

Abstract
We investigated combining a core AD neuropathology measure (plasma amyloid-beta [Aβ] 42/40) with five plasma markers of inflammation, cellular stress, and neurodegeneration to predict cognitive decline. Among 401 participants free of dementia (median [IQR] age, 76 [73–80] years) from the Multidomain Alzheimer Preventive Trial (MAPT), 28 (7.0%) participants developed dementia, and 137 (34.2%) had worsening of clinical dementia rating (CDR) scale over 4 years. In the models utilizing plasma Aβ alone, a tenfold increased risk of incident dementia (nonsignificant) and a fivefold increased risk of worsening CDR were observed as each nature log unit increased in plasma Aβ levels. Models incorporating Aβ plus multiple plasma biomarkers performed similarly to models included Aβ alone in predicting dementia and CDR progression. However, improving Aβ model performance for composite cognitive score (CCS) decline, a proxy of dementia, was observed after including plasma monocyte chemoattractant protein 1 (MCP1) and growth differentiation factor 15 (GDF15) as covariates. Participants with abnormal Aβ, GDF15, and MCP1 presented higher CCS decline (worsening cognitive function) compared to their normal-biomarker counterparts (adjusted β [95% CI], − 0.21 [− 0.35 to − 0.06], p = 0.005). In conclusion, our study found limited added values of multi-biomarkers beyond the basic Aβ models for predicting clinically meaningful cognitive decline among non-demented older adults. However, a combined assessment of inflammatory and cellular stress status with Aβ pathology through measuring plasma biomarkers may improve the evaluation of cognitive performance.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11357-022-00554-y.
Keywords: Alzheimer’s disease, Cognitive decline, Amyloid-beta, Inflammation, Neurodegeneration, Aging
Introduction
Alzheimer’s disease (AD) is a complex and burdening neurodegenerative disorder [1]. Biomarkers of brain amyloid-beta (Aβ) accumulation, a neuropathological hallmark of AD [1], are essential tools for the identification of high-risk individuals [2, 3]. Brain amyloidosis is frequently confirmed by positron emission tomography (PET) or by the increased Aβ levels in cerebrospinal fluid (CSF) [4]. Recently, plasma measures of Aβ using updated techniques showed similar accuracy as classical imaging and CSF markers [5–7] and were prospectively associated with clinical cognitive outcomes [8–10]. However, their associations with cognitive impairment or conversion to AD are still not definitely concluded [10, 11].
AD development has multiple pathophysiologic mechanisms involved. Aggregated Aβ peptides activate an innate immune response in the brain, releasing numerous pro-inflammatory molecules and contributing to disease progression and severity [12]. Indeed, some studies observed that elevated circulating pro-inflammatory markers such as tumor necrosis factor receptor 1 (TNFR1) [13, 14] and interleukin 6 (IL6) [15] were associated with an increased risk of mild cognitive impairment (MCI) or dementia. Furthermore, plasma levels of monocyte chemoattractant protein 1 (MCP1), an important chemokine involved in the chemoattraction of immune cells at the brain lesion site [16], were not only associated with cognitive decline [17, 18] but also showed a significant interaction with Aβ levels on brain neuronal integrity [19]. In addition, higher circulating levels of growth differentiation factor 15 (GDF15), a stress-response cytokine involved in the regulation of inflammation [20] and energy metabolism [21], have been associated with worsening of cognitive function [22] and incident dementia [23]. On the other hand, neurodegeneration is widely accepted as the ultimate stage of AD disease course [3]. Plasma levels of neurofilament light chain (NfL), a structural protein of neurons considered a nonspecific marker for neurodegeneration [24], have been demonstrated to reflect AD severity [25] and to be a potential indicator for longevity [26]. Therefore, concurrently measuring the presence of neurodegeneration with other pathological markers might increase the prediction of AD.
Considering the diverse underlying pathologies of AD, previous studies had proposed that models combining multiple blood-based biomarkers can improve the prediction of AD dementia and longitudinal cognitive function [11, 25]. To the best of our knowledge, one pilot study had investigated the cross-sectional association of AD diagnosis with a plasma signature of Aβ levels and inflammatory factors including TNF-α and IL6 [11]. In this study, the authors demonstrated that using composite biomarker scores, which included plasma Aβ40, Aβ42, TNF-α, and other four inflammatory molecules, better correlated with AD diagnosis than considering age and sex only. However, the predictive abilities of combined plasma biomarker profiles on dementia still need to be evaluated in longitudinal studies.
This study aimed to investigate whether combining measures of plasma Aβ with other important circulating markers potentially involved in AD (i.e., markers of inflammation (TNFR1, IL6, MCP1), cellular stress (GDF15), and neurodegeneration (NfL)) would better determine clinically meaningful cognitive decline, including dementia, worsening of clinical dementia rating (CDR) status, and longitudinal cognitive performance in community-dwelling older adults.
Methods
Data source
This observational study is retrieved data from the Multidomain Alzheimer Preventive Trial (MAPT), a multicenter, 3-year randomized controlled trial whose details have been published elsewhere [27, 28]. Briefly, the MAPT Study failed in showing the protective effect of omega-3 polyunsaturated fatty acid (PUFA) supplementation and multidomain lifestyle interventions (including exercise advice, cognitive training and nutritional counseling) on cognitive decline in community-dwelling older adults [27]. After the 3-year intervention phase, an additional 2-year observation (without any intervention) was performed. The MAPT Study was registered at ClinicalTrials.gov [no.: NCT00672685], approved by the French Ethical Committee located in Toulouse (CPP SOOM II) and authorized by the French Health Authority. All participants signed informed consent.
Study population
The MAPT Study enrolled 1,679 adults aged ≥ 70 years and presented subjective memory concerns. Among them, 427 subjects had available data on all plasma biomarkers investigated in this study (details provided below). We excluded 3 subjects due to CDR scale > 1 (probable dementia) at the time biomarkers were measured (12 months after MAPT Study enrollment); 2 subjects due to diagnosis of dementia before biomarker measures; and 21 subjects due to the lack of longitudinal data during the follow-up period. Finally, 401 subjects were included in this study. Compared to 401 participants included in this study, 21 subjects excluded due to loss to follow-up had higher IL6 levels (Supplementary Table 1). The comparison of baseline characteristics for MAPT participants included and not included in this study is presented in Supplementary Table 2; participants enrolled in the current study have a lower proportion of females than the rest of the MAPT population.
All the data used in this study comes from the time biomarkers were measured (the 12-month visit) onwards (until the last visit at 60 months). The time-point of plasma biomarker measurement (i.e., the 12-month visit) was defined as the baseline in the present study. Supplementary Fig. 1 depicts the data collection timeline for plasma biomarkers and cognitive outcomes.
Measurement of plasma biomarkers
Six biomarkers were measured from the blood samples of participants collected at the 12-month visit: Aβ42/40 ratio, NfL (pg/mL), GDF15 (pg/mL), TNFR1 (pg/mL), IL6 (pg/mL), and MCP1(pg/mL). The details of plasma biomarker assessment were described in supplemental materials. We used the continuous value of plasma biomarkers to build risk prediction models for cognitive decline; all biomarker values were natural log-transformed to correct skewness.
Main outcome measures
Primary outcomes were the incidence of any type of dementia and worsening of CDR status during the 4-year follow-up period. Dementia was determined by an expert committee composed of physicians, psychologists, and intervenors of multidomain intervention in each center, using the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV) and National Institute of Neurological and Communicative Diseases and Stroke/Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) criteria. Each case was then validated by an independent committee of physicians not involved in the MAPT Study. The CDR status was evaluated annually (i.e., at 12, 24, 36, 48, and 60 months) and scored from 0 to 3, with higher values indicating worse cognitive function[29]. Worsening of CDR status was defined as changing from score 0 at 12 months to ≥ 0.5 or from score 0.5 to ≥ 1 during the follow-up.
Considering the few cases of dementia observed in our population (28 out of 401 subjects in the 4-year follow-up period), we used a composite cognitive score (CCS) as an exploratory outcome. The CCS was calculated as a mean Z-score of 4 cognitive tests: free and total recall of the Free and Cued Selective Reminding Test, the 10 orientation items of the Mini-Mental State Examination (MMSE), the Digit Symbol Substitution Test score from the Wechsler Adult Intelligence Scale-Revised, and the Category Naming Test [28]. Lower CCS values, indicating worse cognitive function, are a proxy of future dementia and more sensitive for early cognitive impairment [30] and are therefore used as a main cognitive outcome measure in large randomized controlled trials [28, 31]. The short-term decrement of CCS was considered a clinically relevant and informative outcome of AD, as Coley et al. [32] had suggested that − 0.3 points of CCS change within 1 year was predictive of AD dementia.
Measurement of demographic variables
Several demographic factors were selected due to their potential effects on cognitive outcomes: age, sex, educational level (no diploma or primary school certificate, secondary education, high school diploma, university level), MAPT group allocation (multidomain intervention with omega-3 supplementation, multidomain intervention with placebo, omega-3 supplementation alone, and placebo), and body mass index (BMI, defined as body weight in kg divided by height2 in m2, assessed at 12 months). Given the effect of apolipoprotein E (APOE) ε4 genotype on AD [33], we also measured the APOE ε4 status (defined as having one or two ε4 alleles), to evaluate its impact on model selection in a sensitivity analysis.
Statistical analysis
Descriptive data were presented as median and interquartile ranges (IQRs), or frequencies and percentages, as appropriate. Due to the non-normal distribution of plasma biomarker data, the correlations between each plasma biomarker were analyzed using Spearman rank correlation coefficients. The individual associations between each plasma biomarker and cognitive outcomes were evaluated by Cox proportional hazard regression (with incident dementia or worsening of CDR status as the outcomes) and linear mixed-effect regression (with CCS evolution as the outcome). More details of the Cox regression and linear mixed-effect regression are described below.
For primary outcomes (incident dementia and worsening of CDR status), risk prediction models were constructed by Cox regression. Time to event was defined as the period from the time-point of plasma biomarker measures (the 12-month visit) to either dementia diagnosis or the first visit detecting worsening of CDR status; participants without events were censored at the date of their last visit. We first built an “amyloid model” including plasma Aβ42/40 as a predictor. Then we expanded the amyloid model with other plasma biomarkers (i.e., NfL, GDF15, TNFR1, IL6, MCP1) in 31 different combinations (referred to as “multi-biomarker models”; amyloid model plus other biomarkers). Model performances were assessed using Harrell’s concordant (C) statistic, a higher C index indicated better discrimination [34]. The model performances of multi-biomarker models were compared with the amyloid model, using the “somersd” package in STATA [35]. All models adjusted for demographic covariates mentioned above; initial CDR score (i.e., at 12 months) was also considered when worsening CDR status as the outcome. We also performed sensitivity analyses to test whether adding APOE ε4 genotype as a covariate reduced the effectiveness of multi-biomarker models.
Once the multi-biomarker models with the best performance were identified, the cumulative probability of cognitive outcomes was calculated from these established models using the STATA “survci” command [36]. We estimated specific probabilities for women and men and for individuals with an initial CDR score of 0 or 0.5, under the following conditions: aged 76.7 years (mean value of the study population), BMI as 26.5 kg/m2 (mean value of the study population), no diploma or having primary school certificate (the lowest educational level), and in the placebo group of MAPT Study. We selected the lower quartile as normal and the upper quartile as abnormal for each plasma biomarker; for Aβ, the lower quartile as abnormal and the upper quartile as normal. These values were selected as an example because the models can provide risk estimates for any given value of plasma biomarkers and covariates.
In exploratory analyses, we examined associations of CCS evolution with different biomarker combinations using linear mixed-effect regression with random intercepts and random slopes. The introduction of random intercepts and slopes in the model allows us to improve our estimations considering the variability of individual responses at baseline and throughout the follow-up. In other words, our model’s coefficients for the biomarkers represented the association between biomarkers and CCS at baseline; the time coefficient represented the yearly CCS change; and the coefficients for biomarker-time interaction represented the longitudinal CCS change per unit of biomarker increase. Plasma biomarkers were used as natural log-transformed, and time was measured as the number of years after the baseline. All models were adjusted for demographic covariates: age, sex, education, MAPT group, and BMI. Among our study population, the CCS change presented a quadratic trajectory. Therefore, we considered the quadratic terms for time and their interaction terms with plasma biomarkers.
We followed the same approach previously mentioned to define different multi-biomarker combinations. The best-fitting biomarker model was selected based on the lowest Akaike information criterion (AIC) value, and differences in AIC > 10 between the two models were considered essentially different [37]. Again, a sensitivity analysis with an additional adjustment for APOE ε4 genotype was conducted to test whether we would select the best-fitting model with the same biomarker combination as the main analysis. Once the best-fitting biomarker model was determined, the lower and upper quartile of each biomarker was applied in this selected model to estimate CCS change after 1 and 4 years, respectively. All analyses were performed using SAS version 9.4 (SAS Institute, Inc, Cary, NC) and STATA version 17 (College Station, TX), with a significance level of 0.05.
Results
Characteristics of the study population
Characteristics of the study sample at the 12-month visit, which constitutes the baseline visit for the present work, are presented in Table 1. Of the 401 participants (median [IQR] age, 76 [73 to 80] years), 233 (58.1%) were women, and 225 (56.1%) had an initial CDR score of 0.5 (Table 1). After 4 years of follow-up, 28 participants (7.0%) progressed to dementia (mean [standard deviation, SD] time to event, 1.7 [1.1] years); 137 participants (34.2%) developed worsening of CDR status (mean [SD] time to event, 2.1 [1.1] years), with the majority initially being cognitively normal (122 [69.3%] with initial CDR score 0 vs. 15 [6.7%] with score 0.5; p < 0.001) (Table 1). There is no significant difference in the proportion of cognitive outcomes between APOE ε4 carriers and non-carriers (Supplementary Table 3).
Table 1.
Characteristics of study population
| Total population (n = 401) | Baseline CDR score | |||
|---|---|---|---|---|
| CDR = 0 (n = 176) | CDR = 0.5 (n = 225) | p1 | ||
| Age (year) | 76 (73, 80) | 75 (72, 78) | 77 (73, 80) | 0.002 |
| Female sex | 233 (58.1%) | 115 (65.3%) | 118 (52.4%) | 0.009 |
| Education (n = 397) | ||||
| No diploma or primary school | 101 (25.4%) | 34 (19.4%) | 67 (30.2%) | 0.014 |
| Secondary education | 125 (31.5%) | 55 (31.4%) | 70 (31.5%) | |
| High school | 55 (13.9%) | 22 (12.6%) | 33 (14.9%) | |
| University level | 116 (29.2%) | 64 (36.6%) | 52 (23.4%) | |
| MAPT group | ||||
| MI + omega-3 | 109 (27.2%) | 40 (22.7%) | 69 (30.7%) | 0.270 |
| Omega-3 | 91 (22.7%) | 41 (23.3%) | 50 (22.2%) | |
| MI | 97 (24.2%) | 43 (24.4%) | 54 (24.0%) | |
| Placebo | 104 (25.9%) | 52 (29.6%) | 52 (23.1%) | |
| Body mass index (kg/m2) (n = 399) | 26.2 (23.7, 28.8) | 26.3 (23.6, 29.4) | 26.0 (23.7, 28.3) | 0.344 |
| Composite cognitive score2 (n = 398) | 0.13 (-0.28, 0.55) | 0.39 (0.03, 0.74) | -0.06 (-0.63, 0.44) | < 0.001 |
| APOE ε4 carrier (n = 364) | 96 (26.4%) | 41 (25.5%) | 55 (27.1%) | 0.726 |
| Plasma biomarker | ||||
| Aβ42/40 | 0.113 (0.104, 0.122) | 0.115 (0.105, 0.124) | 0.111 (0.103, 0.120) | 0.018 |
| NfL (pg/mL) | 73.9 (56.7, 94.2) | 73.2 (56.3, 91.0) | 74.1 (56.7, 96.5) | 0.495 |
| GDF15 (pg/mL) | 1144.0 (926.0, 1479.0) | 1087.5 (869.0, 1319.5) | 1197.0 (964.0, 1582.0) | < 0.001 |
| TNFR1 (pg/mL) | 1298 (1075, 1575) | 1276 (1051, 1498) | 1347 (1099, 1617) | 0.049 |
| IL6 (pg/mL) | 2.9 (2.1, 4.2) | 2.8 (2.1, 4.0) | 3.0 (2.2, 4.3) | 0.404 |
| MCP1 (pg/mL) | 224 (185, 270) | 222 (183, 266) | 225 (189, 276) | 0.285 |
| Outcome | ||||
| Incident dementia | 28 (7.0%) | 2 (1.1%) | 26 (11.6%) | < 0.001 |
| Worsening of CDR status3 | 137 (34.2%) | 122 (69.3%) | 15 (6.7%) | < 0.001 |
Values presented in frequency (percentage) for categorical variables or median (IQR) for continuous variables
Aβ, amyloid-beta; APOE, apolipoprotein E; CDR, clinical dementia rating; GDF15, growth differentiation factor 15; IL6, interleukin 6; MAPT, Multidomain Alzheimer Preventive Trial; MCP1, monocyte chemoattractant protein 1; MI, multidomain intervention; NfL, neurofilament light chain; TNFR1, tumor necrosis factor receptor type 1
1p value based on Pearson’s chi-square/Fisher’s exact test for categorical variables and Mann–Whitney U test for continuous variables
2Based on the z score of four cognitive tests: free and total recall of the Free and Cued Selective Reminding test, ten MMSE orientation items, Digit Symbol Substitution Test, and Category Naming Test
3Defined as changing from CDR score 0 to ≥ 0.5, or from score 0.5 to ≥ 1
Correlations between plasma biomarkers
The results of Spearman rank correlation are displayed in Supplementary Table 4. There were significant positive correlations between biomarker pairs of GDF15, TNFR1, IL6, and MCP1 (all p < 0.001, Spearman rank correlation coefficients ranged from 0.28 to 0.60). On the contrary, plasma Aβ42/40 did not show significant correlations with other plasma biomarkers (Supplementary Table 4).
Associations between each plasma biomarker and cognitive outcomes
We first evaluated the associations between each plasma biomarker and the three cognitive outcomes (incident dementia, worsening of CDR status, and CCS evolution). Decreased plasma Aβ42/40 (inverse HR [95% CI], 19.40 [1.34–280.13]; p = 0.030), increased NfL (HR [95% CI], 2.16 [1.20, 3.88]; p = 0.010), and increased GDF15 (HR [95% CI], 3.49 [1.36, 8.93]; p = 0.009) were associated with incident dementia in the separate unadjusted models (Supplementary Table 5). Furthermore, plasma Aβ42/40 was the only marker associated with worsening of CDR status (inverse HR [95% CI], 5.03 [1.27, 20.01]; p = 0.022), after controlling for demographic factors (Supplementary Table 5). Regarding CCS outcome, there was a significant positive association between plasma Aβ42/40 and CCS change over time, indicating that participants with higher plasma Aβ42/40 (a better plasma Aβ profile) had better cognitive function over time (Supplementary Table 6).
Model selection and cumulative probabilities for incident dementia
With dementia as the outcome, 33 models were constructed with demographics, plasma Aβ42/40, NfL, GDF15, TNFR1, IL6, and MCP1 as predictors (Supplementary Table 7). Among all biomarker combinations, the model incorporating Aβ plus TNFR1, IL6, and MCP1 showed the highest Harrell’s C index, but it did not reach statistically significant differences compared to the amyloid model (p = 0.568, Table 2; HRs of plasma biomarkers and covariates present in Fig. 1), indicating that two models performed similarly in predicting dementia. Based on the model enrolled Aβ, TNFR1, IL6, and MCP1, the estimated 4-year progression risks (95% CI) to dementia ranged from 2.2 (0.6%, 8.2%) to 6.6% (2.1%, 19.8%) in women and from 4.0 (1.0%, 16.1%) to 12.0% (3.6%, 35.5%) in men (Supplementary Table 8). Subjects with abnormal values in Aβ, TNFR1, and IL6 tended to have a higher probability of developing dementia compared to other biomarker combinations (Supplementary Table 8).
Table 2.
Comparing model performance for clinically meaningful cognitive outcomes by addition of plasma amyloid-beta and other biomarkers to model with demographic factors (Cox proportional hazard regression)
| Harrell’s C index | p | ||
|---|---|---|---|
| Incident dementia | |||
| Demographic factors: age, sex, education, MAPT group, BMI | 0.8019 | Ref | |
| Plus Aβ42/40 | 0.8075 | 0.684 | Ref |
| Plus Aβ42/40, TNFR1, IL6, MCP11 | 0.8163 | 0.508 | 0.568 |
| Worsening of CDR status | |||
| Demographic factors: age, sex, education, MAPT group, BMI, initial CDR score | 0.8155 | Ref | |
| Plus Aβ42/40 | 0.8169 | 0.726 | Ref |
| Plus Aβ42/40, GDF15, TNFR1, IL61 | 0.8197 | 0.416 | 0.338 |
| Plus Aβ42/40, NfL, GDF15, TNFR1, IL61 | 0.8197 | 0.420 | 0.342 |
Aβ, amyloid-beta; BMI, body mass index; CDR, clinical dementia rating; GDF15, growth differentiation factor 15; IL6, interleukin 6; MAPT, Multidomain Alzheimer Preventive Trial; MCP1, monocyte chemoattractant protein 1; NfL, neurofilament light chain; TNFR1, tumor necrosis factor receptor type 1
1The multi-biomarker combination which showed the highest Harrell’s C index compared to other biomarker models; Harrell’s C indices of models with other biomarker combinations are provided in the supplementary materials
Fig. 1.

Hazard ratios of incident dementia according to the amyloid model (A) and the multi-biomarker model with the highest Harrell’s C index (B). The inverse of the HR point estimate and the 95% confidence interval are presented for plasma Aβ42/40 (HR per unit decrease in the nature log of Aβ42/40 ratio)
Model selection and cumulative probabilities for worsening of CDR status
With worsening CDR as the outcome and adjustment for demographic factors, the amyloid model and multi-biomarker models showed equal discriminative powers (all p > 0.05; Table 2 and Supplementary Table 9). Multi-biomarker models composed of Aβ, GDF15, TNFR1, and IL6 or composed of Aβ, NfL, GDF15, TNFR1, and IL6 showed the highest Harrell’s C index (Table 2). Plasma Aβ significantly increased the hazard ratio of worsening CDR by fivefold (multi-biomarker model with Aβ, GDF15, TNFR1, and IL6: inverse HR, 5.62 [1.37, 23.12]; model with Aβ, NfL, GDF15, TNFR1, and IL6: inverse HR, 5.58 [1.35, 23.06]; Fig. 2). Estimated 1-year and 4-year progression risks according to these two models are provided in Supplementary Table 10.
Fig. 2.
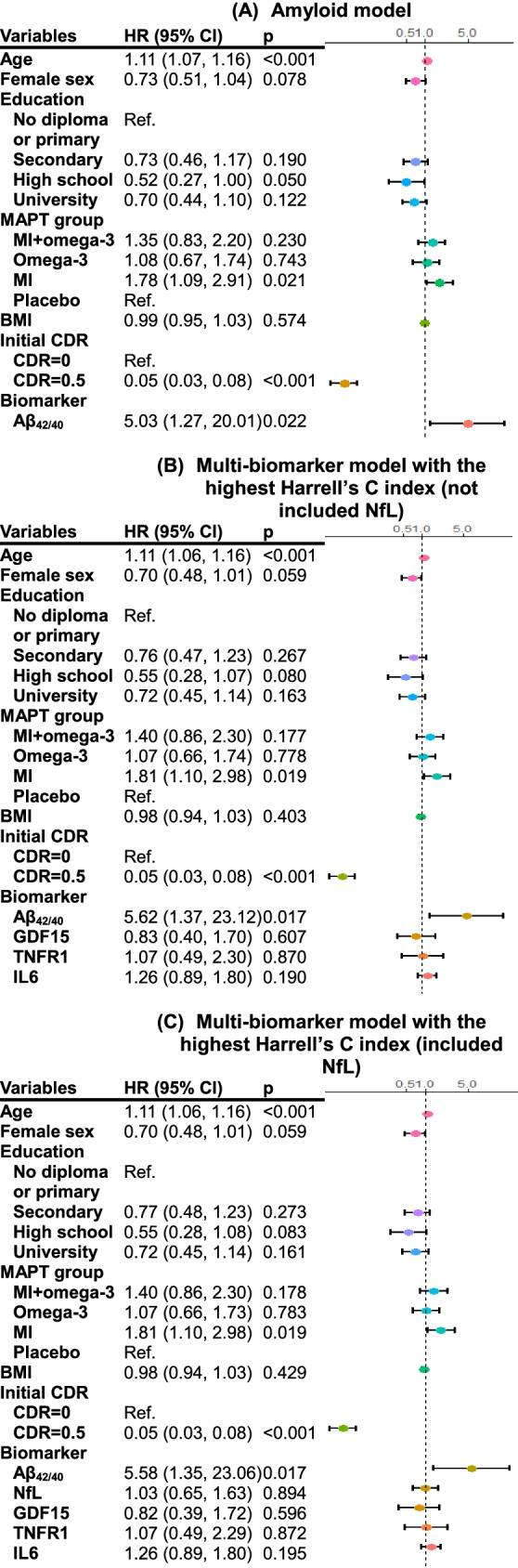
Hazard ratios of worsening CDR according to the amyloid model (A) and the multi-biomarker models with the highest Harrell’s C indices (B and C). The inverse of the HR point estimate and the 95% confidence interval are presented for plasma Aβ42/40 (HR per unit decrease in the nature log of Aβ42/40 ratio)
Model selection and estimated evolution for CCS
Supplementary Table 11 displays the results of linear mixed-effect models for CCS evolution with 33 different biomarker combinations. The multi-biomarker model, which included Aβ, GDF15, MCP1, and demographic covariates, had a significantly higher goodness-of-fit compared to the amyloid model (ΔAIC = − 17.8; Table 3). It is worth noting that plasma GDF15 and MCP1 are only significantly associated with baseline CCS but not with the change of CCS over time (i.e., insignificant plasma biomarker-time interaction terms) (Table 4). On the other hand, plasma Aβ42/40 showed significant association with CCS evolution but not with initial CCS (Table 4). The estimated change of CCS over time according to the best-fitting model that included Aβ, GDF15, and MCP1 was displayed in Fig. 3. Having abnormal Aβ, either combined with other abnormal plasma markers or not, presented a higher decrease in CCS over 4 years compared to the group with normal biomarker values (adjusted between-groups differences: β [95% CI], − 0.14 [− 0.22 to − 0.05], p = 0.002 with Aβ + /GDF15-/ MCP1-; − 0.20 [− 0.33 to − 0.07], p = 0.002 with Aβ + /GDF15 + / MCP1-; − 0.14 [− 0.27 to − 0.02], p = 0.026 with Aβ + /GDF15-/ MCP1 + ; − 0.21 [− 0.35 to − 0.06], p = 0.005 with Aβ + /GDF15 + / MCP1 + ; Fig. 3).
Table 3.
Comparing the goodness-of-fit between linear mixed-effect models for composite cognitive score1 by addition of plasma amyloid-beta and other biomarkers to model with demographic factors
| Model | AIC | ΔAIC | |
|---|---|---|---|
| Demographic factors: age, sex, education, MAPT group, BMI | 2602.2 | Ref | |
| Plus Aβ42/40 | 2592.9 | -9.3 | Ref |
| Plus Aβ42/40, GDF15, MCP12 | 2575.1 | -27.1 | -17.8 |
Aβ, amyloid-beta; BMI, body mass index; GDF15, growth differentiation factor 15; MAPT, Multidomain Alzheimer Preventive Trial; MCP1, monocyte chemoattractant protein 1
1Based on the z score of four cognitive tests: free and total recall of the Free and Cued Selective Reminding test, ten MMSE orientation items, Digit Symbol Substitution Test, and Category Naming Test
2The multi-biomarker combination of the best-fitting linear mixed-effect model; the goodness-of-fit of the models with other biomarker combinations are provided in the supplementary materials
Table 4.
Coefficients of plasma biomarker variables in linear mixed-effect models examining the change of composite cognitive score1 over time
| Biomarker2 | Biomarker2 × time | Biomarker2 × time × time | ||||
|---|---|---|---|---|---|---|
| Coef. (95% CI) | p | Coef. (95% CI) | p | Coef. (95% CI) | p | |
| Amyloid model3 | ||||||
| Aβ42/40 | − 0.15 (− 0.66, 0.35) | 0.546 | 0.60 (0.24, 0.96) | 0.001 | − 0.10 (− 0.18, − 0.01) | 0.026 |
| Multi-biomarker model4 | ||||||
| Aβ42/40 | − 0.11 (− 0.59, 0.38) | 0.671 | 0.60 (0.24, 0.96) | 0.001 | − 0.10 (− 0.18, − 0.01) | 0.028 |
| GDF15 | − 0.53 (− 0.74, − 0.32) | < 0.001 | − 0.07 (− 0.21, 0.07) | 0.325 | 0.01 (− 0.02, 0.04) | 0.583 |
| MCP1 | 0.33 (0.11, 0.55) | 0.003 | 0.04 (− 0.11, 0.20) | 0.606 | − 0.01 (− 0.05, 0.03) | 0.540 |
Aβ, amyloid-beta; GDF15, growth differentiation factor 15; MCP1, monocyte chemoattractant protein 1
1Based on the z score of four cognitive tests: free and total recall of the Free and Cued Selective Reminding test, ten MMSE orientation items, Digit Symbol Substitution Test, and Category Naming Test
2All values of plasma biomarkers were natural log-transformed
3Adjusted for demographic covariates (age, sex, education, Multidomain Alzheimer Preventive Trial (MAPT) group and body mass index (BMI))
4The best-fitting multi-biomarker model among all biomarker combinations; the model was adjusted for demographic covariates (age, sex, education, MAPT group, and BMI). Coefficients of biomarker variables in other multi-biomarker models with different biomarker combinations are provided in supplementary materials
Fig. 3.
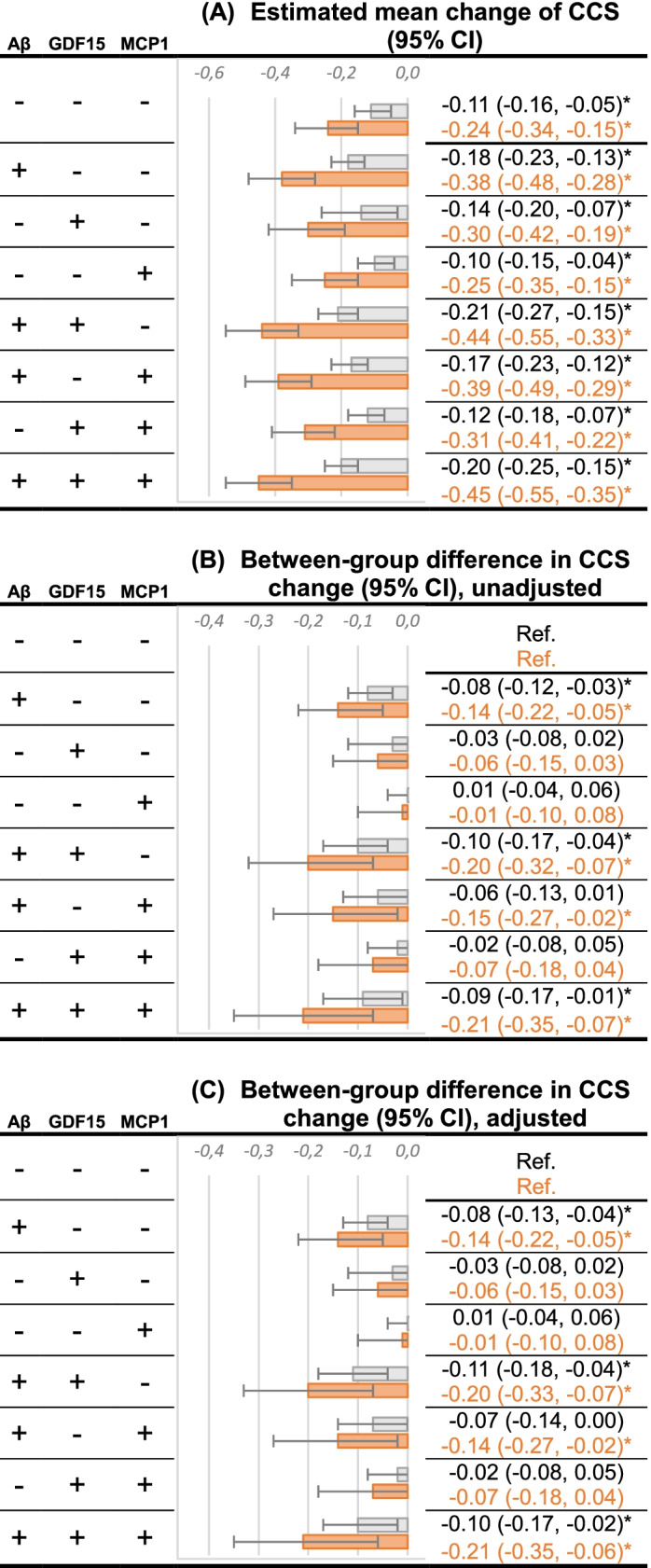
Estimated change of CCS over 1 year (represented by the gray bar) and 4 years (by the orange bar) according to the best-fitting model that included plasma Aβ42/40, GDF15 and MCP1. The lower quartile was selected as normal (−) and the upper quartile as abnormal (+) for GDF15 and MCP1; for Aβ, the upper quartile was selected as normal (−) and the lower quartile as abnormal (+). *p-value < 0.05. (A) Estimated mean change of CCS over time according to the indicated normal (−) / abnormal (+) biomarker values. (B) Unadjusted difference in CCS change compared to the reference group (Aβ-, GDF15-, MCP1-). (C) Difference in CCS change compared to the reference group with adjustment for age, sex, education, MAPT group, and body mass index
Sensitivity analysis: effect of APOE ε4 genotype
After including APOE ε4 status as a covariate, Cox models incorporating multiple biomarkers still demonstrated higher Harrell’s C index but no statistically significant differences compared to the models including plasma Aβ alone, with dementia and worsening of CDR status as the outcomes (Supplementary Table 12). Mixed-effect models evaluating CCS evolution with adjustment for APOE ε4 genotype showed similar results as the main analysis; the multi-biomarker model including Aβ, GDF15, and MCP1 remained with a better goodness-of-fit compared to the amyloid model (ΔAIC = − 11.6; Supplementary Table 12).
Discussion
This study evaluated the combined effects of multiple blood-based biomarkers of or potentially involved in AD on determining longitudinal cognitive changes. We observed the limited added values of multi-biomarkers beyond the basic Aβ model for predicting future dementia onset and CDR progression. Nevertheless, in our exploratory analysis, extending the amyloid model with plasma GDF15 and MCP1 improved the model performance on the change of CCS, suggesting the potential values of combined assessment of inflammatory and cellular stress status with the core AD pathology for evaluating cognitive performance.
Multi-biomarker approaches have been suggested and applied to predict chronic diseases driven by several biological processes, such as heart failure and AD [25, 38, 39]. Compared to focusing on a single marker, combining the assessment of multiple biomarkers covers diverse underlying mechanisms of disease and the potential interaction between different pathophysiological pathways. Furthermore, prognostic models constructed by multiple biomarkers allow risk-stratification of patients [38]. In line with this concept, a multi-biomarker approach including classical pathophysiological markers (e.g., Aβ for AD) and markers derived from the hallmarks of aging [40] could lead to a better understanding of the importance of biological aging in the development of a large variety of chronic age-related diseases; in turn, this would permit the development of more precise treatments according to disease severity and the mechanisms of biological aging involved in the onset/progression of the condition. Indeed, recent studies have suggested that models combined with multiple neuropathological biomarkers improved prediction on AD [25, 41, 42] and longitudinal cognitive decline [25], even though the added marker was not individually associated with cognitive outcomes [25]. Furthermore, the pilot work of Iulita MF et al. emphasized the importance of combining amyloid and inflammatory factors to identify AD dementia [11]. However, their finding was restricted to the cross-sectional level, since individuals’ plasma biomarkers were measured at the same visit of the final AD diagnosis [11]. Our study provides further evidence on this topic by using a 4-year longitudinal design, relatively large sample size, and well-characterized cohort without dementia at baseline and including biomarkers involved in the aging process into the prognostic models, providing a comprehensive measurement of individual factors that potentially influenced the associations between the neuropathological features of AD and the clinical expression of cognitive decline [43].
However, in the present study, we observed limited added value of multiple plasma biomarkers in predicting dementia and CDR worsening. These findings could be explained by our small number of dementia cases and the relatively short follow-up period, considering that progression of MCI to dementia in older adults could take decades [44]. It is worth noting that amyloid biomarkers can be detected as abnormal from 5 to 10 years before dementia onset [45, 46] and become less changed in the later stages of the disease process [3, 47], which might explain the associations of plasma Aβ42/40 per se with the evolution of cognitive performance but not with incident dementia in our results. In addition, our results should be interpreted cautiously since our study population was composed of both cognitively normal individuals (43.9%) and subjects with MCI (56.1%), who had been suggested to present different biomarker profiles[2]. As a result, more large-sample studies applied similar multi-biomarker approaches and focused on cognitively normal, and MCI subjects, respectively, could provide more evidence on this topic.
Our exploratory analysis on changes of CCS revealed an improvement in model fit when extending the amyloid model with plasma GDF15 and MCP1, indicating that both plasma MCP1 and GDF15 might be important variables for evaluating cognitive performance in addition to plasma Aβ and demographic covariates. This result should be interpreted cautiously since both biomarkers were not individually associated with changes of CCS in the model. Plasma Aβ levels remained the main driver of CCS decline. Our findings may support the important role of MCP1 in neuroinflammation in the brain compared to other chemokines and cytokines, including IL6 [48]. However, it is worth mentioning that we measured MCP1 levels in the blood, which we could not completely exclude the influence of MCP1 secreted by peripheral tissues. Elevated levels of GDF15 in blood had been associated with various pathological conditions (e.g., sepsis [49], cardiovascular disease [50], cancer [51], and obesity [51]), serving as an integrative marker of disease severity [52] and recovery after acute illness [53]. GDF15 is induced in inflammation and mitochondrial stress [20], in which higher circulating levels had shown detrimental effects on cognitive performance and brain structure [54]. Our findings were in line with the literature which showed negative effects of GDF15 on cognition, implying a role of impaired mitochondrial function in cognitive decline during aging. Nevertheless, GDF15 has been reported as a neurotrophic factor in some animal models [55]. These conflicting findings reflect the diverse roles of GDF15 depending on the state of cells or their environment [52], and its relationship with neurodegenerative disorders remains to be determined. Given the exploratory portion of this analysis and the single measurement of plasma GDF15 and MCP1, future research with a larger sample size, longitudinal, and multiple time-point of biomarker collection is encouraged to explore the relationship between brain amyloidosis and the underlying pathways of MCP1 and GDF15 at the early phase of cognitive impairment.
As one of the studies to apply multi-biomarker approaches in determining AD-associated cognitive outcomes, we highlight the use of several blood biomarkers, and the assessment of plasma Aβ by a recent and improved measurement technique in a sample of older adults followed longitudinally and well-characterized regarding cognitive-related outcomes and measures. Nevertheless, some limitations should be mentioned. First of all, the main objective of this study was to explore whether combining measures of multiple critical biomarkers of AD played a role in predicting clinically cognitive outcomes rather than proposing new prognostic models on AD dementia. It is worth highlighting that all the models constructed in this study would need calibration and validation (both internal and external) before being used in other study cohorts. Second, we selected the biomarkers of interest based on data availability in the MAPT Study and only measured in a subset of MAPT participants. It is worth mentioning that participants enrolled in the current study had slightly different characteristics compared to the rest of the MAPT population. Finally, three out of four subjects in the current study had received interventions over the first 3 years of follow-up. Although the interventions were not able to prevent or slow cognitive decline [28], they might affect the levels of the biomarkers, which were measured one year after enrollment. In order to minimize this bias, MAPT group allocation was added as a covariate in our analyses.
To conclude, the results of this study do not support the combined effects of multiple biomarkers on predicting dementia onset and CDR evolution. However, promising findings were obtained regarding GDF15 and MCP1 in our exploratory analysis using the CCS, an outcome more sensitive to change. Further investigations on the utility of multi-biomarker approaches in amyloid-associated cognitive decline, particularly enrolling the assessment of inflammatory and cellular stress status, are needed.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We acknowledge Christelle Cantet at CERPOP, UMR1295, Inserm Université Toulouse III, for her thoughtful suggestions and confirmation of our statistical modeling approach and interpretation.
Members of the MAPT/DSA group include:
MAPT/DSA group
MAPT Study group: principal investigator, Bruno Vellas (Toulouse); coordination, Sophie Guyonnet; project leader, Isabelle Carrié; CRA, Lauréane Brigitte; investigators, Catherine Faisant, Françoise Lala, Julien Delrieu, Hélène Villars; psychologists, Emeline Combrouze, Carole Badufle, Audrey Zueras; methodology, statistical analysis, and data management, Sandrine Andrieu, Christelle Cantet, Christophe Morin; multidomain group, Gabor Abellan Van Kan, Charlotte Dupuy, Yves Rolland (physical and nutritional components), Céline Caillaud, Pierre-Jean Ousset (cognitive component), Françoise Lala (preventive consultation) (Toulouse). The cognitive component was designed in collaboration with Sherry Willis from the University of Seattle and Sylvie Belleville, Brigitte Gilbert, and Francine Fontaine from the University of Montreal.
Co-investigators in associated centers, Jean-François Dartigues, Isabelle Marcet, Fleur Delva, Alexandra Foubert, Sandrine Cerda (Bordeaux); Marie-Noëlle-Cuffi, Corinne Costes (Castres); Olivier Rouaud, Patrick Manckoundia, Valérie Quipourt, Sophie Marilier, Evelyne Franon (Dijon); Lawrence Bories, Marie-Laure Pader, Marie-France Basset, Bruno Lapoujade, Valérie Faure, Michael Li Yung Tong, Christine Malick-Loiseau, Evelyne Cazaban-Campistron (Foix); Françoise Desclaux, Colette Blatge (Lavaur); Thierry Dantoine, Cécile Laubarie-Mouret, Isabelle Saulnier, Jean-Pierre Clément, Marie-Agnès Picat, Laurence Bernard-Bourzeix, Stéphanie Willebois, Iléana Désormais, Noëlle Cardinaud (Limoges); Marc Bonnefoy, Pierre Livet, Pascale Rebaudet, Claire Gédéon, Catherine Burdet, Flavien Terracol (Lyon), Alain Pesce, Stéphanie Roth, Sylvie Chaillou, Sandrine Louchart (Monaco); Kristel Sudres, Nicolas Lebrun, Nadège Barro-Belaygues (Montauban); Jacques Touchon, Karim Bennys, Audrey Gabelle, Aurélia Romano, Lynda Touati, Cécilia Marelli, Cécile Pays (Montpellier); Philippe Robert, Franck Le Duff, Claire Gervais, Sébastien Gonfrier (Nice); Yannick Gasnier and Serge Bordes, Danièle Begorre, Christian Carpuat, Khaled Khales, Jean-François Lefebvre, Samira Misbah El Idrissi, Pierre Skolil, Jean-Pierre Salles (Tarbes).
MRI group: Carole Dufouil (Bordeaux), Stéphane Lehéricy, Marie Chupin, Jean-François Mangin, Ali Bouhayia (Paris); Michèle Allard (Bordeaux); Frédéric Ricolfi (Dijon); Dominique Dubois (Foix); Marie Paule Bonceour Martel (Limoges); François Cotton (Lyon); Alain Bonafé (Montpellier); Stéphane Chanalet (Nice); Françoise Hugon (Tarbes); Fabrice Bonneville, Christophe Cognard, François Chollet (Toulouse).
PET scan group: Pierre Payoux, Thierry Voisin, Julien Delrieu, Sophie Peiffer, Anne Hitzel, (Toulouse); Michèle Allard (Bordeaux); Michel Zanca (Montpellier); Jacques Monteil (Limoges); Jacques Darcourt (Nice).
Medico-economics group: Laurent Molinier, Hélène Derumeaux, Nadège Costa (Toulouse).
Biological sample collection: Bertrand Perret, Claire Vinel, Sylvie Caspar-Bauguil (Toulouse).
Safety management: Pascale Olivier-Abbal.
DSA Group: Sandrine Andrieu, Christelle Cantet, Nicola Coley.
Author contribution
Conceptualization, W-HL and PSB; methodology, W-HL and PSB; formal analysis, W-HL; investigation, JEM, AP, GA, ADN, YL, and RJB; data curation, W-HL; writing—original draft preparation, W-HL; writing—review and editing, KVG, JEM, SG, AP, GA, ADN, YL, RJB, BV, and PSB; supervision, BV and PSB; funding acquisition, ADN, RJB, and BV.
Funding
The present work was performed in the context of the Inspire Program, a research platform supported by grants from the Region Occitanie/Pyrénées-Méditerranée (Reference number: 1901175) and the European Regional Development Fund (ERDF) (Project number: MP0022856). This study received funds from Alzheimer Prevention in Occitania and Catalonia (APOC Chair of Excellence – Inspire Program). This study has been partially supported through the grant EUR CARe N°ANR-18-EURE-0003 in the framework of the Programme des Investissements d'Avenir. The plasma measures of this study were supported by institutional gift funds (RJ Bateman, PI), institutional startup funds (AD Nguyen, PI), and National Institute on Aging grants NIH R56AG061900 and RF1AG061900 (RJ Bateman, PI). The MAPT study was supported by grants from the Gérontopôle of Toulouse, the French Ministry of Health (PHRC 2008, 2009), Pierre Fabre Research Institute (manufacturer of the omega-3 supplement), ExonHit Therapeutics SA, and Avid Radiopharmaceuticals Inc. The promotion of this study was supported by the University Hospital Center of Toulouse. The data sharing activity was supported by the Association Monegasque pour la Recherche sur la maladie d’Alzheimer (AMPA) and the INSERM-University of Toulouse III UMR 1295 Unit.
Declarations
Ethics approval
The MAPT Study was approved by the French Ethical Committee located in Toulouse (CPP SOOM II) and authorized by the French Health Authority. All participants signed informed consent.
Conflict of interest
Washington University and Randall Bateman have equity ownership interest in C2N Diagnostics and receive income based on technology (blood plasma assay) licensed by Washington University to C2N Diagnostics. RJB receives income from C2N Diagnostics for serving on the scientific advisory board. Washington University, with RJB as co-inventor, has submitted the US nonprovisional patent application “Plasma Based Methods for Determining A-Beta Amyloidosis.” RJB has received honoraria as a speaker/consultant/advisory board member from Amgen, AC Immune, Eisai, and Hoffman-LaRoche, and reimbursement of travel expenses from AC Immune.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
for the MAPT/DSA Group:
Bruno Vellas, Sophie Guyonnet, Isabelle Carrié, Lauréane Brigitte, Catherine Faisant, Franҫoise Lala, Julien Delrieu, Hélène Villars, Emeline Combrouze, Carole Badufle, Audrey Zueras, Sandrine Andrieu, Christelle Cantet, Christophe Morin, Gabor Abellan Van Kan, Yves Rolland, Charlotte Dupuy, Céline Caillaud, Pierre-Jean Ousset, Franҫoise Lala, Sherry Willis, Sylvie Belleville, Brigitte Gilbert, Francine Fontaine, Jean-François Dartigues, Isabelle Marcet, Fleur Delva, Alexandra Foubert, Sandrine Cerda, Marie-Noëlle Cuffi, Corinne Costes, Olivier Rouaud, Patrick Manckoundia, Valérie Quipourt, Sophie Marilier, Evelyne Franon, Lawrence Bories, Marie-Laure Pader, Marie-France Basset, Bruno Lapoujade, Valérie Faure, Michael Li Yung Tong, Christine Malick-Loiseau, Evelyne Cazaban-Campistron, Franҫoise Desclaux, Colette Blatge, Thierry Dantoine, Cécile Laubarie-Mouret, Isabelle Saulnier, Jean-Pierre Clément, Marie-Agnès Picat, Laurence Bernard-Bourzeix, Stéphanie Willebois, Iléana Désormais, Noëlle Cardinaud, Marc Bonnefoy, Pierre Livet, Pascale Rebaudet, Claire Gédéon, Catherine Burdet, Flavien Terracol, Alain Pesce, Stéphanie Roth, Sylvie Chaillou, Sandrine Louchart, Kristel Sudres, Nicolas Lebrun, Nadège Barro-Belaygues, Jacques Touchon, Karim Bennys, Audrey Gabelle, Aurélia Romano, Lynda Touati, Cécilia Marelli, Cécile Pays, Philippe Robert, Franck Le Duff, Claire Gervais, Sébastien Gonfrier, Yannick Gasnier, Serge Bordes, Danièle Begorre, Christian Carpuat, Khaled Khales, Jean-François Lefebvre, Samira Misbah El Idrissi, Pierre Skolil, Jean-Pierre Salles, Carole Dufouil, Stéphane Lehéricy, Marie Chupin, Jean-François Mangin, Ali Bouhayia, Michèle Allard, Frédéric Ricolfi, Dominique Dubois, Marie Paule Bonceour Martel, Franҫois Cotton, Alain Bonafé, Stéphane Chanalet, Françoise Hugon, Fabrice Bonneville, Christophe Cognard, Franҫois Chollet, Pierre Payoux, Thierry Voisin, Julien Delrieu, Sophie Peiffer, Anne Hitzel, Michèle Allard, Michel Zanca, Jacques Monteil, Jacques Darcourt, Laurent Molinier, Hélène Derumeaux, Nadège Costa, Bertrand Perret, Claire Vinel, Sylvie Caspar-Bauguil, Pascale Olivier-Abbal, and Nicola Coley
References
- 1.Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, Cummings J, van der Flier WM. Alzheimer’s disease. Lancet. 2021;397:1577–1590. doi: 10.1016/S0140-6736(20)32205-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morgan AR, Touchard S, Leckey C, O’Hagan C, Nevado-Holgado AJ, Barkhof F, Bertram L, Blin O, Bos I, Dobricic V, et al. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimer’s Dement. 2019;15:776–787. doi: 10.1016/j.jalz.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bjorkli C, Sandvig A, Sandvig I. Bridging the Gap between fluid biomarkers for Alzheimer’s disease, model systems, and patients. Front Aging Neurosci. 2020;12:272. doi: 10.3389/fnagi.2020.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jack CR, Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ovod V, Ramsey KN, Mawuenyega KG, Bollinger JG, Hicks T, Schneider T, Sullivan M, Paumier K, Holtzman DM, Morris JC, et al. Amyloid β concentrations and stable isotope labeling kinetics of human plasma specific to central nervous system amyloidosis. Alzheimer’s Dement. 2017;13:841–849. doi: 10.1016/j.jalz.2017.06.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Li Y, Gordon BA, Holtzman DM, Morris JC, Benzinger TLS, Xiong C, et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology. 2019;93:e1647–e1659. doi: 10.1212/WNL.0000000000008081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmqvist S, Janelidze S, Stomrud E, Zetterberg H, Karl J, Zink K, Bittner T, Mattsson N, Eichenlaub U, Blennow K, et al. Performance of fully automated plasma assays as screening tests for Alzheimer disease-related β-amyloid status. JAMA Neurol. 2019;76:1060–1069. doi: 10.1001/jamaneurol.2019.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu JL, Lee WJ, Liao YC, Wang SJ, Fuh JL. The clinical significance of plasma clusterin and Aβ in the longitudinal follow-up of patients with Alzheimer’s disease. Alzheimer’s Res Ther. 2017; 9. 10.1186/s13195-017-0319-x. [DOI] [PMC free article] [PubMed]
- 9.Verberk IMW, Hendriksen HMA, van Harten AC, Wesselman LMP, Verfaillie SCJ, van den Bosch KA, Slot RER, Prins ND, Scheltens P, Teunissen CE, et al. Plasma amyloid is associated with the rate of cognitive decline in cognitively normal elderly: the SCIENCe project. Neurobiol Aging. 2020;89:99–107. doi: 10.1016/j.neurobiolaging.2020.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Giudici KV, de Souto Barreto P, Guyonnet S, Li Y, Bateman RJ, Vellas B. Assessment of plasma amyloid-β42/40 and cognitive decline among community-dwelling older adults. JAMA Netw open. 2020;3:e2028634. doi: 10.1001/jamanetworkopen.2020.28634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iulita MF, Ganesh A, Pentz R, Flores Aguilar L, Gubert P, Ducatenzeiler A, Christie S, Wilcock GK, Cuello AC. Identification and preliminary validation of a plasma profile associated with cognitive decline in dementia and at-risk individuals: a retrospective cohort analysis. J Alzheimer’s Dis. 2019;67:327–341. doi: 10.3233/JAD-180970. [DOI] [PubMed] [Google Scholar]
- 12.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [DOI] [PMC free article] [PubMed]
- 13.Diniz BS, Teixeira AL, Ojopi EB, Talib LL, Mendonça VA, Gattaz WF, Forlenza OV. Higher serum sTNFR1 level predicts conversion from mild cognitive impairment to Alzheimer’s disease. J Alzheimer’s Dis. 2010;22:1305–1311. doi: 10.3233/JAD-2010-100921. [DOI] [PubMed] [Google Scholar]
- 14.Gross AL, Walker KA, Moghekar AR, Pettigrew C, Soldan A, Albert MS, Walston JD. Plasma markers of inflammation linked to clinical progression and decline during preclinical AD. Front. Aging Neurosci. 2019; 11. 10.3389/fnagi.2019.00229. [DOI] [PMC free article] [PubMed]
- 15.Darweesh SKL, Wolters FJ, Ikram MA, de Wolf F, Bos D, Hofman A. Inflammatory markers and the risk of dementia and Alzheimer’s disease: a meta-analysis. Alzheimer’s Dement. 2018;14:1450–1459. doi: 10.1016/j.jalz.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 16.Conductier G, Blondeau N, Guyon A, Nahon JL, Rovère C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J Neuroimmunol. 2010;224:93–100. doi: 10.1016/j.jneuroim.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 17.Lee WJ, Liao YC, Wang YF, Lin IF, Wang SJ, Fuh JL. Plasma MCP-1 and cognitive decline in patients with Alzheimer’s disease and mild cognitive impairment: a two-year follow-up study. Sci Rep. 2018; 8. 10.1038/s41598-018-19807-y. [DOI] [PMC free article] [PubMed]
- 18.Bettcher BM, Neuhaus J, Wynn MJ, Elahi FM, Casaletto KB, Saloner R, Fitch R, Karydas A, Kramer JH. Increases in a Pro-inflammatory chemokine, MCP-1, are related to decreases in memory over time. Front Aging Neurosci. 2019; 10. 10.3389/fnagi.2019.00025. [DOI] [PMC free article] [PubMed]
- 19.Bettcher BM, Johnson SC, Fitch R, Casaletto KB, Heffernan KS, Asthana S, Zetterberg H, Blennow K, Carlsson CM, Neuhaus J, et al. Cerebrospinal fluid and plasma levels of inflammation differentially relate to CNS markers of Alzheimer’s disease pathology and neuronal damage. J Alzheimer’s Dis. 2018;62:385–397. doi: 10.3233/JAD-170602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moon JS, Goeminne LJE, Kim JT, Tian JW, Kim SH, Nga HT, Kang SG, Kang BE, Byun JS, Lee YS, et al. Growth differentiation factor 15 protects against the aging-mediated systemic inflammatory response in humans and mice. Aging Cell 2020, 19. 10.1111/acel.13195. [DOI] [PMC free article] [PubMed]
- 21.Chang JY, Hong HJ, Kang SG, Kim JT, Zhang BY, Shong M. The role of growth differentiation factor 15 in energy metabolism. Diabetes Metab J. 2020;44:363–371. doi: 10.4093/dmj.2020.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuchs T, Trollor JN, Crawford J, Brown DA, Baune BT, Samaras K, Campbell L, Breit SN, Brodaty H, Sachdev P, et al. Macrophage inhibitory cytokine-1 is associated with cognitive impairment and predicts cognitive decline - the Sydney Memory and Aging Study. Aging Cell. 2013;12:882–889. doi: 10.1111/acel.12116. [DOI] [PubMed] [Google Scholar]
- 23.McGrath ER, Himali JJ, Levy D, Conner SC, DeCarli C, Pase MP, Ninomiya T, Ohara T, Courchesne P, Satizabal CL, et al. Growth differentiation factor 15 and NT-proBNP as blood-based markers of vascular brain injury and dementia. J Am Heart Assoc. 2020;9:e014659. doi: 10.1161/JAHA.119.014659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, Barro C, Kappos L, Comabella M, Fazekas F, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14:577–589. doi: 10.1038/s41582-018-0058-z. [DOI] [PubMed] [Google Scholar]
- 25.Cullen NC, Leuzy A, Palmqvist S, Janelidze S, Stomrud E, Pesini P, Sarasa L, Allué JA, Proctor NK, Zetterberg H, et al. Individualized prognosis of cognitive decline and dementia in mild cognitive impairment based on plasma biomarker combinations. Nat Aging. 2021;1:114–123. doi: 10.1038/s43587-020-00003-5. [DOI] [PubMed] [Google Scholar]
- 26.Kaeser SA, Lehallier B, Thinggaard M, Häsler LM, Apel A, Bergmann C, Berdnik D, Jeune B, Christensen K, Grönke S, et al. A neuronal blood marker is associated with mortality in old age. Nat Aging. 2021;1:218–225. doi: 10.1038/s43587-021-00028-4. [DOI] [PubMed] [Google Scholar]
- 27.Vellas B, Carrie I, Gillette-Guyonnet S, Touchon J, Dantoine T, Dartigues JF, Cuffi MN, Bordes S, Gasnier Y, Robert P, et al. Mapt study: a multidomain approach for preventing Alzheimer’s disease: design and baseline data. J Prev Alzheimer’s Dis. 2014;1:13–22. doi: 10.14283/jpad.2014.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrieu S, Guyonnet S, Coley N, Cantet C, Bonnefoy M, Bordes S, Bories L, Cufi MN, Dantoine T, Dartigues JF, et al. Effect of long-term omega 3 polyunsaturated fatty acid supplementation with or without multidomain intervention on cognitive function in elderly adults with memory complaints (MAPT): a randomised, placebo-controlled trial. Lancet Neurol. 2017;16:377–389. doi: 10.1016/S1474-4422(17)30040-6. [DOI] [PubMed] [Google Scholar]
- 29.Morris JC. The clinical dementia rating (cdr): Current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 30.Donohue MC, Sperling RA, Salmon DP, Rentz DM, Raman R, Thomas RG, Weiner M, Aisen PS. The preclinical Alzheimer cognitive composite: measuring amyloid-related decline. JAMA Neurol. 2014;71:961–970. doi: 10.1001/jamaneurol.2014.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ngandu T, Lehtisalo J, Solomon A, Levälahti E, Ahtiluoto S, Antikainen R, Bäckman L, Hänninen T, Jula A, Laatikainen T, et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet. 2015;385:2255–2263. doi: 10.1016/S0140-6736(15)60461-5. [DOI] [PubMed] [Google Scholar]
- 32.Coley N, Gallini A, Ousset PJ, Vellas B, Andrieu S. Evaluating the clinical relevance of a cognitive composite outcome measure: an analysis of 1414 participants from the 5-year GuidAge Alzheimer’s prevention trial. Alzheimer’s Dement. 2016;12:1216–1225. doi: 10.1016/j.jalz.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 33.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein e and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harrell FE, Lee KL, Mark DB. Multivariable prognostic models: Issues in developing models, evaluating assumptions and adequacy, and measuring and reducing errors. Stat Med. 1996;15:361–387. doi: 10.1002/(SICI)1097-0258(19960229)15:4<361::AID-SIM168>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 35.Newson RB. Comparing the predictive powers of survival models using Harrell’s C or Somers’ D. Stata J. 2010;10:339–358. doi: 10.1177/1536867X1001000303. [DOI] [Google Scholar]
- 36.Matthew C. Pointwise confidence intervals for the covariate-adjusted survivor function in the Cox model. Stata J. 2011;11:64–81. doi: 10.1177/1536867X19830915. [DOI] [Google Scholar]
- 37.Burnham KP, Anderson DR. Model selection and multimodel inference: a practical information-theoretic approach. 2. New York: Springer; 2002. [Google Scholar]
- 38.Ky B, French B, Levy WC, Sweitzer NK, Fang JC, Wu AHB, Goldberg LR, Jessup M, Cappola TP. Multiple biomarkers for risk prediction in chronic heart failure. Circ Hear Fail. 2012;5:183–190. doi: 10.1161/CIRCHEARTFAILURE.111.965020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richter B, Koller L, Hohensinner PJ, Zorn G, Brekalo M, Berger R, Mörtl D, Maurer G, Pacher R, Huber K, et al. A multi-biomarker risk score improves prediction of long-term mortality in patients with advanced heart failure. Int J Cardiol. 2013;168:1251–1257. doi: 10.1016/j.ijcard.2012.11.052. [DOI] [PubMed] [Google Scholar]
- 40.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Maurik IS, Zwan MD, Tijms BM, Bouwman FH, Teunissen CE, Scheltens P, Wattjes MP, Barkhof F, Berkhof J, Van Der Flier WM. Interpreting biomarker results in individual patients with mild cognitive impairment in the Alzheimer’s Biomarkers in Daily Practice (ABIDE) project. JAMA Neurol. 2017;74:1481–1491. doi: 10.1001/jamaneurol.2017.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Maurik IS, Vos SJ, Bos I, Bouwman FH, Teunissen CE, Scheltens P, Barkhof F, Frolich L, Kornhuber J, Wiltfang J, et al. Biomarker-based prognosis for people with mild cognitive impairment (ABIDE): a modelling study. Lancet Neurol. 2019;18:1034–1044. doi: 10.1016/S1474-4422(19)30283-2. [DOI] [PubMed] [Google Scholar]
- 43.Wallace LMK, Theou O, Godin J, Andrew MK, Bennett DA, Rockwood K. Investigation of frailty as a moderator of the relationship between neuropathology and dementia in Alzheimer’s disease: a cross-sectional analysis of data from the Rush Memory and Aging Project. Lancet Neurol. 2019;18:177–184. doi: 10.1016/S1474-4422(18)30371-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mitchell AJ, Shiri-Feshki M. Rate of progression of mild cognitive impairment to dementia - meta-analysis of 41 robust inception cohort studies. Acta Psychiatr Scand. 2009;119:252–265. doi: 10.1111/j.1600-0447.2008.01326.x. [DOI] [PubMed] [Google Scholar]
- 45.Buchhave P, Minthon L, Zetterberg H, Wallin ÅK, Blennow K, Hansson O. Cerebrospinal fluid levels of β-amyloid 1–42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry. 2012;69:98–106. doi: 10.1001/archgenpsychiatry.2011.155. [DOI] [PubMed] [Google Scholar]
- 46.Chouraki V, Beiser A, Younkin L, Preis SR, Weinstein G, Hansson O, Skoog I, Lambert JC, Au R, Launer L, et al. Plasma amyloid-β and risk of Alzheimer’s disease in the Framingham Heart Study. Alzheimer’s Dement. 2015;11:249–257.e1. doi: 10.1016/j.jalz.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blasko I, Jellinger K, Kemmler G, Krampla W, Jungwirth S, Wichart I, Tragl KH, Fischer P. Conversion from cognitive health to mild cognitive impairment and Alzheimer’s disease: prediction by plasma amyloid beta 42, medial temporal lobe atrophy and homocysteine. Neurobiol Aging. 2008;29:1–11. doi: 10.1016/j.neurobiolaging.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 48.Sokolova A, Hill MD, Rahimi F, Warden LA, Halliday GM, Shepherd CE. Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in alzheimer’s disease. Brain Pathol. 2009;19:392–398. doi: 10.1111/j.1750-3639.2008.00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, Herzog EL, Kang I, Pisani MA, Yu S, et al. GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell. 2019;178:1231–1244.e11. doi: 10.1016/j.cell.2019.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wollert KC. Growth-differentiation factor-15 in cardiovascular disease. Basic Res Cardiol. 2007;102:412–415. doi: 10.1007/s00395-007-0662-3. [DOI] [PubMed] [Google Scholar]
- 51.Baek SJ, Eling T. Growth differentiation factor 15 (GDF15): a survival protein with therapeutic potential in metabolic diseases. Pharmacol Ther. 2019;198:46–58. doi: 10.1016/j.pharmthera.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corre J, Hébraud B, Bourin P. Concise review: growth differentiation factor 15 in pathology: a clinical role? Stem Cells Transl Med. 2013;2:946–952. doi: 10.5966/sctm.2013-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tavenier J, Rasmussen LJH, Andersen AL, Houlind MB, Langkilde A, Andersen O, Petersen J, Nehlin JO. Association of GDF15 with inflammation and physical function during aging and recovery after acute hospitalization: a longitudinal study of older patients and Age-matched controls. Journals Gerontol Ser A. 2021;76:964–974. doi: 10.1093/gerona/glab011. [DOI] [PubMed] [Google Scholar]
- 54.Jiang J, Wen W, Sachdev PS. Macrophage inhibitory cytokine-1/growth differentiation factor 15 as a marker of cognitive ageing and dementia. Curr Opin Psychiatry. 2016;29:181–186. doi: 10.1097/YCO.0000000000000225. [DOI] [PubMed] [Google Scholar]
- 55.Fujita Y, Taniguchi Y, Shinkai S, Tanaka M, Ito M. Secreted growth differentiation factor15 as a potential biomarker for mitochondrial dysfunctions in aging and age-related disorders. Geriatr Gerontol Int. 2016;16:17–29. doi: 10.1111/ggi.12724. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.