

Abstract
The p21 activated kinase 4 (PAK4) is serine/threonine protein kinase that is critical for cancer progression. Guided by X-ray crystallography and structure-based optimization, we report a novel subseries of C-3-substituted 6-ethynyl-1H-indole derivatives that display high potential and specificity towards group II PAKs. Among these inhibitors, compound 55 exhibited excellent inhibitory activity and kinase selectivity, displayed superior anti-migratory and anti-invasive properties against the lung cancer cell line A549 and the melanoma cell line B16. Compound 55 exhibited potent in vivo antitumor metastatic efficacy, with over 80% and 90% inhibition of lung metastasis in A549 or B16-BL6 lung metastasis models, respectively. Further mechanistic studies demonstrated that compound 55 mitigated TGF-β1-induced epithelial-mesenchymal transition (EMT).
Key words: Anti-cancer, PAK4 inhibitor, Kinase selectivity, Cocrystallization, Lung metastasis, EMT
Graphical abstract
6-Ethynyl-1H-indole derivative 55 is a potent and selective PAK4 inhibitor (Ki = 10.2 nmol/L), which effectively inhibits lung metastasis in A549 or B16-BL6 cells.

1. Introduction
The p21 activated kinases (PAKs) are key signaling nodes in small GTPase Cdc42/Rac-PAKs signaling pathways and play pivotal roles in a wide range of cell processes, including cytoskeletal organization, cell motility and morphology1, 2, 3. PAKs are serine/threonine protein kinases that are subdivided broadly into two categories based on their structural homology and regulatory properties: Group I PAKs consisting of PAK1, 2 and 3; and Group II PAKs consisting of PAK4, 5 and 62. The two groups share approximately 50% sequence identity in the GTPase-binding domain (GBD) and kinase domain and differ in tissue expression profiles and subcellular localization. An activation mechanism indicates that they may recognize different substrates and have unique cellular functions4,5.
PAK4, the most extensively studied isoform in group II PAKs, frequently has been found to upregulate at the mRNA or protein level in different types of cancers6. Several studies have indicated that PAK4 is associated with cellular transformation, proliferation and survival, which are required for efficient migration or invasion in prostate, ovarian, pancreatic, breast and lung cancer cell lines7, 8, 9, 10. Subsequent research demonstrated that PAK4 could promote cell growth via ERK- and AKT-regulated signaling11,12, and enhance tumor metastasis through the HGF/LIMK1/cofilin and MEK-1/ERK1/2/MMP2 pathways13,14. In addition, a series of studies showed that PAK4 played an important role in the drug resistance of a variety of tumors15, 16, 17.
Several recent studies have demonstrated that PAK4 also plays an important role in the regulation of antitumor immune responses. Firstly, genetic and pharmacological inhibition of PAK4 disrupts WNT-β-catenin signaling, increases intratumoral T cell infiltration, and sensitizes tumors to PD-1 blockade in a mouse melanoma model18,19. In addition, PAK4 knockout or inhibition induces re-expression of adhesion proteins in endothelial cells, reduces vascular abnormalities, normalizes the vascular microenvironment of tumors, improves T-cell infiltration, and sensitizes glioblastoma to CAR-T immunotherapy20, which provides an additional benefit for PAK4-targeted cancer therapies.
The significance of PAK4 in cancer provides a rationale for the development of PAK4 inhibitors as an antitumor therapeutic strategy, which has resulted in considerable efforts to develop PAK4 inhibitors in recent years, with numerous reported PAK4 inhibitors (Fig. 1)5,21, 22, 23, 24. Up to now, only two PAK4 inhibitors, PF-3758309 (1) and KPT-9274 (2), have reached phase I clinical trial. Unfortunately, the clinical trials of PF-3758309 (NCT00932126) were terminated due to undesirable characteristics25. KPT-9274 (NCT02702492: terminated; NCT04281420: Recruiting), a dual PAK4/NAMPT inhibitor, directly induces PAK4 destabilization instead of inhibiting PAK4 kinase activity, where the degradation mechanisms are yet to be disclosed. KPT-9274 alone or in combination with niacin ER or nivolumab is used for the treatment of advanced solid tumors or non-Hodgkin's lymphoma26, 27, 28. GNE-2861 (3) is a type I 1/2 kinase inhibitor, which was the first reported selective group II PAKs inhibitor (870-fold vs PAK1) based on the unique flexibility of the lipophilic back pocket21. We have also committed to the discovery of selective PAK4 inhibitors as new therapeutic agents for more than a decade29, 30, 31, 32, and a preclinical candidate, CZh-226 (4)29, was finally identified as one of the most potent and selective PAK4 inhibitor (346-fold vs PAK1) in our library thus far.
Figure 1.

The structures of representative PAK4 inhibitors.
GNE-2861 and CZh-226 belong to different types of specific ATP competitive inhibitors, and their selectivity mechanism provides insightful instruction for discovering novel selective PAK4 inhibitors. GNE-2861 targets the back pocket in PAK4 with lipophilic and hydrogen bonding interactions to achieve enhanced selectivity toward PAK4, whereas CZh-226 targets the PAK4 amino acid Asp458 (from the conserved Asp-Phe-Gly [DFG] motif), a conserved amino acid oriented contrary to Asp407 in PAK1. Exquisite selectivity for PAK4 over other PAKs, as well as generally high kinome selectivity, has been achieved by the two series of inhibitors. Furthermore, these approaches provide not only guidance for the design of selective inhibitors, but also are useful tools for understanding the complex biology driven by PAK4.
In this paper, a new series of selective PAK4 inhibitors were developed with the propargyl alcohol group reserved to achieve the isoform selectivity, which was verified by X-ray crystallography diffraction data. Further investigations, including kinase selectivity, mechanism of action, antitumor metastasis effects in vitro and in vivo and preliminary pharmacokinetic characterization, were also carried out for the optimal compound 55.
2. Results and discussion
2.1. Design rationale and structure–activity relationship (SAR)
GNE-2861, a type I 1/2 PAK4 inhibitor, has been reported by Genentech21 to show good selectivity among the kinase family, partially due to the propargyl alcohol substitution. Considering most kinases are unable to accommodate this substituent due to a steric clash with a large gatekeeper residue in their back pocket33, the structural features of GNE-2861 may be a good starting point for discovery of highly selective PAK4 inhibitors. As shown in Fig. 2A, the skeleton 4-(1H-benzo[d]imidazole-1-yl)pyrimidin-2-amine (region 1), forms hydrogen bonds with the PAK4 hinge region and serves as a scaffold for the overall structure. The scaffold structure from the benzimidazole to the indazole or azaindole provides a corresponding position for occupying the ribose pocket of PAK4. Moreover, superposition of GNE-2861/PF-3758309 (Fig. 2B) and GNE-2861/CZh-226 (Fig. 2C) suggests that the hydrophobic pocket is important for recognition of the hydrophobic substitution on the amino group of aminopyrimidine (region 2)22,29, with increased binding affinity for PAK4. Meanwhile, the ribose pocket (region 3), which is also found in various kinase inhibitors29,34, 35, 36, 37, can interact with the 3-position of the core fragment, providing additional interactions to enhance the binding strength. Following this structure-based analysis, our research focused on the modification of the above three areas (Fig. 2A) while keeping the propargyl alcohol unchanged to maintain the kinase selectivity.
Figure 2.

Cocrystal structure of GNE-2861/PAK4 and ATP/PAK4 that guided the structural optimization. (A) Focused regions in the structural optimization and SAR analyses. (B) Superimposition of GNE-2861 (wheat, PDB ID: 4O0V) and PF-3758309 (green, PDB ID: 2X4Z). (C) Superimposition of GNE-2861 (wheat, PDB ID: 4O0V) and CZh-226 (yellow, PDB ID: 5XVG). (D) Superimposition of GNE-2861 (wheat, PDB ID: 4O0V) and ATP (cyan, PDB ID: 4FIE). Related residues are shown as corresponding sticks. Red dashes represent hydrogen bonds.
2.2. The effect of region I
First, we explored the effects of different moieties in region I (Fig. 2A). The 2-aminopyrimidine and propargyl alcohol groups were partially fixed and the benzimidazole was replaced by different azaindoles or indazoles via scaffold hopping. The electron density of the aromatic ring was changed when a heteroatom was introduced into different positions on the aromatic system, which accordingly influenced the biochemical activities, as shown in Table 1. Indole analogue 5 exhibited moderate biochemical potency against PAK4 with a Ki value of 0.066 μmol/L. The indazole analogue 6 and 4-azaindole analogue 7 led to a slight increase in potency, and the 5-azaindole analogue 8 exhibited the most potent biochemical activity (Ki = 0.013 μmol/L) among the bioisosteric groups. However, the 7-azaindole analogue 9 significantly decreased the PAK4 inhibitory activity compared to compound 5. A docking study indicated that the acidic amino acid Asp458 and basic amino acid Lys350 near the 5-position nitrogen atom of 5-azaindole of 8 provided a friendly electrical environment (docking result is not shown). A new analog 5a with a fluorine substituent at the 5-position was prepared, but it was found that a 5-fluoro group was not favorable in this region. Meanwhile, although 8 exhibited the best potency against PAK4, the 1,6-addition of glutathione to the electron-deficient alkyne may provide electron-withdrawing nature to azaindole, which could speed up the metabolic activity of 838,39. Thus, an indole core would be preferable to maintain favorable pharmacokinetic properties in subsequent modifications.
Table 1.
Kinase inhibitory activities of compounds 5a–5d and 5–9 against PAK4.
| Compd. | R1 | PAK4 Ki (μmol/L)a | cLogPb | PSAb | LEc |
|---|---|---|---|---|---|
| 5 |  |
0.066 | 3.85 | 72.53 | 0.39 |
| 6 |  |
0.040 | 3.24 | 85.96 | 0.40 |
| 7 |  |
0.036 | 3.20 | 85.50 | 0.40 |
| 8 |  |
0.013 | 3.21 | 84.39 | 0.42 |
| 9 |  |
0.183 | 3.40 | 84.25 | 0.36 |
| 5a |  |
0.082 | 4.11 | 72.59 | 0.38 |
| 5b |  |
0.370 | 2.91 | 97.82 | 0.33 |
| 5c |  |
0.229 | 3.80 | 72.77 | 0.36 |
| 5d | 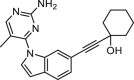 |
0.182 | 4.08 | 72.34 | 0.35 |
| CZh-226 | 0.005 | 2.75 | 99.49 | 0.39 | |
| GNE-2861 | 0.035 (reported 0.003)21 | 3.41 | 103.30 | 0.34 |
PAK4 kinase inhibition was determined using a homogeneous time-resolved fluorescence (HTRF) assay. The Ki values are the average of two independent experiments reported as the mean value.
The clogP values and polar surface area (PSA) were calculated by the Qikprop software.
Ligand efficiency = (−1.4 Log Ki)/(n heavy atoms).
The aminopyrimidine group bound to the hinge region was further explored. Considering the conservative binding mode of the H-bond acceptor-donor-acceptor in the hinge, 5b with a 2,4-diaminopyrimidine substituent was designed. The dramatic decrease in inhibitory activity could be attributed to the fact that one hydrogen bond is essential for the tight binding mode; the number of hydrogen bonds in the hinge-binding region may not be positively correlated with potency40. Compound 5b, an analogue of 5 with a 6-aminopyrimidine substitution, lost most of its PAK4 inhibitory activity, while another 2-amino-5-methylpyrimidine analogue, 5d, showed similar inhibitory activity compared to 5b and 5c but relatively weaker activity than 5. Overall, among the nine compounds, 5, with the indole and 2-aminopyrimidine at region I, was most suitable for the following optimization.
2.3. The effect of region II
There is a hydrophobic cleft orthogonal to the plane of the indole ligands occupied by quinazoline, pyrimidine thiophene and other hydrophobic groups (Fig. 2B and C)22,29,30,41, revealing the spaces which tolerated hydrophobic substituents at the amino group of pyrimidine. As summarized for the generated 20 compounds in Table 2, compounds 10–13 with aromatic methylamine groups showed significantly decreased PAK4 inhibition compared to 5, while the ortho-para-halogen substitutions (11–13) of the benzyl group slightly improved the activity compared to 10. Compounds 15–26, with substituted aniline groups, showed comparable potency to 5. SAR analysis indicated that the meta-substitution of the compound is superior to its ortho- or para-substitution, like meta-chloroaniline 16 and meta-aminobenzenesulfonamide 26. The substitution of the alkyl analogues 27–29 resulted in a significant decrease in the PAK4 inhibitory activity.
Table 2.
Kinase inhibitory activities of compounds 10–29 against PAK4.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | R2 | PAK4 Ki (μmol/L)a | Compd. | R2 | PAK4 Ki (μmol/L)a | Compd. | R2 | PAK4 Ki (μmol/L)a |
| 10 |  |
0.623 | 17 |  |
0.157 | 24 |  |
0.298 |
| 11 | 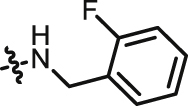 |
0.223 | 18 |  |
0.242 | 25 |  |
0.189 |
| 12 |  |
0.355 | 19 |  |
0.212 | 26 |  |
0.083 |
| 13 |  |
0.291 | 20 |  |
0.396 | 27 | 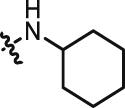 |
>10 |
| 14 |  |
7.455 | 21 |  |
0.173 | 28 |  |
0.680 |
| 15 | 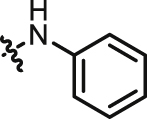 |
0.217 | 22 |  |
0.709 | 29 |  |
1.109 |
|
16 |
 |
0.105 | 23 |  |
0.230 | |||
PAK4 kinase inhibition was determined using a homogeneous time-resolved fluorescence (HTRF) assay. The Ki values are the average of two independent experiments, reported as the mean value.
2.4. The effect of region III
The ribose-binding pocket is surrounded by a series of hydrophobic and hydrophilic amino acids and is partially exposed to the solvent-exposed area, which tolerates a wide range of substituents, including a phenyl group, straight or branched alkyl chains, and cyclic amines. Thus, in the following study, we investigated a series of compounds with substituents at the C-3 position of the indole ring, which allowed the substituents to access the ribose pocket with additional binding affinity towards PAK4.
With the aim of finding suitable substituents that would optimally fit the ribose binding pocket with additional binding affinity toward PAK4, a broad range of substituents were investigated to identify the tolerated groups at the C-3 position. As found in Table 3, not all types of groups are tolerated at the C-3 position. The hydrophobic substituent analogues 30–36 showed a significant decrease in effect against PAK4, while the hydrophilic substitutions of 37–41 resulted in an increase in activity, among which the morpholine analogue 37 maintained similar activity relative to 5, and the N-methylpiperazine analogue 38 resulted in a significant increase in activity. Meanwhile, by comparing 39–41, it was found that, relative to the unsubstituted amide analogue 41 (Ki = 14 nmol/L), there was a gradient of decreased activity with the introduction of the methyl group (40 Ki = 0.044 μmol/L, 39 Ki = 0.082 μmol/L). Through the SAR investigation of these 10 compounds, it can be concluded that the ribose-binding pocket of PAK4 is mainly suitable for hydrophilic group binding, and the introduction of hydrophilic groups in the subsequent modification of the C-3 position is more favorable for affinity enhancement.
Table 3.
Kinase inhibitory activities of compounds 30–41 against PAK4.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compd. | R3 | PAK4 Ki (μmol/L)a | Compd. | R3 | PAK4 Ki (μmol/L)a | Compd. | R3 | PAK4 Ki (μmol/L)a |
| 30 |  |
0.593 | 34 |  |
0.094 | 38 |  |
0.038 |
| 31 |  |
0.804 | 35 |  |
0.376 | 39 |  |
0.082 |
| 32 |  |
0.243 | 36 |  |
0.455 | 40 |  |
0.044 |
| 33 |  |
0.239 | 37 |  |
0.119 | 41 |  |
0.014 |
PAK4 kinase inhibition was determined using a homogeneous time-resolved fluorescence (HTRF) assay. The Ki values are the average of two independent experiments reported as the mean value.
To gain insight into the binding mode of 41, the crystal structure of the PAK4 kinase domain in complex with 41 was solved (Fig. 3). The 2-aminopyrimidine is in proximity to the hinge region of PAK4, forming two key interactions with hinge amino acid Leu398. The propargyl alcohol passes the gatekeeper Met395, forming two hydrogen bonds in the lipophilic back pocket, accepts an H-bond from the backbone NH of Phe459 of the DFG motif and donates an H-bond to Glu366 on the α-C-helix. The amide group at the C-3 position occupies the ribose pocket but does not form any interactions. Analyzing the X-ray structure, we confirmed that the position of C-3 substituents is closer to the solvent-exposed area of the ribose, and the hydrophilic groups provide better binding affinity.
Figure 3.

Cocrystal structure of 41 and PAK4. (A) X-ray cocrystal structure of 41 bound to PAK4 (PDB ID: 7CMB, 2.3 Å resolution) and associated 2|Fo|−|Fc| omit map contoured at 1.0 σ level. (B) PAK4 is shown as a surface. Compound 41 is shown as a cyan stick structure. The key residues are shown as brown sticks. Red dashed lines indicate hydrogen bonds to key amino acids.
Compound 41 was incubated with mouse liver microsomes (MLM) to evaluate its metabolic stability. As shown in Supporting Information Table S3, 41 was rapidly metabolized in liver microsomes with a half-life of 23.7 min and a liver clearance of 105.5 mL/min·kg, suggesting that the stability of 41 needs to be further improved, although it exhibited excellent inhibitory activity.
Based on the insightful understanding of structural information, in particular the acidic Asp444 adjacent to the PAK4 ribose pocket, we hypothesized that the introduction of basic groups would achieve better inhibition by forming electrical complementarity (Table 4). Indeed, the introduction of various basic groups greatly enhanced the activity against PAK4, in particular, the piperazine analogue 42 significantly inhibited PAK4 activity, with a Ki value of 6.6 nmol/L. Compared to the morpholine analogue 37 (Ki = 119 nmol/L), the inhibitory capacity was increased 18-fold simply by replacing the oxygen atom with the nitrogen atom, whereas with replacement of the piperazinyl group with the 2-piperazinone group (56: Ki = 18.1 nmol/L) the introduction of the carbonyl group caused the nitrogen atom to lose its basicity, and the activity was significantly reduced. These observations suggest that the substitution of the base group at the indole C-3 position is very meaningful and potentially capable of forming a salt bond with Asp444, which verifies our hypothesis. In the following modifications, two more substitution groups, 4-aminopiperidin-1-yl group (analogue 43) and N-piperidin-4-yl group (analogue 44), were introduced to extend the distance that the basic amine could penetrate, hopefully to form a stronger interaction with Asp444. The results showed that 43 (Ki = 42.6 nmol/L) had decreased activity compared to 42, while 44 had essentially the same activity (Ki = 8.3 nmol/L).
Table 4.
Kinase inhibitory activities of compounds 42–56 against PAK4.
 | |||||||
|---|---|---|---|---|---|---|---|
| Compd. | R3 | PAK4 Ki (nmol/L)a | PAK1 Ki (nmol/L)b | A549c | cLogPd | PSAd | LEf/LLEg |
| 42 |  |
6.6 | 4990 | 65%h | 3.206 | 124.689 | 0.35/4.97 |
| 43 |  |
42.6 | 1614 | 56% | 3.208 | 124.751 | 0.29/4.16 |
| 44 |  |
8.3 | 4237 | 39% | 2.803 | 127.28 | 0.32/5.28 |
| 45 |  |
25.6 | 2368 | 71% | 2.791 | 117.883 | 0.30/4.80 |
| 46 |  |
48.2 | NDi | 64% | 3.093 | 115.319 | 0.29/4.22 |
| 47 |  |
3.4 | 6986 | 57% | 2.821 | 127.214 | 0.34/5.65 |
| 48 |  |
5.6 | 7130 | 52% | 2.828 | 125.297 | 0.33/5.42 |
| 49 |  |
207.4 | ND | 54% | 2.564 | 129.483 | 0.27/4.12 |
| 50 |  |
27.2 | ND | ND | 3.167 | 115.684 | 0.31/4.40 |
| 51 |  |
25.5 | 6342 | 68% | 3.174 | 115.624 | 0.31/4.42 |
| 52 |  |
30.9 | ND | 63% | 2.026 | 144.108 | 0.31/5.48 |
| 53 |  |
22.7 | 2867 | 63% | 3.176 | 124.552 | 0.31/4.47 |
| 54 |  |
148.3 | ND | ND | 3.301 | 123.455 | 0.27/3.53 |
| 55 |  |
10.2 | 6025 | 32% | 2.835 | 125.298 | 0.32/5.16 |
| 56 |  |
18.1 | 4990 | 73% | 2.575 | 129.268 | 0.31/5.17 |
| GNE-2861 | 35.3 | 3135 | 62%g | 3.41 | 103.298 | 0.34/4.04 | |
PAK4 kinase inhibition was determined using a homogeneous time-resolved fluorescence (HTRF) assay. The Ki values are the average of two independent experiments reported as the mean value.
PAK1 kinase inhibition was determined using a FRET-based Z′-Lyte assay. The Ki values are the average of at least two duplicate experiments. SEM < ±20%.
Effect of compounds on the cell migration of A549 with the concentration of 5 μmol/L. Cell migration was determined by a wound-healing assay. Data are the results of at least two independent assays.
The clogP values and polar surface area were calculated by the Qikprop software.
Ligand efficiency = (−1.4 Log Ki)/(n heavy atoms).
Ligand lipophilicity efficiency = (−Log Ki) − (cLogP).
Effect of compounds on the cell mobility of A549 with a concentration of 20 μmol/L. Cell migration was determined by a wound-healing assay.
ND = not determined.
According to the relative positions of the piperazinyl group of 42 and the carboxyl groups of Asp444, we speculated that introducing an amino group to the 3-position of the piperidine ring would be beneficial for the compound to maintain consistency of the pharmacodynamic conformation and the lowest energy conformation (Table 4). The introduction of chiral 3-aminopiperidine (45–48) validated the conjecture. It could be seen from the comparison of compounds 45 vs 47 and 46 vs 48 that the R-isomer was much more active than the S-isomer and its dominant conformation; the (R)-3-aminopiperidin-ly analogue 47, with a Ki of 3.4 nmol/L (IC50 = 6.3 nmol/L), was the most potent compound. Compound 47 also exhibited an excellent isoform selectivity of 1126-fold over PAK1 (PAK1 Ki = 6.99 μmol/L). After determining its dominant conformation, we then introduced the chiral aminopyrrolidine derivatives 50–53, whose inhibitory potency was approximately at the same level and slightly decreased compared to 42 and 47. The docking and molecular dynamics simulation studies showed that aminopyrrolidine analogues could rotate more freely in the active cavity due to their smaller size and could form compensatory interactions with other amino acids (e.g., Ile327 or Asp458) near the ribose pocket, resulting in similar binding affinities (Supporting Information Fig. S1). The introduction of the (R)-3-hydroxypiperidin-1-yl derivative 49 reduced its activity relative to 47 by a factor of 60, further demonstrating the importance of the basic group for bond strength. The chiral substitution of methylpiperazine was further investigated. The potency against PAK4 of the (S)-3-methylpiperazine derivative 55 exhibited excellent activity (PAK4 Ki = 10.2 nmol/L, IC50 = 18.3 nmol/L). However, the (R)-3-methylpiperazine derivative 54 exhibited approximately 14-fold less potency than 55 against PAK4 (preliminary explanation in Figs. S1E and S1F).
At the same time, to obtain an inhibitor that performs well at both the enzyme and cellular levels, a preliminary inhibitory effect against migration of cancer cells was evaluated based on the wound-healing assay in non-small cell lung cancer (NSCLC) cell line (A549) at 5 μmol/L (Table 4). Compounds 44 and 55 exhibited superior migration inhibitory activity, while most of the other compounds with potent kinase inhibitory activity did not exhibit equivalent cell migration inhibitory capacity. This may be due to the rigid skeleton of the compound and the strong basicity of the aliphatic amines, which make it difficult for this compound to penetrate the membrane.
2.5. Binding modes of compounds 47 and 55 with PAK4
As described above, our efforts succeeded in the identification of two compounds: (S)-3-methylpiperazine derivative 55, with good performance at both the enzyme and cell levels, and (R)-3-aminopiperidin-ly derivative 47 as the most potent and selective PAK4 inhibitor we have synthesized. Their cocrystal structures with PAK4 were solved and diffracted to a resolution of 3.0 Å (47/PAK4, PDB ID: 7CP3) and 2.5 Å (55/PAK4, PDB ID: 7CP4). As shown in Fig. 4, the binding modes of the two compounds are consistent with our hypothesis, where the alkaline amino groups of 47 and 55 pointed toward Asp444 via the twisting of the amide plane and formed strong salt bridge interactions with the deprotonated carboxylic acid side chain. The propargyl alcohol formed two hydrogen bonds with Glu366 and Phe459 in the lipophilic back pocket, which is like that observed in the cocrystal of compound 41. Interestingly, the aminopyrimidine forms H-bond interactions with the hinge region in different crystal morphologies. The aminopyrimidines of 41 and 47 form two H-bonds with the backbone amino and carbonyl group of Leu398; however, the aminopyrimidine of compound 55 forms two H-bonds with the NH of Leu398 and carbonyl group of Glu396 via a torsion angle of −11° (compound 47) to 169° (compound 55). Based on the similar chemical and physiological environments, these results indicated that two conformations coexisted in the active pocket, and the crystal conformation may depend on the initial state of binding to the protein.
Figure 4.

Cocrystal structure of 47/PAK4 and 55/PAK4. (A) X-ray cocrystal structure of 47 bound to PAK4 (PDB ID: 7CP3). Compound 47 is shown as a cyan stick structure. The key amino acids are shown as brown sticks. Red dashed lines indicate hydrogen bonds to key amino acids. (B) X-ray cocrystal structure of 55 bound to PAK4 (PDB ID: 7CP4). Compound 55 is shown as a green stick. The key amino acids are shown as yellow sticks. Red dashed lines indicate hydrogen bonds to key amino acids. (C) QM-torsion angle scans for 4-(6-ethynyl-1H-indol-1-yl)pyrimidin-2-amine (55a as a model molecule in QM-torsion angle scans) under M06-2X/6-311G∗∗. The baseline energy is obtained by subtracting the lowest energy of the compound.
To further understand the rationale for why both conformations exist, QM-torsion angle scan was performed for 55a, which was used as the model molecule without the interference of other substituents. The biaryl torsion angle was scanned in a 10° step, while other degrees of freedom within the ligand were optimized. Fig. 4C depicts the predicted dihedral profile about the rotatable C–N bond of 55a. The biaryl torsion angle of 55a met the low-energy conformations at 0° and ± 160°, and the results indicated that compounds could target either Leu398 or Glu396 (−11° for 47 and 169° for 55) with negligible energy penalty. In the analogue 5d (Table 1), to preferentially target Glu396 by conformational enforcement of this biaryl torsion angle, the inhibitory activity declined only slightly. Thus, a strong inhibitory potency for PAK4 existed whether the small molecule was biased toward hydrogen-bonding with Glu396 or Leu398.
The increased binding affinity demonstrated the rationale of our optimization hypothesis, thereby providing a molecular explanation for the tight binding. Therefore, further investigations, including kinase selectivity, antitumor metastasis effect, in vitro toxicity, and preliminary pharmacokinetic characterization were carried out on compound 55.
2.6. Selectivity profiles of compound 55
With the promising in vitro profiles such as the high enzymatic and moderate cellular inhibitory activity and the good selectivity over PAK1, compound 55 was used to characterize the kinase inhibition profiles against a panel of 108 recombinant human protein kinases via SelectScreen Kinase Profiling (Fig. 5). The results are shown in Supporting Information Table S2. The calculated kinase selectivity S (35) score was 0.08 at single point concentration of 1.0 μmol/L. In addition to PAK4, it also displayed considerable activity against several other kinases, including FES, FER, AMPK (A1B2G2), CAMK1 and the homologue PAK5 (or named PAK7). Importantly, compound 55 exhibited very weak or no inhibitory activity against the other 102 protein kinases.
Figure 5.

KinMap profiling of compound 55. (A) KinMap profiling of 55 at a concentration of 1.0 μmol/L against 108 kinases. Binding activity is expressed as a percentage of the control. (B) The percent control determination of compound 55 against the potential off-target effects.
2.7. Cellular effects of compound 55
Several studies have shown that PAK4 is overexpressed in NSCLC cells A549 and melanoma cells B1618,42, 43, 44. The overexpression of PAK4 was associated with tumor survival, metastasis, and poor prognosis. To further verify the effect of compound 55 on PAK4-associated cells, we performed proliferation, migration and invasion assays utilizing A549 and B16 (B16-F10 and B16-BL6) cells. The antiproliferative effect of compound 55 was first evaluated by MTT and CCK8 assays. As shown in Fig. 6A and Supporting Information Fig. S3A, 55 had little effect against cancer cell viability with A549 and B16 cells even at 20 μmol/L concentration for 24 h, which is similar to the previous observation. Meanwhile, compound 55 showed similar inhibitory effects on normal cells MRC-5 (human embryonic lung fibroblast) and HACAT (human immortal keratinocyte line) as with tumor cells (Fig. S3A). Next, the antimigration and anti-invasion effects of compound 55 were further determined by wound-healing and Transwell assays for the maximum incubation time of 24 h. Results from the wound-healing assay showed that 55 impaired the ability of cells to close a wound in a dose-dependent manner, indicating the inhibition of cellular migration in vitro (Fig. 6B). In Transwell assays the numbers of both migratory and invasive cells were also significantly reduced by compound 55 in a dose-dependent manner as illustrated in Fig. 6D and E. Noteworthily, compound 55 showed stronger inhibitory potency against the more metastatic B16-BL6 than that of B16-F10, suggesting the important role of PAK4 inhibition in tumor metastasis inhibition. Meanwhile, compound 55 showed a stronger blocking effect on the migration and invasion of A549 cells compared with GNE-2861 (Fig. S3). Hence, PAK4 inhibitors block cancer cell metastasis without obvious impairment of cell viability. The relatively low cell growth inhibitory activity and better metastasis inhibitory ability further confirmed that PAK4 may not be the “driving force” in cancer cell proliferation45.
Figure 6.

Compound 55 suppresses the migratory and invasive potential of tumor cells. (A) An MTT assay showed the antiproliferative effect of compound 55 on A549, B16-F10 and B16-BL6 cells. (B) Compound 55 inhibits migration of A549, B16-F10 and B16-BL6 cells in wound healing assays. The migration relative ratio (C) was recorded. Transwell assay was performed to show the effect of indicated concentrations of compound 55 on A549, B16-F10 and B16-BL6 cells migration (D) and invasion (E), and migrated cells were counted, respectively (F, G). (H) Effects of compound 55 on total PAK4, p-PAK4(S474), LIMK1, p-LIMK1(T508) levels with 24 h treatment in A549 cells. The results are presented as the mean ± standard deviation. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs control.
Western blot assay was used to investigate whether compound 55 could affect the PAK4/LIMK1 signaling pathway related to cancer metastasis. As shown in Fig. 6H, phosphorylation of PAK4 and LIMK1 were dose-dependently inhibited by compound 55 treatment without obvious effect on their protein level in A549 cancer cells.
2.8. In vitro metabolic stability, CYP450 inhibition, hERG inhibition, and in vivo pharmacokinetic properties
A preliminary ADMET properties assessment of compound 55 was carried out to evaluate its potential for further development. Compound 55 was incubated with mouse liver microsomes (MLM) and human liver microsomes (HLM) to evaluate metabolic stability. As shown in Table S3, 55 exhibited excellent stability in MLM and HLM, especially in MLM, with a half-life of 124 min (details of compounds 41, 42, 44 and 55 are also displayed in Table S3). Furthermore, CYP450 inhibition was determined, in which compound 55 showed only weak inhibition of CYP2C19, with an IC50 of 42.3 μmol/L, and was without inhibitory activity against other main CYPs (CYP1A2, CYP2C9, CYP2D6 and CYP3A4), indicating a low risk of drug–drug interactions via this metabolic pathway. Blockade of the hERG potassium channels has been identified as an important mechanism of prolonged QT interval and arrhythmia, thereby inducing cardiac risk. Potential hERG inhibitors usually have a pharmacophore of a positively charged group (basic amino group) based on previous studies. Satisfactorily, compound 55 was found to be safe (IC50 > 30 μmol/L) when tested in an hERG patch-clamp assay. With promising in vitro profiles, the preliminary pharmacokinetic characteristics of compound 55 following intravenous infusion into mice was evaluated (Table 5). After intravenous infusion at a dose of 1 mg/kg, compound 55 exhibited a moderate half-life of 1.41 h, and the area under the concentration time curve (AUC0−∞) was 265.63 ng·h/mL. The maximum plasma concentration (Cmax) was 590.87 ng/mL. Unfortunately, compound 55 exhibited relatively high plasma clearance (3764.59 mL/h/kg) and a low bioavailability of 6.26% in oral administration. Overall, further optimization is still needed to improve the pharmacokinetic properties.
Table 5.
PK parameters of 55 in mice.
| Property | 10 mg/kg (po) | 1.0 mg/kg (iv) |
|---|---|---|
| t1/2 (h) | 1.02 | 1.41 |
| Cmax (ng/mL) | 340.26 | 590.87 |
| CL (mL/h/kg) | ‒ | 3764.59 |
| Vss (mL/kg) | ‒ | 2574.92 |
| AUC0−t (ng·h/mL) | 165.32 | 264.27 |
| AUC0−∞ (ng·h/mL) | 168.62 | 265.63 |
| F (%) | 6.26 | ‒ |
Not applicable.
2.9. Inhibition by compound 55 of tumor metastasis in vivo
To confirm in vivo effectiveness, compound 55 was assayed for the ability to inhibit the proliferation and metastasis of A549 cells in zebrafish, an important in vivo model that is widely used for drug screening purposes46,47. Zebrafish embryos were exposed to a range of doses of compound 55 to determine the toxicity by assessment of embryo survival. Based on this screening, a dose of 100 μmol/L did not result in any gross morphological changes after 2 days of exposure (data not shown). Subsequently, red fluorescent dye-labeled (CM-Dil) A549 cells were microinjected into zebrafish embryos. The zebrafish with good tumorigenic consistency were treated with different concentrations of compound 55. As shown in Fig. 7A, 48 h after xenotransplantation, the A549 cells in the control group largely migrated away from the primary site. When treated with compound 55, the metastatic capacity of the A549 cells was profoundly inhibited in a dose-dependent manner, whereas tumor proliferation was not significantly affected, which further demonstrated that compound 55 effectively suppresses tumor metastasis in vivo.
Figure 7.

Compound 55 inhibited lung metastasis in vivo. (A) CM-Dil-labeled A549 cells (red) were microinjected into zebrafish embryos (n = 10 per group) and different concentrations of compound 55 were added. After 48 h, the metastasis and proliferation of A549 cells was imaged under a confocal microscope. (B) Quantification of the fluorescent area of the tumor xenografts, representing A549 cell metastasis. (C) Fluorescence intensity of the tumor xenografts, representing the number of A549 cells. (D) Compound 55 treatment reduced lung metastasis in mice injected with B16-BL6 (n = 6 per group) or A549 cells (n = 5 per group). At 14 days after injection of B16-BL6 cells and 56 days after injection of A549 cells into the tail vein of C57BL/6 mice or BALB/c nude mice, respectively, animals were killed and lungs were examined for metastatic nodules. Tumor nodules formed on the surface of lung from different groups of the B16-BL6 tumor model (E) and A549 tumor model (F). (G) Histological H&E staining performed on lung metastasis samples from the melanoma and lung cancer models. Scale bar: 100 μm. #Total number of metastatic nodules. #P < 0.05, ##P < 0.01, ###P < 0.001. The results are presented as the mean ± SD (n = 5 or 6): ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001.
Given the effective anti-tumor metastatic effect of 55 in vitro and in vivo in zebrafish, and the limited suppression of tumor proliferation, the effect of 55 in rodents was further evaluated using tumor lung metastasis models. Two pulmonary metastasis models of lung cancer A549 (BALB/c nude mice) and melanoma B16-BL6 (C57BL/6 mice) in mice were used to examine the in vivo anti-tumor metastatic activity of 55. At 24 h after tumor cell injection, compound 55 was administered intravenously daily to mice at a dose of 25 or 50 mg/kg based on the PK characteristics, and the control group was given the vehicle alone daily. A group of mice without implanted tumors was set up as a normal group for comparison and background subtraction. The number and size of metastatic nodules in lung were quantitatively evaluated after 15 days of treatment in the melanoma model and after 56 days of treatment in the lung cancer model. As shown in Fig. 7D, the lungs from A549 and B16-BL6 transplanted mice presented apparent metastatic cancer nodules on the surface, in contrast to the smooth surface of the lungs from the normal group. 25 mg/kg of compound 55 significantly reduced the number of nodules in the mice, while the 50 mg/kg dose almost completely prevented lung metastasis. The percentages of mice with metastatic nodules on their lungs was 100% for the vehicle mice and decreased to 44.2% for B16-BL6 xenograft group (34.6% for A549 xenograft group) that received the 25 mg/kg dose, and 9.2% for B16-BL6 xenograft group (19.5% for A549 xenograft group) that received 50 mg/kg (Fig. 7E and F). In addition, the number of nodules larger than 2 mm almost was greatly reduced in the compound 55-treated groups.
The histological analysis qualitatively demonstrated the same results (Fig. 7G), that compound 55 was able to block the metastasis of tumor cells A549 and B16-BL6 cells into lungs. Although treatment with compound 55 inhibited tumor metastasis, there was no significant toxicity as evidenced by similar body weights between the drug-treated mice and normal (Supporting Information Fig. S7).
2.10. Compound 55 suppresses TGF-β1-induced epithelial–mesenchymal transition (EMT) in A549 cells
Among the multitude of different signaling molecules found in the blood, transforming growth factor-beta (TGF-β) is known to promote metastasis by enhancing the EMT of primary cancers48. Furthermore, inhibition of tumor cell responsiveness to TGF-β1 reduces metastatic seeding in the lung49, 50, 51. Multiple studies demonstrate that overexpression of PAK4 is closely associated with TGF-β1-induced EMT in different tumors52, 53, 54, 55, 56. Therefore, given the effective inhibition of tumor metastasis by compound 55 in vitro and in vivo, we investigated its effect on TGF-β1-induced EMT in A549 lung cancer cells.
Treatment with TGF-β1 significantly induced EMT as determined by the increased incidence, and the morphology was found to be aggressive spindle-shape instead of a polygonal shape, along with downregulated protein levels of the epithelial cell biomarker E-cadherin, and upregulated protein levels of the mesenchymal cell marker N-cadherin and vimentin. Unsurprisingly, treatment with compound 55 restored the morphology of cells in a dose-dependent manner (Fig. 8A). Both western blot and immunofluorescence results demonstrated dose-dependent upregulation of the epithelial marker E-cadherin, along with the downregulation of mesenchymal markers N-cadherin and vimentin after treatment with 55 in A549 cells (Fig. 8B and C), suggesting that 55 blocks the TGF-β1-induced EMT process.
Figure 8.

Compound 55 blocks TGF-β1-induced EMT in A549 lung cancer cells. (A) Compound 55 reverses TGF-β1-induced morphological change as observed with an inverted microscope (10 ×). (B) Effect of compound 55 on the expression of the classical EMT markers was evaluated by immunofluorescence assay. Blue represents DAPI (4′,6-diamidino-2-phenylindole) for nucleus; red represents the markers: E-cadherin, vimentin and N-cadherin. Scale bar: 20 μm. (C) Expression of the epithelial proteins E-cadherin, the mesenchymal protein vimentin, and N-cadherin in A549 cells was determined by western blot analysis. (D–F) The levels of E-cadherin, vimentin and N-cadherin. ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs TGF-1 group; #P < 0.05, ##P < 0.01, ###P < 0.001.
2.11. Chemistry
All the compounds in this paper were synthesized following the synthetic routes outlined in Scheme 1, Scheme 2. The synthesis of the compounds 5–29 is described in Scheme 1. The nucleophilic substitution reaction of indole and azaindole with various pyrimidines to afford the intermediates 59, 61, 64 and 66 was carried out in the presence of Cs2CO3. Compound 61 was then converted to the aminopyrimidine 62 in the presence of ammonium hydroxide. The N-modified intermediate 67 was obtained from the reaction of 66 with the corresponding amine in the presence of concentrated hydrochloric acid. Finally, the Sonogashira coupling reaction of the key intermediates with 1-ethynyl-1-cyclohexanol afforded the desired products 5–29.
Scheme 1.

Synthetic route of compounds 5–29. Reagents and conditions: (a) Cs2CO3, DMF, 100 °C; (b) 1-ethynyl-1-cyclohexanol, Pd(PPh3)4, CuI, TEA, DMF, 80 °C; (c) NH4OH, H2O, IPA, 110 °C; (d) amine, HCl, IPA, 110 °C.
Scheme 2.

Synthetic route of compounds 30–56. Reagents and conditions: (a) TFAA, DMF, rt; (b) NaOH, H2O, reflux; (c) (i) oxalyl chloride, DMF, DCM, rt. (ii) KTB, 2-methyl-2-propanol, rt; (d) 2-amino-4-chloropyrimidine, Cs2CO3, DMF, 100 °C; (e) TFA, DCM, rt; (f) 1-ethynyl-1-cyclohexanol, Pd(PPh3)4, CuI, TEA, DMF, 80 °C; (g) amine, DIPEA, HATU, DMF, 0.5–12 h, rt.
Compounds 30–56 were generated following the reaction routes shown in Scheme 2. First, 6-bromo-1H-indole-3-carboxylic acid 69 was obtained through two steps from commercially available 6-bromo-1H-indole. A subsequent esterification reaction with tert-butanol of 69 afforded the corresponding tert-butyl ester derivative 70. A nucleophilic substitution reaction of the intermediate 70 aminopyrimidine afforded the intermediate 71 in the presence of Cs2CO3. The hydrolysis of 71 followed by the Sonogashira coupling reaction afforded the key intermediate 73. Amination reactions of 73 with a variety of amines were carried out in the presence of HATU and DIPEA to provide compounds 30–56.
3. Conclusions
In this investigation a structure-based approach led to the discovery and optimization of compound 55. An interesting phenomenon appears in the cocrystal structures of compounds 41, 47 and 55 with PAK4, where 2-aminopyrimidine exhibits two distinct conformations within the PAK4 pocket, forming two hydrogen bonds with Leu398 or simultaneously with Glu396/Leu398 after a 180° flip. Compound 55 also exhibited good kinome selectivity, inhibiting only a few kinases, including class II PAKs, FES, AMPK and CAMK1. Moreover, we have progressively demonstrated the effectiveness of compound 55 at the molecular, cellular and in vivo levels. Western blot assay results confirmed that 55 effectively inhibited the phosphorylation of PAK4-related substrates in A549 cells. Subsequent experiments showed that compound 55 effectively inhibited cancer cell migration and invasion, with inconspicuous inhibition of the cell viability of A549 and B16 cells in a time- and concentration-dependent manner. In the in vivo efficacy evaluation of lung metastasis models in mice and zebrafish, compound 55 effectively inhibited the metastasis to lung of injected lung cancer cells A549 and melanoma cancer cells B16-BL6. Additionally, compound 55 mitigated TGF-β1-induced the epithelial-mesenchymal transition by inhibiting the PAK4 activity in A549 cells. Compound 55 exhibited good physicochemical properties and a good safety profile, but its in vivo pharmacokinetic properties need further improvement. Taken together, these data suggest that compound 55 could be effective in the treatment of metastatic cancer in man and deserves further research and development.
4. Experimental
4.1. General information
Commercially available and chiral substituents were purchased from Bide Pharmatech Ltd. and used as provided. Unless otherwise indicated, all reagents and solvents were obtained from commercial suppliers and used without further purification. Anhydrous solvents were dried and stored according to standard procedures. Thin layer chromatography (TLC) was performed to monitor reactions (Qingdao Ocean Chemicals). Column chromatography was carried out using silica gel (200–300 mesh, Qingdao Ocean Chemicals). 1H NMR and 13C NMR spectra were obtained with a Bruker ARX-600 NMR or Bruker ARX-400 NMR spectrometers with TMS as the internal standard. High-resolution accurate mass spectrometry (HRMS) results were obtained on a Bruker Micromass Time of Flight mass spectrometer equipped with an electrospray ionization (ESI) detector. All the final compounds were purified to > 95% purity, as determined by high-performance liquid chromatography (HPLC). HPLC analysis was performed on a Waters Breeze system (Waters 1525, binary HPLC pump) equipped with an UV detector (Waters 2487, dual λ absorbance detector) and a WondaSil C-18 Superb column (5 μm, 4.6 mm × 250 mm) using a mobile phase of 70% methanol in water containing 0.08% TFA. The flow-rate was 0.5 mL/min, and the injection volume was 10 μL. Peaks were detected at 254 nm.
4.2. Chemistry
4.2.1. General procedure A for preparation of 59, 61, 64, 66 and 71
A mixture of 57 or 70 (1.0 equiv) and Cs2CO3 (3.0 equiv) in DMF was stirred for 30 min at 0 °C. After the addition of pyrimidine (1.1 equiv), the mixture was heated to 100 °C and stirred for 5 h. After cooling to room temperature, the reaction mixture was diluted with water and extracted with ethyl acetate. The combined organic extracts were dried over Na2SO4 and the solvent was removed under reduced pressure without further purification. The desired products 59, 61, 64, 66 and 71 were obtained with a yield of 45.7%–93.9%.
4.2.2. General procedure B for sonogashira coupling reactions
To a stirred solution of aryl bromide (1.0 equiv), triethylmine (5.0 equiv), catalytic tetrakis (triphenylphosphine)palladium (0.05 equiv) and copper(I) iodide (0.2 equiv) in DMF in a dry round bottom flask under an argon atmosphere, 1-ethynyl-1-cyclohexanol (1.5 equiv) was added and the mixture was refluxed at 80 °C for 10 h. The reaction mixture turned dark orange and was cooled to room temperature, poured into water and extracted with ethyl acetate. The combined organic extracts were dried over Na2SO4 and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel (DCM/Methanol = 100:1 to 10:1) to afford the corresponding product. Yield: 32.5%–63.1%. Compound 73 was added to 15% sodium carbonate solution and washed with ethyl acetate (3 × 50 mL). The aqueous layer was then acidified to pH = 6–6.5 with 6 mol/L HCl aqueous solution, the solid was filtered and dryed under vacuum. The desired product 1-(2-aminopyrimidin-4-yl)-6-((1-hydroxycyclohexyl)ethynyl)-1H-indole-3-carboxylic acid was obtained as a white solid, yield: 61.3%. 1H NMR (600 MHz, DMSO-d6) δ 12.72 (s, 1H), 8.65 (s, 1H), 8.59 (s, 1H), 8.10 (s, 1H), 7.37 (d, J = 6.5 Hz, 1H), 7.14 (d, J = 18.8 Hz, 4H), 5.40 (s, 1H), 1.94–1.46 (m, 10H). MS (ESI) m/z 375.3 [M–H]−.
4.2.3. General procedure C for N-modified intermediate 67
A mixture of 6-bromo-1-(2-chloropyrimidin-4-yl)-1H-indole (1.0 equiv), different amines (1.2 equiv), and concentrated hydrochloric acid (10 μL) in IPA was heated at 110 °C for 4 h. The reaction mixture was cooled to room temperature and the precipitate was collected by filtration and washed with ethanol. The solid was re-crystalized in ethanol to give corresponding amines. The desired product N-benzyl-4-(6-bromo-1H-indol-1-yl)pyrimidin-2-amine was obtained as a white solid, yield: 87.5%. 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.34 (d, J = 5.7 Hz, 1H), 8.23–8.11 (m, 1H), 8.09 (d, J = 3.7 Hz, 1H), 7.57 (d, J = 8.3 Hz, 1H), 7.41 (d, J = 7.6 Hz, 2H), 7.33 (q, J = 7.6, 7.2 Hz, 3H), 7.22 (t, J = 7.4 Hz, 1H), 6.99 (d, J = 5.7 Hz, 1H), 6.80 (d, J = 3.6 Hz, 1H), 4.63 (d, J = 6.3 Hz, 2H). MS (ESI) m/z 378.9 [M + H]+.
4.2.4. 4-(6-Bromo-1H-indol-1-yl)-5-methylpyridin-2-amine (62)
To a solution of 6-bromo-1-(2-chloro-5-methylpyridin-4-yl)-1H-indole (0.4 g, 1.24 mmol) in IPA (8 mL) was added ammonium hydroxide (2 mL) in a Schlenk tube. The mixture was stirred at 110 °C for 11 h. After completion of the reaction (monitored by TLC), the mixture was concentrated under vacuum. The residue was purified on chromatography to afford 0.25 g (66.7%) 62 as a white solid, yield: 53.7%. 1H NMR (600 MHz, DMSO-d6) δ 8.32 (s, 1H), 7.86 (d, J = 1.7 Hz, 1H), 7.71 (d, J = 3.4 Hz, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.29 (dd, J = 8.4, 1.8 Hz, 1H), 6.77 (s, 2H), 6.72 (dd, J = 3.3, 0.8 Hz, 1H), 2.07 (s, 3H). MS (ESI) m/z 303.1 [M–H]−.
4.2.5. 6-Bromo-1H-indole-3-carboxylic acid (69)
The compound 6-bromo-1H-indole (5.0 g, 25.5 mmol) was dissolved in DMF (30 mL). The solution was cooled to 0 °C and trifluoroacetic anhydride (4.3 mL, 30.6 mmol) was added dropwise. The reaction mixture was stirred for 1.5 h at room temperature, then water (100 mL) was added, the solid was filtered and washed with water to afford a pale-yellow solid without further purification. The crude solid was treated with 20% NaOH (100 mL) with reflux overnight. Upon cooling, the solution was washed with ethyl acetate (3 × 30 mL). The aqueous phase was then acidified to pH 4–5 with 6 mol/L HCl aqueous solution, the solid was filtered and dried under vacuum. The desired product 6-bromo-1H-indole-3-carboxylic acid (5.81 g, 24.2 mmol) was obtained as a grey solid, yield: 94.9%. 1H NMR (300 MHz, DMSO-d6): δ 12.07 (br s, 1H), 11.88 (br s, 1H), 8.01 (d, J = 3.0 Hz, 1H), 7.92 (d, J = 8.6 Hz, 1H), 7.64 (d, J = 1.7 Hz, 1H), 7.28 (dd, J = 8.6, 1.7 Hz, 1H); MS (ESI) m/z 239.0 [M–H]−.
4.2.6. tert-Butyl 6-bromo-1H-indole-3-carboxylate (70)
Compound 3 (3.7 g, 15.4 mmol) was dissolved in 25 mL DCM with three drops of DMF, then oxalyl chloride (6.6 mL, 77.1 mmol) was added at room temperature. The mixture was stirred at room temperature for 30 min and concentrated in vacuo to offer the crude chloride as a yellow oil. The oil was dissolved in t-BuOH (25 mL), and potassium tert-butylate (1.9 g, 16.94 mmol) was added. The resulting mixture was stirred at 40 °C for 4 h, diluted with ethyl acetate and washed with saturated NH4Cl aqueous solution and brine. The organic layer was dried over Na2SO4 and concentrated under vacuum. The residue was purified by chromatography to give white solid 70 (4.5 g, 15.3 mmol), yield: 98.8%. 1H NMR (400 MHz, DMSO-d6) δ 11.92 (s, 1H), 7.99 (d, J = 3.0 Hz, 1H), 7.89 (d, J = 8.6 Hz, 1H), 7.65 (dd, J = 1.8, 0.6 Hz, 1H), 7.30 (dd, J = 8.5, 1.8 Hz, 1H), 1.56 (s, 9H). MS (ESI) m/z 296.7 [M–H]−.
4.2.7. 1-(2-Aminopyrimidin-4-yl)-6-bromo-1H-indole-3-carboxylic acid (72)
To a solution of tert-butyl 1-(2-aminopyrimidin-4-yl)-6-bromo-1H-indole-3-carboxylate (9.2 mmol) in DCM (15 mL), TFA (7 mL) was added when the solution was cooled to 0 °C. After consumption of the starting material, the mixture was dried under vacuum and 15% sodium carbonate was added. The aqueous layer was washed twice with DCM and the aqueous layer was then acidified to pH 5–6 with an aqueous solution of 6 mol/L HCl; the residue was filtered and evaporated under vacuum. The desired product 1-(2-aminopyrimidin-4-yl)-6-bromo-1H-indole-3-carboxylic acid (2.77 g, 8.3 mmol) was obtained as an off-white solid, yield: 90.2%. 1H NMR (400 MHz, DMSO-d6) δ 8.90 (d, J = 1.8 Hz, 1H), 8.60 (s, 1H), 8.36 (d, J = 5.5 Hz, 1H), 8.00 (d, J = 8.5 Hz, 1H), 7.51 (dd, J = 8.5, 1.8 Hz, 1H), 7.22–7.05 (m, 3H), 1.60 (s, 9H). MS (ESI) m/z 330.9 [M–H]−.
4.2.8. (S)-(1-(2-Aminopyrimidin-4-yl)-6-((1-hydroxycyclohexyl)ethynyl)-1H-indol-3-yl) (4-methylpiperazin-1-yl)methanone (55)
The carboxyl acid (0.2 g, 0.53 mmol), (S)-(+)-2-methylpiperazine (64.0 mg, 0.64 mmol) and triethylamine (0.22 mL, 1.59 mmol) were dissolved in DMF (3 mL), and HATU (0.22 g, 0.58 mmol) was added to the solution. After 30 min the reaction was complete. The mixture was quenched with water and the aqueous solution was extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography eluting with DCM/methanol (10:1) to give compound 55 as a white solid (109 mg, 45.4%). 1H NMR (600 MHz, DMSO-d6) δ 8.66 (d, J = 1.3 Hz, 1H), 8.33 (d, J = 5.5 Hz, 1H), 8.32 (s, 1H), 7.64 (d, J = 8.2 Hz, 1H), 7.30 (dd, J = 8.1, 1.4 Hz, 1H), 7.06–6.97 (m, 3H), 5.39 (s, 1H), 4.09 (s, 2H), 3.13–2.84 (m, 2H), 2.67 (m, 3H), 1.95–1.42 (m, 10H), 1.27 (dt, J = 3.9 Hz, 1H), 1.05–0.90 (m, 3H). 13C NMR (151 MHz, DMSO-d6) δ 163.9, 163.8, 160.7, 158.9, 134.5, 128.5, 128.2, 126.7, 121.0, 119.0, 118.7, 115.3, 99.3, 95.0, 84.0, 67.4, 49.1, 40.5, 40.4, 25.4, 23.2, 19.5. HRMS (ESI+) Calcd. for C26H31N6O2 [M+H]+: 459.2503. Found: 459.2501.
4.2.9. (S)-(3-Aminopiperidin-1-yl) (1-(2-aminopyrimidin-4-yl)-6-((1-hydroxycyclohexyl)ethynyl)-1H-indol-3-yl)methanone (45)
Carboxyl acid (0.5 g, 1.33 mmol), HATU (0.22 g, 0.58 mmol) and triethylamine (1.9 mL, 14 mmol) were dissolved in DMF (10 mL) and stirred for 10 min at room temperature. (S)-piperidin-3-amine dihydrochloride (1.15 g, 6.25 mmol) was added to the solution and stirring continued for 1 h. After completion of the reaction as indicated by TLC, two product points: 3-aminopiperidin-1-yl derivative (DCM:methanol = 5:1, Rf = 0.35) and piperidin-3-yl-amono derivative (DCM:methanol = 5:1, Rf = 0.1) could be observed. The mixture was quenched with water and the aqueous solution was extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4 and concentrated in vacuo. The crude product was purified by silica gel column chromatography eluting with DCM/methanol (10:1 to 5:1) to give 45 (31.3%) and 46 (17.3%) as canary yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 8.65 (d, J = 1.3 Hz, 1H), 8.33 (d, J = 5.6 Hz, 1H), 8.31 (s, 1H), 7.62 (d, J = 8.2 Hz, 1H), 7.29 (dd, J = 8.2, 1.4 Hz, 1H), 7.01 (s, 2H), 7.00 (s, 1H), 5.38 (s, 1H), 4.02 (s, 2H), 3.05–2.63 (m, 3H), 1.87 (m, 4H), 1.74–1.41 (m, 10H), 1.27–1.22 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 164.1, 163.8, 160.7, 158.9, 134.5, 128.5, 128.0, 126.6, 121.0, 118.9, 118.7, 115.5, 99.2, 94.9, 84.0, 67.4, 48.55, 40.5, 40.2, 33.9, 25.4, 23.2. HRMS (ESI+) Calcd for C26H31N6O2 [M+H]+: 459.2503. Found: 459.2518.
Compounds 45–48 and 50–53 were synthesized in a manner similar to that of 45.
4.3. PAK4 HTRF assay
To test the affinity of PAK4 inhibitors, a biochemical HTRF® KinEASE™-STK kit (Cisbio Bioassays, France) assay was used in white 384-well low volume microplates (Nunc, ThermoFisher Scientific). The compounds were tested at different concentrations with 3-fold serial dilutions in kinase buffer (composed of 5 mmol/L MgCl2, 1 mmol/L DTT and 1X KinEASE enzymatic buffer). The compounds were incubated with PAK4 (41 pmol/L), substrate S2 (1 μmol/L) and ATP (2.5 μmol/L) in 10 μL kinase reaction buffer. After 50 min of incubation at room temperature, 10 μL of a detection solution containing SA-XL665 (0.125 μmol/L) and ATK Ab were added to the wells. After another 1 h incubation at room temperature, the plate was measured on an Infinite® F500 microplate reader (Tecan, Switzerland) using the homogeneous time-resolved fluorescence (HTRF) module (excitation: 337 nm; emission: 620 and 665 nm), and IC50 values were calculated using the Prism GraphPad software. The Ki values reported in this article were calculated by converting the IC50 value to the Ki value by employing the Cheng-Prusoff equation with the XLFIT5 software57.
4.4. Cell proliferation assay
4.4.1. Cell background
Human non-small cell lung cancer (NSCLC) A549 cells were purchased from National Infrastructure of Cell Line Resource (NSTI). Mouse melanoma B16-F10 cells were purchased from Hunan Fenghui Biotechnology Co., Ltd. Mouse melanoma B16-BL6 cells were kindly provided by Mukogawa Women's University, Japan. MRC-5 cells were purchased from Procell Life Science & Technology Co., Ltd. HaCaT cells were obtained from CLS Cell Lines Service.
There are multiple sublines of C57BL/6 mouse melanoma cells B16 with different metastatic potentials despite their same origin. B16-BL6 is more metastatic and aggressive than B16-F10. The low metastatic subline Bl6-F10 was established by intravenous injection of B16 melanoma cells in static lung lesions, whereas the metastatic B16-F10 cells were established by 10 successive selections for metastasis following iv injection. Highly metastatic B16-BL6 cells were established by penetrating mouse bladder membranes with B16-F10 cells.
4.4.2. MTT assay
Lung cancer A549, melanoma cells B16-F10, B16-BL6 and human immortal keratinocyte line HaCaT cells were cultured in DMEM (MCR were cultured in MEM) medium supplemented with 2% fetal bovine serum, 100 U/mL of penicillin, and 100 mg/mL streptomycin at 37 °C in a humidified atmosphere of 5% CO2. The antiproliferative activities of the compounds were determined by MTT (3-[4,5-dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazolium bromide) assay. Briefly, cancer cells (1 × 104 per well) were precultured for 24 h in 96-well plates and then treated with different concentrations of each compound for 24 h. The MTT solution (0.5%) was added to each well. After another 4 h of incubation at 37 °C, the formazan formed was extracted by DMSO. Shaking the plates for 15 min, the absorbance at 495 nm was measured using a Microplate Reader (BIO-RAD).
4.5. Wound healing assay
Plates containing confluent cultures of the cells grown in serum-containing medium were scratched with a sterile 200 μL pipette tip. The cells were cultured in DMEM complete media containing 2% FBS with compound 55 or DMSO at 37 °C for designated times. Images were captured with an inverted fluorescence microscope (Olympus IX71, Olympus Corporation). Distances between the gaps formed by the scratch were measured with Image J software (ImageJ 1.46r). Percentages were calculated relative to the gap size immediately after scratching.
4.6. Transwell assay
Invasion experiments were performed using 24-well Transwell inserts coated with Matrigel (BD Biosciences, USA) at 37 °C for 24 h. A total of 5 × 104 fresh cells in 200 μL serum-free medium were added to the upper layer of the insert. The lower layer was filled with 500 μL DMEM with 10% FBS as the chemoattractant. After incubation in a humidified atmosphere containing 5% CO2 at 37 °C, the Matrigel-coated insert was fixed with 0.4% paraformaldehyde and stained with 0.1% crystal violet. Cells on the upper layer of the inserts were removed with a cotton swab. Pictures were taken with a microscope at 100 × magnification, then decolorized with 33% acetic acid to completely elute the crystal violet, and the OD of the eluate was measured at a wavelength of 570 nm. Randomly selected fields (8–10) were imaged and counted using Image J software (ImageJ 1.46r). Each experiment was repeated three times.
4.7. Western blot analysis
To determine the expression level of relevant proteins, A549 cells were treated with compound 55 for 24 h and cell extracts were prepared from 1 × 106 cells in RIPA lysis buffer (150 mmol/L NaCl, 50 mmol/L Tris/HCl pH 7.4, 0.25% Na deoxycholate, 1 mmol/L EDTA, 1% Nonidet P-40 and protease inhibitor cocktail). Equivalent amounts of protein were separated by SDS-PAGE and transferred to PVDF (polyvinylidene fluoride) membranes. The membranes were blocked with nonfat dry milk in TBS-T (20 mmol/L Tris at pH 7.4, 137 mmol/L NaCl, and 0.05% Tween-20) and incubated at 4 °C with the specific primary antibodies for E-cadherin (3195T), N-cadherin (13116T), vimentin (5741T), PAK4 (62690S), LIMK1 (3842S), phospho-PAK4 (Ser474)/PAK5 (Ser602)/PAK6 (Ser560) (3241S), phospho-LIMK1 (Thr508)/LIMK2 (Thr505) (3841S) from CST (Cell Signaling Technology, MA, USA) overnight. Membranes were washed with TBS-T three times and incubated with secondary antibodies for 2 h at room temperature. Immunoreactive proteins were detected by chemiluminescence (ECL, Thermo Scientific).
4.8. Immunofluorescence
A549 cells were fixed in methanol for 20 min at room temperature and blocked with normal goat serum for 30 min and permeabilized with 0.25% Triton X-100 in PBS for 10 min. Mouse monoclonal anti-N-cadherin (13116T, CST), anti-vimentin (5741T, CST), anti-E-cadherin (3195T, CST) and Goat anti-Rabbit IgG/RBITC (SR134 Solarbio). DNA dye DAPI was used (blue). A confocal analysis system was performed using a laser confocal scanning microscope (C2-si, Nikon) according to established methods, utilizing continuous laser excitation to minimize the possibility of fluorescence emission leakage.
4.9. Pharmacokinetic studies
The pharmacokinetic study of 55 were carried out by Shanghai Medicilon Inc., according to the protocols and guidelines of the institutional care and use committee. All procedures related to mouse handling, care, and treatment in this article were performed in compliance with the Ethics Committee on Laboratory Animal Care and the Guidelines for the Care and Use of Laboratory Animals in Shanghai, China. The pharmacokinetic properties of compound 55 were investigated following single intravenous using ICR mice (3 mice/point, two cohorts). Compound 55 was dissolved in normal saline. The plasma samples were collected into heparinized centrifuge tubes at time points: 0.083, 0.25, 0.5, 1, 2, 4, 8 and 24 h after intravenous administration, and blood concentrations were analyzed by LC−MS/MS. The collected data were analyzed using Phoenix WinNonlin 7.0 (Pharsight, USA) software.
4.10. Antitumor effect on zebrafish embryos
This study was approved by the IACUC (Institutional Animal Care and Use Committee) at Hunter Biotechnology, Inc. and the IACUC approval number was 001458. The zebrafish facility and the laboratory at Hunter Biotechnology, Inc. are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. Zebrafish embryos were used to evaluate the antitumor effects of compound 55. Wild-type (AB strain) zebrafish were obtained from Hangzhou Hunter Biotech Co. Ltd. (Hangzhou, Zhejiang province, China) and raised at 28 °C. The A549 cells (200 cells per zebrafish), labeled with red fluorescence by CM-DiI were injected into the yolk sac of zebrafish embryos at three dpf to establish a zebrafish lung cancer model. The zebrafish embryos were incubated in 6-well plates (30 fish in each well) with or without compound 55 (11.1, 33.3, and 100 μmol/L). Cisplatin (15 μg/mL) was used as a positive control. After five dpf, the effects of compound 55 in lung zebrafish were observed with fluorescence microscopy.
4.11. Antitumor lung metastases test in vivo
All procedures followed the Guiding Principles for the Care and Use of Laboratory Animals approved by Shenyang Pharmaceutical University. Male C57BL/6 mice at 6 weeks of age were obtained from Liaoning Changsheng Biotechnology Co., Ltd., China, and were maintained in an air-conditioned room (23 ± 2 °C and 60 ± 10% humidity) under a 12 h light/dark cycle (lights on 7:00 a.m.). To determine the effect of treatment with 55 on the lung metastasis of B16-BL6 cells, mice were injected intravenously with 5 × 105 cells in the tail vein. Mice were injected intravenously with vehicle alone, compound 55 (25 or 50 mg/kg) daily via tail vein after 24 h. Their body weights were measured weekly. Mice were killed 14 days after cell injection. The lung was excised and fixed in paraformaldehyde neutral buffer solution. Tumor nodules were counted with a 40-fold stereomicroscope and the lung sections (4 μm) were stained with H&E.
Female BALB/c nude mice at 4 weeks of age were obtained from Liaoning Changsheng Biotechnology Co., Ltd., China, and the mice were maintained in an air-conditioned room (23 ± 2 °C and 60 ± 10% humidity) under a 12 h light/dark cycle (lights on 7:00 a.m.). To determine the effect of treatment with compound 55 on the lung metastasis of A549 cells, mice were injected intravenously with 1 × 106 cells in the tail vein. Mice were injected intravenously with vehicle alone, 55 (25 or 50 mg/kg) daily via the tail vein after 24 h. Body weights were measured weekly. Mice were killed 56 days after cell injection. A lung was excised and fixed in paraformaldehyde neutral buffer solution. Tumor nodules were counted with a 40-fold stereomicroscope and the lung sections (4 μm) were stained with H&E.
The data from multiple independent experiments are presented as the mean ± SD. Statistical comparisons were analyzed by one-way ANOVA followed by LSD post-hoc multiple comparison tests using Statistics Package for Social Science software (SPSS 26.0, Chicago, IL, USA), For comparisons between two groups, unpaired t-tests were used for comparison. P < 0.05 was considered to be statistically significant. All statistical charts were completed by GraphPad Prism software 6 (LaJolla, CA, USA).
Acknowledgments
This study was supported by National Natural Science Foundation of China (Grant Nos. 81230077, 81872729 and 22077086), Overseas Expertise Introduction Project for Discipline Innovation (Grant No. D20029, China), Program for Innovative Talents of Higher Education of Liaoning (2012520005, China).
Footnotes
Peer review under responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2022.02.029.
Contributor Information
Mingyu Xia, Email: xmywd@vip.sina.com.
Haitao Li, Email: lht@tsinghua.edu.cn.
Jian Wang, Email: jianwang@syphu.edu.cn.
Maosheng Cheng, Email: mscheng@syphu.edu.cn.
Author contributions
Maosheng Cheng, Jian Wang, and Peilu Song designed this project. Fengjiao Zhang, Dahong Li, Mingyu Xia and Haitao Li assisted in designing this project. Peilu Song and Hanxun Wang performed molecular docking and MD simulation. Peilu Song, Hanxun Wang, Yuan Su, Yujie Wang and Yinli Gao performed the chemical synthesis. Peilu Song performed enzymatic assays of PAK1/4. Jiqiang Qu performed in vivo inhibition assays of pulmonary metastasis models. Jiqiang Qu, Miao Yao and MiaoMiao Zhou performed cell assays. Fan Zhao carried out the culture and analysis of cocrystallizations. Feng Li, Dongmei Zhao and Yu Rao provided valuable comments for the article. Peilu Song, Jian Wang, Dahong Li and Maosheng Cheng contributed to the writing, review and editing of the manuscript. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no competing financial interests.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Knaus U.G., Bokoch G.M. The p21Rac/Cdc42-activated kinases (PAKs) Int J Biochem Cell Biol. 1998;30:857–862. doi: 10.1016/s1357-2725(98)00059-4. [DOI] [PubMed] [Google Scholar]
- 2.Arias-Romero L.E., Chernoff J. A tale of two PAKs. Biol Cell. 2008;100:97–108. doi: 10.1042/BC20070109. [DOI] [PubMed] [Google Scholar]
- 3.Abo A., Qu J., Cammarano M.S., Dan C., Fritsch A., Baud V., et al. PAK4, a novel effector for Cdc42Hs, is implicated in the reorganization of the actin cytoskeleton and in the formation of filopodia. EMBO J. 1998;17:6527–6540. doi: 10.1093/emboj/17.22.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar R., Sanawar R., Li X., Li F. Structure, biochemistry, and biology of PAK kinases. Gene. 2017;605:20–31. doi: 10.1016/j.gene.2016.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudolph J., Crawford J.J., Hoeflich K.P., Wang W. Inhibitors of p21-activated kinases (PAKs) J Med Chem. 2015;58:111–129. doi: 10.1021/jm501613q. [DOI] [PubMed] [Google Scholar]
- 6.Rane C.K., Minden A. p21-activated kinase signaling in cancer. Semin Cancer Biol. 2019;54:40–49. doi: 10.1016/j.semcancer.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Ye D.Z., Field J. PAK signaling in cancer. Cell Logist. 2012;2:105–116. doi: 10.4161/cl.21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radu M., Semenova G., Kosoff R., Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer. 2014;14:13–25. doi: 10.1038/nrc3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costa T.D.F., Zhuang T., Lorent J., Turco E., Olofsson H., Masia-Balague M., et al. PAK4 suppresses RELB to prevent senescence-like growth arrest in breast cancer. Nat Commun. 2019;10:1–18. doi: 10.1038/s41467-019-11510-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dasgupta A., Sierra L., Tsang S.V., Kurenbekova L., Patel T., Rajapakshe K., et al. Targeting PAK4 inhibits Ras-mediated signaling and multiple oncogenic pathways in high-risk Rhabdomyosarcoma. Cancer Res. 2021;81:199–212. doi: 10.1158/0008-5472.CAN-20-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tyagi N., Bhardwaj A., Singh A.P., McClellan S., Carter J.E., Singh S. p-21 activated kinase 4 promotes proliferation and survival of pancreatic cancer cells through AKT- and ERK-dependent activation of NF-kappaB pathway. Oncotarget. 2014;5:8778–8789. doi: 10.18632/oncotarget.2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tabusa H., Brooks T., Massey A.J. Knockdown of PAK4 or PAK1 inhibits the proliferation of mutant KRAS colon cancer cells independently of RAF/MEK/ERK and PI3K/AKT signaling. Mol Cancer Res. 2013;11:109–121. doi: 10.1158/1541-7786.MCR-12-0466. [DOI] [PubMed] [Google Scholar]
- 13.Yang N., Higuchi O., Ohashi K., Nagata K., Wada A., Kangawa K., et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 14.Siu M.K., Chan H.Y., Kong D.S., Wong E.S., Wong O.G., Ngan H.Y., et al. p21-activated kinase 4 regulates ovarian cancer cell proliferation, migration, and invasion and contributes to poor prognosis in patients. Proc Natl Acad Sci U S A. 2010;107:18622–18627. doi: 10.1073/pnas.0907481107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhuang T., Zhu J., Li Z., Lorent J., Zhao C., Dahlman-Wright K., Stromblad S. p21-activated kinase group II small compound inhibitor GNE-2861 perturbs estrogen receptor alpha signaling and restores tamoxifen-sensitivity in breast cancer cells. Oncotarget. 2015;6:43853–43868. doi: 10.18632/oncotarget.6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu X., Feng J., Zeng D., Ding Y., Yu C., Yang B. PAK4 confers cisplatin resistance in gastric cancer cells via PI3K/Akt- and MEK/ERK-dependent pathways. Biosci Rep. 2014;34 doi: 10.1042/BSR20130102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohammad R.M., Li Y., Muqbil I., Aboukameel A., Senapedis W., Baloglu E., et al. Targeting Rho GTPase effector p21 activated kinase 4 (PAK4) suppresses p-Bad-microRNA drug resistance axis leading to inhibition of pancreatic ductal adenocarcinoma proliferation. Small GTPases. 2019;10:367–377. doi: 10.1080/21541248.2017.1329694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abril-Rodriguez G., Torrejon D.Y., Liu W., Zaretsky J.M., Nowicki T.S., Tsoi J., et al. PAK4 inhibition improves PD-1 blockade immunotherapy. Nat Can. 2019;1:46–58. doi: 10.1038/s43018-019-0003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gajewski T.F., Fessler J. PAK4 as a cancer immune-evasion target. Nat Can. 2020;1:18–19. doi: 10.1038/s43018-019-0012-z. [DOI] [PubMed] [Google Scholar]
- 20.Ma W., Wang Y., Zhang R., Yang F., Zhang D., Huang M., et al. Targeting PAK4 to reprogram the vascular microenvironment and improve CAR-T immunotherapy for glioblastoma. Nat Can. 2020;2:83–97. doi: 10.1038/s43018-020-00147-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staben S.T., Feng J.A., Lyle K., Belvin M., Boggs J., Burch J.D., et al. Back pocket flexibility provides group II p21-activated kinase (PAK) selectivity for type I 1/2 kinase inhibitors. J Med Chem. 2014;57:1033–1045. doi: 10.1021/jm401768t. [DOI] [PubMed] [Google Scholar]
- 22.Guo C., McAlpine I., Zhang J., Knighton D.D., Kephart S., Johnson M.C., et al. Discovery of pyrroloaminopyrazoles as novel PAK inhibitors. J Med Chem. 2012;55:4728–4739. doi: 10.1021/jm300204j. [DOI] [PubMed] [Google Scholar]
- 23.Park J.K., Kim S., Han Y.J., Kim S.H., Kang N.S., Lee H., et al. The discovery and the structural basis of an imidazo[4,5-b]pyridine-based p21-activated kinase 4 inhibitor. Bioorg Med Chem Lett. 2016;26:2580–2583. doi: 10.1016/j.bmcl.2016.04.037. [DOI] [PubMed] [Google Scholar]
- 24.Crawford J.J., Hoeflich K.P., Rudolph J. p21-Activated kinase inhibitors: a patent review. Expert Opin Ther Pat. 2012;22:293–310. doi: 10.1517/13543776.2012.668758. [DOI] [PubMed] [Google Scholar]
- 25.Murray B.W., Guo C., Piraino J., Westwick J.K., Zhang C., Lamerdin J., et al. Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth. Proc Natl Acad Sci U S A. 2010;107:9446–9451. doi: 10.1073/pnas.0911863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senapedis W., Landesman Y., Schenone M., Karger B., Wu S., Shacham S., et al. Identification of novel small molecules as selective PAK4 allosteric modulators (PAMs) by stable isotope labeling of amino acids in cells (SILAC) Eur J Cancer. 2014;50:156. [Google Scholar]
- 27.Mitchell S., Zhang P., Cannon M., Beaver L., Lehman A., Harrington B., et al. Anti-tumor NAMPT inhibitor, KPT-9274, mediates gender-dependent murine anemia and nephrotoxicity by regulating SIRT3-mediated SOD deacetylation. J Hematol Oncol. 2021;14:101. doi: 10.1186/s13045-021-01107-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aboukameel A., Muqbil I., Senapedis W., Baloglu E., Landesman Y., Shacham S., et al. Novel p21-activated kinase 4 (PAK4) allosteric modulators overcome drug resistance and stemness in pancreatic ductal adenocarcinoma. Mol cancer ther. 2017;16:76–87. doi: 10.1158/1535-7163.MCT-16-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hao C., Zhao F., Song H., Guo J., Li X., Jiang X., et al. Structure-based design of 6-chloro-4-aminoquinazoline-2-carboxamide derivatives as potent and selective p21-activated kinase 4 (PAK4) inhibitors. J Med Chem. 2018;61:265–285. doi: 10.1021/acs.jmedchem.7b01342. [DOI] [PubMed] [Google Scholar]
- 30.Guo J., Zhao F., Yin W., Zhu M., Hao C., Pang Y., et al. Design, synthesis, structure‒activity relationships study and X-ray crystallography of 3-substituted-indolin-2-one-5-carboxamide derivatives as PAK4 inhibitors. Eur J Med Chem. 2018;155:197–209. doi: 10.1016/j.ejmech.2018.05.051. [DOI] [PubMed] [Google Scholar]
- 31.Hao C., Huang W., Li X., Guo J., Chen M., Yan Z., et al. Development of 2,4-diaminoquinazoline derivatives as potent PAK4 inhibitors by the core refinement strategy. Eur J Med Chem. 2017;131:1–13. doi: 10.1016/j.ejmech.2017.02.063. [DOI] [PubMed] [Google Scholar]
- 32.Song S., Li X., Guo J., Hao C., Feng Y., Guo B., et al. Design, synthesis and biological evaluation of 1-phenanthryl-tetrahydroisoquinoline derivatives as novel p21-activated kinase 4 (PAK4) inhibitors. Org Biomol Chem. 2015;13:3803–3818. doi: 10.1039/c5ob00037h. [DOI] [PubMed] [Google Scholar]
- 33.Zuccotto F., Ardini E., Casale E., Angiolini M. Through the "gatekeeper door": exploiting the active kinase conformation. J Med Chem. 2010;53:2681–2694. doi: 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]
- 34.Hennequin L.F., Allen J., Breed J., Curwen J., Fennell M., Green T.P., et al. N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl) ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy) quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465–6488. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- 35.Ballard P., Bradbury R.H., Harris C.S., Hennequin L.F., Hickinson M., Johnson P.D., et al. Inhibitors of epidermal growth factor receptor tyrosine kinase: novel C-5 substituted anilinoquinazolines designed to target the ribose pocket. Bioorg Med Chem Lett. 2006;16:1633–1637. doi: 10.1016/j.bmcl.2005.12.028. [DOI] [PubMed] [Google Scholar]
- 36.Liao J.J. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J Med Chem. 2007;50:409–424. doi: 10.1021/jm0608107. [DOI] [PubMed] [Google Scholar]
- 37.Crawford J.J., Lee W., Aliagas I., Mathieu S., Hoeflich K.P., Zhou W., et al. Structure-guided design of group I selective p21-activated kinase inhibitors. J Med Chem. 2015;58:5121–5136. doi: 10.1021/acs.jmedchem.5b00572. [DOI] [PubMed] [Google Scholar]
- 38.Blaquiere N., Castanedo G.M., Burch J.D., Berezhkovskiy L.M., Brightbill H., Brown S., et al. Scaffold-hopping approach to discover potent, selective, and efficacious inhibitors of NF-kappaB inducing kinase. J Med Chem. 2018;61:6801–6813. doi: 10.1021/acs.jmedchem.8b00678. [DOI] [PubMed] [Google Scholar]
- 39.Talele T.T. Acetylene group, friend or foe in medicinal chemistry. J Med Chem. 2020;63:5625–5663. doi: 10.1021/acs.jmedchem.9b01617. [DOI] [PubMed] [Google Scholar]
- 40.Hennequin L.F., Allen J., Breed J., Curwen J., Fennell M., Green T.P., et al. N-(5-Chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465–6488. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- 41.Ryu B.J., Kim S., Min B., Kim K.Y., Lee J.S., Park W.J., et al. Discovery and the structural basis of a novel p21-activated kinase 4 inhibitor. Cancer Lett. 2014;349:45–50. doi: 10.1016/j.canlet.2014.03.024. [DOI] [PubMed] [Google Scholar]
- 42.Callow M.G., Clairvoyant F., Zhu S., Schryver B., Whyte D.B., Bischoff J.R., et al. Requirement for PAK4 in the anchorage-independent growth of human cancer cell lines. J Biol Chem. 2002;277:550–558. doi: 10.1074/jbc.M105732200. [DOI] [PubMed] [Google Scholar]
- 43.Yun C.Y., You S.T., Kim J.H., Chung J.H., Han S.B., Shin E.Y., et al. p21-activated kinase 4 critically regulates melanogenesis via activation of the CREB/MITF and β-Catenin/MITF pathways. J Invest Dermatol. 2015;135:1385–1394. doi: 10.1038/jid.2014.548. [DOI] [PubMed] [Google Scholar]
- 44.Cai S., Ye Z., Wang X., Pan Y., Weng Y., Lao S., et al. Overexpression of P21-activated kinase 4 is associated with poor prognosis in non-small cell lung cancer and promotes migration and invasion. J Exp Clin Cancer Res. 2015;34:48. doi: 10.1186/s13046-015-0165-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin A., Giuliano C.J., Palladino A., John K.M., Abramowicz C., Yuan M.L., et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci Transl Med. 2019;11 doi: 10.1126/scitranslmed.aaw8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Osmani N., Goetz J.G. Multiscale imaging of metastasis in zebrafish. Trends Cancer. 2019;5:766–778. doi: 10.1016/j.trecan.2019.10.003. [DOI] [PubMed] [Google Scholar]
- 47.Zon L.I., Peterson R.T. In vivo drug discovery in the zebrafish. Nat Rev Drug Discov. 2005;4:35–44. doi: 10.1038/nrd1606. [DOI] [PubMed] [Google Scholar]
- 48.Oft M., Heider K.-H., Beug H. TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol. 1998;8:1243–1252. doi: 10.1016/s0960-9822(07)00533-7. [DOI] [PubMed] [Google Scholar]
- 49.Biswas S., Guix M., Rinehart C., Dugger T.C., Chytil A., Moses H.L., et al. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest. 2007;117:1305–1313. doi: 10.1172/JCI30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Padua D., Zhang X.H.F., Wang Q., Nadal C., Gerald W.L., Gomis R.R., et al. TGF beta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66–77. doi: 10.1016/j.cell.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siegel P.M., Shu W.P., Cardiff R.D., Muller W.J., Massague J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc Natl Acad Sci U S A. 2003;100:8430–8435. doi: 10.1073/pnas.0932636100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fan Y., Wang X., Li Y., Zhao X., Zhou J., Ma X., et al. PAK4 enhances TGF-beta 1-induced epithelial-mesenchymal transition through activating beta-catenin signaling pathway in renal tubular epithelial cells. Int J Clin Exp Pathol. 2018;11:3026–3035. [PMC free article] [PubMed] [Google Scholar]
- 53.Jin R., Liu W., Menezes S., Yue F., Zheng M., Kovacevic Z., et al. The metastasis suppressor NDRG1 modulates the phosphorylation and nuclear translocation of beta-catenin through mechanisms involving FRAT1 and PAK4. J Cell Sci. 2014;127:3116–3130. doi: 10.1242/jcs.147835. [DOI] [PubMed] [Google Scholar]
- 54.Kesanakurti D., Maddirela D., Banasavadi-Siddegowda Y.K., Lai T.H., Qamri Z., Jacob N.K., et al. A novel interaction of PAK4 with PPARgamma to regulate Nox1 and radiation-induced epithelial-to-mesenchymal transition in glioma. Oncogene. 2017;36:5309–5320. doi: 10.1038/onc.2016.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen T., Wang T., Liang W., Zhao Q., Yu Q., Ma C.M., et al. PAK4 phosphorylates fumarase and blocks TGFbeta-induced cell growth arrest in lung cancer cells. Cancer Res. 2019;79:1383–1397. doi: 10.1158/0008-5472.CAN-18-2575. [DOI] [PubMed] [Google Scholar]
- 56.Huang J., Chen Z., Lai Z., Liu Y., Yu D., Wu L., et al. Kaempferol ameliorates the regulatory effects of PVT1/miR-214 on epithelial-mesenchymal transition through the PAK4/beta-catenin axis in SRA01/04 cells. Future Med Chem. 2021;13:613–623. doi: 10.4155/fmc-2020-0381. [DOI] [PubMed] [Google Scholar]
- 57.Burlingham B.T., Widlanski T.S. An intuitive look at the relationship of Ki and IC50: a more general use for the Dixon Plot. J Chem Educ. 2003;80:214–218. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.