

Abstract
Objectives
Variants of a coronavirus (SARS-CoV-2) have been spreading in a global pandemic. Improved understanding of the infectivity of future new variants is important so that effective countermeasures against them can be quickly undertaken. In our research reported here, we aimed to predict the infectivity of SARS-CoV-2 by using a mathematical model with molecular simulation analysis, and we used phylogenetic analysis to determine the evolutionary distance of the spike protein gene (S gene) of SARS-CoV-2.
Methods
We subjected the six variants and the wild type of spike protein and human angiotensin-converting enzyme 2 (ACE2) to molecular docking simulation analyses to understand the binding affinity of spike protein and ACE2. We then utilized regression analysis of the correlation coefficient of the mathematical model and the infectivity of SARS-CoV-2 to predict infectivity.
Results
The evolutionary distance of the S gene correlated with the infectivity of SARS-CoV-2 variants. The calculated biding affinity for the mathematical model obtained with results of molecular docking simulation also correlated with the infectivity of SARS-CoV-2 variants. These results suggest that the data from the docking simulation for the receptor binding domain of variant spike proteins and human ACE2 were valuable for prediction of SARS-CoV-2 infectivity.
Conclusion
We developed a mathematical model for prediction of SARS-CoV-2 variant infectivity by using binding affinity obtained via molecular docking and the evolutionary distance of the S gene.
Keywords: SARS-CoV-2, COVID-19, Infectivity, Spike protein, Binding affinity, Mathematical model
Abbreviations
- spike protein gene
S gene
- angiotensin-converting enzyme 2
ACE2
- receptor-binding domain
RBD
- molecular dynamics
MD
- Protein Data Bank
PDB
1. Introduction
Variants of the novel coronavirus (SARS-CoV-2 [severe acute respiratory syndrome coronavirus 2]) that are responsible for the worldwide pandemic known as COVID-19 have led to difficulties in enacting countermeasures against infection, because many variants have occurred in succession (Bar-On et al., 2020; Cao et al., 2021; Tao et al., 2021). Infection control methods for SARS-CoV-2 have mainly included wearing face masks, avoiding close contact with other people (such as via lockdowns in urban areas), and providing multiple injections of vaccines (Adil et al., 2021).
The infectivity of SARS-CoV-2 variants has been rapidly changing, so understanding the infectivity of new variants is important for effective responses to the pandemic (Singh et al., 2021). These changes have resulted in higher infectivities of new SARS-CoV-2 variants compared with past variants (Allen et al., 2022; Araf et al., 2022; Chen and Wei, 2022; Davies et al., 2021; Faria et al., 2021; Radvak et al., 2021). These alterations in infectivity indicate that the infectivity of new coronavirus variants may be estimated by utilizing the evolutionary distance of the spike protein gene (S gene) between the wild type and the variants. We previously described a mathematical model in which we used docking simulation results to predict UDP-glucuronosyltransferase 1A1 conjugation capacity (Takaoka et al., 2019). By using the molecular docking simulation analyses, we found a plant leaf extract that inhibited the binding of SARS‑CoV‑2 spike protein to angiotensin-converting enzyme 2 (ACE2) (Hagiyama et al., 2022). Similarly, in silico docking data for a variant of SARS-CoV-2 spike protein and ACE2 may be used to estimate the infectivity of the new variant. In this research here, we choose these two approaches with mathematical models to achieve rapid and better understanding of the infectivity of new SARS-CoV-2 variants.
2. Materials and methods
2.1. Determination of the evolutionary distance between wild-type and variant s genes and infectivities of the variants
We chose six variants of SARS-CoV-2—alpha (Claro et al., 2021), beta (Tegally et al., 2021), gamma (Loconsole et al., 2021), delta (Cherian et al., 2021), omicron BA.1 (Dejnirattisai et al., 2021), and omicron BA.2 (Christensen et al., 2022)]—for this research. Table 1 shows the amino acid substitution of spike protein variants. We used the multiple sequence alignment method for the S gene of the SARS-CoV-2 variants, whose nucleotide sequences were obtained from NCBI (Accession no: MN908947) (National Library of Medicine US, 2022), to perform an analysis via the ClustalW program (Thompson et al., 1994). FastTree (Price et al., 2009) with default parameters then provided the phylogenetic tree and evolutionary distances between each mutant and wild type (Kimura and Ota, 1972). The infectivity of each SARS-CoV-2 variant was obtained from previous research, and we summarized these data and developed an infectivity index for our research (Allen et al., 2022; Araf et al., 2022; Chen and Wei, 2022; Davies et al., 2021; Faria et al., 2021; Radvak et al., 2021).
Table 1.
Amino acid substitutions of spike proteins of SARS-CoV-2 variants.
| SARS-CoV-2 strain | Mutations in spike protein |
|---|---|
| Alpha (B.1.1.7) | H69-V70del, Y144del, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H |
| Beta (B.1.351) | D80A, D215G, L241-A243del, K417N, E484K, N501Y, D614G, A701V |
| Gamma (P.1) | L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I, V1176F |
| Delta (B.1.617.2) | T19R, E156-F157del, R158G, L452R, T478K, D614G, P681R, D950N |
| Omicron BA.1 (B.1.1.529/BA.1) | A67V, H69-V70del, T95I, G142D, V143-Y145del, N211del, L212I, ins214EPE, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F |
| Omicron BA.2 (B.1.1.529/BA.2) | T19I, L24-P26del, A27S, G142D, V213G, G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H, D614G, H655Y, N679K, P681H, N764K, D796Y, Q954H, N969K |
Bold, amino acids in RBD. The information of amino acid substitutions are obtained from the following sources: Alpha, Beta and Gamma, https://covdb.stanford.edu/variants/; Delta, https://covariants.org/variants/21A.Delta; Omicron BA.1, https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-omicron-variant.html; Omicron BA.2, Perumal et al. (J Med Virol, DOI: 10.1002/jmv.27601, 2022).
2.2. Analysis of the three-dimensional structures of SARS-CoV-2 spike protein, analysis of docking of the receptor-binding domain (RBD) and ACE2 protein, and development of a mathematical model
The amino acid sequence of wild-type SARS-CoV-2 spike protein was obtained from UniProt (UniProt ID: P0DTC2) (UniProt Consortium, 2018). The three-dimensional structure of SARS-CoV-2 spike protein (Protein Data Bank [PDB] ID: 6ZGG) (Berman et al., 2000), which is an open state trimer, was used as the template for homology modeling of wild-type and variant SARS-CoV-2 spike proteins. With the amino acid sequences and the template structure, we used Molecular Operating Environment software (Chemical Computing Group, Montreal, Quebec, Canada) to perform the homology modeling. The model structures were then subjected to structural optimization with molecular dynamics (MD) simulation, as described in our previous report (Takaoka et al., 2021). After 10,000-ps MD simulation of each spike protein, docking analysis with ACE2 was performed as follows: The ACE2 structure was obtained from the PDB (PDB ID: 6M0J), an X-ray crystal RBD and ACE2 complex with a high quality among the reported ones (other PDB ID: 6VW1, 6LZG). To reduce the search space for docking, a partial structure of the RBD in the “up” conformation was cut from our simulated structure of the SARS-CoV-2 spike protein trimer. Docking sites were defined on the basis of the crystal structure of SARS-CoV-2 spike protein RBD bound with ACE2 (PDB ID: 6M0J). Two thousand docking runs were performed for each variant by using ZDOCK software (University of Massachusetts Medical School, Worcester, MA) with the following parameters as the docking site: amino acid no 401, 430, 433, 437, 439, 440, 459, 470, 471, 473, 477, 480, 482, 484–486, 489 for RBD and amino acid no 24, 27, 28, 30, 31, 34, 35, 37, 38, 41, 42, 79, 82, 83, 330, 353–355, 357, 393 for ACE2, respectively. To determine the stable complex group among the docking runs, the ZDOCK scores of the resulting complexes were clustered by using the group average clustering algorithm. The upper tail rule Mojena (1977) was applied to determine the number of clusters. We used the most stable ZDOCK score in the docking runs and the average ZDOCK score of the most stable complex group except the one with the most stable score as the binding affinity to derive a mathematical model according to our previous research (Takaoka et al., 2019).
To verify our mathematical model, we used leave-one-out cross-validation for infectivity of SARS-CoV-2 as follows: We removed one of the SARS-CoV-2 strains, determined the constants of the mathematical model by using the rest of the five strains, and then performed calculations to predict the infectivity for the excluded strain. We then confirmed the correlation between the reported infectivity and the results of our cross-validation.
Fig. 1 provides a flow diagram of this research.
Fig. 1.

The method used for molecular simulation analysis.
3. Results
3.1. Evolutionary distances for s gene variants and results of docking of RBD with ACE2 protein
Table 2 shows the evolutionary distances between the wild type and the variants, as well as the binding affinities of RBD with ACE2. After multiple alignments of wild-type and variant S genes by using the ClustalW program, FastTree was used to determine the phylogenetic tree (data not shown) and evolutionary distances. The binding affinity of the SARS-CoV-2 spike protein RBD and ACE2 was determined by using the ZDOCK score as an indicator. ZDOCK scores were utilized in cluster analysis to determine the average ZDOCK score of the most stable cluster and ZDOCK score of the most stable complex for each variant. We used these results to develop a mathematical model to predict infectivities of SARS-CoV-2 variants.
Table 2.
Evolutionary distances and binding affinities of SARS-CoV-2 spike proteins with ACE2.
| Measure | Wild type | Alpha (B.1.1.7) | Beta (B.1.351) | Gamma (P.1) | Delta (B.1.617.2) | Omicron BA.1 (B.1.1.529/BA.1) | Omicron BA.2 (B.1.1.529/BA.2) |
|---|---|---|---|---|---|---|---|
| Evolutionary distance | 0 | 0.00420 | 0.00446 | 0.01077 | 0.01209 | 0.01426 | 0.02600 |
| ZDOCK scores of the most stable complexes | 216.076 | 253.965 | 264.905 | 282.820 | 454.240 | 509.856 | 615.442 |
| ZDOCK scores of the most stable clusters (mean ± SD) | 219.633 ± 50.513 (n = 10) | 247.478 (n = 1) | 260.243 (n = 1) | 270.947 (n = 1) | 428.721 (n = 1) | 417.824 ± 4.936 (n = 5) | 595.340 ± 4.736 (n = 3) |
3.2. Development of a mathematical model to estimate infectivities of SARS-CoV-2 wild type and variants
We developed a mathematical model to predict the infectivities of SARS-CoV-2 wild type and variants according to our previous research (Takaoka et al., 2019). According to Fig. 2 , the relationship between the evolutionary distance and infectivity of each variant can be represented by the following equation:
where x is the evolutionary distance between the wild-type (wild) and a mutant SARS-CoV-2 spike protein. Constant values p and q were estimated by minimizing the sum of squared error between the calculated infectivity and the reported infectivity (ratio per wild type) (Allen et al., 2022; Araf et al., 2022; Chen and Wei, 2022; Davies et al., 2021; Faria et al., 2021; Radvak et al., 2021). We then derived the mathematical model for the binding affinity of RBD and ACE2 by using the results of docking analyses. The binding affinity for the mathematical model was derived from the following equation:
where A is the average ZDOCK score of the most stable cluster and B is the ZDOCK score of the most stable complex. The relationship between the binding affinity and the reported infectivity (Fig. 3 ) can be represented by the following equation:
Fig. 2.

Correlation between evolutionary distance and infectivity of SARS-CoV-2 variants. Collinearity between the evolutionary distance and infectivity was noted. The regression line was indicated by dotted line as an equation of “y = 201.06x + 0.5784″.
Fig. 3.

Correlation between the biding affinity based on our docking simulation analyses and infectivity of SARS-CoV-2 variants. The binding affinity for our mathematical model which is based on the molecular simulation data can be used to predict the infectivity of SARS-CoV-2 variants. The regression curve was indicated by dotted line as an equation of “y = 1.9455ln(x) + 1.2029″.
Constant values s and t were estimated by minimizing the sum of squared error between the calculated infectivity and the reported infectivity (ratio per wild type) (Allen et al., 2022; Araf et al., 2022; Chen and Wei, 2022; Davies et al., 2021; Faria et al., 2021; Radvak et al., 2021). The infectivity of SARS-CoV-2 (P) is defined by the evolutionary distance and the docking result with ACE2:
Constant values u, v, and w were estimated by minimizing the sum of squared error between the calculated infectivity P and the reported infectivity of each SARS-CoV-2 variant V:
where set M represents the SARS-CoV-2 variants whose infectivities are reported. These results suggest that our mathematical model can predict the infectivities of SARS-CoV-2 wild type and variants. Finally, the infectivity of SARS-CoV-2 variants is defined by the following equation:
In addition, the result of the cross-validation of our mathematical model showed that the predicted infectivity of each variant, which was calculated by our model using the data of the other five variants was strongly correlated with the reported infectivity (Fig. 4 ).
Fig. 4.
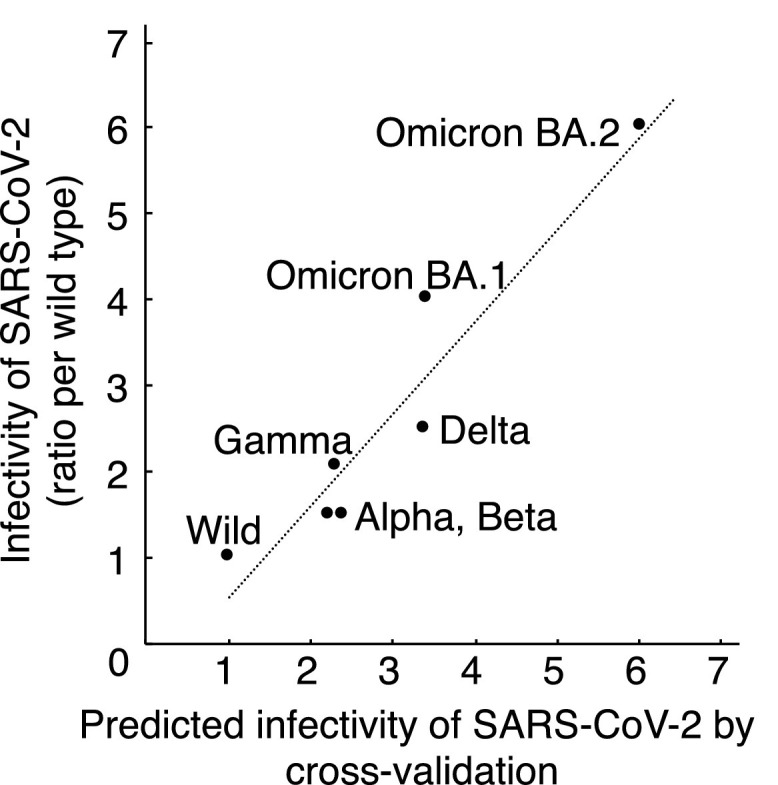
Cross-validation of predicted infectivity of SARS-CoV-2 with reported infectivity. Predicted infectivity of SARS-CoV-2 significantly correlated with reported infectivity (Pearson correlation coefficients R = 0.9527, p = 0.0009).
4. Discussion
Reported infectivities of SARS-CoV-2 variants were correlated with both evolutionary distance of the S gene and the binding affinity for the mathematical model according to our in silico docking data (Figs. 2 and 3). In addition, our mathematical model including these two factors reproduced the reported infectivity (Fig. 4).
In previous research, only the RBD domain of the spike protein was used in the analysis of binding affinity with ACE2: no MD analysis of the structure optimization (Celik et al., 2022; Gan et al., 2021; Shah and Woo, 2021; Sitthiyotha and Chunsrivirot, 2021; Tragni et al., 2022) and/or local optimization of only the RBD domain of the spike protein (Celik et al., 2022; Gan et al., 2021; Kumar et al., 2022; Shah and Woo, 2021; Sitthiyotha and Chunsrivirot, 2021; Tragni et al., 2022; Xue et al., 2021). However, we chose the three-dimensional structure of the complete trimeric spike protein (PDB ID: 6ZGG) in homology modeling in this research. After MD analysis, we confirmed the trajectory of the root mean square deviation and the Ramachandran plot for proper quality of each variant spike protein structure. The RBD domain of the variant trimeric spike protein structures was then provided for the docking analyses with ACE2. Therefore, our experimental strategy achieved high accuracy because we used a molecular simulation method which possibly imitates the behavior of the biomolecules. In addition, our previous research suggested the value of this prediction that utilized our molecular simulation methods (Nakamura et al., 2017; Ohta et al., 2018; Qiang et al., 2017; Sakaeda et al., 2018; Sugawara et al., 2019; Yamamura et al., 2020).
5. Conclusion
We developed a mathematical model for prediction of SARS-CoV-2 variant infectivity. Additional analyses of binding affinities of other spike proteins of SARS-CoV-2 variants with ACE2 may provide greater accuracy, and such experiments are now in progress.
Funding
This work was supported by JSPS Grant-in-Aid for Scientific Research [grant numbers 21K12110 to Y.T. and 19K12202 to A.S].
Author contributions
Y.T. conceived and designed this research. Y.T. and S.A. preformed the analyses and acquired the data. Y.T., S.A., Y.M., and M.O. interpreted the data. Y.T., S.A., and K.M derived mathematical model. Y.T. and S.A. wrote the draft, and all authors reviewed and approved the manuscript.
Ethical approval statement
This research is not applicable because we performed computer analyses by using sequence data obtained from public database.
Author statement
Y.T. conceived and designed this research. Y.T. and S.A. preformed the analyses and acquired the data. Y.T., S.A., Y.M., and M.O. interpreted the data. Y.T., S.A., and K.M derived mathematical model. Y.T. and S.A. wrote the draft, and all authors reviewed and approved the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Not applicable.
Data availability
Data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Adil M.T., Rahman R., Whitelaw D., Jain V., Al-Taan O., Rashid F., et al. SARS-CoV-2 and the pandemic of COVID-19. Postgrad. Med. J. 2021;97(1144):110–116. doi: 10.1136/postgradmedj-2020-138386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen H., Vusirikala A., Flannagan J., Twohig K.A., Zaidi A., Chudasama D., et al. Household transmission of COVID-19 cases associated with SARS-CoV-2 delta variant (B.1.617.2): national case-control study. Lancet Reg. Health Eur. 2022;12 doi: 10.1016/j.lanepe.2021.100252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araf Y., Akter F., Tang Y.D., Fatemi R., Parvez M.S.A., Zheng C., et al. Omicron variant of SARS-CoV-2: genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 2022;94(5):1825–1832. doi: 10.1002/jmv.27588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On Y.M., Flamholz A., Phillips R., Milo R. SARS-CoV-2 (COVID-19) by the numbers. Elife. 2020;9 doi: 10.7554/eLife.57309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. https://www.rcsb.org/ Available from: [Accessed June 14, 2022] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W., Dong C., Kim S., Hou D., Tai W., Du L., et al. Biomechanical characterization of SARS-CoV-2 spike RBD and human ACE2 protein-protein interaction. Biophys. J. 2021;120(6):1011–1019. doi: 10.1016/j.bpj.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik I., Khan A., Dwivany F.M., Fatimawali Wei DQ, Tallei T.E. Computational prediction of the effect of mutations in the receptor-binding domain on the interaction between SARS-CoV-2 and human ACE2. Mol. Divers. 2022 doi: 10.1007/s11030-022-10392-x. [DOI] [PubMed] [Google Scholar]
- Chen J., Wei G.W. Omicron BA.2 (B.1.1.529.2): high potential to becoming the next dominating variant. J. Phys. Chem. Lett. 2022;13:3840–3849. doi: 10.1021/acs.jpclett.2c00469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherian S., Potdar V., Jadhav S., Yadav P., Gupta N., Das M., et al. SARS-CoV-2 spike mutations, L452R, T478K, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. Microorganisms. 2021;9(7) doi: 10.3390/microorganisms9071542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen P.A., Olsen R.J., Long S.W., Snehal R., Davis J.J., Ojeda Saavedra M., et al. Signals of significantly increased vaccine breakthrough, decreased hospitalization rates, and less severe disease in patients with coronavirus disease 2019 caused by the omicron variant of severe acute respiratory syndrome coronavirus 2 in Houston, Texas. Am. J. Pathol. 2022;192(4):642–652. doi: 10.1016/j.ajpath.2022.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claro I.M., da Silva Sales F.C., Ramundo M.S., Candido D.S., Silva C.A.M., de Jesus J.G., et al. Local transmission of SARS-CoV-2 lineage B.1.1.7, Brazil, December 2020. Emerg. Infect. Dis. 2021;27(3):970–972. doi: 10.3201/eid2703.210038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies N.G., Abbott S., Barnard R.C., Jarvis C.I., Kucharski A.J., Munday J.D., et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021;372(6538) doi: 10.1126/science.abg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejnirattisai W., Huo J., Zhou D., Zahradník J., Supasa P., Liu C., et al. Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses. bioRxiv. 2021 doi: 10.1101/2021.12.03.471045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria N.R., Mellan T.A., Whittaker C., Claro I.M., Candido D.D.S., Mishra S., et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science. 2021;372(6544):815–821. doi: 10.1126/science.abh2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan H.H., Twaddle A., Marchand B., Gunsalus K.C. Structural modeling of the SARS-CoV-2 Spike/Human ACE2 complex interface can identify high-affinity variants associated with increased transmissibility. J. Mol. Biol. 2021;433(15) doi: 10.1016/j.jmb.2021.167051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiyama M., Takeuchi F., Sugano A., Yoneshige A., Inoue T., Wada A., et al. Indigo plant leaf extract inhibits the binding of SARS-CoV-2 spike protein to angiotensin-converting enzyme 2. Exp. Ther. Med. 2022;23(4):274. doi: 10.3892/etm.2022.11200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M., Ota T. On the stochastic model for estimation of mutational distance between homologous proteins. J. Mol. Evol. 1972;2(1):87–90. doi: 10.1007/bf01653945. [DOI] [PubMed] [Google Scholar]
- Kumar S., Thambiraja T.S., Karuppanan K., Subramaniam G. Omicron and delta variant of SARS-CoV-2: a comparative computational study of spike protein. J. Med. Virol. 2022;94(4):1641–1649. doi: 10.1002/jmv.27526. [DOI] [PubMed] [Google Scholar]
- Loconsole D., Sallustio A., Centrone F., Casulli D., Ferrara M.M., Sanguedolce A., et al. An autochthonous outbreak of the SARS-CoV-2 P1 variant of concern in Southern Italy, April 2021. Trop. Med. Infect. Dis. 2021;6(3) doi: 10.3390/tropicalmed6030151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojena R. Hierarchical grouping methods and stopping rules: an evaluation. Comput. J. 1977;20(4):359–363. [Google Scholar]
- Nakamura Y., Sugano A., Ohta M., Takaoka Y. Docking analysis and the possibility of prediction efficacy for an anti-IL-13 biopharmaceutical treatment with tralokinumab and lebrikizumab for bronchial asthma. PLoS ONE. 2017;12(11) doi: 10.1371/journal.pone.0188407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Library of Medicine US. National Center for Biotechnology Information (NCBI); Available from: https://www.ncbi.nlm.nih.gov/. [Accessed June 14, 2022].
- Ohta M., Sugano A., Hatano N., Sato H., Shimada H., Niwa H., et al. Co-precipitation molecules hemopexin and transferrin may be key molecules for fibrillogenesis in TTR V30M amyloidogenesis. Transgenic Res. 2018;27(1):15–23. doi: 10.1007/s11248-017-0054-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M.N., Dehal P.S., Arkin A.P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009;26(7):1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang L., Guan Y., Li X., Liu L., Mu Y., Sugano A., et al. CSP-1103 (CHF5074) stabilizes human transthyretin in healthy human subjects. Amyloid. 2017;24(1):42–51. doi: 10.1080/13506129.2017.1308348. [DOI] [PubMed] [Google Scholar]
- Radvak P., Kwon H.J., Kosikova M., Ortega-Rodriguez U., Xiang R., Phue J.N., et al. SARS-CoV-2 B1.1.7 (alpha) and B.1.351 (beta) variants induce pathogenic patterns in K18-hACE2 transgenic mice distinct from early strains. Nat. Commun. 2021;12(1):6559. doi: 10.1038/s41467-021-26803-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaeda T., Kobuchi S., Yoshioka R., Haruna M., Takahata N., Ito Y., et al. Susceptibility to serious skin and subcutaneous tissue disorders and skin tissue distribution of sodium-dependent glucose co-transporter type 2 (SGLT2) inhibitors. Int. J. Med. Sci. 2018;15(9):937–943. doi: 10.7150/ijms.22224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M., Woo H.G. Omicron: a heavily mutated SARS-CoV-2 variant exhibits stronger binding to ACE2 and potently escapes approved COVID-19 therapeutic antibodies. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.830527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S., McNab C., Olson R.M., Bristol N., Nolan C., Bergstrøm E., et al. How an outbreak became a pandemic: a chronological analysis of crucial junctures and international obligations in the early months of the COVID-19 pandemic. Lancet Reg. Health Eur. 2021;398(10316):2109–2124. doi: 10.1016/S0140-6736(21)01897-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitthiyotha T., Chunsrivirot S. Computational design of SARS-CoV-2 peptide binders with better predicted binding affinities than human ACE2 receptor. Sci. Rep. 2021;11(1):15650. doi: 10.1038/s41598-021-94873-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara K., Nomura K., Okada Y., Sugano A., Matsumoto M., Takarada T., et al. In silico and in vitro analyses of the pathological relevance of the R258H mutation of hepatocyte nuclear factor 4α identified in maturity-onset diabetes of the young type 1. J. Diabetes Investig. 2019;10(3):680–684. doi: 10.1111/jdi.12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka Y., Ohta M., Tateishi S., Sugano A., Nakano E., Miura K., et al. In silico drug repurposing by structural alteration after induced fit: discovery of a candidate agent for recovery of nucleotide excision repair in xeroderma pigmentosum group D mutant (R683W) Biomedicines. 2021;9(3) doi: 10.3390/biomedicines9030249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka Y., Takeuchi A., Sugano A., Miura K., Ohta M., Suzuki T., et al. Establishment of the experimental procedure for prediction of conjugation capacity in mutant UGT1A1. PLoS ONE. 2019;14(11) doi: 10.1371/journal.pone.0225244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao K., Tzou P.L., Nouhin J., Gupta R.K., de Oliveira T., Kosakovsky Pond S.L., et al. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021;22(12):757–773. doi: 10.1038/s41576-021-00408-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H., Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J., et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature. 2021;592(7854):438–443. doi: 10.1038/s41586-021-03402-9. [DOI] [PubMed] [Google Scholar]
- Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tragni V., Preziusi F., Laera L., Onofrio A., Mercurio I., Todisco S., et al. Modeling SARS-CoV-2 spike/ACE2 protein-protein interactions for predicting the binding affinity of new spike variants for ACE2, and novel ACE2 structurally related human protein targets, for COVID-19 handling in the 3PM context. EPMA J. 2022:1–27. doi: 10.1007/s13167-021-00267-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium Team. UniProt: the universal protein knowledgebase; 2018. Available from: https://www.uniprot.org/. [Accessed June 14, 2022]. [DOI] [PMC free article] [PubMed]
- Xue X., Shi J., Xu H., Qin Y., Yang Z., Feng S., et al. Dynamics of binding ability prediction between spike protein and human ACE2 reveals the adaptive strategy of SARS-CoV-2 in humans. Sci. Rep. 2021;11(1):3187. doi: 10.1038/s41598-021-82938-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura T., Horinouchi T., Adachi T., Terakawa M., Takaoka Y., Omachi K., et al. Development of an exon skipping therapy for X-linked Alport syndrome with truncating variants in COL4A5. Nat. Commun. 2020;11(1):2777. doi: 10.1038/s41467-020-16605-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data that support the findings of this study are available from the corresponding author upon reasonable request.