

Abstract
Lost Hammer Spring, located in the High Arctic of Nunavut, Canada, is one of the coldest and saltiest terrestrial springs discovered to date. It perennially discharges anoxic (<1 ppm dissolved oxygen), sub-zero (~−5 °C), and hypersaline (~24% salinity) brines from the subsurface through up to 600 m of permafrost. The sediment is sulfate-rich (1 M) and continually emits gases composed primarily of methane (~50%), making Lost Hammer the coldest known terrestrial methane seep and an analog to extraterrestrial habits on Mars, Europa, and Enceladus. A multi-omics approach utilizing metagenome, metatranscriptome, and single-amplified genome sequencing revealed a rare surface terrestrial habitat supporting a predominantly lithoautotrophic active microbial community driven in part by sulfide-oxidizing Gammaproteobacteria scavenging trace oxygen. Genomes from active anaerobic methane-oxidizing archaea (ANME-1) showed evidence of putative metabolic flexibility and hypersaline and cold adaptations. Evidence of anaerobic heterotrophic and fermentative lifestyles were found in candidate phyla DPANN archaea and CG03 bacteria genomes. Our results demonstrate Mars-relevant metabolisms including sulfide oxidation, sulfate reduction, anaerobic oxidation of methane, and oxidation of trace gases (H2, CO2) detected under anoxic, hypersaline, and sub-zero ambient conditions, providing evidence that similar extant microbial life could potentially survive in similar habitats on Mars.
Subject terms: Microbial ecology, Microbial ecology, Next-generation sequencing
Introduction
Axel Heiberg Island in the High Arctic region of Nunavut, Canada, hosts perennial cold saline springs discharging through up to 600 m of continuous permafrost [1, 2]. Lost Hammer (LH) Spring (79° 7′ N, 90° 21′ W) is the most extreme of the springs in terms of temperature and salinity, with liquid water flowing at −5 °C and 24% salinity [3]. LH continuously exudes gas comprised primarily of methane (50%), making it the coldest terrestrial methane seep discovered to date [3]. It emerges in an area of gypsum-anhydrite diapiric uplift through a two-meter-high salt tufa that experiences seasonal emptying and filling; despite this seasonality, conditions in the outflow sediment remain stable. The sediment is nearly anaerobic with trace amounts of O2 (0.25 ppm), highly reducing (−165 mV), and sulfate-rich (1 M) [3].
Microbial life in cryoenvironments, or environments with continuous sub-zero temperatures, is dependent on the presence of liquid water, typically enabled by freezing point depression through high concentrations of salts or other solutes [4]. Such habitats include permafrost ultrathin brine films surrounding soil particles and cryopegs [5, 6], brine veins in sea ice [7], and highly unique habitats like Lake Vida in the Antarctica dry valleys [8]. Microorganisms in these environments contend with multiple stressors, and study of these microorganisms informs our understanding of the cold and saline limits of life on Earth and putative extraterrestrial habitats in the subsurface of Mars and the icy moons Europa and Enceladus [4]. For example, recent evidence obtained through orbital radar sounding indicated the presence of subglacial, hypersaline lakes ~800 m below Mars’ southern ice cap [9, 10]. Recurring slope lineae may be another possible episodic source for liquid water closer to the Martian surface; modeling suggests they may form through hydration and deliquescence of near-surface sulfate-chloride salt brines [11]. Geomorphological features resembling ancient salt springs have also been proposed [12, 13], and as Mars became much colder and drier ~3.7 bya, the last habitable surface environments would likely have been highly saline cryoenvironments; chloride salt deposit imaging suggests surface brines persisted 2–2.5 bya [14]. Indeed, the search for extant and/or past life on Mars, Europa, and Enceladus is a primary driver for current and future planetary science missions [15, 16]. The cold, salty, and anoxic oceans on Europa and Enceladus putatively contain redox components that could support microbial life, including methane in water plumes erupting from Enceladus [17, 18].
Previous studies characterized LH as a low biomass, microbially dominated ecosystem (~105 cells/g sediment) with anaerobic methane-oxidizing archaea (ANME-1) and sulfate-reducing bacteria (SRBs) identified via 16S rRNA gene clone libraries and a 454 pyrosequencing metagenome [3, 19, 20]. However, due to low biomass and high salinity that make nucleic acid extraction difficult, these surveys likely did not capture the complete microbial diversity of the system, as determined by rarefaction curves and low sequencing depth [19, 20]. Sediment incubations identified SRBs capable of metabolism at in situ temperatures, and limited studies have identified putatively active microbiota through sequencing of 16S rRNA (cDNA) transcripts [19, 20]; however, no transcriptome has previously been successfully sequenced.
In this context, the objectives of the present study were to identify the dominant taxonomic and metabolic diversity through genome-resolved metagenomic sequencing combined with metagenome-assembled genome (MAGs) and single-amplified genome (SAG) analyses. In parallel, we attempted to identify the primary active members and metabolisms of the LH spring sediments through metatranscriptomic analyses and to identify adaptations to the polyextreme LH environment focusing on MAGs and SAGs from poorly characterized phylogenetic groups. Lastly, we interpreted the identified active microbial ecosystem members and metabolisms as potential life forms that could exist in very cold and saline environments on other planetary bodies such as Mars.
Materials and methods
Detailed descriptions of the following are in Supplementary Materials.
Site description and sample collection
LH discharges through a precipitated mineral salt tufa as described in previous publications [3, 13, 19, 20] (Fig. S1). Physical and geochemical parameters (Table S1) have remained highly stable since 2005, allowing comparison among samples collected in different years. For this study, sediment samples were collected in July 2017 and July 2019. Sediment from 2017 was used for metagenomic sequencing, and sediment from 2019 was used for RNA and SAG sequencing.
DNA extraction and metagenome analyses
DNA was extracted from two 5 g sediment portions. Resulting DNA from each sample was concentrated and sequenced on a HiSeq2500 (Illumina, San Diego, CA, USA) at The Centre for Applied Genomics (Toronto, ON, Canada). Reads were trimmed with Trimmomatic [21] and classified with Kaiju [22] and phyloFlash [23]. The metagenomes were co-assembled with Megahit [24] and contigs were binned with MetaBAT [25]. Bin quality was assessed with CheckM [26]. The co-assembly was annotated with the Joint Genome Institute’s IMG/M server [27, 28]. Bins were classified with Genome Taxonomy Database Toolkit [29]. Additional annotation is described in Supplementary Materials. Sequencing and assembly statistics are in Tables S2 and S3.
Single cell sorting and SAG analysis
Fluorescence-activated cell sorting and genome amplification was done at the Bigelow Laboratory for Ocean Sciences (East Boothbay, ME, USA). Whole-genome sequencing libraries were prepared and sequenced on a MiSeq (Illumina). Reads were trimmed with BBDuk and contaminant human reads were removed with DeconSeq [30]. Genomes were assembled with SPAdes [31]. Genome annotation was as described for the metagenome.
mRNA sequencing and analysis
RNA was extracted in triplicate, treated to remove contaminating DNA, then pooled and concentrated. Ribosomal RNA was depleted and the generated cDNA library was sequenced on a NovaSeq 6000 at The Center for Applied Genomics. Reads were trimmed with Trimmomatic and rRNA reads were removed with SortMeRNA [32]. Remaining reads were aligned to metagenome and SAG contigs with bowtie2 [33]. Reads aligned to CDS were counted with HTSeq [34] and transcripts per million reads (tpm) was calculated to normalize gene expression values.
Results and discussion
MAGs and SAGs capture novel phylogenetic diversity
We sequenced the metagenome and constructed MAGs in order to characterize microbial diversity within the LH sediment. The majority of classified metagenomic reads were bacterial (98%) (Figs. 1A and S2). The most abundant phyla were Bacteroidota (37%), Proteobacteria (23%), Desulfobacterota (20%), and Campylobacterota (5%). Metagenome binning produced 32 high-quality MAGs (>90% completeness, <5% contamination) and 60 medium-quality MAGs (>50% completeness, <10% contamination) (Table S5). All MAGs were classified as bacterial, representing 12 phyla (Fig. 1B) including the most abundant orders (>1.5%) in the metagenome (Fig. 1A).
Fig. 1. Taxonomic diversity and novelty in MAGs and SAGs.

A Comparison of metagenome taxonomic composition to MAG frequency by order. Metagenome taxonomic composition was determined with phyloFlash, based on classification of metagenomic reads mapping to the SILVA small subunit ribosomal database. B Phylogenomic tree of high- and medium-quality MAGs and SAGs. The tree was constructed in Anvi’o with FastTree using the Bacteria_71 collection of single-copy genes and midpoint-rooted with FigTree. C Level of taxonomic novelty of the MAGs and SAGs by rank. The number of classified and unclassified genomes at each taxonomic level was determined according to its rank assignment and taxonomic placement by GTDB-tk.
SAGs captured additional diversity. Fourteen medium-quality and 53 low-quality SAGs were obtained, of which 33 were bacterial and 34 were archaeal. We intentionally sought to sequence archaeal genomes as no ANME-1 genomes have been previously recovered from LH. These SAGs represented 31 species following identification of closely related genomes based on 16S rRNA gene similarity (>98.6%) and average nucleotide identity (ANI) (>95%). The majority of these species (28 of 31) were found at low-abundance (<1%) in the metagenome; discussion on why this occurred is in Supplementary Materials. Correspondingly, there was little overlap between SAGs and MAGs: ANI analysis classified one low-quality SAG (S38) as the same species as MAGs (M8 and M38). The 14 medium-quality SAGs represented primarily archaeal species, including five ANME-1 SAGs and six SAGs representing QMZS01 (formerly in Candidatus Aenigmarchaeota) and Iainarchaeota (Candidatus Diapherotrites) in the DPANN superphylum (Fig. 1A).
Genome novelty was assessed using assigned rank of taxonomic classification by comparison to the GTDB database, based on placement in a reference tree and whole-genome comparisons to reference genomes [29]. The majority (97%) of MAGs and SAGs were unclassified at the species level, with 23% of genomes unclassified at higher taxonomic ranks (genus (29 genomes), family (5 genomes), and order (1 genome)) (Fig. 1C). Comparatively, a recent compendium of 530 Arctic Ocean MAGs found 83% novelty at the species level [35]. The high level of genomic novelty reflects the uniqueness of LH as well as the relative undersampling of genomes from polar and extreme environments.
Sulfur-cycling Gammaproteobacteria and Desulfobacterota are abundant and active
We identified the active microbial community and metabolisms present in the spring by mapping the metatranscriptome to the metagenome, MAGs, and SAGs. Due to the challenges of extracting RNA from LH sediment, the metatranscriptome had relatively low sequencing depth (4.2 million reads). Therefore, some transcripts were likely not detected in our study and their absence herein does not preclude their presence in the environment. Additionally, the metatranscriptome and metagenome were sequenced from sediment collected in different years; while spring conditions are stable, we cannot exclude the possibility of variation in the sampled microbial communities contributing to observed differences between the two datasets.
The LH sediment contains abundant sulfate (~100,000 mg/kg), and typical sulfur-cycling taxa were among the most abundant in the metagenome, including Desulfobulbales (15%), Campylobacterales (5%), and Halothiobacillales (3.6%) (Fig. 1A). Sulfide oxidation genes were among the most highly expressed metabolic genes (5027 tpm) (Fig. 2; Table S6). Multiple mechanisms of sulfide oxidation were evident, including through elemental sulfur (sqr, fccAB), thiosulfate (sox), and tetrathionate (tsdA) intermediates; elemental sulfur detected in LH is putative evidence of this activity in situ [3, 13, 36]. Relative expression indicated oxidation was largely driven by the versatile Sox multi-enzyme complex (4662 tpm), which can utilize multiple sulfur compounds for complete or partial oxidation [36]. The majority (71%) of total oxidation gene relative expression was attributed to Guyparkeria MAGs M8 and M38 (Table S7) in Halothiobacillales. These MAGs contained complete Sox pathways and also expressed sulfide-oxidizing sqr and fccAB genes, indicating they oxidize sulfide completely to sulfate (Fig. 3; Table S8). Previously cultivated Guyparkeria are characteristically halophilic sulfur-oxidizers, identified in marine and hypersaline waters and sediments [37, 38].
Fig. 2. Key metabolic genes expressed in the LH spring sediment.

Arrow width represents relative expression of genes in transcripts per million reads (tpm). Pie charts represent the phylogenetic classification of relative expression based on the presence of the expressed gene in MAGs or SAGs or phylogenetic classification of the gene for unbinned genes. Complete distribution of tpm in MAGs, SAGs, and unbinned genes can be found in Table S7.
Fig. 3. Metabolic potential and gene expression in high-quality MAGs and medium-quality SAGs.

The identification number for each genome is noted above each column. An ‘X’ indicates the presence of a gene or pathway within the genome; ‘/’ indicates a partial pathway. A complete list of gene IDs and criteria used for denoting the presence of a complete or partial pathway is found in Tables S12 and S13. The square root of transcripts per million reads (tpm) of metatranscriptome reads mapped to the genome is indicated by a color scale within each square. The summed tpm indicates the total tpm of reads mapped to the genome on a logarithmic scale. A corresponding table for medium-quality MAGs is located in Table S8.
Expression of Sox genes was also detected in nine additional Gammaproteobacteria, Alphaproteobacteria, and Campylobacterota MAGs (Fig. 3; Table S8). Variation in partial versus complete Sox pathways and co-expression of other oxidation genes including sqr and rdsrAB was observed in the eleven MAGs with Sox gene expression, suggesting variation in oxidation pathways and substrate utilization as a potential source of niche differentiation, similar to sulfidic environments [39, 40]. Eight MAGs co-expressed cytochrome c oxidases, while one (M37) additionally expressed denitrification genes, suggesting they scavenge for the low amounts of O2 as well as utilize anaerobic electron acceptors; trace nitrite/nitrate has been detected in the sediment (2.87 mg/kg). These MAGs all contained high-affinity cbb3-type cytochrome c oxidases (coxABCD), which enable aerobic respiration in microaerobic environments [41], exclusively or in addition to low-affinity oxidases; five MAGs expressed these high-affinity oxidases.
Expression of reductive sulfur-cycling genes (dsrAB, ttrAB, and psr/phsA) was dominated by Desulfobacterota, with all expression of these genes mapped to MAGs and unbinned genes in the Desulfobulbales order (106 tpm). Complete dissimilatory sulfate reduction (SR) pathways were identified in the three Desulfobulbales MAGs, and expression of reductive dsrAB was identified in BM506 MAG M19 (Table S8). Desulfobacterota, including Desulfobulbales, have previously been characterized in hypersaline and sub-zero ice-covered lakes and subglacial brines [8, 42].
Our previous microcosm experiments detected H2-dependent SR in LH sediments down to −20 °C [19], suggesting hydrogen (~0.65%) exsolving from the spring as a likely electron donor for SRB. The two MAGs that expressed SR genes in our study have unexpressed hydrogen-oxidizing NiFe hydrogenase genes; however, expression was identified for hydrogenase subunits in another Desulfobacterota MAG (SURF-3 sp. M58) (Table S8) and SAG (Desulfurivibrionaceae sp. S23) (Table S9). Thus, hydrogen oxidation is likely coupled with SR for at least some LH SRB, confirming this observed metabolism in situ.
Anaerobic methane oxidation by ANME-1 detected at sub-zero temperatures
Methane seeps have been studied extensively due to their importance in global carbon cycling, most commonly in marine environments where methanotrophy is dominated by anaerobic oxidation of methane (AOM) by anaerobic methane-oxidizing archaea (ANME) [43]. ANME have been detected in hypersaline environments including marine cold seeps and mud volcanoes [44, 45], but are generally absent from sub-zero brines such as cryopegs [46, 47]. ANME have previously been reported in one sub-zero environment (thawing Arctic sub-marine permafrost) where isotopic evidence indicated AOM down to −1 °C [48]. LH continuously exudes methane (11 g/day), with sediment concentrations of ~100 nmol/g [49], and ANME-1 have previously been detected in the sediment [3]. In our study, expression of genes involved in AOM (mcrAB, mtrC, frhB) (165 tpm) was identified in ANME-1 SAGs (Fig. 3), indicating that AOM is occurring under in situ conditions and providing some of the strongest evidence to date that AOM occurs at sub-zero temperatures as low as −5 °C.
AOM is typically coupled with SR in syntrophic consortia with SRB, though associations with iron- and manganese-reducing bacteria also occur [50]. In LH, co-occurrence of active SRB suggests SR-coupled AOM as a putative mechanism. The SRB genomes include Desulfobulbales, which are known to form consortia with ANME [51]. However, our previous CARD-FISH microscopy did not observe close associations of ANME-1 with other cells [3]. Similar observations of unattached ANME, usually ANME-1, suggests they can carry out AOM alone, with potential alternate electron acceptors including metal oxides and humic acids [52]. Decoupled AOM has been shown in nitrate-, iron-, and manganese-reducing ANME-2 species [53–55]. It is unclear what the electron acceptor is for LH ANME-1.
Expression of bacterial methanotrophy marker genes for methane monooxygenase (pmoABC; 102 tpm) was identified in Methylobacter MAG M5 (Fig. 3). Transcripts also mapped to formylmethanofuran dehydrogenase (fmdB; 18 tpm), involved in downstream oxidation of formaldehyde to formate. Methylobacter oxidize methane in environments including seep oxic zones and anoxic lakes [56, 57]. The presence of both cytochrome c oxidase and nitrate reduction genes in MAG M5 suggests possible facultative anaerobic methane oxidation by Methylobacter could be occurring in the micro-oxic sediment although expression of these genes was not detected.
Despite the availability of methane, both ANME-1 and Methylobacter were at low abundance (<1%) in the metagenome. While Methylobacter may be restricted by competition for nitrate and/or oxygen, electron acceptor restriction is unlikely for ANME-1 if coupled with SR given the abundance of sulfate and SRBs. Their activity may be inhibited by the low energy yield of SR-coupled AOM, which is predicted to be unfavorable under in situ LH conditions (6.1 kJ/mol electron−1; Table S10); although ANME-1 have reached abundances of ~50% in hypersaline marine cold seeps [58], they may be less adapted to sub-zero conditions. As ANME are also oxygen-sensitive [59], micro-oxic conditions in LH may be sufficient to restrict their abundance as observed in marine water columns [60] and sediment [61].
Other C and N cycling metabolisms detected
In addition to methane, the gas emitted in LH contains significant amounts of nitrogen (~35%) and carbon dioxide (~10%), as well as trace gases (~<1%) including short-chain alkanes [3]. Expression of aerobic short-chain alkane oxidation genes was detected (180 tpm; Fig. 2), indicating that these gases provide additional energy and carbon sources. Dissimilatory nitrate reduction (napA/narG/nirB; 197 tpm), denitrification (napA/narG/nirKS/norBC/nosZ; 505 tpm), and nitrogen fixation (nifH; 2 tpm) genes were also expressed (Fig. 2). Dissimilatory nitrate reduction and denitrification genes with mapped transcripts were classified as Proteobacteria, Actinobacteriota, and Desulfobacterota; nifH had mapped transcripts in Sulfuricurvum MAG M14 (Campylobacterota) (Figs. 2 and 3). The lack of detected nitrification gene expression represents a gap in the nitrogen cycle, although a small number of bacterial amoA genes in the metagenome suggests nitrification may be carried out by low-abundance taxa. No evidence of anaerobic ammonia oxidation was found.
Photosynthetic gene transcripts were also detected (psaAB/psbABCDTZ; 5337 tpm), mapping to unbinned cyanobacterial (74% of tpm) and eukaryotic genes (Fig. 2). However, months-long winter darkness at LH (~October to February) means this activity is necessarily seasonal. All cyanobacterial 16S rRNA sequences in the metagenome were most similar to chloroplast 16S rRNA, indicating this detected activity may be due to plant or soil matter blown into the spring tufa. Alternatively, Cyanobacteria may be dormant during the winter months; previous metagenomic sequencing found cyanobacterial dormancy genes [20]. Anoxygenic photosynthesis gene expression was also detected by unbinned alphaproteobacterial and eukaryotic genes (pufLMC; 11 tpm), indicating some photosynthetic activity occurs through H2S oxidation.
Lithoautotrophic metabolisms relatively predominant in the active microbial community
We hypothesized that lithoautotrophic metabolisms primarily sustain the spring microbial community, as the potential for photoautotrophy is limited due to long seasonal darkness and heterotrophic metabolism would be limited by low organic C (0.45% TOC) in the sediment. Approximately 40% of the MAGs and SAGs contained CO2-fixation genes, and 73% of MAG and SAG relative expression was attributed to those autotrophic genomes (Fig. S3). However, this did not account for the majority of relative expression (61%) attributed to unbinned genes. In order to estimate the total relative contributions of autotrophs to the active LH community, we multiplied the relative expression in each phylum, including unbinned genes, by its proportion of autotrophic genomes (Fig. 4). Using this method, we estimated 60% of relative expression originated from autotrophic microorganisms. These results suggest the LH microbial community may potentially primarily use lithoautotrophy, though additional in situ evidence (e.g. a bicarbonate mineralization assay) is required to support this finding. Notably, 23% of relative expression was attributed to sulfide-oxidizing Gammaproteobacteria genomes containing Calvin-Benson-Bassham cycle genes, suggesting that they are significant contributors to primary production. Sulfide-oxidizing Gammaproteobacteria are major contributors to carbon fixation in light-limited environments including anoxic coastal sediments, hydrothermal vents, and other hypersaline Axel Heiberg springs [39, 62, 63]; they also comprise major clades at marine cold seeps where methane oxidation and sulfate reduction produce abundant sulfide and CO2 [43].
Fig. 4. Percent relative expression by phylum, including both genes in MAGs and SAGs and unbinned genes classified by JGI.
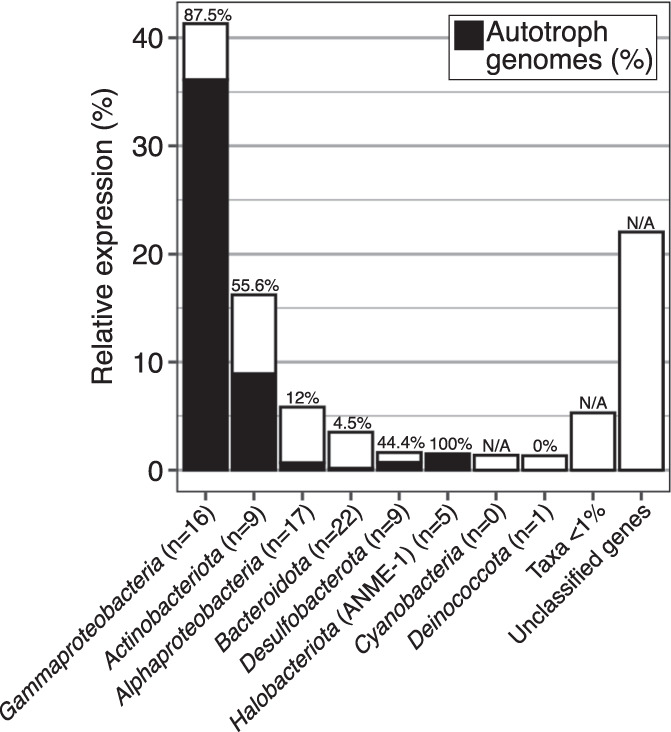
The percentage of autotrophic MAGs and SAGs (containing CO2 fixation genes) in each phylum is indicated in black and noted above each column. The number of MAGs and SAGs in each phylum is indicated in parentheses in each column label. The percentage of relative expression by autotrophic microorganisms was estimated by multiplying the relative expression by the percentage of autotrophic genomes in each taxon.
Mixotrophic growth using trace carbon monoxide (CO) as an energy source is a proposed survival mechanism for heterotrophs under organic carbon limitation [64]. CO dehydrogenase (coxL) genes were present in 11 heterotrophic Alphaproteobacteria MAGs; furthermore, coxL gene expression was detected in three of these MAGs, indicating that mixotrophy is a relevant lifestyle in LH. No measurement of CO has previously been attempted in LH; it may be a previously undetected component of the LH gas, or microorganisms could be utilizing trace atmospheric CO.
Relative expression by Bacteroidota was low (3.5%) compared to their relative abundance in the metagenome (37%) (Figs. 4 and 1B), suggesting their reliance on heterotrophy may limit activity. While these results should not be considered conclusive given low coverage and lack of replicates for the metatranscriptome, this relative decrease was also observed in a previous study involving sequencing of cDNA derived from 16S rRNA [20]. Bacteroidota comprise significant proportions of the microbial community in subglacial brines, sea ice brines, cryopegs, and cold hypersaline marine basins [46, 47, 65], and the most abundant Bacteroidota genera in the metagenome (Gillisia, Salegentibacter, Lutibacter) were found in highly saline soil, subglacial brines, and marine solar salterns [46, 66, 67]. Their putative salt tolerance may enable survival even with low transcriptional activity.
Functional potential and adaptations in poorly characterized taxa
This study utilized two approaches to recovering microbial genomes through assembly of MAGs and SAGs. MAGs are assembled through clustering of metagenomic contigs; accordingly, MAGs often represent “population genomes” of related strains, which may have differing microniches or metabolisms, rather than single genotypes [68]. SAGs are sequenced from individual cells and represent an individual strain rather than population, enabling precise evaluation of amino acid residue substitutions associated with cold and saline adaptation [69]. Our SAGs include genomes for under-studied or candidate taxa, present in low-abundance in LH but with mapped mRNA transcripts indicating metabolic activity. In order to better understand the niches occupied by these microorganisms, the functional potential and gene expression of several SAGs was more closely examined.
ANME-1 genomes reveal potential metabolic flexibility and hypersaline adaptation
Despite extensive study of ANME, much remains unknown due to their recalcitrance to enrichment and culturing. As the LH ANME-1 genomes are the first from species active under ambient sub-zero and hypersaline conditions, we examined them for features enabling their survival. While ANI and marker gene analyses determined that the 17 SAGs (12.3–84.6% completeness) represented multiple species within the ANME-1 family, the genomes are presented together for simplicity.
Two ANME-1 SAGs contained subunits of a Group 1 h NiFe hydrogenase, co-localized with the electron-accepting Hdr2 complex, signifying possible respiratory oxidation of hydrogen. In addition, six SAGs encoded butyryl-CoA dehydrogenase, clustered with mcr genes, and a downstream butyryl-CoA oxidation pathway. This pathway is involved in oxidation of butane and other short-chain alkanes in archaea closely related to ANME (Ca. Syntrophoarchaeum) utilizing mcr-like complexes [70], and has been identified in ANME-1 containing mcr-like sequences [71]. LH butyryl-CoA dehydrogenases were homologous to those from Ca. Syntropharchaeum butanivorans and mcr-like-encoding ANME-1 (40% and 62% amino acid identity, respectively). LH mcrA genes were most similar to canonical ANME-1 mcrA; nevertheless, the presence of this pathway suggests potential alkane oxidation. Taken together, these genes signify potential substrate flexibility in LH ANME-1. As SR-coupled AOM yields little energy [72], alternate electron donors may compensate for energy deficits due to costly hypersaline adaptations. Competition for hydrogen and alkanes (<1% of spring gas) may additionally contribute to low ANME-1 abundance.
The ANME-1 SAGs contained osmotic stress adaptations (Fig. 5), including for uptake and synthesis of compatible solutes and transporters for K+ uptake and Na+ extrusion [73]. Transcripts mapped to a small conductance mechanosensitive channel involved in maintenance of solute concentration [73], suggesting this accumulation occurs in situ. Based on these genes, LH ANME-1 could utilize either a “salt-in” or “compatible solute” osmoregulation strategy, though predicted proteins did not show the typical enrichment of acidic amino acids found in salt-in microorganisms (Fig. S4A). Homologous osmotic stress response genes were identified in related ANME-1 genomes (Table S11), indicating widespread adaptive capabilities and corroborating observed preferential habitation of hypersaline environments by ANME-1 over other ANME clades [45, 50]. Seven SAGs also encoded gas vesicle proteins, proposed to reduce water loss during salt stress [74]. Alternatively, they may enable buoyancy regulation, allowing cells to position themselves more favorably in a spring which experiences seasonal changes in water depth [75]. The presence of chemotaxis proteins and a flagellum suggest ANME may navigate the sediment to find optimal niches or, potentially, SRB for syntrophic partnerships.
Fig. 5. Metabolic reconstruction of ANME-1 SAGs.

Genome contents were based on a composite of 17 medium- (SAGs S10-S14) and low-quality (SAGs S39-S50) ANME-1 SAGs. Genes with mapped transcripts are denoted by red text. Pie charts indicate the number of SAGs containing each gene (out of 17). A complete list of genes can be found in Table S14.
The ANME-1 SAGs contained genes for several multi-heme cytochromes, which have proposed functions in both syntrophic and decoupled AOM [53, 55, 76] (Fig. 5). In syntrophic AOM, direct exchange of electrons is proposed to occur via conductive “nanowires” between ANME and SRB [76]. Examination of the sulfate-reducing MAG M19 as a potential syntrophic partner identified genes with mapped transcripts required for nanowire construction [77] including multi-heme cytochromes, type IV pili, and outer membrane porins. The ANME-1 SAGs additionally contained archaellum genes suggested to facilitate nanowire construction [78]. Syntrophic electron exchange is therefore putatively possible in these microorganisms.
Poorly understood DPANN archaea active in LH
Archaea from the DPANN superphylum were detected with the recovery of Iainarchaeota (Candidatus Diapherotrites) (one SAG, completeness 52.9%) and QMZS01 (formerly part of Aenigmarchaeota) (five SAGs, completeness 57.7–76.2%) SAGs. Within DPANN, these phyla are typically non-extremophilic and have not previously been identified in hypersaline or sub-zero environments [79, 80]. The QMZS01 candidate phylum contains only two other genomes, both MAGs from hydrocarbon- and sulfate-rich Guaymas Basin hot hydrothermal sediments [71].
Due to their resistance to laboratory cultivation, the DPANN superphylum has only recently been defined [81]. DPANN are characterized by small cell and genome sizes and minimal metabolic function [82], but are proposed to have important roles in organic carbon and hydrogen cycling [80, 83]. Characteristically small genomes (<0.7 Mbp, though incomplete), limited anabolic pathways, and acetogenic and hydrogenic fermentation capabilities were observed in LH DPANN SAGs (Figs. S5 and S6), indicating reliance on scavenging for cellular components. All SAGs showed evidence of obligately anaerobic and heterotrophic lifestyles. The Iainarchaeia SAG contained a large subunit form III RubisCO gene (rbcL) (Fig. S7), also identified in Candidatus Iainarchaeum andersonii where it was proposed to take part in the adenosine monophosphate metabolism pathway [79]. The rest of this pathway was identified in the LH SAG (Figure S5), suggesting adenosine degradation to pyruvate as a possible ATP-generating mechanism. The QMZS01 SAGs contained genes involved in polysaccharide degradation (e.g. alpha amylase and endoglucanase) and transcripts mapped to a TrmB-family sugar-specific transcriptional regulator, supporting putative polysaccharide-degradation activity. The two Guaymas Basin QMZS01 MAGs also contain carbohydrate-active enzymes and fermentation genes, indicating similar metabolisms across this phylum. Additionally, LH QMZS01 uniquely contain subunits of group 3b NiFe hydrogenase (hydDA) co-localized with a ferredoxin and an oxidoreductase homologous to hydG and hydB from Pyrococcus furiosis (29% and 40% amino acid identity, respectively), which reversibly couple H2 oxidation with NADPH formation [84]. LH DPANN may thus cycle complex carbon compounds, supplying fermentation products including H2 or acetate to co-occurring hydrogen oxidizers or putative acetate metabolizers including SRB containing the Wood-Ljungdahl pathway or the numerous microorganisms encoding acetate metabolism genes (Tables S12 and S13). Transcripts mapping to DPANN SAGs (2981 tpm) were primarily to non-coding RNA (Table S9). The SAGs also contained stress response genes (Figs. S3 and S4). While the majority of these genes were detected in other Iainarchaeota and QMZS01 genomes (Table S11), LH QMZS01 uniquely contained Na+/H+ antiporter and compatible solute synthesis genes, indicating adaptations to hypersalinity in this clade. DPANN have been detected in High Arctic lakes on Ellesmere Island, including cold (3.5 °C) freshwater Lake Hazen [85] and meromictic ice-covered (0–8 °C) Lake A [86]. DPANN populations in both lakes were primarily Woesearchaeota; however, Iainarcheota and Aenigmarchaeota were detected in the saline (~3%), anoxic, and sulfidic layers of Lake A, where they were similarly theorized to recycle microbial biomass. These results demonstrate that, despite minimal biosynthetic capabilities, DPANN archaea are active in extreme, resource-limited Arctic environments.
Candidate CG03 phylum is likely fermentative
SAG S2 was classified in the phylum CG03, a candidate phylum recently defined by GTDB using their methodology based on phylogenetic tree placement and normalization of taxonomic rank using lineage-specific evolutionary distance [87]. This poorly characterized phylum contains 15 MAGs in the GTDB database, previously deposited in NCBI primarily as unclassified Elusimicrobia and sourced from groundwater and hypersaline sediments; only one study has previously examined some of these CG03 genomes [88]. The LH SAG is the first CG03 genome from a sub-zero environment, although CG03 was recently detected in the anaerobic, methane-rich sulfidic Zodletone spring (22 °C, 1.2% NaCl) [89]. The LH SAG contains a glycolysis/gluconeogenesis pathway, partial TCA cycle, and partial fatty acid oxidation pathway (Figure S8), indicating a likely heterotrophic and anaerobic lifestyle. Interestingly, it encodes partial NADH dehydrogenase and ATP synthase complexes but lacks other electron transport chain proteins, corroborating the previous survey of several CG03 genomes which found near-universal partial ATP synthases. The functionality of these complexes is unclear, but their frequency within the phylum implies an active rather than vestigial role. Regardless, the LH CG03 is likely fermentative, as it contains genes involved in both acetogenic (phosphotransacetylase and acetate kinase) and hydrogenogenic (FeFe Group A hydrogenase) processes. It also encodes homologs to carbon monoxide/xanthine dehydrogenase family proteins. Structure-based functional annotation by I-TASSER [90] confirmed protein similarity to CO dehydrogenase, suggesting possible mixotrophy. These results suggest that, similar to DPANN, CG03 may provide acetate and hydrogen to the microbial community.
Transcripts mapped to the SAG (6718 tpm total) mapped to tRNA genes and a AAA ATPase-family protein, indicating transcriptional activity but providing little indication of metabolic function (Table S9). The genome contained several sodium and potassium transporters, but did not have an enrichment in acidic amino acids in predicted proteins (Fig. S4B), leaving its osmoregulation strategy unclear. It should be noted that the above results are based on a single genome (62.8% completeness) and so should not be considered conclusive.
LH as an analog for relevant microbial metabolisms on Mars
Terrestrial analogs like LH can help constrain and inform theorized habitability and biosignatures in ancient and extant Martian environments. This study provides in situ evidence of an active microbial ecosystem in a sub-zero hypersaline spring supporting Mars-relevant chemolithoautotrophic metabolisms [91, 92]. We identified active ANME-1 putatively capable of AOM, H2 oxidation, CO2 fixation, and N2 fixation. Trace methane (~0.4 ppbv) has been detected in the Mars atmosphere [93] and, while the source is unknown, methane and hydrogen could be produced abiotically in the deep subsurface through rock-water interactions (serpentization; Fischer-Tropsch synthesis) or through putative methanogenesis (Fig. 6) [94]. Regardless of the origin, subsurface gaseous methane or methane clathrates could potentially sustain microbial life by AOM.
Fig. 6. Model for a hypothetical Martian methane cycle, adapted and reproduced with permission from Harris et al. [92].

Putative methane sources, sinks, and reservoirs are noted in orange. Mars-relevant metabolisms identified in Lost Hammer are noted in green.
Sulfates and trace H2 (3) in LH support transcriptionally active H2-oxidizing, SO4-reducing bacteria containing CO2-fixation and N2-fixation genes. Similar conditions for supporting SRBs may exist in the Martian subsurface where sulfate is abundant and may co-localize with H2 and CH4 from serpentinization or methanogenesis [95]. Atmospheric CO (0.075%) on Mars may also support CO-oxidizing bacteria [91], as in Antarctic desert soils where trace CO may support primary production through high-affinity CO dehydrogenases [96]. In this study, CO dehydrogenase genes and transcripts were detected in mixotrophic Alphaproteobacteria MAGs, suggesting Martian CO could support near-surface heterotrophs.
The relatively high abundances of lithoautotrophic sulfide-oxidizing bacteria as well as high expression of S-oxidation genes was somewhat surprising given sulfide-oxidizers are generally aerobic while LH sediment is highly reducing and contains only trace O2. However, extremely low-oxygen environments can sustain aerobic metabolisms [97], including sulfide oxidation in anoxic lakes [98], due to O2 favorability as an electron acceptor. Gibbs energy calculations for LH show redox reactions with O2 are more favorable than those with NO3− or SO42−, including H2S/O2 (−109 kJ/mol electron−1) compared to H2S/NO3− (−93.8 kJ/mol electron−1) (Table S10). While the Martian atmosphere contains 0.1% O2, modeling of perchlorate and sulfate brines under Martian surface conditions demonstrated how O2 can concentrate in near-surface brines allowing for potential micro-oxic and even aerobic metabolisms [99]. While methanogens and SRBs are generally considered the most likely candidate life forms on Mars, this study highlights the capability of lithoautotrophic sulfide-oxidizers to also thrive in a hypersaline, sub-zero, and oxygen-depleted environment.
Conclusion
LH supports an active microbial community including sulfur-cycling Gammaproteobacteria and Desulfobacterota and methane-oxidizing ANME-1, as well as fermentative and heterotrophic populations. Overall, our study provides robust evidence of chemolithoautotrophic metabolisms active in a sub-zero, hypersaline, and anoxic environment similar to those that may exist on Mars.
Supplementary information
Acknowledgements
We thank the Bigelow Single Cell Genomics Center for cell sorting services and support. EM was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) doctoral award (funding reference number CGSD2-534189-2019), Fonds de recherche du Québec—Nature et technologies (FRQNT) doctoral award (funding reference number 272792), and the Northern Scientific Training Program. This research was also supported by the Canada Research Chair Program, the NSERC Discovery and Northern Research Supplement Programs, and the Polar Continental Shelf Project (Arctic logistical support). Field work was carried out with appropriate licensing obtained from the territory of Nunavut and the Nunavut Research Institute (Nunavut Scientific Research License Nos. 02 043 17R-M and 02 051 19N-M).
Author contributions
LGW conceived and supervised the study. EM, IA, and LGW designed the experiments. EM, IA, and CM conducted field work. EM and IA conducted laboratory experiments and sequencing. EM, IA, MAF, YJC, JG, and LGW analyzed and interpreted the data. EM and IA prepared figures. EM, MAF, and LGW wrote the paper. All authors reviewed and edited the paper.
Data availability
Sequencing reads, metagenome, MAGs, and SAGs are in NCBI under BioProject PRJNA699472. JGI metagenome and SAG annotations are available under GOLD Study ID Gs0135943 (SAG IDs in Table S4).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41396-022-01233-8.
References
- 1.Pollard W, Omelon C, Andersen D, McKay C. Perennial spring occurrence in the Expedition Fiord area of western Axel Heiberg Island, Canadian High Arctic. Can J Earth Sci. 1999;36:105–20. doi: 10.1139/e98-097. [DOI] [Google Scholar]
- 2.Andersen DT. Cold springs in permafrost on Earth and Mars. J Geophys Res. 2002;107:4–1-4-7. doi: 10.1029/2000JE001436. [DOI] [Google Scholar]
- 3.Niederberger TD, Perreault NN, Tille S, Lollar BS, Lacrampe-Couloume G, Andersen D, et al. Microbial characterization of a subzero, hypersaline methane seep in the Canadian High Arctic. ISME J. 2010;4:1326–39. doi: 10.1038/ismej.2010.57. [DOI] [PubMed] [Google Scholar]
- 4.Goordial J, Lamarche-Gagnon G, Lay CY, Whyte L. Left out in the cold: life in cryoenvironments. In: Seckbach J, Oren A, Stan-Lotter H, editors. Polyextremophiles. New York: Springer; 2013. p. 335–64.
- 5.Gilichinsky D, Rivkina E, Bakermans C, Shcherbakova V, Petrovskaya L, Ozerskaya S, et al. Biodiversity of cryopegs in permafrost. FEMS Microbiol Ecol. 2005;53:117–28. doi: 10.1016/j.femsec.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 6.Rivkina EM, Friedmann EI, McKay CP, Gilichinsky DA. Metabolic activity of permafrost bacteria below the freezing point. Appl Environ Microbiol. 2000;66:3230–3. doi: 10.1128/AEM.66.8.3230-3233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown MV, Bowman JP. A molecular phylogenetic survey of sea-ice microbial communities (SIMCO) FEMS Microbiol Ecol. 2001;35:267–75. doi: 10.1111/j.1574-6941.2001.tb00812.x. [DOI] [PubMed] [Google Scholar]
- 8.Murray AE, Kenig F, Fritsen CH, McKay CP, Cawley KM, Edwards R, et al. Microbial life at -13 degrees C in the brine of an ice-sealed Antarctic lake. Proc Natl Acad Sci USA. 2012;109:20626–31. doi: 10.1073/pnas.1208607109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orosei R, Lauro SE, Pettinelli E, Cicchetti A, Coradini M, Cosciotti B, et al. Radar evidence of subglacial liquid water on Mars. Science. 2018;361:490–3. doi: 10.1126/science.aar7268. [DOI] [PubMed] [Google Scholar]
- 10.Lauro SE, Pettinelli E, Caprarelli G, Guallini L, Pio Rossi A, Mattei E, et al. Multiple subglacial water bodies below the south pole of Mars unveiled by new MARSIS data. Nat Astron. 2021;5:63–70. doi: 10.1038/s41550-020-1200-6. [DOI] [Google Scholar]
- 11.Bishop JL, Yesilbas M, Hinman NW, Burton ZFM, Englert PAJ, Toner JD, et al. Martian subsurface cryosalt expansion and collapse as trigger for landslides. Sci Adv. 2021;7:1–13. doi: 10.1126/sciadv.abe4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen CC, Oehler DZ. A case for ancient springs in Arabia Terra, Mars. Astrobiology. 2008;8:1093–112. doi: 10.1089/ast.2008.0239. [DOI] [PubMed] [Google Scholar]
- 13.Battler MM, Osinski GR, Banerjee NR. Mineralogy of saline perennial cold springs on Axel Heiberg Island, Nunavut, Canada and implications for spring deposits on Mars. Icarus. 2013;224:364–81. doi: 10.1016/j.icarus.2012.08.031. [DOI] [Google Scholar]
- 14.Leask EK, Ehlmann BL. Evidence for deposition of chloride on Mars from small‐volume surface water events into the Late Hesperian‐Early Amazonian. AGU Adv. 2022;3:1–19. doi: 10.1029/2021AV000534. [DOI] [Google Scholar]
- 15.Howell SM, Pappalardo RT. NASA’s Europa Clipper-a mission to a potentially habitable ocean world. Nat Commun. 2020;11:1–4. doi: 10.1038/s41467-020-15160-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farley KA, Williford KH, Stack KM, Bhartia R, Chen A, de la Torre M, et al. Mars 2020 mission overview. Space Sci Rev. 2020;216:1–41. doi: 10.1007/s11214-020-00762-y. [DOI] [Google Scholar]
- 17.Kargel JS, Kaye JZ, Head JW, Marion GM, Sassen R, Crowley JK, et al. Europa’s crust and ocean: origin, composition, and the prospects for life. Icarus. 2000;148:226–65. doi: 10.1006/icar.2000.6471. [DOI] [Google Scholar]
- 18.Taubner RS, Pappenreiter P, Zwicker J, Smrzka D, Pruckner C, Kolar P, et al. Biological methane production under putative Enceladus-like conditions. Nat Commun. 2018;9:1–11. doi: 10.1038/s41467-018-02876-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamarche-Gagnon G, Comery R, Greer CW, Whyte LG. Evidence of in situ microbial activity and sulphidogenesis in perennially sub-0 degrees C and hypersaline sediments of a high Arctic permafrost spring. Extremophiles. 2015;19:1–15. doi: 10.1007/s00792-014-0703-4. [DOI] [PubMed] [Google Scholar]
- 20.Lay CY, Mykytczuk NC, Yergeau E, Lamarche-Gagnon G, Greer CW, Whyte LG. Defining the functional potential and active community members of a sediment microbial community in a high-arctic hypersaline subzero spring. Appl Environ Microbiol. 2013;79:3637–48. doi: 10.1128/AEM.00153-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menzel P, Ng KL, Krogh A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun. 2016;7:1–9. doi: 10.1038/ncomms11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gruber-Vodicka HR, Seah BKB, Pruesse E. phyloFlash: rapid small-subunit rRNA profiling and targeted assembly from metagenomes. mSystems. 2020;5:1–16. doi: 10.1128/mSystems.00920-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 25.Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:1–15. doi: 10.7717/peerj.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen IA, Chu K, Palaniappan K, Ratner A, Huang J, Huntemann M, et al. The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res. 2020;49:D751–D63. doi: 10.1093/nar/gkaa939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mukherjee S, Stamatis D, Bertsch J, Ovchinnikova G, Sundaramurthi JC, Lee J, et al. Genomes OnLine Database (GOLD) v.8: overview and updates. Nucleic Acids Res. 2020;49:D723–D733. doi: 10.1093/nar/gkaa983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36:1925–7. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmieder R, Edwards R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE. 2011;6:1–11. doi: 10.1371/journal.pone.0017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kopylova E, Noe L, Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012;28:3211–7. doi: 10.1093/bioinformatics/bts611. [DOI] [PubMed] [Google Scholar]
- 33.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Royo-Llonch M, Sanchez P, Ruiz-Gonzalez C, Salazar G, Pedros-Alio C, Sebastian M, et al. Compendium of 530 metagenome-assembled bacterial and archaeal genomes from the polar Arctic Ocean. Nat Microbiol. 2021;6:1561–74. doi: 10.1038/s41564-021-00979-9. [DOI] [PubMed] [Google Scholar]
- 36.Ghosh W, Dam B. Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol Rev. 2009;33:999–1043. doi: 10.1111/j.1574-6976.2009.00187.x. [DOI] [PubMed] [Google Scholar]
- 37.Boden R. Reclassification of Halothiobacillus hydrothermalis and Halothiobacillus halophilus to Guyparkeria gen. nov. in the Thioalkalibacteraceae fam. nov., with emended descriptions of the genus Halothiobacillus and family Halothiobacillaceae. Int J Syst Evol Microbiol. 2017;67:3919–28. doi: 10.1099/ijsem.0.002222. [DOI] [PubMed] [Google Scholar]
- 38.Sorokin DY, Abbas B, van Zessen E, Muyzer G. Isolation and characterization of an obligately chemolithoautotrophic Halothiobacillus strain capable of growth on thiocyanate as an energy source. FEMS Microbiol Lett. 2014;354:69–74. doi: 10.1111/1574-6968.12432. [DOI] [PubMed] [Google Scholar]
- 39.Meier DV, Pjevac P, Bach W, Hourdez S, Girguis PR, Vidoudez C, et al. Niche partitioning of diverse sulfur-oxidizing bacteria at hydrothermal vents. ISME J. 2017;11:1545–58. doi: 10.1038/ismej.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Headd B, Engel AS. Evidence for niche partitioning revealed by the distribution of sulfur oxidation genes collected from areas of a terrestrial sulfidic spring with differing geochemical conditions. Appl Environ Microbiol. 2013;79:1171–82. doi: 10.1128/AEM.02812-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Preisig O, Zufferey R, Thoney-Meyer L, Appleby CA, Hennecke H. A high-affinity cbb3-type cytochrome oxidase terminates the symbiosis-specific respiratory chain of Bradyrhizobium japonicum. J Bacteriol. 1996;178:1532–8. doi: 10.1128/jb.178.6.1532-1538.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mikucki JA, Pearson A, Johnston DT, Turchyn AV, Farquhar J, Schrag DP, et al. A contemporary microbially maintained subglacial ferrous “ocean”. Science. 2009;324:397–400. doi: 10.1126/science.1167350. [DOI] [PubMed] [Google Scholar]
- 43.Ruff SE, Biddle JF, Teske AP, Knittel K, Boetius A, Ramette A. Global dispersion and local diversification of the methane seep microbiome. Proc Natl Acad Sci USA. 2015;112:4015–20. doi: 10.1073/pnas.1421865112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lloyd KG, Lapham L, Teske A. An anaerobic methane-oxidizing community of ANME-1b archaea in hypersaline Gulf of Mexico sediments. Appl Environ Microbiol. 2006;72:7218–30. doi: 10.1128/AEM.00886-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maignien L, Parkes RJ, Cragg B, Niemann H, Knittel K, Coulon S, et al. Anaerobic oxidation of methane in hypersaline cold seep sediments. FEMS Microbiol Ecol. 2013;83:214–31. doi: 10.1111/j.1574-6941.2012.01466.x. [DOI] [PubMed] [Google Scholar]
- 46.Campen R, Kowalski J, Lyons WB, Tulaczyk S, Dachwald B, Pettit E, et al. Microbial diversity of an Antarctic subglacial community and high-resolution replicate sampling inform hydrological connectivity in a polar desert. Environ Microbiol. 2019;21:2290–306. doi: 10.1111/1462-2920.14607. [DOI] [PubMed] [Google Scholar]
- 47.Cooper ZS, Rapp JZ, Carpenter SD, Iwahana G, Eicken H, Deming JW. Distinctive microbial communities in subzero hypersaline brines from Arctic coastal sea ice and rarely sampled cryopegs. FEMS Microbiol Ecol. 2019;95:1–15. doi: 10.1093/femsec/fiz166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winkel M, Mitzscherling J, Overduin PP, Horn F, Winterfeld M, Rijkers R, et al. Anaerobic methanotrophic communities thrive in deep submarine permafrost. Sci Rep. 2018;8:1–13. doi: 10.1038/s41598-018-19505-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lay CY, Mykytczuk NC, Niederberger TD, Martineau C, Greer CW, Whyte LG. Microbial diversity and activity in hypersaline high Arctic spring channels. Extremophiles. 2012;16:177–91. doi: 10.1007/s00792-011-0417-9. [DOI] [PubMed] [Google Scholar]
- 50.Bhattarai S, Cassarini C, Lens PNL. Physiology and distribution of archaeal methanotrophs that couple anaerobic oxidation of methane with sulfate reduction. Microbiol Mol Biol Rev. 2019;83:1–31. doi: 10.1128/MMBR.00074-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kleindienst S, Ramette A, Amann R, Knittel K. Distribution and in situ abundance of sulfate-reducing bacteria in diverse marine hydrocarbon seep sediments. Environ Microbiol. 2012;14:2689–710. doi: 10.1111/j.1462-2920.2012.02832.x. [DOI] [PubMed] [Google Scholar]
- 52.Timmers PH, Welte CU, Koehorst JJ, Plugge CM, Jetten MS, Stams AJ. Reverse methanogenesis and respiration in methanotrophic archaea. Archaea. 2017;2017:1–22. doi: 10.1155/2017/1654237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leu AO, Cai C, McIlroy SJ, Southam G, Orphan VJ, Yuan Z, et al. Anaerobic methane oxidation coupled to manganese reduction by members of the Methanoperedenaceae. ISME J. 2020;14:1030–41. doi: 10.1038/s41396-020-0590-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haroon MF, Hu S, Shi Y, Imelfort M, Keller J, Hugenholtz P, et al. Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature. 2013;500:567–70. doi: 10.1038/nature12375. [DOI] [PubMed] [Google Scholar]
- 55.Cai C, Leu AO, Xie GJ, Guo J, Feng Y, Zhao JX, et al. A methanotrophic archaeon couples anaerobic oxidation of methane to Fe(III) reduction. ISME J. 2018;12:1929–39. doi: 10.1038/s41396-018-0109-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oshkin IY, Wegner CE, Luke C, Glagolev MV, Filippov IV, Pimenov NV, et al. Gammaproteobacterial methanotrophs dominate cold methane seeps in floodplains of West Siberian rivers. Appl Environ Microbiol. 2014;80:5944–54. doi: 10.1128/AEM.01539-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cabrol L, Thalasso F, Gandois L, Sepulveda-Jauregui A, Martinez-Cruz K, Teisserenc R, et al. Anaerobic oxidation of methane and associated microbiome in anoxic water of Northwestern Siberian lakes. Sci Total Environ. 2020;736:1–16. doi: 10.1016/j.scitotenv.2020.139588. [DOI] [PubMed] [Google Scholar]
- 58.Orcutt B, Boetius A, Elvert M, Samarkin V, Joye SB. Molecular biogeochemistry of sulfate reduction, methanogenesis and the anaerobic oxidation of methane at Gulf of Mexico cold seeps. Geochim Cosmochim Acta. 2005;69:4267–81. doi: 10.1016/j.gca.2005.04.012. [DOI] [Google Scholar]
- 59.Knittel K, Losekann T, Boetius A, Kort R, Amann R. Diversity and distribution of methanotrophic archaea at cold seeps. Appl Environ Microbiol. 2005;71:467–79. doi: 10.1128/AEM.71.1.467-479.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schubert CJ, Coolen MJ, Neretin LN, Schippers A, Abbas B, Durisch-Kaiser E, et al. Aerobic and anaerobic methanotrophs in the Black Sea water column. Environ Microbiol. 2006;8:1844–56. doi: 10.1111/j.1462-2920.2006.01079.x. [DOI] [PubMed] [Google Scholar]
- 61.Wang J, Hua M, Cai C, Hu J, Wang J, Yang H, et al. Spatial-temporal pattern of sulfate-dependent anaerobic methane oxidation in an intertidal zone of the East China Sea. Appl Environ Microbiol. 2019;85:1–15. doi: 10.1128/AEM.02638-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dyksma S, Bischof K, Fuchs BM, Hoffmann K, Meier D, Meyerdierks A, et al. Ubiquitous Gammaproteobacteria dominate dark carbon fixation in coastal sediments. ISME J. 2016;10:1939–53. doi: 10.1038/ismej.2015.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perreault NN, Greer CW, Andersen DT, Tille S, Lacrampe-Couloume G, Lollar BS, et al. Heterotrophic and autotrophic microbial populations in cold perennial springs of the high Arctic. Appl Environ Microbiol. 2008;74:6898–907. doi: 10.1128/AEM.00359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cordero PRF, Bayly K, Man Leung P, Huang C, Islam ZF, Schittenhelm RB, et al. Atmospheric carbon monoxide oxidation is a widespread mechanism supporting microbial survival. ISME J. 2019;13:2868–81. doi: 10.1038/s41396-019-0479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nigro LM, Elling FJ, Hinrichs KU, Joye SB, Teske A. Microbial ecology and biogeochemistry of hypersaline sediments in Orca Basin. PLoS ONE. 2020;15:1–25. doi: 10.1371/journal.pone.0231676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rath KM, Fierer N, Murphy DV, Rousk J. Linking bacterial community composition to soil salinity along environmental gradients. ISME J. 2019;13:836–46. doi: 10.1038/s41396-018-0313-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoon JH, Lee MH, Kang SJ, Oh TK. Salegentibacter salinarum sp. nov., isolated from a marine solar saltern. Int J Syst Evol Microbiol. 2008;58:365–9. doi: 10.1099/ijs.0.65448-0. [DOI] [PubMed] [Google Scholar]
- 68.Sangwan N, Xia F, Gilbert JA. Recovering complete and draft population genomes from metagenome datasets. Microbiome. 2016;4:1–11. doi: 10.1186/s40168-016-0154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goordial J, Raymond-Bouchard I, Zolotarov Y, de Bethencourt L, Ronholm J, Shapiro N, et al. Cold adaptive traits revealed by comparative genomic analysis of the eurypsychrophile Rhodococcus sp. JG3 isolated from high elevation McMurdo Dry Valley permafrost, Antarctica. FEMS Microbiol Ecol. 2016;92:1–11. doi: 10.1093/femsec/fiv154. [DOI] [PubMed] [Google Scholar]
- 70.Laso-Perez R, Wegener G, Knittel K, Widdel F, Harding KJ, Krukenberg V, et al. Thermophilic archaea activate butane via alkyl-coenzyme M formation. Nature. 2016;539:396–401. doi: 10.1038/nature20152. [DOI] [PubMed] [Google Scholar]
- 71.Dombrowski N, Teske AP, Baker BJ. Expansive microbial metabolic versatility and biodiversity in dynamic Guaymas Basin hydrothermal sediments. Nat Commun. 2018;9:1–13. doi: 10.1038/s41467-018-07418-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Oren A. Thermodynamic limits to microbial life at high salt concentrations. Environ Microbiol. 2011;13:1908–23. doi: 10.1111/j.1462-2920.2010.02365.x. [DOI] [PubMed] [Google Scholar]
- 73.Gunde-Cimerman N, Plemenitas A, Oren A. Strategies of adaptation of microorganisms of the three domains of life to high salt concentrations. FEMS Microbiol Rev. 2018;42:353–75. doi: 10.1093/femsre/fuy009. [DOI] [PubMed] [Google Scholar]
- 74.Hechler T, Pfeifer F. Anaerobiosis inhibits gas vesicle formation in halophilic. Archaea Mol Microbiol. 2009;71:132–45. doi: 10.1111/j.1365-2958.2008.06517.x. [DOI] [PubMed] [Google Scholar]
- 75.Stokke R, Roalkvam I, Lanzen A, Haflidason H, Steen IH. Integrated metagenomic and metaproteomic analyses of an ANME-1-dominated community in marine cold seep sediments. Environ Microbiol. 2012;14:1333–46. doi: 10.1111/j.1462-2920.2012.02716.x. [DOI] [PubMed] [Google Scholar]
- 76.Wegener G, Krukenberg V, Riedel D, Tegetmeyer HE, Boetius A. Intercellular wiring enables electron transfer between methanotrophic archaea and bacteria. Nature. 2015;526:587–90. doi: 10.1038/nature15733. [DOI] [PubMed] [Google Scholar]
- 77.Skennerton CT, Chourey K, Iyer R, Hettich RL, Tyson GW, Orphan VJ. Methane-fueled syntrophy through extracellular electron transfer: uncovering the genomic traits conserved within diverse bacterial partners of anaerobic methanotrophic archaea. mBio. 2017;8:1–14. doi: 10.3391/mbi.2017.8.1.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krukenberg V, Riedel D, Gruber-Vodicka HR, Buttigieg PL, Tegetmeyer HE, Boetius A, et al. Gene expression and ultrastructure of meso- and thermophilic methanotrophic consortia. Environ Microbiol. 2018;20:1651–66. doi: 10.1111/1462-2920.14077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Youssef NH, Rinke C, Stepanauskas R, Farag I, Woyke T, Elshahed MS. Insights into the metabolism, lifestyle and putative evolutionary history of the novel archaeal phylum ‘Diapherotrites’. ISME J. 2015;9:447–60. doi: 10.1038/ismej.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Castelle CJ, Brown CT, Anantharaman K, Probst AJ, Huang RH, Banfield JF. Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat Rev Microbiol. 2018;16:629–45. doi: 10.1038/s41579-018-0076-2. [DOI] [PubMed] [Google Scholar]
- 81.Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–7. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 82.Dombrowski N, Lee JH, Williams TA, Offre P, Spang A. Genomic diversity, lifestyles and evolutionary origins of DPANN archaea. FEMS Microbiol Lett. 2019;366:1–12. doi: 10.1093/femsle/fnz008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wong HL, MacLeod FI, White RA, 3rd, Visscher PT, Burns BP. Microbial dark matter filling the niche in hypersaline microbial mats. Microbiome. 2020;8:1–14. doi: 10.1186/s40168-020-00910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schut GJ, Nixon WJ, Lipscomb GL, Scott RA, Adams MW. Mutational analyses of the enzymes involved in the metabolism of hydrogen by the hyperthermophilic archaeon Pyrococcus furiosus. Front Microbiol. 2012;3:1–6. doi: 10.3389/fmicb.2012.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ruuskanen MO, Colby G, St. Pierre KA, St. Louis VL, Aris‐Brosou S, Poulain AJ. Microbial genomes retrieved from High Arctic lake sediments encode for adaptation to cold and oligotrophic environments. Limnol Oceanogr. 2020;65:S233–S247. doi: 10.1002/lno.11334. [DOI] [Google Scholar]
- 86.Vigneron A, Cruaud P, Lovejoy C, Vincent WF. Genomic evidence of functional diversity in DPANN archaea, from oxic species to anoxic vampiristic consortia. ISME Commun. 2022;2:1–10. doi: 10.1038/s43705-022-00088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018;36:996–1004. doi: 10.1038/nbt.4229. [DOI] [PubMed] [Google Scholar]
- 88.Meheust R, Castelle CJ, Matheus Carnevali PB, Farag IF, He C, Chen LX, et al. Groundwater Elusimicrobia are metabolically diverse compared to gut microbiome Elusimicrobia and some have a novel nitrogenase paralog. ISME J. 2020;14:2907–22. doi: 10.1038/s41396-020-0716-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hahn CR, Farag IF, Murphy CL, Podar M, Elshahed MS, Youssef NH. Microbial diversity and sulfur cycling in an early earth analogue: from ancient novelty to modern commonality. mBio. 10.1128/mbio.00016-22. (in press). [DOI] [PMC free article] [PubMed]
- 90.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rummel JD, Beaty DW, Jones MA, Bakermans C, Barlow NG, Boston PJ, et al. A new analysis of Mars "Special Regions": findings of the second MEPAG Special Regions Science Analysis Group (SR-SAG2) Astrobiology. 2014;14:887–968. doi: 10.1089/ast.2014.1227. [DOI] [PubMed] [Google Scholar]
- 92.Harris RL, Schuerger AC, Wang W, Tamama Y, Garvin ZK, Onstott TC. Transcriptional response to prolonged perchlorate exposure in the methanogen Methanosarcina barkeri and implications for Martian habitability. Sci Rep. 2021;11:1–16. doi: 10.1038/s41598-020-79139-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Webster CR, Mahaffy PR, Atreya SK, Moores JE, Flesch GJ, Malespin C, et al. Background levels of methane in Mars’ atmosphere show strong seasonal variations. Science. 2018;360:1093–6. doi: 10.1126/science.aaq0131. [DOI] [PubMed] [Google Scholar]
- 94.Oehler DZ, Etiope G. Methane seepage on Mars: where to look and why. Astrobiology. 2017;17:1233–64. doi: 10.1089/ast.2017.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Marlow JJ, Larowe DE, Ehlmann BL, Amend JP, Orphan VJ. The potential for biologically catalyzed anaerobic methane oxidation on ancient Mars. Astrobiology. 2014;14:292–307. doi: 10.1089/ast.2013.1078. [DOI] [PubMed] [Google Scholar]
- 96.Ji M, Greening C, Vanwonterghem I, Carere CR, Bay SK, Steen JA, et al. Atmospheric trace gases support primary production in Antarctic desert surface soil. Nature. 2017;552:400–3. doi: 10.1038/nature25014. [DOI] [PubMed] [Google Scholar]
- 97.Berg JS, Ahmerkamp S, Pjevac P, Hausmann B, Milucka J, Kuypers MMM. How low can they go? Aerobic respiration by microorganisms under apparent anoxia. FEMS Microbiol Rev. 2022;fuac006. 10.1093/femsre/fuac006. [DOI] [PMC free article] [PubMed]
- 98.Berg JS, Pjevac P, Sommer T, Buckner CRT, Philippi M, Hach PF, et al. Dark aerobic sulfide oxidation by anoxygenic phototrophs in anoxic waters. Environ Microbiol. 2019;21:1611–26. doi: 10.1111/1462-2920.14543. [DOI] [PubMed] [Google Scholar]
- 99.Stamenković V, Ward LM, Mischna M, Fischer WW. O2 solubility in Martian near-surface environments and implications for aerobic life. Nat Geosci. 2018;11:905–9. doi: 10.1038/s41561-018-0243-0. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing reads, metagenome, MAGs, and SAGs are in NCBI under BioProject PRJNA699472. JGI metagenome and SAG annotations are available under GOLD Study ID Gs0135943 (SAG IDs in Table S4).