

Abstract
The increasingly frequent outbreaks of pathogenic viruses have underlined the urgent need to improve our arsenal of antivirals that can be deployed for future pandemics. Innate immunity is a powerful first line of defense against pathogens, and compounds that boost the innate response have high potential to act as broad-spectrum antivirals. Here, we harnessed localization-dependent protein-complementation assays (called Alpha Centauri) to measure the nuclear translocation of interferon regulatory factors (IRFs), thus providing a readout of innate immune activation following viral infection that is applicable to high-throughput screening of immunomodulatory molecules. As proof of concept, we screened a library of kinase inhibitors on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and identified Gilteritinib as a powerful enhancer of innate responses to viral infection. This immunostimulatory activity of Gilteritinib was found to be dependent on the AXL-IRF7 axis and results in a broad and potent antiviral activity against unrelated RNA viruses.
Keywords: innate immunity, interferon regulatory factors, drug screening, SARS-CoV-2, emerging viruses, broad-spectrum antivirals
Graphical abstract
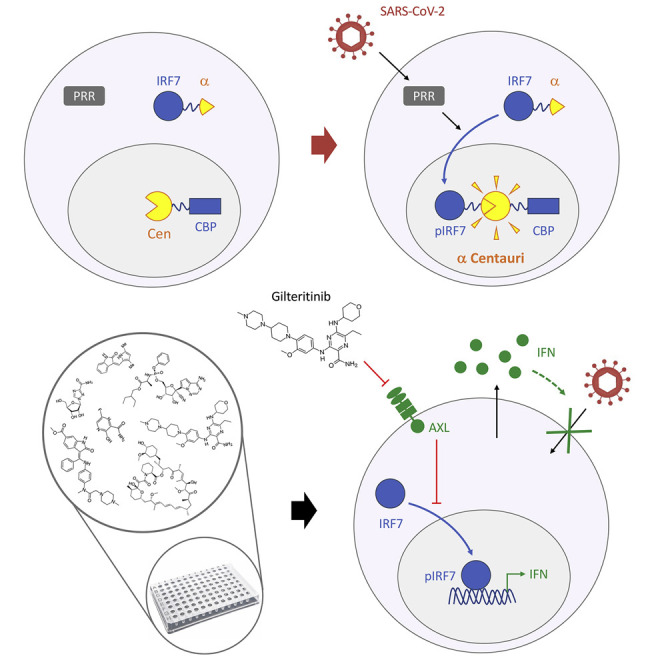
Compounds that enhance innate immunity can constitute potent broad-acting antivirals. Maarifi et al. identify immunostimulatory molecules applying bioluminescence complementation to measure innate immune activation. By assessing kinase inhibitors on SARS-CoV-2-infected cells, they identify Gilteritinib as an enhancer of innate responses with broad-acting antiviral activity.
Introduction
New antiviral treatments are required to address the growing and unpredictable emergence of viruses. Our current arsenal of antiviral therapeutic treatments includes nucleoside analogs (e.g., ribavirin, acyclovir, Favipiravir, Remdesivir), which can be effective across viral families, or molecules that target viral enzymes, which tend to be very specific and favor viral evasion. However, the repositioning of already existing antivirals to target poorly characterized viruses when they emerge in a population, such as West Nile virus in 2002, Zika virus in 2015, and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in 2019, has not scored a high success so far. In the case of SARS-CoV-2, attempts to reposition potent anti-microbials (e.g., Remdesivir, hydroxychloroquine) to treat hospitalized patients with severe coronavirus disease 2019 (COVID-19) have failed (Cao et al., 2020; WHO Solidarity Trial Consortium et al., 2021). There is therefore a pressing need to reconsider our discovery pipelines to identify novel antiviral agents.
In particular, compounds that block viral growth by targeting cellular proteins and pathways instead of the virus itself, so-called host-directed antivirals, are of growing interest because they are less prone to viral evasion and more likely to display broad-spectrum activity (Chitalia and Munawar, 2020). Among these, one strategy is to boost innate defenses by the use of immunostimulatory molecules (Es-Saad et al., 2012; Prussia et al., 2011). Innate immunity is the first and a major line of defense against viral infections (Baum and Garcia-Sastre, 2010; MacMicking, 2012) and a key player in the induction of adaptive immunity. The detection of pathogens relies on a panel of pattern-recognition receptors (PRRs), such as Toll-like receptors (TLRs) or RIG-like receptors (RLRs). These mobilize kinases and ubiquitin-dependent signaling cascades to phosphorylate interferon regulatory factors (IRFs) which, once activated, translocate into the nucleus, where they induce the expression of type I interferons (IFNs; mainly IFN-α and -β). Among the 9 known IRFs, four (IRF1, IRF3, IRF5, and IRF7) are involved in the induction of type I IFN. Moreover, since both IRF1 and IRF5 were shown to be dispensable for type I IFN gene induction by viruses (Matsuyama et al., 1993; Takaoka et al., 2005), only IRF3 and IRF7 are considered the main inducers of type I IFN (Honda et al., 2005; Sato et al., 2000). IRF3 is constitutively expressed, whereas IRF7 is present at low levels in most cells but is potently induced by type I IFNs or virus infection.
Once induced, type I IFNs relay the signal to neighboring cells through binding to their IFN-α/β receptor (IFNAR) at the surface of both infected and bystander cells (Stetson and Medzhitov, 2006; van Boxel-Dezaire et al., 2006). This activates the JAK/STAT signaling cascade and leads to the induction of hundreds of genes, known as IFN-stimulated genes (ISGs), whose products are involved in various antiviral mechanisms.
The concept of enhancing the IFN response to promote a potent antiviral state is not new. The administration of recombinant IFN-α/β by intramuscular injection or inhalation is a commonly prescribed immunomodulatory therapy for many viral infections, cancer, and immune disorders (Friedman and Contente, 2009). However, these treatments are associated with modest efficacy and poor tolerability because of their undesirable systemic effects. The treatment of patients with COVID-19 with IFN-α carries little benefit unless it is applied very early on, and severe COVID-19 cases are characterized by low IFN production but high levels of pro-inflammatory cytokines (Hadjadj et al., 2020). Moreover, although the exogenous use of IFN-λ was initially very promising since its effects are limited to the lung (Galani et al., 2017; Prokunina-Olsson et al., 2020), subsequent studies raised concerns as it was shown to induce damage to the lung epithelial barrier in mice (Broggi et al., 2020). Similarly, although synthetic agonists, such as imiquimod and related imidazoquinolines, have proven successful against some viral infections (Zhang et al., 2014), they turn on the production of IFN and cytokines in all cells and essentially recapitulate the exogenous use of IFNs.
We hypothesized that compounds that potentiate innate immune signaling specifically in cells activated by a pathogen might constitute a more potent and specific approach to enhancing the IFN response therapeutically. Although viruses have evolved mechanisms to evade innate immunity (Bowie and Unterholzner, 2008; Chan and Gack, 2016; Garcia-Sastre, 2017; Rojas et al., 2021), immunostimulatory molecules would give the cells an advantage over the virus by promoting an early or more efficient response. Since all signaling from PRRs converge on the translocation of IRF transcription factors, we devised assays to quantify innate immune signaling pathways based on the nuclear translocation of IRFs in a format that is compatible for high-throughput screening.
Using protein complementation, we report the development of specific and highly quantitative assays to measure the nuclear translocation of IRFs following viral infection. Unexpectedly, we report that different RNA viruses do not activate the same IRFs. Using IRF7 to perform a proof-of-concept screens of compounds that enhance innate sensing of SARS-CoV-2, we identify Gilteritinib as an enhancer of the innate immune response to viral infection active against SARS-CoV-2 and other RNA viruses.
Results
Activation of innate immune signaling can be monitored with the Alpha Centauri assay
Nuclear translocation of key transcription factors is essential to innate immune signaling. We hypothesized that by sequestering a reporter fragment in the nucleus, we could apply protein complementation to measure the nuclear translocation of IRFs following innate sensing, thus providing an easy readout of pathogen sensing applicable to high-throughput screening (Figure 1A). In this assay, termed Alpha Centauri (or AlphaCen), a small fragment of the NanoLuc reporter named Alpha (α; 13 amino acids) is used to tag a protein of interest, while the complementary fragment named Cen is tethered to a subcellular compartment. To test the feasibility of this approach, we tagged IRF3 with the small α fragment, while the complementary large fragment (19 kDa) was fused to the triple nuclear localization signal (nls) of SV40 for tethering in the nucleus (Cen-nls) (Figure 1A). FLAG and hemagglutinin (HA) tags were introduced in all α and Cen constructs, respectively, to monitor their expression and localization. Insertion of the α tag within the IRF3 coding sequence did not disrupt its expression (Figure S1A) nor its ability to translocate into the nucleus upon stimulation with defective-interfering Sendai virus (SeV; Strahle et al., 2006) (Figure 1B). In unstimulated cells, IRF3-α was mainly cytoplasmic, whereas Cen-nls was sequestered in the nucleus, as expected. Stimulation with SeV for 6 h led to the translocation of IRF3-α to the nucleus, where it co-localized partially with Cen (Figure 1B). We performed bioluminescent imaging in SeV-stimulated cells to visualize the localization of the reconstituted NanoLuc reporter. This confirmed a gain of signal that was predominantly nuclear (Figure 1C), and between 2- and 8-fold above unstimulated control cells, in the three cell lines tested (HEK293T, A549, and HeLa) (Figure 1D).
Figure 1.

Measuring activation of IRF3 by IRF-α/nls AlphaCen assay
(A) Schematic representation of the nls-AlphaCen assay, which measures the nuclear translocation of an IRF transcription factor by protein complementation. The NanoLuc reporter is split in two fragments of unequal size, α and Cen, which are fused to an IRF transcription factor and to a nuclear localization signal (nls), respectively. When cells expressing IRF-α and Cen-nls sense a viral infection, activated IRFs are carried into the nucleus, where they encounter Cen-nls. The reporter fragments α and Cen assemble into functional NanoLuc (Nluc), which generates a bioluminescent signal.
(B) The localization of IRF3-α was assessed by confocal imaging in Hela cells, using anti-FLAG and -HA antibodies to detect IRF3-α and Cen-nls, respectively. Scale bar: 20 μm.
(C) Nluc reconstitution in HEK293T cells was assessed by bioluminescent imaging. Images are representative of 4 independent experiments. Scale bar: 20 μm.
(D) IRF3-⍺- and Cen-expressing cells were stimulated with SeV. Nluc signal was measured after 6 h. Results show individual normalized values from 6 independent experiments and mean with range. ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001, as determined by Student’s t test.
The IRF3-α/CBP AlphaCen assay allows the screening of immunomodulatory molecules
Having confirmed that measuring innate immune signaling by protein-complementation assay with α-tagged IRF3 and nucleus-tethered Cen is feasible, we explored ways to establish a reproducible assay for drug screening. First, although defective-interfering SeV is a strong inducer of IFN, its use as an agonist is not optimal for high-throughput screening. Besides the time-cost limitations linked to its production, and the biosafety considerations, there can be considerable variability from one viral stock to the next, particularly in the amount of defective-interfering genomes that trigger sensing (Strahle et al., 2006). Therefore, we replaced SeV with transfection of a 2CARD construct, a constitutively active module of RIG-I. Second, it became apparent from our imaging studies that reconstituted AlphaCen NanoLuc reporter in stimulated cells developed as discrete puncta in the nucleus, reminiscent of cognate promoter hotspots, and therefore that only a minor proportion of nuclear IRF3-α was in proximity to Cen-nls (Figures 1B and 1C). We therefore fused Cen to the transcriptional coactivator CREB-binding protein (CBP) (Figure 2A) and compared signal intensities after immune activation of IRF3-α at 48 h post-transfection (48 hpt), which is the protocol that was used for Cen-nls. Fusing Cen to CBP led to a 5- to 10-fold increase in NanoLuc complementation compared with the nls construct (Figure 2B), despite reasonably similar expression levels (Figure S1B).
Figure 2.

Measuring activation of IRF3 by IRF3-α/CBP AlphaCen assay allows the screening of immunomodulatory molecules
(A) Schematic representation of the CBP AlphaCen assay, where the Cen fragment is fused to murine CREB-binding protein (CBP).
(B) HEK293T cells were transfected with an empty (NS) or a 2CARD-encoding plasmid, together with IRF3-⍺ and either Cen-nls or Cen-CBP. AlphaCen NLuc signal was measured at 48 hpt. Data correspond to means ± SD of a representative experiment performed in triplicate.
(C) 2CARD expression was assessed by anti-FLAG western blot.
(D) HEK293T cells were transfected with IRF3-α and Cen-CBP together with an empty (NS) or a 2CARD-expressing plasmid. AlphaCen NLuc signal was measured at 14, 24, and 39 hpt. Results are from a single experiment performed in triplicate, representative of two independent experiments.
(E) HEK293T cells were transfected with IRF3-α, Cen-CBP, and 2CARD and treated with MRT67307 at the indicated concentrations at 14 hpt. AlphaCen signal was measured at 24 hpt (39 hpt). Data correspond to means ± SD of three independent experiment performed in triplicate. ∗∗∗∗p < 0.0001, as determined by one-way ANOVA with Bonferroni post hoc test.
(F) A panel of 21 kinase inhibitors and 7 SARS-CoV-2 inhibitors were tested with the IRF3-α/CBP AlphaCen readout. A viability cutoff of 50% was applied to remove cytotoxic compounds (indicated by a cross; see Figure S1C). Results were normalized to the 2CARD-stimulated and untreated control (NS, non-stimulated). Results are the mean of 2 independent screens ± SD.
(G) Graphs show the inhibition of the IRF3-α/CBP AlphaCen signal and cell viability for a selection of drugs.
To provide a proof-of-concept screen of our assay, we compared the kinetics of optimal 2CARD expression (24 hpt; Figure 2C) and AlphaCen NanoLuc signal (40 hpt), which allowed us to pinpoint the time of drug addition at 14 hpt (Figure 2D). To confirm that NanoLuc signal resulted from the activation of IRF3, first, we tested the IKKε/TBK1 inhibitor MRT67307 and confirmed a strong dose-dependent reduction in signal (Figure 2E). Next, we tested a panel of 21 additional kinase inhibitors, which we compared with 7 antiviral molecules as control (Figure 2F). A cutoff at 50% viability was used to exclude toxic conditions (Figure S1C). As expected, the two IKKε/TBK1 inhibitors MRT67307 and BX-795 profoundly inhibited the IRF3-α/CBP AlphaCen signal (Figure 2G), thus confirming the specificity of the readout.
Staurosporine, a non-selective kinase inhibitor, Rapamycin, an mTOR kinase inhibitor, and Gilteritinib, a FLT3/AXL inhibitor, also strongly decreased the IRF3-α/CBP AlphaCen signal. Unexpected hits were also obtained at high micromolar concentrations: Remdesivir, an antiviral nucleotide analogue, AG490, a KAK2/STAT3 pathway inhibitor, Nintedanib, a growth factor receptor kinase inhibitor, and two IKK inhibitors, BAY 11–7085 and PS-1145 (Figure 2F).
We concluded that the IRF3-α/CBP AlphaCen assay provides a strong and reproducible readout of innate immune pathway activation within 24 h and is adapted for compound screening in multi-well formats. The signal amplitude is high (around 1 to 2 orders of magnitude) without reaching saturation, thus conceptually allowing the detection of molecules that either inhibit or enhance immune signaling.
Differential activation of IRFs by SARS-CoV-2, influenza A virus, and West Nile virus
Defective-interfering SeV contains plentiful double-stranded RNA (dsRNA) that is sensed by RIG-I. However, since most viruses have developed multiple strategies to antagonize innate signaling in infected cells (Bowie and Unterholzner, 2008; Chan and Gack, 2016; Garcia-Sastre, 2017; Rojas et al., 2021), we anticipated that the IRF-α/nls and /CBP AlphaCen assays might not be effective in detecting replicative virus. SARS-CoV-2, in particular, antagonizes multiple steps of IFN signaling (Sa Ribero et al., 2020). We confirmed that infection with SARS-CoV-2 at an MOI 0.1 was not sufficient to trigger the nuclear translocation of IRF3, unlike SeV stimulation, which was efficient (Figures S2A and S2B), possibly due to viral antagonism of IRF3. As reported previously, however, GRL-0617, an inhibitor of the SARS-CoV-2 papain-like protease (PLP), or a high MOI could overcome antagonism of IRF3 (Blanco-Melo et al., 2020; Shin et al., 2020; Xia et al., 2020) and partially restore nuclear translocation of IRF3 (Figures S2C and S2D). Interestingly, pre-treatment of cells with ivermectin, a ß1-karyopherin inhibitor (Wagstaff et al., 2012), and removal prior to infection improved translocation of IRF3 in infected cells (Figures S2E and S2F). We also noted that pre-treatment with IFN-α, which increases expression of RIG-I and MDA5 (Figure S2G) and has an antiviral effect on SARS-CoV-2 (Lokugamage et al., 2020; Sa Ribero et al., 2020) (Figure S2H), increases its sensing in HEK-ACE2 cells (Figure S2I) and was therefore included in subsequent AlphaCen assays, as indicated.
Since treatments that overcome antagonism of IRF3 by SARS-CoV-2 only marginally improved its detection by AlphaCen assays, we explored the involvement of other IRFs in signal transduction. First, we assessed the ability of SARS-CoV-2 to activate endogenous IRF1, IRF3, IRF5, and IRF7 after 6 h of infection and compared this with defective-interfering SeV and replicative influenza A virus (IAV). SeV was found to trigger the nuclear translocation of IRF1, IRF3, and IRF7 (Figure 3A) but not IRF5, as previously shown (Barnes et al., 2001). IAV, on the other hand, activated only IRF3 and IRF7. SARS-CoV-2 alone was quite poor at activating any of the tested IRFs. However, in the presence of GRL-0617, SARS-CoV-2 activated IRF3, IRF5, and IRF7 quite efficiently (Figures 3A and 3B).
Figure 3.

SARS-CoV-2 and other RNA viruses activate IRFs differentially
(A) Localization of endogenous transcription factors in resting and stimulated A549-ACE2 cells. Cells were infected with SeV, influenza A virus (H1N1 WSN) (IAV) at MOI 1, or SARS-CoV-2 at the indicated MOIs, for 6 h or left uninfected (NI). Alternatively, cells were infected in the presence of the PLP inhibitor GRL-0617 (50 μM). Cells fixed and labeled with anti-IRF1, IRF3, IRF5, or IRF7 were acquired with Airyscan mode. Fluorescence intensities in the nuclear and cytoplasmic regions of interest (ROIs) were measured using FiJi. Each point represents the nucleus/cytoplasm ratios of mean gray values for a single cell. The graph shows the median and interquartile range for ∼30 cells per condition from 3 independent experiments. ∗∗∗∗p < 0.0001, as determined by one-way ANOVA with Bonferroni post hoc test.
(B) Representative images from (A). Scale bar: 5 μm.
Based on these results, we developed and compared IRF1-α, IRF3-α, IRF5-α, and IRF7-α constructs (Figure S3A) and tested them in CBP AlphaCen assays. We routinely assessed transfection efficiency of α and Cen constructs by flow cytometry and applied a cutoff of 30%, below which the cells were not used for experimentation (Figure S3B). First, we confirmed that the stimulation of cells by 2CARD activated all four tested IRFs and led to an appreciable gain of signal in all the IRF-α/CBP AlphaCen systems at 24 hpt (Figure 4A), with some variability in fold changes that reflected differences in transfection efficiencies between experiments. Next, we determined the localization of the different IRF-α transcription factors in resting and stimulated cells. As expected, IRF3-α, IRF5-α, and IRF7-α were all cytoplasmic in uninfected cells, but surprisingly, IRF1-α adopted diffuse nuclear localization in the absence of stimulation (Figure 4B). This is concordant with previous studies identifying a functional nls in IRF1 and showing that ectopic expression of IRF1 alone is sufficient to induce endogenous type I IFN genes (Feng et al., 2021; Schaper et al., 1998). Stimulation of cells with SeV led to the detection of IRF-α transcription factors in the nucleus, often as punctate labelling that coincided with Cen-CBP (Figure 4B).
Figure 4.

Applying different IRF-α/CBP AlphaCen assays to measure the sensing of viral infections
(A) HEK-ACE2 cells were transfected with an empty (NS) or a 2CARD-expressing plasmid, together with Cen-CBP and either IRF1-α, IRF3-α, IRF5-α, or IRF7-α. AlphaCen signal was measured at 39 hpt. Data correspond to individual values and means ± SD from four independent experiments performed in triplicate.
(B) Expression and localization of IRF-⍺ (green) and Cen-CBP (magenta) in HEK293T cells stimulated by SeV for 6 h or unstimulated (NI) and acquired with Airyscan processing. Representative images are shown with artificial coloring. Scale bar: 10 μm.
(C) HEK293T cells were transfected with Cen-CBP and either IRF1-α, IRF3-α, IRF5-α, or IRF7-α. At 24 hpt, cells were infected with SeV, West Nile virus lineage 1 (WNV L1), or IAV (H1N1 WSN) at MOI 1 or left uninfected (NI). AlphaCen signal was measured at 24 hpi. Data correspond to means ± SD of three experiments performed in triplicate. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001, as determined by one-way ANOVA with Bonferroni post hoc test. ns, non-significant.
(D) HEK-ACE2 cells were transfected with Cen-CBP and either IRF1-α, IRF3-α, IRF5-α, or IRF7-α and then treated with IFN-α2 overnight. At 24 hpt, cells were infected with SARS-CoV-2 at the indicated MOIs or left uninfected. Alternatively, cells were infected in the presence of GRL-0617 at 50 μM. AlphaCen signal was measured at 24 hpi. Data correspond to means ± SD of three experiments performed in triplicate. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001, as determined by one-way ANOVA with Bonferroni post hoc test. ns, non-significant.
We then tested the ability of the IRF-α/CBP AlphaCen assays to sense infection by 4 different RNA viruses, which are all known to evade innate immune defenses (Chan and Gack, 2016; Nelemans and Kikkert, 2019): a flavivirus, West Nile virus (WNV), an orthomyxovirus (IAV), a paramyxovirus (SeV), and a beta-coronavirus (SARS-CoV-2). For SARS-CoV-2 infections, ACE2 surface expression was confirmed by flow cytometry (Figure S3C). Results showed strong detection of SeV with all IRF-α transcription factors, which is concordant with the fact that it contains defective-interfering genomes that can strongly activate IFN signaling (Strahle et al., 2006). Surprisingly, the IRF1-α/CBP AlphaCen assay also significantly detected both WNV and IAV (Figure 4C), despite the diffuse nuclear IRF1-α staining already present in unstimulated cells (Figure 4B), confirming that, in the case of IRF1, diffuse nuclear localization is not sufficient to generate AlphaCen signal. Although SARS-CoV-2 could be detected by IRF5-α/CBP AlphaCen, the greatest detection was obtained with the IRF7-α/CBP in the presence of GRL-0617 (Figure 4D). Of note, signal amplitude was greater using CBP as a nuclear tether for Cen compared with nls (Figure S3D). Taken together, these results indicate that the CBP AlphaCen systems are effective at sensing replicative non-defective viral infections but that different IRFs respond to different viruses.
The IRF7-α/CBP AlphaCen assay allows the screening of immunomodulatory molecules in SARS-CoV-2-infected cells
To demonstrate the value of the CBP AlphaCen assay to screen for immunomodulators in virally infected cells, we tested the panel of inhibitors used in Figure 2F on the IRF7-α/CBP system following infection by SARS-CoV-2. AlphaCen measurement was performed at 24 h after infection and treatment (Figure 5A). A cutoff at 50% viability was used to exclude toxic conditions as previously, although we did note that the combination of drug treatment and viral infection led to more toxicity (Figure S4) than the drugs alone (Figures 2G and S1C). The most striking hit was obtained with Gilteritinib, which increased immune signaling in SARS-CoV-2-infected cells by over 300% (Figure 5B). This result was in stark contrast to the inhibition of IRF3 signaling by Gilteritinib observed in the IRF3-α/CBP drug screen with 2CARD stimulation, suggesting that its effect here might be specific to viral infection (Figure 2F). Another molecule that apparently enhanced sensing of SARS-CoV-2 was scutellarein, a naturally occurring flavonoid isolated from traditional Chinese medicine that was shown to inhibit SARS-CoV and SARS-CoV-2 enzymes in vitro (Liu et al., 2021; Yu et al., 2012). Many of the other antivirals reported to be active against SARS-CoV-2 also induced changes in sensing (Figures 5B and 5C). This dual effect on viral replication with concomitant immunomodulatory effects has previously been reported and thought to contribute to the broad-spectrum virustatic activity of these compounds (Mondelli, 2014).
Figure 5.

Screening for modulators of innate immune sensing of SARS-CoV-2 infections using the IRF7-α/CBP AlphaCen assay
HEK-ACE2 were transfected with IRF7-⍺ and Cen-CBP, then treated with IFN-α2 overnight.
(A) Cells were treated with GRL-0617 (50 μM) and infected with SARS-CoV-2 at MOI 1. AlphaCen NLuc signal was acquired at 6 and 24 hpi. Results are a single experiment performed in triplicate.
(B) At 24 hpt, cells were infected with SARS-CoV-2 at MOI 1 in the presence of kinase and SARS-CoV-2 inhibitors at the indicated concentrations. AlphaCen NLuc signal was measured at 24 hpi. Results are normalized for the SARS-CoV-2-infected condition in the absence of tested inhibitor. A viability cutoff of 50% was applied to remove cytotoxic compounds (indicated by a cross; see Figure S4). Results are the mean of triplicate values from 2 independent screens ± SD. ∗∗p < 0.01 and ∗∗∗∗p < 0.0001, as determined by one-way ANOVA with Bonferroni post hoc test.
(C) Graphs show the IRF3-α/CBP AlphaCen signal and cell viability for a selection of drugs. Results are the mean of 2 independent screens ± SD.
Gilteritinib enhances innate signaling in SARS-CoV-2-infected cells and has broad antiviral activity
To assess whether our approach can support the discovery of immunomodulatory molecules that are not direct antivirals, we further investigated Gilteritinib, which was the strongest hit in the IRF7-α/CBP AlphaCen screen (Figures 5B and 5C) and which was also strongly antiviral (Figure 6 ). The anti-SARS-CoV-2 activity of Gilteritinib had a half-maximal inhibitory concentration (IC50) of 0.13 ± 0.05 μM (Figure 6B) and was confirmed by plaque assay (Figure 6C). This antiviral effect was previously reported and tentatively attributed to the inhibition of phosphorylation hotspots on SARS-CoV-2 and/or host cells (Bouhaddou et al., 2020; Stukalov et al., 2021). However, its potent activation of IRF7 suggested that its antiviral effect might be the indirect consequence of IFN stimulation. To establish this, we first demonstrated that Gilteritinib treatment stimulates the expression of IFN-α4, IFN-β, and CXCL10 by SARS-CoV-2-infected cells in a dose-dependent manner but did not affect expression of IFN-γ, interleukin-6 (IL-6), or tumor necrosis factor alpha (TNF-α) (Figures S5A and 6D). Moreover, we showed that a neutralizing anti-IFNAR antibody could reverse the apparent antiviral effect of Gilteritinib on SARS-CoV-2-mNeongreen replication (Figure 6E), thus suggesting that the antiviral effect of Gilteritinib involves type I IFN.
Figure 6.

Testing modulators of innate immune sensing on SARS-CoV-2 replication
(A) HEK-ACE2 cells were treated with kinase and SARS-CoV-2 inhibitors at the indicated concentrations and infected with SARS-CoV-2-mNeonGreen at MOI 0.1 At 24 hpi, cells were fixed, and infection was assessed by flow cytometry for green fluorescence. Live cells were selected by gating on forward scatter (FSC) and side scatter (SSC) parameters. A viability cutoff of 50% was applied to remove cytotoxic compounds (indicated by a cross). Results show the min-to-max values of 2 independent experiments with line at mean.
(B) Inhibition of SARS-CoV-2-mNeonGreen by Gilteritinib. HEK-ACE2 cells were treated with increasing concentrations of Gilteritinib and infected with SARS-CoV-2-mNeonGreen at MOI 0.1. Infection rates were assessed at 24 h post-infection (hpi) by measuring the integrated mean fluorescence intensity (iMFI) of infected cells by flow cytometry. Results are the mean of 2 independent experiments ± SD.
(C) Viral production in supernatants from HEK-ACE2 cells infected with SARS-CoV-2 at MOI 0.1 and treated with the indicated concentrations of Gilteritinib for 48 h was determined by plaque assay on Vero E6 cells. Titers from 2 independent experiments are provided as plaque-forming units (PFU)/mL ± SD.
(D) For the quantification of type I IFN in the culture medium, STING-37 cells were incubated for 24 h with the supernatant of HEK-ACE2 treated with increasing concentrations of Gilteritinib and infected with SARS-CoV-2 at MOI 0.1 for 24 h or with known concentrations of recombinant IFN-α2. Data represent the mean ± SD of three independent experiments performed in duplicate.
(E) HEK-ACE2 cells were untreated or treated with 1 μL of human anti-IFNAR2 and/or Gilteritinib at the indicated concentrations. Cells were then immediately infected with SARS-CoV-2-mNeonGreen at MOI 0.1. Results are the mean of 2 independent experiments ± SD.
(F) A549-ACE2, A549, and HeLa cells were treated with Gilteritinib at the indicated concentrations concomitantly with viral infection with SARS-CoV-2 (A549-ACE2), IAV, WNV (A549), or HIV-1 (HeLa) at MOI 0.1 for 24 h. The effect of Gilteritinib on viral replication was assessed by qRT-PCR detection of viral RNA (SARS-CoV-2, IAV, and WNV) or by measuring luminescence (HIV-1 Luc). Data represent the mean of two independent experiments performed in triplicate ± SD.
(G) HEK-ACE2 cells were transfected with small interfering RNA (siRNA) targeting CypB (CTR), AXL, FLT3, IRF3, or IRF7. At 24 hpt, cells were infected with SARS-CoV-2 at MOI 0.1 for 24 h, and the expression of IFN-α4, IFN-β, CXCL10, and SARS-CoV-2 transcripts was quantified by qRT-PCR. Results show the mean ± SD of 3 experiments performed in duplicate. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05, as determined by two-way ANOVA with Dunnett post hoc test. ns, non-significant.
We reasoned that if Gilteritinib inhibits SARS-CoV-2 replication through an enhancement of the cell’s innate immune response, then it should also block the replication of other viruses that are sensitive to IFN. To test this hypothesis, we investigated the effect of Gilteritinib on unrelated RNA viruses, IAV, WNV, and HIV-1, and found that it also has potent antiviral activity against these viruses, confirming its broad-spectrum antiviral activity (Figure 6F). Furthermore, we note that Gilteritinib had no immunostimulatory effect in the absence of viral infection (Figure 2F), suggesting that it acts as a potentiator of viral sensing rather than as an agonist of IFN response.
To determine the molecular basis for Gilteritinib’s immunostimulatory activity, we knocked down the two main receptor tyrosine kinases targeted by Gilteritinib, FLT3 and AXL (Mori et al., 2017), as well as two downstream IRF transcription factors (Figure S5B). Cells were then treated with increasing doses of Gilteritinib and infected with SARS-CoV-2. Knock down of AXL and IRF7, and, to a lesser extent, IRF3, prevented the dose-dependent expression of IFN-α4, IFN-β, and CXCL10 by Gilteritinib and partially reversed the antiviral effect on SARS-CoV-2 (Figure 6G). Together, these experiments indicated that Gilteritinib activates innate immune responses to infection by blocking AXL, which is an inhibitor of the innate immune response (Rothlin et al., 2007). This immunostimulatory effect is transduced at least in part by IRF7, which turns on the production of type I IFN and activates hundreds of genes encoding antiviral proteins (Barnes et al., 2004). In conclusion, our approach, using protein complementation in the nucleus to quantify the activation of signaling pathways, allowed us to successfully identify an immunomodulatory compound of innate response to viral infection that has broad antiviral activity.
Discussion
There is a growing interest to develop antivirals that target the infected cell rather than the virus. In particular, compounds that specifically promote or enhance protective immune responses in target cells could potentially act as non-specific antivirals that stimulate first-line defenses against emerging pathogens. We reasoned that by promoting the IRF/IFN response, such molecules would promote a beneficial antiviral response, without affecting the nuclear factor κB (NF-κB) pathway associated with deleterious inflammatory responses, as reported in patients COVID-19, for instance (Hadjadj et al., 2020). The specific and sensitive AlphaCen assays that we developed allow us to screen for immunomodulatory drugs in the context of a viral infection. The use of protein-complementation assays to assess the nuclear translocation of transcription factors offers multiple advantages, including a pre-translated reporter system that is not sensitive to the shutdown of the cellular translation machinery, as is frequently observed in viral infections or to genotoxic molecules, and a palette of IRFs that can be extended to other transcription factors to allow for the customized screening of signaling pathways.
All viruses have evolved effective countermeasures to antagonize or evade host-cell immune defenses (Bowie and Unterholzner, 2008; Chan and Gack, 2016; Garcia-Sastre, 2017; Rojas et al., 2021). SARS-CoV-2, in particular, inhibits or evades multiple steps of IRF3 signaling (Sa Ribero et al., 2020; Xia et al., 2020), including antagonizing the translocation of IRF3 or its ISGlation, leading to a poor or delayed IFN response (Rebendenne et al., 2021; Yin et al., 2021). Intriguingly, although inhibiting the SARS-CoV-2 PLP restored the nuclear translocation of IRF3, it did not lead to NanoLuc complementation in the nucleus, suggesting that SARS-CoV-2 might also antagonize IRF-3 after nuclear import, as has been reported for other viruses (Chiang and Liu, 2018).
To bypass the diverse viral evasion mechanisms of host-cell immune defenses, it was necessary to test viruses in multiple IRF-α/nls and/CBP AlphaCen assays to determine those that were least impacted by viral infection and offered the best signal amplitude. Although IRF3 is considered to be the central mediator of the IFN response in most cells, we found that infection triggered different IRFs depending on the virus and the tested cell line. In particular, both IRF5- and IRF7-α/CBP AlphaCen efficiently detected SARS-CoV-2. A recent study also found that IRF5 mediates IFN signaling transduction in response to SARS-CoV-2 infection to the same degree as IRF3; IRF7, however, was not activated, possibly as a result of late-phase suppression of IRF7-mediated IFN response (Yin et al., 2021). Here, we show that IRF7 can indeed be activated by SARS-CoV-2 if it is expressed in cells prior to infection by pre-treatment with IFN-α or ectopic expression. This strengthens the notion that immunostimulatory molecules that promote an early or more efficient IFN response can have broad antiviral action by giving cells the advantage over the virus.
To test this hypothesis, we performed a proof-of-concept screen of a small panel of kinase and SARS-CoV-2 inhibitors, using IRF7-α/CBP AlphaCen as read-out. We thus identified Gilteritinib as a potentiator of the SARS-CoV-2-induced immune response and a broad-acting antiviral molecule. Gilteritinib is a FLT3/AXL tyrosine kinase inhibitor that has been approved for the treatment of acute myeloid leukemia since 2018 (Dhillon, 2019; Mori et al., 2017). Although it inhibits many of its downstream targets (e.g., STAT5, ERK, Akt), which accounts for its inhibitory effect in our IRF3-α/CBP AlphaCen assay, it was shown to promote IRF7 phosphorylation and activation (Mathew et al., 2018), thus providing a possible mechanism for its ability to potentiate innate immunity in the context of viral infection. We confirmed that Gilteritinib’s immunostimulatory effect is transduced at least in part by IRF7 and that this is dependent on inhibition of AXL and not FLT3.
Potent antiviral activity was previously reported for Gilteritinib with an IC50 of 0.807 μM (Bouhaddou et al., 2020; Stukalov et al., 2021), but its mechanism was not known. Here, we confirm a submicromolar IC50 of Gilteritinib in our assays and show that it is an enhancer of IRF7-mediated IFN responses and that its antiviral effect is derived from its ability to induce IFN production in infected cells. Concordantly, we show that Gilteritinib has potent antiviral activity against four unrelated RNA viruses, SARS-CoV-2, WNV, HIV-1, and IAV. Gilteritinib also appears to have an antiviral effect that is independent of IFN since it still has residual antiviral activity in the presence of anti-IFNAR antibodies. This may be explained by the fact that, in addition to type I IFN, IRF7 also activates many genes, known as early ISGs, including ISG15, IFIT1, CXCL10, or Viperin, that can display an IFN-independent antiviral activity and could therefore contribute to the antiviral effect of Gilteritinib.
Our approach can be applied to identify efficient and specific immunomodulators applicable to the treatment of viral infections, cancer, and immune disorders. As such, they can be deployed in an emergency to screen for non-specific antivirals against poorly characterized or emerging viruses. In some cases, increasing the IFN response may not be sufficient to block viral replication. SARS-CoV-2, for instance, inhibits the JAK/STAT pathway downstream of IFN (Chen et al., 2020; Miorin et al., 2020; Sa Ribero et al., 2020). In the case of SARS-CoV-2, it may be more relevant to identify compounds that accelerate the IFN response rather than potentiators, since it was shown to trigger a potent but delayed IFN response (Rebendenne et al., 2021; Yin et al., 2021). Further work will be required to determine whether Gilteritinib or analog molecules can accelerate the virus-induced IFN response to limit replication and spread. This may require us to perform kinetic AlphaCen screens to identify molecules that boost IFN responses before the virus has the opportunity to perform a full replication cycle and start spreading.
Alternatively, the AlphaCen system can also be adapted to screen for inhibitors of innate immunity that can be applicable to chronic infections, as demonstrated by the treatment of chronic lymphocytic choriomeningitis virus (LCMV) in mice (Teijaro et al., 2013) and auto-immune diseases.
Limitations of the study
The screening system to monitor IRF nuclear translocation as an early hallmark of innate immune activation that is described in this article was established in transformed cell lines (e.g., HEK-293T, HeLa, A549). Translating the assay in primary cells that are relevant to innate immune responses following infection will be important. The limitations of the compound screen are that it includes only a small number of compounds and that the potential mechanisms of action for the immune-enhancing effects of the viral RdRp-targeting antiviral drugs ribavirin, Remdesivir, and Favipiravir are not explored. Another limitation concerns the mechanism of action of Gilteritinib, which was shown to involve AXL and IRF7. Further experiments will be required to unravel the connection between AXL and the IFN response following action by Gilteritinib.
Significance
While the world is hotly discussing how to better prepare for the next pandemic, many agree that there is an urgent need to discover original molecules that have broad antiviral properties. Since IFN is a powerful and universal antiviral factor, compounds that can specifically enhance the innate immune response to infection could constitute potent broad-acting antivirals. However, they are intrinsically difficult to screen for, since viral infection interferes with reporter cell lines by disrupting transcription and shutting down translation, and most viruses have evolved to evade the host’s innate immunity.
To address this critical gap in our preparedness, we devised a series of assays based on protein complementation that quantify the nuclear translocation of IRFs following infection by high-throughput screening. This approach offers multiple advantages, including a pre-translated reporter system that is not sensitive to the shutdown of the cellular translation machinery and a palette of IRFs that allows the customized screening of signaling pathways following any viral infection.
We found that different RNA viruses activate IRFs differentially, and we harnessed these differences to perform a proof-of-concept screen of SARS-CoV-2 infection. Using a small library of kinase inhibitors and known antivirals on SARS-CoV-2 infection, we identified Gilteritinib as a potent antiviral that acts by inhibiting AXL and increasing IFN production by infected cells. As a result of its immunostimulatory activity, Gilteritinib inhibited infection by unrelated RNA viruses, such as WNV, IAV, and HIV-1, thus confirming it has broad-spectrum activity.
Our article offers an innovative approach toward the development of efficient host-directed antivirals.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-RIG-I, clone Alme-1 | AdipoGen Life Sciences | Cat# AG-20B-0009; RRID: AB_2490189 |
| Rabbit anti-MDA5, clone D74E4 | Cell Signaling Technology | Cat# 5321; RRID: AB_10694490 |
| Mouse anti-IRF1, clone H8 | Santa Cruz Biotechnology | Cat# sc-74530; RRID: AB_2126826 |
| Rabbit anti-IRF3, clone D6I4C | Cell Signaling Technology | Cat# 11904; RRID: AB_2722521 |
| Rabbit anti-IRF5, clone E7F9W | Cell Signaling Technology | Cat# 76983; RRID: AB_2920569 |
| Rabbit anti-IRF7 | Proteintech | Cat# 22392-1-AP; RRID: AB_2879097 |
| Mouse anti-IRF7, clone G-8 | Santa Cruz Biotechnology | Cat# sc-74472; RRID: AB_2280489 |
| Rabbit anti-Flag, clone D6W5B | Cell Signaling Technology | Cat# 14793; RRID: AB_2572291 |
| Mouse anti-Flag, clone M2 | Sigma-Aldrich | Cat# F3165; RRID: AB_259529 |
| Rat anti-HA, clone 3F10 | Roche | Cat# 11867423001; RRID: AB_390918 |
| Mouse anti-GAPDH, clone 6C5 | Sigma-Aldrich | Cat# MAB374; RRID: AB_2107445 |
| Goat anti-mouse AF488 | Thermo Fisher Scientific | Cat# A-11029; RRID: AB_2534088 |
| Goat anti-rabbit AF488 | Thermo Fisher Scientific | Cat# A-11034; RRID: AB_2576217 |
| Goat anti-rat AF647 | Thermo Fisher Scientific | Cat# A-21247; RRID: AB_141778 |
| Mouse IgG HRP Linked Whole Ab | GE Healthcare | Cat# NA931; RRID: AB_772210 |
| Rabbit IgG HRP Linked Whole Ab | GE Healthcare | Cat# NA934; RRID: AB_772206 |
| Recombinant human anti-IFNAR2, clone REA124 | Miltenyi Biotec | Cat# 130-099-555; RRID: AB_2652222 |
| Bacterial and virus strains | ||
| H4 SeV | (Strahle et al., 2006) | N/A |
| IAV A/WSN/33 (H1N1) | Institut Pasteur, Paris, France | N/A |
| WNV-Tunisia-1997 | Dr I. Leparc Goffart (French National Reference Center on Arboviruses, Marseille, France) | N/A |
| HIV-1 NL4-3 Luc | (He et al., 1995) | N/A |
| SARS-CoV-2 (BetaCoV/France/IDF0372/2020) | Institut Pasteur, Paris, France | N/A |
| SARS-CoV-2-mNeonGreen | (Xie et al., 2020) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Gilteritinib | Chemietek | Cat# CT-GILT CAS: 1254053-43-4 |
| BLZ-945 | MedChemExpress | Cat# HY-12768 CAS: 953769-46-5 |
| Nintedanib | MedChemExpress | Cat# HY-50904 CAS: 656247-17-5 |
| PS-1145 | Selleckchem | Cat# S7691 CAS: 431898-65-6 |
| PD98059 | MedChemExpress | Cat# HY-12028 CAS: 167869-21-8 |
| RO-31-8425.HCl | MedChemExpress | Cat# HY-108136A CAS: 145317-11-9 |
| Semaxinib | MedChemExpress | Cat# HY-10374 CAS: 204005-46-9 |
| BX-795 | MedChemExpress | Cat# HY-10514 CAS: 702675-74-9 |
| Amlexanox | MedChemExpress | Cat# HY-B0713 CAS: 68302-57-8 |
| U-0126-EtOH | MedChemExpress | Cat# HY-12031 CAS: 1173097-76-1 |
| Rapamycin | MedChemExpress | Cat# HY-10219 CAS: 53123-88-9 |
| SB-202190 | MedChemExpress | Cat# HY-10295 CAS: 152121-30-7 |
| BAY 11-7085 | MedChemExpress | Cat# HY-10257 CAS: 196309-76-9 |
| Wortmannin | MedChemExpress | Cat# HY-10197 CAS: 19545-26-7 |
| Staurosporine | MedChemExpress | Cat# HY-15141 CAS: 62996-74-1 |
| BAY 11-7082 | MedChemExpress | Cat# HY-13453 CAS: 19542-67-7 |
| AG 490 | MedChemExpress | Cat# HY-12000 CAS: 133550-30-8 |
| MRT67307 | MedChemExpress | Cat# HY-13018 CAS: 1190378-57-4 |
| BCI - 000209 | BCI PHARMA | N/A |
| BCI - 001736 | BCI PHARMA | N/A |
| BCI - 001775 | BCI PHARMA | N/A |
| Ribavirin | Merck | Cat# R9644 CAS: 36791-04-5 |
| Remdesivir | MedChemExpress | Cat# HY-104077 CAS: 1809249-37-3 |
| Favipiravir | MedChemExpress | Cat# HY-14768 CAS: 259793-96-9 |
| Hydroxychloroquine | Merck | Cat# H0915 CAS: 747-36-4 |
| 4-HPR | Merck | Cat# 390900 CAS: 65646-68-6 |
| Scutellarein | MedChemExpress | Cat# HY-N0752 CAS: 529-53-3 |
| GRL-0617 | MedChemExpress | Cat# HY-117043 CAS: 1093070-16-6 |
| Recombinant Human IFN-α2a | R&D Systems | Cat# 11100-1 |
| Ivermectin | Merck | Cat# I8898 CAS: 70288-86-7 |
| Critical commercial assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7570 |
| Nano-Glo® Luciferase Assay System | Promega | Cat# N1110 |
| Nano-Glo® Live Cell Assay System | Promega | Cat# N2011 |
| Experimental models: Cell lines | ||
| HeLa | ATCC | Cat# CCL-2 |
| A549 | ATCC | Cat# CCL-185 |
| HEK293T/17 | ATCC | Cat# CRL-11268 |
| A549-ACE2 | Olivier Schwartz (Institut Pasteur, Paris, France) | Derived from ATCC Cat# CCL-185 |
| HEK-ACE2 | Olivier Schwartz (Institut Pasteur, Paris, France) | Derived from ATCC Cat# CRL-11268 |
| Vero | ATCC | Cat# CCL-81 |
| Vero E6 | Merck | Cat# 85020206 |
| HCT116 | ATCC | Cat# CCL-247 |
| STING-37 | (Lucas-Hourani et al., 2013) | N/A |
| C6/36 | ATCC | Cat# CRL-1660 |
| Oligonucleotides | ||
| ON-TARGETplus Human PPIB (5479) siRNA SMARTpool | Horizon Discovery | Cat# L-004606 |
| ON-TARGETplus Human IRF3 (3661) siRNA SMARTpool | Horizon Discovery | Cat# L-006875 |
| ON-TARGETplus Human IRF7 (3665) siRNA SMARTpool | Horizon Discovery | Cat# L-011810 |
| ON-TARGETplus Human AXL (558) siRNA SMARTpool | Horizon Discovery | Cat# L-003104 |
| ON-TARGETplus Human FLT3 (2322) siRNA SMARTpool | Horizon Discovery | Cat# L-003137 |
| All qPCR primers are shown in Table S1 | N/A | |
| Recombinant DNA | ||
| pEFBOS(+)-Flag-2CARD | (Yoneyama et al., 2004) | N/A |
| pCenNLS-NLuc | This paper | N/A |
| pCenCBP-NLuc | This paper | N/A |
| pIRF1-α | This paper | N/A |
| pIRF3-α | This paper | N/A |
| pIRF3-α | This paper | N/A |
| pIRF7-α | This paper | N/A |
| Software and algorithms | ||
| Fiji | Open source, GNU General Public License. | N/A |
| FlowJo | Treestar Inc. | N/A |
| FACSDiva | Becton Dickinson | N/A |
| GraphPad Prism | GraphPad Software | N/A |
| Other | ||
| Hoechst 33342 | ThermoFisher Scientific | Cat# H3570 |
| Fluoromount™ Aqueous Mounting Medium | Merck | Cat# F4680 |
| Basticidin | Invivogen | Cat# ant-bl-05 |
| Gibco™ Geneticin™ Selective Antibiotic (G418 Sulfate) | ThermoFisher Scientific | Cat# 11811098 |
| FuGENE® 6 Transfection Reagent | Promega | Cat# E2691 |
| HiPerFect Transfection Reagent | Qiagen | Cat# 301705 |
| Bright-Glo™ Luciferase Assay System | Promega | Cat# E2610 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Sébastien Nisole (sebastien.nisole@inserm.fr).
Materials availability
All requests for resources and reagents should be directed to and will be fulfilled by the lead contact. Alpha Centauri constructs are proprietary and can be obtained through a Materials Transfer Agreement. Other materials will also be available from the lead contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Cell lines and culture
HEK293T (CRL-11268, human, female), A549 (CCL-185, human, male), Vero (CCL-81, African green monkey, female), HeLa (CCL-2, human, female), HCT116 (CCL-247, human, male) and C6/36 (CRL-1660, Aedes albopictus, male) cells were obtained from the American Type Culture Collection (ATCC). Vero E6 (ECACC #85020206, African green monkey, female) were purchased from Merck. HEK293T and A549 stably expressing ACE2 (HEK-ACE2 and A549-ACE2) were kindly provided by Olivier Schwartz (Institut Pasteur, Paris, France). STING-37 cells were kindly provided by Pierre-Olivier Vidalain (CIRI, Lyon, France). All cell lines were cultured in Dulbecco’s modified Eagle Medium (DMEM, Gibco, Cat#61965059) supplemented with 10% fetal bovine serum (Serana, Cat#S-FBS-NL-015), 1% Penicillin/Streptomycin (Gibco, Cat#15070063). All cell types were maintained in 5% CO2 at 37 °C. ACE2 expressing cells were additionally maintained in blasticidin (Invivogen) at 10 μg/mL. When indicated, HEK293T cells and HEK-ACE2 were treated with 250 IU/mL of recombinant human IFN-α2a (R&D systems, Cat#11100–1) for 16 h prior to infection.
Viruses
The strain BetaCoV/France/IDF0372/2020 (SARS-CoV-2) was supplied by the National Reference Centre for Respiratory Viruses hosted by Institut Pasteur (Paris, France) and headed by Sylvie van der Werf. The SARS-CoV-2-mNeonGreen was obtained from Pei-Yong Shi (Department of Biochemistry and Molecular Biology, University of Texas Medical Branch, Galveston, TX, USA) (Xie et al., 2020). Both viruses were amplified on Vero E6 cells (ECACC #85020206) at MOI 0.001. At 3 days post infection, the supernatant was harvested and cleared by centrifugation at 2000 × g for 5 min at 4 °C. The cleared virus-containing supernatant was frozen in 1 mL aliquots at −80°C. For each virus production, a vial was thawed for titration by plaque assay in Vero E6 cells to estimate plaque-forming units per mL of virus (PFU/mL). Viral titers ranged between 3 × 106 and 3 × 107 PFU/mL.
Defective-interfering H4 SeV was provided by Dominique Garcin (Department of Microbiology and Molecular Medicine, University of Geneva, Geneva, Switzerland) and used at 40 hemagglutination units (HAU)/mL (Strahle et al., 2006).
The HIV-1 plasmid pNL4-3.Luc.R-E− was kindly provided by N. Landau (Aaron Diamond AIDS Research Center, The Rockefeller University, New-York, USA) (He et al., 1995). VSV-G pseudotyped luciferase-encoding HIV-1 was produced by transient transfection of HEK293T with pNL4-3.Luc.R-E− and pVSV-G.
The A/WSN/33 (H1N1) virus was kindly provided by Sandie Munier (Unité de Génétique Moléculaire des Virus à ARN, Institut Pasteur, Paris, France). It was produced by reverse genetics and amplified and titrated on Madin-Darby Canine Kidney cells (MDCK) cells.
A lineage 1 clinical strain of WNV was used in this study. The strain was isolated from a human brain during the epidemic that occurred in Tunisia in 1997 and was provided by Isabelle Leparc-Goffart (French National Reference Center on Arboviruses, Marseille, France). The viral stock was produced on the Ae. albopictus cells clone C6/36 and supernatants were collected at 5 days after infection. Viral stock titers were determined on Vero-81 cells.
All cell lines were cultured in Dulbecco’s modified Eagle Medium (DMEM, Gibco, Cat#61965059) supplemented with 10% fetal bovine serum (Serana, Cat#S-FBS-NL-015), 1% Penicillin/Streptomycin (Gibco, Cat#15070063). All cell types were maintained in 5% CO2 at 37°C. ACE2 expressing cells were additionally maintained in blasticidin (Invivogen) at 10 μg/mL. When indicated, HEK293T cells and HEK-ACE2 were treated with 250 IU/mL of recombinant human IFN-α2a (R&D systems) for 16 h prior to infection.
Method details
Cell transfections
All plasmid transfections were performed using FuGENE® 6 Transfection Reagent (Promega) according to the manufacturer’s instructions. The pEFBOS(+)-Flag-2CARD plasmid was provided by M. Si Tahar (Centre d'Étude des Pathologies Respiratoires, Tours, France) (Yoneyama et al., 2004). The Cen-nls plasmid was described previously (Fernandez et al., 2021). The IRF-α and Cen-CBP constructs were synthesized by Genscript. The expression of IRFs and Cen constructs was assessed at 24 h post-transfection (hpt), unless otherwise stated, by flow cytometry, western blotting, or indirect immunofluorescence using anti-Flag and anti-HA antibodies, respectively. Transfections of siRNA were performed using HiPerFect Transfection Reagent (Qiagen) according to the manufacturer’s instructions.
Indirect immunofluorescent labeling and confocal imaging
Cells fixed in 4% paraformaldehyde (Alfa Aesar) for 10 min were permeabilized in 0.5% Triton for 15 min, neutralized with 50 mM NH4Cl for 10 min and blocked with 0.3% BSA for 10 min. Cells were incubated with primary and secondary antibodies for 1 h and 30 min, respectively, at room temperature in a wet chamber. Primary antibodies were mouse and rabbit anti-Flag, rat anti-HA, rabbit anti-IRF1 (Santa Cruz), IRF3 (Cell signalling), IRF5 (Cell signalling), IRF7 (Santa Cruz). Secondary antibodies were goat anti-mouse Alexa 488, anti-rabbit Alexa 555, anti-rat Alexa 647. Nuclei were stained using Hoechst (Invitrogen). All images were acquired using a LSM880 (Zeiss) confocal microscope using a 63× oil immersion objective, in confocal or Airyscan mode (as indicated) and processed using FiJi. Representative images are shown using artificial colouring.
Measuring fluorescence intensity in nuclear and cytoplasmic region of interest (ROIs)
IRF signal intensity was measured in the nuclei and cytoplasms from confocal planes using FiJi. Nuclei were analyzed by automatic particle detection of the Hoechst labelling (with size 40 μm-infinity). Whole cells were delineated using freehand selection and the cytoplasmic space was defined by subtracting ROIs using XOR (exclusive OR) operation. Mean gray values were measured for all ROIs in an average of 30 cells per condition from 3 independent experiments.
RT-qPCR analyses
Total RNA was extracted using a RNeasy Mini kit and submitted to DNase treatment (Qiagen), following the manufacturer’s instructions. RNA concentration and purity were evaluated by spectrophotometry (NanoDrop, 2000c; Thermo Fisher Scientific). In addition, 500 ng of RNA were reverse transcribed with both oligo dT and random primers, using a PrimeScript RT Reagent Kit (Perfect Real Time, Takara Bio Inc.) in a 10 μL reaction. Real-time PCR reactions were performed in duplicate using Takyon ROX SYBR MasterMix blue dTTP (Eurogentec) on an Applied Biosystems QuantStudio 5 (Thermo Fisher Scientific). Transcripts were quantified using the following program: 3 min at 95°C followed by 35 cycles of 15 s at 95°C, 20 s at 60°C, and 20 s at 72°C. Values for each transcript were normalized to expression levels of RPL13A (60S ribosomal protein L13a), using the 2−ΔΔCt method. Primers used for quantification of transcripts by real-time quantitative PCR are listed in Table S1.
Bioluminescent imaging
HEK293T cells were co-transfected with IRF3-⍺ and Cen-nls. After 24 h, cells were transferred to glass-bottomed black 96-well plates (20,000 cells/well). Cells were incubated with NanoGlo Live Cell substrate (Promega) immediately prior to imaging.
Kinase inhibitors and antivirals screened using AlphaCen assays
All chemical compounds were purchased at a purity >99%, resuspended at 10 mM in DMSO and stored at −80°C. To perform the screens, molecules were further diluted in DMEM 0% SVF at a concentration of 50, 10 or 2 μM, in a final concentration of DMSO of 0.5%.
Luminescent plate assays
IRF-⍺ and Cen expressing cells were lysed in NanoGlo substrate (Promega) according to the manufacturer’s instructions. Lysates were transferred in white 96-well plates (50,000 cell equivalents/well) and luminescence was read within 5 min using a Tecan Infinity 200 luminometer.
Cell viability
Cell viability was assessed using the CellTiter assay (Promega) according to the manufacturer’s instructions. Lysates were transferred in black 96-well plates (50,000 cell equivalents/well) and luminescence was read within 10 min using a Tecan Infinity 200 luminometer.
Flow cytometry
For the detection of SARS-CoV-2-mNeonGreen, 5 × 105 HEK-ACE2 cells were treated with the indicated drugs and simultaneously infected with SARS-CoV-2-mNeonGreen at MOI 0.1. When indicated, cells were also treated with 1μL recombinant human anti-IFNAR2 for 50,000 cells (Miltenyi Biotec). At 24 hpi, cells were fixed with 4% formaldehyde for 30 min. For all other experiments, cells were fixed with 2% formaldehyde (Alfa Aesar) for 30 min and permeabilized in a PBS/1% BSA/0.05% saponin solution for 30 min prior to staining with primary antibodies diluted in the permeabilization solution for 1h at 4°C and followed by secondary antibodies for 30 min at 4°C. All acquisitions were performed on a Fortessa cytometer (BD Biosciences), data were collected with FACSDiva software (Becton Dickinson) and were processed with FlowJo software (Treestar Inc., Oregon, USA).
Quantification of secreted IFN
IFN secreted in the culture medium was titrated on STING-37 reporter cells, which correspond to HEK293 cells stably expressing an IFN-stimulated response element (ISRE)-luciferase reporter gene (Lucas-Hourani et al., 2013). A standard curve was established by applying known titers of recombinant human IFN-α2a (R&D Systems) onto STING-37 cells. Luciferase induction in STING-37 cells was determined using the Bright-Glo reagent (Promega), according to the manufacturer’s recommendations.
Western blot
Cell lysates were denatured and loaded on 10% ProSieve gel (Lonza), then subjected to electrophoresis. Chemiluminescent acquisitions were acquired on a Chemidoc™ MP Imager and analyzed using Image Lab™ desktop software (Bio-Rad Laboratories).
Quantification and statistical analysis
All experiments were executed multiple times and independently by different experimentators. The screening and their analyses were performed blindly. No data were excluded from the analyses. Details on quantification are found in figure legends and in Methods details sections. All results are displayed as means ± standard deviation of the means (SD). All the statistical analyses were performed with GraphPad Prism 9 and the statistical significance was calculated using the unpaired Student’s t test, or one-way ANOVA followed by Bonferroni or Dunnett post-hoc test, depending on the experiments. The statistical test used for each experiment is indicated in the respective figure legend. Significance is indicated with ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, or ns = not significant in the corresponding graphs.
Acknowledgments
We thank Pierre-Olivier Vidalain (CIRI, Lyon, France) and Fabien Blanchet (IRIM, Montpellier, France) for helpful discussions. We acknowledge the imaging facility MRI, member of the national infrastructure France-BioImaging supported by the French National Research Agency (ANR-10-INBS-04, “Investissements d’avenir”), and Sylvain de Rossi, in particular, for help with bioluminescence imaging. We thank Christine Chable and Delphine Muriaux at the CEMIPAI BSL-3 facility. We thank Olivier Schwartz (Institut Pasteur, Paris, France), Charles Bodet (LITEC, Université de Poitiers, Poitiers, France), Mustapha Si Tahar (Centre d'Étude des Pathologies Respiratoires, Tours, France), and Sandie Munier (Institut Pasteur) for sharing reagents. We are very grateful to BCI Pharma, in particular to Dominique Surleraux, Philippe Masson, and Remi Guillon, for providing the kinase inhibitor library and sharing their expertise. This work was supported by the Labex EpiGenMed, an Investissements d’avenir program (ANR-10-LABX-12-01 to N.J.A. and S.N.), the Agence Nationale de la Recherche (ANR-20-COVI-000, project Alpha-COV to S.N.), the Occitanie Region (project Alpha-COV to S.N.), and the SATT AxLR (to N.J.A. and S.N.). G.M. is supported by a grant from the Agence Nationale de la Recherche sur le SIDA et les Hépatites virales (ANRS). M.-F.M., J.L., and A.Z. benefited from grants from the Labex EpiGenMed. A.B. is supported by a grant from Sidaction. P.N. and J.F. were supported by grants from the SATT AxLR.
Author contributions
N.J.A. and S.N. conceptualized, designed, and coordinated the study. G.M., M.-F.M, A.Z., A.B., P.N., J.F., J.L., N.J.A., and S.N. conducted the experiments. G.M., M.-F.M, A.Z., J.F., N.J.A., and S.N. analyzed and interpreted the data. D.G. and R.G. provided tools and reagents. G.M., N.J.A., and S.N. wrote and revised the manuscript. All authors read and approved the final manuscript.
Declaration of interests
S.N. and N.J.A. are the inventors of a filed patent for the AlphaCen technology described in this manuscript.
Published: June 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chembiol.2022.05.009.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
References
- Barnes B.J., Moore P.A., Pitha P.M. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J. Biol. Chem. 2001;276:23382–23390. doi: 10.1074/jbc.m101216200. [DOI] [PubMed] [Google Scholar]
- Barnes B.J., Richards J., Mancl M., Hanash S., Beretta L., Pitha P.M. Global and distinct targets of IRF-5 and IRF-7 during innate response to viral infection. J. Biol. Chem. 2004;279:45194–45207. doi: 10.1074/jbc.m400726200. [DOI] [PubMed] [Google Scholar]
- Baum A., García-Sastre A. Induction of type I interferon by RNA viruses: cellular receptors and their substrates. Amino Acids. 2010;38:1283–1299. doi: 10.1007/s00726-009-0374-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D., et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhaddou M., Memon D., Meyer B., White K.M., Rezelj V.V., Correa Marrero M., Polacco B.J., Melnyk J.E., Ulferts S., Kaake R.M., et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell. 2020;182:685–712.e19. doi: 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie A.G., Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broggi A., Ghosh S., Sposito B., Spreafico R., Balzarini F., Lo Cascio A., Clementi N., De Santis M., Mancini N., Granucci F., Zanoni I. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science. 2020;369:706–712. doi: 10.1126/science.abc3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., et al. A trial of lopinavir-ritonavir in adults hospitalized with severe covid-19. N. Engl. J. Med. 2020;382:1787–1799. doi: 10.1056/nejmoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan Y.K., Gack M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016;14:360–373. doi: 10.1038/nrmicro.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.Y., Khan N., Close B.J., Goel R.K., Blum B., Tavares A.H., Kenney D., Conway H.L., Ewoldt J.K., Kapell S., et al. SARS-CoV-2 desensitizes host cells to interferon through inhibition of the JAK-STAT pathway. bioRxiv. 2020 Preprint at. [Google Scholar]
- Chiang H.S., Liu H.M. The molecular basis of viral inhibition of IRF- and STAT-dependent immune responses. Front. Immunol. 2018;9:3086. doi: 10.3389/fimmu.2018.03086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitalia V.C., Munawar A.H. A painful lesson from the COVID-19 pandemic: the need for broad-spectrum, host-directed antivirals. J. Transl. Med. 2020;18:390. doi: 10.1186/s12967-020-02476-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Solidarity Trial Consortium. Pan H., Peto R., Henao-Restrepo A.M., Preziosi M.P., Sathiyamoorthy V., Abdool Karim Q., Alejandria M.M., Al-Bader A.M., Kieny M.P., Murthy S., et al. Repurposed antiviral drugs for covid-19 - interim WHO solidarity trial results. N. Engl. J. Med. 2021;384:497–511. doi: 10.1056/NEJMoa2023184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon S. Gilteritinib: first global approval. Drugs. 2019;79:331–339. doi: 10.1007/s40265-019-1062-3. [DOI] [PubMed] [Google Scholar]
- Es-Saad S., Tremblay N., Baril M., Lamarre D. Regulators of innate immunity as novel targets for panviral therapeutics. Curr. Opin. Virol. 2012;2:622–628. doi: 10.1016/j.coviro.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H., Zhang Y.B., Gui J.F., Lemon S.M., Yamane D. Interferon regulatory factor 1 (IRF1) and anti-pathogen innate immune responses. PLoS Pathog. 2021;17:e1009220. doi: 10.1371/journal.ppat.1009220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez J., Hassen-Khodja C., Georget V., Rose T., Jacob Y., Janin Y.L., Nisole S., Vidalain P.O., Arhel N.J. Measuring the subcellular compartmentalization of viral infections by protein complementation assay. Proc. Natl. Acad. Sci. U S A. 2021;118 doi: 10.1073/pnas.2010524118. e2010524118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman R.M., Contente S. Interferons as therapy for viral and neoplastic diseases: from panacea to pariah to paragon. Pharmaceuticals. 2009;2:206–216. doi: 10.3390/ph2030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galani I.E., Triantafyllia V., Eleminiadou E.E., Koltsida O., Stavropoulos A., Manioudaki M., Thanos D., Doyle S.E., Kotenko S.V., Thanopoulou K., Andreakos E. Interferon-lambda mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity. 2017;46:875–890.e6. doi: 10.1016/j.immuni.2017.04.025. [DOI] [PubMed] [Google Scholar]
- García-Sastre A. Ten strategies of interferon evasion by viruses. Cell Host Microbe. 2017;22:176–184. doi: 10.1016/j.chom.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J., Choe S., Walker R., Di Marzio P., Morgan D.O., Landau N.R. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995;69:6705–6711. doi: 10.1128/jvi.69.11.6705-6711.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K., Yanai H., Negishi H., Asagiri M., Sato M., Mizutani T., Shimada N., Ohba Y., Takaoka A., Yoshida N., Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Liu H., Ye F., Sun Q., Liang H., Li C., Li S., Lu R., Huang B., Tan W., Lai L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J. Enzym. Inhib. Med. Chem. 2021;36:497–503. doi: 10.1080/14756366.2021.1873977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lokugamage K.G., Hage A., de Vries M., Valero-Jimenez A.M., Schindewolf C., Dittmann M., Rajsbaum R., Menachery V.D. Type I interferon susceptibility distinguishes SARS-CoV-2 from SARS-CoV. J. Virol. 2020;94 doi: 10.1128/jvi.01410-20. e01410-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas-Hourani M., Dauzonne D., Jorda P., Cousin G., Lupan A., Helynck O., Caignard G., Janvier G., André-Leroux G., Khiar S., et al. Inhibition of pyrimidine biosynthesis pathway suppresses viral growth through innate immunity. PLoS Pathog. 2013;9:e1003678. doi: 10.1371/journal.ppat.1003678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMicking J.D. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 2012;12:367–382. doi: 10.1038/nri3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew N.R., Baumgartner F., Braun L., O'Sullivan D., Thomas S., Waterhouse M., Müller T.A., Hanke K., Taromi S., Apostolova P., et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat. Med. 2018;24:282–291. doi: 10.1038/nm.4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama T., Kimura T., Kitagawa M., Pfeffer K., Kawakami T., Watanabe N., Kündig T.M., Amakawa R., Kishihara K., Wakeham A., et al. Targeted disruption of IRF-1 or IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell. 1993;75:83–97. doi: 10.1016/s0092-8674(05)80086-8. [DOI] [PubMed] [Google Scholar]
- Miorin L., Kehrer T., Sanchez-Aparicio M.T., Zhang K., Cohen P., Patel R.S., Cupic A., Makio T., Mei M., Moreno E., et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc. Natl. Acad. Sci. U S A. 2020;117:28344–28354. doi: 10.1073/pnas.2016650117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondelli M.U. The multifaceted functions of ribavirin: antiviral, immunomodulator, or both? Hepatology. 2014;60:1126–1129. doi: 10.1002/hep.27186. [DOI] [PubMed] [Google Scholar]
- Mori M., Kaneko N., Ueno Y., Yamada M., Tanaka R., Saito R., Shimada I., Mori K., Kuromitsu S. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Invest. N. Drugs. 2017;35:556–565. doi: 10.1007/s10637-017-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelemans T., Kikkert M. Viral innate immune evasion and the pathogenesis of emerging RNA virus infections. Viruses. 2019;11:961. doi: 10.3390/v11100961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokunina-Olsson L., Alphonse N., Dickenson R.E., Durbin J.E., Glenn J.S., Hartmann R., Kotenko S.V., Lazear H.M., O'Brien T.R., Odendall C., et al. COVID-19 and emerging viral infections: the case for interferon lambda. J. Exp. Med. 2020;217:e20200653. doi: 10.1084/jem.20200653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prussia A., Thepchatri P., Snyder J.P., Plemper R.K. Systematic approaches towards the development of host-directed antiviral therapeutics. Int. J. Mol. Sci. 2011;12:4027–4052. doi: 10.3390/ijms12064027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebendenne A., Valadão A.L.C., Tauziet M., Maarifi G., Bonaventure B., McKellar J., Planès R., Nisole S., Arnaud-Arnould M., Moncorgé O., Goujon C. SARS-CoV-2 triggers an MDA-5-dependent interferon response which is unable to control replication in lung epithelial cells. J. Virol. 2021;95 doi: 10.1128/jvi.02415-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas J.M., Alejo A., Martín V., Sevilla N. Viral pathogen-induced mechanisms to antagonize mammalian interferon (IFN) signaling pathway. Cell. Mol. Life Sci. 2021;78:1423–1444. doi: 10.1007/s00018-020-03671-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothlin C.V., Ghosh S., Zuniga E.I., Oldstone M.B., Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- Sa Ribero M., Jouvenet N., Dreux M., Nisole S. Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog. 2020;16:e1008737. doi: 10.1371/journal.ppat.1008737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M., Suemori H., Hata N., Asagiri M., Ogasawara K., Nakao K., Nakaya T., Katsuki M., Noguchi S., Tanaka N., Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- Schaper F., Kirchhoff S., Posern G., Köster M., Oumard A., Sharf R., Levi B.Z., Hauser H. Functional domains of interferon regulatory factor I (IRF-1) Biochem. J. 1998;335:147–157. doi: 10.1042/bj3350147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., Schulz L., Widera M., Mehdipour A.R., Tascher G., et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson D.B., Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Strahle L., Garcin D., Kolakofsky D. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology. 2006;351:101–111. doi: 10.1016/j.virol.2006.03.022. [DOI] [PubMed] [Google Scholar]
- Stukalov A., Girault V., Grass V., Karayel O., Bergant V., Urban C., Haas D.A., Huang Y., Oubraham L., Wang A., et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature. 2021;594:246–252. doi: 10.1038/s41586-021-03493-4. [DOI] [PubMed] [Google Scholar]
- Takaoka A., Yanai H., Kondo S., Duncan G., Negishi H., Mizutani T., Kano S.i., Honda K., Ohba Y., Mak T.W., Taniguchi T. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature. 2005;434:243–249. doi: 10.1038/nature03308. [DOI] [PubMed] [Google Scholar]
- Teijaro J.R., Ng C., Lee A.M., Sullivan B.M., Sheehan K.C.F., Welch M., Schreiber R.D., Carlos de la Torre J., Oldstone M.B.A. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science. 2013;340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boxel-Dezaire A.H., Rani M.S., Stark G.R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Wagstaff K.M., Sivakumaran H., Heaton S.M., Harrich D., Jans D.A. Ivermectin is a specific inhibitor of importin α/β-mediated nuclear import able to inhibit replication of HIV-1 and dengue virus. Biochem. J. 2012;443:851–856. doi: 10.1042/bj20120150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H., Cao Z., Xie X., Zhang X., Chen J.Y.C., Wang H., Menachery V.D., Rajsbaum R., Shi P.Y. Evasion of type I interferon by SARS-CoV-2. Cell Rep. 2020;33:108234. doi: 10.1016/j.celrep.2020.108234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X., Muruato A., Lokugamage K.G., Narayanan K., Zhang X., Zou J., Liu J., Schindewolf C., Bopp N.E., Aguilar P.V., et al. An infectious cDNA clone of SARS-CoV-2. Cell Host. Microbe. 2020;27:841–848.e3. doi: 10.1016/j.chom.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X., Riva L., Pu Y., Martin-Sancho L., Kanamune J., Yamamoto Y., Sakai K., Gotoh S., Miorin L., De Jesus P.D., et al. MDA5 governs the innate immune response to SARS-CoV-2 in lung epithelial cells. Cell Rep. 2021;34:108628. doi: 10.1016/j.celrep.2020.108628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- Yu M.S., Lee J., Lee J.M., Kim Y., Chin Y.W., Jee J.G., Keum Y.S., Jeong Y.J. Identification of myricetin and scutellarein as novel chemical inhibitors of the SARS coronavirus helicase, nsP13. Bioorg. Med. Chem. Lett. 2012;22:4049–4054. doi: 10.1016/j.bmcl.2012.04.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Chassaing B., Shi Z., Uchiyama R., Zhang Z., Denning T.L., Crawford S.E., Pruijssers A.J., Iskarpatyoti J.A., Estes M.K., et al. Prevention and cure of rotavirus infection via TLR5/NLRC4–mediated production of IL-22 and IL-18. Science. 2014;346:861–865. doi: 10.1126/science.1256999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.