

Abstract
Background:
Polygenic and environmental factors are underlying causes of inflammatory bowel disease (IBD). We hypothesized that integration of the genetic loci controlling a metabolite’s abundance, with known IBD genetic susceptibility loci, may help resolve metabolic drivers of IBD.
Methods:
We measured the levels of 1300 metabolites in the serum of 484 ulcerative colitis (UC) and 464 Crohn’s disease (CD) patients and 365 controls. Differential metabolite abundance was determined for disease status, subtype, clinical and endoscopic disease activity as well as IBD phenotype including disease behavior, location and extent. To inform on the genetic basis underlying metabolic diversity, we integrated metabolite and genomic data. Genetic colocalization and Mendelian randomization (MR) analyses were performed using known IBD risk loci to explore whether any metabolite was causally associated with IBD.
Results:
We found 173 genetically controlled metabolites (mQTL, 9 novel) within 63 non-overlapping loci (7 novel). Furthermore, several metabolites significantly associated with IBD disease status and activity as defined by clinical and endoscopic indexes. This constitutes a resource for biomarker discovery and IBD biology insights. Using this resource, we show that a novel mQTL for serum butyrate levels containing ACADS was not supported as causal for IBD; replicate the association of serum omega-6 containing lipids with the fatty acid desaturase 1/2 locus and identify these metabolites as causal for CD through MR; and validate a novel association of serum plasmalogen and TMEM229B, which was predicted causal for CD.
Conclusion:
An exploratory analysis combining genetics and unbiased serum metabolome surveys can reveal novel biomarkers of disease activity and potential mediators of pathology in IBD.
Keywords: inflammatory bowel disease, metabolome, differential metabolite abundance analysis, Mendelian randomization
Introduction
Inflammatory bowel disease (IBD) affects several million individuals worldwide and includes Crohn’s disease (CD) and ulcerative colitis (UC)1. IBD is a complex mucosal immune response of the gastrointestinal tract triggered by several genetic and environmental factors resulting in a heterogeneous disease at the clinical, genetic and molecular level2. As a result, unbiased multi-omics profiling of IBD patient blood, stool and intestine samples has become common to facilitate unraveling the underlying pathology as well as to facilitate biomarker discovery for patient stratification3,4,5. Importantly, integration across the omics, especially those anchored on genetics, may help resolve which alterations may be causal or secondary to the chronic inflammation in IBD.
Metabolomics is the large-scale quantification of metabolites and arguably provides a closer intermediate phenotype of disease than either genomics or transcriptomics. Indeed, metabolomics surveys of serum from either mouse models of IBD or from IBD patients supports that differences in metabolite abundance are associated with disease state or subtype6,7. For example, several reports have associated intestinal inflammatory responses in IBD with lipid metabolites and their derived signaling molecules (e.g. long-chain polyunsaturated fatty acids (PUFAs) such as arachidonic acid (AA) and eicosanoids). This suggests that metabolomics has the potential to elucidate not only IBD mechanism but also biomarkers for diagnosis and disease activity monitoring.
At present, few, if any, studies have generated comprehensive metabolomics measures in a large-scale cohort of IBD patients, for which detailed clinical and endoscopic phenotyping and genotype information is also available. This has hindered the type of integration strategies that can be applied in order to yield novel and unbiased metabolite-IBD mechanistic insights. To address this limitation, we determined the serum and stool metabolome in a unique and large cohort of UC and CD patients and healthy controls (HCs). Importantly, we integrated the patient metabolite abundances with detailed clinical, endoscopic and genetic data collected in the same individuals either at the same time (for serum) or within several weeks (for stool). Our integration strategy was focused on 1. revealing metabolites associated with clinical and endoscopic phenotypes as a resource of biomarkers and mechanistic insights, and 2. co-localizing previously reported IBD genetic risk loci with present study metabolite QTLs (mQTLs) as a way to test and identify potential metabolic mediators of disease biology. As such, we identified a novel locus and candidate gene associated with plasmalogen levels, which was predicted to causally increase risk of development of CD. On the other hand, we revealed a genetic controller of serum butyrate levels, a short-chain fatty acid (SCFA), which was not associated with IBD status or various IBD traits. Overall our work highlights several genetic bases for serum metabolic diversity that align with IBD genetic risk loci and as such, may be novel mediators of disease pathology.
Methods
Study cohort
Patients with UC (n=484) or CD (n=464) and HCs (ages 18+, n=365) were recruited as part of a cross-sectional study, the Mount Sinai Crohn’s and Colitis Registry (MSCCR), between December 2013 and September 2016 from the outpatient and endoscopy units of the Mount Sinai Hospital. Institutional review board approval and informed consents were obtained. Study participants provided blood samples for serum preparation at the time of their endoscopy. Stool samples were collected as described previously within several weeks (median 55.5 days) of the study endoscopy8. The clinical and demographic information was obtained through a questionnaire (Table 1) and medication use is summarized in Table ST1.
Table 1.
Cohort demographics.
| All (n=1313) | CD (n=464) | UC (n=484) | HC (n=365) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | % | median (min-max) | n | % | median (min-max) | n | % | median (min-max) | n | % | median (min-max) | |
| Age | ||||||||||||
| at endoscopy | 1313 | - | 46 (18 – 82) | 464 | - | 34 (18 – 78) | 484 | - | 44 (20 – 82) | 365 | - | 54 (20 – 82) |
| at diagnosis | 945 | - | 25 (2 – 75) | 462 | - | 24 (2 – 69) | 483 | - | 26 (6 – 75) | 0 | - | |
| Gender | ||||||||||||
| Male | 680 | 51.8 | - | 240 | 51.7 | - | 250 | 51.7 | - | 190 | 52.1 | - |
| Female | 633 | 48.2 | - | 224 | 48.3 | - | 234 | 48.3 | - | 175 | 47.9 | - |
| Metabolomics | ||||||||||||
|
serum
stool |
973 | 74.1 | - | 284 | 61.2 | - | 360 | 74.4 | - | 329 | 90.1 | - |
| 261 | 19.9 | - | 88 | 19 | - | 101 | 20.9 | - | 72 | 19.7 | - | |
| Genotyping | ||||||||||||
| genotyping | 1251 | 95.3 | - | 447 | 96.3 | - | 461 | 95.2 | - | 343 | 94 | - |
| Ancestry | ||||||||||||
| Asian | 16 | 1.3 | - | 4 | 0.9 | - | 4 | 0.9 | - | 8 | 2.3 | - |
| Black | 112 | 9 | - | 21 | 4.7 | - | 20 | 4.3 | - | 71 | 20.7 | - |
| Mexican | 68 | 5.4 | - | 21 | 4.7 | - | 30 | 6.5 | - | 17 | 5 | - |
| Multiple | 114 | 9.1 | - | 34 | 7.6 | - | 21 | 4.6 | - | 59 | 17.2 | - |
| White | 941 | 75.2 | - | 367 | 82.1 | - | 386 | 83.7 | - | 188 | 54.8 | - |
| Clinical disease severity | ||||||||||||
| HBI | 464 | - | 2 (0 – 27) | 464 | - | 2 (0 – 27) | 0 | - | - | |||
| Inactive (0 −4) | 352 | 75.9 | 352 | 75.9 | ||||||||
| Mild (5–7) | 67 | 14.4 | 67 | 14.4 | ||||||||
| Moderate (8–16) | 42 | 9.1 | 42 | 9.1 | ||||||||
| Severe (>16) | 3 | 0.6 | 3 | 0.6 | ||||||||
| SCCAI | 484 | - | 1 (0 – 15) | 0 | - | 484 | - | 1 (0 – 15) | - | |||
| Remission (0–4) | 430 | 88.8 | 430 | 88.8 | ||||||||
| Active (>4) | 54 | 11.1 | 54 | 11.1 | ||||||||
| SESCD | 464 | - | 3 (0 – 41) | 464 | - | 3 (0 – 41) | 0 | - | - | |||
| Inactive(0–2) | 198 | 42.7 | 194 | 42.7 | ||||||||
| Mild (3–6) | 133 | 28.7 | 133 | 28.7 | ||||||||
| Moderate(7–15) | 92 | 19.8 | 92 | 19.8 | ||||||||
| Severe (>15) | 41 | 8.8 | 41 | 8.8 | ||||||||
| Mayo-endo | 482 | - | 1 (0 – 3) | 0 | - | 482 | - | 1 (0 – 3) | - | |||
| Inactive (0) | 234 | 48.5 | 234 | 48.5 | ||||||||
| Mild (1) | 153 | 31.7 | 153 | 31.7 | ||||||||
| Moderate (2) | 64 | 16.3 | 64 | 16.3 | ||||||||
| Severe (3) | 31 | 6.4 | 31 | 6.4 | ||||||||
Genotyping
Blood samples were used for DNA extraction, and genotype data generated using the high-density Illumina Multi-Ethnic Global Array (MEGAEX) and Infinium ImmunoArray-24 v2 BeadChip arrays. We further imputed genotypes using the Michigan Imputation Server2 with the 1000 Genomes reference (supplementary methods).
Metabolome profiling
Serum and stool were stored at −80°C and sent to Metabolon, Inc. (Research Triangle Park, North Carolina) for targeted and untargeted metabolome profiling9,10. Samples were randomized according to disease type and gender. Data for the untargeted platform (semi-quantitative) were normalized according to the minimal level detected for each metabolite/run and reported as log2 signal-to-noise (SNR) ratio. We detected a total of 2177 distinct metabolites in at least one sample.
From the targeted platform, we obtained quantitative values for eight SCFAs measured in both sample types: acetate, propionate, butyrate, isobutyrate, valerate, 2-methylbutyrate, isovalerate, and hexanoate. Unsupervised clustering indicated a major source of variation was surgery. After excluding patients who had undergone IBD surgeries and patients with indeterminate diagnosis, we had 973 serum (284 CD, 360 UC and 329 controls) and 261 stool (88 CD, 101 UC and 72 controls) profiles, respectively. For subsequent statistical analyses, we retained all metabolites present in at least 10% of the samples of a given sample type (serum or stool), yielding 1300 metabolites in serum, and 1443 metabolites in stool, with 786 metabolites shared between the two sample types (Figure 1A). See supplementary methods for details.
Figure 1. Summary of the study workflow.
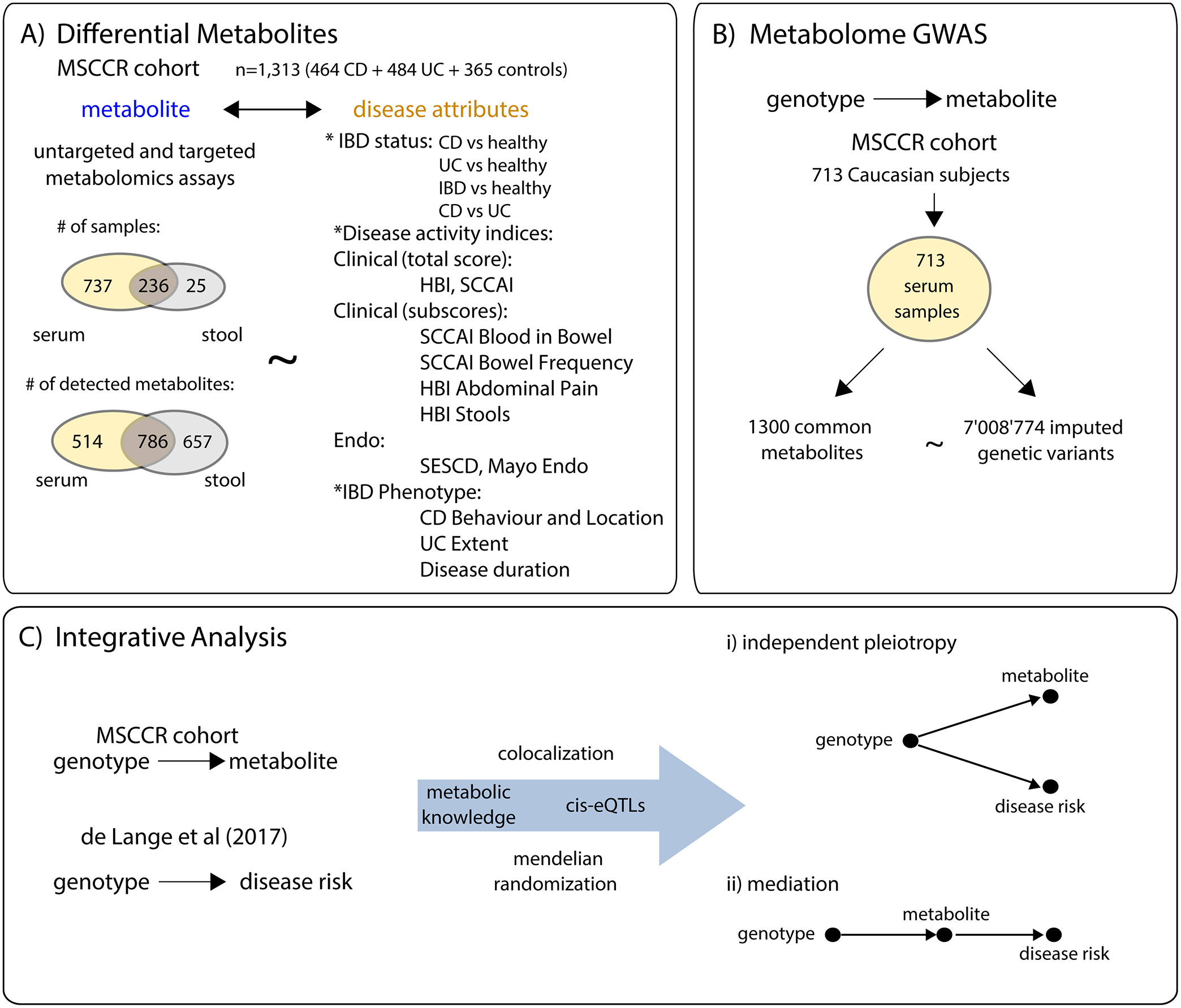
A) An overview of the number of samples and metabolites detected in serum and stool of the MSCCR. B) The metabolite GWAS design. C) Flow chart summarizing the integration of genotype and metabolite information available from the present MSSCR cohort and publicly available IBD meta-analysis11, in order to assess a causal relationship between metabolite changes and IBD.
Statistical Analysis
Differential metabolite abundance analysis (DMA)
We performed DMA through a linear regression model for each metabolite of the form: log2(SNR)~endpoint+covariates, in which we initially considered eight IBD attributes. These included: IBD (vs non-IBD), CD (vs non-IBD), UC (vs non-IBD), CD vs UC as well as the clinical and endoscopic disease activity indices, as continuous variables, including log2(HBI+1) (Harvey-Bradshaw Index, for CD), log2(SCCAI+1) (Simple Clinical Colitis Activity Index, for UC), log2(SESCD+1) (Simple Endoscopic Score for CD), and Mayo endoscopic score (Mayo endo for UC). Covariates for serum were: age, sex, smoking status, storage duration and metabolome profiling batch. Covariates for stool were: age, sex, smoking status, and metabolome profiling batch. Within each sample type and for each IBD attribute tested, we adjusted for multiple testing using the Benjamini-Hochberg (BH) procedure. “Differential Metabolites” (DMs) were all metabolites with a significance level below 10% FDR (false discovery rate).
We used the Fisher’s exact test (FET) to evaluate the overlap between the lists of DMs from the untargeted platform (10% FDR) and the metabolite classifications as provided by Metabolon. Two pathway classifications were used, ‘super pathway’ (10 groups) and ‘subpathway’ (132 groups). Within each sample type, IBD attribute, and pathway level, we adjusted for multiple testing using the BH procedure. We analyzed log2-ratios between pairs of metabolites within the same super-pathway using the same linear model (15,311,568 tests/sample type). All results with p≤0.1 were reported.
We also analyzed metabolite associations with various sub-components of the clinical disease activity indices in addition to their total scores. Also surveyed were associations with IBD phenotypes according to the Montreal sub-classifications. The same regression models and pathway enrichment analysis methods were applied as above (supplementary methods).
Metabolome GWAS
We performed a serum metabolome genome-wide association study (GWAS) by fitting simple linear regressions between the covariates-adjusted metabolite abundances and the imputed effective allele dosages. We used a genome-wide significance cutoff of 5e-8/1292=3.9e-11 (1292 being the number of metabolites tested) for the untargeted platform, and 5e-8/8=6e-9 for the targeted platform. Adjustment covariates were: age, sex, diagnosis (CD, UC or HC), metabolome profiling batch, and the first five genetic principal components. We further restricted the analysis to 713 serum samples from subjects of Caucasian ancestry.
Integrative Analysis
We tested for genetic colocalization between our genetically controlled metabolites (nominal p value < 3.9E-11) and known IBD risk loci11 using the ‘coloc’ method12, as implemented in the coloc R package, version 3.2–1. For each investigated region (the metabolome GWAS peak ± 500Kb), the procedure estimates posterior probabilities for 5 different hypotheses involving the 2 traits under investigation, here IBD (trait 1) and the metabolite level (trait 2) with H0: neither trait has a causal variant; H1: only trait 1 has a causal variant; H2: only trait 2 has a causal variant; H3: traits 1 and 2 have distinct causal variants; H4: trait 1 and trait 2 have a common, shared causal variant.
We estimated causal odds ratios (ORs) of IBD risk per standard deviation (SD)-increase in serum metabolites log-concentrations with a two-samples Mendelian randomization (MR) procedure, specifically the ratio estimator13, using previously published summary statistics11 for the total effect of genotype on disease risk (numerator of the ratio estimator), and the estimated effect of genotype on the standardized metabolite log-concentration (denominator of the ratio estimator) from the Caucasian HCs of the present study (n=184). 95% confidence intervals were derived using Fieller’s method14 (original Fortran code translated to R) and using the delta method, as implemented for the “Inverse Variance Weights” method in the R package “MendelianRandomization”15. Delta method CIs were narrower than those obtained with Fieller’s method in all tests, and always fully contained within Fieller’s CIs for metabolites with strong instruments (p<1E-3): we are thus reporting the more conservative Fieller’s CIs in the main text and in figures.
In vitro validation of a role of TMEM229B in regulating plasmalogen levels
TMEM229B overexpression in HEK293 cells was performed as described in supplementary methods.
GTEx eQTL Analysis
The data used for the analyses described in this manuscript were obtained from the GTEx Portal https://gtexportal.org on 10/30/2020.
Blood Bayesian regulatory gene network generation
The blood Bayesian regulatory gene network was generated as described in the supplementary methods.
Results
Metabolite levels differentiate IBD cases from HCs and associate with disease activity indices
After quality control, we had semi-quantitative values for 1435 stool (345 unknown) and 1292 serum (291 unknown) metabolites, 778 of which were in common. In addition, we obtained the abundance of eight SCFAs in serum and stool. We had 973 (284 CD, 360 UC and 329 HC) serum and 261 (88 CD, 101 UC and 72 HC) stool metabolite profiles (Figure 1A).
DMs in serum and stool (Figure 2A) were obtained for: i) IBD vs HC (436-serum, 692-stool), ii) CD vs HC (240-serum, 478-stool), iii) UC vs HC (442-serum, 609-stool), and iv) CD vs UC (175-serum, none-stool); or that associate with v) HBI (147-serum, 6-stool), vi) SCCAI (296-serum, 69-stool), vii) SESCD (370-serum, 94-stool), and viii) Mayo disease activity scores (202-serum, 64-stool).
Figure 2. Serum and stool metabolites significantly associated with various IBD disease attributes.
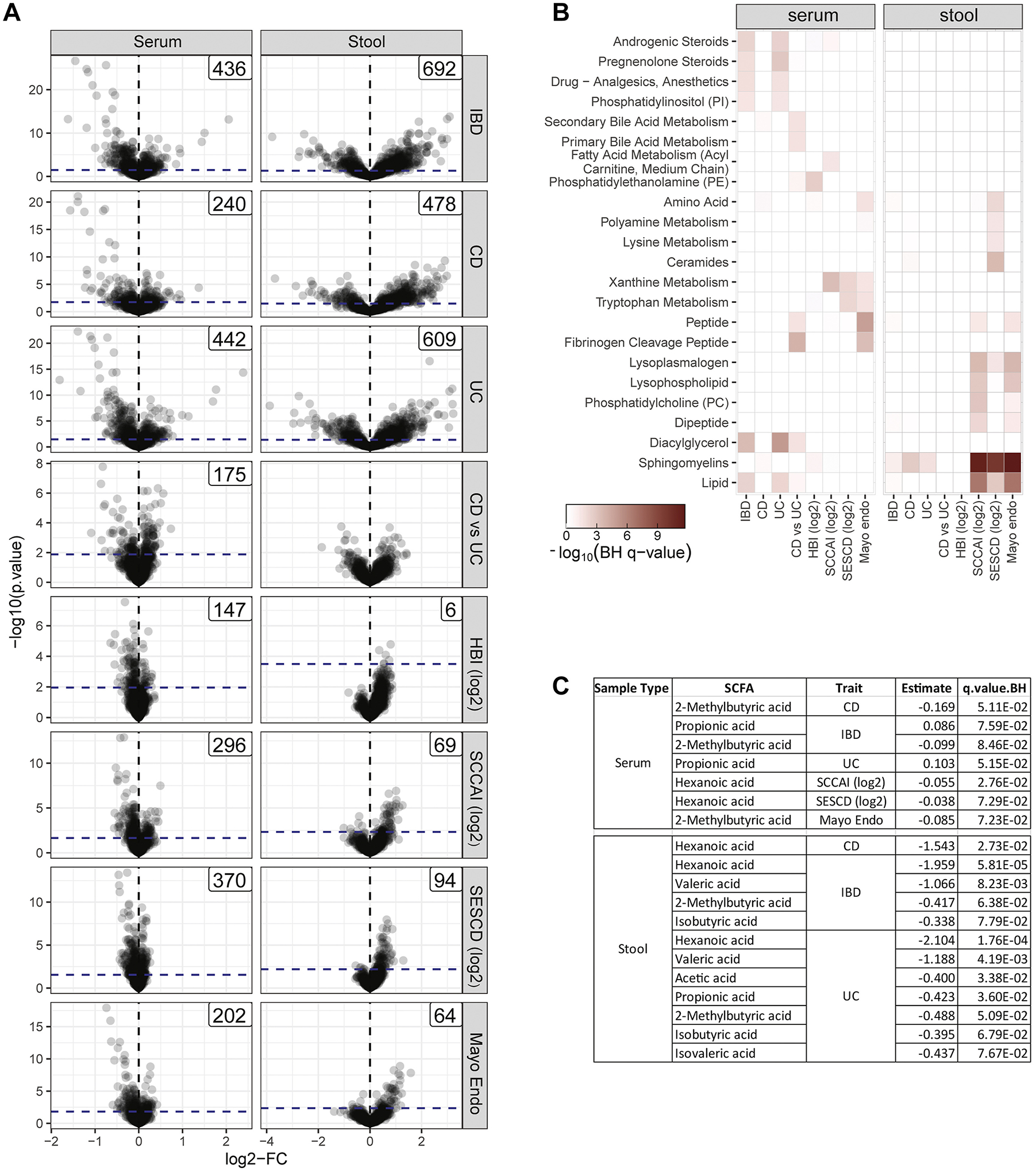
A) Volcano plots showing the DMA of the serum (left panel) and stool samples (right panel), according to the comparisons of interest including: IBD vs control (IBD); CD vs control (CD); UC vs control (UC); and CD vs UC as well as associations to clinical disease activity indices (log2HBI and log2SCCAI) and endoscopy scores (log2SESCD and Mayo_endo). We indicated the 10% FDR threshold as a dashed blue line and the number of DMs). B) A heatmap summarizing the enrichments of the serum and stool DMs according to metabolite classifications. Only classifications that were significantly enriched at FDR <0.05 for at least one endpoint are shown. C) Summary of differentially expressed SCFAs according to endpoint with effect and FDR. Only SCFAs passing FDR <0.10 are shown (Table S2).
These DMs were tested for enrichment according to ‘super’ and ‘sub’-pathway annotations (Figure 2B, ST2–ST3). The stool metabolites associated with disease status or the various disease activity measures were strongly enriched for lipids, sphingomyelins, lysoplasmalogens, lysophospholipids, phosphatidylcholines and dipeptides. In contrast, the serum metabolites associated with disease status or with the various disease activity measures were enriched in fibrinogen cleavage peptides as well as xanthine and tryptophan metabolism products. Serum metabolites distinguishing between CD and UC were strongly enriched in the fibrinogen cleavage peptides and diacylglycerol classes.
SCFAs were also found differentially abundant (Figure 1C) with respect to various IBD attributes. In all cases, the SCFA levels in the stool or serum were found to be lower in cases versus control contrasts, or negatively associated with disease activity indicators. Remarkably, butyrate, which has been previously reported to associate with IBD16, was not significantly associated with any IBD attribute tested (with a q value<0.05), in either the serum or stool.
We also analyzed the ratios between pairs of metabolites17 from the same super pathway, using the same model and endpoints as for the single metabolites analysis. No metabolite ratio reached the nominal p value cutoff of 1E-8. Results up to p=1E-3 (2063 ratios) are reported in ST4.
Metabolite levels associate with various Montreal classifications and patient reported outcomes
We expanded our DMA with the goal of relating metabolite levels to more granular aspects of IBD. We included sub-scores associated with the clinical disease activity scores (SCCAI and HBI) as well as various Montreal sub-classifications, using the same regression models and pathways enrichment analysis as above. We surmised these components may provide for richer basic research perspectives into pathophysiology underlying the different manifestations of disease. The number of DMs for these disease attributes was, however, in general much smaller than observed for disease status of the total disease activity scores, likely related to the reduced sample size per sub-group (SF2 to SF7.) Sub-pathway enrichment analysis, however, did highlight similarly enriched metabolite groups such as serum and stool sphingomyelins, serum fatty acid (FA) metabolism, stool dipeptides and stool lysoplasmalogens. Newly highlighted metabolite sub-groups captured with the more granular IBD attributes included serum sphingosines which associated with CD disease location; serum hemoglobin and porphyrin metabolites that associated with bowel frequency at night; and stool secondary bile acid metabolites that associated with UC extent (E3 vs E1). See supplementary results and SF2 to SF7 and ST5 and ST6 for details.
Metabolite quantitative trait loci (mQTL) analysis reveals novel loci controlling serum metabolite levels
We conducted a GWAS of serum metabolites and identified 173 genetically controlled metabolites (mQTL, at p<3.8e-11) within 63 non-overlapping loci. We compared our mQTLs to those summarized in a recently published review of all genome-wide association and exome- sequencing studies18 as well as selected key papers19–21. Our analysis identified 9 novel mQTLs, 7 of which were on new loci. We observed 80 novel serum metabolite associations at previously identified loci, whereas 93 associations replicated other studies (ST7 and SF8).
Genetically altered serum butyrate levels do not impact risk of IBD
The mQTL analysis for SCFAs revealed a novel association of serum butyrate levels to a region on Chr12 (Figure 3A). Butyrylglycine was a second novel mQTL at this locus known to also associate with ethylmalonate, methylsuccinate and butyrylcarnitine, which were all 3 replicated in our study. Variation in ACADS underlies these associations as all five metabolites can be directly linked to short-chain acyl-CoA dehydrogenase (SCAD) function (Figure 3C). Indeed, SCAD deficiency is biochemically diagnosed by elevated levels of the same metabolites19.
Figure 3. Genetic control of serum butyrate and lack of association with IBD-related traits.
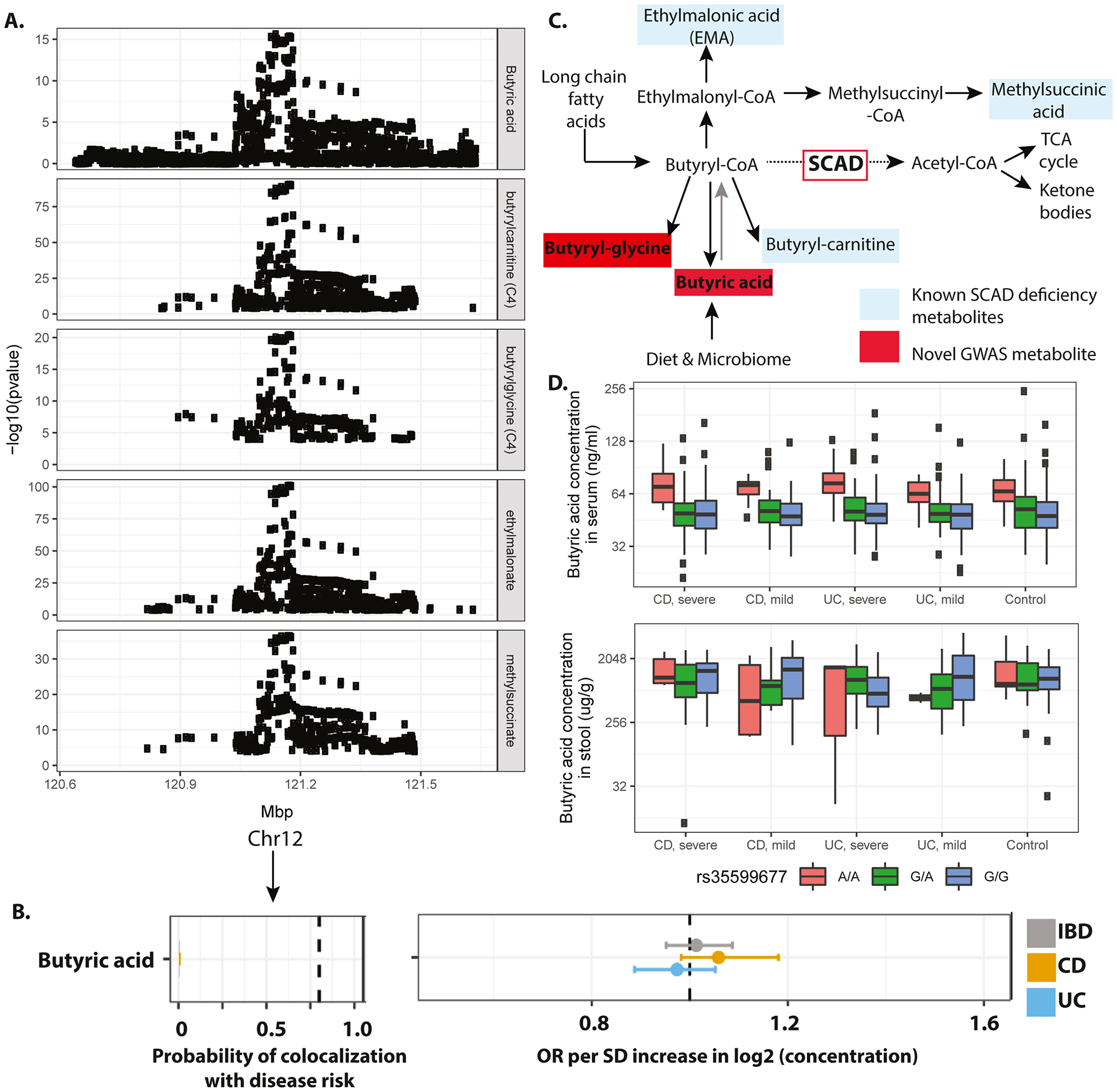
A) Manhattan plots displaying the GWAS results of butyrate and various related metabolites (rows). Genome-wide significance threshold: 5E-8/1300=3.8E-11. B) Results of genetic colocalization between IBD, UC or CD risk and serum butyrate levels as well as results of the MR analysis. Metabolites on the vertical axis, estimates on the horizontal axis, genetic regions in vertical stripes. Posterior colocalization probabilities (left panel), and causal OR (right panel). Within each panel, a dotted vertical line highlights a point of interest: 0.8 posterior probability of colocalization on the left panel, and OR=1 on the right panel. Hatched line indicates position of OR=1, values to the right of the hatched line indicate higher levels of the corresponding metabolite are associated with increased risk and values to the left of the hatched line indicate the opposite. Colocalization and OR estimates were based on summary statistics from the present study (metabolome GWAS) and de Lange et al11 (IBD GWAS). C) Biochemical schema showing metabolic substrates and products of SCAD. Metabolites shaded in red are novel GWAS observations and those in blue are known ACADS GWAS associations. D) Serum (upper panel) and stool (lower panel) butyrate levels grouped by i) genotype at the rs35599677 locus and ii) disease severity as measured by endoscopic assessments. Mayo Scores ≥1 were classified as severe, scores <1 as mild. SESCD ≥3 were classified as severe, <3 as mild.
Given that butyrate-producing bacterial strains are candidate treatments for IBD16, we took advantage of the observed genetic control of serum butyrate levels to unbiasedly assess a potential causal association to IBD. Using the colocalization analysis, we first evaluated if there were any shared genetic variants between serum butyrate levels and IBD risk within the ACADS locus. No SNPs passed significant thresholds suggesting a lack of common genetic origin between IBD and butyrate (Figure 3B). Furthermore, when we segregated the cohort according to genotype at rs35599677 and disease status, we observed a clear genotype effect, but no difference in serum butyrate levels between case and control or with respect to disease severity. We did not observe a significant association between rs35599677 and butyrate levels as measured in the stool where, however, we had a much smaller sample size compared to serum samples (Figure 3D). Overall these results suggest that IBD is not causally influenced by serum butyrate levels.
Integrative analysis of the IBD differential metabolome, IBD GWAS genes and mQTL loci
Colocalization and Mendelian randomization test between IBD risk loci and metabolite GWAS loci
To focus this study’s mQTL knowledge on IBD and in particular on finding metabolites predicted to be causally associated with IBD, we intersected the metabolite controlling loci with known IBD risk loci. We selected 58,400 risk variants in 868 non-overlapping loci with reported p value<1E-4 of association with IBD, CD or UC (de Lange et al11, Jostins et al22 and EBI-NHGRI-GWAS catalog). From the 173 genetically controlled metabolites identified in the present study, 17 could be annotated as also controlled by genetic variants in one of 4 distinct IBD risk loci (Table 2). For these 17 metabolites, we conducted a formal colocalization analysis against IBD risk profiles in each identified locus. We obtained a positive colocalization signal for two loci, one on Chr11 and one on Chr14 (posterior probability of a shared IBD variant ≥ 0.8), which supports evidence for a shared causal variant between risk of IBD and CD and 15 unique serum metabolites (ST8, column ‘p4’). We observed lack of colocalization between IBD risk (all 3 endpoints) and X-11470 metabolite levels (chr6, MSH5, SAPCD1) and fibrinopeptide A (3–15) levels (chr5, CAST, ERAP). Also, none of the molecules controlled by IBD risk variants showed evidence of colocalization with UC risk.
Table 2.
Metabolites controlled by IBD genetic risk variants.
| Analyte | DEM endpoints (10% FDR) | Estimated effect of the IBD risk allele | ||||
|---|---|---|---|---|---|---|
| Serum | Stool | Beta | SE | P value | R2 | |
| locus (hg19): 5:96198479; rsID (prot/risk allele): rs2927614 (C/G); risk allele frequency: 0.78; mapped genes: CAST, ERAP1 | ||||||
| Fibrinopeptide A (3–15) | +CD_vs_UC, +log2SESCD, +Mayo_endo | 0.49 | 0.063 | 3.24E-14 | 7.8% | |
| locus: 6:31709045; rsID (prot/risk allele): rs28381349 (C/T); risk allele frequency: 0.07; mapped genes: ABHD16A, AGER, AIF1, ATF6B, BAG6, C2, C6orf10, C6orf25, CLIC1, CSNK2B, CYP21A1P, CYP21A2, DDAH2, EHMT2, FKBPL, GPANK1, LY6G6C, LY6G6E, LY6G6F, MSH5, MSH5-SAPCD1, NCR3, NOTCH4, PPT2-EGFL8, PRRC2A, PRRT1, RNF5P1, SAPCD1-AS1, SKIV2L, SLC44A4, STK19, TNXB, VARS, VWA7 | ||||||
| X - 11470 | −log2SESCD | −0.75 | 0.100 | 1.47E-13 | 7.4% | |
| locus (hg19): 11:61564299; rsID (prot/risk allele): rs4246215 (G/T); risk allele frequency: 0.32; mapped genes: FADS1, FADS2 | ||||||
| 1-(1-enyl-palmitoyl)-2-arachidonoyl-GPC (P-16:0/20:4)* | −CD, −IBD, −UC | −0.52 | 0.052 | 3.79E-22 | 12.3% | |
| 1,2-dilinoleoyl-GPC (18:2/18:2) | −log2SESCD | 0.37 | 0.054 | 1.65E-11 | 6.2% | |
| 1-arachidonoyl-GPC* (20:4)* | −IBD, −UC | −0.48 | 0.053 | 1.91E-18 | 10.2% | |
| 1-linoleoyl-GPE (18:2)* | 0.39 | 0.054 | 1.93E-12 | 6.7% | ||
| 1-palmitoyl-2-arachidonoyl-GPC (16:0/20:4n6) | −CD, −IBD, −log2SCCAI, −UC | +log2SCCAI | −0.49 | 0.053 | 2.89E-19 | 10.7% |
| 1-stearoyl-2-arachidonoyl-GPC (18:0/20:4) | −CD, −IBD, −log2SCCAI, −UC | −0.62 | 0.051 | 1.10E-31 | 17.6% | |
| docosatrienoate (22:3n6)* | −CD, −IBD | −0.35 | 0.054 | 2.75E-10 | 5.5% | |
| oleoyl-arachidonoyl-glycerol (18:1/20:4) [1]* | +CD_vs_UC, −IBD, −UC | −0.34 | 0.054 | 9.62E-10 | 5.1% | |
| oleoyl-arachidonoyl-glycerol (18:1/20:4) [2]* | −CD, +CD_vs_UC, −IBD, −UC | +CD, +IBD, +log2SCCAI, +log2SESCD, +Mayo_endo, +UC | −0.32 | 0.054 | 4.43E-09 | 4.7% |
| stearoyl-arachidonoyl-glycerol (18:0/20:4) [1]* | −UC | −0.38 | 0.054 | 3.75E-12 | 6.6% | |
| 1-oleoyl-2-linoleoyl-GPE (18:1/18:2)* | +log2HBI | 0.34 | 0.054 | 1.03E-09 | 5.1% | |
| 1-palmitoyl-2-linoleoyl-GPE (16:0/18:2) | +CD_vs_UC, +log2HBI | 0.38 | 0.054 | 5.36E-12 | 6.5% | |
| 1-stearoyl-2-linoleoyl-GPE (18:0/18:2)* | +CD_vs_UC, +log2HBI | 0.37 | 0.054 | 1.86E-11 | 6.2% | |
| locus (hg19): 14:67976325; rsID (prot/risk allele): rs386778467 (T/C); risk allele frequency: 0.49; mapped genes: PLEKHH1, TMEM229B | ||||||
| 1-(1-enyl-palmitoyl)-2-palmitoleoyl-GPC (P-16:0/16:1)* | −CD_vs_UC, −log2HBI, −log2SESCD | 0.44 | 0.050 | 5.78E-18 | 10.0% | |
| 1-(1-enyl-palmitoyl)-2-palmitoyl-GPC (P-16:0/16:0)* | −CD, −IBD, −log2HBI, −log2SESCD | +IBD, +log2SCCAI, +log2SESCD, +Mayo_endo, +UC | 0.46 | 0.050 | 2.22E-19 | 10.8% |
To estimate a putative causal effect of the metabolite levels on disease risk, we utilized Mendelian randomization (MR, Figure 1C). MR uses a genetic instrumental variable (GIV) to unbiasedly estimate the effect of the “exposure” (i.e. metabolite level) on the outcome (IBD, CD and UC risk) so that causality of an observed association (metabolite level and CD risk in this case) can be assessed. To perform the MR, we determined GIVs that fulfil three core assumptions: 1) strong association with the metabolite exposure; 2) no association with potential confounders; and 3) the GIV only acts on disease risk through the metabolite exposure being tested23. First, we chose GIVs for each of the 17 candidate metabolites that were the lead metabolome GWAS variant (by p value) and were also an IBD risk variant. Using the GIVs we then estimated the effect of genotype on metabolite levels within the HCs of Caucasian ancestry from the discovery cohort of the present study (n=184). Given the rarity of IBD (in 2015, prevalence of IBD is estimated at about 1.3% of US adults, www.cdc.gov/ibd, accessed in May 2020), this approximates the genetic effect on metabolites in the general population of European ancestry. Eleven of the 17 tested metabolites showed a highly significant genetic effect (p<1E-3, R2 between 6.9% and 19.7%, ST9), and were thus suitable for MR. To the best of our knowledge, these variants were not associated with confounders beyond those adjusted for (see Methods). Finally, as a working hypothesis, we assumed that the candidate GIVs were not associated with disease risk through pathways other than the metabolite being tested. ST9 summarizes the MR results, namely the estimate of the association of genetically altered metabolite levels on risk of CD using the candidate GIV for each of the 17 candidate metabolites, reported as ORs relative to a 1 standard deviation increase in the serum log-concentration of the metabolite.
Serum omega 6 containing metabolites are causally associated with risk of CD
Two GIVs showed significant ORs in the MR analysis. We estimated a high posterior probability of colocalization between CD risk and omega-6 PUFA containing lipids on a Chr11 locus (Figure 4A). This region contains the FADS (fatty acid desaturase) gene cluster (FADS1/FADS2), for which genetic variants have been previously reported to modify the activity of PUFA desaturation and the lipid composition of serum18,19. We observed that lipids containing omega-6 PUFA products of the FADS enzymes including 1-(1-enyl-palmitoyl)-2-arachidonoyl-GPC (P-16:0/20:4)*, 1-arachidonoyl-GPC (20:4)*, 1-palmitoyl-2-arachidonoyl-GPC (16:0/20:4n6), 1-stearoyl-2-arachidonoyl-GPC (18:0/20:4), and docosatrienoate (22:3n6)* had a protective effect on CD risk (ORs between 0.84 and 0.89)(Figure 4B). Conversely, we estimated a positive effect of linoleic acid containing lipids (the substrate of FADS), such as 1-oleoyl-2-linoleoyl-GPE (18:1/18:2)*, 1-palmitoyl-2-linoleoyl-GPE (16:0/18:2), and 1-stearoyl-2-linoleoyl-GPE (18:0/18:2)* on CD risk (OR between 1.17 and 1.21) (Figure 4B).
Figure 4. Genetic colocalization between essential fatty acids, FADS1/2 and IBD risk.
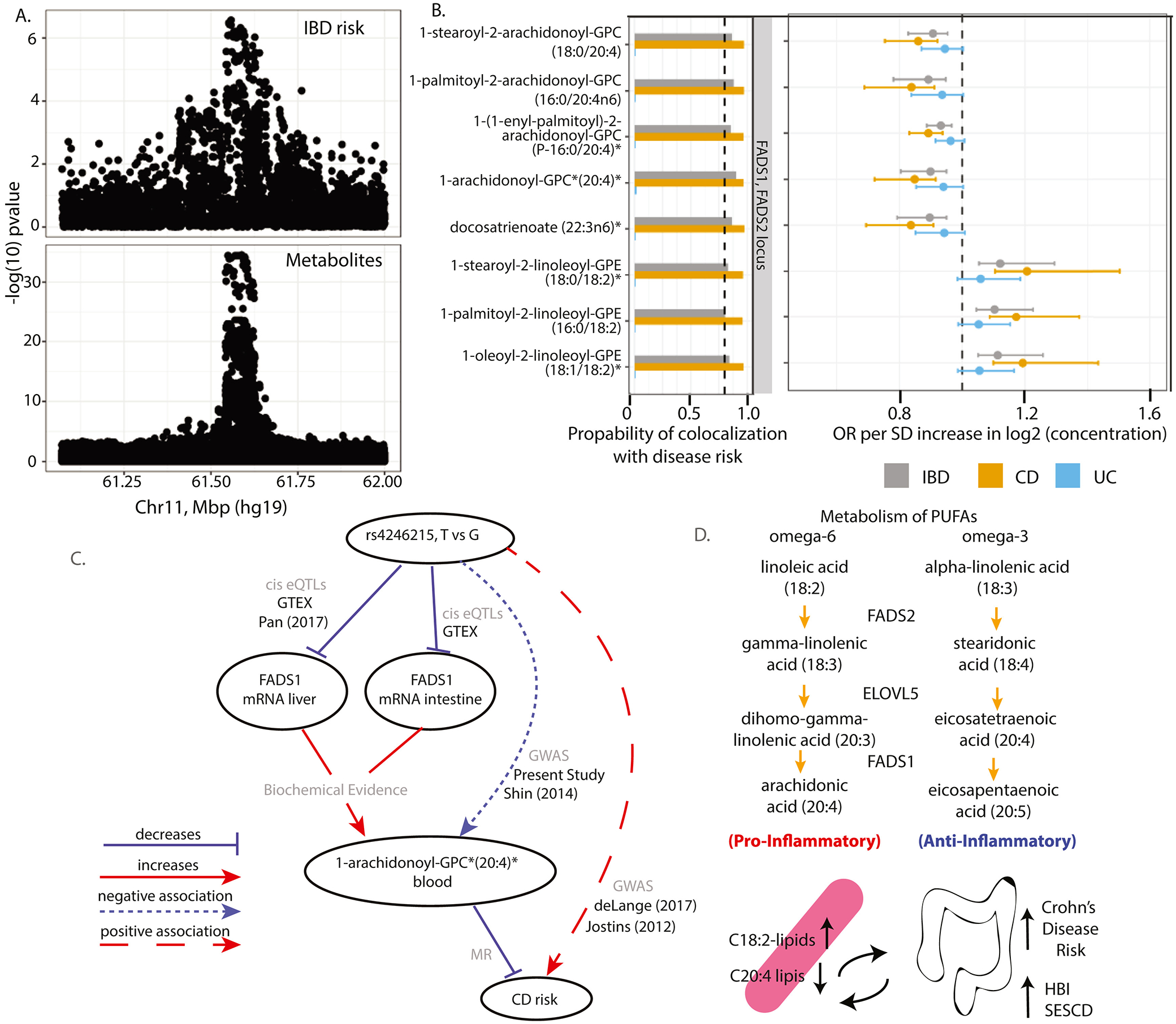
A) Regional association plots of the genetic associations at the Chr11 position which contains the FADS locus indicate co-localization of IBD risk and levels of serum metabolites (ids 33228, 36600, 42449, 42450, 52446, 52462, 52603, 52687, 52689, 54960, 54961, 57450, and 57467). Genome-wide significance threshold: 5E-8/1300=3.8E-11. B) Graph of the results of genetic colocalization analysis, testing for a shared genetic locus controlling serum levels of various omega-6 containing PUFAs with risk of IBD, CD or UC. Also, plotted are the results of the MR analysis including the OR and Fieller’s confidence intervals. Metabolites on the vertical axis, estimates on the horizontal axis, genetic regions in vertical stripes. See Figure Legend 3 for details. C) A schema showing putative causal associations (solid lines) and statistical associations (dashed line) with edges labeled according to the data sources. Evidence for the association between genetic variation and mRNA transcripts was gathered from published eQTL studies or GTEx database searches. D) Biochemical schema of the metabolism of omega-6 and omega-3 PUFAs with the position of FADS1 and FADS2. The bottom image summarizes results of MR which predicts that elevated serum levels of the precursor omega-6 PUFA increase risk of CD, whereas decreased levels of the PUFA end-products are protective.
The lead SNP rs4246215, which in our study associated with serum omega-6 PUFAs, is in linkage disequilibrium with a previously reported FADS1 causal SNP (rs174557-R2=0.68). Pan et al24 showed that the association is caused by an eQTL with the variant allele decreasing FADS1 expression in major lipogenic tissues such as the liver which is a key tissue for the synthesis and metabolism of PUFAs19. The Genotype-Tissue Expression (GTEx) project database also supports a negative association of rs4246215/rs174557 with FADS1 expression in liver and intestine tissues. Our work is thus consistent with these studies, as we observe a negative association between the rs4246215 and serum arachidonoyl-containing lipids, which is reflective of decreased FADS1 activity and therefore decreased conversion of linoleic acid to AA (Figure 4C, D).
A novel causal association of serum plasmalogen levels and the candidate causal gene TMEM229B with CD risk
We report a high posterior probability of colocalization on a region of Chr14 between CD risk and the serum concentration of the plasmalogen 1-(1-enyl-palmitoyl)-2-palmitoyl-GPC (P-16:0/16:0)* (posterior probability of a shared genetic causal variant between CD and serum metabolite levels=98.8%). We estimated a positive causal effect of this metabolite on CD risk (OR=1.24, 95% CI: [1.11–1.64]) (Figure 5A–B). The implicated Chr14 region contains two genes, PLEKHH1 and TMEM229B. Although PLEKHH1 was implicated previously as associated with circulating phospholipid concentrations25, no validation of the true candidate gene in this region has been reported. We found that TMEM229B has a domain which encodes for a putative ABC-transporter type IV. Our Bayesian network model revealed the plasmanylethanolamine desaturase26 (TMEM189) as a neighboring node to TMEM229B (Figure 5C). We also observed a negative association of the rs11158671 CD risk variant with mRNA abundance levels in whole blood GTEx database (Figure 5D).
Figure 5. TMEM229B expression and choline plasmalogens as candidate mediator of CD risk.
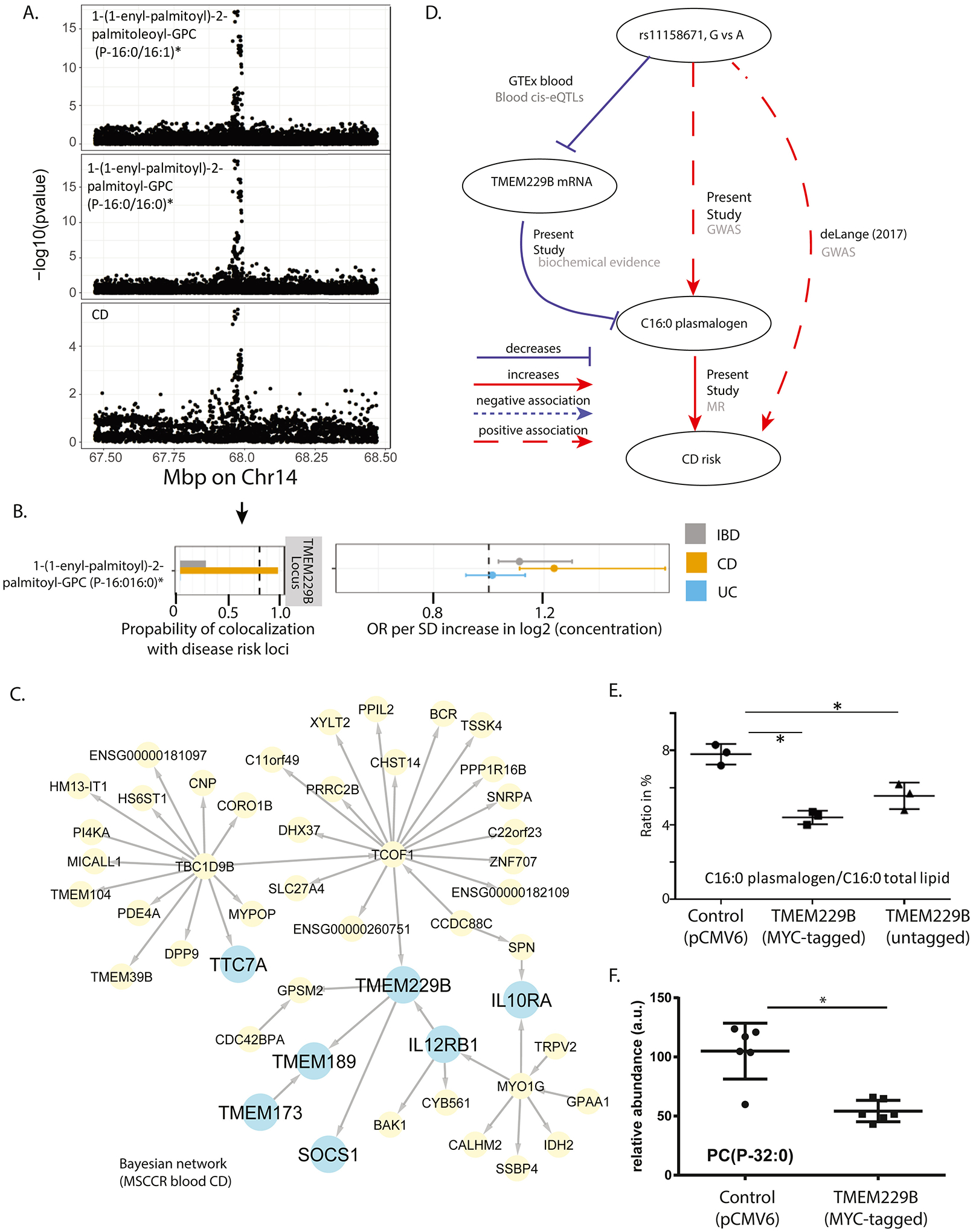
A) Regional association plots of genetic associations at the Chr14 position which contains TMEM229B, with CD risk and levels of serum C16:0 plasmalogen levels. Genome-wide significance threshold: 5E-8/1300=3.8E-11. B) Results of genetic colocalization and MR analysis between CD risk loci and the plasmalogen serum concentration on Chr14. Metabolites on the vertical axis, estimates on the horizontal axis, genetic regions in vertical stripes. See Figure 3 legend for details. C) Bayesian gene regulatory subnetwork built from the blood RNA sequencing data and genotype information on MSCCR CD patients. Arrows indicate predicted causal relationships between genes. Blue colored nodes indicate genes of interest used in discerning TMEM229B’s function or possible role in IBD pathology. Yellow nodes indicate genes belonging to the gene regulatory subnetwork. D) A summary of the evidence on the role of TMEM229B and the plasmalogen on CD risk. Solid lines represent putative causal associations; dashed lines represent statistical associations; edges are further labeled with their data source. E-F) MYC-tagged and non-tagged TMEM229B were overexpressed by transfection into HEK293 cells and cell pellets were collected and subjected to various lipid analyses (ST10). Plotted are the ratio of C16:0 in plasmalogen versus C16:0 in total lipid in E, and relative abundance of the plasmalogen PC(P-32:0) in F (*p<0.05).
We prioritized TMEM229B as the candidate gene for validation. Given low expression in HEK293 cells, we overexpressed tagged and untagged TMEM229B (SF9) and measured total FAs as FA methyl esters. TMEM229B overexpression significantly decreased the ratio of plasmalogen-derived C16 and C18 over C16 and C18 derived from esterified lipids (Figure 5E, SF9). The change in this ratio was caused by decreased levels of plasmalogen-derived C16 and C18, supporting that TMEM229B expression anti-correlates with plasmalogen levels. Additional lipidomic analysis of TMEM229B overexpressing cells (ST10), revealed a prominent decrease in plasmenyl PC species such as 1-(1-enyl-palmitoyl)-2-palmitoyl-GPC (P-16:0/16:0) (Figure 5E).
Discussion
In this study we performed a comprehensive unbiased metabolomics survey in serum and stool samples from a large cohort of UC and CD patients and HCs. Through this survey we identified a variety of metabolites whose abundance was significantly different between cases and controls, IBD subtypes, and that associated with disease activity indices, location and behavior. These data are important as a resource to guide biomarker discovery and for future investigations into the role of metabolism in IBD pathology. Subsequent integration of metabolite abundance in our cohort with previously reported IBD risk loci enabled evaluating if these loci shape disease susceptibility or course via changes in specific metabolites. This analysis revealed four novel insights. 1) Genetically controlled levels of serum butyrate levels do not associate with IBD status; 2) serum linoleoyl-containing lipids are predicted to confer risk for CD; 3) serum arachidonoyl-containing lipids are predicted protective for CD; and 4) increased serum choline plasmalogen levels are predicted to confer risk for CD. Our study highlights the importance of an integrative multi-omic analysis that combines all data to uncover novel insight into the genetic and metabolic factors that cause IBD.
Our study reports numerous novel metabolite:IBD trait associations which we present as a resource with easy to search data tables, as well as a discussion of selected results to emphasize utility. For example, we replicated an overabundance of sphingomyelin levels in the stool of IBD patients relative to controls and a positive association of these sphingomyelins with severity of gut inflammation (i.e. Mayo endo and SESCD scores)27,28, which was explained previously by a dysregulated symbiosis27. Sphingolipid metabolism and IBD association was further emphasized by the association of higher serum S1P levels in CD patients with ileal involvement compared to colonic only disease. These are timely findings as S1P/S1PRs axes are being considered as new therapeutics for targeting aberrant leukocyte migration in IBD29. In our study, serum cleavage products of Fibrinogen A/B, (FPA/B) were found to distinguish CD from UC, elevated in CD versus controls and associated with gut inflammation, data consistent with a previous proteomics study30,31. FPA/B are fibrinogen peptides released by thrombin that initiates fibrin polymerization, as well as potent chemoattractants34. One of our study’s novel loci controlling FPA/B levels was within an IBD GWAS locus, with calpastatin (CAST) as the likely candidate causal gene, however we did not find serum FPA levels to causally associate with IBD risk (Figure S10). However, we suggest that thrombotic events in IBD patients may be the more appropriate endpoint to test than case versus control status.
Correcting gut dysbiosis is considered important for re-equilibrating inflammatory responses in IBD16,32. Clinical remission in anti-TNF responders was associated with restoring intestinal microbial diversity to that of HCs, including phylotypes that are known SCFA producers33,34. Microbiome disturbances are thought to alter metabolite exchanges across bacteria. When compared to responders, anti-TNF non-responders have less frequent exchange of butyrate and substrates involved in butyrate synthesis among bacterial communities. Mechanistically, butyrate has many biological consequences on the gut, including anti-inflammatory effects through histone deacetylase inhibition, modulation of aryl hydrocarbon receptors and G protein coupled receptor activation16. SCAD, is an ubiquitously expressed enzyme involved in butyryl-CoA oxidation and thus a regulator of cellular butyrate levels in many organ systems including the intestine. Indeed, differentiated colonocytes, located at the tops of the crypts, were shown to preferentially oxidize butyrate in order to shield stem/progenitor cells in the stem cell niche from high levels of luminal butyrate, which suppresses their proliferation35. Acads−/− mouse colonocytes showed reduced butyrate oxidation and epithelial proliferation was significantly less in the proliferation zone in the crypt compared to wildtype mice, an effect exaggerated with DSS injury. Butyrate mediated these effects in part through modulating histone acetylation35. The identification of genetic control of butyrate levels placed us in a position to segregate the cohort by ACADS genotype and IBD status and determine if genetically altered levels of serum butyrate impacted disease course. We specifically hypothesized that IBD patients with genetically elevated butyrate would have negative associations with disease risk or activity indices. Surprisingly though, we found no evidence of association or causal connection between serum (or stool) butyrate levels with IBD status or activity (ST2). Thus the cause-effect debate remains. Of note, however, other serum and stool SCFAs were found negatively associated with various IBD traits consistent the notion that IBD is associated with decreased numbers of SCFAs-producing bacteria and a lower amount of fecal SCFAs32,36. As ACADS variants are rather frequent we suggest genetic screening of patients undergoing therapies altering butyrate levels may be important especially in light of the reported inconsistent clinical effects of butyrate treatment in IBD patients37.
Our study replicates the association of omega-6 PUFA containing lipids with the FADS1 locus. For the first time, however, we applied colocalization/MR methods to estimate a potential causal effect. Indeed, we reveal a causal role of omega-6 FA containing lipids on CD, but not UC risk. Serum levels of AA containing lipids, which can be synthesized from the essential omega-6 FA linoleic acid in a pathway that involves FADS1 and FADS2 (Figure 4D), were negatively associated with CD risk, while the linoleic acid containing lipids were positively associated with CD risk. Consistent with published work24, our data suggest that rs4246215 lowers FADS1 mRNA/activity in liver, intestine and other tissues thereby reducing the conversion of linoleic acid into AA leading to the lower serum AA-lipids. Implicating omega-6 PUFAs in CD risk, is in part consistent with literature, showing that children who consumed a higher dietary ratio of omega-6/omega-3 were susceptible to CD, if they were also carriers of mutations in CYP4F3 and FADS238. As both omega-3 and −6 PUFAs are substrates of FADS1/2, altering the omega6/omega3 ratio might affect their metabolism through competition, an effect expected to be even more prominent with limiting FADS1 activity. The negative association of omega-6 products such as AA and positive association of the omega-6 precursor with CD risk is perhaps surprising given that AA can be metabolized into pro-inflammatory prostaglandins, thromboxanes or leukotrienes. This may suggest that the plasma FA composition does not parallel the FA changes in the IBD intestinal mucosa39,40. Indeed, an inverse relationship between plasma and mucosal FAs have been observed previously in IBD patients, with decreased plasma omega-6 and increased omega-6 PUFA end products in mucosa of IBD patients40. Alternatively, prostaglandin E2 (PGE2) supplementation could rescue loss of the mucus layer due to chemical injury and prevent epithelial barrier disruption41 highlighting that not all omega-6 end products are necessarily pathogenic in IBD. Why we did not observe a genetic association with UC patients or omega-3 PUFAs is unclear, however, patient heterogeneity may have diminished signal detection.
Lastly, we made the novel observation that variation on a Chr14 locus, which associated with serum plasmalogen 1-(1-enyl-palmitoyl)-2-palmitoyl-GPC (P-16:0/16:0)* levels, shows evidence of colocalization with CD risk. Through integration of genetic, molecular and clinical data, we observed that carriers of the rs11158671 CD-disease allele have higher levels of this plasmalogen compared to non-carriers. At the same time, however, within each genotype group, CD patients have lower levels of this metabolite. While similar observations of reduced serum plasmalogens were reported in CD versus non-IBD controls (independent of genotype effects) in Fan et al42, our study points out potentially missed complexity when genetics is not interpreted alongside metabolite and disease expression. Interestingly, variation at this locus has been previously reported to associate with various subspecies of ether phospholipids (i.e. plasmalogens), however, the proposed candidate gene, TMEM229B, has not been validated43. Our network analysis also implicated TMEM229B in IBD pathology, given that it is in close association with IL12RB1, IL10RA, SOCS1, TMEM137 and TTC7A, which are known to be involved in IBD pathogenesis44–47. We therefore evaluated overexpression of TMEM229B in HEK293 cells and observed decreased plasmalogens, supporting that TMEM229B inversely regulates plasmalogen levels. We hypothesize TMEM229B may regulate subcellular localization or transport of plasmalogens thus impacting their synthesis or degradation. Why the pool of plasmalogen levels are lower in CD cases compared to HCs is unknown. Secondary plasmalogen deficiency has been demonstrated in various common diseases, such as Alzheimer’s disease, inflammatory conditions and respiratory disorders, due to various roles of plasmalogens from affecting membrane properties; serving as reservoirs for lipid mediators (e.g. AA) and serving as sacrificial oxidants to help terminate lipid oxidation48. The latter function is particularly relevant as IBD is associated with an increase in oxidative stress42, such that the lower serum plasmalogen levels in CD patients may be due to their enhanced oxidative decomposition.
A limitation of our study was the inability to sample multiple time points within the patient’s disease course as well as the patients’ diet, which could affect metabolite levels in addition to their disease status. The effect of diet however, may have been limited as we measured serum metabolomes on endoscopy and as such, patients would have been instructed to undergo a bowel preparation that includes fasting. Also, we were unable to generate an independent IBD cohort with similar metabolomics surveys, genotyping and disease phenotyping in order to replicate our findings. Our results are thus exploratory in nature. However, our mQTL analysis replicated many previous associations and we discuss literature or provide experimental validation wherever possible to augment our study. Finally, we aimed to control for potential confounders by including in our statistical analysis only patients without bowel surgeries with smoking, length of storage of sample type and assay batch effect as covariates. We could however, not account for potential medication effects as this information was unavailable on controls.
In summary, through largescale metabolomics of serum and stool from IBD patients and HCs, we have provided novel insights into how an individual’s metabolite profile is reshaped according to genotype, IBD status and disease activity. This knowledge provides potential understanding to IBD pathology as well as novel biomarkers and emphasizes the value of integrative multi-omic studies.
Supplementary Material
Table S1. Medications use
Table S2. Differential Metabolites
Table S3. DMs – Pathways Enrichment Analysis
Table S4. DRs – Differential metabolites Ratios
Table S5. DMs – IBD Disease activity subscores and phenotype
Table S6. DMs –IBD Disease activity subscores and phenotype – Pathways Enrichment Analysis
Table S7. Metabolome GWAS
Table S8. Colocalization analysis
Table S9. Integrative Analysis.
Table S10. Lipidomics analysis of cell lysates from TMEM229B overexpressing cells
Table S11. Serum untargeted metabolites detected
Table S12. Stool untargeted metabolites detected
Table S13. DMs – Storage Duration
Figure S1. Differential Metabolome Abundance Analysis associated with disease activity subscores – volcano plots. Volcano plots showing the differential metabolome abundance analysis (DMA) of the serum and stool samples, according to patient reported outcomes from the colitis activity index (CAI). Log2-fold changes in the horizontal axis, nominal p values on the vertical axis. The 10% FDR threshold is indicated (dashed blue line) and the total number of significantly differentially expressed metabolites (DMs at <10% FDR) within each panel.
Figure S2. Differential Serum Metabolome Abundance Analysis associated with disease activity subscores and Montreal Classifications– Pathways Enrichment Analysis. A heatmap summarizing the enrichments of the serum DMAs according to metabolite classifications. Only pathways that were significantly enriched at FDR <0.05 for at least one endpoint are shown.
Figure S3. Differential Stool Metabolome Abundance Analysis associated with disease activity subscores and Montreal Classifications – Pathways Enrichment Analysis. A heatmap summarizing the enrichments of the stool DMAs according to metabolite classifications. Only pathways that were significantly enriched at FDR <0.05 for at least one endpoint are shown.
Figure S4. Differential Metabolome Abundance Analysis associated with Montreal subclassifications– volcano plots. Volcano plots showing the differential metabolome abundance analysis (DMA) of the serum and stool samples, according to CD behavior and location subclassifications. Log2-fold changes in the horizontal axis, nominal p values on the vertical axis. The 10% FDR threshold is indicated (dashed blue line) and the total number of significantly differentially expressed metabolites (DMs at <10% FDR) within each panel.
Figure S5. Serum levels of sphingosine metabolism and relationship to CD disease location. A. Box plots showing the serum concentration levels (Signal-to-noise ratio) of sphingosine and sphingosine-1-phosphate according to control, CD and UC diagnosis (upper panels) or by disease location (L1, L2 or L3, lower panels). Nominal p values are indicated in the figure. B. Metabolic schema related to metabolism of sphingosine-1-phosphate, in particular the pathways that can contribute to the pool of ceramide, a precursor of sphingosine. The reactions targeted by three genes, SGPP1 (sphingosine-1-phosphate phosphatase 1), SGPL1 (sphingosine-1-phosphate lyase 1) and SPHK1 (sphingosine kinase 1) are shown that control the generation and irreversible breakdown of sphingosine-1-phosphate. The levels of metabolites labeled in red were also found significantly higher (at adjusted p value <0.10) in L3 vs L2 CD patient serum comparisons. C. A table summarizing the expression levels of 5 known sphingosine 1 phosphate receptors (S1PR1–5) as median transcripts per million (TPM) values in the terminal ileum, transverse colon and sigmoid colon samples from the GTEX database. Cells in table are colored deeper red to indicate where expression is higher.
Figure S6. Differential Metabolome Abundance Analysis associated with subcomponents of the Harvey Bradshaw index (HBI) and indicators of extent of UC Volcano plots showing the differential metabolome abundance analysis (DMA) of the serum and stool samples, according to HBI sub-components as well as UC disease extent indicators (E1, E2 or E3). Log2-fold changes in the horizontal axis, nominal p values on the vertical axis. The 10% FDR threshold is indicated (dashed blue line) and the total number of significantly differentially expressed metabolites (DMs at <10% FDR) within each panel.
Figure S7. Differential Metabolome Abundance Analysis associated with Disease duration (in years) Volcano plots showing the differential metabolome abundance analysis (DMA) of the serum and stool samples, according duration of disease in years of UC or CD. Log2-fold changes in the horizontal axis, nominal p values on the vertical axis. The 10% FDR threshold is indicated (dashed blue line) and the total number of significantly differentially expressed metabolites (DMs at <10% FDR) within each panel.
Figure S8. Metabolome GWAS results. Manhattan plot displaying genome-wide results, separately by metabolite classification (rows). Genome-Wide significance threshold: 5E-8/1300=3.8E-11. Only chromosomes with at least 1 genome-wide significant hit are displayed. Genome-wide significant hits are distributed across 60 non-overlapping regions at least 1Mbp wide.
Figure S9. Western blot demonstrating TMEM229B overexpression in HEK293 cells. HEK293 cells were transfected with plasmids encoding TMEM229B (with or without C-terminal Myc-DDK-tag). A. Western blot analysis of cell lysates with anti-DDK antibody shows a prominent band at ~20kDa, the expected size of full-length TMEM229B. The positions of the molecular weight markers (Mw) are indicated in kDa. Bands are not present in lanes A or B, supporting the specificity of the antibody. These data support successful overexpression of TMEM229B in the HEK293 cells. B. Cell pellets were collected and subjected to phospholipid analyses. Plotted are the ratios of plasmalogen (as C18:0) to total lipid (as C18:0) levels (*p<0.05).
Figure S10. GWAS results for serum fibrinopeptide A and IBD risk loci colocalization. Manhattan plots displaying the genome-wide results, of the association of genetic markers at the Chr5 position which contains the CAST1/ERAP1 genes, calpastatin or endoplasmic reticulum aminopeptidase 1 with IBD risk and levels of serum peptides. Genome-Wide significance threshold: 5E-8/1300=3.8E-11.
Significance.
This is a comprehensive survey of metabolite abundance and their association with IBD. We inform on disease pathology and potential biomarkers of disease activity through 1) performing an unbiased metabolomics survey of serum and stool in a large-scale IBD and control cohort and 2) integrating the patient’s metabolome abundance with their clinical and endoscopic disease activity measures and genotype information to enable multi-scale data interpretation.
Competing interests:
Mount Sinai co-authors (from Genetics and Genomics, Icahn Institute for Data Science and Genomic Technology, Population Health Science and Policy, Division of Gastroenterology, Pediatric GI and Hepatology, Susan and Leonard Feinstein IBD Clinical Center at Icahn School of Medicine at Mount Sinai) were partially funded as part of research alliance between Janssen Biotech and The Icahn School of Medicine at Mount Sinai. RCU has served as an advisory board member or consultant for Eli Lilly, Janssen, Pfizer and Takeda. RCU is supported by an NIH K23 Career Development Award (K23KD111995-01A1). MC, AS, AH, JP and CB are employees at Janssen Research and Development. JRF is a former employee at Janssen Research and Development and is currently employed at Alnylam Pharmaceuticals. MD is a consultant for Janssen. BES discloses consulting fees from 4D Pharma, Abbvie, Allergan, Amgen, Arena Pharmaceuticals, AstraZeneca, BoehringerIngelheim, Boston Pharmaceuticals, Capella Biosciences, Celgene, Celltrion Healthcare, EnGene, Ferring, Genentech, Gilead, Hoffmann-La Roche, Immunic, Ironwood Pharmaceuticals, Janssen, Lilly, Lyndra, MedImmune, Morphic Therapeutic, Oppilan Pharma, OSE Immunotherapeutics, Otsuka, Palatin Technologies, Pfizer, Progenity, Prometheus Laboratories, Redhill Biopharma, Rheos Medicines, Seres Therapeutics, Shire, Synergy Pharmaceuticals, Takeda, Target PharmaSolutions, Theravance Biopharma R&D, TiGenix, Vivelix Pharmaceuticals; honoraria for speaking in CME programs from Takeda, Janssen, Lilly, Gilead, Pfizer, Genetech; research funding from Celgene, Pfizer, Takeda, Theravance Biopharma R&D, Janssen. MCD discloses consulting fees from Abbvie, Allergan, Amgen, Arena Pharmaceuticals, AstraZeneca, BoehringerIngelheim, Celgene, Ferring, Genentech, Gilead, Hoffmann-La Roche, Janssen, Pfizer, Prometheus Biosciences, Takeda, Target PharmaSolutions and research funding from Abbvie, Janssen, Pfizer, Prometheus Biosciences Takeda. Dr. JF Colombel reports receiving research grants from AbbVie, Janssen Pharmaceuticals and Takeda; receiving payment for lectures from AbbVie, Amgen, Allergan, Inc. Ferring Pharmaceuticals, Shire, and Takeda; receiving consulting fees from AbbVie, Amgen, Arena Pharmaceuticals, Boehringer Ingelheim, Celgene Corporation, Celltrion, Eli Lilly, Enterome, Ferring Pharmaceuticals, Geneva, Genentech, Janssen Pharmaceuticals, Landos, Ipsen, Imedex, Medimmune, Merck, Novartis, O Mass, Otsuka, Pfizer, Shire, Takeda, Tigenix, Viela bio; and holds stock options in Intestinal Biotech Development and Genfit. ES, KH, AD, JZ and AK are associated with Sema4. MA receives research support from the Dickler Family Fund, New York Community Trust and the Leona M. and Harry B. Helmsley Charitable Trust Fund for Surveillance of Coronavirus Under Research Exclusion for IBD (SECURE-IBD). CA and ES receive research support from Leona M. and Harry B. Helmsley Charitable Trust and CA was recipient of a Litwin IBD Pioneer’s Award from Crohn’s and Colitis Foundation.
Abbreviations:
- AA
arachidonic acid
- CD
Crohn’s disease
- UC
ulcerative colitis
- HCs
healthy controls
- GWAS
genome-wide association study
- DMA
differential metabolite abundance analysis
- DRA
differential metabolite ratios analysis
- mQTLs
metabolite quantitative trait locus
- GIVs
genetic instrumental variables
References
- 1.Ng SC et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 390, 2769–2778 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Graham DB & Xavier RJ Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 578, 527–539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee HS & Cleynen I Molecular Profiling of Inflammatory Bowel Disease: Is It Ready for Use in Clinical Decision-Making? Cells 8(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seyed Tabib NS et al. Big data in IBD: big progress for clinical practice. Gut 69, 1520–1532 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lloyd-Price J et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Storr M, Vogel HJ & Schicho R Metabolomics: is it useful for inflammatory bowel diseases? Curr Opin Gastroenterol 29, 378–83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scoville EA et al. Alterations in Lipid, Amino Acid, and Energy Metabolism Distinguish Crohn’s Disease from Ulcerative Colitis and Control Subjects by Serum Metabolomic Profiling. Metabolomics 14, 17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Contijoch EJ et al. Gut microbiota density influences host physiology and is shaped by host and microbial factors. Elife 8(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ford L et al. Precision of a Clinical Metabolomics Profiling Platform for Use in the Identification of Inborn Errors of Metabolism. J Appl Lab Med 5, 342–356 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Evans AM et al. High Resolution Mass Spectrometry Improves Data Quantity and Quality as Compared to Unit Mass Resolution Mass Spectrometry in High- Throughput Profiling Metabolomics. Metabolomics 4, 1–3 (2014). [Google Scholar]
- 11.de Lange KM, Moutsianas L, Lee JC. et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nature Genetics 49, 256–261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giambartolomei C et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palmer TM et al. Instrumental variable estimation of causal risk ratios and causal odds ratios in Mendelian randomization analyses. Am J Epidemiol 173, 1392–403 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Dunlap WP & Silver NC Confidence intervals and standard errors for ratios of normal variables. Behavior Research Methods, Instruments, & Computers 18, 469–471 (1986). [Google Scholar]
- 15.Yavorska OO & Burgess S MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol 46, 1734–1739 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deleu S, Machiels K, Raes J, Verbeke K & Vermeire S Short chain fatty acids and its producing organisms: An overlooked therapy for IBD? EBioMedicine 66, 103293 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gieger C et al. Genetics meets metabolomics: a genome-wide association study of metabolite profiles in human serum. PLoS Genet 4, e1000282 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagenbeek FA et al. Heritability estimates for 361 blood metabolites across 40 genome-wide association studies. Nat Commun 11, 39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin SY, Fauman EB, Petersen A, Krumsiek J et al. An atlas of genetic influences on human blood metabolites. Nat Genet 46, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schlosser P, Li Y, Sekula P, et al. Genetic studies of urinary metabolites illuminate mechanisms of detoxification and excretion in humans. Nat Genet 52, 167–176 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Long T et al. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat Genet 49, 568–578 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Jostins L et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Didelez V & Sheehan N Mendelian randomization as an instrumental variable approach to causal inference. Statistical methods in medical research 16, 309–330 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Pan G et al. PATZ1 down-regulates FADS1 by binding to rs174557 and is opposed by SP1/SREBP1c. Nucleic Acids Res 45, 2408–2422 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demirkan A et al. Genome-wide association study identifies novel loci associated with circulating phospho- and sphingolipid concentrations. PLoS Genet 8, e1002490 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Werner ER et al. The TMEM189 gene encodes plasmanylethanolamine desaturase which introduces the characteristic vinyl ether double bond into plasmalogens. Proc Natl Acad Sci U S A 117, 7792–7798 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown EM et al. Bacteroides-Derived Sphingolipids Are Critical for Maintaining Intestinal Homeostasis and Symbiosis. Cell Host Microbe 25, 668–680 e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franzosa EA, Sirota-Madi A, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol 4, 293–305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Danese S, Furfaro F & Vetrano S Targeting S1P in Inflammatory Bowel Disease: New Avenues for Modulating Intestinal Leukocyte Migration. J Crohns Colitis 12, S678–S686 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Bennike T, Birkelund S, Stensballe A & Andersen V Biomarkers in inflammatory bowel diseases: current status and proteomics identification strategies. World J Gastroenterol 20, 3231–44 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meuwis MA, Fillet M, et al. Biomarker discovery for inflammatory bowel disease, using proteomic serum profiling. Biochem Pharmacol 73, 1422–33 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Parada Venegas D et al. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol 10, 277 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aden K, Rehman A, Waschina S et al. Metabolic Functions of Gut Microbes Associate With Efficacy of Tumor Necrosis Factor Antagonists in Patients With Inflammatory Bowel Diseases. Gastroenterology 157, 1279–1292 e11 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Chen J & Vitetta L Butyrate in Inflammatory Bowel Disease Therapy. Gastroenterology 158, 1511 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Kaiko GE, Ryu SH et al. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 165, 1708–1720 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huda-Faujan N et al. The impact of the level of the intestinal short chain Fatty acids in inflammatory bowel disease patients versus healthy subjects. Open Biochem J 4, 53–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vancamelbeke M et al. Butyrate Does Not Protect Against Inflammation-induced Loss of Epithelial Barrier Function and Cytokine Production in Primary Cell Monolayers From Patients With Ulcerative Colitis. J Crohns Colitis 13, 1351–1361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Costea I et al. Interactions between the dietary polyunsaturated fatty acid ratio and genetic factors determine susceptibility to pediatric Crohn’s disease. Gastroenterology 146, 929–31 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Shores DR, Binion DG, Freeman BA & Baker PR New insights into the role of fatty acids in the pathogenesis and resolution of inflammatory bowel disease. Inflamm Bowel Dis 17, 2192–204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esteve-Comas M et al. Plasma polyunsaturated fatty acid pattern in active inflammatory bowel disease. Gut 33, 1365–9 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chuang LS et al. Zebrafish modeling of intestinal injury, bacterial exposures and medications defines epithelial in vivo responses relevant to human inflammatory bowel disease. Dis Model Mech 12(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan F et al. Lipidomic Profiling in Inflammatory Bowel Disease: Comparison Between Ulcerative Colitis and Crohn’s Disease. Inflamm Bowel Dis 21, 1511–8 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Demirkan A, Pool R, Deelen J et al. Genome-wide association study of plasma triglycerides, phospholipids and relation to cardio-metabolic risk factors. bioRxiv, 621334 (2019). [Google Scholar]
- 44.Magyari L et al. Interleukin and interleukin receptor gene polymorphisms in inflammatory bowel diseases susceptibility. World J Gastroenterol 20, 3208–22 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Avitzur Y, Guo C et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology 146, 1028–39 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shouval DS, Kowalik M & Snapper SB The Treatment of Inflammatory Bowel Disease in Patients with Selected Primary Immunodeficiencies. J Clin Immunol 38, 579–588 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Martin GR, Blomquist CM, Henare KL & Jirik FR Stimulator of interferon genes (STING) activation exacerbates experimental colitis in mice. Sci Rep 9, 14281 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Braverman NE & Moser AB Functions of plasmalogen lipids in health and disease. Biochim Biophys Acta 1822, 1442–52 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Medications use
Table S2. Differential Metabolites
Table S3. DMs – Pathways Enrichment Analysis
Table S4. DRs – Differential metabolites Ratios
Table S5. DMs – IBD Disease activity subscores and phenotype
Table S6. DMs –IBD Disease activity subscores and phenotype – Pathways Enrichment Analysis
Table S7. Metabolome GWAS
Table S8. Colocalization analysis
Table S9. Integrative Analysis.
Table S10. Lipidomics analysis of cell lysates from TMEM229B overexpressing cells
Table S11. Serum untargeted metabolites detected
Table S12. Stool untargeted metabolites detected
Table S13. DMs – Storage Duration
Figure S1. Differential Metabolome Abundance Analysis associated with disease activity subscores – volcano plots. Volcano plots showing the differential metabolome abundance analysis (DMA) of the serum and stool samples, according to patient reported outcomes from the colitis activity index (CAI). Log2-fold changes in the horizontal axis, nominal p values on the vertical axis. The 10% FDR threshold is indicated (dashed blue line) and the total number of significantly differentially expressed metabolites (DMs at <10% FDR) within each panel.
Figure S2. Differential Serum Metabolome Abundance Analysis associated with disease activity subscores and Montreal Classifications– Pathways Enrichment Analysis. A heatmap summarizing the enrichments of the serum DMAs according to metabolite classifications. Only pathways that were significantly enriched at FDR <0.05 for at least one endpoint are shown.
Figure S3. Differential Stool Metabolome Abundance Analysis associated with disease activity subscores and Montreal Classifications – Pathways Enrichment Analysis. A heatmap summarizing the enrichments of the stool DMAs according to metabolite classifications. Only pathways that were significantly enriched at FDR <0.05 for at least one endpoint are shown.
Figure S4. Differential Metabolome Abundance Analysis associated with Montreal subclassifications– volcano plots. Volcano plots showing the differential metabolome abundance analysis (DMA) of the serum and stool samples, according to CD behavior and location subclassifications. Log2-fold changes in the horizontal axis, nominal p values on the vertical axis. The 10% FDR threshold is indicated (dashed blue line) and the total number of significantly differentially expressed metabolites (DMs at <10% FDR) within each panel.
Figure S5. Serum levels of sphingosine metabolism and relationship to CD disease location. A. Box plots showing the serum concentration levels (Signal-to-noise ratio) of sphingosine and sphingosine-1-phosphate according to control, CD and UC diagnosis (upper panels) or by disease location (L1, L2 or L3, lower panels). Nominal p values are indicated in the figure. B. Metabolic schema related to metabolism of sphingosine-1-phosphate, in particular the pathways that can contribute to the pool of ceramide, a precursor of sphingosine. The reactions targeted by three genes, SGPP1 (sphingosine-1-phosphate phosphatase 1), SGPL1 (sphingosine-1-phosphate lyase 1) and SPHK1 (sphingosine kinase 1) are shown that control the generation and irreversible breakdown of sphingosine-1-phosphate. The levels of metabolites labeled in red were also found significantly higher (at adjusted p value <0.10) in L3 vs L2 CD patient serum comparisons. C. A table summarizing the expression levels of 5 known sphingosine 1 phosphate receptors (S1PR1–5) as median transcripts per million (TPM) values in the terminal ileum, transverse colon and sigmoid colon samples from the GTEX database. Cells in table are colored deeper red to indicate where expression is higher.
Figure S6. Differential Metabolome Abundance Analysis associated with subcomponents of the Harvey Bradshaw index (HBI) and indicators of extent of UC Volcano plots showing the differential metabolome abundance analysis (DMA) of the serum and stool samples, according to HBI sub-components as well as UC disease extent indicators (E1, E2 or E3). Log2-fold changes in the horizontal axis, nominal p values on the vertical axis. The 10% FDR threshold is indicated (dashed blue line) and the total number of significantly differentially expressed metabolites (DMs at <10% FDR) within each panel.
Figure S7. Differential Metabolome Abundance Analysis associated with Disease duration (in years) Volcano plots showing the differential metabolome abundance analysis (DMA) of the serum and stool samples, according duration of disease in years of UC or CD. Log2-fold changes in the horizontal axis, nominal p values on the vertical axis. The 10% FDR threshold is indicated (dashed blue line) and the total number of significantly differentially expressed metabolites (DMs at <10% FDR) within each panel.
Figure S8. Metabolome GWAS results. Manhattan plot displaying genome-wide results, separately by metabolite classification (rows). Genome-Wide significance threshold: 5E-8/1300=3.8E-11. Only chromosomes with at least 1 genome-wide significant hit are displayed. Genome-wide significant hits are distributed across 60 non-overlapping regions at least 1Mbp wide.
Figure S9. Western blot demonstrating TMEM229B overexpression in HEK293 cells. HEK293 cells were transfected with plasmids encoding TMEM229B (with or without C-terminal Myc-DDK-tag). A. Western blot analysis of cell lysates with anti-DDK antibody shows a prominent band at ~20kDa, the expected size of full-length TMEM229B. The positions of the molecular weight markers (Mw) are indicated in kDa. Bands are not present in lanes A or B, supporting the specificity of the antibody. These data support successful overexpression of TMEM229B in the HEK293 cells. B. Cell pellets were collected and subjected to phospholipid analyses. Plotted are the ratios of plasmalogen (as C18:0) to total lipid (as C18:0) levels (*p<0.05).
Figure S10. GWAS results for serum fibrinopeptide A and IBD risk loci colocalization. Manhattan plots displaying the genome-wide results, of the association of genetic markers at the Chr5 position which contains the CAST1/ERAP1 genes, calpastatin or endoplasmic reticulum aminopeptidase 1 with IBD risk and levels of serum peptides. Genome-Wide significance threshold: 5E-8/1300=3.8E-11.