

Abstract
Over the last several decades, no emerging virus has had a profound impact on the world as the SARS-CoV-2 that emerged at the end of 2019 has done. To know where severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) originated from and how it jumped into human population, we immediately started a surveillance investigation in wild mammals in and around Wuhan when we determined the agent. Herein, coronaviruses were screened in the lung, liver, and intestinal tissue samples from fifteen raccoon dogs, seven Siberian weasels, three hog badgers, and three Reeves’s muntjacs collected in Wuhan and 334 bats collected around Wuhan. Consequently, eight alphacoronaviruses were identified in raccoon dogs, while nine betacoronaviruses were found in bats. Notably, the newly discovered alphacoronaviruses shared a high whole-genome sequence similarity (97.9 per cent) with the canine coronavirus (CCoV) strain 2020/7 sampled from domestic dog in the UK. Some betacoronaviruses identified here were closely related to previously known bat SARS-CoV-related viruses sampled from Hubei province and its neighbors, while the remaining betacoronaviruses exhibited a close evolutionary relationship with SARS-CoV-related bat viruses in the RdRp gene tree and clustered together with SARS-CoV-2-related bat coronaviruses in the M, N and S gene trees, but with relatively low similarity. Additionally, these newly discovered betacoronaviruses seem unlikely to bind angiotensin-converting enzyme 2 because of the deletions in the two key regions of their receptor-binding motifs. Finally, we did not find SARS-CoV-2 or its progenitor virus in these animal samples. Due to the high circulation of CCoVs in raccoon dogs in Wuhan, more scientific efforts are warranted to better understand their diversity and evolution in China and the possibility of a potential human agent.
Keywords: raccoon dog, CCoV, bats, SARS-related coronavirus, Wuhan
1. Introduction
Over the last several decades, no emerging infectious agent has had such a profound impact on the world as severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that emerged at the end of 2019 has done. The disease was first recognized when it appeared in Wuhan of Hubei province, China (Huang et al. 2020; Lu et al. 2020; Wu et al. 2020). A previously unknown coronavirus (Coronaviridae) later termed SARS-CoV-2 was soon determined as the causative agent (Wu et al. 2020). Unfortunately, the virus has caused a severe global pandemic due to its high transmissibility, particularly, having claimed more than six million lives worldwide since its emergence (WHO 2022). Despite extensive research directed toward SARS-CoV-2, crucial questions remain unclear regarding the emergence of this virus, including for where it originated from and when and where it first appeared in humans prior to its initial identification in December 2019 in Wuhan (Zhang and Holmes 2020; Shi et al. 2022).
It is well known that the majority of human viruses have zoonotic origins (Wolfe, Dunavan, and Diamond 2007). The initial research revealed a close evolutionary relationship between SARS-CoV-2 and SARS-related bat viruses (Wu et al. 2020). Especially, the identification of the virus RaTG13 in Rhinolophus affinis bats sampled from Yunnan province of China, which is 96.1 per cent identical to SARS-CoV-2 at the whole-genome sequence level, indicated a probable bat origin of SARS-CoV-2 (Zhou et al. 2020b). Subsequently, other close relatives of SARS-CoV-2 were identified in bats sampled from Yunnan province of China (Zhou et al. 2020a), Japan (Murakami et al. 2020), and Thailand (Wacharapluesadee et al. 2021). Importantly, a virus named BANAL-52 discovered in Rhinolophus malayanus bats from Laos is closer to SARS-CoV-2 than any known viruses and has a potential for infecting humans (Temmam et al. 2022). All these data indicate that bats are a natural reservoir host of SARS-CoV-2. Although SARS-CoV-2-related viruses were also identified in pangolins from Guangdong and Guangxi provinces of China, it shared <92 per cent whole-genome sequence similarity with SARS-CoV-2 (Lam et al. 2020; Liu et al. 2020; Xiao et al. 2020). Additionally, other investigations did not find SARS-CoV-2-related viruses in pangolins (Lee et al. 2020; He et al. 2022). All these data challenge the hypothesis that pangolins are an intermediate host of SARS-CoV-2.
The initial epidemiological investigations targeted their search for early coronavirus disease 2019 (COVID-19) cases with a link to the Huanan Seafood market or its neighborhood in Wuhan (Huang et al. 2020; Lu et al. 2020; The 2019-nCoV Outbreak Joint Field Epidemiology Investigation Team and Li 2020), suggesting that the market might play an important role in SARS-CoV-2 emergence. However, it remains unclear whether the market was the site of a natural spillover or only an amplifier of human transmission in December 2019. Raccoon dogs were found to be susceptible to infection with SARS-CoV-2 and capable of transmitting the virus (Freuling et al. 2020). As various wild animals including raccoon dogs were sold at the market (Xiao et al. 2021; Gao et al. 2022), some studies argued that the emergence of SARS-CoV-2 in Wuhan might be via the live wildlife (particularly, raccoon dogs) at the market (Holmes et al. 2021; Worobey et al. 2022). However, it remains elusive where SARS-CoV-2 emerged in Wuhan derived from Zhang and Holmes (2020), Bloom et al. (2021), and Shi et al. (2022). Here, we immediately performed a surveillance investigation in mammals in and around Wuhan after we identified an unknown coronavirus as the etiologic agent of COVID-19. As a result, canine alphacoronavirus were identified in raccoon dogs, while SARS-CoV-related coronaviruses and recombinant viruses of SARS-related and SARS-CoV-2-related coronaviruses were found in bats. However, no SARS-CoV-2 or the close relatives of SARS-CoV-2 were found in these mammals.
2. Materials and methods
2.1. Sample collection
This study was reviewed and approved by the ethics committee of the National Institute for Communicable Disease Control and Prevention of the China Center for Disease Control and Prevention (CDC), as well as the ethics committee of Shanghai Public Health Clinical Center of Fudan University. All animals were kept alive after capture and treated strictly according to the guidelines for the Laboratory Animal Use and Care from Shanghai Public Health Clinical Center and the Rules for the Implementation of Laboratory Animal Medicine (1998) from the Ministry of Health, China, under the protocols approved by the National Institute for Communicable Disease Control and Prevention of China CDC. All dissections were performed under ether anesthesia, and every effort was made to minimize suffering.
We immediately started a surveillance investigation on the origin of SARS-CoV-2 during 7–18 January 2020. Consequently, the lung, liver, and intestinal tissue samples were collected from mammals, which were captured in the rural area (Changxuanling and Yaoji towns) of Wuhan by three local traders for vendors at animal markets including Huanan Seafood market during 7–18 January. In addition, bats were captured in the surrounding regions (Xiaogan on 7 January and Jingmen on 11 January, respectively) of Wuhan (Fig. 1, Table 1). All animals were identified by sequence analysis of the mitochondrial cytochrome b (mt-Cyt b) gene gene as described previously (Guo et al. 2013) (Supplementary Fig. S1). They were anesthetized with ether before surgery, and all efforts were made to minimize suffering. Tissue samples of heart, liver, spleen, lung, kidney, and brain were collected from bats. All samples were transported and stored at −80℃ for later RNA extraction.
Figure 1.
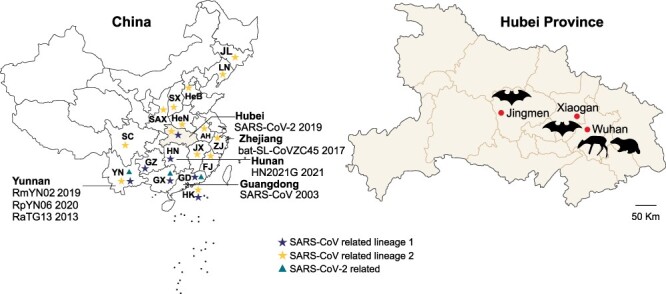
Sampling locations in Wuhan and its surrounding regions in Hubei province, China. SARS-CoV-related and SARS-CoV-2-related viruses identified in China are marked, corresponding to the three lineages in Supplementary Fig. S3. The scattered dots represent Chinese islands. Location abbreviations are as follows: AH, Anhui; FJ, Fujian; GD, Guangdong; GX, Guangxi; GZ, Guizhou; HuB, Hubei; HeB, Hebei; HeN, Henan; HK, Hong Kong; HN, Hunan; JL, Jilin; JX, Jiangxi; LN, Liaoning; SAX, Shaanxi; SC, Sichuan; SX, Shanxi; YN, Yunnan; ZJ, Zhejiang.
Table 1.
Prevalence of coronaviruses in wild animals by species and location in China.
| Family | Species | Wuhan | Xiaogan | Jingmen | Total |
|---|---|---|---|---|---|
| Hipposideridae | Hipposideros armiger | – | – | 0/107 | 0/107 |
| H. pratti | – | 0/3 | – | 0/3 | |
| Rhinolophidae | R. pusillus | – | 7/86 | – | 7/86 |
| R. ferrumequinum | – | 0/4 | – | 0/4 | |
| R. pearsonii | – | 0/2 | – | 0/2 | |
| R. sinicus | – | 2/8 | – | 2/8 | |
| Rhinolophus sp. | – | 0/51 | – | 0/51 | |
| Vespertilionidae | Myotis chinensis | – | 0/2 | – | 0/2 |
| M. petax | – | 0/4 | – | 0/4 | |
| Myotis sp. | – | 0/67 | – | 0/67 | |
| Canidae | N. procyonoides | 8/15 | – | – | 8/15 |
| Cervidae | M. reeves | 0/3 | – | – | 0/3 |
| Mustelidae | Arctonyx collaris | 0/3 | – | – | 0/3 |
| Mustela sibirica | 0/7 | – | – | 0/7 | |
| Total | 14 | 8/28 | 9/227 | 0/107 | 17/362 |
Note: ‘-’ means that no animals were captured.
2.2. DNA and RNA extraction and coronavirus screening
Total DNA was extracted using the DNeasy Blood & Tissue kit (QIAGEN) from tissue samples of animals according to the manufacturer’s protocol. The MT-CYB gene (1,140 bp) was amplified by polymerase chain reaction (PCR) as described previously (Guo et al. 2013).
Total RNA was extracted from tissue samples using TRIzol (Invitrogen, Carlsbad, USA) and RNeasy Plus Universal Mini Kit (QIAGEN, Valencia, USA) according to the manufacturer’s instructions. The RNA was eluted in 50 µl diethyl pyrocarbonate water and was used as the template for reverse transcription PCR (RT-PCR).
Coronavirus screening was performed using a previously published primer set by a pan-CoV-nested PCR targeted to a conserved region of the RNA-dependent RNA polymerase (RdRp) gene as described previously (Wang et al. 2015; Wu et al. 2020). First-round RT-PCR was conducted by using PrimeScript One Step RT-PCR Kit (TaKaRa, Dalian, China). Amplicons 440 bp in length were subjected to direct Sanger sequencing and then searched by basic local alignment search tool based on the National Center for Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov) nucleotide (nt) database. Other coronavirus gene sequences were amplified using the primers designed based on the conserved regions of known genome sequences (Wu et al. 2020). Complete genomes of canine coronaviruses (CCoVs) were amplified using several sets of primers designed by multiple sequence alignments of the conserved regions of known CCoV genome sequences. Additional primers were designed according to the obtained viral sequences recovered in this study. The 5ʹand 3ʹ ends of the genomes of the newly discovered CCoVs were obtained by 5ʹ and 3ʹ rapid amplification of complementary DNA ends (RACE) using a RACE kit (TaKaRa, Dalian, China). RT-PCR amplicons were purified using the QIAquick Gel Extraction kit (Qiagen, Valencia, USA) according to the manufacturer’s recommendations. RT-PCR products of expected size were subject to Sanger sequencing performed by the Sangon corporation (Shanghai, China). Sequences were assembled by SeqMan and manually edited to produce the final viral genomes.
2.3. Phylogenetic analysis and genome recombination analysis
The nucleotide sequences were aligned and found the best substitution model using the MEGA program (Tamura et al. 2011). Phylogenetic analysis was conducted to determine the evolutionary relationship between the newly identified coronaviruses and known coronaviruses using the maximum likelihood (ML) method implemented in PhyML (v3.0) along with 1,000 replicate trees (Guindon et al. 2010).
The full genome alignment of two raccoon dog coronaviruses and other sequences under the Alphacoronavirus-1 were screened for recombination using the RDP, GENECONV, and BootScan methods available within RDP4 (Martin et al. 2010). Potential recombination events were confirmed by Similarity Plot analysis as implemented in Simplot version 3.5.1 (Lole et al. 1999), with a window size of 400 nt and a step size of 40 nt.
3. Results
3.1. Sample collection and coronavirus screening
To investigate the origin of SARS-CoV-2, during 7–18 January 2020, we started to collect fresh lung, liver, and intestinal samples of raccoon dogs, hog badgers, Reeves’s muntjacs, and Siberian weasels captured by local traders in rural areas of Wuhan for vendors in animal markets including the Huanan Seafood market and bats from the surrounding regions of Wuhan (Jingmen and Xiaogan) (Fig. 1) when we identified an unknown coronavirus (Wuhan-Hu-1, later termed SARS-CoV-2) in a patient with severe unknown pneumonia (Wu et al. 2020). These animals included 28 mammals representing four species (Nyctereutes procyonoides, n = 15; Mustela sibirica, n = 7; Arctonyx collaris, n = 3; and Muntiacus reeves, n = 3) sampled from the rural areas of Wuhan, as well as 334 bats (order Chiroptera) comprising ten species of three families (Rhinolophidae, n = 151; Vespertilionidae, n = 73; and Hipposideridae, n = 110) captured in Xiaogan and Jingmen (Table 1, Supplementary Fig. S1). RT-PCR targeting the conserved region of viral RdRp gene was performed to screen coronaviruses as described previously (Wang et al. 2015; Wu et al. 2020). As a result, viral RNA was recovered from a total of seventeen mammals, with an overall detection rate of 4.6 per cent (Tables 1 and Supplementary Table S1). Genetic analysis of all recovered sequences revealed that they were closely related to those of coronavirus, with eight belonging to alphacoronaviruses and nine belonging to betacoronaviruses. All eight alphacoronaviruses were identified in raccoon dogs (N. procyonoides), while nine betacoronaviruses were found in bats (seven Rhinolophus pusillus and two Rhinolophus sinicus) collected from Xiaogan.
To better characterize the newly identified coronaviruses carried by raccoon dogs, two complete viral genome sequences (GH4-2 and GH8-2) were successfully obtained from viral positive samples. Due to 100 per cent genome sequence similarity between the two strains, only the strain GH8-2 was used for genomic feature analysis. For the newly identified bat betacoronaviruses, we also tried to recover the whole-genome sequences of bat coronaviruses, but only five RdRp genes, four S genes, five M genes, and four N genes were obtained from viral positive samples.
3.2. Newly identified raccoon dog alphacoronaviruses
Genetic analysis of all viral sequences recovered from raccoon dogs revealed that they were most closely related to those of the alphacoronaviruses, and the nt similarity between the viruses identified here and previously known alphacoronaviruses ranged from 56.4 per cent to 98.9 per cent. Remarkably, on the phylogenetic tree based on whole genomes (Fig. 2), the coronaviruses (GH4-2 and GH8-2) identified in raccoon dogs here were most closely related to the CCoV 2020/7 detected in feces of a domestic dog from the UK in 2020 (Radford et al. 2021), with a nt similarity of 97.9 per cent (Table 2). However, the evolutionary relationship between the coronaviruses identified here and previously on the trees based on the RdRp, S, M, or N genes is not exactly consistent (Fig. 2). For example, on the RdRp gene tree, both GH4-2 and GH8-2 were closely related to CCoV SD-A1/2021 and CCoV HeB-G1/2021 identified in raccoon dogs from Shandong and Hebei provinces of China (He et al. 2022), as well as to CCoV 2020/7. In contrast, the two viruses were relatively far away from CCoV SD-A1/2021 and CCoV HeB-G1/2021 on both the S and M gene trees, although they remained close to CCoV 2020/7. In addition, on the M gene tree, they were highly similar to CCoV Z19 identified in human urine from Haiti (97.7 per cent), followed by CCoV 2020/7 (Fig. 2). These data suggest the complex evolutionary history of these alphacoronaviruses.
Figure 2.
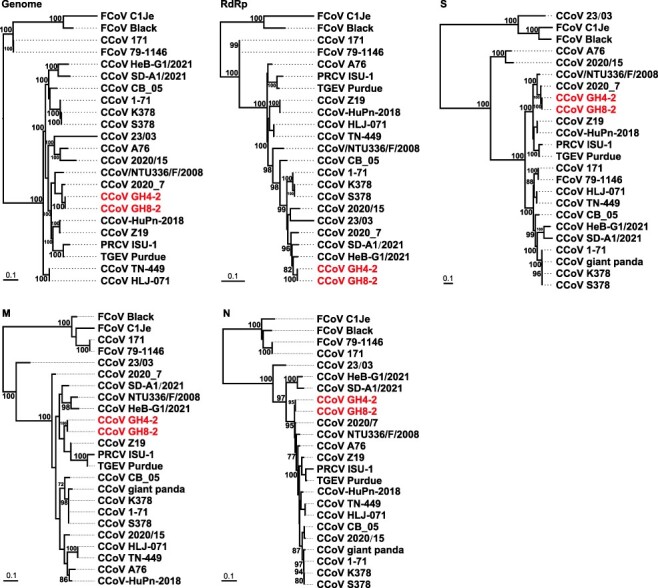
ML phylogenetic trees were constructed based on the nt sequences of the whole genome and RdRp, S, M, and N genes of raccoon dog CoVs and other CoVs. Numbers (>70) above or below branches indicate percentage bootstrap values. The trees were mid-point rooted for clarity only. The scale bar represents the number of substitutions per site.
Table 2.
Percent identities of GH8-2 to Alphacoronavirus-1 reference strains.
| Identity (%) to GH8-2 | |||||||
|---|---|---|---|---|---|---|---|
| AphaCoV-1 | Strain | Accession number | Whole genome (nt) | RdRp (nt/aa) | S (nt/aa) |
M (nt/aa) | N (nt/aa) |
| CCoV-I | 23/03 | KP849472 | 87.3 | 94.6/98.1 | 56.5/47.9 | 87.8/89.0 | 88.1/89.5 |
| CCoV-II | CCoV-HuPn-2018 | MW591993 | 93.4 | 93.8/98.4 | 90.2/93.9 | 92.5/93.9 | 96.0/96.5 |
| CCoV-II | HLJ-071 | KY063616 | 92.1 | 94.1/98.9 | 82.2/83.2 | 92.5/95.4 | 95.6/96.6 |
| CCoV-II | Z19 | MZ420153 | 93.6 | 93.7/98.5 | 90.2/93.7 | 96.7/97.7 | 96.9/99.0 |
| CCoV-II | 1-71 | JQ404409 | 92.8 | 97.1/99.4 | 82.9/83.3 | 94.2/95.8 | 96.8/97.9 |
| CCoV-II | 2020/7 | MT906865 | 97.9 | 97.5/99.4 | 98.3/98.1 | 96.1/96.9 | 99.0/99.0 |
| CCoV-II | 2020/15 | MT906864 | 90.7 | 96.1/99.4 | 70.8/68.7 | 93.0/95.8 | 96.9/97.9 |
| CCoV-II | A76 | JN856008 | 91.0 | 94.3/99.0 | 71.3/68.5 | 92.6/94.3 | 97.3/97.4 |
| CCoV-II | CB/05 | KP981644 | 93.5 | 95.8/98.9 | 83.3/84.0 | 93.7/97.0 | 97.0/97.9 |
| CCoV-II | CCoV/NTU336/F/2008 | GQ477367 | 94.5 | 95.2/99.1 | 93.6/95.9 | 92.6/93.9 | 97.5/99.0 |
| CCoV-II | K378 | KC175340 | 92.6 | 96.9/99.2 | 82.8/82.7 | 94.2/95.8 | 96.7/97.7 |
| CCoV-II | S378 | KC175341 | 92.6 | 96.9/99.2 | 82.8/82.8 | 94.2/95.8 | 96.7/97.7 |
| CCoV-II | SD-A1 | OM451122 | 91.9 | 97.4/99.0 | 79.4/77.6 | 93.5/94.7 | 89.4/89.8 |
| CCoV-II | HeB-G1 | OM451123 | 92.7 | 98.1/99.6 | 80.7/79.9 | 92.6/94.3 | 90.3/91.6 |
| CCoV-II | TN-449 | JQ404410 | 92.1 | 94.2/98.9 | 82.1/83.1 | 92.5/95.4 | 95.6/96.6 |
| CCoV-II | 171 | KC175339 | 84.9 | 92.1/97.4 | 82.4/82.9 | 82.1/85.8 | 79.2/78.6 |
| FCoV-I | C1Je | DQ848678 | 79.2 | 85.9/95.3 | 54.6/46.7 | 81.6/84.0 | 78.9/78.5 |
| FCoV-I | Black | EU186072 | 79.4 | 87.1/95.9 | 54.8/46.5 | 82.3/85.5 | 79.1/78.6 |
| FCoV-II | 79-1146 | NC_002306 | 84.9 | 92.1/97.3 | 82.4/82.9 | 82.1/85.8 | 79.2/78.6 |
| PRCV | ISU-1 | DQ811787 | 91.2 | 93.8/98.3 | 88.8/92.6 | 91.3/95.0 | 92.3/93.2 |
| TGEV | Purdue | AJ271965 | 92.0 | 93.9/98.7 | 89.1/91.5 | 92.1/95.1 | 94.3/94.5 |
3.3. Genome characteristics of alphacoronaviruses in raccoon dogs
The genome of the strain GH8-2 has a size of 29,335 nt, excluding the 3ʹpoly(A) tail, and exhibits a typical genomic organization of Alphacoronavirus-1 (Fig. 3). The 5ʹuntranslated region (UTR) consists of 311 nt including the leader sequence (nt 1–93) and the conserved core 5ʹ-CTAAAC-3ʹ (nt 94–99) of the transcription regulatory sequence (TRS), which is a unique feature of coronaviruses to control continuous and discontinuous RNA synthesis (Sola et al. 2015). Similar TRS signals also precede each of the eight putative mRNA encoding for the structural and nonstructural proteins (Table 3). The 3ʹ end of the viral genome consists of a 275-nt 3ʹUTR that is followed by the poly(A) tail. About two-thirds of the viral genome is occupied by the replicase gene encoding two large polyproteins, pp1a and pp1ab, the latter being synthesized through ribosomal slippage at Position 12,329. The polyproteins of the replicase complex are processed by viral proteinases, the papain-like protease (PLpro) domain of non-structural protein 3 (NSP3) and 3C-like protease (3CLpro) or non-structural protein 5 (NSP5), resulting in sixteen nonstructural proteins (Table 4).
Figure 3.
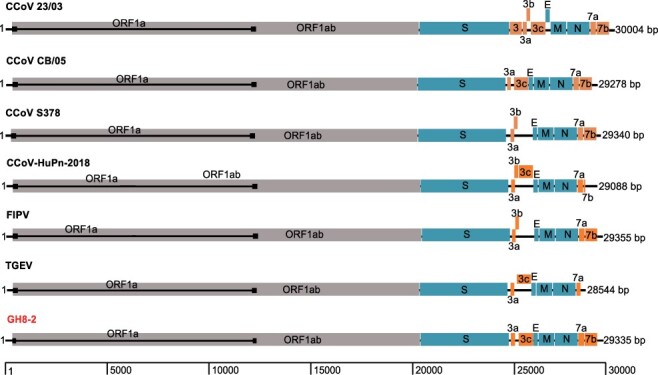
Schematic of the annotated GH8-2 genome in comparison to those of representative alphacoronavirus-1.
Table 3.
Coding of potential and putative transcription regulatory sequences of the GH8-2 genome.
| ORF | Location (nt) | Length(nt) | Length (aa) | TRS location | TRS sequencea |
|---|---|---|---|---|---|
| 1ab | 312-20,359 (shift at 12,329) |
20,049 | 6,682 | 94 | TCGAACTAAACGAAAT |
| S | 20,356-24,729 | 4,374 | 1,457 | 20,324 | AGTTACTAAACTTTGG |
| 3a | 24,793-25,029 | 237 | 78 | 24,785 | AAGAACTAAACTTATG |
| 3c | 25,175-25,909 | 735 | 244 | 24,977 | ATATGCTAAACTTGGT |
| E | 25,896-26,144 | 249 | 82 | 25,853 | CGGTTCTAAACGAAAT |
| M | 26,155-26,943 | 789 | 262 | 26,146 | TTGAACTAAACAAAAT |
| N | 26,956-28,104 | 1,149 | 382 | 26,944 | TATAACTAAACTTCTA |
| 7a | 28,109-28,414 | 306 | 101 | 28,101 | ACGAACTAAACGAATG |
| 7b | 28,419-29,060 | 642 | 213 |
Underlined and bold type indicates the conserved nt in the TRS core sequence.
Table 4.
Prediction of the putative polyprotein pp1ab cleavage sites in GH8-2.
| Cleavage product |
First–last amino acid residues | Protein size (aa) | Putative functional domain(s) |
|---|---|---|---|
| nsp1 | 1Met–Gly110 | 110 | |
| nsp2 | 111Ala–Gly879 | 769 | |
| nsp3 | 880Gly–Gly2386 | 1507 | ADRP, PL1pro, PL2pro |
| nsp4 | 2387Ser–Gln2876 | 490 | |
| nsp5 | 2877Ser–Gln3178 | 302 | 3CLpro |
| nsp6 | 3179Ala–Gln3472 | 294 | |
| nsp7 | 3473Ser–Gln3555 | 83 | |
| nsp8 | 3556Ser–Gln3750 | 195 | |
| nsp9 | 3751Asn–Gln3861 | 111 | |
| nsp10 | 3862Ala–Gln3996 | 135 | |
| nsp11 | 3997Ser–Asp4015 | 19 | Short peptide at the end of ORF1a |
| nsp12 | 3997Ser–Gln4925 | 929 | RdRp |
| nsp13 | 4926Ala–Gln5524 | 599 | Hel |
| nsp14 | 5525Ala–Gln6043 | 519 | ExoN |
| nsp15 | 6044Ser–Gln6382 | 339 | NendoU |
| nsp16 | 6383Ser–Pro6682 | 300 | O-MT |
Four structural proteins were detected downstream of the replicase ORF1ab gene, namely the spike (S), small envelope (E), membrane (M), and nucleocapsid (N) proteins. Finally, the S protein has a size of 1,457 amino acids (aa) with 25 potential N-glycosylation sites predicted by using the NetNGlyc server (http://www.cbs.dtu.dk/services/NetNGlyc/), whereas 28 and 31 potential N-glycosylation sites had been detected in the strains 23/03 and CB/05, respectively (Decaro et al. 2007, 2015).
In addition to a common set of genes for replicase and structural proteins, like other variants of Alphacoronavirus-1, the newly identified raccoon dog coronavirus have some accessory genes between S gene and E gene and downstream of N gene. Analogous to other CCoVs, there are two open reading frames (ORFs, 3a and 3c) in the S–E intergenic region of GH8-2, encoding for products with sizes of 78 and 244 aa, respectively. Additionally, GH8-2 has the ORF3b deletion similar to the strain CB/05 (Fig. 3). Finally, the 3ʹ end accessory genes were ORF7a and ORF7b that encode for 101 and 213 aa, respectively.
Finally, although recombination events were found in known variants of Alphacoronavirus-1, such as feline coronavirus type II (Herrewegh et al. 1998) and CCoV 23/03 (Decaro et al. 2015), a detailed analysis of the whole-genome sequences of GH8-2 did not exhibit any signs for recombination events in the newly identified CCoVs in raccoon dogs.
3.4. Newly identified betacoronaviruses in bats
Genetic analysis of recovered RdRp gene sequences revealed that the newly identified bat coronaviruses showed a closer evolutionary relationship with SARS-CoV and bat SARS-related coronaviruses (>91.7 per cent) than SARS-CoV-2 and SARS-CoV-2-related coronaviruses (<88.3 per cent) (Table 5, Fig. 4 and Supplementary Fig. S2). On the RdRp tree, the newly identified bat sarbecoviruses were divided into two lineages. One lineage included the newly identified viruses XG-104, XG-257, XG-279, and XG-331 all from R. pusillus. They clustered with those (HN2021A and HN2021G) identified in R. pusillus from Hunan province of China (Wu et al. 2022), which is a neighbor of Hubei province, as well as SARS-CoV and other SARS-CoV-related coronaviruses identified in other parts of China (Supplementary Fig. S2). Another lineage included the newly identified virus XG-145 in R. sinicus and known SARS-CoV-related coronaviruses JSB_Rsin (MZ328294.1) and BtRs-BetaCoV/HuB2013 identified in R. sinicus from Hubei (Wu et al. 2016) (Supplementary Fig. S2). Clearly, the RdRp gene of these bat viruses has a common ancestor with known SARS-CoV-related viruses.
Table 5.
Identities of nt and aa sequences of new bat coronaviruses to representative sarbecoviruses.
| RdRp (nt/aa) | S (nt/aa) | M (nt/aa) | N (nt/aa) | |||||
|---|---|---|---|---|---|---|---|---|
| Strain | XG-104 | XG-145 | XG-104 | XG-145 | XG-104 | XG-145 | XG-104 | XG-145 |
| HN2021A | 98.3/99.1 | 92.4/99.2 | 95.6/98.8 | 75.8/82.2 | 98.4/100 | 82.7/89.6 | 95.6/97.9 | 91.4/94.0 |
| HN2021G | 97.6/99.4 | 92.8/99.5 | 96.4/98.7 | 75.4/82.2 | 99.4/99.6 | 83.3/89.6 | 97.5/98.3 | 93.0/94.3 |
| RsSHC014 | 97.7/99.3 | 93.0/99.7 | 73.7/77.9 | 78.4/81.2 | 84.7 /89.6 | 93.2 /96.4 | 91.9/93.6 | 97.2/98.3 |
| WIV1 | 97.6/99.1 | 92.9/99.6 | 74.1/78.0 | 78.5/81.1 | 84.7/89.6 | 93.2 /96.4 | 91.8/93.6 | 97.1/98.3 |
| Rs672/2006 | 97.2/99.4 | 93.1/99.7 | 75.8/81.4 | 87.8/95.2 | 84.5/89.2 | 93.1/96.8 | 91.9/93.6 | 97.7/99.3 |
| SARS-CoV Tor2 | 97.2/99.1 | 93.2/99.5 | 73.5/77.1 | 78.0/80.5 | 84.5/89.6 | 93.4/97.7 | 91.7/93.8 | 96.8/98.6 |
| SZ3 | 97.2/99.0 | 93.2/99.4 | 73.4/76.8 | 77.9/80.2 | 84.5/89.2 | 93.7/97.7 | 91.7/93.8 | 96.8/98.6 |
| Cp/Yunnan2011 | 94.0/98.7 | 92.8/99.2 | 75.4/81.8 | 85.0/92.9 | 86.2/90.5 | 92.3/97.7 | 91.6/93.8 | 97.1/99.3 |
| bat-SL-CoVZC45 | 92.7/98.6 | 92.6/99.0 | 95.3/99.0 | 75.5/82.5 | 97.6/100 | 83.0/89.6 | 95.1/96.7 | 90.7/92.8 |
| JSB_Rsin | 92.4/98.7 | 99.4/99.7 | 76.2/82.6 | 94.6/95.7 | 85.0/90.1 | 95.3/99.1 | 92.5/93.6 | 99.3/99.5 |
| Rf1 | 92.3/98.3 | 92.2/98.6 | 75.5/81.6 | 82.0/88.6 | 83.8/89.2 | 91.9/96.8 | 89.6/91.7 | 94.1/96.9 |
| BtRs-BetaCoV/HuB2013 | 92.2/98.9 | 97.5/99.7 | 76.3/82.2 | 87.7/91.5 | 85.0/90.1 | 95.3/99.1 | 92.0/93.8 | 98.3/99.8 |
| HKU3-1 | 91.7/98.3 | 92.4/98.8 | 76.2/83.1 | 85.4/93.9 | 84.7/90.1 | 95.0/99.1 | 91.7/92.6 | 96.2/97.9 |
| SARS-CoV-2 Wuhan-Hu-1 | 88.3/96.2 | 87.3/96.2 | 77.2/82.1 | 72.7/76.6 | 94.5/98.7 | 83.8/90.5 | 89.4/92.9 | 88.6/91.4 |
| RaTG13 | 88.0/96.2 | 86.9/96.2 | 77.5/81.8 | 73.0/76.5 | 94.3 /99.1 | 83.2 /89.6 | 89.4/93.6 | 88.2/91.6 |
| bat/Yunnan/RpYN06/2020 | 88.2/96.2 | 87.0/96.2 | 92.4/98.0 | 75.3/82.1 | 93.2/99.1 | 83.0/89.6 | 90.0/93.3 | 88.7/91.9 |
| RacCS203 | 87.8/96.1 | 86.9/96.1 | 74.9/78.4 | 73.2/78.9 | 94.3/98.6 | 83.5/89.6 | 91.1/95.2 | 88.9/93.1 |
| BM48-31 | 87.8/97.6 | 87.4/98.2 | 68.6/71.9 | 71.0/75.9 | 76.8/87.4 | 79.9/91.4 | 77.5/88.5 | 78.7/88.7 |
| Rc-o319 | 86.9/96.6 | 86.4/96.7 | 72.9/77.6 | 71.0/75.9 | 85.9/91.5 | 85.6/93.7 | 86.7/89.7 | 87.1/90.6 |
| PCoV_GX-P2V | 86.4/96.0 | 86.4/96.0 | 76.8/81.5 | 73.4/76.4 | 91.5 /98.2 | 82.1/89.6 | 89.5/92.3 | 88.1/93.0 |
| MP789 | 85.1/96.1 | 84.6/96.1 | 80.4/86.8 | 72.1/76.6 | 94.2/99.6 | 82.9/89.2 | 89.7/93.1 | 88.2/91.9 |
Figure 4.

ML phylogenetic trees were based on the nt sequences of the RdRp, S, M, and N genes of sarbecoviruses. Numbers (>70) above or below branches indicate percentage bootstrap values. The trees were mid-point rooted for clarity only. The scale bar represents the number of substitutions per site. All viruses found in this study are labeled in red. SARS-CoV and SARS-CoV-related virus groups are labeled in red, and SARS-CoV-2 and SARS-CoV-2 related virus group are labeled in green. The GenBank and GISAID accession numbers are available in Supplementary Table S3.
Interestingly, although the newly identified coronavirus XG-145 was still closely related to SARS-CoV-related bat viruses on the M, N, and S gene trees, the coronaviruses (XG-104, XG-257, XG-279, and XG-331) clustered together with SARS-CoV-2-related bat coronaviruses rather than SARS-CoV-related bat coronaviruses (Fig. 4, Supplementary Table S2). Particularly, for all the four genes, these bat viruses were most closely related to the viruses (HN2021A and HN2021G) identified in R. pusillus from Hunan province of China (Fig. 4, Supplementary Table S2), which were characterized as recombinants of SARS-CoV-related and SARS-CoV-2-related viruses (Wu et al. 2022). In addition to the viruses HN2021A and HN2021G, they also showed a closer evolutionary relation with the virus bat-SL-CoVZC45 identified also in R. pusillus from an island (Zhoushan) of Zhejiang province (Hu et al. 2018), which is ∼1,000 km away from Xiaogan. Interestingly, these viruses (XG-104, HN2021A, HN2021G, and bat-SL-CoVZC45) showed a 92.4 per cent S gene similarity with bat/Yunnan/RpYN06/2020, but lower (<77.7 per cent) S gene similarity with other SARS-CoV-related coronaviruses (Supplementary Table S2). Finally, the variation of these viruses on the RdRp gene and other three gene trees also suggest that they are probably originated through the recombination between SARS-CoV-related and SARS-CoV-2-related viruses. Unfortunately, as no whole-genome sequences were obtained, we could not perform a recombinant analysis.
SARS-CoV and SARS-CoV-2, as well as some bat SARS-CoV-related viruses, are known to use angiotensin-converting enzyme 2 (ACE2) as the receptor to enter human cells (Li et al. 2003; Hu et al. 2017; Zhou et al. 2020b), whereas other bat SARS-CoV-related viruses are unable to bind to ACE2 (Ren et al. 2008; Hu et al. 2017). To know the potential of the newly identified bat coronaviruses to infect human, we further compared the receptor-binding domain of the spike protein of these viruses with those of other sarbecoviruses (Supplementary Fig. S3). As shown in Supplementary Fig. S3B, XG-104 and XG-145 had similar aa deletions at Positions 444–449 and 478–491 as the bat SARS-CoV-related viruses (e.g. bat-SL-CoVZC45 and Rf1), which lack capability to use human ACE2 because of the deletions in the key regions of receptor-binding motif (Starr et al. 2022). Hence, the newly identified viruses seem not to be able to infect human cell although infection experiments are needed for confirmation.
4. Discussion
Like the majority of human viruses (Wolfe, Dunavan, and Diamond 2007), all previous human coronaviruses have zoonotic origins (Holmes et al. 2021). The emergence of SARS-CoV at the end of 2002 in Guangdong province of China was believed to be associated with markets selling live animals, and the progenitor virus of SARS-CoV was identified in palm civets and raccoon dogs sampled from live animal markets in Guangzhou with extensively high detection rates (Guan et al. 2003; Kan et al. 2005). Hence, the palm civets and raccoon dogs were believed to be the intermediate hosts of SARS-CoV. As some of the early cases were linked to the Huanan Seafood market (Huang et al. 2020; Lu et al. 2020), in which various animals including raccoon dogs and hog badgers were sold (Xiao et al. 2021; Gao et al. 2022), the emergence of SARS-CoV-2 in Wuhan was considered to bear the similar signatures of SARS-CoV (Holmes et al. 2021). Importantly, an experimental study demonstrated that raccoon dogs were susceptible to SARS-CoV-2 infection and can transmit the virus to direct in-contact animals (Freuling et al. 2020). Hence, the Huanan Seafood market was also speculated to be an epicenter of SARS-CoV-2 emergence, probably via the live wildlife at the market (Holmes et al. 2021; Worobey et al. 2022). However, SARS-CoV-2 was identified in environmental samples but not in animal samples collected at the market at the beginning of COVID-19 (Gao et al. 2022). Importantly, there has been no consensus on the origin of SARS-CoV-2 in Wuhan so far (Bloom et al. 2021). Herein, we screened SARS-CoV-2 and related coronaviruses in the lung, liver, and intestinal tissue samples of raccoon dogs, hog badgers, Siberian weasels, and Reeves’s muntjacs, which were collected at the beginning of COVID-19 emergence from the local people who captured these wild animals in rural areas of Wuhan for vendors at animal markets including the Huanan Seafood market. Interestingly, coronaviruses were only identified in raccoon dogs with a very high detection rate (53.3 per cent), all of which belong to alphacoronaviruses. No SARS-CoV-2 or the close relatives of both SARS-CoV and SARS-CoV-2 were found in these animal samples, in keeping with a recent surveillance investigation in game animals in China (He et al. 2022). As these animal samples were collected at the beginning of COVID-19 emergence in Wuhan, our data suggest that SARS-CoV-2 or SARS-CoV-2-related viruses are not present in these local wild animals. Considering that the sample size and diversity of wild animals analyzed here are relatively small, more epidemiologic investigations with a large sample size and wide geography will be helpful to determine whether SARS-CoV-2 or closely related viruses are present or not in wild and farmed raccoon dogs and other related mammals (e.g. hog badgers and minks) and whether they act as intermediate hosts or not.
CCoVs have been identified in a range of domestic and wild animals (Islam et al. 2021). Different genotypes of CCoVs of Alphacoronavirus-1 species cause moderate-to-severe enteric disease in dogs (Decaro and Buonavoglia 2011). Remarkably, the discovery of a CCoV (termed CCoV-HuPn-2018) in children with pneumonia in Malaysia highlights the significance of the virus in public health (Vlasova et al. 2021). CCoVs in raccoon dogs sampled from China were reported recently (He et al. 2022). Notably, the CCoVs identified in raccoon dogs from China shared a higher whole-genome sequence similarity (97.9 per cent) with the virus strain 2020/7 identified in domestic dogs in the UK than other CCoVs (CCoV HeB-G1/2021 and CCoV SD-A1/2021) found in raccoon dogs from China (Fig. 2), suggesting a possibility of the wide geographical distribution of these highly similar CCoVs through pet trade. However, the evolutionary relationship between the newly identified CCoVs and the strain 2020/7 and those from other regions of China varied on different gene trees, indicating a complex evolutionary history of CCoVs. Finally, a high similarity in the S protein among the newly identified CCoVs and those causing disease in humans or domestic animals also calls for more surveillance to CCoVs in China.
Bats are a natural reservoir host of coronaviruses. Since the discovery of SARS-CoV-related viruses in bats from China in 2005 (Li et al. 2005), extensive researches have been performed into the diversity and circulation of coronaviruses in bats globally (Cui, Li, and Shi 2019; Lin et al. 2017, Van Brussel and Holmes 2022). The discovery of close relatives of SARS-CoV-2 (RaTG13, RmYN02, Rc-o319, and RacCS203) in Rhinolophus bats sampled in and outside of China indicates a probable bat origin of SARS-CoV-2 (Murakami et al. 2020; Zhou et al. 2020a, 2020b; Wacharapluesadee et al. 2021). Recently, the betacoronaviruses, which were identified in Rhinolophus bats sampled from Laos, shared a higher homology with SARS-CoV-2 than any known viruses including RaTG13 (Temmam et al. 2022). Importantly, they seem to be potentially infectious for humans. Hence, these data provide strong evidence that SARS-CoV-2 has a natural origin via bats (Mallapaty 2021; Temmam et al. 2022). Herein, the bats were collected around Wuhan (Xiaogan and Jingmen) and included ten species of three families (Table 1). Although five species of Rhinolophus bats are in circulation in the surrounding regions of Wuhan, we did not find the close relatives of SARS-CoV-2 as reported previously (Murakami et al. 2020; Zhou et al. 2020a, 2020b; Wacharapluesadee et al. 2021; Temmam et al. 2022). However, the betacoronaviruses (XG-104, XG-257, XG-279, and XG-331), which were more similar to SARS-CoV in some parts of the genome but more similar to SARS-CoV-2 in other parts, were identified in the bats sampled from Xiaogan (Fig. 1). Notably, similar viruses were also reported recently in bats sampled from Hunan (Wu et al. 2022) and an island of Zhejiang (Hu et al. 2018). The wide geographic distribution and geographic difference of these viruses suggest that they have a relatively long evolutionary history. Considering the large difference between these bat recombinant viruses and SARS-CoV-2, the virus emergence in humans in Wuhan at the end of 2019 is impossible to originate directly from these bat viruses.
In summary, CCoVs were identified in wild raccoon dogs sampled from the rural areas of Wuhan, while SARS-CoV-related viruses and recombinant coronaviruses were found in bats sampled around Wuhan. However, SARS-CoV-2 or its progenitor virus had not been identified in these animals. Hence, more scientific efforts are needed to determine from where and how SARS-CoV-2 appeared in Wuhan at the end of 2019. Due to the identification of the close relatives of SARS-CoV-2 in bats in multiple regions of the Indochinese Peninsula, it is still noteworthy to perform more investigations to find the progenitor virus of SARS-CoV-2 in bats and other animals in the regions.
Supplementary Material
Contributor Information
Wen Wang, Shanghai Public Health Clinical Center, Shanghai key laboratory of organ transplantation of Zhongshan Hospital, State Key Laboratory of Genetic Engineering, School of Life Sciences and Human Phenome Institute, Fudan University, No. 2901 Caolang Road, Jinshan district, Shanghai 200000, China; Department of Zoonosis, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, No. 155 Changbai Road, Changping district, Beijing 102206, China.
Jun-Hua Tian, Hubei Key Laboratory of Resources Utilization and Sustainable Pest Management, College of Plant Science and Technology, Huazhong Agricultural University, No. 1 Shizishan Street, Hongshan district, Wuhan 430000, China; Wuhan Center for Disease Control and Prevention, No. 288 Machang Road, Jianghan district, Wuhan 430000, China.
Xiao Chen, College of Marine Sciences, South China Agricultural University, No. 483 Wushan Road, Tianhe district, Guangzhou 510000, China.
Rui-Xue Hu, Shanghai Public Health Clinical Center, Shanghai key laboratory of organ transplantation of Zhongshan Hospital, State Key Laboratory of Genetic Engineering, School of Life Sciences and Human Phenome Institute, Fudan University, No. 2901 Caolang Road, Jinshan district, Shanghai 200000, China.
Xian-Dan Lin, Wenzhou Center for Disease Control and Prevention, No. 41 Xincheng Road, Lucheng district, Wenzhou 325000, China.
Yuan-Yuan Pei, Shanghai Public Health Clinical Center, Shanghai key laboratory of organ transplantation of Zhongshan Hospital, State Key Laboratory of Genetic Engineering, School of Life Sciences and Human Phenome Institute, Fudan University, No. 2901 Caolang Road, Jinshan district, Shanghai 200000, China.
Jia-Xin Lv, Shanghai Public Health Clinical Center, Shanghai key laboratory of organ transplantation of Zhongshan Hospital, State Key Laboratory of Genetic Engineering, School of Life Sciences and Human Phenome Institute, Fudan University, No. 2901 Caolang Road, Jinshan district, Shanghai 200000, China.
Jiao-Jiao Zheng, Shanghai Public Health Clinical Center, Shanghai key laboratory of organ transplantation of Zhongshan Hospital, State Key Laboratory of Genetic Engineering, School of Life Sciences and Human Phenome Institute, Fudan University, No. 2901 Caolang Road, Jinshan district, Shanghai 200000, China.
Fa-Hui Dai, Shanghai Public Health Clinical Center, Shanghai key laboratory of organ transplantation of Zhongshan Hospital, State Key Laboratory of Genetic Engineering, School of Life Sciences and Human Phenome Institute, Fudan University, No. 2901 Caolang Road, Jinshan district, Shanghai 200000, China.
Zhi-Gang Song, Shanghai Public Health Clinical Center, Shanghai key laboratory of organ transplantation of Zhongshan Hospital, State Key Laboratory of Genetic Engineering, School of Life Sciences and Human Phenome Institute, Fudan University, No. 2901 Caolang Road, Jinshan district, Shanghai 200000, China.
Yan-Mei Chen, Shanghai Public Health Clinical Center, Shanghai key laboratory of organ transplantation of Zhongshan Hospital, State Key Laboratory of Genetic Engineering, School of Life Sciences and Human Phenome Institute, Fudan University, No. 2901 Caolang Road, Jinshan district, Shanghai 200000, China.
Yong-Zhen Zhang, Shanghai Public Health Clinical Center, Shanghai key laboratory of organ transplantation of Zhongshan Hospital, State Key Laboratory of Genetic Engineering, School of Life Sciences and Human Phenome Institute, Fudan University, No. 2901 Caolang Road, Jinshan district, Shanghai 200000, China.
Data availability
The two whole-genome sequences of CCoV obtained in this study have been deposited in GenBank under accession numbers OM950728–OM950729. The viral sequences of bat SARS-CoV sequences are under accession numbers ON012454–ON012471. The MT-CYB gene sequences of mammals are under accession numbers ON012473–ON012508.
Supplementary data
Supplementary data are available at Virus Evolution online.
Funding
National Natural Science Foundation of China (Grants 32041004, 31930001, 32130002, 81861138003, and 81672057). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest:
None declared.
References
- Bloom J. D. et al. (2021) ‘Investigate the Origins of COVID-19’, Science, 372: 694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J., Li F., and Shi Z. L. (2019) ‘Origin and Evolution of Pathogenic Coronaviruses’, Nature Reviews Microbiology, 17: 181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N. et al. (2007) ‘Molecular Characterisation of the Virulent Canine Coronavirus CB/05 Strain’, Virus Research, 125: 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2015) ‘Full-length Genome Analysis of Canine Coronavirus Type I’, Virus Research, 210: 100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaro N., and Buonavoglia C. (2011) ‘Canine Coronavirus: Not Only an Enteric Pathogen’, Veterinary Clinics of North America: Small Animal Practice, 41: 1121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freuling C. M. et al. (2020) ‘Susceptibility of Raccoon Dogs for Experimental SARS-CoV-2 Infection’, Emerging Infectious Diseases, 26: 2982–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao G. F. et al. (2022) Surveillance of SARS-CoV-2 in the Environment and Animal Samples of the Huanan Seafood Market.doi: 10.21203/rs.3.rs-1370392/v1. [DOI]
- Guan Y. et al. (2003) ‘Isolation and Characterization of Viruses Related to the SARS Coronavirus from Animals in Southern China’, Science, 302: 276–8. [DOI] [PubMed] [Google Scholar]
- Guindon S. et al. (2010) ‘New Algorithms and Methods to Estimate Maximum-likelihood Phylogenies: Assessing the Performance of PhyML 3.0’, Systematic Biology, 59: 307–21. [DOI] [PubMed] [Google Scholar]
- Guo W. P. et al. (2013) ‘Phylogeny and Origins of Hantaviruses Harbored by Bats, Insectivores, and Rodents’, PLoS Pathogens, 9: e1003159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W. T. et al. (2022) ‘Virome Characterization of Game Animals in China Reveals a Spectrum of Emerging Pathogens’, Cell. 185: 1117–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A. A. et al. (1998) ‘Feline Coronavirus Type II Strains 79-1683 and 79-1146 Originate from a Double Recombination between Feline Coronavirus Type I and Canine Coronavirus’, Journal of Virology, 72: 4508–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes E. C. et al. (2021) ‘The Origins of SARS-CoV-2: A Critical Review’, Cell, 184: 4848–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B. et al. (2017) ‘Discovery of a Rich Gene Pool of Bat SARS-Related Coronaviruses Provides New Insights into the Origin of SARS Coronavirus’, PLoS Pathogens, 13: e1006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D. et al. (2018) ‘Genomic Characterization and Infectivity of a Novel SARS-like Coronavirus in Chinese Bats’, Emerging Microbes & Infections, 7: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. et al. (2020) ‘Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China’, The Lancet, 395: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam A. et al. (2021) ‘Evolutionary Dynamics and Epidemiology of Endemic and Emerging Coronaviruses in Humans, Domestic Animals, and Wildlife’, Viruses, 13: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan B. et al. (2005) ‘Molecular Evolution Analysis and Geographic Investigation of Severe Acute Respiratory Syndrome Coronavirus-like Virus in Palm Civets at an Animal Market and on Farms’, Journal of Virology, 79: 11892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam T. T. et al. (2020) ‘Identifying SARS-CoV-2-related Coronaviruses in Malayan Pangolins’, Nature, 583: 282–5. [DOI] [PubMed] [Google Scholar]
- Lee J. et al. (2020) ‘No Evidence of Coronaviruses or Other Potentially Zoonotic Viruses in Sunda Pangolins (Manis javanica) Entering the Wildlife Trade via Malaysia’, Ecohealth, 17: 406–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 2019-nCoV Outbreak Joint Field Epidemiology Investigation Team and Li Q. (2020) ‘An Outbreak of NCIP (2019-ncov) Infection in China - Wuhan, Hubei Province, 2019-2020’, China CDC Weekly, 2: 79–80. [PMC free article] [PubMed] [Google Scholar]
- Li W. et al. (2003) ‘Angiotensin-converting Enzyme 2 Is a Functional Receptor for the SARS Coronavirus’, Nature, 426: 450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2005) ‘Bats are Natural Reservoirs of SARS-like Coronaviruses’, Science, 310: 676–9. [DOI] [PubMed] [Google Scholar]
- Lin X. D. et al. (2017) ‘Extensive Diversity of Coronaviruses in Bats from China’, Virology, 507: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P. et al. (2020) ‘Are Pangolins the Intermediate Host of the 2019 Novel Coronavirus (SARS-CoV-2)?’, PLoS Pathogens, 16: e1008421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lole K. S. et al. (1999) ‘Full-length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-infected Seroconverters in India, with Evidence of Intersubtype Recombination’, Journal of Virology, 73: 152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R. et al. (2020) ‘Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding’, The Lancet, 395: 565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallapaty S. (2021) ‘Laos Bats Host Closest Known Relatives of Virus behind COVID’, Nature, 597: 603. [DOI] [PubMed] [Google Scholar]
- Martin D. P. et al. (2010) ‘RDP3: A Flexible and Fast Computer Program for Analyzing Recombination’, Bioinformatics, 26: 2462–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S. et al. (2020) ‘Detection and Characterization of Bat Sarbecovirus Phylogenetically Related to SARS-CoV-2, Japan’, Emerging Infectious Diseases, 26: 3025–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford A. D. et al. (2021) ‘Outbreak of Severe Vomiting in Dogs Associated with a Canine Enteric Coronavirus, United Kingdom’, Emerging Infectious Diseases, 27: 517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W. et al. (2008) ‘Difference in Receptor Usage between Severe Acute Respiratory Syndrome (SARS) Coronavirus and SARS-like Coronavirus of Bat Origin’, Journal of Virology, 82: 1899–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M. et al. (2022) ‘Total Infectome Characterization of Respiratory Infections in pre-COVID-19 Wuhan, China’, PLoS Pathogens, 18: e1010259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola I. et al. (2015) ‘Continuous and Discontinuous RNA Synthesis in Coronaviruses’, Annual Review of Virology, 2: 265–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T. N. et al. (2022) ‘ACE2 Binding Is an Ancestral and Evolvable Trait of Sarbecoviruses’, Nature, 603: 913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K. et al. (2011) ‘MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods’, Molecular Biology and Evolution, 28: 2731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temmam S. et al. (2022) ‘Bat Coronaviruses Related to SARS-CoV-2 and Infectious for Human Cells’, Nature, 604: 330–6. [DOI] [PubMed] [Google Scholar]
- Van Brussel K., and Holmes E. C. (2022) ‘Zoonotic Disease and Virome Diversity in Bats’, Current Opinion in Virology, 52: 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlasova A. N. et al. (2021) ‘Novel Canine Coronavirus Isolated from a Hospitalized Pneumonia Patient, East Malaysia’, Clinical Infectious Diseases: An Official Publication of the Infectious Diseases Society of America, 74: 446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacharapluesadee S. et al. (2021) ‘Evidence for SARS-CoV-2 Related Coronaviruses Circulating in Bats and Pangolins in Southeast Asia’, Nature Communications, 12: 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. (2015) ‘Discovery, Diversity and Evolution of Novel Coronaviruses Sampled from Rodents in China’, Virology, 474: 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . (2022), WHO Coronavirus (COVID-19) Dashboard | WHO Coronavirus (COVID-19) Dashboard with Vaccination Data. <https://covid19.who.int/> accessed 9 Jun 2022.
- Wolfe N. D., Dunavan C. P., and Diamond J. (2007) ‘Origins of Major Human Infectious Diseases’, Nature, 447: 279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worobey M. et al. (2022), The Huanan Market Was the Epicenter of SARS-CoV-2 Emergence. ZENOBO. <https://zenodo.org/record/6299116> accessed 9 Jun 2022.
- Wu F. et al. (2020) ‘A New Coronavirus Associated with Human Respiratory Disease in China’, Nature, 579: 265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. et al. (2016) ‘Deciphering the Bat Virome Catalog to Better Understand the Ecological Diversity of Bat Viruses and the Bat Origin of Emerging Infectious Diseases’, The ISME Journal, 10: 609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2022) ‘A Comprehensive Survey of Bat Sarbecoviruses across China for the Origin Tracing of SARS-CoV and SARSCoV-2’, Research Square. <https://www.researchsquare.com/article/rs-885194/v1> accessed 9 Jun 2022. [Google Scholar]
- Xiao K. et al. (2020) ‘Isolation of SARS-CoV-2-related Coronavirus from Malayan Pangolins’, Nature, 583: 286–9. [DOI] [PubMed] [Google Scholar]
- Xiao X. et al. (2021) ‘Animal Sales from Wuhan Wet Markets Immediately Prior to the COVID-19 Pandemic’, Scientific Reports, 11: 11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Z., and Holmes E. C. (2020) ‘A Genomic Perspective on the Origin and Emergence of SARS-CoV-2’, Cell, 181: 223–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. et al. (2020a) ‘A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein’, Current Biology, 30: 3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P. et al. (2020b) ‘A Pneumonia Outbreak Associated with A New Coronavirus of Probable Bat Origin’, Nature, 579: 270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The two whole-genome sequences of CCoV obtained in this study have been deposited in GenBank under accession numbers OM950728–OM950729. The viral sequences of bat SARS-CoV sequences are under accession numbers ON012454–ON012471. The MT-CYB gene sequences of mammals are under accession numbers ON012473–ON012508.