

Abstract
Glioma is a primary aggressive brain tumor with high recurrence rate. The poor efficiency of chemotherapeutic drugs crossing the blood‒brain barrier (BBB) is well-known as one of the main challenges for anti-glioma therapy. Moreover, massive infiltrated tumor-associated macrophages (TAMs) in glioma further thwart the drug efficacy. Herein, a therapeutic nanosystem (SPP-ARV-825) is constructed by incorporating the BRD4-degrading proteolytic targeting chimera (PROTAC) ARV-825 into the complex micelle (SPP) composed of substance P (SP) peptide-modified poly(ethylene glycol)-poly(d,l-lactic acid)(SP-PEG-PDLLA) and methoxy poly(ethylene glycol)-poly(d,l-lactic acid) (mPEG-PDLLA, PP), which could penetrate BBB and target brain tumor. Subsequently, released drug engenders antitumor effect via attenuating cells proliferation, inducing cells apoptosis and suppressing M2 macrophages polarization through the inhibition of IRF4 promoter transcription and phosphorylation of STAT6, STAT3 and AKT. Taken together, our work demonstrates the versatile role and therapeutic efficacy of SPP-ARV-825 micelle against glioma, which may provide a novel strategy for glioma therapy in future.
Key words: Glioma, BRD4, ARV-825, Micelle, M2 macrophage
Graphical abstract
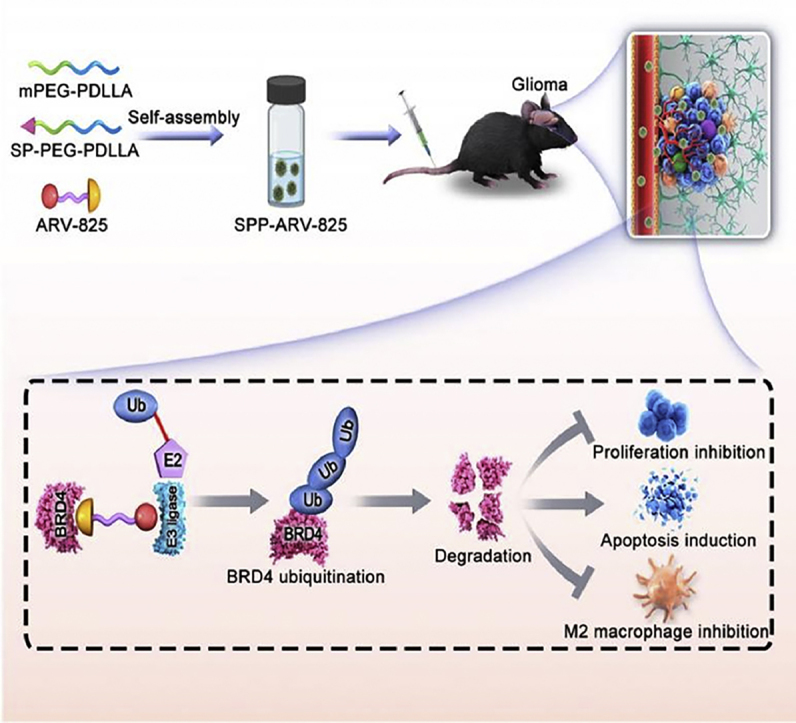
This work introduces a targeting drug delivery system (SPP-ARV-825) consisting of mPEG-PDLLA, SP-PEG-PDLLA and BRD4 PROTAC drug ARV-825 for glioma therapy. The SPP-ARV-825 efficiently induces BRD4 degradation and attenuates glioma tumor growth. The antitumor mechanisms of SPP-ARV-825 involve cells proliferation inhibition, apoptosis induction and M2 macrophages reduction.
1. Introduction
Glioma is a primary intracranial malignant tumor derived from glial cells or precursor cells, accounting for 25.5% of central nervous system tumors1. Clinically, the treatment of glioma primarily depends on surgical resection, supplemented by postoperative chemoradiotherapy2,3. However, gliomas are characterized by infiltrative growth, strong invasiveness and vague boundary with surrounding tissue, leading to the failure of complete surgical resection4, 5, 6. Therefore, chemotherapy has become a significant adjuvant therapy for glioma treatment, but chemotherapy is still confronted with formidable challenges due to the blood‒brain barrier (BBB)7,8. At the same time, glioma can promote self-growth, metastasis and invasion by recruiting microglia and peripheral macrophages as well as inducing M2 macrophages polarization, thus leading to the development of drug resistance and immunosuppression9,10. Thereupon, to improve the efficiency of drug delivery and find new therapeutic targets are crucial for glioma chemotherapy.
Recent studies have revealed that targeting epigenetics process in tumor could not only regulate tumor proliferation but also manage tumor microenvironment11,12. Bromodomain containing protein 4 (BRD4), one of the bromodomain and extraterminal domain (BET) family members, is a vital epigenetic regulator and transcription cofactor. Moreover, BRD4 is closely related to tumors, which is highly expressed in tumors, enriched in the promoter region of oncogenes and involved in tumor cell proliferation13, 14, 15, 16. BRD4 is also found to be a vital immunomodulatory factor17, 18, 19. Due to the important role of BRD4 in tumor progression, many BRD4 small molecular inhibitors have been developed for tumor therapy, but their clinic application is limited by drug resistance and side effects20,21. Proteolytic targeting chimera (PROTAC) technology, a novel protein blocking technology based on the ubiquitination‒proteasome system (UPS) to target and induce protein degradation, has potential advantages in terms of dosage, side effects and drug resistance in drug discovery22,23. The action form of "PROTAC" consists of the E3 ubiquitin ligase ligand and the target protein ligand, and the two active ligands are linked together by a specially designed "Linker" structure. The PROTAC protein-target ligand binds to the target protein, and the E3 ubiquitin ligand binds to the substrate binding region of the E3 ubiquitin ligase, enabling the UPS system to degrade the target protein23,24. ARV-825, a BRD4 degrader based on PROTAC technology, can ubiquitinate BRD4 protein via recruiting E3 ubiquitin ligase, further leading to rapid, effective and continuous degradation of BRD4 protein, thereby exerting a powerful anti-tumor effect25. PROTAC-based drugs exhibit several benefits such as a wider range of action, improved selectivity, higher activity, and can target "non-generic" targets as well as overcome drug resistance. Compared with traditional occupancy-driven inhibitors such as JQ1, ARV-825 possesses high activity with only a small amount, which is conducive to the reduction of drug resistance26,27. It is reported that BRD4-targeting PROTACs has potential therapeutic benefit for hematological malignancies including multiple myeloma28, secondary AML29, lymphoma30,31, and castration-resistant prostate cancer32 in preclinical studies. However, the large molecular weight and low water solubility of ARV-825 restrict its further antitumor activity.
Another difficult challenge in glioma treatment is to overcome the BBB, which could be resolved with the advent of nano-based drug delivery system (NDDS)33, 34, 35, 36. Polymer micelle is a commonly used nano-delivery system, which can enhance the water solubility of drugs, reduce the phagocytosis of the reticuloendothelial system and prolong drug circulation time37, 38, 39, 40. Furthermore, based on the highly-expressed receptors in vascular endothelial cells and gliomas, such as neurokinin receptor (NK-R), αVβ3 integrin, low-density lipoprotein receptor associated protein 1 (LRP1), transferrin receptor, ligand modification of drug-loading micelles is implemented to realize tumor targeted release of drugs, so as to enhance the therapeutic effect of glioma41, 42, 43, 44. Herein, the substance P (SP) peptide composed of 11 amino acids, which can specifically bind to neurokinin 1 receptor (NK-1R), is applied to the design of a glioma-targeting nanodrug delivery system45.
Above all, we used BRD4 as a new target against glioma and developed a tumor-targeting amphiphilic micelle loading ARV-825 to treat glioma (Scheme 1). Subsequently, the effects and mechanisms of drug-loaded micelle on glioma cells proliferation, apoptosis and M2 macrophages polarization were further explored. At present, there are few reports on the treatment of glioma with BRD4 PROTAC nanodrug. We hope the study will bring forward a novel approach for glioma therapy and provide research basis for clinical drug investigation.
Scheme 1.

Schematic illustration of brain-targeting SPP-ARV-825 micelle against glioma. The mPEG-PDLLA, SP-PEG-PDLLA and ARV-825 were used for the preparation of SPP-ARV-825 micelle with self-assembly way. SPP-ARV-825 was administrated intravenously and reached tumor site. Subsequently, released ARV-825 acted on BRD4 and induced BRD4 ubiquitination and degradation, further exerting anti-tumor function by cells proliferation inhibition, apoptosis induction and M2 macrophages polarization suppression.
2. Materials and methods
2.1. Preparation and characterization of SPP-ARV-825
The thin-film hydration method was adopted to prepare SPP-ARV-825. ARV-825 (10 mg), SP-PEG-PDLLA (15 mg) and mPEG-PDLLA (75 mg) were dissolved in acetone, and the mixed solution was evaporated to form film under reduced pressure on a rotary evaporator at 55 °C. A certain amount of normal saline was added to make ARV-825 and SP-PEG-PDLLA/mPEG-PDLLA self-assemble to form SPP-ARV-825 micelle. Non-targeted micelle PP-ARV-825 (mass ratio, mPEG-PDLLA:ARV-825 = 9:1) and blank micelle PP (mass ratio, mPEG-PDLLA:ARV-825 = 10:0) were prepared by the same method.
The particle size, distribution coefficient and zeta potential of SPP-ARV-825 were measured by the laser particle size analyzer (Brookhaven Instruments, USA). The morphology of SPP-ARV-825 was observed by the transmission electron microscope (TEM) (FEI, USA). The release of ARV-825 and SPP-ARV-825 were studied in PBS containing 0.5% Tween 80 at 37 °C. In brief, 0.9 mL of ARV-825 or SPP-ARV-825 were dialyzed (MWCO 3.5K) against 20 mL of PBS containing 0.5% Tween 80. At predesignated time points (2, 4, 6, 8, 12, 24, 48, 72 and 96 h), 4 mL of release medium outside the dialysis bag was taken out as sample for HPLC analysis, and 4 mL of fresh release medium was replenished to keep 20 mL of release medium.
2.2. Cell viability assay
GL261 cells and U87 cells were respectively seeded in 96-well plates (GL261: 2 × 103 cells per well, U87: 1.2 × 104 cells per well) and cultured overnight. Cells were treated with different concentrations of ARV-825, PP-ARV-825 and SPP-ARV-825 for 48 h. Untreated cells served as a control group. Cell viability was evaluated by MTT assay. Briefly, 20 μL MTT (5 mg/mL) was added to incubate for 3–4 h at 37 °C. The medium was then discarded and 150 μL dimethyl sulfoxide (DMSO) was added to dissolve crystal for 10 min. The spectroscopic value was measured at 570 nm with a microplate reader (Thermo Fisher Scientific, USA).
2.3. FCM analysis of cell cycle
GL261 and U87 cells were planted on 6 well plates. After being cultured for 24 h, cells were incubated with media containing ARV-825 or PP-ARV-825 or SPP-ARV-825. At the predetermined time, cells were collected and washed once with PBS followed by adding 1 mL precooled 70% ethanol to fixed cells for 12–24 h at 4 °C. After being washed once with PBS, fixed cells were stained for 30 min with propidium iodide (PI) staining solution at 37 °C in dark. The change of cell cycle was detected by flow cytometry (FCM, ACEA NovoCyte, USA).
2.4. FCM detection of cells apoptosis
GL261 cells were seeded to 6 well plates at a density 1 × 105 per well. After cultured overnight, cells were incubated in the presence of ARV-825 or PP-ARV-825 or SPP-ARV-825. The untreated cell was regarded as a negative control. After being incubated for 48 h, cells were washed once with PBS and stained with 100 μL binding buffer containing 5 μL Annexin V-FITC and 5 μL PI for 15 min followed by adding 400 μL binding buffer to detect cell apoptosis with FCM.
2.5. Subcutaneous xenograft model
C57BL/6 mice were subcutaneously inoculated with GL261 cells at the density of 2 × 106. When the tumor grew to 5 mm × 5 mm, the mice were randomly divided into 5 groups: normal saline (NS), PP, ARV-825, PP-ARV-825 and SPP-ARV-825. The dose of 20 mg/kg ARV-825 was administered to mice intravenously every two days, for a total of 5 times, and the body weight and tumor volume of the mice were recorded each time. After the treatment, tumors of mice in each group were dissected to photograph and weigh. Furthermore, tumors were fixed with 4% paraformaldehyde, embedded in paraffin and sliced, and Ki67 staining, CD31 staining and TUNEL assay were performed to determine the effect of SPP-ARV-825 on tumor cell proliferation, angiogenesis and apoptosis, respectively. The vital organs (heart, liver, spleen, lung, and kidney) and serum were separately collected for hematoxylin-eosin (HE) staining and blood biochemical assay to evaluate safely of SPP-ARV-825. All animal experiments were approved by the Animal Experimental Ethics Committee of State Key Laboratory Biotherapy (SKLB), Sichuan University (SKLB 2020-1021) and conducted according to the Institutional Animal Care and Use Guidelines.
2.6. Glioma orthotopic xenograft model
C57BL/6 mice and BALB/c nude mice anesthetized with chloral hydrate were fixed with brain stereotactic apparatus. The scalp was disinfected and cut open to find the anterior fontanel. The puncture point was located 0.5 mm behind the coronal suture and 2.5 mm to the right of the sagittal suture. The microinjector was used to enter the needle vertically at the puncture point to 4.5 mm, and then withdraw the needle for 1 mm. At this position, the microinjector was slowly pushed to inject 5 μL GL261-Luc cell suspension (2 × 105 cells) or U87-Luc cell suspension (2 × 106 cells) and the injection was completed in 5 min. After 3 min of needle retention, the needle was slowly withdrawn for 2 min. The head skin of mice was sutured with medical suture, disinfected with iodophor, and all postoperative mice were put back into the cage. The mice were randomly divided into 5 groups (NS, PP, ARV-825, PP-ARV-825 and SPP-ARV-825), 4 days after inoculation with GL261-Luc or 12 days after inoculation with U87-Luc. The mice were treated with 20 mg/kg of drugs by tail vein for 5 times. GL261 mice received treatments every two days and U87 mice received treatments every three days. At the end of treatment, the mice were anesthetized with isoflurane and intraperitoneally injected with 15 mg/mL d-fluorescein potassium solution at a dose of 10 μL/g. After 10 min, the mice were put into the small animal imaging instrument to observe the luminescence and evaluate therapeutic effect of tumor in situ. Subsequently, the mice were sacrificed and the whole brain was isolated and soaked in 4% paraformaldehyde for fixation, which was used for HE staining to observe the tumor in brain tissue. All animal experiments were approved by the Animal Experimental Ethics Committee of State Key Laboratory Biotherapy (SKLB), Sichuan University (SKLB 2020-1021) and conducted according to the Institutional Animal Care and Use Guidelines.
2.7. FCM analysis of M2-like macrophages
C57BL/6 mice were subcutaneously inoculated with GL261 cells (2 × 106). When the tumor grew to 5 mm × 5 mm, the mice were randomly divided into two groups: NS and ARV-825 (20 mg/kg). After the mice were treated for three consecutive days, the tumor cells were obtained and stained with M2 type macrophage related antibodies (anti-CD45-PerCP-Cy5.5, anti-CD11b-PE, anti-F4/80-APC, anti-CD206-FITC) (BD Bioscience, USA) for 30 min at 4 °C, followed by washing and resuspending with PBS to perform FCM analysis.
The bone marrow derived macrophages (BMDMs) isolated from healthy female mice (6–8 weeks) were assigned to four groups (0, IL4, DMSO + IL4 and ARV-825 + IL4). Cells of DMSO + IL4 group and ARV-825 + IL4 group were respectively pretreated with DMSO and ARV-825 for 12 h. IL4 was then added to incubate for 24 h. The IL4 group and DMSO + IL4 group were the positive control groups and cells in 0 group were untreated. At the predetermined time point, cells were stained with mouse anti-CD45-PerCP-Cy5.5, anti-CD11b-FITC, anti-F4/80-PE and anti-CD206-APC antibodies (BD Bioscience, USA) for 30 min at 4 °C. Subsequently, stained cells were washed once and resuspended with PBS for FCM detection.
2.8. RNA sequencing
The BMDMs were given different treatments (0, IL4, DMSO + IL4, DRUG + IL4) for the predesigned time followed by completely removing medium in wells and adding 1 mL trizol to pipet repeatedly. And total RNA collected was sent to Liebing Technology Biomedical Co., Ltd. (China) for sequencing analysis.
2.9. Dual-luciferase reporter assay
The genomic DNA from mouse peritoneal macrophages was extracted as a PCR template to amplify the promoter of IRF4 (forward primer: 5′-GCGTGCTAGCCCGGGCTCGAGTTGGCTGAGGAAAATGTTGAAA-3′; reverse primer: 5′-CAGTACCGGAATGCCAAGCTTTGCCACCAGCCTCACACTCC-3′), and subsequently the PCR product was purified to obtain the IRF4 promoter gene. The promoter of IRF4 gene was constructed on the vector PGL3-Basic to form IRF4-Luc gene expressing firefly luciferase. RAW264.7 cells were seeded in 12 well plates. After 24 h, the plasmid PRL-SV40 expressing renilla luciferase and the plasmid IRF4-Luc were cotransfected into cells with lipofectamine reagent 3000 (Invitrogen, USA). After 4 h, solution in wells was replaced with DMEM complete medium with or without 50 ng/mL ARV-825, and placed in the incubator for 48 h. The activity of luciferase was detected with the double-luciferase reporter assay kit (TransGen Biotech, China).
2.10. Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed via the ChIP Assay Kit (Cell Signaling Technology, USA) according to the manufacturer's instructions. In brief, RAW264.7 macrophages were incubated with 50 ng/mL ARV-825 and equal volume of DMSO respectively for 48 h, followed by fixing with formaldehyde to crosslink targeted proteins and DNA to form chromatin complexes. After breaking cells with ultrasonic, the chromatin complexes were immunoprecipitated via anti-BRD4 antibody (Cell Signaling Technology, USA) or negative control IgG, and subsequently the protein G magnetic beads were used to capture the antibody–chromatin complexes formed in front, followed by elution and de-crosslinking. Finally, the purified DNA samples were detected by qRT-PCR. The primers are listed in Supporting Information Table S3.
2.11. Statistical analysis
In this study, data were presented as mean ± standard error of measurement (SEM). The statistical differences of data were analyzed with the unpaired two-tailed Student t test for two groups comparison and ordinary one-way analysis of variance for multiple groups comparison in GraphPad Prism 9.0 software. The P value of more than 0.05 indicated no significant difference and was marked with ns. P value of less than 0.05 was considered statistically significant and were presented as ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 and ∗∗∗∗P < 0.0001.
3. Results and discussion
3.1. Preparation and characterization of SPP-ARV-825 micelle
Given the hydrophobicity and the poor availability of ARV-825 drug, we utilized amphiphilic mPEG-PDLLA to deliver ARV-825. To identify the interaction process of mPEG-PDLLA and ARV-825, molecular dynamics simulation was performed to investigate their interaction in water environment (pH 7.0) and tumor-mimicking environment (pH 6.5). In both environments, ARV-825 presented constant change in position and conformations while interacting with mPEG-PDLLA within 10 ns, and ARV-825 was gradually encapsulated in hydrophobic PDLLA block of mPEG-PDLLA, ultimately forming a stable drug-loaded micelle (Fig. 1A and B and Supporting Information Fig. S1A and B). Then, we synthesized PEG-PDLLA with ring-opening polymerization and modified SP peptide to PEG-PDLLA to form targeted SP-PEG-PDLLA (Fig. S2A and B), whose structure was confirmed via 1H NMR analysis (Supporting Information Fig. S3). Subsequently, drug loaded micelle, termed as SPP-ARV-825, was fabricated via self-assembly of mPEG-PDLLA, SP-PEG-PDLLA and ARV-825. Detection of dynamic lighting scatter (DLS) showed that SPP-ARV-825 had a small particle size (26.3 ± 0.7 nm) and polydispersity index (0.157 ± 0.035), and zeta potential of −13.3 ± 8.0 mV (Fig. 1C and D). SPP-ARV-825 presented a uniformly distributed spherical structure by the observation of transmission electron microscope (TEM) (Fig. 1E). Subsequently, drug release behavior of ARV-825 from SPP-ARV-825 was investigated in PBST containing 0.5% Tween 80. As shown in Fig. 1F, ARV-825 was continuously released from SPP-ARV-825 and cumulative release ratio was 26.68% at 96 h, indicating that SPP improved the release characteristics of ARV-825.
Figure 1.

Characterizations of SPP-ARV-825 micelle. Molecular dynamic simulations of the interaction between mPEG-PDLLA and ARV-825 in (A) water environment (pH = 7) and (B) tumor-mimicking environment (pH = 6.5). Conformations (I), (II), (III), (IV), (V), and (VI) represented the snapshots of the interaction between mPEG-PDLLA and ARV-825 at 0, 2, 4, 6, 8 and 10 ns, respectively. (C) Particle size distribution of SPP-ARV-825. (D) Zeta potential of SPP-ARV-825. (E) TEM image of SPP-ARV-825 (scale bar = 50 nm). (F) The cumulative release of ARV-825 from SPP-ARV-825 micelle (the black line indicated free agent of ARV-825).
3.2. SPP-ARV-825-induced cells proliferation inhibition and cells apoptosis in vitro
We then assessed the cytotoxicity of SPP-ARV-825 against glioma GL261 cells and U87 cells through MTT assay. The results showed that glioma cells viability was inhibited with increased drug concentrations, and SPP-ARV-825 treatment induced the strongest cell cytotoxicity compared with ARV-825 and PP-ARV-825 (Fig. 2A and Supporting Information Fig. S4). Meanwhile, the similar inhibitory phenomenon was observed in clone formation assay of GL261 cells treated with ARV-825, PP-ARV-825 and SPP-ARV-825, respectively (Fig. 2B and C), indicating that SPP-ARV-825 exhibited a stronger proliferation inhibition effect than ARV-825 on glioma cells. The improved effect might be due to the reason that SPP-ARV-825 could bind to NK-1R on glioma cells, further relying on receptor-mediated endocytosis to increase the cellular uptake of ARV-825. Subsequently, the effect of SPP-ARV-825 on glioma cell cycle was analyzed by flow cytometry (FCM). As shown in Fig. 2D and E, GL261 cells incubated with ARV-825, PP-ARV-825 and SPP-ARV-825 were evidently blocked in G0/G1 phase. The consistent result was also observed in U87 cells (Supporting Information Fig. S5). Furthermore, the GL261 cells and U87 cells were stained by EdU and detected by FCM, which showed that the proliferating cells were significantly decreased after 24 and 48 h of SPP-ARV-825 incubation (Fig. 2F and G and Supporting Information Fig. S6), which further verified the inhibitory effect of SPP-ARV-825 on glioma cells proliferation.
Figure 2.

Inhibitory effect of SPP-ARV-825 on GL261 cells proliferation in vitro. (A) Cell viabilities of GL261 cells treated with ARV-825, PP-ARV-825 and SPP-ARV-825 at different concentrations for 48 h (n = 3). (B) Representative images of colony formation of GL261 cells incubated with 200 ng/mL ARV-825, PP-ARV-825 and SPP-ARV-825. (C) Quantification of colony formation (n = 3). (D) GL261 cells cycle distribution determined by flow cytometry after treatment with 400 ng/mL ARV-825, PP-ARV-825 and SPP-ARV-825 for 24 h. (E) Quantification of cell cycle distribution (n = 3). (F) Flow cytometry detection of GL261 cells proliferation after treatment with 400 ng/mL SPP-ARV-825 for 24 and 48 h. (G) Quantification of Edu positive GL261 cells (n = 3). (H) BRD4 expression detection by Western blot analysis after incubation of GL261 cells with different concentrations of SPP-ARV-825 for 48 h and with 400 ng/mL SPP-ARV-825 for different time. (I, J) Representative Western blot analysis of (I) cyclin D1, cyclin E1 and β-action and (J) P-AKT, AKT, P-ERK1/2, ERK1/2, P-STAT3, STAT3 and GAPDH in GL261 cells treated with different concentrations of SPP-ARV-825 for 24 h. GAPDH or β-action served as a loading control. Data are presented as mean ± SEM. ∗P < 0.05, ∗∗∗P < 0.001 and ∗∗∗∗P < 0.0001.
BRD4 is an epigenetic reader that regulates gene transcription and participates in the process of tumorigenesis and development. Recently, Tao et al. found that in glioma, highly-expressed BRD4 could regulate Notch 1 transcription, further increasing carcinogenicity46. ARV-825, one of the BRD4 inhibitors, has been used in the BRD4 degradation of various tumors to achieve therapeutic function47. In our study, SPP-ARV-825 also exhibited an effective BRD4 degradation capacity in glioma GL261 and U87 cells (Fig. 2H and Supporting Information Fig. S7). Interestingly, BRD4 was obviously degraded in GL261 cells treated with 400 ng/mL SPP-ARV-825 for 48 h, while a significant reduction of BRD4 protein was observed in U87 cells treated with 6.25 ng/mL for only 4–6 h. This might be related to the heterogeneity of tumor cells. In addition, ARV-825 also exhibited an inhibitory function on cycle-related proteins in triple negative breast cancer48. As determined by Western blot (WB) analysis, SPP-ARV-825 could decrease the expressions of G0/G1 phase proteins, including cyclin D1 and cyclin E1 in GL261 cells and cyclin D1, CDK4, cyclin E1 and CDK2 in U87 cells (Fig. 2I and Supporting Information Fig. S8), which was consistent with our FCM analysis results on cell cycle. The above results implied that SPP-ARV-825 could restrain proliferation of glioma cells, but the specific pathway of SPP-ARV-825 inhibiting proliferation was unknown. According to previous literature reports, the tumor cells proliferation inhibition might be caused by restrained phosphorylation of AKT, MAPK and STAT349,50. Thus, we supposed that the inhibitory effect of SPP-ARV-825 on glioma cells proliferation might also be due to blocking of the AKT, MAPK or STAT3 pathway. To verify the point, the expression of related proteins from glioma cells treated with different concentrations SPP-ARV-825 for 24 h was analyzed by WB analysis. As illustrated in Fig. 2J, the expression levels of phosphorylated AKT, ERK1/2 and STAT3 all descended in a concentration-dependent manner, suggesting that SPP-ARV-825 hindered the proliferation of GL261 cells through inhibiting AKT, ERK1/2 and STAT3-related pathways. Similarly, decreased phosphorylation levels of AKT and ERK1/2 were also found in U87 cells (Supporting Information Fig. S9).
Inducing cells apoptosis is one of the important strategies to treat tumor. The effect of SPP-ARV-825 on GL261 cells apoptosis was detected by FCM. As shown in Fig. 3A and B, compared with untreated cells, increased apoptosis were observed in cells incubated with ARV-825, PP-ARV-825 and SPP-ARV-825, particularly SPP-ARV-825 group, indicating that targeted nano delivery systems enhanced antitumor activity of ARV-825. GL261 cells apoptosis induced by SPP-ARV-825 was further confirmed by the fact that mitochondrial membrane potential yielded a decline after SPP-ARV-825 incubation for 48 h (Fig. 3C and D). To investigate the mechanism of SPP-ARV-825 inducing apoptosis, WB analysis was used to detect expression levels of apoptosis-related proteins. As demonstrated in Fig. 3E, SPP-ARV-825 up-regulated the pro-apoptosis protein Bax, down-regulated anti-apoptosis proteins MCL-1 and BCL-2, and induced cleavage of caspase 8, caspase 3 and PARP. The results were accordant with previous studies on BRD4 inhibitor inducing tumor cells apoptosis51,52.
Figure 3.

SPP-ARV-825-mediated cells apoptosis in vitro. (A) Flow cytometry detection of GL261 cells apoptosis after different treatments for 48 h. (B) Statistical analysis of apoptosis cells (n = 3). (C) Flow cytometry analysis of mitochondrial membrane potential levels of GL261 cells stained with JC-1 after treatment with 400 ng/mL SPP-ARV-825 for 48 h. Untreated cells were used as the control. (D) Statistical analysis of the red/green fluorescence signal ratio of JC-1 (n = 3). (E) Expressions of apoptosis related proteins (Bax, MCL-1, BCL-2, pro-caspase 8, cleaved caspase 8, pro-caspase 3, cleaved caspase 3, PARP) and the loading control GAPDH were determined by Western blot analysis after treatment of GL261 cells with different concentrations of SPP-ARV-825. Data are presented as mean ± SEM. ∗∗∗∗P < 0.0001.
3.3. Antitumor efficacy and biosafety evaluation of SPP-ARV-825 in vivo
To assess the therapeutic effect of SPP-ARV-825 in vivo, we constructed a subcutaneous GL261 glioma mice model and tumor-bearing mice were intravenously treated with normal saline (NS), PP, ARV-825, PP-ARV-825 and SPP-ARV-825 for five times at a time interval of 1 day (Fig. 4A). At the endpoint, ARV-825, PP-ARV-825 and SPP-ARV-825 all caused the inhibition of tumor growth compared with PP or NS, and SPP-ARV-825 exhibited the strongest tumor inhibition capability, reflecting in the minimum of tumor weight and volume (Fig. 4B‒D). Moreover, SPP-ARV-825 group mice had no obvious body weight change compared with other group mice (Fig. 4E). The results suggested that SPP-ARV-825 possessed a better therapeutic effect than other treatments, which might be attributed to both passive and active targeting of SPP, allowing more ARV-825 to accumulate at the tumor site and enhance anti-tumor effect.
Figure 4.

Antitumor effect of SPP-ARV-825 in GL261 glioma subcutaneous model and orthotopic model. (A) Schedule of experimental design in mouse subcutaneous model. (B) The representative image of tumors excised from mice treated with NS, PP, ARV-825, PP-ARV-825 and SPP-ARV-825 at ARV-825 dose of 20 mg/kg, respectively. (C) Tumor growth curves for GL261 tumor-bearing mice treated with different formulation (n = 8). (D) Tumor weights of mice receiving different treatments (n = 8). (E) Changes of body weight of mice in different group during treatment (n = 8). (F) Schedule of experimental design in mouse orthotopic model. (G) Body weight changes of mice with orthotopic tumor during treatment (n = 6). (H) In vivo imaging of GL261-Luc tumor-bearing mice at end of different treatments. (I) Representative H&E staining images of brain tissues from different therapeutic groups (scale bar = 1000 μm). Red circles in images show tumors. Data are presented as mean ± SEM. No significant difference is marked with ns. ∗P < 0.05, ∗∗P < 0.01 and ∗∗∗∗P < 0.0001.
We further established orthotopic GL261-Luc and U87-Luc glioma mice model to verify the anti-glioma effect of SPP-ARV-825 (Fig. 4F and Supporting Information Fig. S10A). Similarly, tumor-bearing mice were randomly divided into five groups (NS, PP, ARV-825, PP-ARV-825 and SPP-ARV-825) and received corresponding treatment. During the treatment, there were no significant changes in the body weight of mice among all treatment groups (Figs. 4G and S10B), which demonstrated that SPP-ARV-825 had no evident toxicity and the result was consistent with that of in the subcutaneous mice model. After the treatment, in vivo imaging results showed highest biofluorescence intensity in NS group and PP group, moderate intensity in ARV-825 and PP-ARV-825 and the lowest intensity in SPP-ARV-825, indicating that SPP-ARV-825 significantly inhibited tumor growth (Figs. 4H and S10C). In addition, brain tissue slices from each group mice were stained with hematoxylin-eosin (HE) dye to assess tumor size. The results demonstrated that brain tumor foci in SPP-ARV-825 group was significantly smaller than that in the other four groups (Figs. 4I and S10D), which was in accord with the in vivo imaging results.
To investigate the potential anti-tumor mechanisms of SPP-ARV-825 in vivo, tumor tissue sections from subcutaneous glioma mice model were stained with Ki67, CD31 and TUNEL. As shown in Fig. 5, abundant Ki67 positive cells and CD31 positive vessels and few TUNEL positive cells were observed in NS and PP groups, whereas SPP-ARV-825 treatment significantly decreased Ki67 and CD31 expression and increased TUNEL positive cells. These results implied that ARV-825 encapsulated in the targeted SPP micelle can inhibit tumor cells proliferation and angiogenesis more effectively, induce more tumor cells apoptosis, and further exerting a stronger antitumor effect in vivo.
Figure 5.

SPP-ARV-825 enhanced inhibition of cells proliferation and angiogenesis, and induction of apoptosis in GL261 subcutaneous tumor. (A) Representative Ki67 staining images of tumors form different therapeutic groups (scale bar = 50 μm). (B) Representative CD31 staining images of tumors form different therapeutic groups (scale bar = 50 μm). (C) Representative TUNEL staining images of tumors from different therapeutic groups (blue: cell nuclei; green: apoptosis cells, scale bar = 50 μm). (D–F) Statistical analysis of (D) Ki67-positive cells, (E) microvessels number per field and (F) apoptosis cells in different groups (n = 4). Data are presented as mean ± SEM. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 and ∗∗∗∗P < 0.0001.
In addition to efficacy, safety is also very important for nano-based drug delivery system. The above results had shown that SPP-ARV-825 administration had no significant influence on mice body weight. We further evaluated the toxicity of SPP-ARV-825 through HE staining of major organs (heart, liver, spleen, lung and kidney) and the blood biochemical assays. As shown in Supporting Information Fig. S11, SPP-ARV-825 treatment, similar with other treatments, has no obvious pathological toxicity to the major organs. Meanwhile, various blood biochemical indexes exhibited no significant abnormalities and no significant differences among groups (Supporting Information Fig. S12), suggesting that SPP-ARV-825 treatment had no obvious liver and kidney function damage.
3.4. Inhibition of ARV-825 on M2 macrophage polarization
Tumor-associated macrophages (TAM) are closely related to the occurrence and development of tumors, and participate in the processes of tumor angiogenesis, invasion and metastasis and immune escape. The phenotype of tumor-associated macrophage is mostly M2-like macrophage, a polarized macrophage phenotype activated by IL4, IL-13 and TGF-β, which is involved in tissue repair, immunosuppression and angiogenesis, further exerting pro-tumor effects53. At present, several studies showed that BRD4 had a close relationship with macrophages. BRD4 is revealed to participate in macrophage inflammatory response and activate tumor-associated macrophages to promote tumor growth54. Comparatively, BRD4 inhibitor can reduce the expression of PD-L1 and CSF1 in tumor, ultimately blocking tumor immunosuppression and hindering tumor growth55,56. However, BRD4 has not been report as a target to regulate the polarization of macrophages in gliomas and thus affecting glioma growth. To further understand the anti-glioma mechanism of ARV-825, we performed FCM analysis of M2 macrophages in subcutaneous tumors of mice treated with ARV-825 and NS. Results showed that compared with NS groups, the proportion of M2 macrophages (F4/80+CD206+) in ARV-825 group was significantly reduced (Fig. 6A and B), indicating that ARV-825 could decrease M2-like macrophages in tumors.
Figure 6.

Effect of ARV-825 on macrophages in vitro and in vivo detected by flow cytometry. (A) Flow cytometry detection of M2-type macrophages in tumor tissues from mice receiving NS and ARV-825 treatment. (B) Statistical analysis of M2-type macrophages (F4/80+CD206+) in vivo (n = 3). Data are presented as mean ± SEM. (C) Flow cytometry detection of M2-type macrophages in BMDM after different treatments in vitro. (D) Statistical analysis of M2-type macrophages (F4/80+CD206+) in BMDM (n = 3). Data are presented as mean ± SEM. ∗∗P < 0.01 and ∗∗∗∗P < 0.0001.
Based on the above result of tumor microenvironment, further experiment was conducted to confirm if ARV-825 could inhibit M2 polarization of macrophages in vitro. Firstly, we selected the concentration of 50 ng/mL ARV-825 for treatment in subsequent experiments. Under this concentration, macrophages viability was not affected and BRD4 could be degraded (Supporting Information Fig. S13). Then, bone marrow derived macrophages (BMDMs) were treated by different agents (0, IL4, DMSO + IL4 and ARV-825 + IL4). In the IL4 induced M2 macrophage polarization model, IL4 markedly augmented the proportion of M2 macrophage compared with untreated cells, whereas the ARV-825 and IL4 co-treatment group showed a significant reduction of M2-type macrophages compared to DMSO and IL4 co-treatment group, suggesting that ARV-825 could inhibit IL4-induced M2 polarization of macrophages in vitro (Fig. 6C and D). Macrophages polarization is often accompanied by changes in cell phenotypes including IRF4, Ym1, Arg1, CCL17, CCL22 and Mrc1, etc. To explore whether ARV-825 changed the transcription of M2 macrophages related genes, we treated BMDMs with ARV-825 in the presence of IL4 and subsequently implemented RNA-seq analysis. As shown in Fig. 7A and B, ARV-825 significantly down-regulated genes associated with M2 macrophages such as IRF4, Ym1, Arg1 and CCL22. To further verify RNA-seq analysis results, RT-qPCR was performed to detect the effect of ARV-825 on the transcription level of M2-like macrophage markers. As demonstrated in Fig. 7C, ARV-825 obviously reduced mRNA expression levels of M2-like macrophage markers (Fizz1, Ym1, Arg1, IRF4, Mrc1, CCL17, CCL22, IL10), which was consistent with RNA-seq analysis results. In addition, a significant increase of protein expression levels of M2-like macrophage markers (Fizz1, Ym1+Ym2, IRF4, Arg1), determined by WB assay, was observed in BMDMs after IL4 treatment, while these proteins were substantially restrained by ARV-825 treatment (Fig. 7D). The results were in accord with the mRNA levels detected by RT-qPCR, which further validated the inhibitory effect of ARV-825 on the IL4-induced M2 polarization of macrophages and implied that inhibition of ARV-825 on M2 macrophage polarization might be another mechanism of its anti-tumor activity.
Figure 7.

ARV-825-mediated suppression of IL4-induced M2 macrophages polarization. (A) Heatmap of statistically significant (FDR < 0.05) differentially gene expression in different treated macrophages by RNA-seq profiling. (B) Heatmap of statistically significant (FDR < 0.05) differentially gene expression related to M2 macrophages in different treatments. (C) RT-qPCR detection of mRNA expression related to M2 macrophages (Fizz1, Ym1, Arg1, IRF4, Mrc1, CCL17, CCL22, IL10) in BMDMs after different treatments (n = 3). (D) Western blot analysis of M2 macrophages-related proteins expression (Fizz1, Ym1, IRF4 and Arg1). Data are presented as mean ± SEM. ∗∗P < 0.01, ∗∗∗P < 0.001 and ∗∗∗∗P < 0.0001.
3.5. Study on the mechanism of ARV-825 inhibiting M2 macrophages polarization
Through analysis of the TIMER database, we found that increased BRD4 gene expression, high-density macrophages infiltration and high-density M2 macrophages infiltration were associated with poor prognosis of glioma patients, and BRD4 expression was positively correlated with macrophage infiltration level in gliomas (Supporting Information Fig. S14). This phenomenon, together with the inhibition of ARV-825 on M2 macrophage polarization mentioned above, implied that BRD4 may be associated with M2 macrophages. Meanwhile, in IL4-induced M2 macrophage polarization model, IL4 treatment enhanced the mRNA and protein expression level of BRD4, yet the BRD4 expression was impaired in the presence of ARV-825 (Supporting Information Fig. S15). The polarization of macrophages is regulated by gene transcription. The transcription factors that promote the M1 macrophage polarization are STAT1, IRF5, AP-1 and NF-κB, etc., while the transcription factors that promote the polarization of M2 macrophage are STAT6, IRF4, PPARγ and C/EBPβ, etc.57, 58, 59. BRD4, as a regulator of gene transcription, might participate in macrophages polarization through regulating the transcription of related gene. Previous results showed that ARV-825 could significantly down-regulate the phenotypic gene IRF4 of M2 macrophages. Whereupon, we speculated that BRD4 might regulate the transcription factor IRF4 to promote M2 polarization of macrophages, and ARV-825 could inhibit this effect (Fig. 8A). To verity this point, IRF4-Luc gene was constructed by inserting the IRF4 gene promoter sequence (−1562 to +122) into the luciferase reporter vector pGL3-Basic (Fig. 8B), and double luciferase activity detection was performed subsequently. As shown in Fig. 8C, in RAW264.7 macrophages, compared with the only IRF4-Luc transfection, the ARV-825 and IRF4-Luc transfection co-treatment significantly reduced the luciferase activity transcribed by IRF4 promoter. Furthermore, as confirmed by chromatin immunoprecipitation PCR (ChIP-PCR), BRD4 showed a binding effect on the promoter of IRF4, whereas ARV-825 inhibited this effect (Fig. 8D). These results suggested that inhibiting IRF4 transcription was a mechanism of suppression of ARV-825 on M2 macrophage polarization, and BRD4 might participate in the macrophages polarization process through interaction with IRF4 in gliomas.
Figure 8.

Mechanisms of ARV-825 repressing M2 macrophages polarization. (A) The diagram of BRD4 acting on the IRF4 gene promoter. (B) The diagram of ARV-825 reducing luciferase gene transcription initiated by the IRF4 promoter through inhibiting BRD4. (C) Relative IRF4 luciferase reporter activity in RAW264.7 macrophages treated with or without ARV-825 for 48 h detected by dual-luciferase reporter assay (n = 3). (D) ChIP-qPCR analysis of IRF4 promoter in DMSO or ARV-825 treated RAW264.7 macrophages for 48 h using IgG or anti-BRD4 (n = 3). (E) Western blot analysis of proteins (P-STAT6, STAT6, P-AKT, AKT, P-STAT3 and STAT3) related to M2 macrophages polarization pathways. Data are presented as mean ± SEM. No significant difference is mark with ns. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 and ∗∗∗∗P < 0.0001.
Additionally, M2 polarization of macrophages also involves increased phosphorylation levels of STAT6, STAT3 and AKT60,61. We found that ARV-825 reduced the phosphorylation levels of STAT6, STAT3 and AKT (Fig. 8E), implying that ARV-825 might also attenuate IL4-induced M2 macrophages polarization by inhibiting the phosphorylation of STAT6, STAT3 and AKT.
Herein, the small size brain-targeting SPP-ARV-825 drug micelle was developed for glioma therapy. Nanoplatform delivery system has been widely used for drug delivery due to its advantages of good bioavailability, sustained drug release and reduced side effects62, 63, 64. As a promising anti-tumor drug candidate, the low water solubility of ARV-825 limits its application in cancer therapy. The mPEG-PDLLA-based micelles greatly improved the bioavailability of ARV825 in the study. The hydrophobic PDLLA incorporated insoluble ARV-825 inside the core structure, and the PEGylated shell structure improved systemic delivery of drug and reduced potential toxicity65. SP peptide modification endowed drug micelles with the ability to actively target to the NK-1R overexpressed in endothelial cells and gliomas43,45, meanwhile the unique enhanced permeability and retention (EPR) effects of nanoparticles enabled passive retention of drugs at tumor sites66,67. Therefore, the SPP-micelles were able to deliver ARV-825 drug through BBB and reach tumor site for glioma therapy. In vitro, SPP-ARV-825 had shown remarkable effect on suppressing glioma cell proliferation, blocking cell cycle in G0/G1 phase and inducing cell apoptosis involving multiple pathways. In vivo, SPP-ARV-825 efficiently attenuated glioma subcutaneous tumor and xenograft tumor growth, exhibiting stronger anti-tumor effect than PP-ARV-825 and ARV-825. Tumor proliferation and angiogenesis were significantly suppressed while tumor apoptosis was promoted by SPP-ARV-825 treatment. Intriguingly, SPP-ARV-825 provided great benefits for the inhibition of M2 macrophages polarization via attenuating IRF4 promoter transcription and the phosphorylation pathways of STAT6, STAT3 and AKT, suggesting that BRD4 might participate in M2 macrophages polarization by acting on IRF4 promoter and SPP-ARV-825 might further exert anti-glioma effect by reducing M2 macrophages in tumor. Thus, our findings suggest SPP-ARV-825 plays a versatile anti-tumor role including altering tumor biological behaviors and regulating tumor-associated macrophages. Taken together, SPP-ARV-825 would be a promising drug candidate for glioma therapy in future.
4. Conclusions
In summary, we constructed a BRD4 degrader-loaded micelle with brain targetability and intensively explored its anti-glioma efficacy and mechanisms in the study. Results demonstrated that the SPP-ARV-825 micelle could achieve effective anti-tumor effect by glioma cells proliferation impediment, apoptosis induction and M2 macrophages polarization reduction in glioma therapy. Overall, this work proposes a potential strategy for targeted therapy of glioma and may have a significant impact on BRD4-targeted cancer treatment.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 82172630, 81972347 and 82003493), the Key R&D Projects of the Science and Technology Department of Sichuan Province (No. 2020YFS0213, China) and the 1·3·5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (No. ZYJC21022, No. ZYYC21001 and 2019HXFH017, China).
Author contributions
Xiang Gao conceived and supervised the work. Tingting Yang, Yuzhu Hu and Junming Miao participated in the design and implementation of the experiment. Jing Chen carried out data processing and analysis. Jiagang Liu and Yongzhong Cheng revised the manuscript. All authors discussed and approved the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.02.009.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Ostrom Q.T., Patil N., Cioffi G., Waite K., Kruchko C., Barnholtz-Sloan J.S. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013‒2017. Neuro Oncol. 2020;22(12 Suppl 2):iv1–iv96. doi: 10.1093/neuonc/noaa200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan A.C., Ashley D.M., López G.Y., Malinzak M., Friedman H.S., Khasraw M. Management of glioblastoma: state of the art and future directions. CA A Cancer J Clin. 2020;70:299–312. doi: 10.3322/caac.21613. [DOI] [PubMed] [Google Scholar]
- 3.Bell E.H., Zhang P., Shaw E.G., Buckner J.C., Barger G.R., Bullard D.E., et al. Comprehensive genomic analysis in NRG oncology/RTOG 9802: a phase III trial of radiation versus radiation plus procarbazine, lomustine (CCNU), and vincristine in high-risk low-grade glioma. J Clin Oncol. 2020;38:3407–3417. doi: 10.1200/JCO.19.02983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quinn J.A., Pluda J., Dolan M.E., Delaney S., Kaplan R., Rich J.N., et al. Phase II trial of carmustine plus O6-benzylguanine for patients with nitrosourea-resistant recurrent or progressive malignant glioma. J Clin Oncol. 2002;20:2277–2283. doi: 10.1200/JCO.2002.09.084. [DOI] [PubMed] [Google Scholar]
- 5.Cuddapah V.A., Robel S., Watkins S., Sontheimer H. A neurocentric perspective on glioma invasion. Nat Rev Neurosci. 2014;15:455–465. doi: 10.1038/nrn3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alifieris C., Trafalis D.T. Glioblastoma multiforme: pathogenesis and treatment. Pharmacol Ther. 2015;152:63–82. doi: 10.1016/j.pharmthera.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 7.van Tellingen O., Yetkin-Arik B., de Gooijer M.C., Wesseling P., Wurdinger T., de Vries H.E. Overcoming the blood‒brain tumor barrier for effective glioblastoma treatment. Drug Resist Updates. 2015;19:1–12. doi: 10.1016/j.drup.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Han L., Jiang C. Evolution of blood‒brain barrier in brain diseases and related systemic nanoscale brain-targeting drug delivery strategies. Acta Pharm Sin B. 2021;11:2306–2325. doi: 10.1016/j.apsb.2020.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen P., Zhao D., Li J., Liang X., Li J., Chang A., et al. Symbiotic macrophage-glioma cell interactions reveal synthetic lethality in PTEN-null glioma. Cancer Cell. 2019;35:868–884.e6. doi: 10.1016/j.ccell.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hambardzumyan D., Gutmann D.H., Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19:20–27. doi: 10.1038/nn.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gangoso E., Southgate B., Bradley L., Rus S., Galvez-Cancino F., McGivern N., et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell. 2021;184:2454–2470.e26. doi: 10.1016/j.cell.2021.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L., Cui J., Zhao Y., Liu X., Chen L., Xia Y., et al. KDM6A-ARHGDIB axis blocks metastasis of bladder cancer by inhibiting Rac1. Mol Cancer. 2021;20:77. doi: 10.1186/s12943-021-01369-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohnken R., Wen J., Mundy-Bosse B., McConnell K., Keiter A., Grinshpun L., et al. Diminished microRNA-29b level is associated with BRD4-mediated activation of oncogenes in cutaneous T-cell lymphoma. Blood. 2018;131:771–781. doi: 10.1182/blood-2017-09-805663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu L., Chen Z., Lin X., Tian L., Su Q., An P., et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. 2020;27:255–268. doi: 10.1038/s41418-019-0353-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Echevarría-Vargas I.M., Reyes-Uribe P.I., Guterres A.N., Yin X., Kossenkov A.V., Liu Q., et al. Co-targeting BET and MEK as salvage therapy for MAPK and checkpoint inhibitor-resistant melanoma. EMBO Mol Med. 2018;10 doi: 10.15252/emmm.201708446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donati B., Lorenzini E., Ciarrocchi A. BRD4 and cancer: going beyond transcriptional regulation. Mol Cancer. 2018;17:164. doi: 10.1186/s12943-018-0915-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheung K.L., Zhang F., Jaganathan A., Sharma R., Zhang Q., Konuma T., et al. Distinct roles of Brd2 and Brd4 in potentiating the transcriptional program for Th17 cell differentiation. Mol Cell. 2017;65:1068–1080.e5. doi: 10.1016/j.molcel.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong X., Hu X., Bao Y., Li G., Yang X.D., Slauch J.M., et al. Brd4 regulates NLRC4 inflammasome activation by facilitating IRF8-mediated transcription of Naips. J Cell Biol. 2021;220 doi: 10.1083/jcb.202005148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang N., Wu R., Tang D., Kang R. The BET family in immunity and disease. Signal Transduct Target Ther. 2021;6:23. doi: 10.1038/s41392-020-00384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amorim S., Stathis A., Gleeson M., Iyengar S., Magarotto V., Leleu X., et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3:e196–e204. doi: 10.1016/S2352-3026(16)00021-1. [DOI] [PubMed] [Google Scholar]
- 21.Shu S., Wu H.J., Ge J.Y., Zeid R., Harris I.S., Jovanović B., et al. Synthetic lethal and resistance interactions with BET bromodomain inhibitors in triple-negative breast cancer. Mol Cell. 2020;78:1096–1113.e8. doi: 10.1016/j.molcel.2020.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan S., Zhang X., Lv D., Zhang Q., He Y., Zhang P., et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat Med. 2019;25:1938–1947. doi: 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burslem G.M., Crews C.M. Proteolysis-targeting chimeras as therapeutics and tools for biological discovery. Cell. 2020;181:102–114. doi: 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y., Jiang X.Y., Feng F., Liu W.Y., Sun H.P. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B. 2020;10:207–238. doi: 10.1016/j.apsb.2019.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saenz D.T., Fiskus W., Qian Y., Manshouri T., Rajapakshe K., Raina K., et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia. 2017;31:1951–1961. doi: 10.1038/leu.2016.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J., Qian Y., Altieri M., Dong H., Wang J., Raina K., et al. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piya S., Mu H., Bhattacharya S., Lorenzi P.L., Davis R.E., McQueen T., et al. BETP degradation simultaneously targets acute myelogenous leukemia stem cells and the microenvironment. J Clin Invest. 2019;129:1878–1894. doi: 10.1172/JCI120654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang X.H., Lee H.C., Shirazi F., Baladandayuthapani V., Lin H., Kuiatse I., et al. Protein targeting chimeric molecules specific for bromodomain and extra-terminal motif family proteins are active against pre-clinical models of multiple myeloma. Leukemia. 2018;32:2224–2239. doi: 10.1038/s41375-018-0044-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saenz D.T., Fiskus W., Manshouri T., Mill C.P., Qian Y.M., Raina K., et al. Targeting nuclear beta-catenin as therapy for post-myeloproliferative neoplasm secondary AML. Leukemia. 2019;33:1373–1386. doi: 10.1038/s41375-018-0334-3. [DOI] [PubMed] [Google Scholar]
- 30.Sun B., Fiskus W., Qian Y., Rajapakshe K., Raina K., Coleman K.G., et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia. 2018;32:343–352. doi: 10.1038/leu.2017.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain N., Hartert K., Tadros S., Fiskus W., Havranek O., Ma M.C.J., et al. Targetable genetic alterations of TCF4 (E2-2) drive immunoglobulin expression in diffuse large B cell lymphoma. Sci Transl Med. 2019;11 doi: 10.1126/scitranslmed.aav5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Raina K., Lu J., Qian Y.M., Altieri M., Gordon D., Rossi A.M.K., et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2016;113:7124–7129. doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan J., Duan X., Zhang F., Ban X., Mao J., Cao M., et al. Theranostic nanomedicine for synergistic chemodynamic therapy and chemotherapy of orthotopic glioma. Adv Sci. 2020;7:2003036. doi: 10.1002/advs.202003036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Mascolo D., Palange A.L., Primavera R., Macchi F., Catelani T., Piccardi F., et al. Conformable hierarchically engineered polymeric micromeshes enabling combinatorial therapies in brain tumours. Nat Nanotechnol. 2021;16:820–829. doi: 10.1038/s41565-021-00879-3. [DOI] [PubMed] [Google Scholar]
- 35.Ju X., Miao T., Chen H., Ni J., Han L. Overcoming Mfsd2a-mediated low transcytosis to boost nanoparticle delivery to brain for chemotherapy of brain metastases. Adv Healthc Mater. 2021;10 doi: 10.1002/adhm.202001997. [DOI] [PubMed] [Google Scholar]
- 36.Khan N., Ni J., Ju X.F., Miao T.T., Chen H.Y., Han L. Escape from abluminal LRP1-mediated clearance for boosted nanoparticle brain delivery and brain metastasis treatment. Acta Pharm Sin B. 2021;11:1341–1354. doi: 10.1016/j.apsb.2020.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z., Guo J., Liu X., Sun J., Gao W. Temperature-triggered micellization of interferon alpha-diblock copolypeptide conjugate with enhanced stability and pharmacology. J Control Release. 2020;328:444–453. doi: 10.1016/j.jconrel.2020.08.065. [DOI] [PubMed] [Google Scholar]
- 38.Xu F., Huang X., Wang Y., Zhou S. A size-changeable collagenase-modified nanoscavenger for increasing penetration and retention of nanomedicine in deep tumor tissue. Adv Mater. 2020;32 doi: 10.1002/adma.201906745. [DOI] [PubMed] [Google Scholar]
- 39.Gupta M.K., Martin J.R., Dollinger B.R., Hattaway M.E., Duvall C.L. Thermogelling, ABC triblock copolymer platform for resorbable hydrogels with tunable, degradation-mediated drug release. Adv Funct Mater. 2017;27:1704107. doi: 10.1002/adfm.201704107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L., Huang X., You X., Yi T., Lu B., Liu J., et al. Nanoparticle enhanced combination therapy for stem-like progenitors defined by single-cell transcriptomics in chemotherapy-resistant osteosarcoma. Signal Transduct Target Ther. 2020;5:196. doi: 10.1038/s41392-020-00248-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y., Zhang M., Jin H., Li D., Xu F., Wu A., et al. Glioma dual-targeting nanohybrid protein toxin constructed by intein-mediated site-specific ligation for multistage booster delivery. Theranostics. 2017;7:3489–3503. doi: 10.7150/thno.20578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang C.W., Chuang C.P., Chen Y.J., Wang H.Y., Lin J.J., Huang C.Y., et al. Integrin αβ-targeting ferritin nanocarrier traverses the blood‒brain barrier for effective glioma chemotherapy. J Nanobiotechnol. 2021;19:180. doi: 10.1186/s12951-021-00925-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Królicki L., Bruchertseifer F., Kunikowska J., Koziara H., Królicki B., Jakuciński M., et al. Safety and efficacy of targeted alpha therapy with Bi-DOTA-substance P in recurrent glioblastoma. Eur J Nucl Med Mol Imag. 2019;46:614–622. doi: 10.1007/s00259-018-4225-7. [DOI] [PubMed] [Google Scholar]
- 44.Schonberg D.L., Miller T.E., Wu Q., Flavahan W.A., Das N.K., Hale J.S., et al. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell. 2015;28:441–455. doi: 10.1016/j.ccell.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruan C., Liu L., Lu Y., Zhang Y., He X., Chen X., et al. Substance P-modified human serum albumin nanoparticles loaded with paclitaxel for targeted therapy of glioma. Acta Pharm Sin B. 2018;8:85–96. doi: 10.1016/j.apsb.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tao Z., Li X., Wang H., Chen G., Feng Z., Wu Y., et al. BRD4 regulates self-renewal ability and tumorigenicity of glioma-initiating cells by enrichment in the Notch1 promoter region. Clin Transl Med. 2020;10:e181. doi: 10.1002/ctm2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Minko T. Nanoformulation of BRD4-degrading PROTAC: improving druggability to target the 'undruggable' MYC in pancreatic cancer. Trends Pharmacol Sci. 2020;41:684–686. doi: 10.1016/j.tips.2020.08.008. [DOI] [PubMed] [Google Scholar]
- 48.Noblejas-López M.D.M., Nieto-Jimenez C., Burgos M., Gómez-Juárez M., Montero J.C., Esparís-Ogando A., et al. Activity of BET-proteolysis targeting chimeric (PROTAC) compounds in triple negative breast cancer. J Exp Clin Cancer Res. 2019;38:383. doi: 10.1186/s13046-019-1387-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong P.P., Muñoz-Félix J.M., Hijazi M., Kim H., Robinson S.D., De Luxán-Delgado B., et al. Cancer burden is controlled by mural cell-β3-integrin regulated crosstalk with tumor cells. Cell. 2020;181:1346–1363. doi: 10.1016/j.cell.2020.02.003. e21. [DOI] [PubMed] [Google Scholar]
- 50.Hoxhaj G., Manning B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20:74–88. doi: 10.1038/s41568-019-0216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zong D., Gu J., Cavalcante G.C., Yao W., Zhang G., Wang S., et al. BRD4 levels determine the response of human lung cancer cells to BET degraders that potently induce apoptosis through suppression of Mcl-1. Cancer Res. 2020;80:2380–2393. doi: 10.1158/0008-5472.CAN-19-3674. [DOI] [PubMed] [Google Scholar]
- 52.Shi C., Ye Z., Han J., Ye X., Lu W., Ji C., et al. BRD4 as a therapeutic target for nonfunctioning and growth hormone pituitary adenoma. Neuro Oncol. 2020;22:1114–1125. doi: 10.1093/neuonc/noaa084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmieder A., Michel J., Schönhaar K., Goerdt S., Schledzewski K. Differentiation and gene expression profile of tumor-associated macrophages. Semin Cancer Biol. 2012;22:289–297. doi: 10.1016/j.semcancer.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 54.Dey A., Yang W., Gegonne A., Nishiyama A., Pan R., Yagi R., et al. BRD4 directs hematopoietic stem cell development and modulates macrophage inflammatory responses. EMBO J. 2019;38 doi: 10.15252/embj.2018100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu H., Bengsch F., Svoronos N., Rutkowski M.R., Bitler B.G., Allegrezza M.J., et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16:2829–2837. doi: 10.1016/j.celrep.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yin M., Guo Y., Hu R., Cai W.L., Li Y., Pei S., et al. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat Commun. 2020;11:1833. doi: 10.1038/s41467-020-15290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmidt S.V., Krebs W., Ulas T., Xue J., Baßler K., Günther P., et al. The transcriptional regulator network of human inflammatory macrophages is defined by open chromatin. Cell Res. 2016;26:151–170. doi: 10.1038/cr.2016.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kaikkonen M.U., Spann N.J., Heinz S., Romanoski C.E., Allison K.A., Stender J.D., et al. Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell. 2013;51:310–325. doi: 10.1016/j.molcel.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lawrence T., Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 60.Huang S.C.C., Smith A.M., Everts B., Colonna M., Pearce E.L., Schilling J.D., et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity. 2016;45:817–830. doi: 10.1016/j.immuni.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tao S., Chen Q., Lin C., Dong H. Linc00514 promotes breast cancer metastasis and M2 polarization of tumor-associated macrophages via Jagged1-mediated notch signaling pathway. J Exp Clin Cancer Res. 2020;39:191. doi: 10.1186/s13046-020-01676-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Izci M., Maksoudian C., Manshian B.B., Soenen S.J. The use of alternative strategies for enhanced nanoparticle delivery to solid tumors. Chem Rev. 2021;121:1746–1803. doi: 10.1021/acs.chemrev.0c00779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang X., Duan X., Hu Y., Tang Z., Miao C., Tao W., et al. One-step and facile synthesis of peptide-like poly(melphalan) nanodrug for cancer therapy. Nano Today. 2021;37:101098. [Google Scholar]
- 64.You X., Wang L., Wang L., Wu J. Rebirth of aspirin synthesis by-product: prickly poly (salicylic acid) nanoparticles as self-anticancer drug carrier. Adv Funct Mater. 2021;31:2100805. [Google Scholar]
- 65.Suk J.S., Xu Q.G., Kim N., Hanes J., Ensign L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99:28–51. doi: 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fang J., Islam W., Maeda H. Exploiting the dynamics of the EPR effect and strategies to improve the therapeutic effects of nanomedicines by using EPR effect enhancers. Adv Drug Deliv Rev. 2020;157:142–160. doi: 10.1016/j.addr.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 67.Shi Y., van der Meel R., Chen X., Lammers T. The EPR effect and beyond: strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics. 2020;10:7921–7924. doi: 10.7150/thno.49577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.