

Abstract
Until now, renin-angiotensin system (RAS) hyperactivity was largely thought to result from angiotensin II (Ang II) dependent stimulation of the Ang II type 1 receptor (AT1R). Here we assessed the role of soluble (pro)renin receptor (sPRR), a product of site-1 protease-mediated cleavage of (pro)renin receptor (PRR), as a possible ligand of the AT1R in mediating: (i) endothelial cell dysfunction in vitro; and (ii) arterial dysfunction in mice with diet-induced obesity. Primary human umbilical vein endothelial cells (HUVECs) treated with a recombinant histidine-tagged sPRR (sPRR-His) exhibited IκBα degradation concurrent with NF-κB p65 activation. These responses were secondary to sPRR-His evoked elevations in Nox4-derived H2O2 production that resulted in inflammation, apoptosis and reduced NO production. Each of these sPRR-His-evoked responses was attenuated by AT1R inhibition using Losartan (Los) but not ACE inhibition using captopril (Cap). Further mechanistic exploration revealed that sPRR-His activated AT1R downstream Gq signaling pathway. Immunoprecipitation coupled with autoradiography experiments and radioactive ligand competitive binding assays indicate sPRR directly interacts with AT1R via Lysine199 and Asparagine295. Important translational relevance was provided by findings from obese C57/BL6 mice that sPRR-His evoked endothelial dysfunction was sensitive to Los. Besides, sPRR-His elevated blood pressure in obese C57/BL6 mice, an effect that was reversed by concurrent treatment with Los but not Cap. Collectively, we provide solid evidence that the AT1R mediates the functions of sPRR during obesity-related hypertension. Inhibiting sPRR signaling should be considered further as a potential therapeutic intervention in the treatment and prevention of cardiovascular disorders involving elevated blood pressure.
Keywords: soluble (pro)renin receptor, Angiotensin II type 1 receptor, endothelial dysfunction, hypertension
Introduction
In 2002, Nguyen et al. cloned a novel receptor for prorenin and renin, termed it the (pro)renin receptor (PRR), and assigned a renin-regulatory function to this receptor.(1) On the basis of structure, PRR belongs to the type I transmembrane receptor family, consisting of a large N-terminal extracellular domain, a single transmembrane protein, and a short cytoplasmic domain.(2) The soluble (pro)renin receptor (sPRR) is generated by protease-mediated cleavage in the Golgi apparatus and is released to plasma or urine.(3) Although furin and ADAM19 were initially thought to be the cleavage enzyme, independent studies by Nakagawa et al. (4) and us (5) have consistently identified site-1 protease (S1P) as being the predominant source of sPRR. PRR/sPRR are considered as new members of the RAS, one of the most important hormonal systems for homeostatic control of fluid, electrolytes, vasculature and blood pressure as well as for the pathogenesis of cardiovascular disease.(6, 7)
The prevalence of obesity-related disorders such as atherosclerosis, hypertension and other cardiovascular diseases is increasing at an alarming rate.(8) Endothelial dysfunction is a common factor, which plays an important role in the development of cardiovascular complications of obesity.(9) Vascular endothelial cells play a major role in maintaining cardiovascular homeostasis in health. Endothelium contributes to the regulation of vascular function and blood pressure largely by releasing NO.(10) Increased serum sPRR levels have been demonstrated in patients with obesity-related disorders, such as atherosclerosis(11), heart failure(12) and hypertension(13). While each of these conditions is associated with endothelial dysfunction, the influence of sPRR on endothelial cells has not been determined.
Within the vasculature, PRR is abundantly and specifically expressed in the vascular smooth muscle cells (VSMCs) but not endothelial cells.(1) We have reported that Ang II increased PRR expression in VSMCs and blood pressure in vivo, and stimulated sPRR release in cultured rat VSMCs in vitro, and each effect could be attenuated by PRR inhibition.(6) Therefore, we speculate that VSMC-derived sPRR may act in a paracrine fashion to induce endothelial dysfunction, which plays an important role in the development of hypertension.
NF-κB (nuclear factor kappa light chain enhancer of activated B cells) is a family of highly conserved transcription factors involved in regulating inflammatory responses as well as other cellular functions.(14) NF-κB activation is an important precursor to vascular inflammation and apoptosis, resulting in the development of several cardiovascular diseases.(15) The p65 protein of NF-κB is localized to the cytosol under basal conditions by the inhibitor IκBα. When IκBα is degraded, p65 becomes phosphorylated and translocates to the nucleus.(14) Thus, IκBα status indirectly reflects the translocation and activation of NF-κB.(16)
Physiological levels of the Ang II play an essential role in homeostatic regulation of fluid, electrolytes, vasculature and blood pressure by activating AT1R. Overstimulation of AT1R, however, has been implicated in many human diseases that include but are not limited to hypertension, atherosclerosis, heart failure, aortic aneurysm, Marfan’s syndrome, and age-related aortic stiffness.(17–22) These pathologies often are effectively treated with AT1R blockers i.e., ARBs, even though evidence exists that these conditions may exist in the presence of physiological concentrations of circulating renin or Ang II . While tissue levels of Ang II might be elevated, it is certainly possible that activation of AT1R during cardiovascular disease might be Ang II-independent.
A major goal of the present study was to test the hypothesis that sPRR may induce inflammatory and apoptotic response in vascular endothelial cells, contributing to obesity-related endothelial dysfunction and hypertension. During the course of this investigation, we observed that the vascular effect of sPRR occurred in the absence of elevated Ang II but was abolished by an AT1R blocker losartan, raising a possibility that sPRR may directly activate AT1R independently of the activity of canonical RAS. Therefore, we further examined the direct interaction between sPRR and AT1R in the context of vascular endothelial injury.
Materials and Methods
Plasmid Construction and sPRR Protein Purification
The cDNA of PRR (Gen-Bank accession no. NM_001007091.1; also known as “ATP6AP2”) was subcloned into the pMD-18T vector (Takara, Kusatsu city, Shiga prefecture, Japan). sPRR, a solubilized form of PRR (residues 17–274) lacking the transmembrane domain at the C terminus, was combined with an eight-histidine tag in the C terminus (sPRR-His) generated by PCR from the PRR expression construct, and subsequently cloned into pcDNA3.1. sPRR-His was created using a mammalian 293 cell system and was purified by binding to IDA-Sephadex G-25 (GE Healthcare) (XBIO, Shanghai, China). sPRR-His was separated as a single 29.6-kDa band in 12% SDS/PAGE gel, which proves the quality and the purity of the his-tagged sPRR recombinant protein.(23)
Materials
Endothelial cell medium (ECM) (cat. no.1001) was purchased from ScienCell (Carlsbad, CA, USA). The dual Nox1/4 inhibitor GKT137892 was a gift from Genkyotex SA (Geneva, Switzerland). The NF-κB inhibitor ammonium pyrrolidinedithiocarbamate (PDTC; cat. no. ab141406), the anti-Nox4 antibody (cat. no. ab133303) and the anti-PKC antibody (cat. no. ab23511) were purchased from Abcam (Cambridge, Massachusetts, USA). Monoclonal antibodies against phosphorylated-NF-κB p65 (cat. no.3033), NF-κB p65 (cat. no.8242), IκBα (cat. no.4812), cleaved caspase-3 (Asp175) (cat. no.9664), p-ERK1/2 (cat. no.9101), t-ERK1/2 (cat. no.9102), p-PKC (cat. no.9371), p-eNOSS1177 (cat. no.9570) and eNOS (cat. no.9586) were obtained from Cell Signaling Technology (Danvers, Massachusetts, USA). The AT1R inhibitor Losartan (HY-17512) was obtained from MedChem Express (New Jersey, USA). Other chemicals were obtained from Sigma (St. Louis, MO, USA).
Cell culture
The primary human umbilical vein endothelial cells (HUVECs) were gifts from Dr. Guoquan Gao and Dr. Xia Yang (Sun Yat-sen University, Guangzhou, China). All procedures were approved by the Ethics Committee of Sun Yat-sen University in accordance with the Declaration of Helsinki. HUVECs were isolated and cultured as described previously.(24) Briefly, cells were grown at 37°C in a humidified atmosphere containing 5% CO2/95% oxygen. All treatments were performed at passages 3–6 in HUVECs grown to 70–90% confluence. Cells were serum starved and pretreated with a serum-free medium that contained 10μM GKT137892, 50μM PDTC, 10μM Losartan or 10μM Captopril for one hour. Next, cells were incubated with a serum-free medium that contained 50nM sPRR-His. An equal volume of blank buffer served as a vehicle (Veh) control for sPRR-His and an equal volume of DMSO or PBS was used as the Veh control for GKT137892, PDTC, Losartan and Captopril. 200μM hydrogen peroxide (H2O2) served as a positive control treatment.(25)
Small interfering RNA (siRNA) transfection
Nox4 siRNA (cat. no.AM16708) and scrambled siRNA (cat. no.AM4636) were purchased from Invitrogen (Carlsbad, California, USA). The AT1R siRNA was designed and purchased from GenePharma (Shanghai, China). At 50–70% confluence, HUVECs were transfected with Nox4 siRNA, AT1R siRNA or scrambled siRNA using HiPerFect Transfection Reagent (cat. no.301707, QIAGEN, Dusseldorf, Germany). All siRNA transfected cells were harvested for RNA analyses after 48 hours, and for western blot and ELISA procedures after 72 hours.(25, 26)
Western blot analysis
HUVECs were lysed for 30-min on ice with a RIPA lysis buffer (Beyotime, Shanghai, China) including protease inhibitor cocktail (REF 04693116001, Roche, Basel, Switzerland). Next, whole cell lysates were collected, sonicated, and centrifuged for 10 min × 12000 rpm × 4°C. The supernatant was used to detect protein (BCA Protein Assay Kit; cat. no.23225, Thermo, Rockford, USA). An equal amount of protein was loaded onto SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore, Massachusetts, USA). After blocking with 5% skimmed milk for 1 h at room temperature, membranes were incubated with the appropriate primary antibodies overnight at 4°C. Next, membranes were washed with TBST (NaCl 8.0 g, Tris 6.0 g, Tweeen-20 1.0 %), and secondary antibodies including HRP-linked anti-mouse or HRP-linked anti-rabbit antibody were incubated at room temperature for 1 h. β-Actin (1:10000, Sigma) was used as a loading control. Antibody labeling was visualized by addition of chemiluminescence substrate for detection of HRP (cat. no.WBKLS0500, Millipore).(5, 26)
Quantitative RT-PCR
Total RNA was extracted using Trizol (Invitrogen) and then reverse transcribed to cDNA using the Transcriptor First Strand cDNA Synthesis Kit (REF 04897030001, Roche). Real-time PCR was performed on the AB Applied Biosystems using the Fast Start Universal SYBR Green Master (ROX) (REF 04913914001, Roche). Values were normalized using GAPDH content. The gene primers used in this study are shown in Supplementary Table S1.(25, 26)
Enzyme-linked immunosorbent assay
To evaluate the secretion of IL-6, IL-8 and Angiotensin II, cellular supernatant was collected subsequent to each treatment, aliquoted into eppendorf tubes, and stored at −80°C. IL-6, IL-8 and Angiotensin II concentrations were quantified using commercially available ELISA kits (Human IL-6 Platinum ELISA: BMS213/2, eBioscience, Tasmania, Australia; Human IL-8 Platinum ELISA: BMS204/3, eBioscience; Angiotensin II ELISA kit: ADI-900–204, Enzo Life Sciences, Farmingdale, USA; Bradykinin ELISA kit: ADI-900–206, Enzo Life Sciences) according to procedures recommended by the manufacturer.(5, 26)
Measurement of H2O2
H2O2 was measured using the ROS-Glo H2O2 Assay kit (cat. no. G8820, Promega, Wisconsin, USA) according to the manufacturer’s instructions. The assay is based on a luminescent signal generated by a chemical reaction of H2O2 and its substrate. Luminescence was measured using a FLUOstar OPTIMA microplate reader (BMG Labtech, Ortenberg, Germany) and the value was normalized to protein content.(25)
Flow cytometry
Apoptosis was quantified with the Annexin V-FITC/PI Apoptosis Detection Kit (Beyotime, Shanghai, China) according to the manufacturer’s instructions. At the end of the treatment, cells were trypsinized and then carefully transferred to a new centrifuge tube with corresponding supernatant. After centrifugation (1000 rpm) for 5 min, the cell pellet was washed with cold phosphate buffer saline (PBS), and resuspended in Annexin V-FITC/PI binding buffer to a concentration of 105 cells/195μl. The cell suspension was stained with 5μl Annexin V-FITC and 10μl propidium iodide (PI) and then was incubated for 20 min at room temperature in the dark. Samples were collected for flow cytometry (Bechman Coulter Epics XL-MCL Flow Cytometer, Miami, Florida, USA) and data were analyzed using FlowJo (Ashland, Oregon, USA) software.(26)
Ang II-I125 radioactive ligand competitive binding assay
HUVECs were pretreated with 1nM, 10nM, 50nM sPRR-His or 10μM Losartan for 30min followed by exposure to 1μM Ang II-I125 (NEX105050UC, perkinelmer, Massachusetts, Waltham, USA). To remove the unbound Ang II-I125, the cells were washed by PBS twice after removing cell medium. Next, HUVECs were lysed with a RIPA lysis buffer (Beyotime, Shanghai, China) including protease inhibitor cocktail (REF 04693116001, Roche, Basel, Switzerland). Whole cell lysates were collected, mixed, transferred to polystyrene tubes, and incubated at room temperature for 1 hour, followed by the measurement of radioactivity using a gamma counter (SN-6105, Shanghai He Suo Ri Huan photoelectric instrument co., LTD, Shanghai, China). Non-specific binding (control group cells without Ang II-I125 treatment) was subtracted from all measured values. The measured CPM represent Ang II-I125 that is combined with cellular AT1R. All values were normalized to protein content of the sample.(27)
Construction of plasmids and immunoprecipitation
The pcDNA3.1-sPRR-His plasmid was constructed by subcloning rat sPRR cDNA into the pcDNA3.1 vector. sPRR, a solubilized form of PRR (residues 17–274) lacking the transmembrane domain at the C terminus, was combined with a six-histidine tag in the C terminus (sPRR-His), was generated by PCR from the PRR expression construct, and then was cloned into pcDNA3.1 vector. pCMV6-AT1 plasmid was purchased from Origene, its four different mutants: pCMV6-AT1S109A, pCMV6-AT1K199A, pCMV6-AT1N295A and pCMV6-AT1S109A, K199A, N295A were also generated by PCR to introduce site-mutation or site-mutations into pCMV6-AT1 plasmid (The primers are shown in Supplementary Table S1). All generated plasmids were verified by DNA sequencing (DNA sequencing core, University of Utah).
35S-labeled proteins were synthesized by T7 TNT in vitro transcription and in vitro translation system (Promega). These mammalian-based systems express soluble, functional proteins that are post-translationally modified. Expressed proteins can be used directly after expression; no additional protein purification needed. Following instructions from the manual, 1 μg of pcDNA3.1 vector, pcDNA3.1-sPRR-His, pCMV6-AT1 plasmid or its mutants with 2μl of 35S-methionine (10 μCi/μl, PerkinElmer) were added to 40μl of T7 TNT in vitro transcription and translation system at 30°C for 2 hours. To test the protein interaction, 0.5μg of recombinant human AT1 protein (Novus Biologicals) or sPRR-His protein (XBIO, Shanghai, China) was incubated with the in vitro translation mixture at 4°C overnight. The bound proteins to the unlabeled AT1 protein or sPRR-His protein were precipitated by incubating the mixture with 2μg of the rabbit anti AT1 antibody (Santa Cruz Biotechnology, Inc.) pelleted with protein G agarose (Roche), or by incubating with 2 μg of PRR antibody (Sigma) against mouse PRR 32–45 AA (Thermo fisher Com.), antibody against mouse PRR 219–235 AA (Thermo fisher Com.) and then pelleted with protein A agarose (Thermo fisher Com.). The same amount of rabbit IgG was used as the control for these antibodies. Those pelleted products were dissolved in SDS loading buffer and separated by SDS-PAGE (polyacrylamide) gel. The radioactive signals were captured by a phosphor imager (LS6500 Multipurpose Scintillation Counter, Beckman Coulter, Indianapolis, USA).
35S-sPRR radioactive ligand competitive binding assay
When HUVECs reached 50% confluence, 0, 5, 10 or 20 μM Losartan was added into the medium. One hour later, 35S-sPRR-His was added to media at a dilution of 1: 500. As mentioned earlier, 35S-sPRR-His was obtained by in vitro translation. To eliminate free 35S-methionine, the in vitro translation mixture was centrifuged with a 10 kD filter at 4200 Revolutions Per Minute (rpm) for 30 min. The free 35S-methionine was filtered, and 35S-sPRR-His was obtained in the unfiltered solution. Then the unfiltered solution was diluted with culture medium (Lifeline) and added to the cultured cells. Thirty min, 3 h, or 15 h after adding the 35S-sPRR-His, the medium was removed, cells were washed with PBS five times, and then treated with lysis buffer (Sigma Aldrich). The radioactivity of the cell lysate was measured with a multi-purpose scintillation counter (LS 6500 Beckman Coulter).
Nitric oxide measurement
Nitric oxide generation was estimated via 4-amino-5-methylamino-2′, 7′-difluorofluorescein diacetate (DAF-FM Diacetate; Life technologies, D-23844) according to procedures recommended by the manufacturer. HUVECs treated with 500μM of 2-(N, N-Diethylamino)-diazenolate-2-oxide diethylammonium salt (DEA NoNoate; Cayman Chemicals, 82100) were used as a positive control, whereas cells treated with 500μM of NG-monomethyl L-arginine citrate (LNMMA; Sigma Aldrich, M7033) were used to inhibit NO generation.(28)
Immunofluorescence staining
To test the localization of NF-κB, HUVECs were treated with sPRR-His (50nM) for different times. Cells on glass coverslips were fixed with 4% paraformaldehyde for 15 minutes and permeabilized with 0.2% Triton X-100 for 15 minutes. Coverslips were blocked in 1% BSA for 1 h and were then incubated with the anti-NF-κB p65 antibody (1:100 dilution; cat. no.8242, Cell Signaling Technology) overnight at 4°C. After washing, cells were incubated for 1 h at room temperature with Alexa Fluor® 555 Donkey anti-Rabbit IgG (H+L) (1:200, cat. no. A31572, Life Technologies, Grand Island, NY, USA). After washing off the secondary antibody, nuclei were stained in VECTASHIELD® Hard Set Mounting Medium with DAPI (cat. no.H-1500, Vector Laboratories, Burlingame, CA, USA). Images were captured using a Leica DMI4000B fluorescence microscope (Wetzlar, Germany).(7) (28)
Nuclear and cytosolic separation
Nuclear proteins were isolated using a commercially available kit (Nuclear Extraction Kit, item No.10009277, Cayman, Ann Arbor, Michigan, USA) according to manufacturer’s instructions. Briefly, adherent cells were dislodged following the respective treatments, collected in pre-chilled tubes, and centrifuged at 300 × g for 5-min at 4°C. The pellet was resuspended in 5 ml of ice-cold PBS/Phosphatase Inhibitor Solution, centrifuged at 300 × g for 5-min at 4°C, and this step was repeated. Next, 500μl ice-cold 1X Complete Hypotonic Buffer was added to the pellet, and the cells incubated on ice for 15 min to allow cells to swell. After adding 100μl of 10% NP-40 Assay Reagent to the mixture, the solution was centrifuged for 30 s at 4°C. The supernatant containing the cytosolic fraction was transferred to a new tube. The pellet was resuspended in 100μl ice-cold Complete Nuclear Extraction Buffer, vortexed vigorously for 15–30 s, placed on ice on a rocker for 15-min, and centrifuged at 14000 × g for 10 min at 4°C. The resulting supernatant contained the nuclear fraction. Protein concentration was quantified using Cayman’s Protein Determination Kit (Item No.704002). The presence of the cytosolic reference protein β-Tubulin or β-Actin in the cytosolic but not the nuclear fraction was used to confirm successful fractionation.(23)
NF-κB transcription factor activity assay
After the respective treatments, nuclear and cytosolic proteins were isolated as described earlier. Binding of NF-κB in the nuclear extract to the NF-κB response element was detected with NF-κB p65 Transcription Factor Assay Kit (Cayman, Item No.10007889). Briefly, 10 μl of nuclear extract was added to wells coated with a specific double-stranded DNA sequence. Next, 90 μl of complete transcription factor buffer was added to blank wells, a positive control, non-specific binding wells, and competitor dsDNA wells. After overnight incubation at 4°C, wells were washed five times and incubated with NF-κB (p65) antibody (except for blanks) for 1 h at room temperature. Wells were subsequently washed five times and incubated with an HRP-conjugated secondary antibody (except for blanks) for 1 h at room temperature. Each well was subsequently washed, and 100 μl of developing solution was added for 45-min until 100μl stop solution was added.
Measurement of intracellular ROS
The formation of intracellular ROS was measured via monitoring the increasing fluorescence of 2′, 7′-dichlorofluorescein (DCF). The cell-permeant 2′, 7′-dichlorodihydrofluorescein diacetate (H2DCF-DA, cat. no. D399, Thermo Fisher Scientific, Grand Island, USA) enters the cell where intracellular esterases cleave off the diacetate group. The resulting H2DCF is retained in the cytoplasm and oxidized to DCF by ROS. HUVECs were seeded into each well of a 96-well plate and allowed to attach overnight. Subsequently, the cells were washed once with HBSS and treated with 50nM sPRR-His. After the incubation for different time periods from 30min to 3h, cells were washed with HBSS, loaded with 50μM H2DCF-DA for 30 min and washed again with HBSS. After a further incubation in HBSS for 1 h the fluorescence intensities were measured at 492–495 nm excitation and 517–527 nm emission in the fluorescence reader (TECAN infinite M200 PRO, TECAN, Switzerland).(28)
Measurement of the intracellular free calcium concentration (Ca2+)
The intracellular Ca2+ concentration was measured using Fluo-3-AM (Abcam, ab145254). HUVECs were washed three times with PBS and incubated with 10μM Fluo-3-AM for 30 min at 37 °C in 5% CO2. After proper rinsing, Ca2+ fluorescence was recorded and quantified by Automatic living cell imaging analysis system (BioTek lionheart FX). Images were captured every 10s after sPRR-His stimulation.(27)
Animal care
All animal protocols were approved by the Animal Care and Use Committee at the University of Utah. All animal work took place in the Tianxin Yang’s lab at University of Utah. All procedures conform to the NIH Guide for the Care and Use of Laboratory Animals. All animals were cage-housed and maintained in a temperature-controlled room with a 12:12-h light-dark cycle, with free access to tap water and standard chow. The euthanasia for animals was done by inhalant isoflurane with anesthetic vaporizer.
Animal model
Male C57/BL6 mice were purchased from Taconic (Rensselaer, New York). At one month of age, animals were placed on a high-fat diet for 9 months i.e., 60% kcal high fat irradiated diet (Cat#D12492, Research Diets, New Brunswick, NJ). At 33 weeks of high fat diet, all animals were anesthetized with isoflurane by using inhalation anesthesia machine (Induction 3–5% in O2, maintenance 1.5–2% in O2), radiotelemetric devices (DSI) were impanated through catheterization of left carotid artery and they were allowed to recover for 1 week. After collection of baseline data for 2 days, the mice were infused with sPRR-His at 30 μg/kg/d via jugular vein catheterization connected to a separate osmotic minipump (Alzet model 1002, Alza, Palo Alto, CA) alone or combined treatment with subcutaneous infusion of Losartan at 5 mg/kg/d or Captopril at 50 mg/kg/d (29–31) via another minipump for 14 days.
Vascular reactivity
24 h after collecting blood pressure and HR data for 15 days, mice were anesthetized using 2–5% isoflurane, the chest was opened, and the heart was excised. Mesenteric arteries were dissected free from adherent tissue, and their reactivity were evaluated ex vivo using isometric procedures.
Segments of mesenteric artery (~ 150μm, internal diameter) were mounted on a wire myograph system while immersed in iced NPSS. After a 30-min equilibration period wherein the vessel chamber was heated to 37°C, 100 mg tension was placed on the vessel at 10-min intervals until 400 mg tension was achieved. Next, a series of internal circumference-active tension curves was completed to determine the vessel diameter that evoked the greatest tension development (Lmax) to 100mM potassium chloride (KCl). Four dose-response curves then were performed, each separated by 30-min. Non-receptor mediated and receptor mediated vasoconstrictive responses to KCl (20–100 mM) and phenylephrine (PE, 10−8-10−5 M), respectively, were assessed. After arteries were pre-contracted to ~65% of maximal PE-induced contraction and tension was stable, responses to acetylcholine (ACh, 10−8-10−6 M; to determine endothelium-dependent vasorelaxation) and sodium nitroprusside (SNP, 10−9-10−4 M; to determine endothelium-independent vasorelaxation) were evaluated. All tension data were recorded continuously using an analog-to-digital interface card (Biopac Systems Inc., Santa Barbara, CA) that allowed for subsequent off-line quantitative analyses. Vascular reactivity was assessed in two segments of mesenteric artery per mouse per group.(32–34)
Statistical analyses
Data are presented as mean ± standard error of the mean. Significance was accepted when p<0.05. A test to determine normality of distribution for each data set was performed by GraphPad Prism software. For comparison among three or more mean values, if the data were distributed normally, a one-way ANOVA was performed to determine whether significant differences exist among groups. If significance was obtained, a Tukey’s post hoc test was used to identify the location of the differences. If the data were not distributed normally, a Kruskall-Wallis ANOVA was performed. If significance was obtained, a Dunn’s post hoc test was used to identify the location of the differences. For comparison among two mean values, a paired or unpaired t-test was performed as appropriate.(28, 35)
Results
sPRR-His activates NF-κB in HUVECs.
In primary HUVECs, sPRR-His reduced IκBα expression (Fig. S1A), increased NF-κB p-p65 protein (Fig. S1B), and elevated nuclear NF-κB p65 transcription factor activity (Fig. S1C) at 2h and 3h. Each response was maximal at 2h. Our ability to successfully separate the cytosolic and nuclear fractions is shown in Fig. S2A. Immunofluorescence staining supported these data i.e., 50nM sPRR-His activated NF-κB p65 maximally at 2h (Fig. S1D).
sPRR-His activates NF-κB secondary to increasing intracellular Nox4-derived H2O2.
The formation of intracellular ROS was measured via monitoring the increasing fluorescence of 2’7’-dichlorofluorescein (DCF), and results showed that sPRR-His increased intracellular ROS generation at 30-min (Fig. S2C). Subsequently, this result has been validated by using an independent method, e.g. the ROS-Glo H2O2 assay for measurement of H2O2 (Fig. S2D). Evidence exists that ROS are capable of NF-κB activation.(36) Therefore, we investigated whether sPRR-His leads to NF-κB activation by increasing cellular H2O2. In support of this notion, the important role of ROS was confirmed by findings that exogenous H2O2 recapitulated sPRR-His treatment concerning IκBα degradation and NF-κB p-p65 up-regulation (Fig. S3D). This is an evidence that H2O2 is an important mediator of sPRR-His-induced NF-κB activation in HUVECs.
We next sought to determine the source of sPRR-His evoked H2O2 production. Nox4 is an NADPH oxidase subunit capable of generating H2O2 in endothelial cells.(15) sPRR-His only elevated Nox4 mRNA and protein expression at 1, 2, and 3h, with a maximal response observed at 2h (Fig. S3, A–B). A pharmacological and genetic approach were used next to assess the involvement of Nox4 in sPRR-His-induced H2O2 generation and NF-κB activation. The dual Nox1 and Nox4 inhibitor GKT137892 (GKT) attenuated sPRR-His evoked H2O2 generation (Fig. S3C), IκBα degradation and NF-κB p-p65 up-regulation (Fig. S3D). Further, HUVECs with efficacious knockdown of Nox4 mRNA (Fig. S2B) were refractory to sPRR-His-induced H2O2 generation (Fig. S3E), IκBα degradation and NF-κB p-p65 up-regulation (Fig. S3F). Taken together, these results indicate sPRR-His-induced H2O2 generation is derived from Nox4 and is capable of activating NF-κB.
sPRR-His evokes inflammation.
We sought to determine whether sPRR-His activates NF-κB to an extent that evokes inflammation in HUVECs. sPRR-His increased IL-6, IL-8, VCAM-1 and ICAM-1 mRNA expression levels at 2h, 3h and 6h (Fig. S4A). The maximal response was observed at 3h, remained elevated at 6h, but decreased to basal levels by 9h and 12h (Fig. S4A). Moreover, sPRR-His dose-dependently evoked inflammation, the noticeable changes of cytokine expression were observed at as low as 1 nM and maximal responses were seen at 50 nM (Fig. S4B). Importantly, sPRR-His evoked mRNA indices of inflammation after 3-h treatment were attenuated by the dual Nox1 and Nox4 inhibitor GKT, Nox4 siRNA and NF-κB inhibition using PDTC (Fig. S4, C–E). Likewise, while sPRR-His elevated protein indices of inflammation i.e., IL-6 and IL-8 expression, these responses were inhibited by Nox1/4 inhibition, Nox4 siRNA and NF-κB blockade (Fig. S5, A–C). Collectively, sPRR-His evokes inflammation that is secondary to Nox4/H2O2/NF-κB activation in HUVECs.
sPRR-His increases apoptosis.
Oxidative stress contributes importantly to apoptosis(37) and this was evaluated in the context of the current experimental conditions using two approaches. sPRR-His elevated the protein abundance of cleaved caspase-3 at 3h (Fig. S6A), a response that was recapitulated by exogenous H2O2 (Fig. S6B). Of note, the ability of sPRR-His to increase cleaved caspase-3 was attenuated by GKT, siRNA-mediated knockdown of Nox4 and PDTC (Fig. S6, B–D). These findings were substantiated by quantitative analysis of apoptosis using flow cytometry. In this regard, sPRR-His increased the apoptotic population of cells at 3h, and this result was reiterated by administering exogenous H2O2 (Fig. S7A). Importantly, sPRR-His was unable to evoke apoptosis in cells transfected with Nox4 siRNA, or pharmacological inhibition of Nox1/Nox4 and NF-κB (Fig. S7, A–C). Quantitative analysis of the percentage of total apoptotic cells is shown in Fig. S8. These results support the statement that sPRR-His increases apoptosis by activating the Nox4/H2O2/NF-κB pathway in HUVECs.
The AT1R mediates the functions of sPRR-His.
To this point our data indicate that sPRR-His evokes inflammation and apoptosis by activating the Nox4/H2O2/NF-κB pathway. The neutralizing antibody against sPRR (Ab) markedly attenuated sPRR-His-evoked Nox4 activation, IκBα degradation, NF-κB p-p65 up-regulation and cleaved caspase-3 up-regulation (Fig. S9A), confirming specificity of the action of sPRR-His. We next explored whether these responses are secondary to sPRR binding to the AT1R. First we observed that AT1R blockade using Los (Fig. 1, A–B) or AT1R siRNA knockdown (Fig. 1, C–D) attenuated sPRR-His evoked Nox4 activation, H2O2 generation, IκBα degradation, NF-κB p-p65 up-regulation and cleaved caspase-3 up-regulation. Second, sPRR-His elevated mRNA (Fig. 2, A–B) and protein (Fig. 2, C–D) indices of inflammation in the absence but not the presence of Los or AT1R siRNA knockdown. Finally, quantitative analyses using flow cytometry indicated that Los inhibited sPRR-His-induced apoptosis (Fig. 2, E–F). These results support the statement that sPRR-His-induced inflammation and apoptosis are secondary to AT1R activation in HUVECs.
Fig. 1. sPRR-His activates the AT1R.
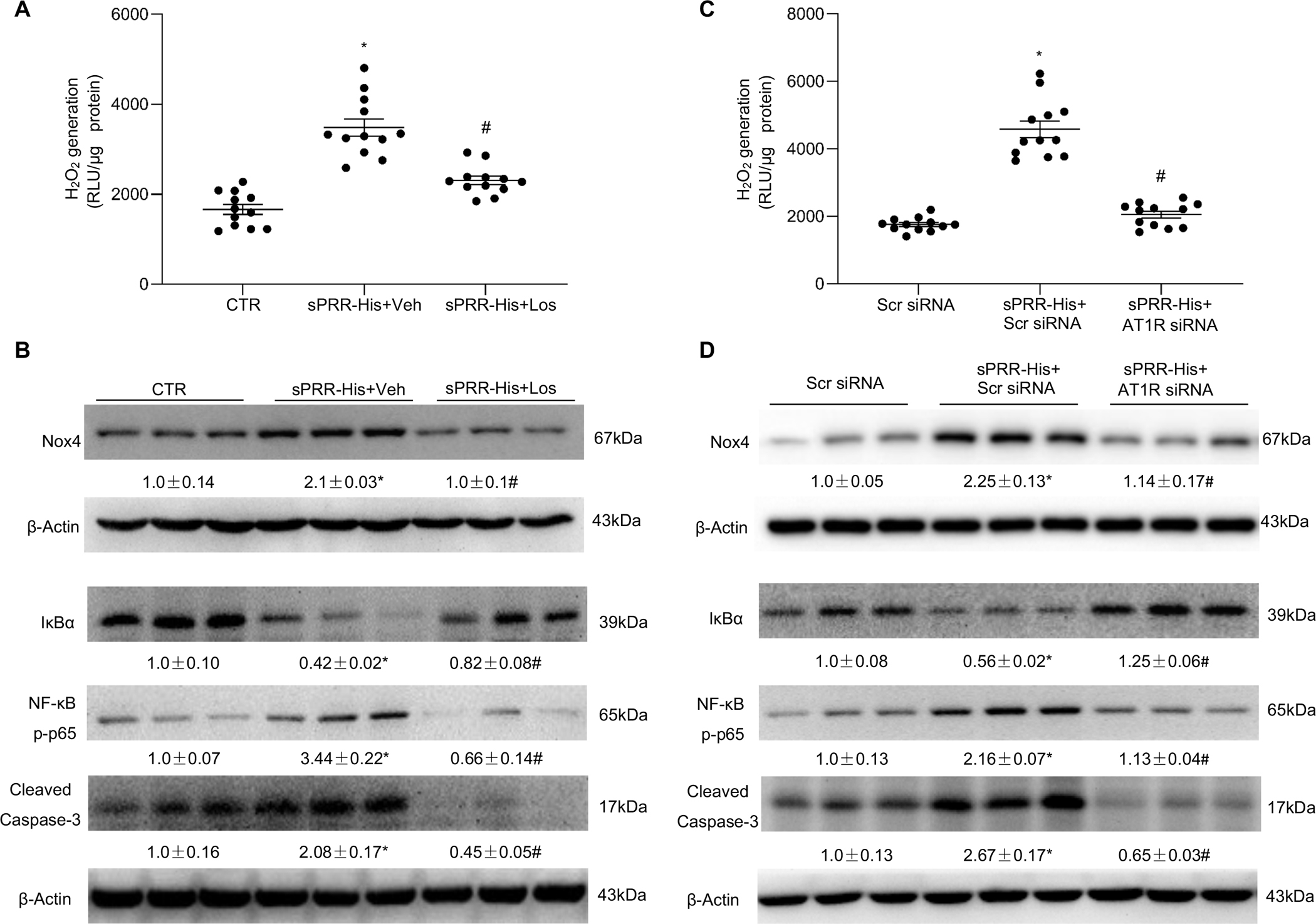
(A-B) HUVECs were pretreated with 10μM Losartan (Los) or vehicle (Veh) for 1h followed by exposure to sPRR-His. (A) Effect of Los on sPRR-His-induced H2O2 generation. (B) Effect of sPRR-His on protein expression of Nox4, IκBα, NF-κB p-p65 and cleaved caspase-3 in the presence or absence of 10μM Los. (C-D) HUVECs were transfected with scrambled small interfering RNA (Scr siRNA) or AT1R siRNA, followed by exposure to sPRR-His. (C) Effect of AT1R siRNA knockdown on sPRR-His-induced H2O2 generation. (D) Effects of AT1R siRNA knockdown on protein expression of Nox4, IκBα, NF-κB p-p65 and cleaved caspase-3 after sPRR-His treatment. The value beneath the image indicates the densitometry of the protein normalized to β-Actin for 3 separate experiments. N=9–12 (N represents the number of samples in each group, and the repetitions of separate experiments are 3–4). Statistical significance was determined by using one-way ANOVA with the Bonferroni test for multiple comparisons. *P<0.01, compared with the CTR or Scr siRNA; #P<0.01, compared with the sPRR-His or sPRR-His+Scr siRNA. Data are mean ± SEM.
Fig. 2. The AT1R mediates the effects of sPRR-His activation.
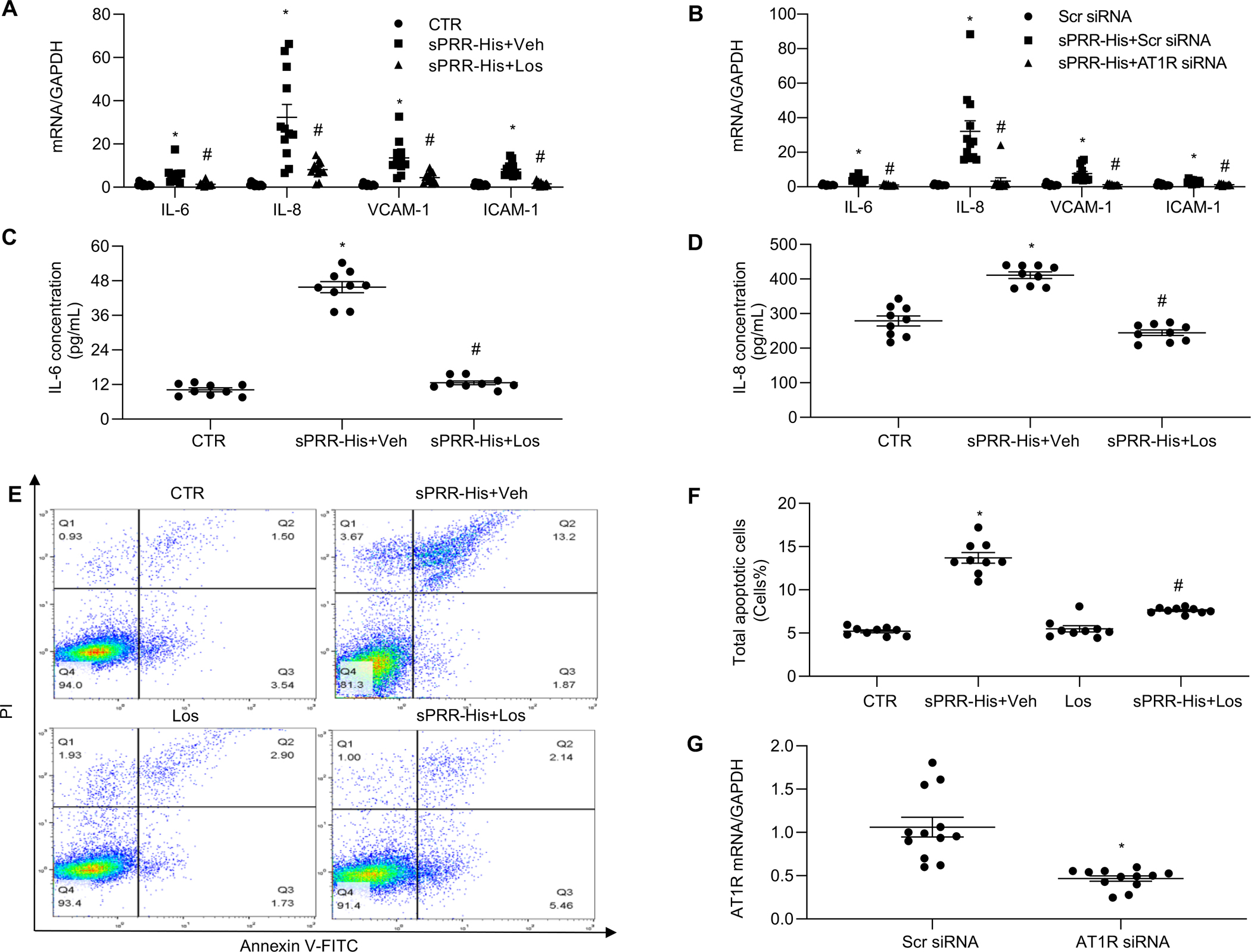
Effects of 10μM Los (A) or AT1R siRNA knockdown (B) on mRNA expression of IL-6, IL-8, VCAM-1 and ICAM-1 after sPRR-His 3-h treatment. Effect of 10μM Los on protein levels of IL-6 (C) and IL-8 (D) in the medium of HUVECs after 50nM sPRR-His treatment. (E-F) Effects of 10μM Los on sPRR-His-induced apoptosis. (E) The apoptotic populations were determined by Annexin V-FITC/PI flow cytometry analysis. (F) Quantitative analysis of the percentage of total apoptotic cells. The early apoptotic cells percentage added to the late apoptotic cells percentage equals the total apoptotic cells percentage. (G) Validation of AT1R knockdown by qRT-PCR. N=9–12 (N represents the number of samples in each group, and the repetitions of separate experiments are 3–4). Statistical analysis was performed by using one-way ANOVA with the Bonferroni test for multiple comparisons or by using unpaired Student’s t test for two comparisons. *P<0.01, compared with the CTR or Scr siRNA; #P<0.01, compared with the sPRR-His or sPRR-His+Scr siRNA. Data are mean ± SEM.
sPRR-His directly interacts with AT1R.
We next sought to elucidate the mechanism whereby sPRR-His activates the AT1R. Ang II was assessed in cell medium before and after HUVECs were treated with sPRR-His. Because Ang II concentrations were refractory to sPRR-His treatment (Fig. 3A), we suspected that sPRR-His might act in a manner that was independent of the canonical RAS. As such, we explored whether sPRR-His directly interacts with the AT1R. To test this, Ang II-I125 radioactive ligand competitive binding assays were completed in the presence or absence of different concentrations of sPRR-His with or without Los, to determine the amount (i.e., counts per min; CPM) of Ang II-I125 that combined with cellular AT1R in each condition. Notably, sPRR-His binding to the AT1R out-competed Ang II-I125 binding to the AT1R in a dose-dependent manner that was equivalent to Los at 50 nM (Fig. 3B). These data indicate that sPRR-His is a competitive ligand of Ang II. To strengthen these findings, we conducted 35S-sPRR radioactive ligand competitive binding assays in the presence or absence of Los. Findings indicate that Los dose-dependently inhibits the uptake of 35S-sPRR by cultured HUVECs with a maximal effect at 20 μM (Fig. 3C). Subsequently, a direct protein-protein interaction was examined by radioisotope labeling coupled with immunoprecipitation. 35S-AT1R or 35S-sPRR was generated by in vitro translation in the presence of 35S-methionine. 35S-AT1R was incubated with sPRR-His, followed by immunoprecipitation with each of the 3 anti-PRR antibodies. Autoradiography revealed a 35S-AT1R band with each of the three anti-PRR antibodies but the absence of a band with IgG control (Fig. 3D). Similarly, the interaction of 35S-sPRR and full-length AT1R protein was confirmed following anti-AT1R antibody-mediated immunoprecipitation and autoradiography analysis of 35S-sPRR (Fig. 3D).
Fig. 3. sPRR-His directly interacts with the AT1R.
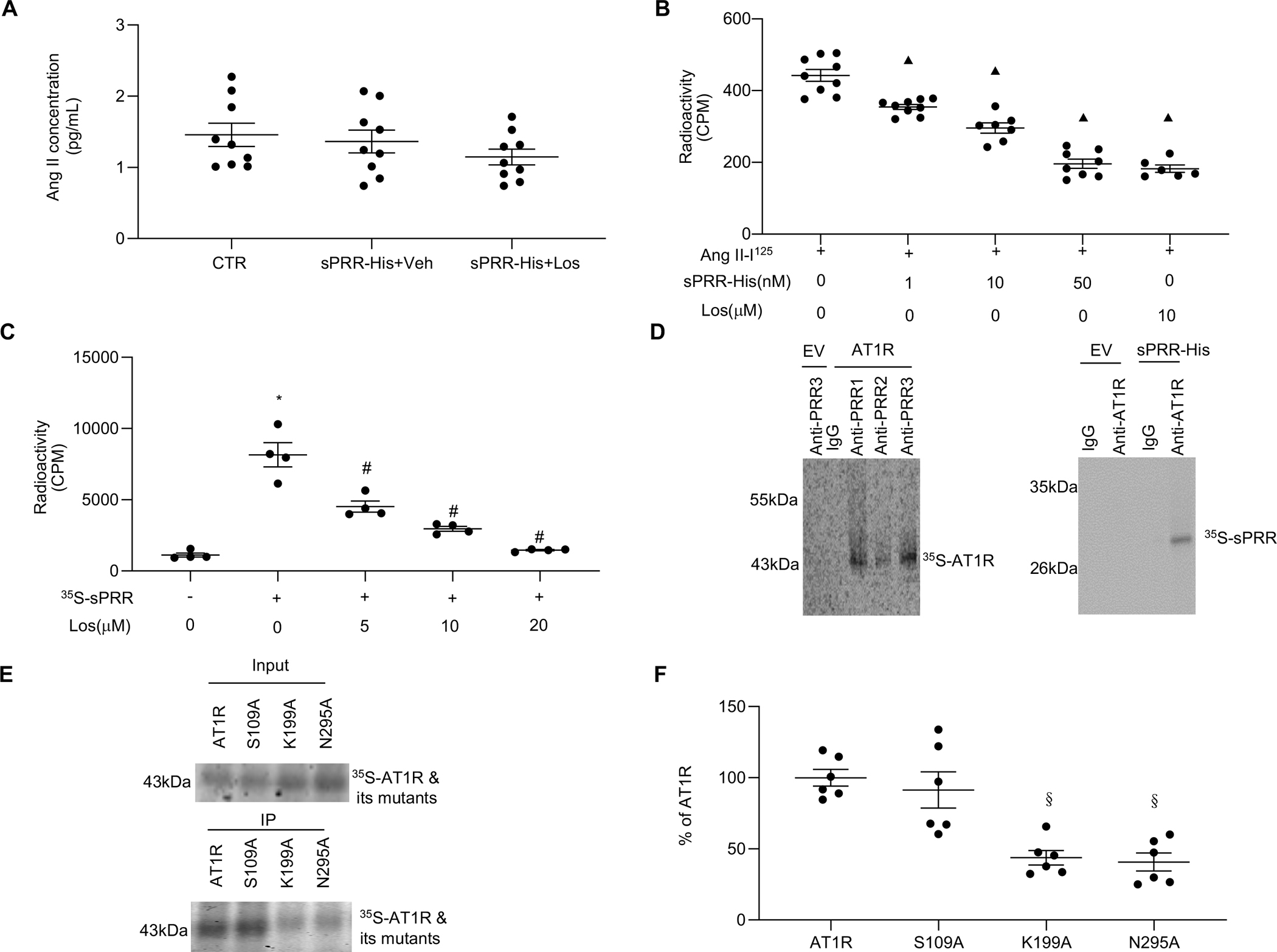
(A) Effect of 10μM Los on protein levels of Ang II in the medium of HUVECs after 50nM sPRR-His treatment. (B) HUVECs were pretreated with 1nM, 10nM, 50nM sPRR-His or 10μM Los for 1h followed by exposure to 1μM Ang II-I125. A radioactive ligand competitive binding assay then was performed. (C) HUVECs were incubated with 35S-labeled sPRR (35S-sPRR) overnight after one-hour pretreatment with Los at different dosages. A radioactive ligand competitive binding assay then was performed. The radioactive counts per minute (CPM) were measured in each group. (D) In vitro protein-protein interactions between AT1R and sPRR-His were detected. The AT1R or sPRR-His was labeled with 35S-methionine during in vitro translation. The commercial sPRR-His or AT1R proteins and its respective antibodies were added to immunoprecipitate the 35S-labeled proteins. EV: empty vector control; Anti-PRR1: PRR antibody (Sigma); Anti-PRR2: antibody against mouse PRR AA 32–45; Anti-PRR3: antibody against mouse PRR AA 219–235. (E) In vitro protein-protein interactions between sPRR-His and 35S-AT1R or 35S-AT1R site-mutations were detected. The immunoprecipitation experiment with anti-PRR antibody following incubation of sPRR-His and 35S-AT1R or its mutants was performed. The quantitative analysis is shown in (F). S109A: 35S-AT1R site-mutation in Serine109; K199A: 35S-AT1R site-mutation in Lysine199; N295A: 35S-AT1R site-mutation in Aspargine295. N=6–9. Statistical analysis was performed by using one-way ANOVA with the Bonferroni test for multiple comparisons or by using unpaired Student’s t test for two comparisons. ▲P < 0.01, compared with the Ang II-I125; *P < 0.01, compared with the CTR; #P < 0.01, compared with the 35S-sPRR; §P < 0.01, compared with the AT1R group. Data are mean ± SEM.
We further attempted to identify potential amino acid residues involved in AT1R binding with the sPRR. Although multiple amino acid residues in the transmembrane domain of AT1R constitute Los binding site, the involvement of Serine109 and Asparagine295 is relatively better defined.(38) Similarly, Lysine199 of AT1R is heavily involved in interaction with Ang II.(38) To this end, we conducted mutagenesis of each of these three residues of 35S-AT1R to Alanine and examined the consequences via immunoprecipitation coupled with autoradiography. 35S-AT1RS109A precipitated with anti-PRR antibody showed similar band intensity on autoradiography as wild type 35S-AT1R. In contrast, the band intensity of immunoprecipitated 35S-AT1RL199A or 35S-AT1RN295A was significantly reduced (Fig. 3, E–F). In addition, no 35S-AT1R and its mutants were precipitated with an IgG control. These results suggest that Lysine199 and Asparagine295 but not Serine109 are required for the protein-protein interaction between AT1R and sPRR.
sPRR-His directly activates AT1R-Gq signaling.
Next we explored the downstream signaling events subsequent to sPRR-His AT1R binding in HUVECs. sPRR-His phosphorylates ERK1/2 and PKC rapidly (i.e., within 10-min), and activates intracellular Ca2+ signaling within 90s, and all responses are sensitive to Los or AT1R siRNA knockdown (Fig. S10, A–D).
AT1R activation by sPRR-His reduces NO generation.
Nox4-mediated ROS production inhibits activating phosphorylation of serine 1177 on the eNOS enzyme i.e., p-eNOSS1177.(39) As would be expected based on results presented thus far, sPRR-His reduced p-eNOSS1177 : total eNOS and NO generation at 3h (Fig. 4A). Los pretreatment (Fig. 4, B–C) or AT1R siRNA-mediated knockdown (Fig. 4, D–E) prevented each response, substantiating the importance of the AT1R. Taken together, these results indicate sPRR-His reduces NO generation by activating AT1R.
Fig. 4. sPRR-His reduces NO generation by activating AT1R.
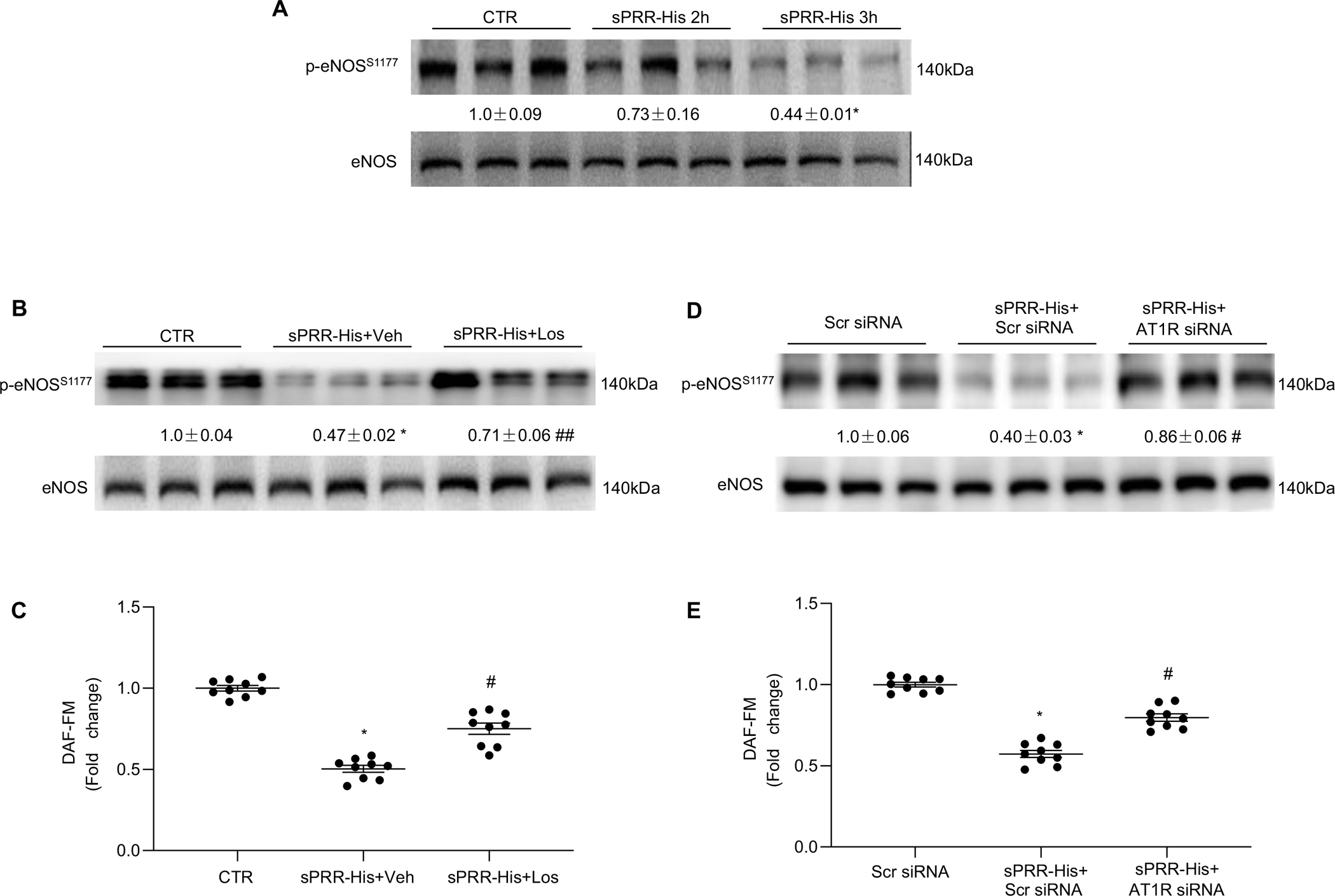
(A) Relative protein expression levels of p-eNOSS1177 and eNOS after 50nM sPRR-His 2-h and 3-h treatment were measured by Western blotting. (B) Effect of 50nM sPRR-His on relative protein expression levels of p-eNOSS1177 and eNOS in the presence or absence of 10μM Los. (C) Effect of 50nM sPRR-His on NO generation in the presence or absence of 10μM Los. (D) Effect of 50nM sPRR-His on relative protein expression levels of p-eNOSS1177 and eNOS in the presence or absence of AT1R siRNA knockdown. (E) Effect of 50nM sPRR-His on NO generation in the presence or absence of AT1R siRNA knockdown. The value beneath the image indicates the densitometry of the protein normalized to eNOS for 3 separate experiments. N=9 (N represents the number of samples in each group, and the repetitions of separate experiments are 3). Statistical analysis was performed by using one-way ANOVA with the Bonferroni test for multiple comparisons or by using unpaired Student’s t test for two comparisons. *P<0.01, compared with the CTR or Scr siRNA; #P<0.01, compared with the sPRR-His or sPRR-His+Scr siRNA; ##P<0.05, compared with the sPRR-His or sPRR-His +Scr siRNA. DAF (diaminofluorescein)-FM indicates 4-amino-5-methylamino-2′, 7′-difluorofluorescein diacetate. Data are mean ± SEM.
sPRR-His induces hypertension and endothelial dysfunction by activating AT1R in obese C57/BL6 mice.
Next we sought to discern the translational relevance of our findings in HUVECs to intact animals. One-month old C57/BL6 mice consumed high-fat chow for 36 weeks to produce diet-induced obesity (DIO). At 33 weeks of feeding, radiotelemetry devices were implanted for the eventual measure of blood pressure and heart rate (HR) in conscious mice. At 34 weeks of feeding, mice were subdivided to receive sPRR-His (DIO+sPRR-His) or sPRR-His+Los (DIO+sPRR-His+Los) for the last 2 weeks while blood pressure and HR were assessed. High fat diet induced an increase in plasma sPRR levels from 3.86 ng/ml to 6.16 ng/ml in C57/BL6 mice (P<0.01, N=5–10 per group). Administering sPRR-His increased circulating sPRR concentrations from 6.16 ng/ml to 13.38 ng/ml in obese C57/BL6 mice (P<0.01, N=5–10 per group). The mean arterial pressure (MAP), systolic blood pressure (SBP) and diastolic blood pressure (DBP) were higher in DIO+sPRR-His vs. DIO+sPRR-His+Los treated mice (Fig. 5, A–C). As would be expected in mice with intact baroreceptor function, HR was lower in DIO+sPRR-His treated vs. DIO+sPRR-His+Los treated animals (Fig. 5D). Moreover, we performed additional experiments to examine the effect of sPRR-His in lean C57/BL6 mice. Radiotelemetry demonstrated that sPRR-His infusion to C57/BL6 mice at the same dosage over 2 wks didn’t significantly affect blood pressure and HR (Fig. S11, A–D). It is possible that sPRR-His may play a specific role in mediating obesity-induced hypertension.
Fig. 5. The effects of sPRR-His with or without Losartan or Captopril on blood pressure and heart rate in DIO mice.
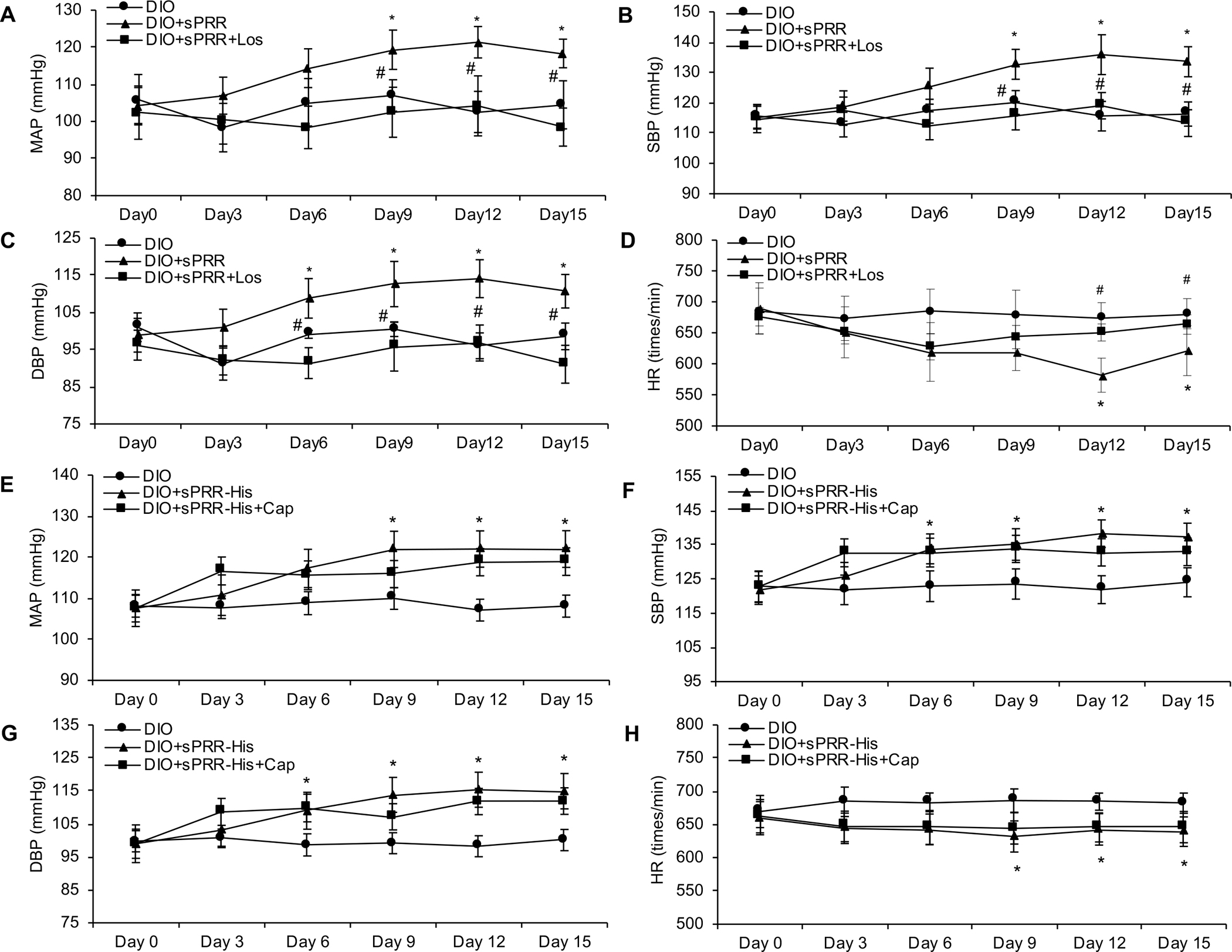
Radiotelemetry was performed to record (A&E) mean arterial pressure (MAP), (B&F) systolic blood pressure (SBP), (C&G) diastolic blood pressure (DBP) and (D&H) heart rate (HR). N=5 per group. Statistical significance was determined by using one-way ANOVA with the Bonferroni test for multiple comparisons. *P<0.05, compared with DIO; #P<0.05, compared with DIO+ sPRR-His. Los: Losartan, Cap: Captopril. Data are mean ± SEM.
After measuring blood pressure and HR for the final two weeks of the 36-weeks feeding regimen, mice were anesthetized and mesenteric arterial function was assessed. We sought to determine if hypertension observed in DIO+ sPRR-His treated mice was secondary to endothelial dysfunction, and whether AT1R blockade might be protective. Acetylcholine-evoked vasorelaxation was greater in mesenteric arteries from Los-treated vs. sPRR-His-treated DIO mice, while responses to sodium nitroprusside were similar among groups (Fig. 6, A–B). Moreover, acetylcholine-evoked vasorelaxation was similar in arteries from Los-treated vs. sPRR-His-treated lean mice, and responses to sodium nitroprusside were not different among groups (Fig. 6, C–D). However, vascular smooth muscle responses were similar among groups (Fig. S12, A–D). These data suggest that endothelial function is preserved by AT1R blockade, and it is not unreasonable to speculate that this contributed importantly to normalizing arterial blood pressure.
Fig. 6. The effects of sPRR-His and Losartan on vasorelaxation function in DIO mice or lean mice.
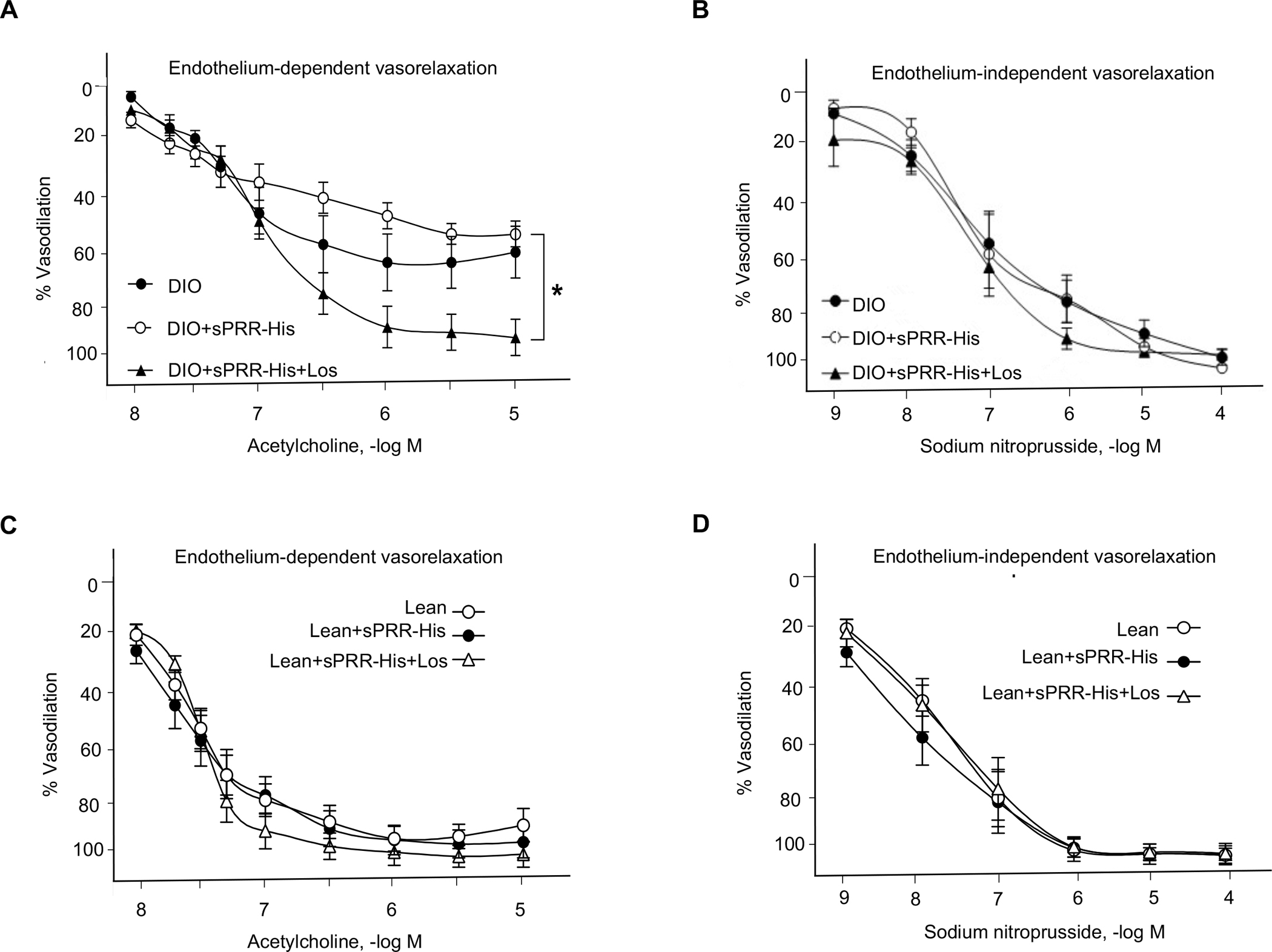
Percentage (%) vasorelaxation to acetylcholine (A&C) and sodium nitroprusside (B&D) were measured using isometric tension procedures. Comparison of one time point among groups was made using one-way ANOVA. Comparison of multiple time points among groups was made using one-way or two-way repeated-measures ANOVA. Tukey post hoc tests were performed when significant main effects were obtained. *P<0.05, compared with DIO+ sPRR-His. For A and B, N=6–8 per group. For C and D, N=8 per group. Data are mean ± SEM.
ACE inhibition using captopril does not affect sPRR-His responses in vitro or in vivo.
Concern might be raised that sPRR-His facilitates Ang II generation, which may mediate the action of sPRR-His. To address this, experiments were completed using the angiotensin converting enzyme inhibitor (ACEi) captopril (Cap). Cap did not attenuate sPRR-His-evoked Nox4 activation, IκBα degradation, NF-κB p-p65 up-regulation and cleaved caspase-3 up-regulation in HUVECs (Fig. S9B). Furthermore, radiotelemetry data concerning arterial blood pressure indicated that Cap did not significantly affect sPRR-His-induced hypertension in DIO mice (Fig. 5, E–H). In order to test the effective dosage of Cap, we measured plasma Ang II and bradykinin levels by ELISA. The plasma Ang II level was significantly reduced from 7462 to 2492 pg/ml in DIO+sPRR-His vs. DIO+sPRR-His+Cap treated mice (P<0.01, N=5 per group), and the plasma bradykinin level was increased from 31.61 to 61.26 pg/ml in DIO+sPRR-His vs. DIO+sPRR-His+Cap treated mice (P<0.01, N=5 per group), confirming the effectiveness of Cap. Together, these data suggest that the prohypertensive action of sPRR-His infusion in DIO mice is mediated by direct activation of AT1R, independent of generation of Ang II.
Discussion
For many decades AT1R activation was thought to completely depend on its sole physiological ligand, Ang II. Findings from the present study challenge this notion. Here we report for the first time that sPRR directly binds to and activates the AT1R in an Ang II-independent manner to suppress NO generation by endothelial cells and we provide compelling evidence for a novel mechanism that is responsible. In addition, we provide important translational evidence concerning the (patho) physiological relevance of this signaling pathway in the context of hypertension and arterial dysfunction that exists in obese mice. To our knowledge no prior studies have reported the biological function of sPRR in the vasculature or it’s signaling through AT1R. We believe AT1R mediates the functions of sPRR during obesity-related hypertension and sPRR should be considered further as a viable therapeutic target in the treatment and prevention of cardiovascular disorders.
Treatment of HUVECs with nanomolar concentrations of sPRR-His for 2 h degraded cytosolic IκBα to an extent that facilitated nuclear translocation of NF-κB p65 and increased NF-κB p65 transcription factor activity. Because ROS are a common stimulus for NF-κB activation in general, and H2O2 can initiate nuclear translocation of NF-κB in endothelial cells in particular,(39) we examined these intracellular messengers in the context of the current experimental conditions.
Next, the source of ROS was determined. Because Nox4 expression is 100-fold higher than Nox1, Nox2, or Nox5 in endothelial cells,(40) we suspected Nox4 to be a major source of H2O2 in response to sPRR-His. Supporting this, sPRR-His evoked a robust increase in Nox4 mRNA and protein expression, H2O2 generation, and NF-κB activation in HUVECs transfected with scrambled but not Nox4 siRNA, and in HUVECs treated with vehicle but not the dual Nox1 and Nox4 inhibitor GKT. These findings substantiate the importance of Nox4-derived H2O2 in response to sPRR-His. This notion agrees well with a significant number of studies showing pro-inflammatory and pro-apoptotic role of Nox4 in the vasculature.(41) It’s somewhat puzzling however that some recent studies suggest a vasoprotective role of Nox4.(42) The reason for the controversy is unknown and needs to be addressed in the future.
Because NF-κB upregulates the expression of inflammatory cytokines and adhesion molecules e.g., IL-6, IL-8, VCAM-1 and ICAM-1,(15) we were prompted to determine whether sPRR-His evokes inflammation and adhesion via this pathway. Indeed, sPRR-His precipitated increases in IL-6, IL-8, VCAM-1 and ICAM-1 mRNA expression, and IL-6 and IL-8 protein expression. These data provide solid evidence that elevated sPRR-His promotes inflammation/adhesion in endothelial cells. Inflammation plays an important role in the development and pathophysiology of hypertension.(43)
Substantial evidence exists that Ang II induces inflammation and apoptosis by activating the AT1R/NADPH/ROS/NF-κB pathway.(44, 45) These signaling events are similar to those we report here for sPRR-His. Moreover, the preliminary evidence suggests that AT1R and PRR can form a heterodimer that is functionally active to enhance ERK phosphorylation in PC12W cells.(46) These findings provided rationale for our exploration into whether the AT1R directly mediates the signaling events precipitated by elevated sPRR. Of significant interest to us, results from immunoprecipitation experiments and radioactive ligand competitive binding assays indicated that sPRR directly interacts with the AT1R. In particular, the direct protein-protein interaction between sPRR and AT1R was validated in a cell-free system by immunoprecipitation coupled with autoradiography. Furthermore, site-directed mutagenesis was conducted to map the binding site. The key amino acid residues of AT1R are Serine109 and Asparagine295 for Los binding and Lysine199 for Ang II binding.(38,47) We found that mutation of Lysine199 and Asparagine295 but not Serine109 significantly reduced sPRR binding to the AT1R. Therefore, sPRR may share partial or complete binding sites of AT1R with Ang II or Los. In addition to Lysine199, other residues including Tryptophan253, Phenylalanine259, Asparagine263, and Arginine167 also interact with Ang II (38,47) but their involvement concerning sPRR binding has not been investigated. What’s more, we observed that sPRR-His stimulated AT1R mediated Gq signaling, confirming that sPRR can directly activate AT1R. The current study is the first to report the direct interaction between sPRR and AT1R in general, and in endothelial cells in particular.
To determine whether our findings in vitro are relevant to the intact animal, we studied the effects of sPRR-His on vascular function and blood pressure in obese mice. Results using isometric tension techniques revealed that endothelium-dependent vasodilation displayed by arteries from obese mice treated with sPRR-His was improved by Los. Because vascular smooth muscle responses were similar between groups, it is not unreasonable to suggest that Los improved an endothelium-dependent defect evoked by sPRR-His. Endothelial dysfunction plays an important role in the development and pathophysiology of hypertension.(48) Indeed, we observed elevated blood pressure in obese mice treated with sPRR-His, an effect that was reversed by concurrent treatment with Los but not ACEi using Cap. The distinct effects of Los versus Cap strongly support that sPRR induces hypertension via direct activation of AT1R as opposed to the generation of Ang II. Cap significantly reduced plasma Ang II level and increased plasma bradykinin level in DIO+sPRR-His treated mice, confirming the effectiveness of ACE inhibition. While captopril effectively inhibited the production of Ang II, the radiotelemetry data showed no significant effect of captopril on sPRR-His-induced hypertension in DIO mice, indicating that the increased Ang II was not the reason for sPRR-His-induced hypertension in DIO mice. Of note, in agreement with the present study, Gatineau et al. reported a similar prohypertensive action of sPRR in DIO mice (49). However, discrepancy exists in that Gatineau et al. found no effect of losartan on the prohypertensive action of sPRR. The exact reason for the discrepancy is unclear but may be related to differences in experimental models (36 versus 21 weeks of high fat diet) and protocols. It might be possible that sPRR-mediated activation of AT1R in the vasculature may predominate during prolonged high fat diet treatment whereas baroreflex dysfunction may be relatively more important during regular high fat diet treatment (49). Indeed, the mechanism of obesity-related hypertension is complex, involving endothelial dysfunction, Na+ retention, increased leptin levels and baroreflex dysfunction (50). Relative importance of each of the underlying mechanisms may vary at various stages of the hypertension development (51, 52).
We have examined the property of sPRR-His in cultured vascular smooth muscle cells as well as isolated mesentery arteries from the normal control lean mouse. To our surprise, sPRR-His did not induce activation of AT1R signaling pathway such as Gq (data not shown). In agreement with this observation, while potent Ang II-induced vasoconstriction was observed in mesenteric arteries as examined ex vivo, sPRR-His was without effect (Fig. S13). It is likely that within the vasculature sPRR-His acts specifically on endothelial but not vascular smooth muscle cells and such specificity may be determined by some unknown co-receptor (s) rather than AT1R. An example is that fibroblast growth factor 23 (FGF-23) via its receptor FGFR acts in a wide variety of tissues and its specific action in the kidney for regulation of tubular phosphate transport is rather determined by its co-receptor Klotho.
The AT1R is a core member in the RAS that promotes myriad signaling pathways contributing to the pathogenesis of cardiovascular diseases.(53) In the classic sense, Ang II has been considered as the exclusive ligand for the AT1R. Here we provide compelling in vitro and in vivo evidence that sPRR serves as an alternative activator of the AT1R, and that activation via this pathway contributes to cardiovascular complications (e.g., arterial dysfunction and hypertension) associated with diet-induced obesity. Taken together, our results provide solid support for sPRR to be considered further as a predictive marker and/or therapeutic target for cardiovascular related pathologies.
Supplementary Material
Clinical perspectives.
Obesity is a major risk factor of endothelial dysfunction and hypertension with unclear mechanism.
The present study for the first time demonstrates that sPRR directly interacts with AT1R to lead to NF-κB activation, inflammation, apoptosis, and attenuated p-eNOSS1177 in primary HUVECs. Besides, the interaction between sPRR and AT1R contribute to endothelial dysfunction and hypertension in DIO mice.
These findings offer a novel mechanism of endothelial dysfunction and hypertension associated with obesity. sPRR can be considered further as a predictive marker and / or therapeutic target for cardiovascular disorders associated with endothelial dysfunction.
Acknowledgments
We thank Dr. Guoquan Gao and Dr. Xia Yang (Sun Yat-sen University) for providing the primary human umbilical vein endothelial cells (HUVECs).
Funding
This work was supported by the and National Institutes of Health Grants HL139689, DK104072, HL135851, and VA Merit Review I01BX00481 from the Department of Veterans Affairs and National Natural Science Foundation of China Grant No. 81930006. J. David Symons was supported by the American Heart Association (AHA:16GRNT31050004) and National Institutes of Health (NIH:RO3AGO52848; NIH:RO1HL141540). T. Yang is Senior Research Career Scientist in Department of Veterans Affairs (IK6BX005223).
Nonstandard Abbreviations
- PRR
(pro)renin receptor
- sPRR
soluble (pro)renin receptor
- HUVECs
Human umbilical vein endothelial cells
- VSMCs
Vascular smooth muscle cells
- ROS
Reactive oxygen species
- NF-κB
Nuclear factor kappa B
- IκB
Inhibitory kappa B
- Veh
vehicle
- PDTC
ammonium pyrrolidinedithiocarbamate
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- IL-6
Interleukin 6
- IL-8
Interleukin 8
- VCAM-1
Vascular Cell Adhesion Molecule-1
- ICAM-1
Intercellular Adhesion Molecule-1
- AT1R
Angiotensin II type 1 receptor
- ACEi
Angiotensin converting enzyme inhibitor
- Ang II
Angiotensin II
- GKT
GKT137892
- Los
Losartan
- Cap
Captopril
- MAP
Mean arterial pressure
- SBP
Systolic blood pressure
- DBP
Diastolic blood pressure
- HR
Heart rate
Footnotes
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.
References
- 1.Nguyen G, Delarue F, Burcklé C, Bouzhir L, Giller T, and Sraer J-D (2002) Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. Journal of Clinical Investigation 109, 1417–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burckle C, and Bader M (2006) Prorenin and Its Ancient Receptor. Hypertension 48, 549–551 [DOI] [PubMed] [Google Scholar]
- 3.Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L, and Nguyen G (2009) Soluble Form of the (Pro)Renin Receptor Generated by Intracellular Cleavage by Furin Is Secreted in Plasma. Hypertension 53, 1077–1082 [DOI] [PubMed] [Google Scholar]
- 4.Nakagawa T, Suzuki-Nakagawa C, Watanabe A, Asami E, Matsumoto M, Nakano M, Ebihara A, Uddin MN, and Suzuki F (2017) Site-1 protease is required for the generation of soluble (pro)renin receptor. Journal of biochemistry 161, 369–379 [DOI] [PubMed] [Google Scholar]
- 5.Fang H, Xu C, Lu A, Zou CJ, Xie S, Chen Y, Zhou L, Liu M, Wang L, Wang W, and Yang T (2017) (Pro)renin receptor mediates albumin-induced cellular responses: role of site-1 protease-derived soluble (pro)renin receptor in renal epithelial cells. American journal of physiology. Cell physiology 313, C632–c643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang F, Lu X, Liu M, Feng Y, Zhou SF, and Yang T (2015) Renal medullary (pro)renin receptor contributes to angiotensin II-induced hypertension in rats via activation of the local renin-angiotensin system. BMC medicine 13, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu C, Lu A, Lu X, Zhang L, Fang H, Zhou L, and Yang T (2017) Activation of Renal (Pro)Renin Receptor Contributes to High Fructose-Induced Salt Sensitivity. Hypertension (Dallas, Tex. : 1979) 69, 339–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bray GA (2004) Medical consequences of obesity. The Journal of clinical endocrinology and metabolism 89, 2583–2589 [DOI] [PubMed] [Google Scholar]
- 9.Avogaro A, and de Kreutzenberg SV (2005) Mechanisms of endothelial dysfunction in obesity. Clinica chimica acta; international journal of clinical chemistry 360, 9–26 [DOI] [PubMed] [Google Scholar]
- 10.Busse R, Edwards G, Félétou M, Fleming I, Vanhoutte PM, and Weston AH (2002) EDHF: bringing the concepts together. Trends in pharmacological sciences 23, 374–380 [DOI] [PubMed] [Google Scholar]
- 11.Amari Y, Morimoto S, Nakajima F, Ando T, and Ichihara A (2016) Serum Soluble (Pro)Renin Receptor Levels in Maintenance Hemodialysis Patients. PloS one 11, e0158068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukushima A, Kinugawa S, Homma T, Masaki Y, Furihata T, Abe T, Suga T, Takada S, Kadoguchi T, Okita K, Matsushima S, and Tsutsui H (2013) Increased plasma soluble (pro)renin receptor levels are correlated with renal dysfunction in patients with heart failure. International Journal of Cardiology 168, 4313–4314 [DOI] [PubMed] [Google Scholar]
- 13.Morimoto S, Ando T, Niiyama M, Seki Y, Yoshida N, Watanabe D, Kawakami-Mori F, Kobori H, Nishiyama A, and Ichihara A (2014) Serum soluble (pro)renin receptor levels in patients with essential hypertension. Hypertension Research 37, 642–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruland J (2011) Return to homeostasis: downregulation of NF-kappaB responses. Nat Immunol 12, 709–714 [DOI] [PubMed] [Google Scholar]
- 15.Xu X, Guo H, Jing Z, Yang L, Chen C, Peng L, Wang X, Yan L, Ye R, Jin X, and Wang Y (2016) N-Oleoylethanolamine Reduces Inflammatory Cytokines and Adhesion Molecules in TNF-alpha-induced Human Umbilical Vein Endothelial Cells by Activating CB2 and PPAR-alpha. Journal of cardiovascular pharmacology 68, 280–291 [DOI] [PubMed] [Google Scholar]
- 16.Ghosh S, and Karin M (2002) Missing pieces in the NF-kappaB puzzle. Cell 109 Suppl, S81–96 [DOI] [PubMed] [Google Scholar]
- 17.Vejakama P, Thakkinstian A, Lertrattananon D, Ingsathit A, Ngarmukos C, and Attia J (2012) Reno-protective effects of renin-angiotensin system blockade in type 2 diabetic patients: a systematic review and network meta-analysis. Diabetologia 55, 566–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan BV (2011) The effect of amlodipine besylate, losartan potassium, olmesartan medoxomil, and other antihypertensives on central aortic blood pressure and biomarkers of vascular function. Therapeutic advances in cardiovascular disease 5, 241–273 [DOI] [PubMed] [Google Scholar]
- 19.Lee M, Saver JL, Hong KS, Hao Q, Chow J, and Ovbiagele B (2012) Renin-Angiotensin system modulators modestly reduce vascular risk in persons with prior stroke. Stroke 43, 113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pepe G, Giusti B, Sticchi E, Abbate R, Gensini GF, and Nistri S (2016) Marfan syndrome: current perspectives. The application of clinical genetics 9, 55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu ML, Sun RL, Zhang HY, Zhao FR, Pan GP, Zhang C, Song P, Li P, Xu J, Wang S, and Yin YL (2019) Angiotensin II type 1 receptor blockers prevent aortic arterial stiffness in elderly patients with hypertension. Clinical and experimental hypertension (New York, N.Y. : 1993) 41, 657–661 [DOI] [PubMed] [Google Scholar]
- 22.Vijayaraghavan K, and Deedwania P (2011) Renin-angiotensin-aldosterone blockade for cardiovascular disease prevention. Cardiology clinics 29, 137–156 [DOI] [PubMed] [Google Scholar]
- 23.Lu X, Wang F, Xu C, Soodvilai S, Peng K, Su J, Zhao L, Yang KT, Feng Y, Zhou SF, Gustafsson JA, and Yang T (2016) Soluble (pro)renin receptor via beta-catenin enhances urine concentration capability as a target of liver X receptor. Proceedings of the National Academy of Sciences of the United States of America 113, E1898–1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baudin B, Bruneel A, Bosselut N, and Vaubourdolle M (2007) A protocol for isolation and culture of human umbilical vein endothelial cells. Nature Protocols 2, 481–485 [DOI] [PubMed] [Google Scholar]
- 25.Lu X, Wang F, Liu M, Yang KT, Nau A, Kohan DE, Reese V, Richardson RS, and Yang T (2016) Activation of ENaC in collecting duct cells by prorenin and its receptor PRR: involvement of Nox4-derived hydrogen peroxide. American journal of physiology. Renal physiology 310, F1243–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su J, Liu X, Xu C, Lu X, Wang F, Fang H, Lu A, Qiu Q, Li C, and Yang T (2017) NF-kappaB-dependent upregulation of (pro)renin receptor mediates high-NaCl-induced apoptosis in mouse inner medullary collecting duct cells. American journal of physiology. Cell physiology 313, C612–c620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li T, Yu B, Liu Z, Li J, Ma M, Wang Y, Zhu M, Yin H, Wang X, Fu Y, Yu F, Wang X, Fang X, Sun J, and Kong W (2018) Homocysteine directly interacts and activates the angiotensin II type I receptor to aggravate vascular injury. Nature communications 9, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bharath LP, Cho JM, Park SK, Ruan T, Li Y, Mueller R, Bean T, Reese V, Richardson RS, Cai J, Sargsyan A, Pires K, Anandh Babu PV, Boudina S, Graham TE, and Symons JD (2017) Endothelial Cell Autophagy Maintains Shear Stress-Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to Endothelial Nitric Oxide Synthase. Arteriosclerosis, thrombosis, and vascular biology 37, 1646–1656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sonsalla PK, Coleman C, Wong LY, Harris SL, Richardson JR, Gadad BS, Li W, and German DC (2013) The angiotensin converting enzyme inhibitor captopril protects nigrostriatal dopamine neurons in animal models of parkinsonism. Experimental neurology 250, 376–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Omobowale TO, Oyagbemi AA, Ogunpolu BS, Ola-Davies OE, Olukunle JO, Asenuga ER, Ajibade TO, Adejumobi OA, Afolabi JM, Falayi OO, Ashafa A, Adedapo AA, and Yakubu MA (2019) Antihypertensive Effect of Polyphenol-Rich Fraction of Azadirachta indica on Nomega-Nitro-L-Arginine Methyl Ester-Induced Hypertension and Cardiorenal Dysfunction. Drug research 69, 12–22 [DOI] [PubMed] [Google Scholar]
- 31.Leckie BJ (2001) The action of salt and captopril on blood pressure in mice with genetic hypertension. Journal of hypertension 19, 1607–1613 [DOI] [PubMed] [Google Scholar]
- 32.Symons JD, McMillin SL, Riehle C, Tanner J, Palionyte M, Hillas E, Jones D, Cooksey RC, Birnbaum MJ, McClain DA, Zhang QJ, Gale D, Wilson LJ, and Abel ED (2009) Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circulation research 104, 1085–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Symons JD, Hu P, Yang Y, Wang X, Zhang QJ, Wende AR, Sloan CL, Sena S, Abel ED, and Litwin SE (2011) Knockout of insulin receptors in cardiomyocytes attenuates coronary arterial dysfunction induced by pressure overload. American journal of physiology. Heart and circulatory physiology 300, H374–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petersen C, Bharat D, Cutler BR, Gholami S, Denetso C, Mueller JE, Cho JM, Kim JS, Symons JD, and Anandh Babu PV (2018) Circulating metabolites of strawberry mediate reductions in vascular inflammation and endothelial dysfunction in db/db mice. International journal of cardiology 263, 111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang H, Deng M, Zhang L, Lu A, Su J, Xu C, Zhou L, Wang L, Ou JS, Wang W, and Yang T (2018) Role of (pro)renin receptor in albumin overload-induced nephropathy in rats. American journal of physiology. Renal physiology 315, F1759–f1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao QD, Viswanadhapalli S, Williams P, Shi Q, Tan C, Yi X, Bhandari B, and Abboud HE (2015) NADPH oxidase 4 induces cardiac fibrosis and hypertrophy through activating Akt/mTOR and NFkappaB signaling pathways. Circulation 131, 643–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang X, Wang Y, Zhang Z, Wang Y, Chen X, Wang Y, and Gao Y (2017) Ophiopogonin D and EETs ameliorate Ang II-induced inflammatory responses via activating PPARα in HUVECs. Biochemical and Biophysical Research Communications 490, 123–133 [DOI] [PubMed] [Google Scholar]
- 38.Yamano Y, Ohyama K, Kikyo M, Sano T, Nakagomi Y, Inoue Y, Nakamura N, Morishima I, Guo DF, Hamakubo T, and et al. (1995) Mutagenesis and the molecular modeling of the rat angiotensin II receptor (AT1). The Journal of biological chemistry 270, 14024–14030 [DOI] [PubMed] [Google Scholar]
- 39.Xu C, Tang F, Lu M, Yang J, Han R, Mei M, Hu J, and Wang H (2016) Pretreatment with Astragaloside IV protects human umbilical vein endothelial cells from hydrogen peroxide induced oxidative stress and cell dysfunction via inhibiting eNOS uncoupling and NADPH oxidase - ROS - NF-kappaB pathway. Can J Physiol Pharmacol, 1–9 [DOI] [PubMed] [Google Scholar]
- 40.Li XX, Ling SK, Hu MY, Ma Y, Li Y, and Huang PL (2019) Protective effects of acarbose against vascular endothelial dysfunction through inhibiting Nox4/NLRP3 inflammasome pathway in diabetic rats. Free radical biology & medicine 145, 175–186 [DOI] [PubMed] [Google Scholar]
- 41.Langbein H, Brunssen C, Hofmann A, Cimalla P, Brux M, Bornstein SR, Deussen A, Koch E, and Morawietz H (2016) NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. European heart journal 37, 1753–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bautista LE, Vera LM, Arenas IA, and Gamarra G (2005) Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. Journal of human hypertension 19, 149–154 [DOI] [PubMed] [Google Scholar]
- 43.Liu J, Zhang FF, Li L, Yang J, Liu J, Guan YY, and Du YH (2013) ClC-3 deficiency prevents apoptosis induced by angiotensin II in endothelial progenitor cells via inhibition of NADPH oxidase. Apoptosis : an international journal on programmed cell death 18, 1262–1273 [DOI] [PubMed] [Google Scholar]
- 44.Maneesai P, Bunbupha S, Kukongviriyapan U, Senggunprai L, Kukongviriyapan V, Prachaney P, and Pakdeechote P (2017) Effect of asiatic acid on the Ang II-AT1R-NADPH oxidase-NF-kappaB pathway in renovascular hypertensive rats. Naunyn-Schmiedeberg’s archives of pharmacology 390, 1073–1083 [DOI] [PubMed] [Google Scholar]
- 45.Miller FJ Jr., Chu X, Stanic B, Tian X, Sharma RV, Davisson RL, and Lamb FS (2010) A differential role for endocytosis in receptor-mediated activation of Nox1. Antioxidants & redox signaling 12, 583–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quadri Syed Siraj Ahmed, Caixia Li, Silas Culver, and Siragy Helmy M. Abstract P099: Angiotensin Type I Receptor (AT1R) and (Pro)renin Receptor (PRR) Functional Heterodimer Mediates ERK Phosphorylation. Originally published 3 Nov 2015. Hypertension. 2015;66:AP099 10.1161/hyp.66.suppl_1.p099 [DOI] [Google Scholar]
- 47.Bhuiyan MA, Ishiguro M, Hossain M, Nakamura T, Ozaki M, Miura S, and Nagatomo T (2009) Binding sites of valsartan, candesartan and losartan with angiotensin II receptor 1 subtype by molecular modeling. Life sciences 85, 136–140 [DOI] [PubMed] [Google Scholar]
- 48.Cibor D (2016) Endothelial dysfunction in inflammatory bowel diseases: Pathogenesis, assessment and implications. World Journal of Gastroenterology 22, 1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gatineau E, Gong MC, and Yiannikouris F (2019) Soluble Prorenin Receptor Increases Blood Pressure in High Fat-Fed Male Mice. Hypertension 74, 1014–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gino S, and Guido G (2016) Sympathetic Nervous System, Hypertension, Obesity and Metabolic Syndrome. High Blood Press Cardiovasc Prev 23, 175–9 [DOI] [PubMed] [Google Scholar]
- 51.Alexandre A, Jussara C, John D, and John H (2009) The role of the sympathetic nervous system in obesity-related hypertension. Curr Hypertens Rep 11, 206–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alfredo G, Rocío F, Sachin P, Ginnie F, Andre D, and Italo B (2016) Autonomic Blockade Reverses Endothelial Dysfunction in Obesity-Associated Hypertension. Hypertension 68, 1004–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawai T, Forrester SJ, O’Brien S, Baggett A, Rizzo V, and Eguchi S (2017) AT1 receptor signaling pathways in the cardiovascular system. Pharmacol Res 125, 4–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.