

Abstract
Background
Transcription factor Wilms’ tumor gene 1 (WT1) is an ideal tumor target based on its expression in a wide range of tumors, low-level expression in normal tissues and promoting role in cancer progression. In clinical trials, WT1 is targeted using peptide-based or dendritic cell-based vaccines and T-cell receptor (TCR)-based therapies. Antitumor reactivities were reported, but T-cell reactivity is hampered by self-tolerance to WT1 and limited number of WT1 peptides, which were thus far selected based on HLA peptide binding algorithms.
Methods
In this study, we have overcome both limitations by searching in the allogeneic T-cell repertoire of healthy donors for high-avidity WT1-specific T cells, specific for WT1 peptides derived from the HLA class I associated ligandome of primary leukemia and ovarian carcinoma samples.
Results
Using broad panels of malignant cells and healthy cell subsets, T-cell clones were selected that demonstrated potent and specific anti-WT1 T-cell reactivity against five of the eight newly identified WT1 peptides. Notably, T-cell clones for WT1 peptides previously used in clinical trials lacked reactivity against tumor cells, suggesting limited processing and presentation of these peptides. The TCR sequences of four T-cell clones were analyzed and TCR gene transfer into CD8+ T cells installed antitumor reactivity against WT1-expressing solid tumor cell lines, primary acute myeloid leukemia (AML) blasts, and ovarian carcinoma patient samples.
Conclusions
Our approach resulted in a set of naturally expressed WT1 peptides and four TCRs that are promising candidates for TCR gene transfer strategies in patients with WT1-expressing tumors, including AML and ovarian carcinoma.
Keywords: CD8-Positive T-Lymphocytes; Immunotherapy, Adoptive
WHAT IS ALREADY KNOWN ON THIS TOPIC
In current WT1-targeting vaccine and TCR gene therapy studies, T-cell reactivity is hampered by self-tolerance to WT1 and limited number of WT1 peptides.
WHAT THIS STUDY ADDS
With newly identified WT1 peptides, derived from the HLA class I associated ligandome of primary acute myeloid leukemia and ovarian carcinoma samples, we identified potent and specific WT1 TCRs from the allo-HLA T-cell repertoire. No T cells with potent antitumor reactivity were identified for previously used WT1 peptides.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
Our set of TCRs and naturally expressed WT1 peptides are expected to improve WT1-targeting therapies.
Introduction
Transcription factor Wilms’ tumor gene 1 (WT1) is expressed in a broad range of cancers, especially in ovarian carcinomas (OVCA), mesotheliomas, and acute myeloid leukemia (AML).1 2 These tumor types have low 5 year survival rates of 49%, 12% and 29%, respectively, and there is a strong need for additional treatment options.3 4 In patients with OVCA and AML, WT1 expression is associated with poor prognosis.5 6 This corresponds to the promoting role of WT1 in cancer progression through the induction of tumor angiogenesis and metastasis formation.7 Among the numerous tumor associated antigens, WT1 is an ideal tumor target based on its expression in a wide range of hematological malignancies and solid tumors but low-level expression in normal tissues, as well as the tumor promoting characteristics. WT1 was even ranked by the US National Cancer Institute as the most promising tumor target for cancer vaccination.8 Currently, most clinical studies targeting WT1 use peptide-based or dendritic cell-based vaccines. Mainly hematological malignancies are included in these trials, but also several solid tumor types such as gynecological malignancies and sarcomas are treated.9–11 More recently, WT1 has been targeted by adoptive T-cell based therapies as well. In this rapidly growing therapy field promising clinical results have already been achieved in especially B-cell malignancies.12–14 For WT1, HLA class I restricted WT1-specific T cells were previously identified,15 16 and patients with AML and myelodysplastic syndrome (MDS) were treated with WT1-reactive CD8+ T cells or WT1-TCR engineered T cells (TCR-T cells) in clinical trials.17–19 In these clinical studies, antileukemic reactivity and persistence of WT1-specific T cells have been reported. However, there are still many issues to be solved to improve avidity and increase killing potential of tumor cells by vaccine activated T cells or infused WT1-specific T-cell products.20
WT1 is expressed during embryogenesis and involved in the embryonic development of kidneys, gonads, and several organs lined by the mesothelium. In adults, WT1 is involved in homeostasis processes for tissue maintenance and recovery, resulting in expression in renal podocytes, epicardial cells, Sertoli cells, granulosa cells, and hematopoietic stem cells.21–23 The expression levels in adults are expected to be sufficient to induce negative selection of high-avidity WT1-specific T cells during T-cell development, as mechanism to centrally delete self-reactive T cells. As a consequence, only the remaining low-avidity WT1-specific T cells present in the T-cell repertoire can be activated by WT1 peptide vaccines. In addition, the WT1-specific T cells and subsequently WT1-specific TCR-T cells transferred into patients were of low-avidity and demonstrated low antitumor reactivity.24 Strategies to circumvent self-tolerance to WT1 have been developed. Increased reactivity was demonstrated by stimulating T cells with synthetic analog peptides derived from the WT1 protein25 26 or by using affinity enhanced WT1-specific TCRs.27 Another strategy to circumvent self-tolerance is by searching for WT1-specific T cells in the allogeneic-HLA (allo-HLA) repertoire. Since allo-HLA reactivity of T cells is not subjected to negative selection, high-affinity allo-HLA-restricted TCRs recognizing self-antigens are present in the TCR repertoire. This approach previously allowed the identification of high-affinity TCRs specific for several B-cell restricted antigens.28–30 Also for WT1, T cells recognizing HLA-A*02:01 restricted peptides were successfully isolated using this approach.16 31
Besides the relative low-affinity of WT1-specific TCRs used in clinical studies, the selection of WT1-specific peptides used for peptide vaccination studies and identification of TCRs can most likely also be optimized. WT1 peptides were thus far selected based on HLA peptide binding algorithms, and although these peptides efficiently bind to HLA class I, it is unknown whether these peptides are efficiently processed and presented in HLA class I at the cell surface of tumor cells. Furthermore, the thus far selected WT1 peptides were presented only in either HLA-A*02:01 or HLA-A*24:02, whereas more HLA alleles are necessary to cover a larger part of the patient population. In this study, WT1 peptides were identified from the HLA class I ligandome of primary leukemia and OVCA patient samples, and a large-scale search was performed to identify high-avidity WT1-specific CD8+ T cells targeting these peptides from the allo-HLA T-cell repertoire of healthy donors. Using broad panels of malignant cells and healthy cell subsets, we selected potent and specific WT1-reactive T-cell clones. The TCR sequences of these T-cell clones were analyzed and TCR gene transfer into CD8+ T cells installed antitumor reactivity against WT1-expressing solid tumor cell lines, as well as primary AML and ovarian carcinoma samples.
Material and methods
WT1 expression by real-time quantitative polymerase chain reaction
WT1 expression was quantified by real-time quantitative polymerase chain reaction (RT-qPCR). Total RNA was isolated using the RNAqueous-Micro Kit (Ambion) or ReliaPrep RNA Cell Miniprep System (Promega). First strand cDNA synthesis was performed with Moloney murine leukemia virus reverse transcriptase and Oligo (dT) primers (Invitrogen by Thermo Fisher Scientific). RT-qPCR was performed using Fast Start TaqDNA Polymerase (Roche) and EvaGreen (Biotium), and gene expression was measured on the Lightcycler 480 (Roche). Expression was calculated as percentage relative to the average of housekeeping genes GUSB, VPS29, and PSMB4, which was set at 100%. All samples and genes were run in triplicate with 10 ng cDNA per reaction. The following primers were used: WT1 (forward: AGACCCACACCAGGACTCAT, reverse: GATGCATGTTGTGATGGCGG), GUSB (forward: ACTGAACAGTCACCGACGAG, reverse: GGAACGCTGCACTTTTTGGT), PSMB4 (forward: GTTTCCGCAACATCTCTCGC, reverse: CATCAATCACCATCTGGCCG), VPS29 (forward: TGAGAGGAGACTTCGATGAGAATC, reverse: TCTGCAACAGGGCTAAGCTG).
Sample collection for peptide elution
To identify T-cell epitopes derived from the WT1 gene, in total 37 tumor samples were collected. Cell pellets (2×109–610×109 cells) were made of 11 acute lymphoblastic leukemia (ALL) samples, 15 AML samples, 1 hairy cell leukemia (HCL) sample, and 2 OVCA cell lines. Also seven solid primary OVCA samples (2.3 –30 g) were included and one ascites primary OVCA sample (6×109 cells). The OVCA samples were residual material and collected anonymously. Solid OVCA tumors were sliced into small pieces and dead, and clotted or non-tumor material was removed. The small tumor-pieces were added to a C-tube (Miltenyi Biotec) with ice cold buffer without detergent and cOmplete Protease Inhibitor (Sigma-Aldrich), to prevent protein degradation. Using a gentleMACS (Miltenyi Biotec) procedure, the small tumor-pieces were dissociated until an almost homogenous cell solution. Benzonase (Merck) was added in a concentration of 125 IU/mL to remove DNA/RNA complexes during lysis. HLA typing was performed of all samples and WT1 expression was analyzed by RT-qPCR.
HLA class I-peptide elution procedure, fractionation and mass spectrometry
Peptide elution was performed as outlined previously.32 In short, the cell pellets were lysed and subjected to an immunoaffinity column to collect bound peptide-HLA complexes, with either an HLA class-I antibody (W6/32, ATCC) or an HLA-A*02:01 antibody (BB7.2, ATCC). To separate the peptides, bound peptide-HLA complexes were dissociated with 10% acetic acid and filtrated using a 10 kDa membrane. Eluted peptide pools were either fractionated by strong cation exchange chromatography (SCX)32 or by high pH reversed phase fractionation (High pH-RP).33 SCX and high pH-RP peptide fractions were lyophilized, dissolved in 95/3/0.1 water/ acetonitrile/formic acid v/v/v and subsequently analyzed by data-dependent MS/MS on either an LTQ FT Ultra equipped with a nanoflow liquid chromatography 1100 HPLC system (Agilent Technologies) or a Q Exactive mass spectrometer equipped with an easy-nLC 1000 (Thermo Fisher Scientific). Proteome Discoverer V.2.1 (Thermo Fisher Scientific) was used for peptide and protein identification, using the mascot search node for identification (mascot V.2.2.04) and the UniProt Homo Sapiens database (UP000005640; Jan 2015; 67,911 entries). All unique WT1-derived peptides with a length between 8 and 14 amino acids, a minimal Best Mascot Ion (BMI) score of 20, a mass accuracy of 10 ppm and predicted to bind to a common HLA molecule (HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*24:02, HLA-B*07:02, HLA-B*35:01, HLA-C*07:01, HLA-C*07:02) according to the netMHC peptide binding algorithm34 were selected as candidate for peptide synthesis and validation.
Peptide synthesis and pMHC-multimer production
Eight peptides met all the criteria and their synthetic peptides were in-house synthesized using standard Fmoc chemistry. By mass spectrometry, the tandem mass spectra of the eluted peptides were validated with synthetic peptides (online supplemental data 2). Of in total 12 WT1-derived peptides, pMHC-multimer complexes were generated with minor modifications.35 In short, monomers consisting of the selected HLA allele heavy chain, human beta-2 microglobulin (B2M) light chain and selected peptide were purified by gel-filtration high-performance liquid chromatography and biotinylated. Subsequently, pMHC-multimers were generated by adding PE-conjugated streptavidin (Invitrogen, Thermo Fisher Scientific).
jitc-2021-004409supp002.pdf (1.1MB, pdf)
Cell culture
T cells were cultured in T-cell medium (TCM) composed of Iscove’s Modified Dulbecco’s Medium (IMDM) (Lonza), 5% heat-inactivated fetal bovine serum (FBS) (Gibco, Thermo Fisher Scientific), 5% human serum (Sanquin Reagents), 1.5% 200 mM L-glutamine (Lonza), 1% 10,000 U/mL penicillin/streptomycin (Pen/Strep; Lonza) and 100 IU/mL IL-2 (Novartis Pharma). Every 10–14 days, 0.2×106 T cells were (re)stimulated with 1×106 irradiated (35 Gy) PBMCs, 0.1×106 irradiated (55 Gy) EBV-LCLs and 0.8 µg/mL phytohemagglutinin (PHA) (Oxoid Microbiology Products, Thermo Fisher Scientific).
Most tumor cell lines were cultured in IMDM, 10% FBS, 1.5% L-glutamine, and 1% Pen/Strep. OVCA cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM, high glucose 4.5 g/L, NEAA) (Gibco), 8% FBS, 2% 200 mM L-glutamine, and 1% 10,000 U/mL Pen/Strep. Using the PlasmoTest Mycoplasma Detection Kit (InvivoGen), all cell lines were found to be mycoplasma negative. Of all cell lines, HLA typing was performed and WT1 expression was analyzed by RT-qPCR. If needed, the WT1 gene or an HLA allele was introduced by retroviral transduction. These genes were expressed in MP71 retroviral backbone vectors with marker genes nerve growth factor receptor (NGF-R), green fluorescent protein, CD34, or mouse CD19 (mCD19). Target cells were enriched for marker gene expression via magnetic-activated cell sorting (MACS) or fluorescence-activated cell sorting (FACS) and purity was confirmed by FACS.
Primary AML samples were thawed 1 day before being used as target cells in screening experiments. Cells were cultured overnight at 37° and 5% CO2 in IMDM containing 10% human serum and if needed, live cells were isolated using Ficoll gradient separation. Blast percentage of the AML samples was on average 83% (range 40%–99%), as determined by FACS expression (CD13, CD33, and CD34). Primary OVCA cells were thawed 3 days before being used as target cells in screening experiments. Cells were cultured in IMDM, 10% FBS, 1.5% 200 mM L-glutamine and 1% 10,000 U/mL Pen/Strep, on FBS precoated plates.
Healthy cell subset isolation
Hematopoietic healthy cell subsets were isolated from PBMCs of healthy donors, either HLA-A*01:01 and HLA-B*35:01 positive or HLA-A*02:01 positive. CD14, CD19, and CD34 positive cells were enriched by MACS using anti-CD14 MicroBeads (Miltenyi Biotec/200-070-118), anti-CD19 MicroBeads (Miltenyi Biotec/130-060-301) or anti-CD34 MicroBeads (Miltenyi Biotec/130-046-702). Activated CD19+ B cells were generated by coculturing CD19+ cells on CD40L-transduced irradiated (70 Gy) mouse fibroblasts for 7 days in IMDM supplemented with 2 ng/mL IL-4 (Schering-Plough) and 10% human serum. Immature and mature CD14-derived dendritic cells (DCs) were differentiated in vitro from isolated CD14+ cells. Immature DCs were generated by culturing 0.1×106 cells/mL for 3 days in IMDM supplemented with 100 ng/mL GM-CSF (Sandoz Novartis Pharma), 500 IU/mL IL-4 (Schering-Plough), and 10% human serum. Mature DCs were generated by culturing immature DCs for an additional 3 days in IMDM supplemented with 100 ng/mL GM-CSF, 10 ng/mL TNFalpha (CellGenix), 10 ng/mL IL-1b (Bioscource Invitrogen), 10 ng/mL IL-6 (Sandoz Novartis Pharma), 1 µg/mL PGE-2 (Sigma Aldrich), 500 IU/mL IFN-γ (Boehringer Ingelheim), and 10% human serum. Isolated CD34+ hematopoietic precursor cells were directly used after isolation. Purity of the isolated and generated cells was assessed using FACS analysis. Fibroblasts from skin biopsies were cultured in DMEM with 1 g/L glucose (Lonza BE12-708F) and 10% FBS. Keratinocytes from skin biopsies were cultured in keratinocyte serum free medium (Thermo Fisher Scientific 17 005–059) supplemented with 30 µg/mL bovine pituitary extract and 2 ng/mL epithelial growth factor (both Thermo Fisher Scientific 37 000–015).
Antibodies and FACS analysis
FACS was performed on an LSR II flow cytometer (BD Biosciences) and data were analyzed using FlowJo software (TreeStar). T cells were stained with the following conjugated antibodies: CD4 FITC (BD/555346), CD14 FITC (BD/555397), CD19 FITC (BD/555412), CD8 AF700 (Invitrogen/MHCD0829), murine TCR-β (mTCR-β) APC (BD/553174), and pMHC-multimers PE. Target cells with transduced WT1 or HLA-alleles were stained with: NGF-R/CD271 APC (Sanbio/CL10013APC), CD34 APC (BD/555824), murine CD19 PE (BD/557399), and HLA-A2 PE (BD/558570). Non-malignant hematopoietic subsets with: CD14 FITC (BD/555397), CD19 FITC (BD/555412), CD34 APC (BD/555824), CD80 PE (BD/557227), and CD86 PE (BD/555658). AML samples with: CD13 PE (BD/347406), CD33 FITC (BD/555626), and CD34 APC (BD/555824).
Isolation of WT1-specific T cells by pMHC-multimer enrichment
Buffy coats of healthy donors negative for HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*24:02, or HLA-B*35:01 were collected after informed consent (Sanquin). PBMCs were isolated using Ficoll gradient separation and incubated with a selection of WT1-specific pMHC-multimers for 1 hour at 4°C or 15 min at 37°C. pMHC-multimers were only included if the healthy donor was negative for the restricted HLA allele. pMHC-multimer bound cells were MACS enriched using anti-PE MicroBeads (Miltenyi Biotec/130-048-801). The positive fraction was stained with AF700-conjugated antibody against CD8 and FITC-conjugated antibodies against CD4, CD14, and CD19. pMHC-multimer and CD8 positive cells were single-cell sorted using an Aria III cell sorter (BD Biosciences) in a 96-well round bottom plate containing 5×104 irradiated PBMCs (35 Gy) and 5×103 EBV-JY cells (55 Gy) in 100 uL TCM with 0.8 µg/mL PHA. T-cell recognition was assessed 10–14 days after stimulation, followed by restimulation or storage of the selected T-cell clones.
T-cell reactivity assays
T-cell recognition was measured by an IFN-y ELISA (Sanquin). 5000 T cells were cocultured overnight with target cells in various effector-to-target (E:T) ratios in 60 µL TCM in 384-well flat-bottom plates (Greiner Bio-One). To upregulate HLA expression, all adherent target cells were treated for 48 hours with 100 IU/mL IFN-y (Boehringer Ingelheim) before coculture. All T cells and target cells were washed thoroughly before coculture to remove expansion-related cytokines. Supernatants were transferred during the ELISA procedure using the Hamilton Microlab STAR Liquid Handling System (Hamilton company) and diluted 1:5, 1:25, and/or 1:125 to quantify IFN-y production levels within the area of the standard curve. The Hamilton System was also used to split 96-well T-cell cultures into 4 wells of 384-well flat bottom plates during our large-scale T-cell search in 28 healthy donors, making it feasible to screen the T cells against different combinations of Raji cells loaded with and without peptides (1 µM).
T-cell mediated cytotoxicity was measured in a 6-hour 51chromium release assay. Target cells were labeled with 100 µCi 51chromium (PerkinElmer) for 1 hour at 37°C, washed, and cocultured with T cells at various E:T ratios in 100 µL TCM per well in 96-well U-bottom culture plates (Costar). Spontaneous and maximum 51Cr release for all targets were measured in separate plates with per well 100 µL TCM or 100 µL TCM with 1% Triton-X 100 (Sigma-Aldrich), respectively. After 6 hours of coculture, 25 µL supernatant was harvested and transferred to 96-well LumaPlates (PerkinElmer) and 51chromium release was measured in counts per minute on a 2450 Microbeta2 plate counter (PerkinElmer). The percentage of killed target cells was calculated with the following formula = ((experimental release – spontaneous release)/(maximum release – spontaneous release)) *100.
TCR identification and production of retroviral supernatants
TCR α and β chains of the selected T-cell clones were identified by sequencing with minor modifications, as previously described.36 mRNA was isolated by the Dynabeads mRNA DIRECT Kit (Invitrogen) or total RNA was isolated by the ReliaPrep RNA cell Miniprep System (Promega). TCR cDNA was generated using TCR constant α and β primers, a SA.rt anchor template-switching oligonucleotide, and SMARTScribe Reverse Transcriptase (Takara, Clontech).37 The TCR α and β products were generated in a first PCR using Phusion Flash (Thermo Fisher Scientific), followed by a second PCR that was used to include 2-sided barcode sequences for the different T-cell clones. Barcoded TCR PCR products were pooled and TCR sequences were identified by HiSeq or NovaSeq (GenomeScan). The Vα and Vβ families were determined of the NGS data using the MiXCR software and ImMunoGeneTics (IMGT) database.38 The TCR chains were codon optimized, synthesized, and cloned in MP71-TCR-flex retroviral vectors by Baseclear. The MP71-TCR-flex vector contains codon-optimized and cysteine-modified murine TCRαβ constant domains and P2A sequence to link TCR chains, resulting in optimized TCR expression and increased preferential pairing.39 Apart from the WT1-specific TCRs, a murinized CMV-specific TCR (NLVPMVATV peptide presented in HLA-A*02:01) was included as a negative control. Phoenix-AMPHO (ATCC) cells were transiently transfected with the created constructs and after 48 hours retroviral supernatants were harvested and stored at −80°C.
TCR gene transfer to healthy donor CD8+ T cells
CD8+ T cells were isolated from PBMCs of different healthy individuals/donors by MACS using anti-CD8 MicroBeads (Miltenyi Biotech/130-045-201). CD8+ T cells were stimulated with irradiated autologous feeders (40 Gy) and 0.8 µg/mL PHA in 24-well flat-bottom culture plates (Costar). Two days after stimulation, CD8 +T cells were transferred to 24-well flat-bottom suspension culture plates (Greiner Bio-One) for retroviral transduction. These plates were first coated with 30 µg/mL retronectin (Takara, Clontech) and blocked with 2% human serum albumin. Retroviral supernatants were added, and plates were centrifuged at 3000 g for 20 min at 4°C. After removal of the retroviral supernatant, 0.3×106 CD8+ T cells were transferred per well. After O/N incubation, CD8+ T cells were transferred to 24-well flat-bottom culture plates (Costar). Seven days after stimulation, CD8+ T cells were MACS enriched for the murine TCR, using mTCR-β APC antibody (BD/553174) and anti-APC MicroBeads (Miltenyi Biotec/130-090-855). Ten days after stimulation, CD8+ T cells were functionally tested and purity was checked by FACS.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software (V.9.0.1.). The statistical test used is indicated in the figure legend, and p<0.05 was considered significant. Significance levels are indicated as *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Study approval
AML patient samples were used from the Leiden University Medical Center Biobank for Hematological Diseases. This study was approved by the Institutional Review Board of the Leiden University Medical Center (approval number 3.4205/010/FB/jr) and the METC-LDD (approval number HEM 008/SH/sh). Materials from patients and healthy individuals were collected after written informed consent according to the Declaration of Helsinki. The OVCA samples were residual material and collected anonymously.
Results
Identification of WT1-derived peptides in ovarian carcinoma and leukemia samples
To identify WT1-derived peptides that are efficiently expressed in HLA class I at the cell surface of tumors, patient samples were selected based on WT1 expression levels. By RT-qPCR, gene expression was measured in primary OVCA, ALL, and AML samples and healthy cell subsets. WT1 expression (≥5% relative expression compared with housekeeping genes) was observed in all OVCA samples (16/16), most ALL samples (3/4) and in more than half of the AML samples (18/30) (figure 1A). In addition, WT1 was widely expressed across a variety of tumor cell lines and, although not significant, on average, the highest WT1 expression was observed in OVCA, ALL, and AML cell lines (figure 1B). These results indicate that tumor samples of patients with OVCA, ALL, and AML could be used for the identification of naturally expressed WT1 peptides.
Figure 1.

High WT1 expression in primary OVCA, ALL, and AML samples, low or absent expression in various healthy cell subsets. WT1 mRNA expression was measured by RT-qPCR. WT1 expression is shown as percentage relative to the three housekeeping genes GUSB, VPS29, and PSMB4, which was set at 100%. The minimum gene expression is set at 0.01%. Dots illustrate in (A) primary tumor samples, or healthy cell subsets, and in (B) tumor cell lines. The vertical bars indicate mean, and number of samples/cell lines per group are shown between brackets. Differences were tested by a Kruskal-Wallis test followed by a Dunn’s multiple comparisons test. The WT1 expression levels in the primary tumor samples in (A) were significantly different compared with the healthy cell subsets combined (p<0.043), and no significant differences were observed between any of the tumor cell line types in (B). ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; APL, acute promyelocytic leukemia; iDCs and mDCs, immature and mature dendritic cells; OVCA, primary ovarian carcinoma; PBECs, primary bronchus epithelial cells; PTECs, proximal tubular epithelial cells.
The HLA class I associated ligandome was determined of 11 patients with ALL, 15 patients with AML, 1 patient with HCL, and 8 patients with OVCA, as well as 2 OVCA cell lines. All WT1 peptides with a length between 8 and 14 amino acids, a minimal BMI score of 20, and a mass accuracy of 10 ppm were selected as candidate for peptide synthesis and validation. Peptides were synthesized if they were predicted to bind common HLA molecules (HLA-A*01:01, -A*02:01, -A*03:01, -A*24:02, -B*07:02, -B*35:01, -C*07:01, -C*07:02) according to netMHC peptide binding algorithm,34 and matched the HLA typing of the material from which the peptides originated (online supplemental data 1). Eight peptides were identified and the sequences of these peptides were validated by comparing mass spectra of eluted peptides with the mass spectra of synthetic peptides (online supplemental data 2). WT1 peptides frequently studied and used in clinical trials; RMFPNAPYL (HLA-A*02:01 restricted),19 CMTWNQMNL (HLA-A*02:01 and HLA-A*24:02 restricted)17 31 and RWPSCQKKF (HLA-A*24:02 restricted)40 were not identified in our large HLA-ligandome database, whereas 20 and 9 of the eluted tumor samples expressed HLA-A*02:01 or HLA-A*24:02, respectively. Considering the frequent use of these peptides, we added them to our final set of WT1 peptides, and in total 12 PE-labeled peptide-MHC (pMHC)-multimer complexes were generated (table 1).
Table 1.
Overview of WT1 peptides included in this study
| Nr | Peptide | HLA | Sample/cell line source | BMI |
| 1 | WTEGQSNHSTGY | A*01:01 | OVCA-G1, ALL-1833, AML-4443, AML-10197 | 43 |
| 2 | VLDFAPPGASAY | A*01:01 | AML-4443 | 36 |
| 3 | ALLPAVPSL | A*02:01 | OVCA-L23 | 31 |
| 4 | VLDFAPPGA | A*02:01 | OVCA-L23 | 26 |
| 5 | FGPPPPSQA | A*02:01 | OVCA-L23, AML-10197 | 42 |
| 6 | AQFPNHSFK | A*03:01 | Cell line-COV362.4 | 20 |
| 7 | HAAQFPNHSF | B*35:01 | HCL-4512 | 36 |
| 8 | TPYSSDNLY | B*35:01 | ALL-2184 | 41 |
| 9 | RMFPNAPYL | A*02:01 | x | x |
| 10 | CMTWNQMNL | A*02:01 | x | x |
| 11 | RWPSCQKKF | A*24:02 | x | x |
| 12 | CMTWNQMNL | A*24:02 | x | x |
Overview of the 12 WT1 peptides included in this study. For the eight WT1 peptides identified in our HLA ligandome analyses, the source and best Mascot ion score (BMI) is listed. The four peptides not identified in this study, but previously described in literature, were added at the end of the table.
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; BMI, Best Mascot Ion; HCL, hairy cell leukemia; OVCA, primary ovarian carcinoma.
Large-scale WT1-reactive T-cell search in the allo-HLA T-cell repertoire of 28 healthy donors
With the pMHC-multimers, we searched for WT1-reactive T cells, with the ultimate goal to identify high-affinity WT1-specific TCRs. PBMCs of 28 healthy HLA typed donors were incubated with pMHC-multimers, pMHC-multimer positive CD8+ T cells were enriched by MACS, and single-cell sorted (online supplemental data 3). pMHC-multimers were only included if the donor was negative for the HLA allele, enabling us to search within the allo-HLA T-cell repertoire and thereby circumventing self-tolerance. On average, 650*10∧6 PBMCs per donor were used and between 20 and 658 pMHC-multimer positive CD8+ T cell clones were expanded after single-cell sorting (online supplemental data 3). Two weeks after clonal expansion, a peptide specificity screening was conducted on 7916 isolated T-cell clones. Burkitt lymphoma cell line Raji, negative for WT1, transduced with HLA-alleles of interest, were pulsed with the WT1 peptide pool and used as stimulator cells. In total, 461 T-cell clones (6%) were WT1 peptide specific, recognizing WT1 peptide pool pulsed Raji cells, whereas non-pulsed Raji cells were not recognized. The other T-cell clones were either reactive against all stimulator cells, nonreactive, or reactive against one specific HLA-allele, independent of added peptides. The peptide-specific T-cell clones were subsequently tested against the individual peptides and screened for recognition of endogenously processed and presented WT1. Of the 461 T-cell clones, 71 (15%) clones recognized WT1 transduced Raji cells. The peptide specificity of these T-cell clones is summarized in online supplemental data 4, T-cell clones were identified for 9 out of 12 WT1 peptides.
Identification of five potent and specific WT1-reactive T-cell clones
To select the most potent and specific WT1-reactive T-cell clones, the 71 T-cell clones were analyzed in additional screenings. This selection approach is illustrated in online supplemental data 5 for seven T-cell clones recognizing the VLDFAPPGA peptide in HLA-A*02:01. T cells were tested against a tumor cell line panel composed of WT1 positive and negative tumor cell lines that were positive or transduced with the corresponding HLA restriction molecule (online supplemental data 5A). The T-cell clones recognizing at least two WT1 positive tumor cell lines, combined with no recognition of WT1 negative tumor cell lines were selected (28/71 T-cell clones). To avoid allo-HLA cross-reactivity, selected T-cell clones were additionally screened against a panel of 25 Epstein-Barr virus transformed lymphoblastoid cell lines (EBV-LCL), expressing all HLA alleles with an allele frequency >1% present in the Caucasian population (online supplemental data 5B). Eight T-cell clones were excluded that showed HLA cross-reactivity against one or several prevalent HLA alleles, exemplified by clone 23.2E4. Those T-cell clones that recognize only one non-prevalent HLA allele were not excluded, exemplified by clone 20.3D10. Finally, to investigate clinical potential of the remaining 20/71 T-cell clones, we analyzed the antitumor reactivity against primary AML and OVCA patient samples expressing variable levels of WT1 (online supplemental data 5C).
Using this selection strategy, we identified 5/71 promising WT1-specific T-cell clones that demonstrated the most potent antitumor reactivity against WT1 positive primary AML and/or OVCA patient samples. The recognition patterns of these T-cell clones, recognizing five different WT1 peptides in the context of three different HLA class I molecules, are shown in figure 2. Clone 20.3D10 is specific for VLDFAPPGA in HLA-A*02:01 (VLD/A2), clone 22.1H1 is specific for ALLPAVPSL also in HLA-A*02:01 (ALL/A2), clone 12.5H9 is specific for VLDFAPPGASAY in HLA-A*01:01 (VLD/A1), clone 17.2G4 is specific for TPYSSDNLY in HLA-B*35:01 (TPY/B35), and clone 17.2D6 is specific for HAAQFPNHSF also in HLA-B*35:01 (HAA/B35). All five WT1-specific clones demonstrated antitumor reactivity against WT1 positive tumor cell lines. Only those tumor cell lines with a WT1 expression below 15% were more variable recognized (figure 2A). As depicted in figure 2B, two of the five T-cell clones showed HLA cross-reactivity against one non-prevalent HLA allele. The global frequencies of these HLA-alleles are low (HLA-A*33:01: 1.77% and HLA-A*02:05: 1.17%), implicating that when one of these TCRs would be used for clinical practice, only a small group of patients would not be suitable candidates for this particular TCR-based therapy.41 Apart from the recognized EBV-LCLs, no other target cells used in the assays expressed these HLA-alleles. Furthermore, primary AML and OVCA patient samples with WT1 expression were recognized by the different WT1-specific T-cell clones (figure 2C). Interestingly, not all recognition patterns correlated with WT1 expression levels. For example, clone 12.5H9VLD/A1 showed the highest recognition of AML-4443 (13% relative WT1 expression), from which the VLD/A1 peptide was eluted (table 1), whereas the other AML samples with WT1 expression between 23% and 27% were less well recognized. In addition, clone 20.3D10VLD/A2 and the other VLD/A2 reactive T-cell clones (online supplemental data 5) did not recognize AML-6588 (10%) and AML-4716 (3%), while the 22.1H1ALL/A2 clone recognized both. This also accounts for some of the HLA-B*35:01 positive primary AML patient samples. Clone 17.2G4TPY/B35 recognized AML-5905 (44%), whereas clone 17.2D6HAA/B35 did not, while both recognized the other two primary AML patient samples. This could not be explained by differences in antigen presenting capacity of the primary AML patient samples, since similar reactivity by allo-HLA reactive T-cell clones directed against these different AML samples was observed (online supplemental data 6A, B).
Figure 2.

Recognition patterns of the five most promising WT1-specific T-cell clones. Recognition patterns based on IFN-ƴ production (ng/mL) after overnight coculture assays with three screening panels. (A) Panel with WT1+ and WT- tumor cell lines (E:T=1:6). (B) Panel with 25 EBV-LCLs, expressing all frequent HLA alleles (with an allele frequency >1%) present in the Caucasian population (E:T=1:6). The HLA-allele is depicted if only one HLA-allele is recognized by the T-cell clone, meeting the requirement that all EBV-LCLs with this HLA-allele are recognized. (C) Panel with primary AML samples (E:T=1:16) and OVCA patient samples (E:T=1:6). Blast percentage of the AML samples was on average 83% (range 40%–99%), as determined by FACS expression (CD13, CD33, and CD34). All cell lines in panels (A) and (C) express the HLA-allele that presents the targeted peptide, either wildtype (wt) or the HLA-allele was introduced by transduction (+A2, +A1, +B35). Percentage relative WT1 expression is depicted, as determined by RT-qPCR. Dark gray bars depict high WT1+ targets and light gray bars the WT1- targets. Bars represent averaged duplicate values and are representative of two independent experiments. AML, acute myeloid leukemia; EBV-LCL, Epstein-Barr virus transformed lymphoblastoid cell lines; FACS, fluorescence-activated cell sorting; OVCA, primary ovarian carcinoma.
Notably, none of the selected T-cell clones were specific for the peptides that were previously identified based on HLA peptide binding algorithms, including the most frequently used RMFPNAPYL peptide presented in HLA-A*02:01 (RMF/A2). Although high numbers of T-cell clones specific for RMF/A2 peptide were identified, of which a limited number were reactive against overexpressed WT1 transduced Raji cells (546% relative WT1 expression), all these T-cell clones were not reactive against WT1 positive tumor cell lines and primary AML patient samples. To illustrate the limited reactivity, RMF/A2 and VLD/A2 reactive T-cell clones isolated from five donors were compared. In figure 3A, it is shown that 12% of the RMF/A2-specific T-cell clones were reactive against WT1 transduced cells, whereas over 33% of the VLD/A2-specific T-cell clones were reactive against WT1 transduced cells. The most potent T-cell clones for both peptides were selected and no difference in peptide sensitivity was observed in a peptide titration (figure 3B). However, reactivity against WT1 positive tumor cell lines with 19%–230% relative WT1 expression was very limited by all RMF/A2 clones (figure 3C), and none of the primary AML patient samples with 6%–44% relative WT1 expression were recognized (figure 3D). In contrast, VLD/A2-specific T-cell clones were highly reactive against these WT1 positive tumor cell lines as well as primary AML patient samples (figure 3C–D).
Figure 3.
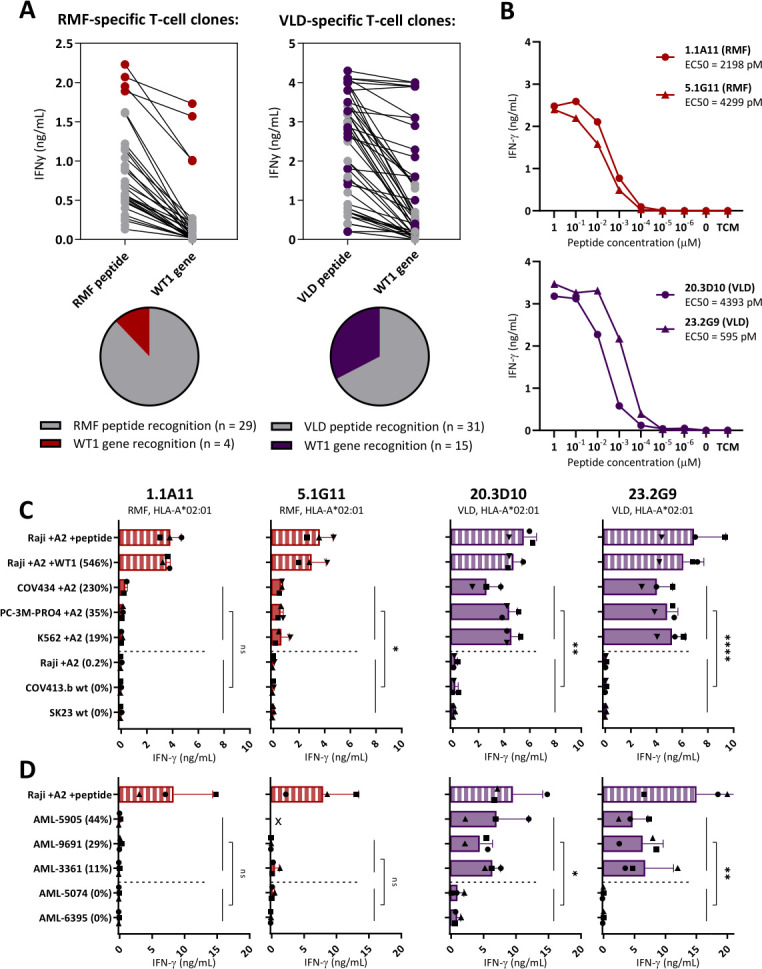
Limited WT1-specific reactivity of the RMF-specific T-cell clones. Comparison of T-cell clones recognizing the RMFPNAPYL (RMF) and VLDFAPPGA (VLD) peptide, both presented in HLA-A*02:01. (A) IFN-γ production (ng/mL) of T-cell clones recognizing Raji pulsed with RMF/A2 or VLD/A2 peptide (1 µM) identified in five healthy donors. T-cell clones also recognizing Raji transduced with WT1 (WT1 gene) on a similar level (>50%), are depicted with colored dots. (B) The four most potent T-cell clones stimulated overnight with Raji pulsed with titrated peptide. EC50 values represent the peptide concentrations needed to induce 50% of the maximum cytokine production, and values were calculated based on sigmoidal curves. (C) T-cell clones stimulated overnight with tumor cell lines (E:T=1:6). All cell lines express the HLA-A*02:01, either wildtype (wt) or introduced by transduction (+A2). Percentage relative WT1 expression is depicted, as determined by RT-qPCR. (D) T-cell clones stimulated overnight with primary AML patient samples (E:T=1:16). (x=Recognition of AML-5905 is not shown for clone 5.1G11, since this sample is HLA-B*35:01 positive and the 5.1G11 clone demonstrated allo-HLA reactivity against this allele in an EBV-LCL panel). (C, D) Symbols represent averaged duplicate values from three independent experiments. Mean and SD are depicted. WT1+ and WT1- targets were grouped, and reactivity of WT1 T-cell clones directed against these groups were compared using an unpaired t-test (two-sided). AML, acute myeloid leukemia; EBV-LCL, Epstein-Barr virus transformed lymphoblastoid cell lines; ns, not significant.
TCR gene transfer in CD8+ T cells installs potent WT1-specific reactivity without on-target off-tumor toxicity
Next, to test whether the TCRs of the selected T-cell clones can be used for TCR gene therapeutic strategies, we analyzed their TCRs in more detail. We continued with four of the five T-cell clones: 20.3D10VLD/A2, 22.1H1ALL/A2, 12.5H9VLD/A1, and 17.2G4TPY/B35 based on potency. The TCR α and β chains were identified by sequencing and transferred using retroviral vectors into CD8+ T cells of four different donors. TCR-engineered T cells (TCR-T cells) were enriched based on murine TCR (mTCR) expression and pMHC-multimer staining demonstrated that the TCR-T cells efficiently express the TCR at the cell surface (online supplemental data 7A). Functional reactivity of the TCR-T cells measured by peptide titration experiments was quite comparable to their parental T-cell clones (online supplemental data 7B).
To investigate the antitumor potential of the WT1 TCR-T cells, they were screened against multiple tumor panels, assessed for IFN-ƴ production and compared with CMV TCR-T cells as negative control. The four WT1 TCRs demonstrated effective antitumor reactivity, recognizing WT1 positive tumor cell lines, whereas WT1 negative cell lines were not recognized (figure 4A). The included WT1 positive primary AML and OVCA patient samples were recognized (figure 4B–C). The level of recognition of these primary tumor samples correlated with the level of WT1 expression, except for TCR-T 12.5H9VLD/A1 that only recognized the AML-4443 sample in which the VLD/A1 peptide was initially eluted (table 1).
Figure 4.

WT1-specific TCR-T cells recognize tumor cell lines and primary tumor samples, without recognition of healthy cell subsets. IFN-ƴ production (ng/mL) of purified TCR-T cells (CD8+) from four different donors cocultured overnight with (A) tumor cell lines (E:T=1:6), (B) primary AML samples (E:T=1:16), (C) primary OVCA samples (E:T=1:6), and (D) several healthy cell subsets (E:T=1:4 for keratinocytes, fibroblasts and CD14+, 1:6 for CD19+ cells, and 1:12 for CD34 +cells). The different symbols represent the TCR- T cells from four different donors, transduced with either the WT1-TCRs (colored bars, black symbols) or CMV-TCR (white bars, gray symbols). OVCA-L23 was ascites material passage 0 (wildtype HLA-A*02:01+) and OVCA-L25 was ascites material passage 3 transduced with HLA-A*02:01 (35%). The healthy cell subsets used in (D) were isolated either from an HLA-A*02:01+donor (for VLD/A2 and ALL/A2 TCRs) or from an HLA-A*01:01+ and HLA-B*35:01+ donor (for VLD/A1 and TPY/B35 TCRs). Also, the other targets express the HLA-allele that presents the targeted peptide, either wildtype (wt) or the HLA-allele was introduced by transduction (+A2, +A1, +B35). In all figures, the percentage relative WT1 expression is depicted, as determined by RT-qPCR. (A–D) Data represent averaged duplicate values from four different donors. Mean and SD are depicted, and WT1-TCR and CMV-TCR T cells were compared using two-way ANOVA, followed by Bonferroni posthoc analysis. Only significant results are shown. ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; ANOVA, analysis of variance; OVCA, primary ovarian carcinoma; TCR, T-cell receptor.
To investigate the safety profile of the TCR-T cells, the engineered T cells were tested against a variety of different healthy cell subsets. Keratinocytes, fibroblasts, and several hematopoietic cell subsets, including CD34+ hematopoietic precursor cells, CD14+-derived immature and mature DCs, and B cells were tested for recognition by the TCR-T cells. No WT1 expression was detected in these healthy cell subsets, only in CD34+ hematopoietic precursor cells limited WT1 expression (<0.5%) was observed (figure 1A). The TCR-T cells demonstrated no reactivity against the different healthy cell subsets, even CD34+ hematopoietic precursor cells were not or very limited (20.3D10VLD/A2 TCR) recognized, indicating that these TCR-T cells do not exhibit on-target off-tumor reactivity (figure 4D). Reactivity against all targets was observed by the allo-HLA reactive T-cell clones (online supplemental data 6C–F). In conclusion, TCR gene transfer of WT1-TCRs into CD8+ T cells installed high WT1-specific reactivity against WT1 positive tumor cells, without indications of on-target off-tumor reactivity.
WT1-specific TCR-T cells efficiently kill primary AML samples and OVCA cell lines
Finally, to investigate clinical potential of the four selected TCR-T cells, cytotoxic capacity against primary AML and OVCA patient samples and OVCA cell lines was measured in a 6-hour 51chromium release assay (figure 5A–C). Raji pulsed with WT1 peptide were included as control (figure 5D). Both 20.3D10VLD/A2, 22.1H1ALL/A2, and 17.2G4TPY/B35 TCR-T cells efficiently killed WT1 positive primary AML samples as well as OVCA patient samples and OVCA cell lines. The 12.5H9VLD/A1 TCR exhibited limited killing of AML samples but efficient killing of WT1 positive OVCA cell lines. Killing of all targets was observed by the allo-HLA reactive T-cell clones (online supplemental data 6G–I).
Figure 5.

WT1-specific TCR-T cells efficiently kill primary AML and OVCA samples. Purified TCR-T cells (CD8+) were tested for cytotoxic capacity in a 6-hour 51Cr-release assay against (A, B) primary AML patient samples, (C) primary OVCA samples and OVCA cell lines, and (D) Raji pulsed with WT1 peptide (0.2 µM). Percentage of killed cells is indicated on y-axis. (B–D) Cytotoxic capacity of the different WT1-TCR T cells was compared with CMV-TCR T cells. For VLD/A2, ALL/A2 TCRs killing capacity is depicted at E:T (effector-to-target) ratio of 30:1 and for VLD/A1 and TPY/B35 TCRs at E:T ratio of 10:1. All targets express the HLA-allele that presents the targeted peptide, either wildtype or the HLA-allele was introduced in cell lines by transduction. OVCA-L23 was ascites material passage 0 (wildtype HLA-A*02:01+). Percentage relative WT1 expression is depicted, as determined by RT-qPCR. (A) Mean and SD of technical triplicates are depicted for donor 1, and WT1+ and WT1- AML samples were grouped and killing capacity of the WT1-TCR against these groups were compared using an unpaired t-test (two-sided). (B–D) Mean and SD of technical triplicates are depicted for four donors, WT1-TCR and CMV-TCR T cells were compared using a paired t-test (two-sided). Only significant results are shown. AML, acute myeloid leukemia; OVCA, primary ovarian carcinoma; TCR, T-cell receptor.
Overall, these results demonstrate that TCR-T 20.3D10VLD/A2, 22.1H1ALL/A2, and 17.2G4TPY/B35 results in efficient killing of primary AML and OVCA patient samples and OVCA cell lines. To conclude, these TCRs are promising candidates for TCR gene transfer strategies for the treatment of patients with AML and OVCA. Given the lack of sufficient killing of primary AML samples, TCR 12.5H9VLD/A1 is only a potential candidate for TCR gene transfer strategies for patients with OVCA.
Discussion
In this study, we describe eight WT1 peptides that were identified from the HLA class I associated ligandome of primary leukemia and OVCA patient samples. In a large-scale search for WT1-specific TCRs present in the allo-HLA repertoire, T-cell clones directed against five different WT1 peptides presented in three different HLA class I molecules were identified. No WT1-specific T-cell clones reactive against tumor cells could be identified that recognized peptides previously identified based on HLA peptide binding algorithms. By gene transfer of four of the five WT1-TCRs into CD8+ T cells, we analyzed the antitumor potential as well as the safety profile of the TCR-T cells, and our results demonstrated that these WT1-TCRs are promising candidates for TCR gene transfer strategies in patients with WT1-expressing tumors, including AML and/or ovarian carcinoma.
The HLA class I restriction molecules for TCR 20.3D10VLD/A2, TCR 22.1H1ALL/A2, TCR 12.5H9VLD/A1, and TCR 17.2G4TPY/B35 are common. Of the global population 39% expresses HLA-A*02:01, 17% expresses HLA-A*01:01, and 8% expresses HLA-B*35:01.41 WT1-TCR-T cells induced potent killing of WT1 positive primary AML and OVCA patient samples and/or OVCA cell lines, without reactivity against healthy cell subsets and WT1 negative tumor cells. These results indicate that WT1-TCR-T cells do not exhibit off-target or on-target off-tumor toxicity. TCR 20.3D10VLD/A2 and TCR 22.1H1ALL/A2 demonstrated some cross-reactivity against HLA-A*33:01 and HLA-A*02:05, respectively. The frequencies of these HLA-alleles are however below 2%, implicating that only a small group of patients will not be suitable candidates for this particular TCR-based therapy.41
Our results demonstrate the relevance of establishing the HLA class I associated ligandome of tumors, since T-cell clones reactive against naturally WT1-expressing tumor cells were only isolated for peptides that were identified in the HLA class I associated ligandome. The most commonly used WT1 peptide is the RMF peptide presented in HLA-A*02:01. To our knowledge, this peptide has never been found in peptide-elution databases of tumor samples42 and since we also failed to elute this peptide in our large set of WT1-expressing HLA-A*02:01 tumor samples, we question whether this peptide is efficiently processed and presented in WT1 positive tumors. Although high numbers of T-cell clones recognizing the RMF/A2 peptide were identified, recognition of naturally WT1-expressing tumor cell lines and primary AML patient samples was absent or very low. In contrast, VLD/A2 T cells were highly reactive against all WT1 positive tumor cell lines and primary AML patient samples (figure 3). Only WT1 transduced Raji cells (546% relative WT1 expression) were highly recognized by the RMF/A2 T cells, demonstrating that the RMF/A2 peptide is processed and presented if WT1 is artificially overexpressed. These data indicate that limited antitumor reactivity can be expected of RMF/A2-specific T cells and this corresponds to observations of Jaigirdar et al that also suggested that the RMF/A2 peptide is not a suitable target for T-cell based immunotherapies.24 They demonstrated that a high-affinity RMF/A2-specific TCR, reactive against peptide loaded and WT1-transfected target cells, was not reactive against naturally WT1-expressing tumor cells due to absence of immunoproteasomes, resulting in no processing and presentation of the RMF/A2 peptide.24 Moreover, RMF/A2-specific T cells were easily found in the autologous-HLA (auto-HLA) T-cell repertoire of most healthy individuals, but also no antitumor reactivity was observed.43 Finally, our data also correspond to the limited antitumor effects found in (pre)clinical studies targeting the RMF/A2 peptide.11 44 Overall, our data suggest that better clinical results may be achieved with the WT1 peptides that were identified in the HLA ligandome and for which potent T-cell clones were found.
The variation in antitumor reactivity against primary AML patient samples by the 12.5H9VLD/A1 TCR-T cells suggest variation in processing and presentation of HLA class I peptides between different samples, hypothetically due to differences in immunoproteasome expression. The 12.5H9VLD/A1 TCR-T cells efficiently recognize primary patient sample AML-4443, from which initially the VLD/A1 peptide was eluted (table 1), suggesting optimal processing and presentation of this peptide in sample AML-4443. The lack of recognition of the other two AML samples, with higher WT1 expression, could be an indication of limited processing and presentation of this 12 amino acids long VLD/A1 peptide. It could also be an indication of alternative splicing or somatic mutations in the WT1 gene, which occur in 6%–15% of de novo AML.45 46 Furthermore, also for the HLA-A*02:01 and HLA-B*35:01 positive primary AML patient samples, a variety in antitumor reactivity was observed which could not be explained by WT1 expression. Both the ALL/A2 and TPY/B35 TCR-T cells and T-cell clones were reactive against all WT1 positive primary AML patient samples, whereas the VLD/A2 and HAA/B35 TCR-T cells and T-cell clones were not reactive against two samples (figure 2). Our data therefore suggest that the ALL/A2 and TPY/B35 peptides are processed and presented in more AML samples and therefore might be preferred targets to treat more AML patients. Nevertheless, more patient samples are needed to support this theory.
In this paper, we demonstrate that potent and specific WT1-reactive T cells can be identified from the allo-HLA repertoire. Especially since these T cells were not subjected to the negative selection, we carefully evaluated the safety of our final TCRs. No on-target and off-target toxicity was observed. Still, to eliminate unexpected serious adverse events of the WT1-TCR therapy, an additional clinical safety approach may be required. By co-transducing the WT1-TCR engineered T cells with a suicide switch, such as the inducible caspase-9 gene, prompt elimination of the engineered T cells can be induced. This approach was demonstrated for high-affinity PRAME-TCR transduced T cells in vivo.47
The identified WT1-TCRs demonstrated potent antitumor reactivity against AML and OVCA tumors. By RT-qPCR, we confirmed WT1 expression in primary OVCA, AML as well as ALL patient samples. Notably, not all AML patients are suitable candidates for WT1-TCR therapy, since only 60% of the AML patient samples expressed WT1. WT1 is additionally expressed in a broad variety of other tumors,48 indicating that also other solid tumors can be treated with WT1-TCR therapy. Besides the broad expression in various different tumors, several characteristics make WT1 an interesting target. WT1 promotes cancer progression through the induction of tumor angiogenesis and metastasis formation.7 In addition, WT1 is a strong predictor of leukemia relapse and is used as marker for minimal residual disease.49 Also in patients with MDS, overexpression of WT1 is associated with a higher risk for disease progression and AML transformation.50 Finally, in solid tumors, WT1 expression is also associated with poor prognosis, and this is among others related to increased epithelial-to-mesenchymal transition.51
In summary, we identified the 20.3D10VLD/A2, 22.1H1ALL/A2, 12.5H9VLD/A1, and 17.2G4TPY/B35 TCRs in a large-scale search for potent and specific WT1 TCRs present in the allo-HLA repertoire. We expect these TCRs to be a more potent option than the currently used WT1-TCRs from the auto-HLA repertoire. Also, the naturally expressed WT1 peptides identified from the HLA class I associated ligandome of primary leukemia and patients with OVCA are expected to improve future vaccine and TCR gene therapy studies for WT1-expressing tumors.
jitc-2021-004409supp001.pdf (1.9MB, pdf)
Acknowledgments
The authors thank the operators of the LUMC Flow cytometry Core Facility (LUMC, Leiden, The Netherlands) for providing expert technical assistance in flow cytometric cell sorting and Els M.E. Verdegaal (Department of Medical Oncology, Leiden University Medical Center, Leiden, The Netherlands) for providing primary OVCA patient samples and OVCA cell lines.
Footnotes
Contributors: RAvA designed, performed, analyzed, and interpreted all experiments and wrote the manuscript. RSH determined TRAV and TRBV usage and constructed retroviral expression vectors. DFGR performed RT-qPCR and constructed retroviral expression vectors. AKW, DCA and MvdM performed experiments. DMvdS and MGDK generated and analyzed peptide elution data and produced pMHC-multimers. AHdR performed and analyzed mass spectrometry experiments. MG provided mRNAseq data of the primary AML patient samples. PAvV produced and analyzed MS data. JHFF critically revised the manuscript and supervised the study. MHMH designed, analyzed, and interpreted all experiments, conceptualized and supervised the study, and wrote the manuscript. All authors reviewed the manuscript.
Funding: The research in this study was funded by Bellicum Pharmaceuticals (unrestricted grant) and Health Holland (grant number LSHM17002).
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
This study involves materials from human participants and was approved by Institutional Review Board of the Leiden University Medical Center (approval number 3.4205/010/FB/jr) and the METC-LDD (approval number HEM 008/SH/sh). Materials from patients and healthy individuals were collected after written informed consent.
References
- 1.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 4.Surveillance Research Program, N.C.I . SEER*Explorer: an interactive website for seer cancer statistics. Available: https://seer.cancer.gov/explorer/ [Accessed 11 Nov 2021].
- 5.Carter JH, Deddens JA, Mueller G, et al. Transcription factors WT1 and p53 combined: a prognostic biomarker in ovarian cancer. Br J Cancer 2018;119:462–70. 10.1038/s41416-018-0191-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nomdedéu JF, Hoyos M, Carricondo M, et al. Bone marrow WT1 levels at diagnosis, post-induction and post-intensification in adult de novo AML. Leukemia 2013;27:2157–64. 10.1038/leu.2013.111 [DOI] [PubMed] [Google Scholar]
- 7.Wagner K-D, Cherfils-Vicini J, Hosen N, et al. The Wilms' tumour suppressor WT1 is a major regulator of tumour angiogenesis and progression. Nat Commun 2014;5:5852. 10.1038/ncomms6852 [DOI] [PubMed] [Google Scholar]
- 8.Cheever MA, Allison JP, Ferris AS, et al. The prioritization of cancer antigens: a national cancer Institute pilot project for the acceleration of translational research. Clin Cancer Res 2009;15:5323–37. 10.1158/1078-0432.CCR-09-0737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maslak PG, Dao T, Bernal Y, et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv 2018;2:224–34. 10.1182/bloodadvances.2017014175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Driessche A, Berneman ZN, Van Tendeloo VFI. Active specific immunotherapy targeting the Wilms' tumor protein 1 (WT1) for patients with hematological malignancies and solid tumors: lessons from early clinical trials. Oncologist 2012;17:250–9. 10.1634/theoncologist.2011-0240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Di Stasi A, Jimenez AM, Minagawa K, et al. Review of the results of WT1 peptide vaccination strategies for myelodysplastic syndromes and acute myeloid leukemia from nine different studies. Front Immunol 2015;6:36. 10.3389/fimmu.2015.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park JH, Rivière I, Gonen M, et al. Long-Term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med Overseas Ed 2018;378:449–59. 10.1056/NEJMoa1709919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rapoport AP, Stadtmauer EA, Binder-Scholl GK, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 2015;21:914–21. 10.1038/nm.3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munshi NC, Anderson LD, Shah N, et al. Idecabtagene Vicleucel in relapsed and refractory multiple myeloma. N Engl J Med 2021;384): :705–16. 10.1056/NEJMoa2024850 [DOI] [PubMed] [Google Scholar]
- 15.Ohminami H, Yasukawa M, Fujita S. HLA class I-restricted lysis of leukemia cells by a CD8(+) cytotoxic T-lymphocyte clone specific for WT1 peptide. Blood 2000;95:286–93. 10.1182/blood.V95.1.286 [DOI] [PubMed] [Google Scholar]
- 16.Gao L, Bellantuono I, Elsässer A, et al. Selective elimination of leukemic CD34(+) progenitor cells by cytotoxic T lymphocytes specific for WT1. Blood 2000;95:2198–203. 10.1182/blood.V95.7.2198 [DOI] [PubMed] [Google Scholar]
- 17.Tawara I, Kageyama S, Miyahara Y, et al. Safety and persistence of WT1-specific T-cell receptor gene-transduced lymphocytes in patients with AML and MDS. Blood 2017;130:1985–94. 10.1182/blood-2017-06-791202 [DOI] [PubMed] [Google Scholar]
- 18.Chapuis AG, Egan DN, Bar M, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med 2019;25:1064–72. 10.1038/s41591-019-0472-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chapuis AG, Ragnarsson GB, Nguyen HN, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med 2013;5:ra27. 10.1126/scitranslmed.3004916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang Y, Lv X, Ge X, et al. Wilms tumor gent 1 (WT1)-specific adoptive immunotherapy in hematologic diseases. Int Immunopharmacol 2021;94:107504. 10.1016/j.intimp.2021.107504 [DOI] [PubMed] [Google Scholar]
- 21.Wilm B, Muñoz-Chapuli R. The role of WT1 in embryonic development and normal organ homeostasis. Methods Mol Biol 2016;1467: :23–39. 10.1007/978-1-4939-4023-3_3 [DOI] [PubMed] [Google Scholar]
- 22.Hastie ND. Wilms' tumour 1 (WT1) in development, homeostasis and disease. Development 2017;144:2862–72. 10.1242/dev.153163 [DOI] [PubMed] [Google Scholar]
- 23.Wagner N, Ninkov M, Vukolic A, et al. Implications of the Wilms' tumor suppressor WT1 in cardiomyocyte differentiation. Int J Mol Sci 2021;22. 10.3390/ijms22094346. [Epub ahead of print: 21 Apr 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaigirdar A, Rosenberg SA, Parkhurst M. A high-avidity WT1-reactive T-cell receptor mediates recognition of peptide and processed antigen but not naturally occurring WT1-positive tumor cells. J Immunother 2016;39:105–16. 10.1097/CJI.0000000000000116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pinilla-Ibarz J, May RJ, Korontsvit T, et al. Improved human T-cell responses against synthetic HLA-0201 analog peptides derived from the WT1 oncoprotein. Leukemia 2006;20:2025–33. 10.1038/sj.leu.2404380 [DOI] [PubMed] [Google Scholar]
- 26.Tsuboi A, Oka Y, Udaka K, et al. Enhanced induction of human WT1-specific cytotoxic T lymphocytes with a 9-mer WT1 peptide modified at HLA-A*2402-binding residues. Cancer Immunol Immunother 2002;51:614–20. 10.1007/s00262-002-0328-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmitt TM, Aggen DH, Stromnes IM, et al. Enhanced-affinity murine T-cell receptors for tumor/self-antigens can be safe in gene therapy despite surpassing the threshold for thymic selection. Blood 2013;122:348–56. 10.1182/blood-2013-01-478164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jahn L, van der Steen DM, Hagedoorn RS, et al. Generation of CD20-specific TCRs for TCR gene therapy of CD20low B-cell malignancies insusceptible to CD20-targeting antibodies. Oncotarget 2016;7:77021–37. 10.18632/oncotarget.12778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jahn L, Hombrink P, Hagedoorn RS, et al. TCR-based therapy for multiple myeloma and other B-cell malignancies targeting intracellular transcription factor BOB1. Blood 2017;129:1284–95. 10.1182/blood-2016-09-737536 [DOI] [PubMed] [Google Scholar]
- 30.Meeuwsen MH, Wouters AK, Jahn L, et al. A broad and systematic approach to identify B cell malignancy-targeting TCRs for multi-antigen-based T cell therapy. Mol Ther 2022;30:564-578. 10.1016/j.ymthe.2021.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bellantuono I, Gao L, Parry S, et al. Two distinct HLA-A0201-presented epitopes of the Wilms tumor antigen 1 can function as targets for leukemia-reactive CTL. Blood 2002;100:3835–7. 10.1182/blood.V100.10.3835 [DOI] [PubMed] [Google Scholar]
- 32.van der Lee DI, Reijmers RM, Honders MW, et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J Clin Invest 2019;129:774–85. 10.1172/JCI97482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Waard AA, Verkerk T, Hoefakker K, et al. Healthy cells functionally present TAP-independent SSR1 peptides: implications for selection of clinically relevant antigens. bioRxiv 2020;146449. 10.2139/ssrn.3641946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 2016;32:511–7. 10.1093/bioinformatics/btv639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burrows SR, Kienzle N, Winterhalter A, et al. Peptide-MHC class I tetrameric complexes display exquisite ligand specificity. J Immunol 2000;165:6229–34. 10.4049/jimmunol.165.11.6229 [DOI] [PubMed] [Google Scholar]
- 36.van Bergen CAM, van Luxemburg-Heijs SAP, de Wreede LC, et al. Selective graft-versus-leukemia depends on magnitude and diversity of the alloreactive T cell response. J Clin Invest 2017;127:517–29. 10.1172/JCI86175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koning MT, Kiełbasa SM, Boersma V, et al. Artisan PCR: rapid identification of full-length immunoglobulin rearrangements without primer binding bias. Br J Haematol 2017;178:983–6. 10.1111/bjh.14180 [DOI] [PubMed] [Google Scholar]
- 38.Lefranc MP, Giudicelli V, Ginestoux C, et al. IMGT, the International immunogenetics database. Nucleic Acids Res 1999;27:209–12. 10.1093/nar/27.1.209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linnemann C, Heemskerk B, Kvistborg P, et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat Med 2013;19:1534–41. 10.1038/nm.3359 [DOI] [PubMed] [Google Scholar]
- 40.Azuma T, Makita M, Ninomiya K, et al. Identification of a novel WT1-derived peptide which induces human leucocyte antigen-A24-restricted anti-leukaemia cytotoxic T lymphocytes. Br J Haematol 2002;116:601–3. 10.1046/j.0007-1048.2001.03329.x [DOI] [PubMed] [Google Scholar]
- 41.Bui H-H, Sidney J, Dinh K, et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics 2006;7:153. 10.1186/1471-2105-7-153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vita R, Mahajan S, Overton JA, et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res 2019;47:D339–43. 10.1093/nar/gky1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roex MCJ, Hageman L, Veld SAJ, et al. A minority of T cells recognizing tumor-associated antigens presented in self-HLA can provoke antitumor reactivity. Blood 2020;136:455–67. 10.1182/blood.2019004443 [DOI] [PubMed] [Google Scholar]
- 44.Rezvani K, Yong ASM, Mielke S, et al. Repeated PR1 and WT1 peptide vaccination in Montanide-adjuvant fails to induce sustained high-avidity, epitope-specific CD8+ T cells in myeloid malignancies. Haematologica 2011;96:432–40. 10.3324/haematol.2010.031674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hou H-A, Huang T-C, Lin L-I, et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood 2010;115:5222–31. 10.1182/blood-2009-12-259390 [DOI] [PubMed] [Google Scholar]
- 46.King-Underwood L, Renshaw J, Pritchard-Jones K. Mutations in the Wilms' tumor gene WT1 in leukemias. Blood 1996;87:2171–9. 10.1182/blood.V87.6.2171.bloodjournal8762171 [DOI] [PubMed] [Google Scholar]
- 47.Orlando D, Miele E, De Angelis B, et al. Adoptive immunotherapy using PRAME-Specific T cells in medulloblastoma. Cancer Res 2018;78:3337–49. 10.1158/0008-5472.CAN-17-3140 [DOI] [PubMed] [Google Scholar]
- 48.Naitoh K, Kamigaki T, Matsuda E. Immunohistochemical analysis of WT1 antigen expression in various solid cancer cells. Anticancer Res 2016;36:3715–24. [PubMed] [Google Scholar]
- 49.Pozzi S, Geroldi S, Tedone E, et al. Leukaemia relapse after allogeneic transplants for acute myeloid leukaemia: predictive role of WT1 expression. Br J Haematol 2013;160:503–9. 10.1111/bjh.12181 [DOI] [PubMed] [Google Scholar]
- 50.Rautenberg C, Germing U, Pechtel S, et al. Prognostic impact of peripheral blood WT1-mRNA expression in patients with MDS. Blood Cancer J 2019;9:86. 10.1038/s41408-019-0248-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Artibani M, Sims AH, Slight J, et al. Wt1 expression in breast cancer disrupts the epithelial/mesenchymal balance of tumour cells and correlates with the metabolic response to docetaxel. Sci Rep 2017;7:45255. 10.1038/srep45255 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2021-004409supp002.pdf (1.1MB, pdf)
jitc-2021-004409supp001.pdf (1.9MB, pdf)
Data Availability Statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.