

Abstract
Despite the evidences of elevated expression of Mer tyrosine kinase (MerTK) in multiple human cancers, mechanisms underlying the oncogenic roles of MerTK in hepatocellular carcinoma (HCC) remains undefined. We explored the functional effects of MerTK and N-Glycosylated MerTK on HCC cell survival and tumor growth. Here, we show that MerTK ablation increases reactive oxygen species (ROS) production and promotes the switching from glycolytic metabolism to oxidative phosphorylation in HCC cells, thus suppressing HCC cell proliferation and tumor growth. MerTK is N-glycosylated in HCC cells at asparagine 294 and 454 that stabilizes MerTK to promote oncogenic transformation. Moreover, we observed that nuclear located non-glycosylated MerTK is indispensable for survival of HCC cells under stress. Pathologically, tissue microarray (TMA) data indicate that MerTK is a pivotal prognostic factor for HCC. Our data strongly support the roles of MerTK N-glycosylation in HCC tumorigenesis and suggesting N-glycosylation inhibition as a potential HCC therapeutic strategy.
Keywords: MerTK, N-Glycosylation, Hepatocellular Carcinoma, Oxidative phosphorylation, Warburg Effect
Graphical abstract
Highlights
-
•
MerTK promotes the switching from oxidative phosphorylation to glycolytic metabolism in HCC cells.
-
•
MerTK is N-glycosylated in HCC cells at asparagine 294 and 454 that stabilizes MerTK to promote HCC tumor growth.
-
•
The nuclear located non-glycosylated MerTK is indispensable for survival of HCC cells under stress.
-
•
MerTK is a pivotal prognostic factor for HCC and its N-glycosylation inhibition is a potential HCC therapeutic strategy.
1. Introduction
Hepatocellular carcinoma (HCC) is one of the most prevalent malignancies of liver and occurs predominantly in patients with underlying chronic liver disease and cirrhosis [[1], [2], [3]]. HCC is the third leading cause of cancer-related deaths worldwide in 2020, with over 900,000 people affected and 830,000 deaths, and the incidence is highest in Asia and Africa [[1], [2], [3]]. The incidence of HCC in the United States is rising because of the increased prevalence of hepatitis C virus (HCV) infection [[4], [5], [6]], and the overall 5-year survival rate for all stages of HCC combined is only 16% [7]. The poor prognosis of HCC is due to the fact that it is diagnosed at an advanced stage and lack of efficient therapy, as well as the highly aggressive and proliferative capacity of HCC cells [8,9]. HCC is a heterogeneous disease, that is usually not amenable to standard chemotherapy and is resistant to radiotherapy. Despite the recent advances in the diagnosis, staging and treatment of HCC, surgical resection and liver transplantation remain the only curative treatment options for advanced stage HCC patients. Therefore, understanding of molecular mechanisms of HCC and development of novel and effective HCC therapies is crucial as they have the potential to dramatically improve the outcomes of HCC patients.
MerTK is a member of the TYRO3, AXL, and MERTK receptors (TAMs) family of RTKs expressed predominantly on differentiated myeloid-derived hematopoietic and epithelial cells [[10], [11], [12], [13]]. Aberrant expression of MerTK leads to a transformed phenotype in fibroblasts and cytokine-independent growth in lymphocytes [14,15]. MerTK is overexpressed in a number of hematologic and solid malignancies, which leads to more aggressive and invasive malignant behavior [[16], [17], [18], [19], [20]]. In addition, MerTK protein levels are significantly upregulated in both cell lines from different malignancies and patient-derived xenografts treated with AXL inhibitors, while the inhibition of MerTK could resensitize tumors to the inhibitors in preclinical HNSCC, TNBC, and NSCLC models [21]. Our previous study showed that MerTK provides a mechanistic link between cancer progression and efferocytosis in a manner that overexpression of MerTK in epithelial cells drives efferocytosis in a gain-of-function capacity [22]. In the liver tissue of non-alcoholic fatty liver disease patients with F2–F4 fibrosis and in mouse models of fibrogenesis, MerTK is dramatically up-regulated [23]. Interestingly, MerTK signaling in liver macrophages has been shown to promote liver fibrosis in non-alcoholic steatohepatitis [24,25]. Additionally, MerTK ligand GAS6 stimulates cell migration and induces procollagen expression in cultured human hepatic stellate cells [23]. MerTK plays a critical role in promoting inflammatory response resolution following acute liver injury [26]. More importantly, allele-specific downregulation of MerTK was demonstrated to prevent HCV-induced liver fibrosis [27]. These evidences highlighted the essential function of MerTK in the process of liver pathological lesions. However, the role of MerTK in HCC tumorigenesis remains unclear. The importance of N-glycosylation of Tyrosine Kinase (TK) receptors were widely investigated such as epidermal growth factor receptor (EGFR) and ErbB2 [[28], [29], [30]], VEGFR-2 [31], PDGF [32], M-CSF [33], Src [34], etc. In HCC, N-glycosylation of CD147 is crucial for the tumor promotive activity of CD147 [35]. Here, we investigate a potential role of MerTK and mechanism in regulating HCC tumorigenesis with focus on the regulation of N-glycosylation of MerTK and stability of MerTK in facilitating HCC development.
2. Materials and methods
2.1. Patients and tissue specimens
Sixty-three paired HCC tissue samples including both tumor and matched normal tissue from the same patient were obtained from the First Affiliated Hospital of Wenzhou Medical University (Wenzhou, China) from February 2015 to August 2016. The tissues were immediately frozen in liquid nitrogen after surgical resection, and were then stored at −80 °C until use. The HCC tumors were graded and classified according to the criteria of the 2004 edition of the World Health Organization (WHO). The carcinomas were staged in accordance to the sixth edition International Union-American Joint Committee on Cancer (AJCC)/Union International Contre Cancer (UICC) tumor-node-metastasis (TNM) classification system. All patients who participated in this study provided written informed consents in accordance with the Declaration of Helsinki.
2.2. Tissue microarrays and immunohistochemistry assay
Tissue microarray (TMA) containing a total of 111 formalin-fixed, paraffin-embedded tissue sections were constructed as described previously [36,37]. TMA specimens were subjected to immunohistochemistry (IHC) analysis as described previously [37].
2.3. Cell lines and cell culture
In this study, cell lines were used as follows: human HCC cell lines (MHCC97H, HCCLM3, Huh7), a human hepatoblastoma cell line HepG2, normal liver cell line (LO2) and low-passage HEK293T cells were purchased from the Cell Bank of Shanghai Institute of Cell Biology (Shanghai, China). The HLE cell line was kindly provided by Prof. Qichao Huang (Fourth Military Medical University, China). All cells were cultured in DMEM Medium (Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Life Technologies, Grand Island, NY, USA) and antibiotics (100U /mL penicillin and 100 μg/mL streptomycin) at 37 °C, in a 95% air, 5% CO2 humidified incubator. All cell lines were confirmed mycoplasma free and authenticated by the Cell Bank of the Chinese Academy of Sciences before use. Cell lines were also routinely tested and confirmed for mycoplasma free during this study.
2.4. Reagents and antibodies
Diethylnitrosamine (DEN, 73861), CCl4 (1601168), Oligomycin (O4876), Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, C2920), antimycin A (A8674), rotenone (R8875), 2-deoxy-d-glucose (2-DG, D8375), PNGase F (G5166) and Swainsonine (S9263) and D-(+)-Glucose (G7021) were obtained from Sigma (St. Louis, MO, USA). SYBR green was purchased from Bio-Rad (Hercules, CA, USA). Horseradish peroxidase (HRP)-conjugated anti-rabbit, anti-mouse immunoglobulin G were obtained from Beyotime (Haimen, Jiangsu, China). Glucose deficient DMEM medium and Trizol Reagent were obtained from Life Technologies (Carlsbad, CA, USA). Tunicamycin (B7417) was obtained from ApexBio Technology (Houston, TX, USA). UNC-2250 (S7342) was purchased from Selleck Chemicals (Houston, TX, USA). The antibodies used in this study are: MerTK (abcam; ab52968), Phospho-MerTK (abcam; ab14921), Akt (Cell Signaling Technology; 4691), Phospho-Akt (Ser473) (Cell Signaling Technology; 4060), Phospho-GSK3β (Cell Signaling Technology; 5558S), PGK1 (ZN; 501965), LDHA (ZN; 501146), PFKM (ZN; 505477), PKM2 (ZN; 505477), PDHK1 (Cell Signaling Technology; 3820S), Lamin B (Proteintech; 12987-1-AP), GAPDH (Abmart; M20028) and β-Actin (Proteintech; 60008-1-1 g).
2.5. Western blot
Western blotting was performed as described previously [38].
2.6. RNA extraction and qRT-PCR
Total RNA was prepared from cell samples and tissues using Trizol Reagent according to the manufacturer's protocol, and 2 μg of total RNA of each sample was used for reverse transcription using Primescript™ RT reagent kit with gDNA Eraser (Takara, Dalian, China) according to the manufacturer's instructions. qRT-PCR was performed using the CFX Connect™ real-time system (Bio-Rad, Hercules, CA, USA) with SYBR Green kit (Bio-RAD, Hercules, CA, USA) according to the manufacturer's protocol as follows: 95 °C for 10 min, 45 cycles of denaturation at 95 °C for 10 s and extension at 60 °C for 30 s. The threshold cycle number (CT) was recorded for each reaction. Primers for MERTK gene amplification were as follows: forward primer: 5′-CGAGCTCGGATCTCTGTTCA-3’; reverse primer: 5′-GAGGGGGCATAATCTACCCA-3’.
2.7. Plasmids
The human MERTK clone was obtained from the mRNA of 293T cell line by reverse transcription-Polymerase Chain Reaction (RT-PCR) subsequently cloned into pLVX-Puro lentiviral expression vector to establish MerTK overexpression cell line. Using the pLVX-Puro/MerTK expression vector as a template, MerTK mutants (N294Q, N395Q, N442Q, N454Q and 3Y) were developed by performing a site directed mutagenesis. PLKO.1-shRNA vector expressing shRNA targeting endogenous MERTK mRNA to interfere MERTK level. The asparagine and tyrosine point mutation primers and shRNA sequences of MERTK are shown inTables S2 and S3.
2.8. Generation of MerTK knockdown and cDNA-overexpression cell lines
MerTK stable knockdown or overexpression of WT and various mutants of MerTK MHCC97H, HCCLM3, and Huh7 cells lines were constructed by lenti-virus packaged plasmid infection. 24 h post infection, 1.5 μg/mL of puromycin (Calbiochem, San Diego, CA) was applied to the culture medium and selected clones were validated for loss or overexpression of MerTK via Western blot and qPCR analysis. The lentiviral vector delivery efficiency was optimized as follows. Firstly, centrifugal sedimentation was used to increase the virus titer. Secondly, Multiple rounds of transduction was employed to enhance cell infection ratio. Thirdly, polybrene (1:1000) was added into the culture medium to reduce the repulsion force between cells and viruses and thereby to promote the binding of retroviral particle to the cell surface resulting in a higher efficiency of transduction.
2.9. Cell proliferation assay
MerTK stable knockdown or overexpression MHCC97H, HCCLM3, and Huh7 cells were seeded into 12-well cell culture plates, and cultured at 37 °C, 5% CO2. Cell numbers were counted at following time points: 1, 2, 3, 4, 5, 6 days.
2.10. Tumor xenograft assay
Male BALB/c mice were purchased from Shanghai Laboratory Animal Center, CAS (Shanghai, China) and housed in a specific pathogen-free environment. For in vivo tumorigenesis analysis, control and MerTK stable knockdown or overexpression MHCC97H, HCCLM3, Huh7and HepG2 cells (2.5 × 106 cells in 50 μl medium mixed with 50 μl Matrigel) were injected subcutaneously into the flanks of 5-week-old nude mice (n = 5–8). Tumor length (L) and width (W) were measured every 2 days with calipers, and the tumor volume (V) was calculated (V = 1/2(L × W2)). Before the diameter of the tumor reached 1.5 cm, the mice were sacrificed, and the tumors were dissected out and weighted. Tumor tissues were homogenized and lysed for western blotting analysis. All animal procedures were conducted under the guidelines approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University.
2.11. DEN and CCl4-induced HCC mouse model
The DEN and CCl4-induced HCC mouse model was established as previously described [39,40]. Briefly, a single dose of DEN (25 mg/kg body weight in PBS) were injected into 15-day-old male mice intraperitoneally (i.p.) to initiate tumor formation. At 4 weeks of age, mice were injected i. p. Once a week with CCl4 (0.5 mL/kg body weight in corn oil) for an additional 16 weeks. All mice were sacrificed at 30 weeks of age. Livers were removed and the number and size of HCC were recorded, and individual lobes were snap frozen in paraffin embedded and subjected to histological and immunochemical analyses.
2.12. Colony formation assay
MerTK stable depleted or overexpressed MHCC97H and HCCLM3 cells were counted, and a total of 400 cells per well were seeded evenly into 6-well plates and incubated at 37 °C for 14 days. Cells were washed with pre-warmed PBS three times, fixed with 4% Paraformaldehyde, stained with Giemsa solution for 15 min, then washed with pre-warmed PBS three times, and counted by two independent investigators.
2.13. Metabolic flux assays
Real time intact cellular oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using the Seahorse XF96 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA) as previously described [41]. Briefly, MerTK stable knockdown or overexpression MHCC97H and HCCLM3 cells were seeded into the seahorse 96-well cell culture plate at a density of 2.5 × 104 and 2 × 104 cells/80μl/well, respectively, and cultured at 37 °C, 5% CO2 incubator overnight. Prepare the assay medium for OCR (primary medium 49 mL, 1 mL sodium pyruvate, 0.225 g glucose, pH7.4) and ECAR (primary medium 49 mL, 0.0164 g l-glutamine, pH 7.4). Add 140 μl calibrate solution into the calibrate plate and incubated at non-CO2 incubator overnight. The next day, replace the calibrator microplate with cell culture plate and start the protocol as we optimized previously to measure the OCR and ECAR [41].
2.14. Apoptosis and mitochondrial membrane potential analysis by FACS
For apoptosis analysis, cells were collected and incubated with Annexin V-FITC/PI (BD, San Jose, CA), followed by incubation in the dark at room temperature for 20 min. Apoptosis rate was measured immediately using a BD Accuri™ C6 Plus flow cytometer (BD, Franklin Lakes, NJ).
Mitochondrial membrane potential was measured by FACS as previously described [41].
2.15. Glucose uptake
MerTK stable knockdown or overexpression MHCC97H, HCCLM3 Cells were collected and incubated for 20 min at 37 °C with 100 nM 2-NBDG (2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxyglucose), prior to FACS analysis.
2.16. Lactate production
Lactate levels secreted into the media were measured by Fluorescent colorimetric assays. The assay was performed as per the manufacturer's instructions utilizing Amplite™ Fluorimetric l-Lactate Assay Kit (AAT Bioquest).
2.17. Preparation of nuclear extracts
To isolate nuclear component from cytoplasm, cells were collected and treated with a nuclear protein extraction kit (Beyotime Biotechnology, Haimen, Jiangsu, China) following the manufacturer's instructions. Briefly, the cell samples were harvested and homogenated in the plasma protein extraction reagent A containing 1 mM PMSF, vortexed in the highest speed for 5s and then bathed on ice for 10–15 min, after which plasma protein extraction reagent B were added. The samples were vortexed in the highest speed for 5 s and bathed on ice for 1 min, then centrifuged at 13,000 rpm at 4 °C for 5 min. The supernatants, corresponding to the cytosolic fraction, were transferred to fresh tube, and measured for protein content by using Pierce BCA Protein Assay Kit. Further, the nuclei pellet was suspended in nuclear protein extraction reagent, vortexed in the highest speed for 15–30 s. After which the nuclear components were bathed on ice for 1–2 min, vortexed in the highest speed for 15–30 s for every 1–2 min lasting 30 min and centrifuged at 12,000–16,000 × g at 4 °C for 10 min. The supernatants, corresponding to the nuclear fraction, were transferred to a fresh tube, and measured for protein content by using Pierce BCA Protein Assay Kit.
2.18. Blue native PAGE (BNG) gel
BNG was performed as previously published protocol with slight modifications [42]. Briefly, 40 μl of Buffer A (50 mM NaCl, 50 mM imidazole, 2 mM 6-aminohexanoic, and 1 mM EDTA, pH 7.0) was added to 10 mg of pelleted cells. Then 12 μl digitonin was added (20% (w/v)) and allowed to solubilization for 10–20 min at on ice. After centrifugation for 60 min at 20,000 g, supernatants were collected. Then 50% (v/v) glycerol and 5% (w/v) Coomassie blue G-250 dye stock was mixed to yield a glycerol/dye ratio of 2. Next, the mixture was added to the samples to yield a samples/mixture ratio of 5 (v/v). Finally, the samples were run on 3–11% acrylamide gradient gels at 4 °C. The gel was run 60 min at 45 V for spacer gel and 150 min at 180 V for separation gel.
2.19. Immunoprecipitation and ubiquitination assay
To detect ubiquitination status of MerTK in vivo, MHCC97H and HCCLM3 cells were lysed with lysis buffer containing protein inhibitor for 30 min on ice. Then cell lysates were centrifuged at 13,000 rpm for 10 min at 4 °C and the supernatant were transferred to a fresh tube. The content of these cell lysates was measured as per the manufacturer's instructions by using Pierce BCA Protein Assay Kit. 30 μl Protein A beads were added in 1 mg cell lysates for 4 h with rotation at 4 °C and then centrifuged for 5 min at 12,000 rpm. Supernatant was incubated with 4 μg MerTK primary antibody overnight, after which 50 μl Protein A beads were added for an additional 4 h. Immunoprecipitates were washed three times with PBS. Proteins were eluted in 1.25 × SDS-Loading buffer and analyzed by Western blot with anti-ubiquitin antibody.
2.20. Statistical analysis
Statistical significance was determined using GraphPad Prism software. The χ2 test was performed to evaluate the relationship between the clinical-pathological features and MerTK expression in the IHC (TMA) results. Kaplan–Meier log-rank tests and Cox regression analysis were used for survival analysis. All data were presented as mean ± SD unless specified. Student's t-test was used for two-group comparisons. One-way ANOVA with Bonferroni post-test was used to analyze multiple groups with only one variable tested. Statistical significance is displayed as *P < 0.05, **P < 0.01, ***P < 0.001, n. s., not significant.
3. Results
3.1. MerTK is overexpressed in HCC tissues and MerTK high expression is associated with poor outcomes in HCC patients
To understand the extent of MerTK protein comparative expression in human liver cancer patients as compared with adjacent normal liver tissue, we assessed 63 HCC patient biopsies by Western blot analysis and 37 HCC patient biopsies by qRT-PCR (Fig. 1A–C). Of the 63 or 37 HCC patient biopsies, the immunoblotting results showed a significantly increased MerTK expression at protein level, however, there were no differences in MERTK mRNA level between tumor and adjacent normal liver tissues. Bioinformatics analysis further confirmed that MERTK mRNA remains unchanged in normal and HCC tumor tissues (Figs. S1A and B). We also performed immunohistochemistry-based TMA (n = 111) analysis to validate MerTK expression in HCC tumor tissue sections. MerTK expression was significantly increased in HCC tumor tissues from patients with advanced stages or high-grade HCC compared with normal liver tissues (Fig. 1D). To further assess the clinical relevance of MerTK expression, we analyzed the survival of our cohort of HCC patients. Our data showed that increased MerTK expression was significantly correlated to the poorer overall survival (OS) with median 10 months versus 50 months, respectively, for patients with higher and lower MerTK expressions of tumors (P = 0.0162; Fig. 1E).
Fig. 1.

Aberrant MerTK expression in HCC and high expression of MerTK were associated with poor outcomes in HCC Patients. (A) Relative MerTK protein expression in HCC tumor adjacent normal (N) and matched tumor tissues (T) was examined by Western blot (n = 63). (B) Relative MerTK protein expression of Western blot analyses was quantified using Image J and normalized to the β-Actin (n = 63). Data are presented as mean ± SD, statistical significance was determined by paired Student's t-test. ***P < 0.001. (C) MERTK mRNA expression in HCC tumor adjacent normal (N) and matched tumor tissues was detected by using qRT-PCR (n = 37). Data are presented as mean ± SD, statistical significance was determined by paired Student's t-test. n. s., not significant. (D) IHC staining of MerTK protein in HCC tumor tissues. Representative MerTK IHC staining photomicrographs (400 × ) of normal liver tissue, Grade 1 (G1), Grade 2 (G2), Grade 3 (G3) and Grade 4 (G4) HCC tumor tissues (upper panel), and TNM stage I, II, III and IV HCC tumor tissues (lower panel), are shown. (E) Kaplan-Meier analysis of the OS of HCC patients with high (n = 35) or low MerTK (n = 76) expression. (F) Kaplan-Meier overall survival analysis of HCC patients diagnosed with I-II (n = 58) or III-IV (n = 53) stage according to MerTK protein expression. (G) Kaplan-Meier OS curve analysis of HCC patients diagnosed with Cirrhosis (n = 87) according to MerTK protein expression.
To further evaluate the relationship between MerTK expression level and OS, we analyzed OS of HCC patients diagnosed with I-II or III-IV stages and G1-G2 or G3-G4 grades. Our data suggest that MerTK expression level is not closely related to OS in group I-II (Fig. 1F, left panel), G1-G2 and G3-G4 (Fig. S1D); however, in group III-IV, higher MerTK expression indicates a significantly poor OS, with median 8 months versus 23.5 months, respectively (P = 0.015; Fig. 1F, right panel). Moreover, we evaluated prognostic value of MerTK in patients diagnosed with or without hepatitis virus B (HBV) infection, and we found the survival time was much shorter in MerTK high group regardless of HBV infection (Fig. S1E). In addition, we evaluated whether MerTK correlated with cirrhosis caused poor outcomes of HCC patients. We found that higher MerTK expression indicated short OS compared to lower MerTK expression group, with median 12.5 months versus 40 months, respectively (Fig. 1G). However, there was no significant difference in survival between HCC patients with lower and higher MERTK transcripts (Fig. S1C). Finally, we evaluated the correlation between MerTK expression and various clinical-pathological factors of HCC patients. Higher MerTK was significantly correlated with age and cirrhosis occurrence (P = 0.010 and P = 0.036, respectively; Table S1). The COX regression model was applied to evaluate the hazard index of MerTK. As shown in Table S2, MerTK could be considered as an independent prognostic factor of HCC. Taken together, these data indicate a strong positive correlation of MerTK expression in HCC and aggressive behavior of the tumor progression and OS of the patients.
3.2. MerTK is critical for HCC cell proliferation and migration
To better understand the role of MerTK in human liver cancer, we performed loss- and gain-of-function experiments. First, we assessed MerTK expression by Western blotting in four HCC cell lines HLE, Huh7, HCCLM3 and MHCC97H, as well as HepG2 cell, a human hepatoblastoma cell line, and normal liver cell line LO2. Our results revealed that MerTK protein was predominantly expressed in indicated human liver cancer cell lines, while it was barely detected in LO2 cells (Fig. 2A). As uncontrolled cell proliferation leads to tumor initiation and progression, we further investigate the role of MerTK in HCC cell proliferation. We constructed lentivirus packaged shRNA plasmid targeting MERTK (shMerTK#1 and shMerTK#2), as well as the negative control plasmid (shCont). HCC cell lines MHCC97H, HCCLM3 and Huh7 were infected with lentivirus packaged plasmids and the endogenous MerTK was successfully silenced (Fig. 2B). MerTK ablation caused significant inhibition of cell proliferation and migration in HCC cells (Fig. 2C; Fig. S2A). Conversely, over-expression of exogenous MerTK enhanced cell proliferation in both LO2 cells and HCC cells as well as cell migration in HCC cells in vitro (Fig. 2D and E; Fig. S2A). MerTK ablation also increased cell apoptosis (Figs. S2B and C) as well as reduced mitochondrial membrane potential in HCC cells (Fig. S2D). Moreover, we found that MerTK knockdown inhibited HCC cell colony formation (Fig. 2F and G). Reversely, overexpressed MerTK increased colony number of HCC cell (Fig. 2F and G). As it is wildly demonstrated that MerTK is a pivotal regulator of ERK, Akt, NF-κB, and Src family kinases [[43], [44], [45], [46], [47]], we then investigated the Akt-GSK3α/β signaling in MerTK depleted or overexpressed HCC cells. We found that MerTK ablation and overexpression reversibly regulates the phosphorylation of Akt, GSK3β (Fig. 2H and I). Together, these results indicate a role for MerTK in conferring proliferative and migratory advantage in HCC partially by regulating Akt, GSK3β.
Fig. 2.

MerTK is crucial for HCC cell proliferation. (A) Western blot analysis and quantification of MerTK expression in total cell extracts in LO2, HLE, Huh7, HepG2, HCCLM3, and MHCC97H cells. β-Actin was used as a loading control. Data are presented as mean ± SD of three independent experiments, statistical significance was assessed by One-way ANOVA with Bonferroni post-test, ***P < 0.001. (B) Western blot analysis demonstrating MerTK stable knockdown in the indicated HCC cells transfected with shRNA specific to MERTK mRNA. (C) Cell proliferation of MerTK stable knockdown MHCC97H, HCCLM3, and Huh7 cells lines was measured by cell counting. Data are presented as mean ± SD of three independent experiments, statistical significance was assessed by One-way ANOVA with Bonferroni post-test, ***P < 0.001. (D and E) Western blot analysis demonstrating MerTK overexpression in LO2 cells and the indicated HCC cells (D). Cell proliferation of MerTK overexpression LO2, MHCC97H, HCCLM3, and Huh7 cells lines were measured by cell counting (E). Cont, refers to cells transduced with empty vector and hereafter; MerTK OE, refers to cells transduced with plasmid overexpressing MerTK and hereafter. Data are presented as mean ± SD of three independent experiments, statistical significance was determined by Student's t-test, *P < 0.05. (F and G) Representative images (F) and quantification (G) of colony formation assay of MerTK stable knockdown or overexpression MHCC97H and HCCLM3 cells. Data are presented as mean ± SD, statistical significance was assessed by One-way ANOVA with Bonferroni post-test and Student's t-test, *P < 0.05, **P < 0.01. (H and I) Immunoblots of tumor growth related proteins p-GSK3β, p-Akt, Akt in MerTK stable knockdown (H) or overexpression (I) MHCC97H and HCCLM3 cells. β-Actin was used as a loading control.
3.3. MerTK modulates the warburg effect in HCC cells
Metabolic reprogramming is a critical feature of cancer cells [48]. The liver serves as a central metabolic organ being responsible for detoxification, storing glycogen, biosynthesis of urea and several secreted protein and biotransformation. Consequently, patients suffering with HCC also have an altered metabolic pattern of liver displaying enhanced addiction to glucose, and enhanced aerobic glycolysis, like many tumor cells undergoing the Warburg effect [49]. To decipher the role of MerTK in regulating cellular metabolism, we measured Extracellular Acidification Rate (ECAR) and Oxygen Consumption Rate (OCR) in the indicated HCC cells. MerTK depletion caused repression of aerobic glycolysis in MHCC97H and HCCLM3 cells (Fig. 3A; Fig. S3A), while OCR was increased (Fig. 3B; Fig. S3B). In contrast, overexpression of MerTK enhanced the aerobic glycolysis (Fig. 3C; Fig. S3C) and repressed mitochondrial respiration (Fig. 3D; Fig. S3D). We also found MerTK depletion caused excessive ROS generation while overexpression of MerTK partially eliminated ROS (Fig. S3E). To further confirm the effect of MerTK ablation in switching of the Warburg effect, we measured cell glucose uptake and lactate production in MerTK depleted or overexpressed HCC cells, and found that relative glucose uptake and lactate production were significantly decreased in MerTK knockdown cells while increased by MerTK overexpression, respectively (Fig. 3E and F). Similarly, we found that MerTK regulated the expression of glycolytic enzymes PGK1, LDHA, PFKM, PKM2 and PDHK1, in a manner that knockdown or overexpression reversibly regulated the expression levels of these indicated proteins (Fig. 3G and H). Collectively, our data implicate that MerTK facilitates the Warburg effect through regulating key glycolytic enzymes.
Fig. 3.

MerTK modulates bioenergetics metabolism through promoting the Warburg Effect and suppressing OXPHOS in HCC cells. (A and C) The intact cellular ECAR of control, MerTK stable knockdown (A) and overexpression (C) MHCC97H and HCCLM3 cells were measured in real time using the Seahorse XF96 Extracellular Flux Analyzer. Data were analyzed by Seahorse XF-96 Extracellular Flux Analyzer. ECAR is reported in mpH/minute, the results were normalized to cell number. Data are presented as mean ± SD, statistical significance was assessed by One-way ANOVA with Bonferroni post-test, *P < 0.05, **P < 0.01. (B and D) The intact cellular OCR of control, MerTK stable knockdown (B) and overexpression (D) MHCC97H and HCCLM3 cells were measured in real time using the Seahorse XF96 Extracellular Flux Analyzer. Data were analyzed by Seahorse XF-96 Extracellular Flux Analyzer. OCR is reported in pmols/minute, the results were normalized to cell number. Data are presented as mean ± SD, statistical significance was assessed by One-way ANOVA with Bonferroni post-test, *P < 0.05, **P < 0.01. (E) Glucose uptake of MerTK stable knockdown or overexpression MHCC97H, HCCLM3 by 2-NBDG incorporation by FACS. Data are presented as mean ± SD, statistical significance was assessed by One-way ANOVA with Bonferroni post-test and Student's t-test, *P < 0.05, **P < 0.01. (F) Lactate production of MerTK stable knockdown or overexpression MHCC97H, HCCLM3 cells were measured following manufacturer's instructions. Data are presented as mean ± SD, statistical significance was assessed by One-way ANOVA with Bonferroni post-test and Student's t-test, *P < 0.05, **P < 0.01. (G and H) Immunoblots of glycolysis related enzymes PGK1, LDHA, PFKM, PKM2, PDHK1 in MerTK stable knockdown (G) or overexpression (H) MHCC97H and HCCLM3 cells. β-Actin was used as a loading control.
3.4. MerTK promotes HCC cell growth in vivo
To further demonstrate the essential role of MerTK in promoting HCC cell growth, we subcutaneously injected control and MerTK knockdown MHCC97H, HCCLM3, Huh7 and HepG2 cells into BALB/c nude mice, respectively. MerTK downregulation significantly suppressed HCC cell growth in vivo (Fig. 4A–C). Stable Knockdown of MerTK protein in tumor of nude mice was confirmed by Western blot (Fig. 4D). We further assessed the levels of key enzymes of Akt-GSK3α/β signaling which involve in regulating glycolysis in cancer cells. Strikingly, Akt and GSK3β were all inactivated and PGK1, LDHA, PFKM, PKM2 and PDHK1 were significantly decreased due to MerTK knockdown (Fig. 4E). Consistently, overexpression of MerTK in MHCC97H cells promoted tumor growth in vivo (Fig. 4F–H). MerTK overexpression in tumor of nude mice was also confirmed by Western blot (Fig. 4I). Moreover, we demonstrated that MerTK expression was significantly increased in the tumor tissue of DEN and CCl4-induced primary HCC mice model (Figs. S4A and B). In addition, the phosphorylation of key enzymes of Akt-GSK3α/β signaling (Akt and GSK3β) were remarkably upregulated (Figs. S4B and C). The glycolytic enzyme PGK1, LDHA, PFKM, PKM2 and PDHK1 were also significantly increased (Figs. S4B and D). These data further confirmed an indispensable role of MerTK in promoting HCC cell growth through regulating the Warburg effect.
Fig. 4.

MerTK promotes HCC cell growth in vivo. (A) Representative dissected tumors derived from control and MerTK knockdown MHCC97H, HCCLM3, Huh7 and HepG2 cells are shown. (B) Growth of tumors derived from control and MerTK knockdown cells are shown. n = 8 (MHCC97H and HCCLM3 cells) or 5 (Huh7 and HepG2 cells). Data are presented as mean ± SEM, statistical significance was determined by Student's t-test, **P < 0.01, ***P < 0.001. (C) Tumor weights at the end of the experiment are measured. Data are presented as mean ± SEM, statistical significance was determined by Student's t-test, ***P < 0.001. (D) MerTK expression in the lysates of tumor tissue (A) are shown. (E) Immunoblots of tumor growth related proteins p-GSK3β, p-Akt and glycolysis related enzymes PGK1, LDHA, PFKM, PKM2, PDHK1 in tumor tissues derive from nude mice. β-Actin is used as a loading control. (F) Representative dissected tumors derived from control and MerTK overexpression MHCC97H cells are shown. (G and H) Tumors growth were measured once in two days (G) and tumor weights are measured at the end of the experiment (H). n = 8. Data are presented as mean ± SEM, statistical significance was assessed by Student's t-test, *P < 0.05, **P < 0.01. (I) MerTK expression in the lysates of tumor tissue (F) are shown.
3.5. Mertk deletion suppresses chemical-induced liver tumorigenesis
To investigate the physiological and pathological functions of Mertk deletion suppresses liver tumorigenesis, we generated a Mertk knockout (Mertk−/−) mice by crossing Mertk± with Mertk± mice. Western blotting analysis revealed that Mertk was knockout in all tissues, including heart, liver, spleen, lung, kidney, and skeletal muscle (Fig. 5A). To further investigate the contributions of MerTK to HCC development, the mouse liver tumors were induced by DEN and CCl4 in male mice of two genotypes, Mertk+/+ and Mertk−/− (Fig. 5B). The tumor burden (number and size) was then recorded at 30 weeks of age (Fig. 5C and D). Strikingly, our results showed that the tumor numbers detected in the Mertk−/− mice were much less than those in the Mertk+/+ control littermates, and the tumor size was also significantly smaller. Immunohistochemical analysis confirmed that MerTK was expressed in livers of the Mertk+/+ mice, but not in Mertk−/− mice (Fig. 5E). Histological examination revealed extensive liver pathology associated with severe steatosis in the DEN and CCl4-treated Mertk+/+ mice (Fig. 5F). Moreover, the tumor lesions were positive for alpha-fetoprotein (AFP), an established liver tumor marker (Fig. 5G and H). These findings suggest that Mertk deletion suppresses chemical-induced liver tumorigenesis.
Fig. 5.

Deletion of MerTK Inhibits DEN and CCl4-Induced HCC Formation
(A) Lysates of all tissues from Mertk+/+ and Mertk−/− mice were analyzed by Western blotting with antibodies against MerTK or GAPDH at 30 weeks of age. GAPDH served as a loading control. (B) Schematic representation of DEN and CCl4-treated Mertk+/+ and Mertk−/− mice. At 15 days of age, mice were injected with DEN. At 4 weeks of age, mice were injected with CCl4 once a week for 16 weeks, mice were sacrificed at 30 weeks. (C) Representative macroscopic pictures of livers from Mertk+/+ and Mertk−/− mice at 30 weeks of age. Arrowhead indicates tumor nodules. (D) Quantification of liver tumors in Mertk+/+ and Mertk−/− mice at 30 weeks of age. Data are presented as mean ± SD, statistical significance was determined by Student's t-test, ***P < 0.001. (E) Immunohistochemical analysis of liver sections for MerTK expression from Mertk+/+ and Mertk−/− mice at 30 weeks of age. Images were obtained at 40 × magnification. (F) Histological analysis (H&E) of livers from Mertk+/+ and Mertk−/− mice at 30 weeks of age. Images were obtained at 40 × magnification. (G) Immunohistochemical analysis of liver sections for AFP expression from Mertk+/+ and Mertk−/− mice at 30 weeks of age. Images were obtained at 40 × magnification. (H) Quantification of positive areas per field in (F) was determined by Image J software. Data are presented as mean ± SD, statistical significance was determined by Student's t-test, **P < 0.01.
3.6. N-glycosylated MerTK is essential for HCC cell growth
MerTK was found to be heavily glycosylated on multiple asparagines residues which may regulate MerTK activity [50]. Thus, we asked whether N-glycosylation of MerTK is pivotal for the proto-oncogenic property in HCC. We used tunicamycin (TM), a GlcNAc phosphotransferase inhibitor which blocks the formation of N-glycosidic linkages, and swainsonine which can completely inhibit mammalian Golgi α-mannosidase II which is critical in the formation of N-glycosylation of proteins, as well as PNGase F which cleaves between the innermost GlcNAc and asparagine residues of high mannose, hybrid, and complex oligosaccharides. Expectedly, due to TM, swainsonine and PNGase F treatment, we found the molecular weight of MerTK was largely reduced which indicates that the higher molecular weight form is indeed N-glycosylated MerTK. We further treated the cells with or without glucose and found the molecular weight of MerTK was gradually reduced with time, which suggests that N-glycosylation was reduced due to glucose starvation (Fig. 6A). Next, we asked which asparagine site(s) of MerTK is/are glycosylated. According to the bioinformatics analysis, we mutated the N294, N395, N442, N454 (N was replaced by Q) and Y747, 753 and 754 (hereafter named 3Y, Y was replaced by A) in MERTK, and found N294 and N454 were crucial sites for N-glycosylation, but not N395 and N442 residues. Intriguingly, we found triple tyrosine mutant also deprived N-glycosylation of MerTK (Fig. 6B and C). To further understand the N-glycosylation of MerTK on HCC cell growth, LO2, MHCC97H and HCCLM3 cells were transfected with control, N294Q, N395Q, N442Q, N454Q and 3Y mutant plasmids. Cell growth was not enhanced by expression of N294Q, N454Q and 3Y mutants in vitro, whereas N395Q and N442Q mutants in which MerTK were still glycosylated exhibited pro-growth activity in these three cell lines (Fig. 6D). Moreover, we determined the ECAR and OCR alteration due to MerTK point mutation. N395Q- and N442Q-MerTK could effectively upregulate glycolytic rate, while mitochondrial oxidative phosphorylation (OXPHOS) was suppressed (Fig. 6E and F; Figs. S5A and B). The impacts of MerTK mutants on cellular bioenergetics were confirmed by glucose uptake only increased in N395Q- and N442Q-MerTK overexpressed LO2 cells (Fig. 6G). Together, the results indicate that MerTK is exclusively N-glycosylated at N-294 and N454, which is 3Y dependent, and N-glycosylated MerTK promotes both HCC cells and normal liver cells (LO2) growth.
Fig. 6.

N-Glycosylation of MerTK is Essential for HCC Cell Growth. (A) Western blot analysis of MHCC97H, HCCLM3 treated with or without Tunicamycin (TM), PNGase F, Swainsonine and glucose starvation. (B) Structure of MerTK and the modification site of N294Q, N454Q and 3Y. (C and D) MerTK expression was detected by Western blot in LO2, MHCC97H and HCCLM3 cells transfected with the N294Q, N395Q, N442Q, N454Q and 3Y mutant plasmids, respectively (C). Cell proliferation was analyzed by cell counting (D). Data are presented as mean ± SD of three independent experiments, statistical significance was assessed by One-way ANOVA with Bonferroni post-test, *P < 0.05. (E and F) The intact cellular ECAR and OCR of WT and indicated MerTK mutant LO2 Cells were measured in real time using the Seahorse XF96 Extracellular Flux Analyzer. Data are presented as mean ± SD, statistical significance was assessed by One-way ANOVA with Bonferroni post-test, *P < 0.05. (G) Glucose uptake of WT and MerTK indicated mutant LO2 Cells by 2-NBDG incorporation by flow cytometry. Data are presented as mean ± SD, statistical significance was assessed by One-way ANOVA with Bonferroni post-test, *P < 0.05, n. s., not significant.
3.7. N-glycosylation is crucial for MerTk stability
Next, we asked whether deglycosylated MerTK is sensitive to ubiquitination-dependent degradation. We treated LO2, MHCC97H and HCCLM3 cells with protease inhibitor MG132. Deglycosylated MerTK was increased in MG132 treated LO2 cells, but not in MHCC97H and HCCLM3 cells (Fig. 7A). However, MerTK was increased upon MG132 treatment when MerTK was deglycosylated by TM (Fig. 7B and C). We co-immunoprecipitated MerTK in TM or both TM and MG132 treated MHCC97H and HCCLM3 cells, and the immunoprecipitates were examined by Western blot with an anti-ubiquitin antibody. The data showed that ubi-MerTK level in MHCC97H and HCCLM3 cells was significantly increased following TM treatment (Fig. 7D). Importantly, MG132 treatment caused much more ubi-MerTK accumulation than the control (Fig. 7D), suggesting N-glycosylation is crucial for MerTK stability. To understand whether these mutants (N294Q, N454Q and 3Y) would affect MerTK stability, we treated MHCC97H cells with Cycloheximide (CHX, 125 μg/mL) at indicated intervals to examine MerTK turnover. N294, N454 and 3Y mutants of MerTK exhibited shorter half-life than G-MerTK (Fig. 7E). These data indicated that deglycosylated at N294 and N454 of MerTK is more sensitive to degradation. Intriguingly, dephosphorylation at Y749, Y753 and Y754 is crucial for N-glycosylation of MerTK which could also regulate the ubiquitin-dependent degradation. Previous work has demonstrated that ligand activated MerTK homodimerization is essential for autophosphorylation and subsequently stimulated multiple signals transduction as well as enhanced MerTK stability. We further asked whether N-glycosylation is indispensable for stabilizing MerTK. To clarify our hypothesis, we analyzed the dimer status of wild type MerTK and various mutant MerTK. MerTK mutant of N294Q, N454Q and 3Y impaired the MerTK dimerization (Fig. 7F). Strikingly, glucose deprivation and TM treatment caused deglycosylation of MerTK and almost completely abolished the ability to form dimer (Fig. 7G). Taken together, these data indicated that N-glycosylation and phosphorylation are essential for MerTK stability.
Fig. 7.

N-glycosylation modification stabilizes MerTK. (A) Western blot analysis of LO2, MHCC97H and HCCLM3 treated with indicated concentration of MG132 for 24 h followed by Western blot analysis with indicated antibodies. (B) MHCC97H and HCCLM3 cells were treated with TM (1 μg/ml) and/or indicated concentration of MG132 for 24 h followed by Western blot analysis with indicated antibodies. (C) Western blot analysis of MHCC97H and HCCLM3 cells treated with TM (1 μg/mL) and/or MG132 (125 μg/mL) (left panel) and relative DG-MerTK expression level was quantified by Image J and normalized to the β-Actin (right panel). Data are presented as mean ± SD of three independent experiments, statistical significance was assessed by Student's t-test, **P < 0.01. (D) MHCC97H and HCCLM3 cells with TM (1 μg/mL) and/or MG132 (125 μg/mL) treatment were subjected to MerTK immunoprecipitation (IP) followed by Western blot analyses with anti-ubiquitin. (E) WT and indicated MerTK mutants expressing in MHCC97H cells were treated with 125 μg/mL CHX at indicated intervals and detected by Western blot analysis. The intensity of MerTK protein was quantified using Image J and normalized to the β-Actin. Data are presented as mean ± SD of three independent experiments, statistical significance was assessed by One-way ANOVA with Bonferroni post-test, **P < 0.01. (F) SDS-PAGE and Blue Native PAGE of WT and indicated MerTK mutants expressing in MHCC97H and HCCLM3 cells to detect the MerTK dimerization. (G) SDS-PAGE and Blue Native PAGE of endogenous MerTK in MHCC97H cells treated with or without TM or in presence or absence of glucose (left panel). Proposed model of N-glycosylation and phosphorylation modification modulating MerTK homodimerization (right panel).
3.8. Nuclear located non-glycosylated MerTK is indispensable for HCC cell survival under stress
To further investigate the essential role of MerTK in promoting HCC cell survival, we examined cell death in response to glucose starvation. Intriguingly, we found glucose starvation induced gradually deglycosylation of MerTK as well as upregulated MerTK protein expression (Fig. 8A, upper panel). Furthermore, glucose starvation increased MERTK mRNA level in a time dependent manner (Fig. 8A, lower panel). To investigate pro-survival activity of MerTK under stress, we cultured LO2 cells that express low MerTK in glucose-free medium and collected the cells at 0, 6, 12 and 24 h followed by Western blot analysis. We found that MerTK expression was not affected under glucose starvation (MHCC97H used as a MerTK positive control to know the location of MerTK in blot) (Fig. 8B, left panel). We compared cell apoptosis rate of LO2, MHCC97H and HCCLM3 with or without glucose for 24 h, and found apoptosis was significantly induced in LO2 but only modestly in MHCC97H and HCCLM3 cells (Fig. 8B, right panel). Previous studies have demonstrated that prolonged Gas6 addition can promote MerTK translocation from cell surface to nucleus, which is related to the MerTK deglycosylation [51]. Hence, we further evaluated the sub-cellular location of N-glycosylated and deglycosylated MerTK. Our data shows a modest nucleus translocation of deglycosylated MerTK in both LO2 and MHCC97H cells transfected with N294Q- and N454Q-MerTK (Fig. 8C). Furthermore, we found that TM treatment induces a higher nuclear translocation of total and phosphorylated MerTK in MHCC97H cells (Fig. 8D, lane 8). Indeed, when phosphorylation was inhibited by UNC-2250, nucleus located MerTK was suppressed (Fig. 8D, lane 9), and cytoplasmic MerTK was also reduced significantly (Fig. 8D, lane 6). We also found UNC-2250 treatment reduced MerTK level (Fig. 8E) as well as facilitated cell apoptosis initiated by glucose starvation or TM treatment in MHCC97H cells (Fig. 8F). Further work is required to validate the downstream signaling induced by nucleus-located MerTK. Collectively, these results suggest that phosphorylation of deglycosylated MerTK is also required for nucleus translocation to facilitate HCC cell survival under stress condition.
Fig. 8.

MerTK is indispensable for HCC cell survival under glucose starvation stress or N-glycosylation blocked by TM. (A) MHCC97H and HCCLM3 cells were cultured in the presence or absence of glucose at indicated intervals. MerTK protein (upper panel) and mRNA (lower panel) levels were analyzed by Western blot and qRT-PCR, respectively. (B) Western blot analysis of MerTK expression in LO2 cells under glucose starvation at indicated intervals (left panel). LO2, MHCC97H and HCCLM3 cells were cultured with or without glucose for 24 h, and cell apoptosis was measured by FACS (right panel). Data are presented as mean ± SD of three independent experiments, statistical significance was assessed by One-way ANOVA with Bonferroni post-test, **P < 0.01. (C) Western blot analysis of nuclear and cytoplasmic located MerTK in LO2 and MHCC97H cells transfected with WT and various MerTK mutants. (D) Western blot analysis of nuclear and cytoplasmic located MerTK in MHCC97H cells treated with TM (1 μg/mL) and/or UNC-2250 (16 μM) for 24 h. (E and F) Western blot analysis of MerTK and p-MerTK in MHCC97H cells treated with TM (1 μg/mL) and/or UNC-2250 (16 μM) or in presence or absence of glucose (E). Apoptosis rate was analyzed by FACS (F). Data are presented as mean ± SD, statistical significance was assessed by One-way ANOVA with Bonferroni post-test, **P < 0.01, ***P < 0.001.
4. Discussion
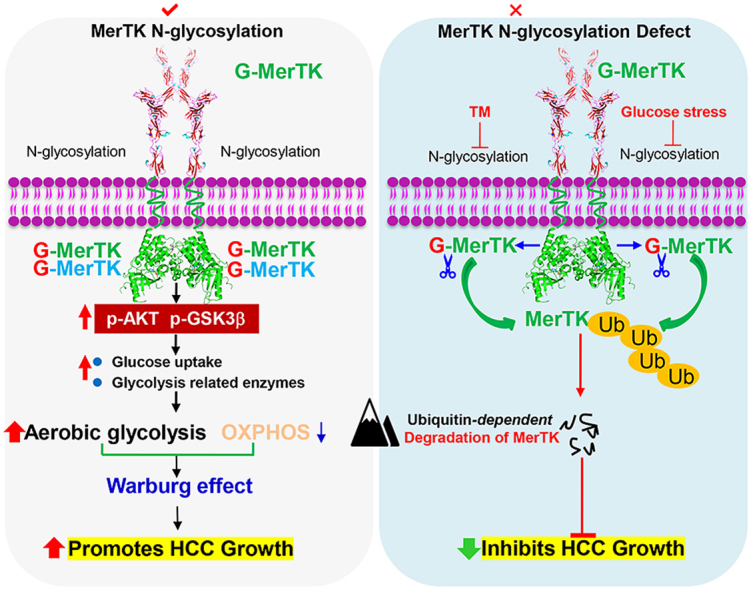
In this study, we report the pro-tumorigenic activity of MerTK in HCC through regulating Akt-GSK3α/β signaling as well as cellular bioenergetics. We also demonstrate that N-glycosylation is critical for MerTK homodimerization and stability. Importantly, we found tyrosine phosphorylation is indispensable for nucleus translocation of deglycosylated MerTK. These findings renovate current understanding of MerTK promoted tumorigenesis.
Aberrantly upregulated MerTK was widely found in multiple cancer, and MerTK inhibition probably caused cell growth arrest as well as increased cell death. Previous work has demonstrated MerTK is a pivotal regulator of certain tumor associated signal transduction, such as Akt, Src, p38, ERK1/2, GSK3α/β, MEK1/2, AMPK, STAT5. CHK-2, focal adhesion kinase (FAK) and STAT6 [49,50]. Consistently, we show that MerTK depletion dramatically suppresses HCC cell growth in vitro and in vivo. In HCC, we found MerTK functions as a metabolic modulator by integrating aerobic glycolysis and OXPHOS. PGK1, LDHA, PFKM, PKM2 and PDHK1 are key downstream factors of MerTK in regulating the Warburg effect (Fig. 9). Our study unravels a novel role of MerTK in modulating cellular bioenergetics which enriches the metabolic regulator property of MerTK. However, further investigation will be needed to understand whether Akt may also regulate PFKM phosphorylation and stabilization in HCC cells.
Fig. 9.

Proposed model for the role of MerTK in HCC tumorigenesis. N-glycosylation modification stabilizes MerTK that promotes HCC cell growth through facilitating the Warburg effect and activating Akt/GSK3β signaling. Glucose starvation or blockade of glycosylation leads to vast non-glycosylated MerTK translocated into the nucleus, therefore promoting HCC cell survival under stress.
Several studies have demonstrated and highlighted the glycosylation (both O-glycosylation and N-glycosylation) in regulating crucial TK receptors in carcinogenesis [52]. N-glycosylation of EGFR is essential for the stability and resistant to TK inhibition and N-glycosylation is also indispensable for the oncogenic property of c-Met [53,54]. N-linked glycosylation is a highly conserved post-translational modification via the addition of carbohydrates to regulate functions of various proteins, including protein folding, assembly, intracellular trafficking. As a transmembrane molecule, N-glycosylation commonly occurs at certain asparagine of MerTK. We identified two asparagines at N294 and N454 are important for MerTK N-glycosylation. Intriguingly, 3Y mutants also disturbed MerTK glycosylation. We demonstrated deglycosylation of MerTK deprived pro-tumor growth and metabolic reprogramming properties, which emphasized the significance of MerTK N-glycosylation. Additionally, partial deglycosylated MerTK could be stabilized in response to MG132 treatment in LO2 cells, but not in MHCC97H and HCCLM3 cells. However, TM caused non-glycosylated MerTK could also be stabilized by MG132 in MHCC97H and HCCLM3 cells. We validated that N-glycosylation is indispensable for MerTK to form dimmers which contributes to the increased stabilization. We also found under glucose starvation or TM treatment, MerTK fails to form homodimerization efficiently. Since MerTK, similar to other RTKs, was found to form dimer and activate auto-phosphorylation upon ligand stimulation which is crucial for several downstream signals’ activation. N-glycosylation modification enhanced MerTK stability maybe the reason why MerTK protein level was increased in HCC tissues while the mRNA transcripts remained unchanged. In this context, we found that MerTK depletion increased the sensitivity of HCC cells to cellular stress such as glucose starvation. Under glucose starvation, increased MERTK transcription was exhibited in a time-dependent manner. Previous work demonstrated that in certain instances, TAM may heterodimerize or cross-phosphorylate [55], which may serve as the signaling for the nuclear translocation of MerTK. We proposed that TM treatment caused MerTK deglycosylation, therefore, MerTK may interact with other TAM or kinases to cross-phosphorylate or be phosphorylated. Furthermore, our results showed that decreased nucleus-located MerTK was more sensitive to glucose starvation or TM treatment which highlighted the pro-survival roles of nuclear MerTK. But whether nucleus located MerTK could bind to chromatin subsequently to activate MERTK transcription or regulate other potential transcriptional factors to promote MERTK transcription need more evidences. Complete understanding of molecular mechanism of MerTK in promoting HCC tumorigenesis maybe important for future developing effective therapeutic strategies in the HCC treatment.
5. Conclusions
In summary, we elucidated the critical role of MerTK in regulating HCC cell growth and revealed the N-glycosylation is essential for protein stability and tumor-promoter activity of MerTK. Moreover, phosphorylated MerTK could translocate to nucleus that renders HCC cells a cell survival response. Our present study provides insights into the molecular mechanism of MerTK in promoting HCC tumorigenesis that maybe important for targeting MerTK drug development and developing effective therapeutic strategies in the treatment of HCC.
Funding
This work was supported by grants from National Natural Science Foundation of China (31771534, 31570772 to B.L.), Wenzhou Municipal Science and Technology Bureau grant (no. Y2020178) to Y. L, and the Scientific Research Foundation of University of South China (211RJC002 to B.L.).
Author contributions
B.L. conceived the project and designed the experimental protocols; Y.L., L.L., Y.-J.L., J.L., L.H., Y.D., M.F., J.-W. L., F·S., J.-P.L., J.W., L.W., and K.H. performed most of the experiments; Y.-J.L., and L.H. performed the DEN and CCl4-induced HCC mouse model; L.H. and F.S. assisted the animal experiments; Y.L., L.L., Y.-J.L., L.H., J.-W. L., and B. L. analyzed the data. L.L., Y.-J.L., and J.-W. L. contributed to the statistical analyses of the data. B.L. supervised the study and wrote the manuscript.
Declaration of competing interest
The authors declare that they have no competing interests.
Acknowledgements
We thank Prof. Yi Wu (Soochow University, China) for providing Mer tyrosine receptor knockout mice (Mertk−/−) mice. We also thank Dr. Jianhong Zhu (Wenzhou Medical University, China) for the comments on the manuscript and members of the B. L. Lab for technical support and valuable discussions.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102366.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Forner A., Llovet J.M., Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D., Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 4.Tagger A., Donato F., Ribero M.L., Chiesa R., Portera G., Gelatti U., Albertini A., Fasola M., Boffetta P., Nardi G. Case-control study on hepatitis C virus (HCV) as a risk factor for hepatocellular carcinoma: the role of HCV genotypes and the synergism with hepatitis B virus and alcohol. Brescia HCC Study. Int. J. Cancer. 1999;81(5):695–699. doi: 10.1002/(sici)1097-0215(19990531)81:5<695::aid-ijc4>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 5.Tsukuma H., Hiyama T., Tanaka S., Nakao M., Yabuuchi T., Kitamura T., Nakanishi K., Fujimoto I., Inoue A., Yamazaki H., et al. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N. Engl. J. Med. 1993;328(25):1797–1801. doi: 10.1056/NEJM199306243282501. [DOI] [PubMed] [Google Scholar]
- 6.El-Serag H.B., Mason A.C. Rising incidence of hepatocellular carcinoma in the United States. N. Engl. J. Med. 1999;340(10):745–750. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- 7.Piratvisuth T., Tanwandee T., Thongsawat S., Sukeepaisarnjaroen W., Esteban J.I., Bes M., Köhler B., He Y., Swiatek-de Lange M., Morgenstern D., et al. Multimarker panels for detection of early stage hepatocellular carcinoma: a prospective, multicenter, case-control study. Hepatol Commun. 2022;6(4):679–691. doi: 10.1002/hep4.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siegel R.L., Miller K.D., Jemal A. Cancer statistics. 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 9.El-Serag H.B., Rudolph K.L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 10.Graham D.K., Dawson T.L., Mullaney D.L., Snodgrass H.R., Earp H.S. Cloning and mRNA expression analysis of a novel human protooncogene, c-mer. Cell Growth Differ. 1994;5(6):647–657. [PubMed] [Google Scholar]
- 11.Lai C., Gore M., Lemke G. Structure, expression, and activity of Tyro 3, a neural adhesion-related receptor tyrosine kinase. Oncogene. 1994;9(9):2567–2578. [PubMed] [Google Scholar]
- 12.O'Bryan J.P., Frye R.A., Cogswell P.C., Neubauer A., Kitch B., Prokop C., Espinosa R., 3rd, Le Beau M.M., Earp H.S., Liu E.T. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell Biol. 1991;11(10):5016–5031. doi: 10.1128/mcb.11.10.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linger R.M., Keating A.K., Earp H.S., Graham D.K. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008;100:35–83. doi: 10.1016/S0065-230X(08)00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ling L., Kung H.J. Mitogenic signals and transforming potential of Nyk, a newly identified neural cell adhesion molecule-related receptor tyrosine kinase. Mol. Cell Biol. 1995;15(12):6582–6592. doi: 10.1128/mcb.15.12.6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Georgescu M.M., Kirsch K.H., Shishido T., Zong C., Hanafusa H. Biological effects of c-Mer receptor tyrosine kinase in hematopoietic cells depend on the Grb2 binding site in the receptor and activation of NF-kappaB. Mol. Cell Biol. 1999;19(2):1171–1181. doi: 10.1128/mcb.19.2.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graham D.K., Salzberg D.B., Kurtzberg J., Sather S., Matsushima G.K., Keating A.K., Liang X., Lovell M.A., Williams S.A., Dawson T.L., et al. Ectopic expression of the proto-oncogene Mer in pediatric T-cell acute lymphoblastic leukemia. Clin. Cancer Res. 2006;12(9):2662–2669. doi: 10.1158/1078-0432.CCR-05-2208. [DOI] [PubMed] [Google Scholar]
- 17.Krause S., Pfeiffer C., Strube S., Alsadeq A., Fedders H., Vokuhl C., Loges S., Waizenegger J., Ben-Batalla I., Cario G., et al. Mer tyrosine kinase promotes the survival of t(1;19)-positive acute lymphoblastic leukemia (ALL) in the central nervous system (CNS) Blood. 2015;125(5):820–830. doi: 10.1182/blood-2014-06-583062. [DOI] [PubMed] [Google Scholar]
- 18.Linger R.M., Cohen R.A., Cummings C.T., Sather S., Migdall-Wilson J., Middleton D.H., Lu X., Barón A.E., Franklin W.A., Merrick D.T., et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene. 2013;32(29):3420–3431. doi: 10.1038/onc.2012.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.von Mässenhausen A., Sanders C., Thewes B., Deng M., Queisser A., Vogel W., Kristiansen G., Duensing S., Schröck A., Bootz F., et al. MERTK as a novel therapeutic target in head and neck cancer. Oncotarget. 2016;7(22):32678–32694. doi: 10.18632/oncotarget.8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yi J.H., Jang J., Cho J., Do I.G., Hong M., Kim S.T., Kim K.M., Lee S., Park S.H., Park J.O., et al. MerTK is a novel therapeutic target in gastric cancer. Oncotarget. 2017;8(57):96656–96667. doi: 10.18632/oncotarget.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDaniel N.K., Cummings C.T., Iida M., Hülse J., Pearson H.E., Vasileiadi E., Parker R.E., Orbuch R.A., Ondracek O.J., Welke N.B., et al. MERTK mediates intrinsic and adaptive resistance to AXL-targeting agents. Mol. Cancer Therapeut. 2018;17(11):2297–2308. doi: 10.1158/1535-7163.MCT-17-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen K.Q., Tsou W.I., Calarese D.A., Kimani S.G., Singh S., Hsieh S., Liu Y., Lu B., Wu Y., Garforth S.J., et al. Overexpression of MERTK receptor tyrosine kinase in epithelial cancer cells drives efferocytosis in a gain-of-function capacity. J. Biol. Chem. 2014;289(37):25737–25749. doi: 10.1074/jbc.M114.570838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petta S., Valenti L., Marra F., Grimaudo S., Tripodo C., Bugianesi E., Cammà C., Cappon A., Di Marco V., Di Maira G., et al. MERTK rs4374383 polymorphism affects the severity of fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2016;64(3):682–690. doi: 10.1016/j.jhep.2015.10.016. [DOI] [PubMed] [Google Scholar]
- 24.Cai B., Dongiovanni P., Corey K.E., Wang X., Shmarakov I.O., Zheng Z., Kasikara C., Davra V., Meroni M., Chung R.T., et al. Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell Metabol. 2020;31(2):406–421. doi: 10.1016/j.cmet.2019.11.013. e407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pastore M., Grimaudo S., Pipitone R.M., Lori G., Raggi C., Petta S., Marra F. Role of myeloid-epithelial-reproductive tyrosine kinase and macrophage polarization in the progression of atherosclerotic lesions associated with nonalcoholic fatty liver disease. Front. Pharmacol. 2019;10:604. doi: 10.3389/fphar.2019.00604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Triantafyllou E., Pop O.T., Possamai L.A., Wilhelm A., Liaskou E., Singanayagam A., Bernsmeier C., Khamri W., Petts G., Dargue R., et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut. 2018;67(2):333–347. doi: 10.1136/gutjnl-2016-313615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cavalli M., Pan G., Nord H., Wallén Arzt E., Wallerman O., Wadelius C. Genetic prevention of hepatitis C virus-induced liver fibrosis by allele-specific downregulation of MERTK. Hepatol. Res. 2017;47(8):826–830. doi: 10.1111/hepr.12810. [DOI] [PubMed] [Google Scholar]
- 28.Azimzadeh Irani M., Kannan S., Verma C. Role of N-glycosylation in EGFR ectodomain ligand binding. Proteins. 2017;85(8):1529–1549. doi: 10.1002/prot.25314. [DOI] [PubMed] [Google Scholar]
- 29.Contessa J.N., Bhojani M.S., Freeze H.H., Rehemtulla A., Lawrence T.S. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 2008;68(10):3803–3809. doi: 10.1158/0008-5472.CAN-07-6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sethi M.K., Kim H., Park C.K., Baker M.S., Paik Y.K., Packer N.H., Hancock W.S., Fanayan S., Thaysen-Andersen M. In-depth N-glycome profiling of paired colorectal cancer and non-tumorigenic tissues reveals cancer-, stage- and EGFR-specific protein N-glycosylation. Glycobiology. 2015;25(10):1064–1078. doi: 10.1093/glycob/cwv042. [DOI] [PubMed] [Google Scholar]
- 31.Chandler K.B., Leon D.R., Meyer R.D., Rahimi N., Costello C.E. Site-specific N-glycosylation of endothelial cell receptor tyrosine kinase VEGFR-2. J. Proteome Res. 2017;16(2):677–688. doi: 10.1021/acs.jproteome.6b00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaetzel D.M., Jr., Morgan D., 3rd, Reid JDt, Fenstermaker R.A. Site-directed mutagenesis of the N-linked glycosylation site in platelet-derived growth factor B-chain results in diminished intracellular retention. Biochim. Biophys. Acta. 1996;1298(2):250–260. doi: 10.1016/s0167-4838(96)00136-7. [DOI] [PubMed] [Google Scholar]
- 33.Góra-Sochacka A., Redkiewicz P., Napiórkowska B., Gaganidze D., Brodzik R., Sirko A. Recombinant mouse granulocyte-macrophage colony-stimulating factor is glycosylated in transgenic tobacco and maintains its biological activity. J. Interferon Cytokine Res. 2010;30(3):135–142. doi: 10.1089/jir.2009.0053. [DOI] [PubMed] [Google Scholar]
- 34.Kazui A., Ono M., Handa K., Hakomori S. Glycosylation affects translocation of integrin, Src, and caveolin into or out of GEM. Biochem. Biophys. Res. Commun. 2000;273(1):159–163. doi: 10.1006/bbrc.2000.2903. [DOI] [PubMed] [Google Scholar]
- 35.Li J.H., Huang W., Lin P., Wu B., Fu Z.G., Shen H.M., Jing L., Liu Z.Y., Zhou Y., Meng Y., et al. N-linked glycosylation at Asn152 on CD147 affects protein folding and stability: promoting tumour metastasis in hepatocellular carcinoma. Sci. Rep. 2016;6 doi: 10.1038/srep35210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nocito A., Bubendorf L., Tinner E.M., Süess K., Wagner U., Forster T., Kononen J., Fijan A., Bruderer J., Schmid U., et al. Microarrays of bladder cancer tissue are highly representative of proliferation index and histological grade. J. Pathol. 2001;194(3):349–357. doi: 10.1002/1096-9896(200107)194:3<349::AID-PATH887>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 37.Liu Y., Lan L., Huang K., Wang R., Xu C., Shi Y., Wu X., Wu Z., Zhang J., Chen L., et al. Inhibition of Lon blocks cell proliferation, enhances chemosensitivity by promoting apoptosis and decreases cellular bioenergetics of bladder cancer: potential roles of Lon as a prognostic marker and therapeutic target in baldder cancer. Oncotarget. 2014;5(22):11209–11224. doi: 10.18632/oncotarget.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qi J., Zhou N., Li L., Mo S., Zhou Y., Deng Y., Chen T., Shan C., Chen Q., Lu B. Ciclopirox activates PERK-dependent endoplasmic reticulum stress to drive cell death in colorectal cancer. Cell Death Dis. 2020;11(7):582. doi: 10.1038/s41419-020-02779-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park E.J., Lee J.H., Yu G.Y., He G., Ali S.R., Holzer R.G., Osterreicher C.H., Takahashi H., Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Uehara T., Pogribny I.P., Rusyn I. The DEN and CCl4 -induced mouse model of fibrosis and inflammation-associated hepatocellular carcinoma. Curr. Protoc. Pharmacol. 2014;66 doi: 10.1002/0471141755.ph1430s66. 14.30.11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xie D., Wu X., Lan L., Shangguan F., Lin X., Chen F., Xu S., Zhang Y., Chen Z., Huang K., et al. Downregulation of TFAM inhibits the tumorigenesis of non-small cell lung cancer by activating ROS-mediated JNK/p38MAPK signaling and reducing cellular bioenergetics. Oncotarget. 2016;7(10):11609–11624. doi: 10.18632/oncotarget.7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wittig I., Braun H.P., Schagger H. Blue native PAGE. Nat. Protoc. 2006;1(1):418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 43.Nishi C., Yanagihashi Y., Segawa K., Nagata S. MERTK tyrosine kinase receptor together with TIM4 phosphatidylserine receptor mediates distinct signal transduction pathways for efferocytosis and cell proliferation. J. Biol. Chem. 2019;294(18):7221–7230. doi: 10.1074/jbc.RA118.006628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crittenden M.R., Baird J., Friedman D., Savage T., Uhde L., Alice A., Cottam B., Young K., Newell P., Nguyen C., et al. Mertk on tumor macrophages is a therapeutic target to prevent tumor recurrence following radiation therapy. Oncotarget. 2016;7(48):78653–78666. doi: 10.18632/oncotarget.11823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ammoun S., Provenzano L., Zhou L., Barczyk M., Evans K., Hilton D.A., Hafizi S., Hanemann C.O. Axl/Gas6/NFκB signalling in schwannoma pathological proliferation, adhesion and survival. Oncogene. 2014;33(3):336–346. doi: 10.1038/onc.2012.587. [DOI] [PubMed] [Google Scholar]
- 46.Graham D.K., DeRyckere D., Davies K.D., Earp H.S. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer. 2014;14(12):769–785. doi: 10.1038/nrc3847. [DOI] [PubMed] [Google Scholar]
- 47.Cummings C.T., Deryckere D., Earp H.S., Graham D.K. Molecular pathways: MERTK signaling in cancer. Clin. Cancer Res. 2013;19(19):5275–5280. doi: 10.1158/1078-0432.CCR-12-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 49.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 50.Schlegel J., Sambade M.J., Sather S., Moschos S.J., Tan A.C., Winges A., DeRyckere D., Carson C.C., Trembath D.G., Tentler J.J., et al. MERTK receptor tyrosine kinase is a therapeutic target in melanoma. J. Clin. Invest. 2013;123(5):2257–2267. doi: 10.1172/JCI67816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Migdall-Wilson J., Bates C., Schlegel J., Brandão L., Linger R.M., DeRyckere D., Graham D.K. Prolonged exposure to a Mer ligand in leukemia: Gas6 favors expression of a partial Mer glycoform and reveals a novel role for Mer in the nucleus. PLoS One. 2012;7(2) doi: 10.1371/journal.pone.0031635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mehta A., Herrera H., Block T. Glycosylation and liver cancer. Adv. Cancer Res. 2015;126:257–279. doi: 10.1016/bs.acr.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yen H.Y., Liu Y.C., Chen N.Y., Tsai C.F., Wang Y.T., Chen Y.J., Hsu T.L., Yang P.C., Wong C.H. Effect of sialylation on EGFR phosphorylation and resistance to tyrosine kinase inhibition. Proc. Natl. Acad. Sci. U. S. A. 2015;112(22):6955–6960. doi: 10.1073/pnas.1507329112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen R., Li J., Feng C.H., Chen S.K., Liu Y.P., Duan C.Y., Li H., Xia X.M., He T., Wei M., et al. c-Met function requires N-linked glycosylation modification of pro-Met. J. Cell. Biochem. 2013;114(4):816–822. doi: 10.1002/jcb.24420. [DOI] [PubMed] [Google Scholar]
- 55.Brown J.E., Krodel M., Pazos M., Lai C., Prieto A.L. Cross-phosphorylation, signaling and proliferative functions of the Tyro3 and Axl receptors in Rat2 cells. PLoS One. 2012;7(5) doi: 10.1371/journal.pone.0036800. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.