

Abstract
We evaluated the effect of the antidiabetic drug metformin on patients enrolled in the ADNI study considering patients with mild cognitive impairment (MCI) due to Alzheimer’s disease (AD). Employing data from this observational study, we performed a principal component analysis focusing on the cognitive sphere by evaluating data from neuropsychological tests included in a modified version of the Alzheimer’s Disease Cooperative Study-Preclinical Alzheimer Cognitive Composite (ADCS-PACC). Second, we included the levels of amyloid-β, tau, and phosphorylated tau in CSF. We found that MCI metformin-treated patients were globally characterized as subjects with a better cognitive performance and CSF biomarkers profile than the mean population of MCI patients. On the other hand, control subjects and type 2 diabetes patients (T2D) were paired by age, gender, ApoE allele, and years of education, defining three groups: MCI, MCI + T2D, and MCI + T2D + metformin. We evaluated the effect of T2D and metformin treatment employing the PACC score and composites defined from standardized ADNI variables to evaluate the memory and learning function. We found that MCI + T2D patients had a worse cognitive performance than MCI patients, but this deleterious effect was not observed in MCI + T2D + metformin patients. These cognitive variations were associated with changes in cortical thickness and hippocampal volume. Finally, no differences were found in metabolic plasmatic parameters (glycemia, cholesterol, triglycerides). Our study—employing different strategies for data analysis from the global study ADNI—shows a beneficial effect of metformin treatment on cognitive performance, CSF biomarkers profile, and neuroanatomical measures in MCI due to AD patients.
Keywords: Alzheimer’s disease, Metformin, Type 2 diabetes
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease that causes dementia and progressive cognitive decline. AD accounts for more than half of the cases of dementia around the world and is characterized by the aberrant accumulation of intraneuronal tau aggregates and extracellular plaques mainly composed of amyloid-β peptides (Association 2018). Tau and amyloid pathology are the key features considered by the classical amyloid cascade hypothesis proposed in 1992, which acted as the main framework for the search of therapeutic targets [32]. Currently, only few treatments targeting tau or amyloid have shown signs of partial effectiveness and the problem remains unsolved. At day, tau and amyloid pathology need to be detected in post-mortem brains in order to confirm the diagnosis of AD. However, in living subjects, neuroimaging techniques and the detection of these proteins in cerebrospinal fluid (CSF) are frequently considered and recommended for the diagnosis. In this case, AD progression is associated with an increase in tau and pTau levels in CSF, probably due to neuronal death and release of these proteins to the extracellular medium. On the other hand, AD progression is associated with a reduction of β-amyloid levels in CSF, probably due to its accumulation in parenchimatic amyloid plaques [2, 3, 8]
Besides amyloid and tau pathology, AD is also characterized by neuroinflammation, decreased brain metabolism, oxidative stress, and loss of protein homeostasis in the brain, both in patients and animal models. In fact, it is a complex and multifactorial pathology associated with several non-modifiable and modifiable risk factors. The first group includes aging and the presence of the ε4 allele for ApoE, causing an altered brain lipid and glucose metabolism [16]. Among modifiable risk factors, one of the most relevant is the presence of metabolic pathologies, including cardiovascular risk factors. Concordantly, these risk factors have also been found in population-based studies [36] and their treatment has been associated with decreased progression to dementia in at-risk patients [23].
Remarkably, type 2 diabetes mellitus (T2D) causes a 73% increased risk of developing AD, affecting 5–9% of the global population and its incidence accelerates during aging [32]. After a meta-analysis involving more than 17 studies, Zhang et al.have concluded that subjects with diabetes had a significantly higher incidence of AD than those without diabetes [28]. Therefore, AD and T2D are distinct diseases showing potential overlapping metabolic dysfunction that precedes the cognitive decline [14].
The emergence of the type 3 diabetes concept—AD being regarded as a metabolic disease—was supported 15 years ago by evidencing a progressive decline in the expression and function of insulin and insulin-like growth factor (IGF) signaling in the brain [42, 44]. Moreover, AD progression is associated with brain insulin resistance and insulin deficiency [1, 25], brain oxidative stress and mitochondrial dysfunction [7], among other alterations related to brain metabolic activity [45]. Shared mechanisms are impaired in both pathologies, as it was recently reviewed by Vinuesa et al. [27]. During T2D and AD progression, insulin resistance and chronic inflammation are directly linked to loss of synaptic plasticity and neuronal survival, impaired protein degradation pathways such as autophagy, and enhanced expression of cytokines and amyloid-related genes [26, 27]. Brain glucose metabolism is also compromised during AD pathogenesis, and recent findings suggest that untreated diabetes is directly associated with an enhanced tau pathology [31].
Therefore, metabolism could be a novel shared link between AD and T2D, emphasizing the opportunity to explore common therapeutic targets [8, 9]. During the last years, a strategy called drug repositioning was proposed as an alternative for the search of a pharmacological intervention for some complex and multifactorial diseases, like AD [17]. It implies the re-purposing of “old” de-risked drugs that have shown to be effective in the treatment of a particular pathology to treat a new related disease. Considering the abovementioned, drugs related to T2D treatment are interesting candidates for drug repositioning in the search for a therapeutic approach for AD.
Metformin is a synthetic derivative of biguanide that has been used for decades as the first-line choice for the treatment of T2D because of its clinical efficacy and high safety. The well-known glucose-lowering effect of this drug is primarily attributable to its ability to regulate energy metabolism, including reduction of glucose absorption, inhibition of gluconeogenesis in the liver, and increase of glucose utilization in peripheral tissues [3]. Despite some controversy regarding its mechanism of action, most of its therapeutic effects seem to be related to indirect AMPK activation, which is a master regulator of cellular metabolism controlling lipid and protein homeostasis [13]. Metformin treatment is linked to an improvement in the lipid profile in addition to the exertion of anti-oxidant, anti-apoptotic, and anti-inflammatory properties, reducing inflammatory cell adhesion to endothelium [2].
In the brain, AMPK activation in rodents and other experimental models was associated with enhanced autophagic flux, decreased neuroinflammation, enhanced neuronal survival, and preserved cognitive status, indicating that AMPK activation is an interesting target for intervention in AD [30, 38, 43]. In humans, a meta-analysis by Campbell et al. (2018) concluded that metformin administration was associated with a reduced risk of developing dementia or AD in T2D patients, but did not find evidence in favor of a neuroprotective function in non-diabetic patients [15]. In the same line, a recent prospective observational study by Samaras and collaborators showed that metformin-receiving diabetic patients from the Sydney Memory and Ageing Study exhibited a reduced cognitive decline and lower risk of dementia compared to patients receiving other antidiabetic drugs [39].
However, there is only limited evidence at the moment concerning the potential beneficial effects of metformin on patients diagnosed with AD, and parameters particularly related to AD pathogenesis. In 2017, Koenig et al. published results from a preliminary randomized study in the USA, where metformin administration to patients diagnosed with MCI due to AD showed a significant improvement in executive function and on memory and learning tasks [24]. Conversely, other authors suggest that long-term treatment with metformin could be slightly detrimental on AD patients [18, 34]. Regarding preclinical studies, in a paper published by Ou et al., authors employed a transgenic mouse exhibiting amyloid-β overproduction as an experimental model of AD, and demonstrated that a treatment with metformin on aged mice improves cognition, reduces amyloid pathology, and drastically diminishes the neuroinflammatory reaction (Abbatecola et al. 2004). Moreover, the direct administration of metformin on cultured neurons seems to activate a pro-amyloidogenic pathway [41]. The existence of this controversy in the literature suggests that a pertinent study is needed in order to assess the potential beneficial effect of metformin on AD patients.
The main aim of this study was to evaluate the impact of metformin treatment on cognition and AD-related parameters in patients diagnosed with MCI due to AD. To address that particular question, we employed data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) study, which recompiles information from clinical centers all around the world in a systematic way, allowing us to test our hypothesis in a representative sample of a general population, considering the controversial and partial information that was previously reported in the bibliography.
Methods
Participants
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public–private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer’s disease (AD).
ADNI collects data from sites in the USA, Europe, Japan, Korea, China, India, and Argentina. Considering its different phases, at the time of this study, the ADNI database included 6937 patients that were 55–90 years old at baseline. Subjects were recruited from 127 centers all around the world. Exclusion criteria of ADNI were inadequate visual and auditory acuity, sign of infection, infarction, or other focal lesion evidenced by MRI at baseline, signs of major depression, alcohol, or substance abuse, and history of schizophrenia. In this study, patients diagnosed as MCI due to AD (MCIAD) in the ADNI database were considered. The inclusion criteria for MCI patients were as follows: subjects must have a subjective memory concern, abnormal memory function documented by scoring within the education adjusted ranges on the Logical Memory II subscale, Mini Mental State Exam score between 24 and 30 (inclusive), and Clinical Dementia rating must have been 0.5, where Memory Box score must have been at least 0.5. In these patients, general cognition and functional performance were sufficiently preserved such that a diagnosis of AD could not be made. Patients with any other significant neurologic disease different than suspected incipient AD were excluded from the study.
Principal component analysis: study design
This study was performed in order to evaluate whether patients receiving metformin had a similar distribution in terms of cognition-associated parameters and biomarkers profile than other MCIAD subjects. We identified 810 patients diagnosed as MCIAD according to the criteria mentioned above from the ADNI database. ADNI also includes a complete record for all medications, durations of use, and dosage at baseline, which allowed us to identify that 55 of these 810 participants were receiving metformin at baseline.
For neurocognitive characterization, we constructed a modified version of the ADCS-PACC which is a neuropsychological battery that combines tests that assess episodic memory, attention, and global cognition and that reliably measure the first signs of cognitive decline in early AD. To construct our modified version, we included from the ADNI’s neuropsychological battery the tests that assess the same cognitive domains as those included in the original version. In the memory domain, we replaced the California Verbal Test II (CVLT) with the Rey Auditory Verbal Learning Test delayed (RAVLT delayed), and in the attentional domain, we replaced the Digit Symbol Substitution Test with the Trail Making Test A (TMTA). General cognition and paragraph memory were assessed with the original tests from the ADCS-PACC, the Mini Mental State Examination (MMSE), and the Logical Memory delayed, respectively. The scores obtained in the TMTA were inverted in order to easily interpret the meaning of the coefficients obtained.
Additionally, in an independent analysis, we considered the CSF levels for AD biomarkers: Amyloid-β, tau, and phosphorylated tau.
Principal component analysis: statistics
Each principal component analysis was performed employing InfoStat software (Universidad Nacional de Córdoba, Argentina). In each analysis, we considered every principal component that explained more than 20% of the variance in the population. Then, we performed a one-sample t-test to evaluate if the score of these principal components was significantly different than zero in patients receiving metformin, considering a significance level of 5% (p-value < 0.05). If patients receiving metformin had a similar distribution to the whole population of MCIAD subjects, we expected that the mean standardized score obtained for these subjects would not be significantly different than zero, which is the mean of the population.
Paired analysis: study design and statistics
This study was designed to control confounding factors that could affect cognition and other biomarkers in the principal component analysis detailed above. Considering that subjects receiving metformin were diagnosed with T2D, we also wanted to evaluate if the presence of this metabolic pathology was affecting the parameters considered in this study. We identified 17 MCIAD patients that were also diagnosed with T2D but were not receiving metformin at baseline, constituting the so-called MCIAD + T2D group. These patients were compared with a group of patients that were not diagnosed with T2D—mentioned as MCIAD group—and a group of 17 patients receiving metformin, called MCIAD + T2D + met. These last two groups were constituted by 17 patients that were paired with each patient from the MCIAD + T2D group considering the following demographic criteria: same age (± 2 years), same years of education (± 2 years), and same gender and genotype for ApoE, when it was possible. We then corroborated that the three groups that were defined employing this strategy were not different between them in terms of these demographic variables, employing one-way ANOVA. Table 1 shows relevant information about diabetic patients considered for paired analysis, including years since diabetes diagnosis and time under treatment with metformin. There were no significant differences in years since diabetes diagnosis between these groups (p = 0.40, one-tailed t-test). The mean time that patients were treated with metformin was 8.7 years until the moment they were included in ADNI baseline.
Table 1.
Time since diabetes diagnosis and metformin treatment for diabetic groups included in the paired analysis. Data is expressed in years. (*) The columns “subject” correspond to the ID assigned for each patient in ADNI database. “NI” means “not informed”
| MCIAD + T2D group | MCIAD + T2D + met group | |||
|---|---|---|---|---|
| Subject (*) | Time since diabetes diagnosis (years) | Subject (*) | Time since diabetes diagnosis (years) | Time treated with metformin (years) |
| 42 | 13 | 339 | 3 | 3 |
| 60 | 3 | 1408 | 0 | 0 |
| 87 | 4 | 1423 | 3 | 3 |
| 219 | 44 | 2063 | 8 | 8 |
| 273 | 3 | 2072 | 15 | 15 |
| 294 | 2 | 2193 | 10 | 10 |
| 633 | NI | 2263 | 8 | 1 |
| 669 | 15 | 4077 | 5 | 5 |
| 702 | 0 | 4432 | 10 | 5 |
| 790 | 3 | 4653 | NI | 4 |
| 836 | 3 | 4675 | 27 | 22 |
| 1066 | 4 | 4689 | 14 | 14 |
| 1114 | 46 | 4765 | 42 | 42 |
| 1201 | 1 | 4803 | NI | 8 |
| 1244 | 5 | 4919 | 5 | 3 |
| 1260 | 1 | 6463 | NI | 1 |
| 1343 | 18 | 6535 | NI | 4 |
Statistical analyses were performed considering all patients with available data retrieved from ADNI database (http://adni.loni.ucla.edu/) on August 2020, using R software version 3.6.3 (http://www.r-project.org). We used linear mixed-effects models to assess the neuropsychological and neuroanatomical differences between groups (MCIAD, MCIAD + T2D, MCIAD + T2D + met). According to the demographic criteria previously described, we set paired patients as the random variable. We applied a type III analysis of variance with Satterthwaite’s method and Kenward-Roger method for denominator degrees of freedom for F-test, followed by a Tukey post hoc test with pairwise comparisons. All statistical significance was set at p ≤ 0.05.
Paired analysis: cognitive parameters
Composites were designed as measures of cognitive function by combining individual test scores from the ADNI battery to create a domain-specific cognitive composite score. The goal of the composite scores was to optimize their capacity to detect changes in cognition and avoid alpha errors. The use of composites allowed us to consolidate type 1 error into a single outcome. To calculate a composite outcome, scores for each contributing test were converted to z-scores by dividing the differences between individual scores from the control group mean at baseline by the control group standard deviation. Z-scores were sorted so that positive scores reflected a better performance and negative scores reflected a worse performance. The ADCS-PACC-M [12] was calculated from scores obtained in RAVLT delayed recall, TMTA, Logical Memory delayed recall, and MMSE for each patient. The Learning Composite was calculated including scores obtained by patients in the RALVT 5 trials total score, the Logical Memory (immediate recall), and question number 1 from the Alzheimer’s Disease Assessment Scale-Cognitive (ADAS-Cog). The Memory Composite was calculated including the Logical memory delayed recall, Rey Auditory Verbal Learning Test delayed recall, and question number 4 from the ADAS-Cog.
Paired analysis: MRI and metabolic parameters
Cortical thickness and hippocampal volume were obtained from structural MRI and informed in the ADNI database. We evaluated cortical thickness though AD signature [5], defined as the mean of a set of regions particularly affected during the prodromal stage of AD. To evaluate this parameter, we summed up the thickness informed for left and right medial temporal cortex, inferior temporal gyrus, superior parietal lobule, temporal pole, and precuneus. We used right and left hippocampal volume to asses atrophy.
We also included metabolic parameters in order to assess the impact of metformin treatment in these patients. Data regarding plasmatic levels of glucose, triglycerides, and total cholesterol were compared between groups.
Results
Subjects receiving metformin had a better cognitive performance and biomarker profile than other patients in the prodromal stage of Alzheimer’s disease
For neurocognitive characterization, 810 MCI due to Alzheimer’s disease (MCIAD) patients were selected from the ADNI database according to the diagnostic criteria employed in that study. The selected population had an average age of 73.4 years with a standard deviation of 6.2 years. Among these subjects, 51.9% carried the ε4 allele for ApoE and 39.9% were female. The average period of education was 16.0 years with a standard deviation of 2.8 years. Principal component analysis was performed in order to describe the structure of this population, and to evaluate if the 55 subjects that received metformin had a similar distribution to the general population. The variables considered were the score obtained in the RAVLT, TMTA (inverted), Logical Memory delayed recall and Mini mental state exam.
Figure 1A shows the biplot obtained in the principal components analysis, where black dots represent the subjects receiving metformin at the baseline of the study (MCIAD-metformin patients). The principal component 1 explained 45% of the variance in the MCIAD population. Interestingly, the mean of the MCIAD-metformin patients in the principal component 1 was significantly positive (Fig. 1B), indicating that the subpopulation of patients receiving metformin presents a structure that was not comparable to the mean of MCIAD patients. The standardized coefficients obtained in this study were all positive (Fig. 1C), suggesting that the subpopulation of MCIAD-metformin patients had a better performance in these cognitive tests compared to the total MCIAD group, in general terms
Fig. 1.

Metformin use was associated with a lesser cognitive decline in patients with prodromal Alzheimer’s disease. A Biplot obtained from a principal component analysis performed on 810 MCIAD subjects. Black dots represent patients receiving metformin. B Box plots indicating the mean value of principal component 1 and principal component 2 for MCIAD-metformin patients (n = 55). *p < 0.05; ns, non-significant differences (one-sample t-test compared with a theoretical mean of zero). C Standardized coefficient obtained for each variable included in the principal component analysis. Note that the values from the Trail Making Test A used for this analysis were inverted
In a similar way, the Aβ, tau, and pTau levels in cerebrospinal fluid (CSF) were employed in an independent principal component analysis. These measures were determined in 455 MCIAD subjects from the ADNI database, where 31 received metformin at the baseline of ADNI study. The mean age of MCIAD patients were 73.2 years with a standard deviation of 6.0 years. From these patients, 38.2% were female and 54.7% carried the ε4 allele for ApoE. The average in years of education was 16.1. These demographic parameters were considerably similar to those of the subjects included in the analysis above.
MCIAD-metformin patients were considerably located in the left part of the biplot shown in Fig. 2A. As indicated, the mean value for the principal component 1—which explained 59% of the variance—was significantly negative for this subpopulation (Fig. 2B), suggesting that its characteristics were different from the whole MCIAD population. According to the standardized coefficients obtained in this analysis (Fig. 2C), MCIAD-metformin patients had, in general terms, higher levels of Aβ and lower levels of tau and pTau in CSF than the mean of MCIAD subjects.
Fig. 2.

Treatment with metformin is associated with a healthier biomarker profile in patients with prodromal Alzheimer’s disease. A Biplot obtained from a principal component analysis performed on 455 MCIAD subjects. Black dots represent patients receiving metformin. B Box plots indicating the mean value of principal components 1 and 2 for MCIAD-metformin patients (n = 31). *p < 0.05; ns, non-significant differences (one-sample t-test compared with a theoretical mean of zero). C Standardized coefficients obtained for each variable included in the principal component analysis
Taken together, these principal component analyses indicate that the subpopulation of MCIAD patients receiving metformin had a globally better cognitive performance, associated with a better biomarker profile compared to the mean of MCIAD population.
Metformin treatment abolished the deleterious effects of diabetes on cognition in patients with prodromal Alzheimer’s disease
Taking into account that ADNI is an observational study and that MCIAD-metformin patients could have different demographic characteristics than other MCIAD patients, we performed a paired analysis that allowed us to control several confounding factors, like the presence of T2D in these patients. We identified 17 MCIAD patients that received an antidiabetic treatment different than metformin (MCIAD + T2D group). Then, employing demographic variables as the criteria for pairing, we assigned to each case a non-diabetic patient (MCIAD group) and a patient receiving metformin, who was also diagnosed with T2D (MCIAD + T2D + Met group). As a result, we defined three groups from this observational study with similar demographic characteristics (see Table 2).
Table 2.
Demographic characteristics of the included ADNI participants
| MCIAD | MCIAD + T2D | MCIAD + T2D + Met | |
|---|---|---|---|
| Number of subjects | 17 | 17 | 17 |
| Age (Years ± SE) | 74.55 ± 6.62 | 74.92 ± 7.00 | 74.53 ± 6.97 |
| Gender (%Male/%Female) | 70,6/29,4 | 76.9/23.1 | 76.9/23.1 |
| Education (Years ± SE) | 15.65 ± 2.26 | 14.00 ± 3.46 | 15.65 ± 2.26 |
| ApoE genotype (Ɛ4 alleles) | |||
| 0 | 11 | 8 | 11 |
| 1 | 5 | 8 | 5 |
| 2 | 1 | 1 | 1 |
As the PACC is standardized with the population of cognitively normal subjects in ADNI, the mean value obtained for each MCI group was negative, as expected. MCIAD + T2D patients exhibited a severe cognitive impairment compared to MCIAD patients, but the treatment with metformin abolished this effect in MCIAD + T2D + Met group (Fig. 3A). In these patients, metformin treatment increased the PACC score by 43.9% (Cohen’s d: 1.06). Next, we defined a composite to evaluate the memory function, employing the results obtained by patients in the Logical Memory delayed recall, RAVLT test delayed recall, and question number 4 from the Alzheimer’s Disease Assessment Scale-Cognitive (ADAS-Cog). We found that T2D negatively affects the memory function in these patients, while this effect was not present in diabetic patients treated with metformin (Fig. 3B). Employing the same methodology, we constructed a composite to evaluate the learning function, including results from the RAVLT (trials 1–5), Logical Memory immediate recall, and question number 1 from the Alzheimer’s Disease Assessment Scale-Cognitive (ADAS-Cog). In a similar way to the memory function, we found that metformin treatment abolished the deleterious effect of T2D on learning function in MCIAD patients (Fig. 3C), causing an increment of 35.4% in MCIAD + T2D group (Cohen’s d: 0.81). Finally, we evaluated the results obtained by patients in consecutive trials of the learning phase of the RALVT. We detected that MCIAD patients exhibited a better performance in this test during consecutive trials, while MCIAD + T2D obtained lower scores compared to other groups starting from trial 2 (Fig. 3D). Interestingly, MCIAD + T2D + Met group had a performance that was not significantly different to MCIAD patients, denoting an improvement in this learning parameter.
Fig. 3.
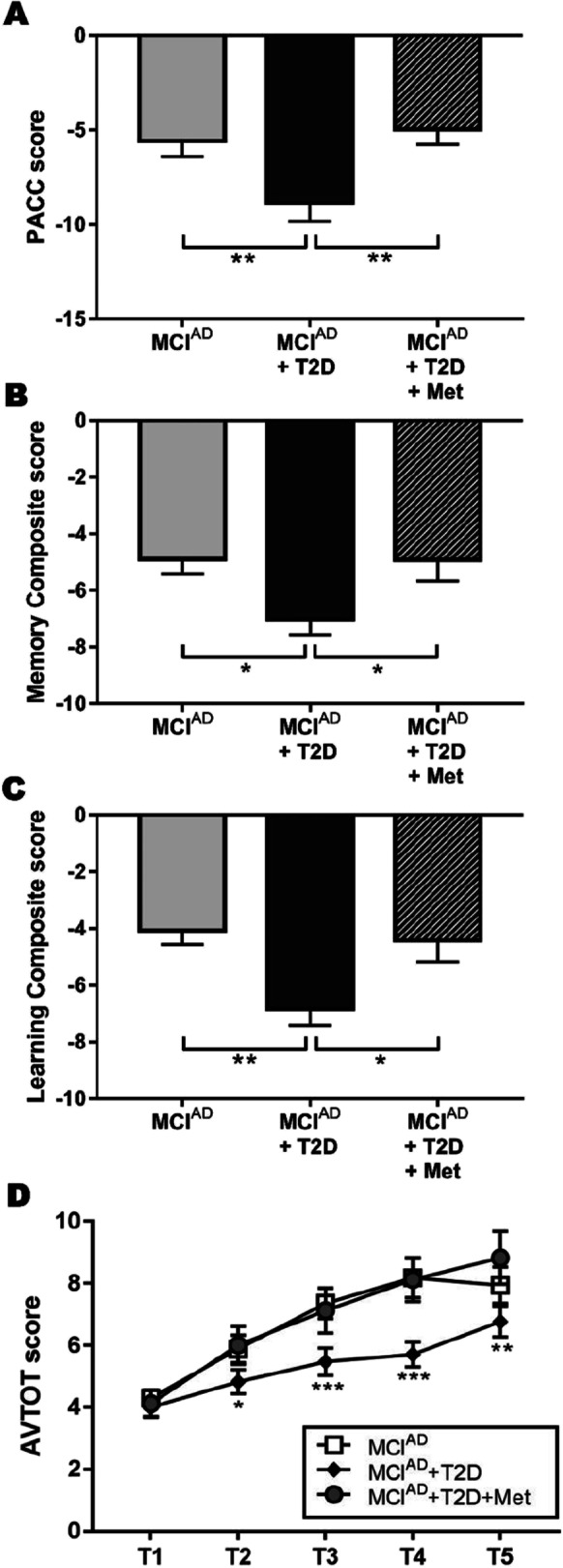
Metformin ameliorated the cognitive impairment caused by diabetes in patients with Alzheimer’s disease. Mean PACC score (A), mean Memory Composite score (B), and mean Learning Composite score (C) are expressed in relation to cognitively normal subjects. (D) Rey Auditory Verbal Learning Test (AVTOT) score in consecutive trials (T1–T5). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, and ***p < 0.001 (one-way ANOVA with pairwise comparisons)
Diabetic MCI patients receiving metformin exhibited preserved neuroanatomical measures with slight changes in metabolic parameters
Despite the neurocognitive effect of metformin, we evaluated the association of its administration with changes in neuroanatomical integrity. Cortical thickness was estimated from MRI—as informed in the ADNI database—and the sum of cortical thickness was calculated considering the region of the cortex included in the “AD signature” (Bakkour, 2009). These regions (including medial temporal, inferior temporal gyrus, and superior parietal cortex which magnitude of atrophy correlates with dementia progression) were considered particularly affected in early stages of AD. Diabetic patients receiving a treatment different than metformin had a significant reduction for this parameter in comparison to other MCIAD patients (Fig. 4A). However, patients receiving metformin showed a total cortical thickness significantly higher than MCIAD + T2D patients (Cohen’s d: 1.55). In a similar way, data obtained from MRI allowed us to find that diabetes caused a significant reduction in total hippocampal volume compared to MCIAD group, while MCIAD + T2D + Met patients exhibited no differences compared to non-diabetic patients in the prodromal stage of Alzheimer’s disease (Fig. 4B).
Fig. 4.
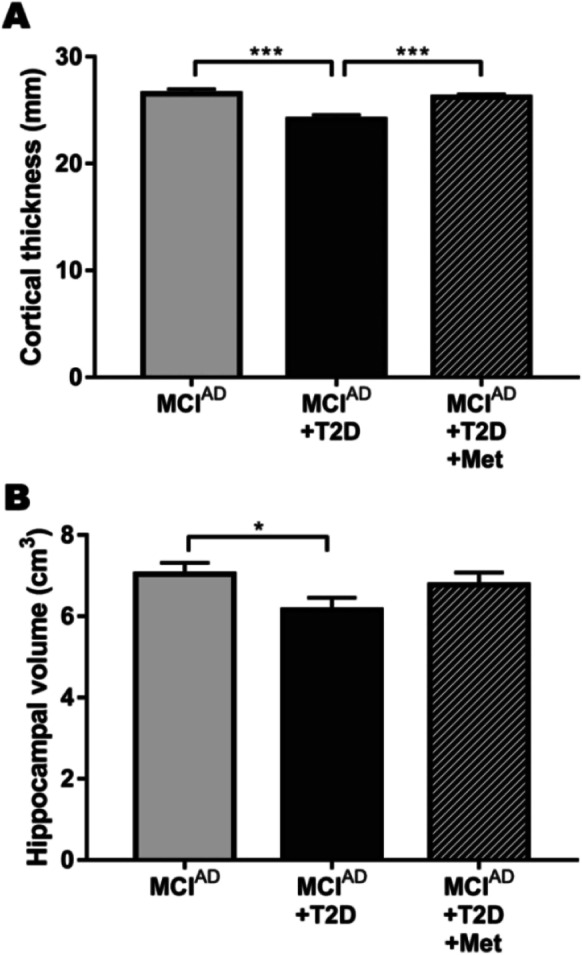
Metformin treatment diminished the neuroanatomical alterations exhibited by diabetic patients with prodromal Alzheimer’s disease. A Cortical thickness of the AD signature (expressed as the sum of the thickness for each area measured in mm). B Total hippocampal volume (expressed as the sum of both left and right hippocampal volume in cm3). *p < 0.05 and ***p < 0.001 (one-way ANOVA)
Considering that diabetes is a metabolic disease and that metabolism regulation by metformin administration could be mediating the effects on cognition, we analyzed metabolic parameters informed in the ADNI database. As shown in Fig. 5A, we found that diabetic patients had increased levels of plasma glucose compared to non-diabetic patients. Metformin seemed to be more efficient in reducing glycemia than other anti-diabetic treatments; however, there were no significant differences between MCIAD + T2D and MCIAD + T2D + Met groups. In these patients, metformin treatment caused no effect on plasma triglycerides and plasma cholesterol compared to other MCIAD subjects (Fig. 5B–C).
Fig. 5.

Metformin-receiving patients exhibited a metabolic profile that was similar to non-diabetic MCI patients. Levels of plasmatic glucose (A), triglycerides (B), and total cholesterol (C) measured in mg/100 ml. Data are presented as mean ± SEM. **p < 0.01 (one-way ANOVA with pairwise comparisons)
Taken together, these findings suggest that the neurocognitive effects of metformin on diabetic MCIAD patients were associated with anatomical changes in the brain. These effects could not be explained by changes in the metabolic parameters evaluated in the ADNI study.
Discussion
The vast social and economic impact of diabetes and AD and the fact that there is no effective treatment for AD available to this day support the necessity to redefine the strategy followed by researchers [4]. One of the most recent propositions is drug repositioning, which implies that some drugs that were proven to be safe could be used for the treatment of other pathologies [17]. Considering the link existing between T2D and AD, many researchers in the field are focusing on the potential impact that metformin—the main drug approved for the treatment of T2D—could have during AD progression and aging [15, 35].
Metformin, found in a traditional herbal medicine more than 300 years ago and re-discovered in the 1940s as an antimalarial agent, was finally approved by the Food and Drug Administration as an antidiabetic in 1995. Nowadays, it is the most widely prescribed oral antihyperglycemic medication for T2D treatment and prevention. But apart from its major role in the management of T2D, metformin has pleiotropic actions that are well documented. Several reports, employing different animal models, have linked metformin with longevity [10, 33]. Moreover, metformin was associated with improved cardiovascular treatment response and nephroprotective effects probably interfering with key inflammatory-molecules [21, 29, 40]. Importantly, there is emerging data from in vitro and in vivo experiments showing a strong anti-inflammation effect, beyond its glucose control capability [2, 6]. Its anti-inflammatory function is particularly relevant in this scenario, as neuroinflammation and glial reaction are chronically present during aging, and particularly exacerbated during AD [22]. These findings present the possibility of boosting the therapeutic potential of metformin on brain pathophysiological aging, emerging as a leading candidate in clinical trials [37]. Lately, an interesting “geroscience perspective” has been suggested [20].
Our aim here was to obtain evidence about the effect of metformin on cognitive impairment due to Alzheimer’s disease from an observational study that includes a heterogeneous population—in terms of demographic and clinical factors—more comparable to the global population, as the global research study ADNI. We consider that evaluating this kind of hypothesis should start by estimating a global trend—i.e., employing multivariate statistics on large samples—to further single out the impact of particular factors by controlling them. We applied a settled mathematical method for reducing data dimensionality but retaining variation as possible, that is to say, the principal component analysis. We conclude that this general approach is particularly relevant in this scenario, where previous evidence suggests that the effect of metformin could be dependent on mentioned factors, which are not controlled in this type of observational studies. We then performed a paired analysis controlling for some confounding factors—like demographic variables affecting cognition and AD progression—to evaluate our hypothesis in a more controlled scenario. Even though the number of patients included in the paired analysis was low, the fact that the main confounding factors were controlled was manifested in the magnitude of the effects we found.
Using these two strategies for data analysis, we consistently found that patients receiving metformin had a better cognitive performance compared to other patients, particularly in a selection of neuropsychological tests designed to measure cognitive functions affected in prodromal AD and AD patients, like memory and learning capabilities. Moreover, it is important to emphasize that the positive impact of metformin treatment was not restricted to the cognitive sphere, as it is supported by biological changes in terms of biomarkers associated with AD progression. In the principal component analysis, we found that patients receiving metformin exhibited also a healthier profile of biomarkers in the CSF, associated with AD progression, globally suggesting a diminished impact of the pathology in this subpopulation. In the paired analysis, we found that cognitive improvement in metformin-receiving patients was associated with reduced atrophy in the hippocampus and cortex, considering sub-areas that are particularly affected in the prodromal stage of AD.
One of the elemental questions arising from our results is whether metformin is acting on the damage caused by diabetes or caused by AD. It is particularly difficult to answer this question in an observational study, where metformin was administered only to diabetic patients. So, in our study, all the patients with MCI due to AD receiving metformin showed metabolic alterations, and thus, we were not able to analyze the effect of metformin in each scenario separately. Qualitatively, data from our paired analysis showed that metformin partially reduced the cognitive impairment and neuroanatomical alterations in diabetic patients into values that were very similar to non-diabetic subjects. This suggests that metformin could be acting on the brain damage caused only by diabetes. Although this study was not designed to answer this particular question, Samaras et al. [39] recently reported an investigation similar to our paired analysis on cognitively normal subjects and diabetic patients with no sign of dementia from the Sydney Memory and Ageing Study. They found that metformin has a strong effect on reverting or preventing cognitive damage due to T2D in this particular demographic. So, our results confirm these previous findings, and extend this conclusion to a global population and to patients with prodromal AD.
However, we cannot discard the fact that metformin is also acting on the brain damage caused by AD. At this point, it is important to note that the impact of metformin on diabetic-MCI patients could be underestimated in our analysis. As patients were assigned to different groups according to their diagnosis in the baseline of ADNI, it is possible that some MCI patients that were receiving metformin in the past reverted their diagnosis to cognitively normal, so they are not included in the MCI group. If that was the case, we would be excluding from our analysis the cases where metformin has the strongest effect on damage caused by both diabetes and AD. This underestimation arises from the fact that patients in ADNI are diagnosed regarding the neuropsychological parameters. In recent years, these diagnostic criteria have been debated, and several authors support the idea that diagnosis should be based on biological parameters, as it was claimed in 2018 by the NIA-AA Research Framework [19]. The AT(N) system is the most accepted criteria within its field, and is based on biomarkers for amyloid and tau pathology, including biomarkers for neurodegeneration. Even if it would be very informative to redefine our groups in terms of the AT(N) system, a large number of metformin-receiving patients included in ADNI have no measures for biomarkers related to amyloid or tau pathology, so the number of individuals could be drastically lower.
Our paired analysis is also limited by the low number of patients included in each group. This limitation arises from the fact that most diabetic patients in ADNI, as expected, were treated with metformin, limiting the number of subjects included in the MCIAD + T2D group and, by pairing, the others groups. Because of this limitation, we decided to include both male and female patients, even if males are considerably overrepresented in the groups and the risk of AD is quite lower in male than females. The low number of subjects and the overrepresentation of males in the paired analysis could also be responsible for the lack of significant differences between patients receiving metformin and non-diabetic MCI patients. In this case, the principal component analysis that includes a greater number of subjects seems to be a more powerful tool to discriminate the effect of metformin in MCI patients. It is important to note that in the principal components analysis, the original variables make a differential contribution in the discrimination of the subjects, being the Logical Memory delayed recall (a memory test) the most important to explain these differences. In the paired analysis, however, PACC was designed so each one of the original variables makes an equal contribution to the total PACC score.
On the other hand, the group of patients receiving metformin was defined by subjects that were receiving metformin as a concurrent medication at baseline. That means—in terms of ADNI criteria—that these individuals were receiving metformin during at least the last 3 months before baseline. Therefore, we are not able to know if this drug was administered before or after the appearance of cognitive impairment caused by T2D or AD. So, one of the limitations of the present study is that it is not able to confirm if metformin is preventing or reverting the deleterious effects of diabetes and/or AD. However, the results that we obtained exhibited a preserved cortex and hippocampal integrity in these patients, which suggest that metformin could be acting by preventing pathological changes, at least in terms of brain atrophy. Moreover, we already know by data informed in Table 1 that most of the patients included in the paired analysis were treated with metformin during several years. This means that our study contains mainly patients that were treated with this drug in a long-term period, probably even prior to MCI diagnosis. It is also consistent with the findings by Samaras et al. in cognitively normal people, as they informed a reduced risk of dementia associated with metformin treatment after 5 years [39].
At this point, it is interesting to discuss if the effects of metformin on cognition, CSF biomarkers, and neuroanatomy are mediated by an improvement in metabolic status or by a direct effect on the brain. We wanted to address this question by including a partial characterization of the metabolic profile of the patients with relevant variables evaluated in ADNI, which was not designed to have a comprehensive understanding of these parameters. Even though metformin is considered the drug of choice for the treatment of T2D, we have not found a great effect on parameters other than plasmatic glucose, where metformin is slightly effective. Although we cannot discard that these or other metabolic parameters affected by metformin might have a major impact on brain and cognition, we think that metformin is probably acting directly upon brain function and maintenance. Findings in several experimental models of AD support this claim. For instance, Ou et al. (Abbatecola et al. 2004) demonstrated that metformin administration on APP/PS1 mice—an animal model of familiar AD—attenuates the spatial memory and learning deficits showcased by these mice. The authors also suggest a direct activation of glial AMPK by metformin, causing a decrease in the neuroinflammatory response. More recently, Chen and collaborators [11] employed the same murine model of AD and proved that metformin administration reduces amyloid and tau pathology in association to an increase in the capacity of glial cells to degrade extracellular amyloid. So, if it cannot be disproved that metformin could be acting indirectly on the brain by regulating the metabolic status, metformin could be also acting directly upon the brain by promoting amyloid and tau degradation and preserving neurons from chronic neuroinflammation.
To conclude, we report that metformin administration was associated with improved cognition and preserved brain status in diabetic patients with prodromal AD. Future interventional studies should be directed to evaluate the potential benefits of the treatment with metformin on AD patients without diabetes. As ADNI is a multicenter study that includes patients from all around the world, it would be interesting to replicate the analysis we performed here with data obtained from other observational studies, to identify the peculiarity of each population. It would also be desirable to compile additional data regarding the metabolic profile and biomarkers for neuroinflammation.
Acknowledgements
The authors gratefully acknowledge the scientific contribution of Ezequiel Surace, PhD; Juan Beauquis, MD, PhD; Angeles Vinuesa, PhD; and Patricio Chrem Mendez MD.
Funding
This work was supported by Williams, René Barón, and Florencio Fiorini Foundations, ANPCyT PICT Grants: 2016–1046, 2016–1572, 2019–3419 and UBACyT 2018 Grant. The funding sources had no involvement in the study design nor the collection, analysis and interpretation of data. CP is recipient of CONICET Fellowship. FS is CONICET Researcher.
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12–2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuroimaging at the University of Southern California.
Footnotes
Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Abbatecola AM, Paolisso G, Lamponi M, Bandinelli S, Lauretani F, et al. Insulin resistance and executive dysfunction in older persons. Journal of the American Geriatrics Society. 2004;52:1713–8. doi: 10.1111/j.1532-5415.2004.52466.x. [DOI] [PubMed] [Google Scholar]
- 2.Andreasen N, Blennow K. Beta-amyloid (Abeta) protein in cerebrospinal fluid as a biomarker for Alzheimer's disease. Peptides. 2002;23:1205–1214. doi: 10.1016/s0196-9781(02)00056-6. [DOI] [PubMed] [Google Scholar]
- 3.Arai H, Terajima M, Miura M, Higuchi S, Muramatsu T, et al. Tau in cerebrospinal fluid: a potential diagnostic marker in Alzheimer's disease. Ann Neurol. 1995;38:649–652. doi: 10.1002/ana.410380414. [DOI] [PubMed] [Google Scholar]
- 4.Alzheimer’s Association. Alzheimer’s disease facts and figures. 2018. https://www.alz.org/media/HomeOffice/Facts%20and%20Figures/facts-and-figures.pdf.
- 5.Bakkour A, Morris JC, Dickerson BC. The cortical signature of prodromal AD: regional thinning predicts mild AD dementia. Neurology. 2009;72:1048–1055. doi: 10.1212/01.wnl.0000340981.97664.2f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bharath LP, Agrawal M, McCambridge G, Nicholas DA, Hasturk H, et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell metabolism. 2020;32:44–55 e6. doi: 10.1016/j.cmet.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biancardi VC, Son SJ, Ahmadi S, Filosa JA, Stern JE. Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood-brain barrier. Hypertension. 2014;63:572–579. doi: 10.1161/HYPERTENSIONAHA.113.01743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blennow K, Zetterberg H. Biomarkers for Alzheimer's disease: current status and prospects for the future. J Intern Med. 2018;284:643–663. doi: 10.1111/joim.12816. [DOI] [PubMed] [Google Scholar]
- 9.Criado-Marrero M, Smith TM, Gould LA, Kim S, Penny HJ, et al. FKBP5 and early life stress affect the hippocampus by an age-dependent mechanism. Brain, behavior, & immunity - health. 2020;9:100143. doi: 10.1016/j.bbih.2020.100143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen J, Ou Y, Li Y, Hu S, Shao LW, Liu Y. Metformin extends C. elegans lifespan through lysosomal pathway. eLife 2017;6. [DOI] [PMC free article] [PubMed]
- 11.Chen Y, Zhao S, Fan Z, Li Z, Zhu Y, et al. Metformin attenuates plaque-associated tau pathology and reduces amyloid-beta burden in APP/PS1 mice. Alzheimer's research & therapy. 2021;13:40. doi: 10.1186/s13195-020-00761-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donohue MC, Sperling RA, Salmon DP, Rentz DM, Raman R, et al. The preclinical Alzheimer cognitive composite: measuring amyloid-related decline. JAMA Neurol. 2014;71:961–970. doi: 10.1001/jamaneurol.2014.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duarte AI, Moreira PI, Oliveira CR. Insulin in central nervous system: more than just a peripheral hormone. Journal of aging research. 2012;2012:384017. doi: 10.1155/2012/384017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farr SA, Roesler E, Niehoff ML, Roby DA, McKee A, Morley JE. Metformin improves learning and memory in the SAMP8 mouse model of Alzheimer's disease. Journal of Alzheimer's disease : JAD. 2019;68:1699–1710. doi: 10.3233/JAD-181240. [DOI] [PubMed] [Google Scholar]
- 15.Ghasemi R, Haeri A, Dargahi L, Mohamed Z, Ahmadiani A. Insulin in the brain: sources, localization and functions. Mol Neurobiol. 2013;47:145–171. doi: 10.1007/s12035-012-8339-9. [DOI] [PubMed] [Google Scholar]
- 16.Han J, Li Y, Liu X, Zhou T, Sun H, et al. Metformin suppresses retinal angiogenesis and inflammation in vitro and in vivo. PloS one. 2018;13:e0193031. doi: 10.1371/journal.pone.0193031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ihara M, Saito S. Drug repositioning for Alzheimer's disease: finding hidden clues in old drugs. Journal of Alzheimer's disease : JAD. 2020;74:1013–1028. doi: 10.3233/JAD-200049. [DOI] [PubMed] [Google Scholar]
- 18.Imfeld P, Bodmer M, Jick SS, Meier CR. Metformin, other antidiabetic drugs, and risk of Alzheimer's disease: a population-based case-control study. J Am Geriatr Soc. 2012;60:916–921. doi: 10.1111/j.1532-5415.2012.03916.x. [DOI] [PubMed] [Google Scholar]
- 19.Jack CR, Jr, Bennett DA, Blennow K, Carrillo MC, Dunn B, et al. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimer's Dementia. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Justice JN, Gubbi S, Kulkarni AS, Bartley JM, Kuchel GA, Barzilai N. A geroscience perspective on immune resilience and infectious diseases: a potential case for metformin. GeroScience. 2021;43:1093–1112. doi: 10.1007/s11357-020-00261-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim D, Lee JE, Jung YJ, Lee AS, Lee S, et al. Metformin decreases high-fat diet-induced renal injury by regulating the expression of adipokines and the renal AMP-activated protein kinase/acetyl-CoA carboxylase pathway in mice. Int J Mol Med. 2013;32:1293–1302. doi: 10.3892/ijmm.2013.1508. [DOI] [PubMed] [Google Scholar]
- 22.Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer's disease. Alzheimers Dement (N Y) 2018;4:575–590. doi: 10.1016/j.trci.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kivipelto M, Mangialasche F, Snyder HM, Allegri R, Andrieu S, et al. World-Wide FINGERS Network: a global approach to risk reduction and prevention of dementia. Alzheimer's Dementia. 2020;16:1078–1094. doi: 10.1002/alz.12123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koenig AM, Mechanic-Hamilton D, Xie SX, Combs MF, Cappola AR, et al. Effects of the insulin sensitizer metformin in Alzheimer disease: pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis Assoc Disord. 2017;31:107–113. doi: 10.1097/WAD.0000000000000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee S, Kang BM, Kim JH, Min J, Kim HS, et al. Real-time in vivo two-photon imaging study reveals decreased cerebro-vascular volume and increased blood-brain barrier permeability in chronically stressed mice. Sci Rep. 2018;8:13064. doi: 10.1038/s41598-018-30875-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li MZ, Zheng LJ, Shen J, Li XY, Zhang Q, et al. SIRT1 facilitates amyloid beta peptide degradation by upregulating lysosome number in primary astrocytes. Neural Regen Res. 2018;13:2005–2013. doi: 10.4103/1673-5374.239449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Chhipa RR, Nakano I, Dasgupta B. The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol Cancer Ther. 2014;13:596–605. doi: 10.1158/1535-7163.MCT-13-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lv WS, Wen JP, Li L, Sun RX, Wang J, et al. The effect of metformin on food intake and its potential role in hypothalamic regulation in obese diabetic rats. Brain Res. 2012;1444:11–19. doi: 10.1016/j.brainres.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 29.Ma R, Yi B, Riker AI, Xi Y. Metformin and cancer immunity. Acta Pharmacol Sin. 2020;41:1403–1409. doi: 10.1038/s41401-020-00508-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacVicar BA, Newman EA. Astrocyte regulation of blood flow in the brain. Cold Spring Harbor Perspect Biol 2015;7. [DOI] [PMC free article] [PubMed]
- 31.Manaenko A, Chen H, Kammer J, Zhang JH, Tang J. Comparison Evans Blue injection routes: intravenous versus intraperitoneal, for measurement of blood-brain barrier in a mice hemorrhage model. J Neurosci Methods. 2011;195:206–210. doi: 10.1016/j.jneumeth.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Markowicz-Piasecka M, Sikora J, Szydlowska A, Skupien A, Mikiciuk-Olasik E, Huttunen KM. Metformin - a future therapy for neurodegenerative diseases: Theme: Drug Discovery, Development and Delivery in Alzheimer's Disease Guest Editor: Davide Brambilla. Pharm Res. 2017;34:2614–2627. doi: 10.1007/s11095-017-2199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore EM, Mander AG, Ames D, Kotowicz MA, Carne RP, et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care. 2013;36:2981–2987. doi: 10.2337/dc13-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ng TP, Feng L, Yap KB, Lee TS, Tan CH, Winblad B. Long-term metformin usage and cognitive function among older adults with diabetes. Journal of Alzheimer's disease : JAD. 2014;41:61–68. doi: 10.3233/JAD-131901. [DOI] [PubMed] [Google Scholar]
- 36.Ngandu T, Lehtisalo J, Solomon A, Levalahti E, Ahtiluoto S, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385:2255–2263. doi: 10.1016/S0140-6736(15)60461-5. [DOI] [PubMed] [Google Scholar]
- 37.Ping F, Jiang N, Li Y. Association between metformin and neurodegenerative diseases of observational studies: systematic review and meta-analysis. BMJ Open Diabetes Res Care 2020;8. [DOI] [PMC free article] [PubMed]
- 38.Ronaldson PT, Davis TP. Blood-brain barrier integrity and glial support: mechanisms that can be targeted for novel therapeutic approaches in stroke. Curr Pharm Des. 2012;18:3624–3644. doi: 10.2174/138161212802002625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samaras K, Makkar S, Crawford JD, Kochan NA, Wen W, et al. Metformin use is associated with slowed cognitive decline and reduced incident dementia in older adults with type 2 diabetes: the Sydney Memory and Ageing Study. Diabetes Care. 2020;43:2691–2701. doi: 10.2337/dc20-0892. [DOI] [PubMed] [Google Scholar]
- 40.Soraya H, Clanachan AS, Rameshrad M, Maleki-Dizaji N, Ghazi-Khansari M, Garjani A. Chronic treatment with metformin suppresses toll-like receptor 4 signaling and attenuates left ventricular dysfunction following myocardial infarction. Eur J Pharmacol. 2014;737:77–84. doi: 10.1016/j.ejphar.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP. Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci. 2008;11:309–317. doi: 10.1038/nn2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vinuesa A, Bentivegna M, Calfa G, Filipello F, Pomilio C, et al. Early exposure to a high-fat diet impacts on hippocampal plasticity: implication of microglia-derived exosome-like extracellular vesicles. Mol Neurobiol. 2019;56:5075–5094. doi: 10.1007/s12035-018-1435-8. [DOI] [PubMed] [Google Scholar]
- 43.Wang YW, He SJ, Feng X, Cheng J, Luo YT, et al. Metformin: a review of its potential indications. Drug Des Dev Ther. 2017;11:2421–2429. doi: 10.2147/DDDT.S141675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White MD, Angiolini JF, Alvarez YD, Kaur G, Zhao ZW, et al. Long-lived binding of Sox2 to DNA predicts cell fate in the four-cell mouse embryo. Cell. 2016;165:75–87. doi: 10.1016/j.cell.2016.02.032. [DOI] [PubMed] [Google Scholar]
- 45.Yuan SY, Liu J, Zhou J, Lu W, Zhou HY, et al. AMPK mediates glucocorticoids stress-induced downregulation of the glucocorticoid receptor in cultured rat prefrontal cortical astrocytes. PloS one. 2016;11:e0159513. doi: 10.1371/journal.pone.0159513. [DOI] [PMC free article] [PubMed] [Google Scholar]