

Summary
While somatic mutations are known to be enriched in genome regions with non-canonical DNA secondary structure, the impact of particular mutagens still needs to be elucidated. Here, we demonstrate that in human cancers, the APOBEC mutagenesis is not enriched in direct repeats, mirror repeats, short tandem repeats, and G-quadruplexes, and even decreased below its level in B-DNA for cancer samples with very high APOBEC activity. In contrast, we observe that the APOBEC-induced mutational density is positively associated with APOBEC activity in inverted repeats (cruciform structures), where the impact of cytosine at the 3’-end of the hairpin loop is substantial. Surprisingly, the APOBEC-signature mutation density per TC motif in the single-stranded DNA of a G-quadruplex (G4) is lower than in the four-stranded part of G4 and in B-DNA. The APOBEC mutagenesis, as well as the UV-mutagenesis in melanoma samples, are absent in Z-DNA regions, owing to the depletion of their mutational signature motifs.
Subject areas: cancer, cancer mutagenesis
Graphical abstract
Highlights
-
•
APOBEC mutagenesis is not enriched in most non-canonical DNA structures
-
•
Inverted repeats (cruciform structures) show increased APOBEC mutagenesis
-
•
G-quadruplex’s unstructured strand has low APOBEC-induced mutation density
-
•
Decrease of APOBEC mutagenesis in non-B DNA possibly associated with PrimPol
Cancer; Cancer mutagenesis.
Introduction
Recent studies of human cancer genomes revealed a significant role of the APOBEC (Apolipoprotein B mRNA Editing Catalytic polypeptide-like) family cytidine deaminases in cancer mutagenesis (Burns et al., 2013a; Nik-Zainal et al., 2012; Roberts et al., 2012). The APOBEC-family enzymes are a part of the human immune system acting against viruses and transposable elements (Salter et al., 2016). APOBEC cytidine deaminases change the substrate DNA by binding to single-stranded DNA regions and deaminating cytosines in the TpC context, leading to C→T and C→G substitutions (Shi et al., 2017). Positional clusters of somatic mutations with the APOBEC mutational signature were found in many types of cancers, in particular, breast, lung, bladder, head/neck, and cervical cancers (Alexandrov et al., 2013; Burns et al., 2013b; Roberts et al., 2013). It has been suggested that these mutation clusters arise when APOBEC binds to and slides along single-stranded DNA (ssDNA) accessible during replication, transcription, or double-strand breaks (Yang et al., 2017). Recent studies support a link between these ssDNA-generated processes and APOBEC mutagenesis in the human cell (Saini and Gordenin, 2020).
Several types of mutational heterogeneity along the genome, presumably associated with replication and transcription (Haradhvala et al., 2016), were identified for APOBEC mutagenesis. An increased density of APOBEC-induced mutations was found in early-replicating genome regions (Kazanov et al., 2015), as opposed to other types of cancer mutagenesis, and in highly transcribed genes (Chervova et al., 2021). Higher rate of APOBEC-induced mutations was also observed on the lagging replicating DNA strand (Seplyarskiy et al., 2016) and the non-transcribed DNA strand (Chervova et al., 2021).
It is known that ssDNA, a preferred APOBEC substrate, can adopt diverse local conformations such as hairpins, loops, and pseudoknots (Zhang et al., 2001). A recent study has shown that cytosine at the 3’-end of a hairpin loop is a hotspot of APOBEC and can even be targeted by APOBEC while not being preceded by thymine (Langenbucher et al., 2021), essentially forming a second type of the APOBEC signature, dependent on the ssDNA secondary structure. Various forms of the ssDNA secondary structure have been found in the human genome regions with non-canonical DNA structure, that is, distinct from the right-handed DNA double-helix (Zhao et al., 2010).
To date, about ten types of non-B DNA structures are known, including hairpins/cruciform, triplexes (H-DNA), tetraplexes, slipped DNA, and Z-DNA. The cruciform structures are formed by inverted repeats (Brázda et al., 2011; Gordenin et al., 1993) that base-pair, forming an intrastrand hairpin stem and looping out the spacer between the repeat copies as ssDNA (Figure 1). Thus, the cruciform structure consists of two hairpin-loop arms and a four-way junction. The triplex DNA (H-DNA) structures can form at mirror repeats (Frank-Kamenetskii and Mirkin, 1995), where ssDNA can bind in the major groove of the underlying DNA duplex forming a three-stranded helix (Figure 1). The four-stranded G-quadruplex structure is a co-planar array of four guanines formed by guanine-rich DNA stretches (Spiegel et al., 2020) (Figure 1). The slipped strand DNA structures are formed when one strand of one copy of direct repeat pairs with the complementary strand of another copy of a direct repeat (Sinden et al., 2007), yielding looped-out ssDNA (Figure 1). Sequences with an abundance of alternating purines and pyrimidines may form the double helix with a left-handed zigzag pattern called Z-DNA (Ravichandran et al., 2019) (Figure 1). A-phased repeats are segments of consecutive adenines or thymines separated by 10 nucleotides, associated with DNA bending (Figure 1). As can be seen, most non-B DNA structures contain stretches of ssDNA, which might be expected to be an efficient substrate for the APOBEC enzymes.
Figure 1.

Enrichment of somatic single-base substitutions (SBS) observed in various types of non-canonical DNA structure genome regions and different cancers
Single dot corresponds to a particular cancer sample, with the vertical position indicating the log-ratio of the mutational density in non-B DNA structure genome regions to the mutational density in B-DNA genome regions.
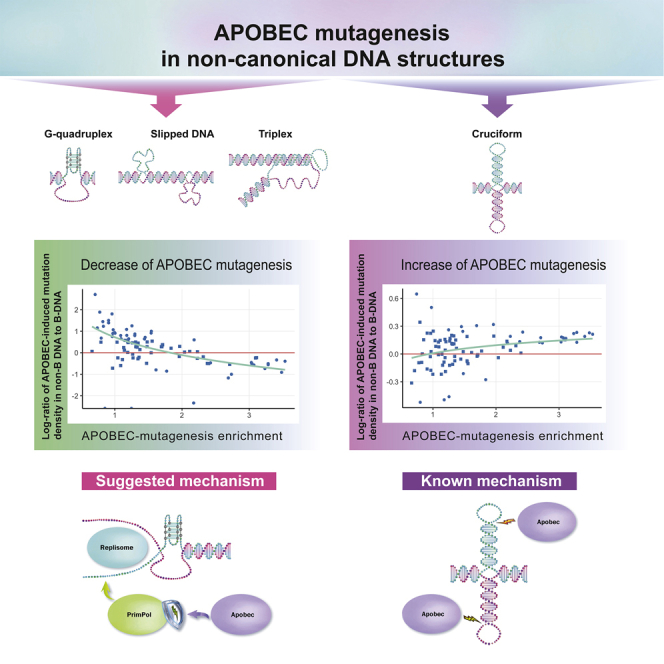
Studies on cancer mutagenesis in non-B DNA genome regions showed an increased density of somatic mutations compared to the genome regions with conventual double-helix DNA structure (Georgakopoulos-Soares et al., 2018). Recent advances in the research on cancer mutagenesis have demonstrated that it is possible to assess the impact of particular mutagens using their mutational signatures. Here, we used the known mutational signature of APOBEC enzymes and analyzed the impact of the APOBEC-related mutagenesis in various types of non-B DNA genome structures, focusing on ssDNA regions. We found that, despite the presence of ssDNA in most non-B DNA structures, the APOBEC mutagenesis is not enriched in most non-B DNA structures, an exception being inverted repeats, and even is relatively lower in cancer samples with very high APOBEC activity.
Results
Single-base substitutions are generally enriched in non-B DNA structures in human cancers
To obtain a general view of somatic mutagenesis in non-B DNA structures in human cancer and use it further as a baseline for comparison with the APOBEC mutagenesis, we first calculated the densities of all somatic single-base substitutions (SBS) in non-B and B-DNA genome regions using mutational data from the Pan-Cancer Analysis of Whole Genomes (PCAWG) project (Campbell et al., 2020). Seven types of Non-B DNA structures were considered using genomic coordinates of non-B DNA motifs from the non-B DB (Cer et al., 2013): direct repeats (DR), inverted repeats (IR), mirror repeats (MR), short tandem repeats (STR), G-quadruplexes (G4), A-phased repeats (APR), and Z-DNA. As was observed earlier for a smaller cancer dataset (Georgakopoulos-Soares et al., 2018), the density of somatic SBS is usually enriched in non-B DNA structures. Here, we used more than 1500 cancer samples from 41 cancer types and observed a similar enrichment with some exceptions (Figure 1). From the non-B DNA motif axis, the most prominent exception is that A-phased repeats are not associated with the enrichment of somatic SBS density: the mean fold enrichment among cancers was not statistically different from the zero enrichment (the Mann-Whitney-Wilcoxon test, p-value = 0.34). The same trend was observed earlier for germline mutations (Guiblet et al., 2021). From the cancer axis, the most striking deviation was the decrease of mutational density in the Z-DNA motif for melanoma: by a factor of 2.16 (p = 9.1 × 10−7) for the MELA cancer type and by a factor of 3.06 (p = 4.8 × 10−8) for the SKCM cancer type. We have analyzed these deviations in more detail later in discussion (see the respective subsection). In all other cases, the mutational density was enriched in non-B DNA motifs: the mean of the mean fold enrichment over all cancer types was 3.0 for DR (p = 1.4×10−14), 2.7 for G4 (p = 7.1×10−14), 1.4 for IR (p = 1.4×10−14), 2.1 for MR (p = 1.4×10−14), 2.4 for STR (p = 1.4×10−13), and 1.9 for Z-DNA (p = 4.2 × 10−9). We also calculated enrichment in non-B DNA structures across all cancers separately for mutations attributed to the APOBEC signature TpC motif (Figure S1). The most noticeable difference compared to all mutations was the statistical insignificance of the enrichment (p = 0.06) in IR genome regions.
APOBEC mutagenesis is not enriched in non-canonical DNA structures, unlike other types of mutagenesis, inverted repeats being an exception
To elucidate the characteristics of APOBEC mutagenesis in non-canonical DNA genome regions, we estimated the density of APOBEC-induced mutations for each type of non-canonical DNA structure in each cancer sample. We used the known APOBEC mutational signature TpC to extract mutations presumably associated with the APOBEC enzymes, excluding CpG-island regions, where APOBEC mutagenesis can overlap with the hypermutation of methylated CpG sites. We first calculated the number of APOBEC-signature motifs, i.e., potential APOBEC targets, in each type of non-B DNA structure (Figure S2A). The fraction of TpC motifs for most types of non-B DNA structures was in the range of 0.07–0.12 except for Z-DNA, where this fraction was negligibly small, 0.008. Thus, we observed a depletion of the APOBEC-signature motif, TpC, in Z-DNA and have not further analyzed the characteristics of the APOBEC mutagenesis in this type of non-B DNA structure.
For the remaining six types of non-canonical DNA structures, we calculated the density of APOBEC-induced mutations in cancer samples and compared it with the density of APOBEC-induced mutations in B-DNA genome regions in these samples. In four out of six non-B DNA motif types—DR, STR, MR, and G4—we observed characteristics of APOBEC mutagenesis, which are different from the ones observed for other types of mutagenesis: the APOBEC-signature mutation density in non-canonical DNA genome regions was approximately equal to the density in B-DNA genome regions with the increase of APOBEC activity. Moreover, some cancer samples with a very high level of APOBEC mutagenesis demonstrated a lower density of APOBEC-signature mutations in non-canonical DNA genome regions compared to B-DNA genome regions. We also found that most APOBEC-associated mutations in these samples satisfied the APOBEC3A-like mutational signature (Campbell et al., 2017; Chan et al., 2015) (Figure S11).
More specifically, Figures 2A and S3 show how the log-ratio of the density of APOBEC-induced mutations within particular non-canonical DNA structure genome regions to the density of APOBEC-induced mutations in B-DNA genome regions depends on the activity of APOBEC mutagenesis in the samples. The activity of APOBEC mutagenesis for each cancer sample was estimated by the APOBEC enrichment as before (Roberts et al., 2013). It can be seen that samples with low APOBEC activity have an increased density of APOBEC-signature mutations in comparison with its density in B-DNA. This agrees with the fact that the density of somatic mutations in human cancer in non-canonical DNA motifs is generally higher than in canonical regions, as confirmed by PCAWG data in the previous section. It should be noted that most mutations with the APOBEC signature in these samples are likely associated with non-APOBEC mutagenesis, as the absence of APOBEC enrichment reflects a low level of APOBEC mutagenesis. Meanwhile, Figures 2A and S3 show that as the APOBEC activity increases, the APOBEC-signature mutation density in the four non-canonical DNA genome regions (DR, STR, MR, and G4) decreases to the level of the APOBEC-induced mutational density in B-DNA genome regions and to even lower level for cancer samples highly enriched in the APOBEC-signature mutations. The strongest effect was observed for the G4 structure.
Figure 2.

Comparison of the activity of APOBEC mutagenesis in non-B and B-DNA genome regions
(A) Dependence of the log-ratio of APOBEC-induced mutation densities in non-B DNA to B-DNA genome regions on the activity of APOBEC mutagenesis in cancer samples. Point shape—round or square—corresponds to significant or insignificant statistical differences between two densities in a particular cancer sample, respectively.
(B) Difference in the distribution of the log-ratio of APOBEC-induced mutation densities in non-B DNA to B-DNA genome regions in cancer samples with low (APOBEC enrichment < 2.0) and high (APOBEC enrichment > 2.0) APOBEC activity. Data are represented as box plots displaying minimum, first quartile, median, third quartile, and maximum values. (3) APOBEC-induced and other mutation densities in non-B and B-DNA genome regions for bladder carcinoma (BLCA) samples. Wilcoxon-Mann-Whitney test notation: ∗∗∗ – p-value < 0.001, ∗∗ – p-value < 0.01, ∗ – p-value < 0.05.
The behavior of APOBEC mutagenesis in IR regions was completely different. Figures 2A and S3 show that the density of APOBEC-induced mutations in IR genome regions increased with increasing APOBEC activity in cancer samples faster than the density of APOBEC mutagenesis in B-DNA genome regions. The effects observed for five of the considered non-canonical DNA structure types were statistically significant (Figure 2B). In the remaining APR motif, we observed no enrichment of the APOBEC mutagenesis compared to B-DNA genome regions at all levels of activity of APOBEC mutagenesis in cancer samples (Figure S3). To assess the observed differences between the APOBEC mutation density in the non-canonical DNA structures and in B-DNA across the cancer samples, we calculated and visualized these densities in each sample (Figures 2C, S5 and S6).
We also compared the distribution of APOBEC-induced mutation clusters (Sakofsky et al., 2019) in non-B and B-DNA genome regions and did not find any statistically significant differences (data not shown). Additionally, the total size of APOBEC-enriched mutation clusters in a cancer sample could serve as an estimate of the fraction of hypermutable ssDNA in the genome formed during the repair of double-strand breaks (DSB) (Sakofsky et al., 2019). Thus, we analyzed the dependence of the density of APOBEC-induced mutations in non-B genome regions on the total size of APOBEC-enriched mutation clusters. As the total cluster size increased, we observed the same trends as described above: relative decrease of APOBEC-induced mutation density in non-B DNA structures to the level in B-DNA and even below for DR, STR, MR, G4, and increase of the APOBEC-induced mutation density in IR (Figure S4).
Observed effects are not the result of the heterogeneity of APOBEC mutagenesis and non-B DNA structures along the genome
Then, we verified that the observed effects did not result from the known heterogeneity of APOBEC-induced mutations along the replication timing (Kazanov et al., 2015), that is, the increased density of APOBEC-induced mutations in early-replicating regions and the decreased density in late-replicated regions. First, we calculated the distribution of APOBEC targets (TpC motifs) contained in the considered non-B DNA structures along the replication timing (Figure 3A). All types of non-B DNA structures, except for A-phased repeats, showed either an almost uniform distribution (MR and IR) or a distribution skewed towards an increased fraction of TpC motifs, residing in non-canonical DNA structures, in early-replicating genome regions (DR, STR, G4). In contrast, TpC motifs contained in A-phased repeats were enriched in late-replicating regions.
Figure 3.

Independence of the observed effects on the replication timing domains
(A) Distribution of the non-B DNA structures along the replication timing. Data are represented as mean ± SEM.
(B) Dependence of the log-ratio of APOBEC-induced mutation densities in G-quadruplex to B-DNA genome regions on the activity of APOBEC mutagenesis in different replication timing bins for the BRCA cancer.
If the effects for DR, STR, MR, and G4, described in the previous section, were the result of APOBEC mutagenesis heterogeneity relative to the replication timing, we should expect a reduction in the number of non-B structures in the regions of enriched APOBEC mutagenesis, i.e., in early-replicating genome regions. However, we did not observe that for these four types of non-B DNA structures. For additional evidence, we calculated the APOBEC-signature mutation density in non-canonical DNA genome regions dividing the genome into seven separate replication timing bins, from the early to the late replication timing. Figure 3B shows the results for G-quadruplexes, which have the most skewed distribution toward early-replicating regions, in the BRCA cancer, the cancer type having the largest number of samples. In this example and for the majority of other non-B DNA structures (data not shown), the observed effect of relative decrease of the APOBEC mutagenesis in most non-B regions as the APOBEC activity increases, is visible for each replication bin, i.e., in all sets of genome regions with approximately same replication timing. Thus, we conclude that the observed effects are not a consequence of the mutational heterogeneity of the APOBEC mutagenesis along the replication timing.
Increase of APOBEC mutagenesis in inverted repeats, apparently associated with DNA secondary structure
To understand possible causes of the observed characteristics of APOBEC mutagenesis in non-B DNA genome regions, we analyzed the distribution of APOBEC-induced mutations relative to the secondary structure of non-B DNA motifs. We first analyzed the distribution of APOBEC-induced mutations in IR motifs. IR genome regions form hairpin-loop arms on both DNA strands. We stratified detected APOBEC-induced mutations into the ones that occur in the hairpin stem and in the hairpin loop. Following recent reports on the propensity of APOBEC enzymes toward cytosine located at the 3’-end of the hairpin loop (Buisson et al., 2019; Langenbucher et al., 2021) we also further divided the hairpin loop mutations into two categories, ones occurring in the cytosine at the 3’-end of a loop (here and further 3’LEC (3’-loop end cytosine)) and all other positions of the hairpin loop (Figure 4A).
Figure 4.

APOBEC mutagenesis in different parts of the cruciform structure
(A) Different parts of the IR motif’s secondary structure: stem, loop, and the cytosine at the 3’-end of the hairpin loop.
(B and C) Distribution of APOBEC-induced mutation densities at TpC motifs in different parts of the IR secondary structure in cancer samples with high (B) and low (C) APOBEC activity. Data are represented as box plots displaying minimum, first quartile, median, third quartile, and maximum values.
(D) Contribution of mutations at the cytosine at the 3’-end of the IR hairpin loop to the overall IR motif APOBEC-induced mutation load. Plots for different cancer types demonstrate the dependency of the APOBEC-induced mutation density per base pair on the activity of APOBEC mutagenesis for real and simulated data. In simulated data, the APOBEC-induced mutation density at the cytosine at the 3’-end of the IR hairpin loop was replaced by the average APOBEC-induced mutation density in the IR hairpin stem and the rest of the hairpin loop. Wilcoxon-Mann-Whitney test notation: ∗∗∗ – p-value < 0.001, ∗∗ – p-value < 0.01, ∗ – p-value < 0.05.
Figure 4B shows that the density of APOBEC-induced mutations in IR genome regions in the 3’LECs is significantly higher than the density in the loops and stems observed in samples with high APOBEC activity (APOBEC enrichment > 2.0). Figure 4C shows that this effect is also less prominent but still observable in samples with low APOBEC activity (APOBEC enrichment <2.0). Surprisingly, we still observed this effect when we extended the analysis to cancer types not associated with APOBEC mutagenesis (Figure S7). This finding may reflect a presence of APOBEC mutagenesis in these cancer types, although at a low level.
To elucidate the impact of APOBEC-induced mutagenesis at the 3’-end of IR hairpin loops on the APOBEC-mutagenesis of the whole IR genome regions, we recalculated the mutation density by removing mutations at 3’LEC positions and replaced them with the average density of APOBEC-signature mutations in the IR hairpin loop and stem. Figure 4D shows that the increased density of APOBEC-signature mutagenesis at 3’LEC significantly impacts the accumulation of APOBEC-induced mutations in IR genome regions (p-values of the linear regression coefficients <0.05 for four out of six cancer types). Thus, if the density of APOBEC-induced mutations at 3’LEC were not increased, then the overall density of APOBEC mutagenesis in IR regions would be approximately the same as in B-DNA regions (coefficients of the linear regression are not significantly different from zero), while actually it increases in comparison with the APOBEC-induced density in B-DNA with increasing of APOBEC activity.
According to (Buisson et al., 2019), the 3’LEC can be mutated by APOBEC even when it is not preceded by thymine, i.e., outside of the APOBEC’s mutational signature TpC. Therefore, we repeated the analysis for С->T and С->G mutations in the VpС motifs, comparing the mutation densities between non-B and B-DNA regions. We found much less prominent, but still observable trends for IR and other non-B DNA structures (Figure S10).
Single-stranded DNA complementary to the G-quadruplex four-stranded structure is not a favorable target of APOBEC enzymes
We also compared the frequency of APOBEC-induced mutations in G-quadruplex genome regions in the DNA strand with a four-stranded guanine-rich structure and in the complementary, unstructured DNA strand. Firstly, we calculated the density of TpC motifs in these two strands (Figure 5A). As expected, the DNA strand of G4 with a four-stranded structure showed a reduced number of TpC motifs in comparison with the average density of TpC motifs in B-DNA genome regions, as a larger fraction of the nucleotide sequence was guanines. On the other hand, the density of TpC motifs on the opposite strand was almost two-fold higher than the average density of TpC motifs in B-DNA. This increase was obviously owing to the stretches of cytosines complementary to guanines in the guanine-rich strand.
Figure 5.

APOBEC mutagenesis in different parts of the G-quadruplex structure
(A) Comparison of the densities of TpC motifs per base pair on both strands of G-quadruplex structures and B-DNA.
(B and C) Distribution of APOBEC-induced mutational densities at TpC motifs on both strands of G-quadruplex structures in cancer samples with high (B) and low (C) APOBEC activity. Data are represented as box plots displaying minimum, first quartile, median, third quartile, and maximum values.
(D) Impact of both strands of G-quadruplex structures on the APOBEC-induced mutation density per base pair in G4 genome regions.
Then, we calculated the densities of APOBEC-induced mutations in TpC motifs in both G4-motif strands. Surprisingly, the density of APOBEC-induced mutations in TpC motifs in the unstructured DNA strand of G4, which seemed to be a favorable APOBEC substrate, turned out to be smaller than the density in the guanine-rich strand. Notably, these differences in densities were observed in samples with both high activity of APOBEC mutagenesis (APOBEC enrichment >2.0, Figure 5B) and low activity (APOBEC enrichment <2.0, Figure 5C), and hence possibly represent an effect independent of the activity of the APOBEC mutagenesis. Moreover, in samples with high APOBEC activity, the density of APOBEC-induced mutations in the unstructured DNA strand of G4 was constantly smaller than the density in canonical double-strand DNA (Figures 5B and 5C), which also showed that this single-stranded DNA was not an optimal substrate of the APOBEC enzymes. At the same time, owing to a small number of TpC motifs in the guanine-rich strand, its impact on the overall APOBEC-induced mutational density per base pair in G4 genome regions is small in comparison with the impact of the unstructured DNA strand of G4 (Figure 5D).
According to several studies (Barzak et al., 2019; Byeon et al., 2016), APOBEC3A – a member of the APOBEC family, which was repeatedly associated with cancer mutagenesis – is also capable of deaminating the 3’-end cytosine in the CCC motif (the mutated nucleotide is underlined). To estimate the presumable APOBEC3A activity in C-rich sequences, including the unstructured strand of G-quadruplexes, we calculated the mutation densities of C→T or C→G substitutions in CCCA and CCCG motifs (Figures S13 and S14). We have not found any increase in the mutation density in C-rich regions of G-quadruplexes in samples with the prevalence of APOBEC3A-like mutations, which also supports the observed reduction of APOBEC mutagenesis in most non-B DNA structures.
We have also checked whether the APOBEC-induced mutation density in G4-motif strands depends on the direction of DNA replication (Figure S9). This analysis was possible for G4 as this non-B DNA structure is the only asymmetric structure among the ones considered in this study. We did not find a statistically significant dependence (chi-squared test) between the APOBEC-induced mutational density in the G4 structure and the DNA replication strand that is were different from the known replication strand bias of APOBEC-induced mutations in B-DNA (Seplyarskiy et al., 2016).
Following the reports on the localization of G-quadruplexes (Qiao et al., 2017; Xu et al., 2020) of another member of the AID/APOBEC family enzymes - activation-induced cytidine deaminase (AID) - we have also analyzed the AID-signature mutation density in G4 and other non-B DNA genome regions (Figure S12). We found an increased AID-signature mutation density in direct repeats for the AID-signature WRCY motif and in short tandem repeats for the sub-motif TACY, but not in the G4 structures.
Purine-pyrimidine alternations in Z-DNA lead to the absence of the APOBEC- and UV-mutageneses in this DNA structure
As described above, we observed a depletion of TpC motifs in Z-DNA genome regions (Figure S2A) apparently owing to the purine-pyrimidine alternations specific to the Z-DNA sequence (Ravichandran et al., 2019; Rich and Zhang, 2003). The latter leads to a nearly complete absence of the APOBEC-mutagenesis in Z-DNA genome regions. Although analyzing the density of somatic mutations in non-B regions in human cancers (Figure 1), we also found a prominent absence of the enrichment of somatic mutations in Z-DNA of skin cancers (MELA, SKCM). Using the UV-mutagenesis mutational signature, we calculated the density of UV-target motifs (TpC, CpC) and UV-associated C→T mutations. Similar to the APOBEC mutagenesis, we found that the apparent reason for the absence of UV-associated mutagenesis in Z-DNA is a depletion of UV-signature targets (Figure S2B). Indeed, the fractions of TpC and CpC di-nucleotides in Z-DNA genome regions were 14- and 7-fold smaller than the fractions of the same di-nucleotides in B-DNA, respectively. Moreover, the density of UV-associated mutations per UV-target motifs was also decreased in Z-DNA in comparison with B-DNA (Figure S2C, PMELA, TC = 2.2×10−10, PMELA, CC = 2.1×10−15, PSKCM, TC = 3.7 × 10−5, PSKCM, TC = 1.5 × 10−7).
Discussion
Rapid accumulation of mutation data for cancer genomes provides an excellent opportunity to study mutational processes in human cancer and their heterogeneity along the genome. Understanding the mutational heterogeneity along the genome is an essential component in computational methods for the identification of cancer-associated genes (Lawrence et al., 2013). It has been found earlier that genome regions with non-canonical DNA structures such as G-quadruplex, cruciform, various types of repeats, and Z-DNA, which cover together about 10% of the human genome, usually have an increased density of somatic(Georgakopoulos-Soares et al., 2018) and germline (Guiblet et al., 2021) mutations. At the same time, the classification of mutations into mutagen-related classes based on their nucleotide context using so-called mutational signatures in many cases allows for deciphering the individual impact of endogenous and exogenous mutagens. Here, we analyzed the activity of mutagenesis induced by APOBEC enzymes in non-B DNA genome regions, compared its level with the APOBEC mutagenesis in B-DNA, and elucidated the impact of structural parts of non-B DNA structures, focusing on ssDNA regions.
Using the large PCAWG dataset of mutations in cancer genomes, we confirmed that, in general, the density of somatic mutations is relatively larger in non-B DNA genome regions, as was found in other datasets of cancer mutagenesis (Georgakopoulos-Soares et al., 2018; McKinney et al., 2020). With most of the mutagenic sources, mutations in cancer genomes originate from either misincorporation during DNA copying or from unrepaired lesions in dsDNA. Within this paradigm, the increased density of mutations in non-B DNA could be explained by a higher rate of replication errors or/and by lower access to error-prone DNA repair systems. We unexpectedly found that for four out of seven considered non-B DNA structures (DR, STR, MR, G4), the density of APOBEC-signature mutations decreases to the level in B-DNA as the APOBEC activity in cancer samples increases. Moreover, in samples with very high activity of APOBEC mutagenesis, the APOBEC mutational density in these non-B DNA structures was even lower than the density in B-DNA genome regions. We found that, in these samples, the largest fraction of APOBEC-associated mutations has the APOBEC3A-like mutational signature, suggesting that the observed effect is possibly APOBEC3A-specific.
We speculate that the observed tendency to a similarity in APOBEC-induced mutation densities in B- and non-B DNA genome regions can be explained by the unique association of APOBEC mutagenesis with ssDNA, which can be generated during replication, double-strand break repair, and transcription (Saini and Gordenin, 2020). Once APOBEC deaminates a cytosine in ssDNA, it can be accurately returned to nonmutant sequence by base-excision repair if the complementary strand is available as a repair template. However, if ssDNA is persistent, i.e., does not have access to the complementary template for accurate repair, the deaminated cytosine will be fixed into mutation in the next rounds of replication (Saini and Gordenin, 2020). We propose that both non-B DNA and B-DNA sequences are equally prone to generating persistent ssDNA. In the case of non-B DNA structures, unwinding could be aided by special helicases (Sharma, 2011).
Moreover, a lower density of APOBEC mutations in non-B-DNA as compared to B-DNA regions that we observed in cancer samples with very high levels of APOBEC mutagenesis can also be explained in connection with the requirement of persistent ssDNA substrate for mutagenesis if persistent ssDNA formed in non-B DNA regions is less accessible by APOBEC deaminases compared to ssDNA formed in B-DNA. It is well established that non-B DNA structures cause replication stalling and require special systems aiding to replicate these regions (Wang and Vasquez, 2017). This could lead to higher recruitment of replication protein A (RPA), which binds to ssDNA and is known to counteract APOBEC deamination (Brown et al., 2021; Wong et al., 2021). An alternative or an additional explanation could be recruiting for the replicating of non-B DNA regions the specific DNA polymerase called Primase-Polymerase (PrimPol). This polymerase is known to shield DNA from APOBEC/AID mutagenesis (Pilzecker et al., 2016). PrimPol is known for its implication in eukaryotic DNA damage tolerance (Bailey et al., 2019), displaying both translesion synthesis and (re)-priming properties (Bainbridge et al., 2021). PrimPol is required for replicating G-quadruplexes (Schiavone et al., 2016) and presumably other types of non-canonical DNA structures (Šviković et al., 2019). It has been shown that PrimPol prevents mutagenesis of abasic sites induced by APOBEC/AID by repriming downstream of AP-sites on the leading strand, prohibiting error-prone TLS, and simultaneously stimulating error-free homology-directed repair (Pilzecker et al., 2016).
Despite this observed general trend, we found the opposite effect for the APOBEC mutagenesis in IR genome regions—the density of APOBEC-induced mutations increased with increasing APOBEC activity in cancer samples. We showed that a substantial part of this increase could be attributed to the mutagenesis in cytosine at the 3′-end of the IR hairpin loop, which was recently identified as the APOBEC’s hotspot (Buisson et al., 2019). As this report also provides the evidence that, at this hotspot, APOBEC can mutate cytosine not preceded by thymine, we repeated our analysis for the VpC instead of TpC motif and found similar but much less prominent trends for inverted repeats and other non-B DNA motifs. This supports the assumption that VpC motifs also harbor APOBEC-induced mutations at 3’LEC-based hotspots in IRs and suggest the existence of other types of APOBEC-hotspots in VpC motifs. Furthermore, our analysis of cancer types not associated with APOBEC mutagenesis detected a substantially higher density of mutations in 3’LEC hotspots, possibly reflecting some level of APOBEC mutagenesis in these cancer types.
We speculate that the increase of APOBEC mutagenesis in IR regions may be associated with double-strand breaks (Roberts et al., 2012) induced by the replication stalling at cruciform DNA structures (Lu et al., 2015), as it is known that 5’ ends of these breaks are resected to generate 3’-protruding ssDNA regions for subsequent repair process (Ceccaldi et al., 2016).
Apart from considering non-B DNA regions as a whole, we also analyzed when possible the distribution of mutation densities between the dsDNA and ssDNA parts of non-canonical DNA structures. Thus, in the cruciform structure (IR motif), the cytosine at the 3’-end of the IR hairpin loop showed the largest APOBEC-induced mutational density in comparison with the densities in the IR hairpin stem and other parts of the loop. We also focused on the large stretches of presumably single-stranded DNA opposite to the G-quadruplex's guanine-rich strand. Surprisingly, we found that APOBEC-induced mutational density per TpC motif in this ssDNA is less than the density both in the guanine-rich strand of the G-quadruplex and in B-DNA. We suggest that a reason for that could be the occupation of this ssDNA by other single strand-binding proteins (Kang et al., 2014; Lacroix et al., 2000) or formation of cell cycle-depending alternative secondary structure from the stacked cytosines, called the intercalated motif (i-motif) (Abou Assi et al., 2018). Additionally, we verified that ssDNA genome regions detected in vivo (Kouzine et al., 2017) actually intersected with computationally predicted G4 regions used in this study (Figure S8).
The analysis of APOBEC-induced mutation density in A-phased repeats did not show any statistically significant differences in comparison with B-DNA. Z-DNA was excluded from the analysis as we found a negligible number of TpC motifs in these genome regions, as Z-DNA is formed by purine-pyrimidine alternating sequences. Interestingly, we have observed that this also decreases the relative level of UV-mutagenesis in Z-DNA genome regions and hence provides one more example of reduced cancer mutagenesis in non-B DNA genome regions.
Overall, we have observed unexpectedly low densities of APOBEC-induced somatic mutations in non-canonical DNA structures in human cancers, which, in contrast to mutations caused by other mutagens, is approximately equal, or even smaller than the level of APOBEC-mutagenesis in B-DNA, despite the presence of ssDNA in most of non-B DNA structures. Elucidation of the mechanistic basis of these observations requires further research.
Limitations of the study
The limitations of this study are linked to the properties of the input data. Although the information on non-canonical DBA structures that we obtained from the non-B DB database (Cer et al., 2013) is comprehensive and widely used in many studies, there are new experimental data on non-B DNA structures scattered over diverse publications, and these additional data are difficult to combine and normalize. Indeed, there is a need for a shared repository that consolidates experimental data and computational predictions on all types of non-canonical DNA structures.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| PCAWG project data on somatic mutations | Campbell et al.2020 | https://dcc.icgc.org/pcawg |
| Data on non-canonical DNA structures | Cer et al., 2013 | https://nonb-abcc.ncifcrf.gov/apps/site/default |
| Replication timing data | Ding et al., 2021 | https://www.thekorenlab.org/data |
| APOBEC-induced mutational clusters data | Sakofsky et al., 2019 | https://journals.plos.org/plosbiology/article/file?type=supplementary&id=10.1371/journal.pbio.3000464.s011 |
| CpG islands annotation | Kent et al., 2002 | http://genome.ucsc.edu/cgi-bin/hgTrackUi?hgsid=1331196707_EAILpAgyAr0ebJaZbcbAHajFpaOb&c=chr1&g=cpgIslandExt |
| Genome-wide data on single-stranded DNA | Kouzine et al., 2017 | SRA: SRA072844 |
| Software and algorithms | ||
| Samtools | Danecek et al., 2021 | http://www.htslib.org/ |
| Bedtools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Marat Kazanov (m.kazanov@skoltech.ru).
Materials availability
This study did not generate new unique reagents.
Method details
Somatic mutations were taken from the Pan-Cancer Analysis of Whole Genomes project (Campbell et al., 2020). Six cancer types BLCA (Bladder Urothelial Cancer), BRCA (Breast Cancer), HNSC (Head and Neck Squamous Cell Carcinoma), LUAD (Lung Adenocarcinoma), LUSC (Lung Squamous Cell Carcinoma), CESC (Cervical Squamous Cell Carcinoma), each having a substantial number of samples enriched with the APOBEC mutagenesis signature (APOBEC-mutagenesis enrichment >2.0, calculated as in (Roberts et al., 2013)), were selected for the analysis. Non-B DNA regions were downloaded from Non-B DB (Cer et al., 2013). Seven non-B DNA motifs were considered: G-quadruplex, inverted repeats, mirror repeats, direct repeats, A-phased repeats, short tandem repeats, and Z-DNA. Information on non-B DNA motif secondary structure was obtained from the Non-B DB annotations. The inverted repeats had a minimum length of six nucleotides and an unlimited maximum length. The maximum length of the spacer separating the arms of the repeat was 100 nucleotides (Cer et al., 2013). Human genome assembly GRCh37/hg19 was used as the reference. The mutation density DAPOBEC of the APOBEC mutagenesis per target in a particular genome region was calculated as the number of single-base substitutions C→T or C→G in the TpC motif on both, direct and complementary strands, divided by the total number of the TpC motifs in this region: DAPOBEC = NAPOBEC / NTCN. The same density per base pair was calculated as the number of single-base substitutions C→T or C→G in the TpC motif divided by the genome region size. The density of AID-signature mutations was calculated similarly for the WRCY motif. Cancer samples were classified into high and low APOBEC activity samples using the APOBEC enrichment metric introduced in (Roberts et al., 2013) with the threshold value 2.0. The classification of samples into APOBEC3A-like and APOBEC3B-like mutation signature groups was taken from (Campbell et al., 2017). The direction of DNA replication was identified using replication timing profiles obtained from (Ding et al., 2021). We identified left- and right-replicating regions based on the sign of the first derivative of the replication profile as in (Haradhvala et al., 2016; Seplyarskiy et al., 2016).
Single strand DNA-sequencing data was taken from ref. (Kouzine et al., 2017). Short reads were aligned to the human (GRCh37/hg19) genome using Bowtie2 (ver. 2.3.0) (Langmead and Salzberg, 2012). Only unique mappings were kept using samtools (Danecek et al., 2021). Sequence reads with a mapping quality of less than 30 were filtered out. SAM-file was converted to the BEDGRAPH format using bedtools (Quinlan and Hall, 2010). Data on APOBEC-induced mutational clusters was taken from (Sakofsky et al., 2019). Coordinates of CpG islands were taken from the UCSC Genome Browser (Kent et al., 2002).
Quantification and statistical analysis
Statistical significance of the difference in the mutation density between genome regions was calculated by the 10000-fold random shuffling of mutation positions in each chromosome of each sample. The statistical significance of differences in the log-ratio mutation density distributions was estimated by Wilcoxon-Mann-Whitney test. The statistical significance of differences between coefficients of two regression linear models was estimated by combining both datasets and introducing the interaction term.
Acknowledgments
We thank Dmitry Gordenin for thoughtful discussion, critical reading of the article, and insightful suggestions, Irina Ponomareva for the non-B DNA structures and graphical abstract artworks. This study was supported by Russian Science Foundation (grant 22-14-00132) to M.D.K. The authors would like to acknowledge the dbGaP repository for providing access to the TCGA dataset (the accession number is phs000178.v11.p8).
Author contributions
M.D.K. conceived the study. G.V.P., B.F., V.A.N., R.A., E.S., N.L., A.A.D, and A.A.C. performed the calculations. All authors contributed to the data analysis. M.S.G. and M.D.K. wrote the article.
Declaration of interests
The authors declare no competing interests.
Published: July 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104535.
Supplemental information
Data and code availability
-
•
Data used for this research was published previously by Campbell et al. (2020).
-
•
A custom R and C++ scripts developed for this project is available at https://github.com/KazanovLab/nonBDNA.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Abou Assi H., Garavís M., González C., Damha M.J. i-Motif DNA: structural features and significance to cell biology. Nucleic Acids Res. 2018;46:8038–8056. doi: 10.1093/nar/gky735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A.J.R., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.L., et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey L.J., Bianchi J., Doherty A.J. PrimPol is required for the maintenance of efficient nuclear and mitochondrial DNA replication in human cells. Nucleic Acids Res. 2019;47:4026–4038. doi: 10.1093/nar/gkz056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge L.J., Teague R., Doherty A.J. Repriming DNA synthesis: an intrinsic restart pathway that maintains efficient genome replication. Nucleic Acids Res. 2021;49:4831–4847. doi: 10.1093/nar/gkab176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barzak F.M., Harjes S., Kvach M.V., Kurup H.M., Jameson G.B., Filichev V.V., Harjes E. Selective inhibition of APOBEC3 enzymes by single-stranded DNAs containing 2′-deoxyzebularine. Org. Biomol. Chem. 2019;17:9435–9441. doi: 10.1039/c9ob01781j. [DOI] [PubMed] [Google Scholar]
- Brázda V., Laister R.C., Jagelská E.B., Arrowsmith C. Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 2011;12:33. doi: 10.1186/1471-2199-12-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A.L., Collins C.D., Thompson S., Coxon M., Mertz T.M., Roberts S.A. Single-stranded DNA binding proteins influence APOBEC3A substrate preference. Sci. Rep. 2021;11:1–13. doi: 10.1038/s41598-021-00435-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R., Langenbucher A., Bowen D., Kwan E.E., Benes C.H., Zou L., Lawrence M.S. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science. 2019;80:364. doi: 10.1126/science.aaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M.B., Lackey L., Carpenter M.A., Rathore A., Land A.M., Leonard B., Refsland E.W., Kotandeniya D., Tretyakova N., Nikas J.B., et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M.B., Temiz N.A., Harris R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon I.J.L., Byeon C.H., Wu T., Mitra M., Singer D., Levin J.G., Gronenborn A.M. Nuclear magnetic resonance structure of the APOBEC3B catalytic domain: structural basis for substrate binding and DNA deaminase activity. Biochemistry. 2016;55:2944–2959. doi: 10.1021/acs.biochem.6b00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P.J., Getz G., Korbel J.O., Stuart J.M., Jennings J.L., Stein L.D., Perry M.D., Nahal-Bose H.K., Ouellette B.F.F., Li C.H., et al. Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. doi: 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P.J., Getz G., Stuart J.M., Korbel J.O., Stein L.D., Net, - ICGC/TCGA pan-cancer analysis of whole genomes Pan-cancer analysis of whole genomes. bioRxiv. 2017;3 doi: 10.1101/162784. Preprint at. [DOI] [Google Scholar]
- Ceccaldi R., Rondinelli B., D’Andrea A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cer R.Z., Donohue D.E., Mudunuri U.S., Temiz N.A., Loss M.A., Starner N.J., Halusa G.N., Volfovsky N., Yi M., Luke B.T., et al. Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res. 2013;41:94–100. doi: 10.1093/nar/gks955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K., Roberts S.A., Klimczak L.J., Sterling J.F., Saini N., Malc E.P., Kim J., Kwiatkowski D.J., Fargo D.C., Mieczkowski P.A., et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet. 2015;47:1067–1072. doi: 10.1038/ng.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervova A., Fatykhov B., Koblov A., Shvarov E., Preobrazhenskaya J., Vinogradov D., Ponomarev G.V., Gelfand M.S., Kazanov M.D. Analysis of gene expression and mutation data points on contribution of transcription to the mutagenesis by APOBEC enzymes. NAR Cancer. 2021;3:1–12. doi: 10.1093/narcan/zcab025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10 doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q., Edwards M.M., Wang N., Zhu X., Bracci A.N., Hulke M.L., Hu Y., Tong Y., Hsiao J., Charvet C.J., et al. The genetic architecture of DNA replication timing in human pluripotent stem cells. Nat. Commun. 2021;12:1–18. doi: 10.1038/s41467-021-27115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Kamenetskii M.D., Mirkin S.M. Triplex DNA structures. Annu. Rev. Biochem. 1995;64:65–95. doi: 10.1146/annurev.bi.64.070195.000433. [DOI] [PubMed] [Google Scholar]
- Georgakopoulos-Soares I., Morganella S., Jain N., Hemberg M., Nik-Zainal S. Noncanonical secondary structures arising from non-B DNA motifs are determinants of mutagenesis. Genome Res. 2018;28:1264–1271. doi: 10.1101/gr.231688.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordenin D.A., Lobachev K.S., Degtyareva N.P., Malkova A.L., Perkins E., Resnick M.A. Inverted DNA repeats: a source of eukaryotic genomic instability. Mol. Cell Biol. 1993;13:5315–5322. doi: 10.1128/mcb.13.9.5315-5322.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiblet W.M., Cremona M.A., Harris R.S., Chen D., Eckert K.A., Chiaromonte F., Huang Y.F., Makova K.D. Non-B DNA: a major contributor to small- and large-scale variation in nucleotide substitution frequencies across the genome. Nucleic Acids Res. 2021;49:1497–1516. doi: 10.1093/nar/gkaa1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haradhvala N.J., Polak P., Stojanov P., Covington K.R., Shinbrot E., Hess J.M., Rheinbay E., Kim J., Maruvka Y.E., Braunstein L.Z., et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell. 2016;164:538–549. doi: 10.1016/j.cell.2015.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H.-J., Kendrick S., Hecht S.M., Hurley L.H. The transcriptional complex between the BCL2 i-motif and hnRNP LL is a molecular switch for control of gene expression that can be modulated by small molecules. J. Am. Chem. Soc. 2014;136:4172–4185. doi: 10.1021/ja4109352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanov M.D., Roberts S.A., Polak P., Stamatoyannopoulos J., Klimczak L.J., Gordenin D.A., Sunyaev S.R. APOBEC-induced cancer mutations are uniquely enriched in early-replicating, gene-dense, and active chromatin regions. Cell Rep. 2015;13:1103–1109. doi: 10.1016/j.celrep.2015.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler a.D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzine F., Wojtowicz D., Baranello L., Yamane A., Nelson S., Resch W., Kieffer-Kwon K.R., Benham C.J., Casellas R., Przytycka T.M., Levens D. Permanganate/S1 nuclease footprinting reveals non-B DNA structures with regulatory potential across a mammalian genome. Cell Syst. 2017;4:344–356.e7. doi: 10.1016/j.cels.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix L., Liénard H., Labourier E., Djavaheri-Mergny M., Lacoste J., Leffers H., Tazi J., Hélène C., Mergny J.L. Identification of two human nuclear proteins that recognise the cytosine-rich strand of human telomeres in vitro. Nucleic Acids Res. 2000;28:1564–1575. doi: 10.1093/nar/28.7.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langenbucher A., Bowen D., Sakhtemani R., Bournique E., Wise J.F., Zou L., Bhagwat A.S., Buisson R., Lawrence M.S. An extended APOBEC3A mutation signature in cancer. Nat. Commun. 2021;12:1–11. doi: 10.1038/s41467-021-21891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A., et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S., Wang G., Bacolla A., Zhao J., Spitser S., Vasquez K.M. Short inverted repeats are hotspots for genetic instability: relevance to cancer genomes. Cell Rep. 2015;10:1674–1680. doi: 10.1016/j.celrep.2015.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney J.A., Wang G., Mukherjee A., Christensen L., Subramanian S.H.S., Zhao J., Vasquez K.M. Distinct DNA repair pathways cause genomic instability at alternative DNA structures. Nat. Commun. 2020;11:1–12. doi: 10.1038/s41467-019-13878-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S., Alexandrov L.B., Wedge D.C., Van Loo P., Greenman C.D., Raine K., Jones D., Hinton J., Marshall J., Stebbings L.A., et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilzecker B., Buoninfante O.A., Pritchard C., Blomberg O.S., Huijbers I.J., Van Den Berk P.C.M., Jacobs H. PrimPol prevents APOBEC/AID family mediated DNA mutagenesis. Nucleic Acids Res. 2016;44:4734–4744. doi: 10.1093/nar/gkw123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Q., Wang L., Meng F.L., Hwang J.K., Alt F.W., Wu H. AID recognizes structured DNA for class switch recombination. Mol. Cell. 2017;67:361–373.e4. doi: 10.1016/j.molcel.2017.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran S., Subramani V.K., Kim K.K. Z-DNA in the genome: from structure to disease. Biophys. Rev. 2019;11:383–387. doi: 10.1007/s12551-019-00534-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich A., Zhang S. Timeline: Z-DNA: the long road to biological function. Nat. Rev. Genet. 2003;4:566–572. doi: 10.1038/nrg1115. [DOI] [PubMed] [Google Scholar]
- Roberts S.A., Lawrence M.S., Klimczak L.J., Grimm S.A., Fargo D., Stojanov P., Kiezun A., Kryukov G.V., Carter S.L., Saksena G., et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts S.A., Sterling J., Thompson C., Harris S., Mav D., Shah R., Klimczak L.J., Kryukov G.V., Malc E., Mieczkowski P.A., et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell. 2012;46:424–435. doi: 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini N., Gordenin D.A. Hypermutation in single-stranded DNA. DNA Repair. 2020;91–92:102868. doi: 10.1016/j.dnarep.2020.102868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky C.J., Saini N., Klimczak L.J., Chan K., Malc E.P., Mieczkowski P.A., Burkholder A.B., Fargo D., Gordenin D.A. Repair of multiple simultaneous double-strand breaks causes bursts of genome-wide clustered hypermutation. PLoS Biol. 2019;17 doi: 10.1371/journal.pbio.3000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter J.D., Bennett R.P., Smith H.C. The APOBEC protein family: united by structure, divergent in function. Trends Biochem. Sci. 2016;41:578–594. doi: 10.1016/j.tibs.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavone D., Jozwiakowski S.K., Romanello M., Guilbaud G., Guilliam T.A., Bailey L.J., Sale J.E., Doherty A.J. PrimPol is required for replicative tolerance of G quadruplexes in vertebrate cells. Mol. Cell. 2016;61:161–169. doi: 10.1016/j.molcel.2015.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seplyarskiy V.B., Soldatov R.A., Popadin K.Y., Antonarakis S.E., Bazykin G.A., Nikolaev S.I. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016;26:174–182. doi: 10.1101/gr.197046.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. Non-B DNA secondary structures and their resolution by RecQ helicases. J. Nucleic Acids. 2011;2011:1–15. doi: 10.4061/2011/724215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K., Carpenter M.A., Banerjee S., Shaban N.M., Kurahashi K., Salamango D.J., McCann J.L., Starrett G.J., Duffy J.V., Demir Ö., et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017;24:131–139. doi: 10.1038/nsmb.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinden R.R., Pytlos-Sinden M.J., Potaman V.N. Slipped strand DNA structures. Front. Biosci. 2007;12:4788–4799. doi: 10.2741/2427. [DOI] [PubMed] [Google Scholar]
- Spiegel J., Adhikari S., Balasubramanian S. The structure and function of DNA G-quadruplexes. Trends Chem. 2020;2:123–136. doi: 10.1016/j.trechm.2019.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šviković S., Crisp A., Tan-Wong S.M., Guilliam T.A., Doherty A.J., Proudfoot N.J., Guilbaud G., Sale J.E. R-loop formation during S phase is restricted by PrimPol mediated repriming. EMBO J. 2019;38:1–19. doi: 10.15252/embj.201899793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G., Vasquez K.M. Effects of replication and transcription on DNA Structure-Related genetic instability. Genes. 2017;8:17. doi: 10.3390/genes8010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong L., Vizeacoumar F.S., Vizeacoumar F.J., Chelico L. APOBEC1 cytosine deaminase activity on single-stranded DNA is suppressed by replication protein A. Nucleic Acids Res. 2021;49:322–339. doi: 10.1093/nar/gkaa1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.-Z., Jenjaroenpun P., Wongsurawat T., Byrum S.D., Shponka V., Tannahill D., Chavez E.A., Hung S.S., Steidl C., Balasubramanian S., et al. Activation-induced cytidine deaminase localizes to G-quadruplex motifs at mutation hotspots in lymphoma. NAR Cancer. 2020;2:9–12. doi: 10.1093/narcan/zcaa029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B., Li X., Lei L., Chen J. APOBEC: from mutator to editor. J. Genet. Genomics. 2017;44:423–437. doi: 10.1016/j.jgg.2017.04.009. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Zhou H., Ou-Yang Z.C. Stretching single-stranded DNA: interplay of electrostatic, base-pairing, and base-pair stacking interactions. Biophys. J. 2001;81:1133–1143. doi: 10.1016/S0006-3495(01)75770-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Bacolla A., Wang G., Vasquez K.M. Non-B DNA structure-induced genetic instability and evolution. Cell. Mol. Life Sci. 2010;67:43–62. doi: 10.1007/s00018-009-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data used for this research was published previously by Campbell et al. (2020).
-
•
A custom R and C++ scripts developed for this project is available at https://github.com/KazanovLab/nonBDNA.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.