

Abstract
In Gram-negative pathogens, the stringent response regulator DksA controls the expression of hundreds of genes, including virulence-related genes. Interestingly, Pseudomonas aeruginosa has two functional DksA paralogs: DksA1 is constitutively expressed and has a zinc-finger motif, while DksA2 is expressed only under zinc starvation conditions and does not contain zinc. DksA1 stimulates the production of virulence factors in vitro and is required for full pathogenicity in vivo. DksA2 can replace these DksA1 functions. Here, the role of dksA paralogs in P. aeruginosa tolerance to H2O2-induced oxidative stress has been investigated. The P. aeruginosa dksA1 dksA2 mutant showed impaired H2O2 tolerance in planktonic and biofilm-growing cultures and increased susceptibility to macrophages-mediated killing compared to the wild type. Complementation with either dksA1 or dksA2 genes restored the wild type phenotypes. The DksA-dependent tolerance to oxidative stress involves, at least in part, the positive transcriptional control of both katA and katE catalase-encoding genes. These data support the hypothesis that DksA1 and DksA2 are eco-paralogs with indistinguishable function but optimal activity under different environmental conditions, and highlight their mutual contribution to P. aeruginosa virulence.
Subject terms: Bacterial genetics, Bacteriology, Pathogens
Introduction
Bacteria constantly contend with toxic reactive oxygen species (ROS) such as hydrogen peroxide (H2O2), superoxide (O2−), hypochlorous acid (HOCl) and the hydroxyl free radical (·OH), which cause DNA damage, lipid peroxidation and negatively affect protein structure and functionality. These molecules are ubiquitous in the environment, and some of them are also generated by aerobically growing bacterial cells. During infection, pathogenic bacteria must cope with immune response cells, which produce particularly high levels of ROS that are either released in the extracellular milieu or used to kill engulfed bacteria1. After phagocytosis of bacteria, professional phagocytic cells, including macrophages, activate the NOX2 NADPH oxidase, a multicomplex enzyme assembled in the phagosomal membrane, which catalyses the reduction of molecular oxygen to superoxide. The latter can directly kill the bacteria within phagosomes and is converted to H2O2 by superoxide dismutases (SODs)2,3. Interestingly, bacterial SODs contribute to the production of microbicidal H2O2 within macrophages, as recently observed in Pseudomonas aeruginosa4. H2O2 can be further converted to hydroxyl free radical via Fenton reaction or halogenated ROS. This quick ROS production in the phagosome, a process known as oxidative burst, plays a key role in the host innate immune response5,6. Bacteria must also cope with extracellular ROS. As an example, the oxidases DUOX1 and DUOX2 generate H2O2 on the extracellular side of the apical membrane of the airway epithelium7.
P. aeruginosa is an opportunistic human pathogen responsible for life-threatening acute and chronic infections in immunocompromised, hospitalized, and cystic fibrosis patients5. The increasing prevalence of multi-drug resistant P. aeruginosa strains in the hospital setting increases the risk of antimicrobial treatment failure. Therefore, P. aeruginosa belongs to the ESKAPE group of bacterial pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, P. aeruginosa, Enterobacter spp.) for which novel treatment approaches are urgently needed8,9.
In addition to a wide arsenal of virulence factors, P. aeruginosa expresses an array of ROS-scavenging enzymes to counteract endogenous and exogenous ROS, including the four catalases KatA, KatB, KatE and KatN5,6,10–12. The KatA and KatB catalases are crucial for P. aeruginosa adaptation to the high levels of H2O2 encountered in the host11,13,14. Expression of katA is constitutive and increases in the presence of H2O2. Conversely, the expression of katB is induced only upon exposure to exogenous H2O210,15. Up to now, it is unclear if also the KatE (also named KatC) and KatN (also named KatM) catalases are involved in H2O2 detoxification in P. aeruginosa.
The bacterial stringent response regulator DksA contributes to ROS tolerance in bacterial pathogens such as Salmonella enterica and Haemophilus ducreyi16–19. DksA also plays a key role in the control of virulence-related genes. In some pathogenic bacteria (e.g., Escherichia coli, H. ducreyi, P. aeruginosa, S. enterica, Vibrio cholerae and A. baumannii) dksA mutants are less virulent than the wild type both in vitro and in vivo18,20–25.
Up to now, P. aeruginosa is the only bacterium known to express two functional DksA paralogs, encoded by the dksA1 and dksA2 genes. The DksA1 and DksA2 proteins share a similar overall structure containing: a RNAP-interacting coiled-coil domain, a globular domain and a C-terminal α-helix. However, the globular domain of DksA1 contains a canonical zinc-finger structure, with a four-cysteine motif (4-Cys) for zinc binding, while the DksA2 globular domain has only two cysteine residues (2-Cys), and does not coordinate zinc26,27. The constitutively expressed DksA1 protein broadly affects the P. aeruginosa transcriptome, and positively controls virulence genes expression23,24. Conversely, DksA2 is exclusively expressed under conditions of zinc starvation28.
By artificially inducing DksA2 expression in Luria–Bertani Broth (LB), our group has recently shown that the DksA2 protein can replace DksA1 in the control of about sixteen hundred genes, including those involved in virulence and biofilm formation24. Only a small number of functionally unrelated genes seems to be exclusively controlled by each one of the two DksA paralogues under our artificial conditions24. Since the zinc-containing 4-Cys motif is required for the proper folding and functionality of P. aeruginosa DksA1, our study supports the previous hypothesis that the structural stability and functionality of DksA1 could be impaired in zinc-poor environments, where the zinc-free DksA2 protein could replace or adjuvate DksA1 function26,28.
A difference in the functionality of P. aeruginosa DksA1 and DksA2 proteins has also been proposed by Crawford et al.19, who showed that a S. enterica ∆dksA mutant complemented with the P. aeruginosa dksA2 gene was less resistant to H2O2 and more susceptible to macrophage-mediated killing than the same strain complemented with the P. aeruginosa dksA1 gene. Although intriguing, the differential role of DksA1 and DksA2 in oxidative stress tolerance has not yet been tested in P. aeruginosa.
The objective of this study has been to investigate the role of DksA1 in P. aeruginosa tolerance to H2O2 and macrophages-mediated killing, and to verify to which extent DksA2 can replace DksA1 function. Evidence is here provided that DksA1 is required for H2O2 tolerance in both planktonic and biofilm growing cells, and that this protein protects against macrophages-mediated killing. One of the mechanisms involved in DksA-dependent resistance to oxidative stress implies the positive control of katA and katE genes expression. Finally, in the homologous P. aeruginosa system, DksA2 fully replaced the protective function of DksA1 against oxidative stress, in contrast with previous observations in the heterologous Salmonella system19.
Results
DksA1 contributes to hydrogen peroxide tolerance, and DksA2 can replace DksA1 function
In P. aeruginosa, the dksA1 gene is constitutively expressed while the dksA2 gene is strongly repressed by Zur in the presence of available zinc24,28. To achieve comparable expression levels of the two dksA paralogs in a rich laboratory medium, such as LB, we previously generated a set of four P. aeruginosa PAO1 recombinant strains: the wild type strain (PAO1) and the dksA1 dksA2 double mutant (∆dksA1-2), both carrying the pME6032 empty vector, and the ∆dksA1-2 mutant carrying either pDksA1 or pDksA2. The latter are pME6032-derivative plasmids for isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible expression of dksA1 and dksA2, respectively (Supplementary Table S1). As shown previously, the deletion of dksA paralogs or the presence of pME6032 derivatives or IPTG addition did not affect growth in LB. In addition, IPTG induction drove similar levels of dksA paralogs expression in PAO1 ∆dksA1-2 carrying pDksA1 or pDksA224. Here, the same experimental framework has been used to investigate the role of DksA1 and DksA2 in P. aeruginosa response to oxidative stress caused by H2O2.
The minimum inhibitory concentration (MIC) of H2O2 determined for planktonic P. aeruginosa cells in LB supplemented with 0.1 mM IPTG was 1 mM for all the tested strains. Subsequently, the P. aeruginosa tolerance to H2O2, i.e., the ability to survive transient exposure to 50X MIC H2O2 concentrations, was determined. Results indicate that the percentage of bacteria survival after 30-min treatment with 50 mM H2O2 was fivefold higher in PAO1(pME6032) than in the ∆dksA1-2(pME6032) mutant (Fig. 1). In addition, complementation with either dksA1 or dksA2 genes in ΔdksA1-2(pDksA1) and ΔdksA1-2(pDksA2) restored H2O2 tolerance, with no significant difference between the two dksA paralogs.
Figure 1.
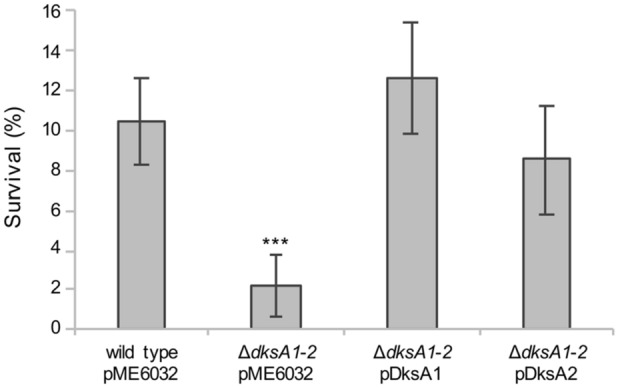
Effect of dksA paralogs on H2O2 tolerance in P. aeruginosa planktonic cultures. Survival of the indicated strains grown in LB supplemented with 0.1 mM IPTG to the late exponential phase (OD600 ≈ 2.5) after treatment with 50 mM H2O2 for 30 min. Survival is expressed as percentage of the CFU counts of the treated samples relative to the CFU counts of the corresponding untreated controls. The average values from five independent experiments are reported with standard deviations. ***p < 0.001 versus wild type (ANOVA).
The role of dksA1 and dksA2 in P. aeruginosa biofilm tolerance to H2O2 was also investigated. To this purpose, the MIC of H2O2 was determined in M9-glu-CAA, a chemically-defined medium suited for P. aeruginosa biofilms studies24. In M9-glu-CAA, the MIC of H2O2 was 0.1 mM for all the tested strains, thus tenfold lower than that determined in LB. Hence, pre-formed P. aeruginosa biofilms were challenged with supra-MIC concentrations of H2O2, ranging from 50 to 200X MIC, and compared to the respective untreated controls.
In the absence of H2O2 treatment, higher levels of biofilm biomass were detected by crystal violet (CV) staining in the ∆dksA1-2(pME6032) mutant compared to PAO1(pME6032) and the ΔdksA1-2(pDksA1) and ΔdksA1-2(pDksA2) complemented strains (Fig. 2a), in accordance with our previous study24. However, the fluorescein diacetate (FDA) assay showed that the metabolic activity of PAO1(pME6032) and ∆dksA1-2(pME6032) biofilms were comparable in the absence of H2O2 (Fig. 2b), suggesting that in the ∆dksA1-2(pME6032) mutant there could be an increased production of extracellular biofilm matrix components compared to PAO1(pME6032). After challenging with H2O2, a dose–response biofilm disrupting effect was detected for all the tested strains by using both CV and FDA detection methods, though ∆dksA1-2(pME6032) biofilms were significantly more reduced by the treatment compared to the wild type and complemented strains (Fig. 2). As observed in liquid cultures (Fig. 1), complementation with either dksA1 or dksA2 genes in the ΔdksA1-2 mutant restored similar levels of H2O2 tolerance.
Figure 2.
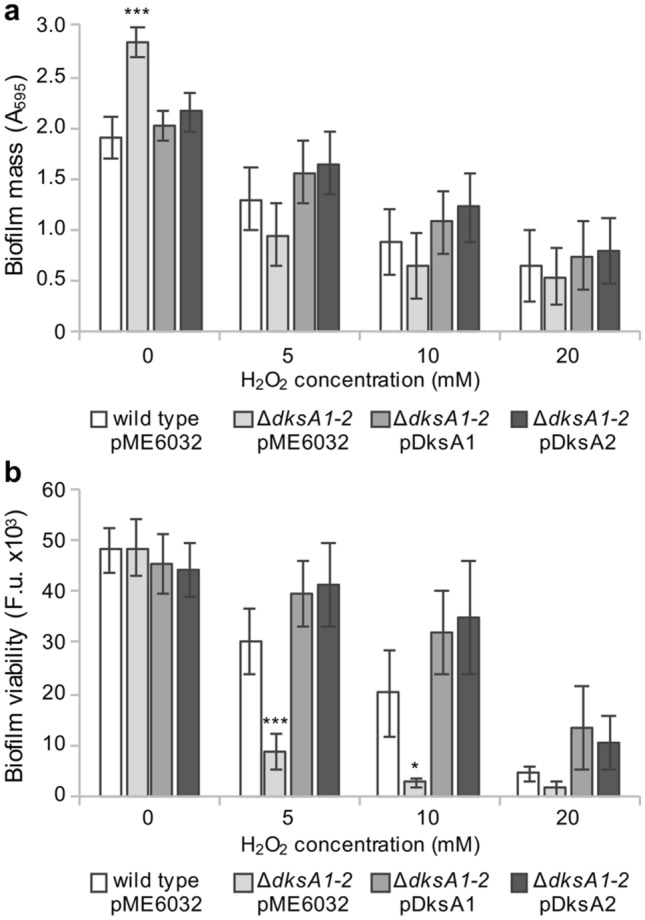
Effect of dksA paralogs on H2O2 tolerance in P. aeruginosa biofilm. Biofilm biomass (a) and viability (b; expressed as Fluorescence units, F. u.) of the indicated strains grown in M9-glu-CAA supplemented with 0.1 mM IPTG, challenged for 6 h with 5, 10 or 20 mM H2O2. The average values from five independent experiments performed on multiple wells per condition are reported with standard deviations. ***p < 0.001 versus untreated wild type (a; ANOVA); *p < 0.05 and ***p < 0.001 versus wild type challenged with 10 and 5 mM H2O2, respectively (b; ANOVA).
Overall, since dksA2 expression is shut-off in PAO1 grown in the zinc-proficient media, such as LB and M9-glu-CAA24, the decreased H2O2 tolerance of the ∆dksA1-2 mutant, relative to the PAO1 parent strain, should in principle be attributable to the lack of dksA1 expression. Hence, the above results suggest that DksA1 is required for H2O2 tolerance of both planktonic and biofilm cultures of P. aeruginosa, and that dksA2 can replace dksA1 function when artificially expressed in zinc-containing media.
DksA1 contributes to macrophages-mediated killing tolerance, and DksA2 can replace DksA1 function
As mentioned in the introduction, the oxidative burst is the first microbicidal mechanism activated by macrophages to kill engulfed bacteria5,6. Thus, we analysed the role of DksA1 and DksA2 in counteracting oxidative stress within macrophages. First, RAW macrophages were infected for 15 and 30 min with PAO1 or the ∆dksA1-2 mutant, both carrying the pUCP30T-GFPmut3 plasmid for constitutive expression of the green fluorescent protein (GFP). Flow cytofluorimetry analysis showed that the percentage of fluorescent macrophages was similar in both infections, revealing that the ∆dksA1-2 double mutation has no impact on P. aeruginosa internalization by macrophages (Supplementary Fig. S1).
Hence, the susceptibility to macrophages-mediated killing was compared in P. aeruginosa PAO1 and its isogenic recombinant strains. After 30 min from the infection, the number of live bacteria recovered from RAW macrophages was higher for PAO1(pME6032) than for the ∆dksA1-2(pME6032) mutant (Fig. 3). Taking into consideration that these strains are similarly phagocytosed, this result indicates that the ∆dksA1-2 mutant is killed more efficiently than the wild type strain. The intra-macrophage survival defect of the ∆dksA1-2 double mutant was complemented by pDksA1- or pDksA2-driven expression of either dksA1 or dksA2, respectively (Fig. 3), providing evidence that DksA2 can replace DksA1 function even inside macrophages. Overall, these results indicate that both DksA paralogs can protect P. aeruginosa against NADPH-oxidase-dependent macrophages oxidative burst.
Figure 3.
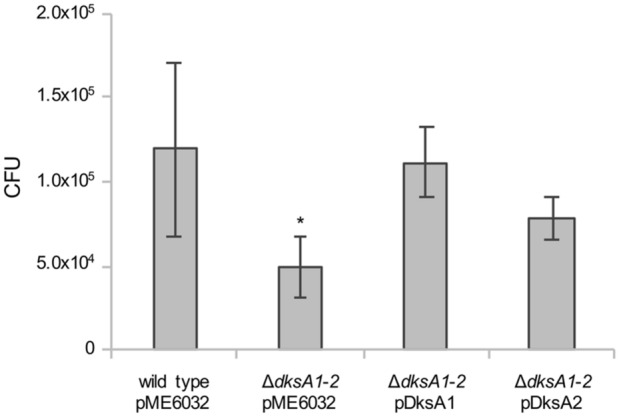
Intracellular survival of P. aeruginosa strains in RAW macrophages. Macrophages were infected for 30 min with the indicated strains, treated with gentamycin and lysed. Total live bacteria in cell lysates were recovered and quantified by CFU counts on PIA plates. The average CFUs per well from four independent experiments, each in duplicate, are reported with standard deviations. *p < 0.05 versus wild type (ANOVA).
DksA1 modulates endogenous ROS content by controlling catalases expression, and DksA2 can replace DksA1 function
The results presented above show that both P. aeruginosa dksA paralogs restore wild type levels of tolerance to H2O2 exposure or macrophage oxidative burst. However, the DksA paralogs are also involved in the fine modulation of several central metabolic pathways, and an unbalanced metabolism could also affect endogenous ROS levels29. In agreement with this hypothesis, endogenous ROS levels were about 36% lower in PAO1(pME6032) than in ∆dksA1-2(pME6032), both grown in LB (Fig. 4a). Hence, DksA1 contributes to the homeostasis of the intracellular ROS levels naturally produced by cellular metabolism during aerobic growth, even in the absence of exogenous oxidative stress and nutrient starvation. An enzymatic assay revealed that ∆dksA1-2(pME6032) disclosed a 44% reduction of catalase activity compared to PAO1(pME6032) (Fig. 4b), suggesting that the increased ROS levels measured in the ∆dksA1-2 mutant could be related, at least in part, to a decreased expression of the H2O2 detoxifying enzymes. Finally, the expression of either DksA1 or DksA2 in the ΔdksA1-2(pDksA1) and ΔdksA1-2(pDksA2) fully restored wild type catalase activity and ROS levels, providing evidence that also in this case DksA2 can replace DksA1.
Figure 4.
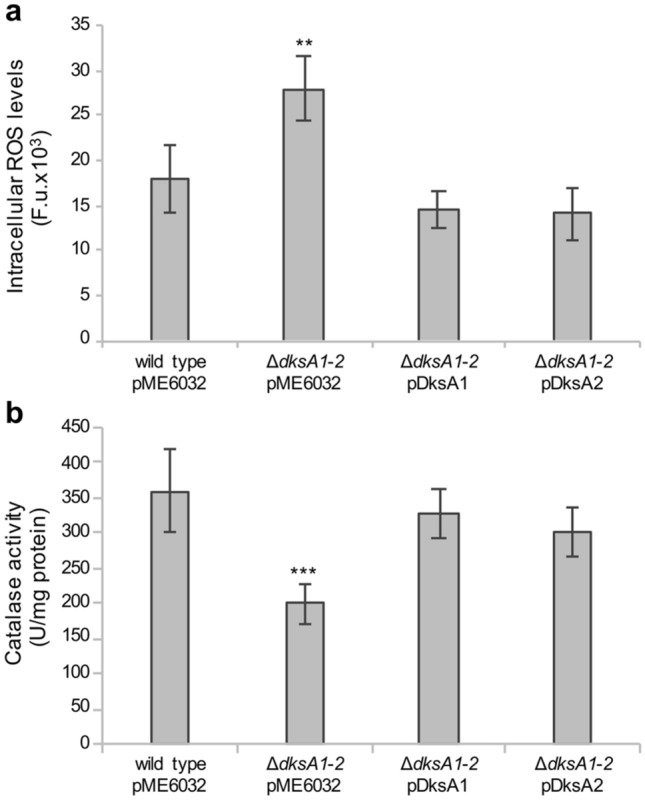
Effect of dksA1 and dksA2 on endogenous ROS levels and catalase activity. Intracellular levels of ROS (expressed as Fluorescence Units, F. u.) (a) and catalase activity (b) were determined in the indicated strains grown to the late exponential phase (OD600 ≈ 2.5) in LB supplemented with 0.1 mM IPTG. The average values from three independent experiments are reported with standard deviations. **p < 0.01, ***p < 0.001 versus wild type (ANOVA).
To study the impact of the dksA gene products on the expression of different catalase-encoding genes, relative mRNA levels of katA, katB, katE and katN were investigated by means of Real Time PCR analyses in P. aeruginosa cultures grown in LB containing 0.1 mM IPTG, treated or not with 1 mM H2O2 (1X MIC) for 20 min, to measure the expression of the H2O2-inducible genes. Preliminary experiments showed that the expression of dksA1 and dksA2 was not affected in PAO1 in response to 1 mM H2O2 (Supplementary Fig. S2).
As previously reported10,15, the mRNA levels of both katA and katB increased in PAO1 exposed to H2O2 (Fig. 5). Interestingly, the dksA1 dksA2 double deletion had opposite effect on the H2O2-dependent expression of these genes. Indeed, katA and katB mRNA levels decreased and increased in ∆dksA1-2(pME6032) relative to PAO1(pME6032) in the presence of H2O2, respectively (Fig. 5a,b).
Figure 5.
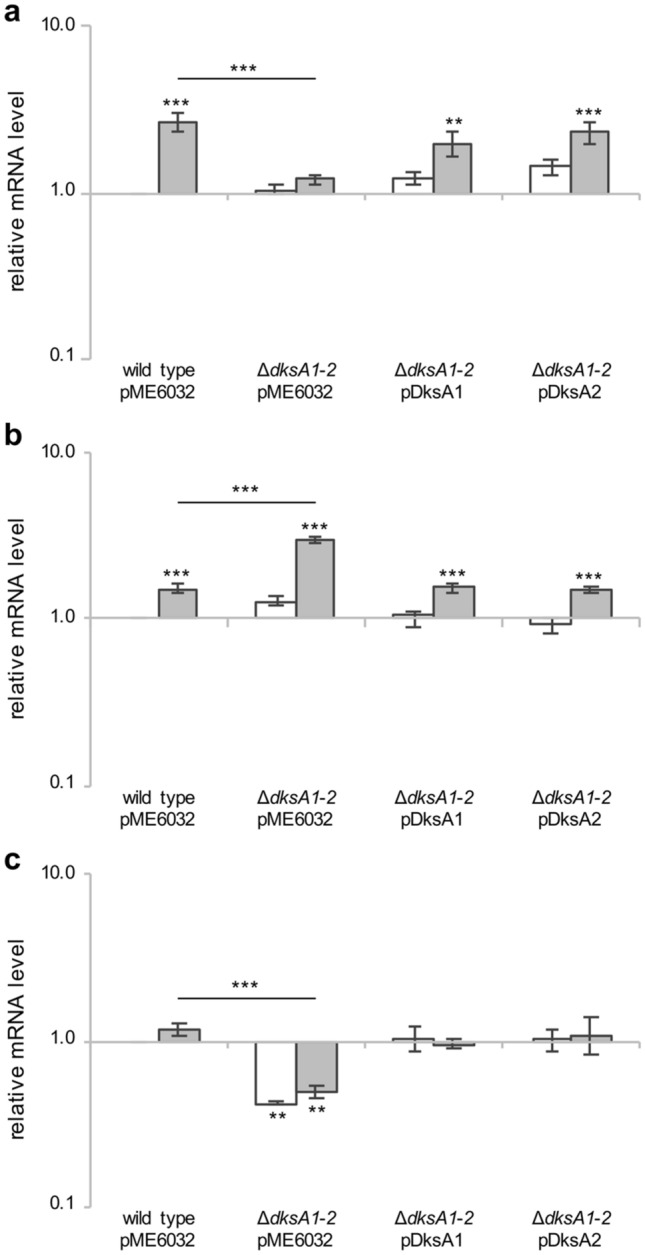
Expression analysis of oxidative stress response genes. Bacterial cultures were grown to the exponential phase (OD600 ≈ 2.5) in LB supplemented with 0.1 mM IPTG, then incubated with or without 1 mM H2O2 for 20 min. The mRNA levels of katA (a), katB (b) and katE (c) were determined by Real Time PCR analysis. Gene expression in the indicated untreated (white bars) or treated (grey bars) strains was reported as fold change in gene expression relative to the level of each gene in the untreated wild type PAO1(pME6032) strain. The average of two independent analyses, each performed on three technical replicates, is reported with standard deviations. Asterisks above the horizontal lines refer to p values versus wild type challenged with 1 mM H2O2; asterisks above the bars refer to p values versus the untreated wild type; **p < 0.01, ***p < 0.001 (ANOVA).
Concerning the other two P. aeruginosa catalase-encoding genes, katN expression was not induced by H2O2 and not affected by the dksA1 dksA2 double deletion (Supplementary Fig. S3). Conversely, katE mRNA levels were downregulated in ∆dksA1-2(pME6032) relative to PAO1(pME6032), irrespective of H2O2 presence (Fig. 5c).
Since DksA2 is expressed at low-basal levels in PAO1(pME6032) grown in LB, the above results show that at least DksA1 is required for the H2O2-dependent transcriptional upregulation and downregulation of katA and katB, respectively. In addition, DksA1 is also required for full expression of katE, irrespective of H2O2.
Finally, the IPTG-driven expression of either DksA1 or DksA2 in the ΔdksA1-2(pDksA1) and ΔdksA1-2(pDksA2) strains restored wild type mRNA levels for all the tested genes (Fig. 5), demonstrating that DksA2 can replace the function of DksA1 in promoting katA and katE expression, and in down-regulating katB expression.
Discussion
The stringent response controls bacterial adaption to nutrient starvation and other stressful conditions. In Gram-negative bacteria, (p)ppGpp binds the ß’ subunit of RNAP at the interface with the ω subunit and in a second site, sandwiched between the ß′ subunit and the DksA protein, which is strictly required for this interaction30,31. Hence, both (p)ppGpp and DksA are required for full stringent response, even if the two factors can also work independently20,32–35. While previous studies showed that (p)ppGpp positively affects P. aeruginosa tolerance to H2O236,37, the contribution of DksA1 and DksA2 to this process remained unknown so far.
In the culture media used in this and our previous study24, dksA2 expression is strongly repressed by zinc in the P. aeruginosa PAO1 wild type strain. Hence, the comparison of the wild type and dksA1 dksA2 double mutant phenotypes only reveals the effect of DksA1 depletion. However, complementation of the double mutant with either DksA1 or DksA2 expressed by the corresponding gene under the control of an IPTG-inducible promoter allows comparing the activity of the two DksA paralogs. Overall, the dksA1 and dksA2 expression levels obtained by this approach are adequate to restore wild type levels of all the tested phenotypes.
While the dksA1 dksA2 double deletion does not affect P. aeruginosa long-term exposure to H2O2 in both LB and M9-glu-CAA (i.e., the MIC of H2O2 is identical for wild type PAO1 and the ΔdksA1-2 mutant), DksA1 increases P. aeruginosa tolerance to transient H2O2 exposure, both in planktonic and in biofilm growing cultures. In line with these results, the deletion of both dksA1 and dksA2 undermines P. aeruginosa survival within macrophages. The dksA1 dksA2 double deletion leads to an increase in P. aeruginosa biofilm biomass, as already shown in our previous study by using different biofilm models (i.e., CV staining, pellicle formation and Congo-Red binding assays)24. In the same study, it was shown that c-di-GMP levels were lower in the ΔdksA1-2 mutant compared to the wild type, indicating that DksA1 negatively controls biofilm formation through a c-di-GMP-independent pathway. Furthermore, the RNA-Seq analysis showed that some of the known P. aeruginosa biofilm-related genes were up-regulated and others were down-regulated in the ΔdksA1-2 mutant compared to the wild type strain24. This study showed that the biofilm cells of the ΔdksA1-2 mutant are less viable after H2O2 exposure relative to wild type biofilm cells, despite the increased biofilm biomass. This suggests a defect in the ΔdksA1-2 biofilm matrix composition and development. In the future, it could be interesting to further investigate this issue.
In the absence of exogenous H2O2, the ΔdksA1-2 mutant showed an increase in the endogenous ROS content and a decrease in the overall catalase activity, compared to wild type PAO1. This could be due to the reduced expression of katE in the ΔdksA1-2 strain, since the expression of the other P. aeruginosa catalase genes was not affected by dksA1 dksA2 double deletion in the absence of oxidative stress. However, since DksA1 controls more than one thousand genes, including primary metabolism pathways23,24, the ROS increase measured in the ΔdksA1-2 strain could also be due to a generalized metabolic effect. Moreover, in the presence of exogenous H2O2, katA and katE genes were downregulated while katB was up-regulated in the double mutant compared to the wild type PAO1.
Overall, it is plausible that the defective expression of katA and katE genes, combined with an altered metabolism leading to increased levels of endogenous ROS, could overcome the effects of the increased katB expression, ultimately reducing the tolerance of the ΔdksA1-2 mutant to exogenous H2O2 and macrophages-mediated killing, relative to wild type PAO1. In this context, it is to note that the role of the KatE catalase in P. aeruginosa H2O2 tolerance has not been clarified yet. Indeed, KatE seems not to be involved in H2O2 detoxification in the PA14 strain11, while a protective role of KatE against oxidative stress only at elevated temperatures was described in the PAO6049 strain38.
Interestingly, a P. aeruginosa mutant lacking the relA and spoT genes, required for (p)ppGpp synthesis, showed several phenotypes similar to those caused by the dksA1 dksA2 double deletion, i.e., increased susceptibility to H2O2, impaired overall catalase activity and high levels of endogenous ROS with respect to the parental strain36,37,39. However, different from what we observed for DksA1, (p)ppGpp positively controls the expression of both katA and katB genes. Interestingly, this (p)ppGpp-dependent regulation is independent of exogenous H2O2 for katA and H2O2-dependent for katB36,37.
By combining our data with previous findings, it can be argued that both (p)ppGpp and DksA1 play a major role in P. aeruginosa tolerance to endogenous and exogenous oxidative stress, even if they differently regulate the expression of H2O2-scavenger enzymes. Further studies should be carried out to unravel the regulatory mechanisms underlying the different effects of DksA1 and (p)ppGpp on katA, katB and katE genes expression, as well as the impact of KatE-mediated H2O2 detoxification in P. aeruginosa PAO1.
To our knowledge, the importance of DksA in oxidative stress tolerance has so far been documented only for S. enterica and H. ducreyi. Each one of these pathogens expresses only one DksA protein, containing a zinc finger domain, as it is for DksA116,18. Hence the possibility that DksA2 could replace DksA1 deserves special attention, also considering that a previous study showed that a S. enterica ∆dksA mutant expressing the P. aeruginosa dksA2 gene had an impaired tolerance to H2O2 in vitro, reduced survival in macrophages, and reduced virulence in mice relative to the S. enterica ∆dksA mutant expressing the P. aeruginosa dksA1 gene. The different behaviour of DksA1 and DksA2 in the heterologous host Salmonella was supported by experiments carried out with the purified proteins, showing that DksA1 was more resistant to oxidation than DksA2, likely due to the presence of zinc in the globular domain19. Different from what observed in S. enterica, we showed by multiple experimental strategies that DksA2 replaces all DksA1 functions related to P. aeruginosa H2O2 tolerance when expressed in its isogenic background, at least under our experimental conditions.
Certainly, the possibility that dksA2 overexpression may mask a higher sensitivity of DksA2 to ROS than DksA1 cannot be rule out, and this issue should be further studied under conditions of zinc starvation, mimicking the environmental conditions where dksA2 is naturally expressed. In the wild type genetic background, under zinc-limiting conditions, both dksA1 and dksA2 genes are expressed from their natural promoters28 and they may have a different impact on P. aeruginosa oxidative stress response. However, while dksA2 expression levels are related to the extent of zinc limitation, DksA1 activity might be impaired under conditions of extreme zinc deficiency, due to incorrect folding of the Zn-finger domain28.
Zinc depletion is recognised as an innate immunity mechanism faced by pathogens during the infection. Interestingly, dksA2 is expressed in the sputum of cystic fibrosis patients colonized by P. aeruginosa40,41, supporting a role for DksA2 at least in this kind of infection. While further studies should be carried out to better define the relative importance of DksA1 and DksA2 in the protection from H2O2 exposure under zinc starvation conditions, this study provides the evidence that DksA2 has the intrinsic ability to replace the dksA1 gene product under oxidative stress conditions.
Overall, the results of this study are in line with our previous observation that DksA2 can replace DksA1 function in controlling the expression of almost all the DksA1-regulated genes and DksA1-dependent virulence phenotypes24 strengthening the hypothesis that DksA1 and DksA2 are eco-paralogs as defined by Sanchez-Perez et al.42, i.e., paralogs with the same overall function, having optimal activity under different environmental conditions.
Materials and methods
Bacterial strains and growth conditions
Bacterial strains used in this study are listed in Supplementary Table S1. P. aeruginosa strains were routinely grown at 37 °C in shaking conditions in LB43, or LB supplemented with 1.5% (w/v) agar. When required, the media were supplemented with 0.1 mM IPTG or 100 µg/mL tetracycline (Tc). M9 medium [0.77% (w/v) Na2HPO4·2H2O; 0.3% (w/v) KH2PO4; 0.05% (w/v) NaCl; 0.025% (w/v) MgSO4·7H2O; 0.002% (w/v) CaCl2]43,44 supplemented with 0.39% (w/v) glucose and 0.5% (w/v) casamino acids (CAA) as carbon sources (M9-glu-CAA)24 was used for specific assays. H2O2 [stock 30% (v/v), Sigma Aldrich] was added to the media at the concentrations indicated in the text.
MIC assays
The MIC of H2O2 was evaluated as previously detailed45 with the standard microdilution method, according to the Clinical and Laboratory Standards Institute guidelines46. Cultures of P. aeruginosa were incubated at 37 °C with shaking in LB containing 100 μg/mL Tc or in M9-glu-CAA containing 50 μg/mL Tc, both supplemented with 0.1 mM IPTG. After overnight growth, cultures were diluted to an optical density at 600 nm wavelength (OD600) of ≈ 0.0005 (ca. 5 × 105 CFU/mL) in 100 μL of LB or M9-glu-CAA, both supplemented with 0.1 mM IPTG, in the presence of increasing concentrations of H2O2. The MIC values were evaluated after 24 h of static incubation at 37 °C.
H2O2 killing assays
H2O2 killing assays were performed as previously described36, with minor modifications. Late exponential phase cultures (OD600 ≈ 2.5) were normalized to an OD600 of 0.5 in LB supplemented with 0.1 mM IPTG and 50 mM H2O2 before incubation at 37 °C in shaking conditions. At the same time, untreated cultures were incubated at 37 °C as a control. After 30 min, 0.2% (w/v) sodium thiosulfate was added to neutralize residual H2O2. Colony forming units (CFU) were determined by the standard microdilution technique on LB agar plates. Survival was reported as percentage of CFU counted in the treated sample relative to the CFU counted in the untreated sample.
Biofilm assays
The crystal violet (CV) binding assay and the fluorescein diacetate (FDA) assay were performed in microtiter plates as already detailed in previous studies47–49, with slight changes. Bacterial cells were grown in M9-glu-CAA with 50 µg/mL Tc and 0.1 mM IPTG for 8 h in shaking conditions and subsequently diluted to OD600 ≈ 0.015 in fresh M9-glu-CAA medium supplemented with 0.1 mM IPTG. Aliquots of 100 µL were transferred to sterile 96-well polystyrene microtiter plates and incubated at 30 °C for 15 h in static conditions. After the removal of the planktonic phase, biofilms were refreshed with 100 μL of fresh medium supplemented with 5, 10 or 20 mM H2O2 (treated biofilms) or without the addition of H2O2 (untreated biofilms). Microtiter plates were incubated at 30 °C for 6 h. After removing the liquid phase, biofilm mass was quantified by CV binding assay, while biofilm viability was determined by FDA assay. Briefly, CV binding assay was performed staining the attached cells with 1% (w/v) CV for 15 min. Hence, all wells were washed four times with distilled water and air dried. Finally, the biofilm-bound dye was solubilized with 200 µL of ethanol for 10 min and measured as A595 in an automated luminometer-spectrometer plate reader (Tecan Spark 10 M). To perform the FDA assay, the FDA stock solution (10 mg/mL, prepared in acetone; Sigma Aldrich) was diluted 1:100 in 100 mM 3-(N-morpholino)-propanesulfonic acid (MOPS, pH 7.0; FDA working solution) and 200 μL of FDA working solution were dispensed in all wells. The microtiter was incubated in the dark at 37 °C in static condition. Fluorescence was measured at 485 nm excitation and 535 nm emission wavelengths after 1 h through an automated luminometer-spectrometer plate reader (Tecan Spark 10 M).
Macrophages infection assays
Murine macrophages RAW264.7 (ATCC TIB71) were grown in Dulbecco’s modified Eagle’s medium (DMEM; Corning) high glucose supplemented with 10% (v/v) fetal bovine serum and 1% (w/v) glutamine (all from EuroClone, Italy) at 37 °C in 5% CO2. For the infection assay, the day before infection 5 × 104 cells/well were seeded in 48-well plates and incubated over-night in antibiotic-free medium. Each well was infected with different PAO1 strains at a multiplicity of infection (MOI) of 10. After 30 min of incubation at 37 °C, extracellular bacteria were killed by gentamycin (600 µg/mL) treatment during 15 min of incubation. For counting intracellular bacteria, infected RAW264.7 cells were washed with phosphate-buffered saline (PBS), lysed with 1% (v/v) Triton X-100, and dilutions plated on Pseudomonas isolation agar (PIA).
For the phagocytosis assay, macrophages were infected as described above, using GFP expressing bacteria obtained by transformation of PAO1 and ∆dksA1-2 with the pUCP30T-GFPmut3 plasmid (Supplementary Table S1)50. After either 15 or 30 min of infection, infected macrophages were washed twice with PBS, detached from the wells, recovered by centrifugation and finally resuspended in 300 µL PBS for flow cytometry (BD FACSCalibur, BD Biosciences, France). Phagocytosis was evaluated by the fraction of GFP positive cells in the bulk populations. Data were analysed using the CellQuest software and images processed using FlowJo.
Measurement of intracellular ROS levels
The intracellular ROS levels were measured as previously described51, with minor modifications. P. aeruginosa strains were grown in LB with 0.1 mM IPTG to the late exponential phase (OD600 of ≈ 2.5). Then, cells were collected by centrifugation, washed with PBS, and resuspended in PBS at an OD600 of about 1.25. Bacteria were incubated with 10 µM 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA; stock 2 mM, dissolved in dimethyl sulfoxide [DMSO]; Sigma Aldrich) for 20 min at 37 °C in the dark. Cells treated with an equal volume of DMSO were used as a negative control. Aliquots (200 µL) of each bacterial suspension were dispensed in a 96-well black microtiter plate. Fluorescence was measured at 485 nm excitation and 535 nm emission wavelengths with an automated luminometer-spectrometer plate reader (Tecan Spark 10 M) and normalized to the OD600 of each sample.
Catalase activity assay
Catalase activity was measured as described previously52, with minor modifications. Bacteria were grown in LB with 0.1 mM IPTG to the late exponential phase (OD600 of ≈ 2.5). Hence, cells were collected by centrifugation, resuspended in 50 mM potassium-phosphate buffer (PPB, pH 6.8), lysed by sonication and subsequently centrifugated to collect the supernatants. Total proteins from the soluble fraction were quantified by the Bradford assay53, with bovine serum albumin as the standard. Aliquots containing 10 µg of proteins were added to 50 mM PPB, and catalase activity was monitored by following the decomposition of 20 mM H2O2 in 50 mM PPB at 240 nm (OD240) for 1 min (readings every 10 s) by using UV-transparent disposable cuvettes. One unit of catalase activity corresponded to 1 μmol of H2O2 hydrolysed per min by 1 mg of total proteins at 25 °C.
RNA extraction and expression profiling experiments
P. aeruginosa strains were grown at 37 °C with shaking at 200 rpm in 10 mL of LB supplemented with 0.1 mM IPTG until reaching an OD600 of ≈ 2.5. Exponential phase cells were incubated with or without 1 mM H2O2 for 20 min in shaking conditions and 1 mL of each culture was mixed with 2 mL of RNA Protect Bacteria Reagent (Qiagen) for RNA extraction. Total RNA was extracted as previously described24,54. Briefly, RNA isolation was performed using RNeasy Mini Kit (Qiagen), including the on-column DNase I digestion step, followed by 1 h treatment at 37 °C with TURBO DNase (0.2 U per μg of RNA; Ambion) and SUPERase-In (0.4 U per µg of RNA; Ambion), and subsequent purification with the RNeasy Column Purification Kit (Qiagen). After confirmation of the lack of contaminating chromosomal DNA by PCR with the primer pair FWPpqsL and RVPpqsL (Supplementary Table S2), 1 μg of total RNA was used to synthesize cDNA using the iScript Reverse Transcription Supermix for RT-qPCR kit (BioRad). Real Time PCR analyses were performed using the iTaq Universal SYBR Green Supermix (BioRad), the AriaMx Real-Time PCR system (Agilent Technologies; software version 1.0) and the target-specific primers obtained by means of the Primer-Blast designing tool (www.ncbi.nlm.nih.gov/tools/primer-blast; Supplementary Table S2). The thermal cycling protocol was denaturation for 2 min at 95 °C, followed by 40 cycles of amplification at 95 °C for 15 s and 60 °C for 45 s. Fluorescence was registered in the last 15 s of the 60 °C step. The relative fold change in gene expression was calculated by the 2−ΔΔCt method using rpoD as the housekeeping gene. The average data and standard deviations (SD) were calculated from two independent experiments each performed on three technical replicates.
Statistical analysis
Statistical analysis was performed with the software GraphPad Prism 6.01, using one-way analysis of variance (ANOVA) followed by Tukey–Kramer multiple comparison test. Statistical analysis of macrophages infections used two-way ANOVA. A p value of < 0.05 was considered statistically significant.
Supplementary Information
Acknowledgements
This work was supported by the Italian Ministry of Education, University and Research (MIUR) with the following Grants: Excellence Departments (art. 1, commi 314–337 Legge 232/2016) to the Department of Science, Roma Tre University; PRIN 2017 (Prot. 20177J5Y3P) to P.V.; PRIN 2020 to L.L. (Prot. 202089LLEH). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author contributions
A.F., D.C., V.S. and V.P. performed the experiments; L.L., G.R., F.A. and P.V. designed the experiments; L.L. and F.A. conceived the study; A.F. and D.C. wrote the first draft of the manuscript; L.L., P.V. and F.A. contributed reagents and materials; all authors analyzed the data, corrected/amended the draft of the manuscript and approved the submitted version.
Data availability
All data generated or analysed during this study are included in this published article (and relative Supplementary Information file).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-14635-7.
References
- 1.Johnson LA, Hug LA. Distribution of reactive oxygen species defense mechanisms across domain bacteria. Free Radic. Biol. Med. 2019;140:93–102. doi: 10.1016/j.freeradbiomed.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 2.Cifani N, et al. Reactive-oxygen-species-mediated P. aeruginosa killing is functional in human cystic fibrosis macrophages. PLoS ONE. 2013;8:e71717. doi: 10.1371/journal.pone.0071717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riazanski V, Sui Z, Nelson DJ. Kinetic separation of oxidative and non-oxidative metabolism in single phagosomes from alveolar macrophages: impact on bacterial killing. iScience. 2020;23:101759. doi: 10.1016/j.isci.2020.101759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavinato L, et al. Escaping the phagocytic oxidative burst: the role of SODB in the survival of Pseudomonas aeruginosa within macrophages. Front. Microbiol. 2020;11:326. doi: 10.1128/jb.177.22.6536-6544.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malhotra S, Hayes D, Jr, Wozniak DJ. Cystic fibrosis and Pseudomonas aeruginosa: The host–microbe interface. Clin. Microbiol. Rev. 2019;32:e00138–e218. doi: 10.1128/CMR.00138-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.da Cruz Nizer WS, et al. Oxidative stress response in Pseudomonas aeruginosa. Pathogens. 2021;10:1187. doi: 10.3390/pathogens10091187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarr D, Tóth E, Gingerich A, Rada B. Antimicrobial actions of dual oxidases and lactoperoxidase. J. Microbiol. 2018;56:373–386. doi: 10.1007/s12275-018-7545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rice LB. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect. Dis. 2008;197:1079–1081. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 9.Boucher HW, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 10.Palma M, DeLuca D, Worgall S, Quadri LE. Transcriptome analysis of the response of Pseudomonas aeruginosa to hydrogen peroxide. J. Bacteriol. 2004;186:248–252. doi: 10.1128/JB.186.1.248-252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee JS, Heo YJ, Lee JK, Cho YH. KatA, the major catalase, is critical for osmoprotection and virulence in Pseudomonas aeruginosa PA14. Infect. Immun. 2005;73:4399–4403. doi: 10.1128/IAI.73.7.4399-4403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi YS, et al. Identification of Pseudomonas aeruginosa genes crucial for hydrogen peroxide resistance. J. Microbiol. Biotechnol. 2007;17:1344–1352. [PubMed] [Google Scholar]
- 13.Brown SM, Howell ML, Vasil ML, Anderson AJ, Hassett DJ. Cloning and characterization of the katB gene of Pseudomonas aeruginosa encoding a hydrogen peroxide-inducible catalase: purification of KatB, cellular localization, and demonstration that it is essential for optimal resistance to hydrogen peroxide. J. Bacteriol. 1995;177:6536–6544. doi: 10.1128/jb.177.22.6536-6544.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim BO, Chung IY, Cho YH. Differential expression of the major catalase, KatA in the two wild type Pseudomonas aeruginosa strains, PAO1 and PA14. J. Microbiol. 2019;57:704–710. doi: 10.1007/s12275-019-9225-1. [DOI] [PubMed] [Google Scholar]
- 15.Ochsner UA, Vasil ML, Alsabbagh E, Parvatiyar K, Hassett DJ. Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, and ahpC-ahpF. J. Bacteriol. 2000;182:4533–4544. doi: 10.1128/JB.182.16.4533-4544.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henard CA, Bourret TJ, Song M, Vázquez-Torres A. Control of redox balance by the stringent response regulatory protein promotes antioxidant defenses of Salmonella. J. Biol. Chem. 2010;285:36785–36793. doi: 10.1074/jbc.M110.160960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henard CA, et al. The 4-cysteine zinc-finger motif of the RNA polymerase regulator DksA serves as a thiol switch for sensing oxidative and nitrosative stress. Mol. Microbiol. 2014;91:790–804. doi: 10.1111/mmi.12498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holley CL, et al. DksA and (p)ppGpp have unique and overlapping contributions to Haemophilus ducreyi pathogenesis in humans. Infect. Immun. 2015;83:3281–3292. doi: 10.1128/IAI.00692-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crawford MA, et al. Redox-active sensing by bacterial DksA transcription factors is determined by cysteine and zinc content. MBio. 2016;7:e02161-15. doi: 10.1128/mBio.02161-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Åberg A, Fernández-Vázquez J, Cabrer-Panes JD, Sánchez A, Balsalobre C. Similar and divergent effects of ppGpp and DksA deficiencies on transcription in Escherichia coli. J. Bacteriol. 2009;191:3226–3236. doi: 10.1128/JB.01410-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pal RR, Bag S, Dasgupta S, Das B, Bhadra RK. Functional characterization of the stringent response regulatory gene dksA of Vibrio cholerae and its role in modulation of virulence phenotypes. J. Bacteriol. 2012;194:5638–5648. doi: 10.1128/JB.00518-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Azriel S, Goren A, Rahav G, Gal-Mor O. The stringent response regulator DksA is required for Salmonella enterica serovar Typhimurium growth in minimal medium, motility, biofilm formation, and intestinal colonization. Infect. Immun. 2016;84:375–384. doi: 10.1128/IAI.01135-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Min KB, Yoon SS. Transcriptome analysis reveals that the RNA polymerase-binding protein DksA1 has pleiotropic functions in Pseudomonas aeruginosa. J. Biol. Chem. 2020;295:3851–3864. doi: 10.1074/jbc.RA119.011692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fortuna A, Bähre H, Visca P, Rampioni G, Leoni L. The two Pseudomonas aeruginosa DksA stringent response proteins are largely interchangeable at the whole transcriptome level and in the control of virulence-related traits. Environ. Microbiol. 2021;23:5487–5504. doi: 10.1111/1462-2920.15693. [DOI] [PubMed] [Google Scholar]
- 25.Kim N, et al. Global regulator DksA modulates virulence of Acinetobacter baumannii. Virulence. 2021;12:2750–2763. doi: 10.1080/21505594.2021.1995253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perederina A, et al. Regulation through the secondary channel-structural framework for ppGpp-DksA synergism during transcription. Cell. 2004;118:297–309. doi: 10.1016/j.cell.2004.06.030. [DOI] [PubMed] [Google Scholar]
- 27.Furman R, et al. DksA2, a zinc-independent structural analog of the transcription factor DksA. FEBS Lett. 2013;587:614–619. doi: 10.1016/j.febslet.2013.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blaby-Haas CE, Furman R, Rodionov DA, Artsimovitch I, de Crécy-Lagard V. Role of a Zn-independent DksA in Zn homeostasis and stringent response. Mol. Microbiol. 2011;79:700–715. doi: 10.1111/j.1365-2958.2010.07475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imlay JA. Where in the world do bacteria experience oxidative stress? Environ. Microbiol. 2019;21:521–530. doi: 10.1111/1462-2920.14445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ross W, et al. ppGpp binding to a site at the RNAP-DksA interface accounts for its dramatic effects on transcription initiation during the stringent response. Mol. Cell. 2016;62:811–823. doi: 10.1016/j.molcel.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Myers AR, Thistle DP, Ross W, Gourse RL. Guanosine tetraphosphate has a similar affinity for each of its two binding sites on Escherichia coli RNA polymerase. Front. Microbiol. 2020;11:587098. doi: 10.3389/fmicb.2020.587098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magnusson LU, Gummesson B, Joksimović P, Farewell A, Nyström T. Identical, independent, and opposing roles of ppGpp and DksA in Escherichia coli. J. Bacteriol. 2007;189:5193–5202. doi: 10.1128/JB.00330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dalebroux ZD, Yagi BF, Sahr T, Buchrieser C, Swanson MS. Distinct roles of ppGpp and DksA in Legionella pneumophila differentiation. Mol. Microbiol. 2010;76:200–219. doi: 10.1111/j.1365-2958.2010.07094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Łyżeń R, et al. The dual role of DksA protein in the regulation of Escherichia coli pArgX promoter. Nucleic Acids Res. 2016;44:10316–10325. doi: 10.1093/nar/gkw912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das B, Bhadra RK. (p)ppGpp metabolism and antimicrobial resistance in bacterial pathogens. Front. Microbiol. 2020;11:563944. doi: 10.3389/fmicb.2020.563944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khakimova M, Ahlgren HG, Harrison JJ, English AM, Nguyen D. The stringent response controls catalases in Pseudomonas aeruginosa and is required for hydrogen peroxide and antibiotic tolerance. J. Bacteriol. 2013;195:2011–2020. doi: 10.1128/JB.02061-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sampathkumar, G., Khakimova, M., Chan, T. & Nguyen, D. The stringent response and antioxidant defences in Pseudomonas aeruginosa. in Stress and Environmental Regulation of Gene Expression and Adaptation in Bacteria. (de Bruijn, F.J., Ed.). 10.1002/9781119004813.ch46 (2016).
- 38.Mossialos D, Tavankar GR, Zlosnik JE, Williams HD. Defects in a quinol oxidase lead to loss of KatC catalase activity in Pseudomonas aeruginosa: KatC activity is temperature dependent and it requires an intact disulphide bond formation system. Biochem. Biophys. Res. Commun. 2006;341:697–702. doi: 10.1016/j.bbrc.2005.12.225. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen D, et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science. 2011;334:982–986. doi: 10.1126/science.1211037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mastropasqua MC, et al. Efficient zinc uptake is critical for the ability of Pseudomonas aeruginosa to express virulence traits and colonize the human lung. J. Trace Elem. Med. Biol. 2018;48:74–80. doi: 10.1016/j.jtemb.2018.03.009. [DOI] [PubMed] [Google Scholar]
- 41.Rossi E, Falcone M, Molin S, Johansen HK. High-resolution in situ transcriptomics of Pseudomonas aeruginosa unveils genotype independent patho-phenotypes in cystic fibrosis lungs. Nat. Commun. 2018;9:3459. doi: 10.1038/s41467-018-05944-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez-Perez G, Mira A, Nyiro G, Pasić L, Rodriguez-Valera F. Adapting to environmental changes using specialized paralogs. Trends. Genet. 2008;24:154–158. doi: 10.1016/j.tig.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 43.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor; 1989. [Google Scholar]
- 44.Bertani I, Rampioni G, Leoni L, Venturi V. The Pseudomonas putida Lon protease is involved in N-acyl homoserine lactone quorum sensing regulation. BMC Microbiol. 2007;7:71. doi: 10.1186/1471-2180-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baldelli V, et al. Identification of FDA-approved antivirulence drugs targeting the Pseudomonas aeruginosa quorum sensing effector protein PqsE. Virulence. 2020;11:652–668. doi: 10.1080/21505594.2020.1770508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clinical and Laboratory Standards Institute (CLSI). Reference method for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, approved standard, 9th ed. M07-A9. CLSI, Wayne, PA (2012).
- 47.Merritt JH, Kadouri DE, O’Toole GA. Growing and analysing static biofilms. Curr. Protoc. Microbiol. 2005 doi: 10.1002/9780471729259.mc01b01s00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peeters E, Nelis HJ, Coenye T. Comparison of multiple methods for quantification of microbial biofilms grown in microtiter plates. J. Microbiol. Methods. 2008;72:157–165. doi: 10.1016/j.mimet.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 49.Paiardini A, et al. A novel bacterial l-arginine sensor controlling c-di-GMP levels in Pseudomonas aeruginosa. Proteins. 2018;86:1088–1096. doi: 10.1002/prot.25587. [DOI] [PubMed] [Google Scholar]
- 50.Barbier M, Damron FH. Rainbow vectors for broad-range bacterial fluorescence labeling. PLoS ONE. 2016;11:e0146827. doi: 10.1371/journal.pone.0146827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pasqua M, et al. Ferric uptake regulator Fur is conditionally essential in Pseudomonas aeruginosa. J. Bacteriol. 2017;199:e00472–e517. doi: 10.1128/JB.00472-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beers RF, Jr, Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952;195:133–140. doi: 10.1016/S0021-9258(19)50881-X. [DOI] [PubMed] [Google Scholar]
- 53.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 54.Rampioni G, et al. Unravelling the genome-wide contributions of specific 2-Alkyl-4-Quinolones and PqsE to quorum sensing in Pseudomonas aeruginosa. PLoS Pathog. 2016;12:e1006029. doi: 10.1371/journal.ppat.1006029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analysed during this study are included in this published article (and relative Supplementary Information file).