021.
- 59.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1 and S2. Download aac.00290-22-s0001.pdf, PDF file, 0.07 MB (73.2KB, pdf)
Table S3. Download aac.00290-22-s0002.xlsx, XLSX file, 0.06 MB (60.5KB, xlsx)
Data Availability Statement
Raw data from sequencing have been deposited in the NCBI Sequence Read Archive under BioProject accession number PRJNA798964.