

Abstract
Various BRAF kinase inhibitors were developed to treat cancers carrying the BRAFV600E mutation. First-generation BRAF inhibitors could lead to paradoxical activation of the MAPK pathway, limiting their clinical usefulness. Here, we show the development of two series of BRAFV600E-targeting PROTACs and demonstrate that the exchange of the inhibitor scaffold from vemurafenib to paradox-breaker ligands resulted in BRAFV600E degraders that did not cause paradoxical ERK activation.
Novel BRAFV600E PROTACs were developed that maintain target degradation while sparing paradoxical activation of the MAPK pathway in BRAFwt cells.
Introduction
Kinases catalyse phosphorylation reactions of target substrates that are critical for controlling intra- and extracellular signalling pathways. Dysregulation of these enzymes can lead to enhanced cellular proliferation and contribute to cancer growth. In particular, regulatory disturbance of the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) axis is frequently observed in cancer. Accordingly, kinase inhibitors directed against the ERK signalling pathway have received considerable attention in drug discovery.1,2 One compound class was developed to target BRAF harbouring a point mutation at position 600 (BRAFV600E), the most prevalent oncogenic protein mutation, and a critical driver of malignant melanoma. This modification enhances spontaneous RAF homo- and heterodimerisation, leading to the uncontrolled activity of the kinase.3 BRAF inhibitor (BRAFi) research enjoyed vast success and culminated in several generations of RAF inhibitors, including clinically approved drugs such as vemurafenib (PLX4032) and dabrafenib (Fig. 1A).4–6 However, initial accomplishments were mitigated by the rapid development of drug resistance and most melanoma patients relapse within a short time.4 Surprisingly, these BRAFi did not inhibit ERK signalling in tumours possessing additional mutations in RAS or its upstream signalling receptors.7 BRAFV600E inhibitors such as vemurafenib fail to prevent BRAF-CRAF heterodimers.4,8 As RAF dimerization and activation is allosterically regulated, one inhibitor-bound protomer can still transactivate the other component of a heterodimer and induce proliferative MAPK signalling, thus promoting paradoxical ERK activation.9
Fig. 1. (A) Structures of the clinically approved BRAFV600E inhibitors vemurafenib (PLX4720) and dabrafenib as well as paradox-breakers PLX7904 and PLX8394. (B) Selected BRAF-targeting PROTACs.

To overcome the issues associated with paradoxical activation, further development of drugs that inhibit BRAF and evade resistance mechanisms is needed. Next-generation BRAFV600E inhibitors PLX7904 and PLX8394 (Fig. 1A) were aimed at restricting RAF dimerisation by shifting critical amino acid side chains located at the interface responsible for dimer formation (Fig. 2).7,10 An alternative approach is focused at depleting kinases at the proteome level by applying the proteolysis-targeting chimeras (PROTACs) technique.11–13 In this new modality, chimeric degrader molecules induce the destruction of target proteins by modulating the substrate scope of E3 ligases through ternary PROTAC:E3:target complexes. The ligase-mediated ubiquitination of proteins of interest can ultimately induce their degradation via the proteasome machinery.14–16 Multiple teams have attempted to degrade BRAF proteins using degraders derived from rigosertib,17 dabrafenib,18 vemurafenib,19,20 or other RAF inhibitors.18 A selection of vemurafenib-based PROTACs are shown in Fig. 1B. Kinase degraders may have intrinsic advantages over classical inhibitors as they silence both catalytic and non-catalytic functions of BRAF. However, PROTAC response may depend on the RAS/RAF mutational context,12 as well as the expression levels of the hijacked E3 ligase.21 Furthermore, the so-called “hook effect”14 could lead to disadvantageous features of BRAFV600E PROTACs and stimulate RAF dimerisation when productive ternary complexes are prevented.22
Fig. 2. BRAFV600E in complex with vemurafenib (cyan, PDB 3OG7) and PLX7904 (orange, PDB 4XV1). The surface of the N-methyl group of PLX7904 illustrates the reason for the Leu505 shift, which affects the conformation of the conserved RKTR motif (shown with sticks) at the dimer interface. Arg506 is particularly important for the stabilization of dimeric complexes. In the case of binding of a paradox-breaker, Arg506 in the αC-helix displays an outward movement which reduces the transactivation of ERK signalling. The protein surface of chains A and B is shown for 3OG7 only.

In this study, we designed a series of von Hippel–Lindau (VHL)-based PROTACs targeting BRAFV600Evia PLX-derived ligands. Particular attention was paid to the BRAFi functions of the bivalent molecules. By addressing the paradoxical activation in a particular mutational context, we demonstrate an advantageous feature of these PROTACs, which arises from the introduction of next-generation paradox-breaker ligands. Such a conceptual approach may lead to a more effective generation of BRAFV600E-targeting PROTACs.
Results and discussion
Vemurafenib was selected as the BRAFV600E binding moiety to design bifunctional degrader molecules. Visual inspection of the crystal structures of BRAFV600E in complex with vemurafenib (PDB 3OG7) or with a homodimeric derivative (PDB 5JT2)26 revealed that the phenyl ring at the azaindole is solvent-exposed and suitable for linker attachment (Fig. S1†). To exploit this exit vector for PROTAC design, synthetic access to a modified vemurafenib scaffold was necessary. Key intermediate 3 (Scheme 1) was synthesised from the acid 1 and 5-bromo-7-azaindole (2) by an optimised Friedel–Crafts acylation procedure (Table S1†). The synthetic entry deviated from the initially published route.27 The correct structure of the acyl product 3 was confirmed by means of NMR spectroscopy and X-ray crystallography (Fig. S2†).28 Its subsequent transformation in microwave-assisted Suzuki cross-couplings afforded vemurafenib (4) and functionalised BRAFV600E inhibitors 5–7. An indirect access to 8via the aldehyde 7 and subsequent reductive amination proceeded in significant better yields compared to the direct use of the appropriate boronic acid leading to 8 from 3.
Scheme 1. Modular design and synthesis of vemurafenib-derived BRAFV600E PROTACs.

VHL-binding E3 ligase ligands were readily available, as described in our recent work on the improved synthetic pathway towards the amine 9 and the phenolic derivative 10.29 Notably, these distinct functional groups allowed the installation of orthogonally protected linkers 11 and 12 (ref. 30 and 31) on two differently oriented exit vectors of the E3 ligands.
For the first series of VHL-based PROTACs, the Cbz protecting group of linkers 11 was removed hydrogenolytically, and the amine group was coupled to the carboxyl group released from 6. The resulting vemurafenib-linker conjugates were deprotected at the terminal linker moiety and subjected to a further amide formation with VHL ligand 9. The final PROTACs 15a to 15c cover a moderate range of lipophilicity (Table 1) and span linker lengths between 10 and 20 atoms. In the second series of PROTACs, the linker was tethered to the VHL ligand by alkylating the phenolic group in 10. The protecting groups at the VHL-linker conjugates 14 were removed, and intermediates were coupled to 6 or 8 after their respective deprotection. In PROTACs 16a–16c, which represent differently oriented VHL-based degraders, lipophilicity and linker length fall within a similar range as in the first series.
Overview on physicochemical properties as well as kinase inhibition and degradation potencies of BRAFV600E-targeting PROTACs.
| Cmpd | Linker atoms | TPSAa | elog Db | %PPBc | IC50d (nM) | D BRAF e |
|---|---|---|---|---|---|---|
| GW5074 | — | 49 | 2.3 | n.d.f | 5.8 | n.d. |
| 15a | 10 | 288 | 2.3 | 95% | 8.5 | >95% |
| 15b | 16 | 297 | 2.4 | 95% | 0.31 | >95% |
| 15c | 20 | 288 | 3.7 | 96% | 48 | >95% |
| 16a | 17 | 288 | 3.6 | 96% | 10 | 77% |
| 16b | 13 | 297 | 2.5 | 96% | 3.4 | 73% |
| 16c | 16 | 297 | 2.7 | 96% | 3.8 | 77% |
Topological polar surface area given in Å2.
Experimental distribution coefficient at pH 7.4.23
Protein binding values were estimated by an HPLC-based method.24
In vitro BRAFV600E inhibition employing a radiometric assay using 10 μM [33P]-ATP and 1 μM substrate peptide,25 see also Fig. S8.†
Degradation indicated as remaining BRAFV600E levels after 4 h treatment with 1 μM of each compound (as determined by densitometric analysis of Western blot assays).
Not determined.
Next, we investigated our set of putative BRAFV600E degraders in an in vitro BRAF inhibition assay. The RAF inhibitor GW5074 was used as a control.32 All compounds retained BRAF mutant inhibitory properties, but apparent differences were observed within the two subseries (Table 1). Surprisingly, compounds 15c and 16a, possessing the most extended and hydrophobic linkers, had the highest IC50 values. Compounds were then tested in SK-MEL-28 cells (BRAFV600E/NRASwt) for their ability to induce RAF degradation. After a short treatment period of 4 h and concentrations between 0.1 and 10 μM, all compounds significantly reduced phospho-ERK (p-ERK) levels, but only compounds 16 caused moderate target degradation (Fig. S3† and Table 1). However, prolonged treatment with 10 μM of the PROTACs did not result in more efficient BRAFV600E degradation (Fig. S4†). Given these in vitro and in cellulo features, it is likely that the compounds did not efficiently form ternary complexes needed for the ubiquitination step of the PROTAC's mode of action. In line with this, the recently described BRAF degraders SJF-0628 and cmpd 12 (Fig. 1B) contain considerably shorter linkers, underscoring the potential need for positive cooperativity to efficiently degrade BRAF.33
For comparison, the established BRAF PROTACs SJF-0628 and cmpd 12 were included in our studies.19,20 The BRAFV600E degrader SJF-0628 caused significant degradation of its target protein in SK-MEL-28 cells (Fig. S5A†) between 0.1 and 10 μM. As expected, the control compound SJF-0661, which is tailored to be inoperative at the VHL-binding unit, did not cause BRAFV600E degradation. The CRBN-based PROTAC cmpd 12 caused only modest target degradation in our assay. All compounds provoked a fundamental reduction of pERK levels which may be attributed to either degradation or inhibition of BRAFV600E.
To pinpoint the encoding of kinase inhibitor functions in their derived degraders, we adapted the privileged linker design in SJF-0628 for the syntheses of paradox-breaker PROTACs (Scheme 2). Their synthetic entry proceeded via the nitro derivative 18, which was subsequently reduced to 19. Sulfamoylation with N-ethyl-N-methylsulfamoyl chloride gave azaindole 20, which was further subjected to Suzuki cross-coupling reactions with appropriate boronic acid pinacol esters. Products 21 and 22 represent close analogues of the paradox-breakers PLX7683 and PLX7904, respectively.7 However, the solvent-exposed aryl moieties were substituted with an N-Boc-piperazine heterocycle to install a linker handle. Subsequent alkylation with tert-butyl bromoacetate and coupling to the VHL ligands 10 or 27 yielded putative paradox-breaking PROTACs 25 and 26. The latter bears an additional, stereochemically defined methyl group at the VHL ligand, which was reported to enhance degrader efficacy.23,34 This modification balanced the physicochemical properties of 26 (Table 2). However, the polar surface area is increased due to the pyrimidine moiety present in this BRAFi ligand.
Scheme 2. Synthesis of PROTACs possessing paradox-breaker properties.

Overview on physicochemical properties as well as BRAFV600E degradation potencies of a PLX4032-based PROTAC and paradox-breaking PROTACs 25 and 26.
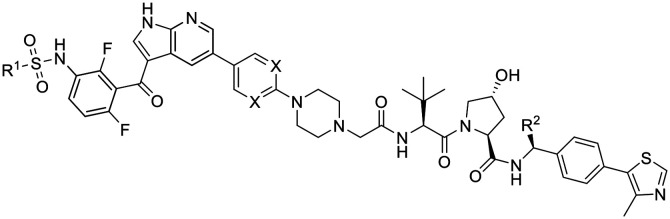
| |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | X | TPSAa | elog Db | %PPBc | D BRAF d |
| SJF-0628 |

|
H | CH | 247 | 2.9 | 96% | 46% |
| 25 (CST905) |

|
H | CH | 250 | 3.2 | 96% | 52% |
| 26 |

|
Me | N | 276 | 3.2 | 96% | >95% |
Topological polar surface area given in Å2.
Experimental distribution coefficient at pH 7.4.23
Protein binding values were estimated by an HPLC-based method.24
Degradation indicated as remaining BRAFV600E levels after 4 h treatment with 1 μM of each compound (as determined by densitometric analysis of Western blot assays).
Degraders 25 and 26 were then tested in SK-MEL-28 cells for BRAFV600E degradation (Fig. S5B†). Surprisingly, only 25 (CST905) showed distinct target degradation. Its effects on protein degradation (Fig. 3A) and cell viability reduction (Fig. S6†) were comparable to SJF-0628 and significantly more pronounced compared to the control SJF-0661, which can only function as a BRAFV600E inhibitor due to the mitigated ligase binding portion. Next, paradox-breakers 25 and 26 were investigated in the neuroblastoma cell line SK-N-AS possessing RAF wild-type and RAS mutant status. As expected, neither wild-type RAF nor pERK levels were affected by these compounds (Fig. S7†). A head-to-head comparison with SJF-0628 in this cell line (Fig. 3B) revealed the undesired paradoxical activation of ERK signalling induced by the vemurafenib-derived PROTAC SJF-0628. The paradox-breaking PROTAC 25, also referred to as development compound CST905, remedied this undesirable feature.
Fig. 3. (A) Head-to-head comparison of vemurafenib-based PROTACs (cmpd 12, SJF-0628), the negative control SJF-0661 with paradox-breaker PROTACs 25 and 26. SK-MEL-28 cells were treated with 10 μM compounds for 24 h; (B) in the NRAS mutant cell line SK-N-AS, SJF-0628 but not 25 (CST905) induced paradoxical ERK-signalling. Cells were treated for 4 h at the dose indicated. Quantified values for BRAFV600E, pERK1/2, and pMEK1/2 refer to mean of duplicates.

Conclusions
We disclosed a new series of BRAFV600E-targeting PROTACs in which the replacement of vemurafenib with a paradox-breaking BRAF ligand caused substantially different cellular outcomes. Herein, we introduce CST905, an exceptionally potent BRAFV600E degrader with a DC50 value of 18 nM and a maximal degradation concentration of 50 nM after a short treatment period of 4 h (Fig. S8†). In the BRAFV600E/NRASwt cell line SK-MEL-28, CST905 significantly reduced ERK phosphorylation (IC50 = 31 nM), and did not lead to paradoxical MAPK activation in a RAS-mutant cell line. Dissociating BRAFV600E depletion from paradoxical activation of MAPK pathway might lead to compounds with significantly more durable efficacy and a safer profile in comparison to first-generation BRAF inhibitors. Furthermore, this study pinpoints the need to track inhibitor functions of target ligands incorporated in PROTACs.
Author contributions
M. G. and C. S. designed the study; D. S. J. M., G. S., and C. S. performed experiments; S. A. V., I. S., and C. S. synthesised compounds. M. P. created computational illustrations. All authors analysed and interpreted data; O. W. R., M. G., I. C., and C. S. supervised the project; C. S. wrote the manuscript with contributions from all co-authors. All authors have approved the final article.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We thank C. Ennenbach, Prof. A. C. Filippou, M. Schneider (University of Bonn, Bonn, Germany), and M. Frelih (University of Ljubljana, Ljubljana, Slovenia) for their support and technical assistance. D. S. J. M., O. R. and I. C. acknowledge funding from Cancer Research UK programme grant C309/11566, and from The Institute of Cancer Research. I. S. acknowledges support by the Slovenian Research Agency (ARRS, program P1-0208 and grant J1-2485).
Electronic supplementary information (ESI) available: Supplementary Table 1, Supplementary Figures S1–S9; biological, chemical and physicochemical methods; synthetic procedures; structures, 1H NMR, 13C NMR and MS data; Western blots. CCDC 2143596. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2md00064d
Notes and references
- Ferguson F. M. Gray N. S. Nat. Rev. Drug Discovery. 2018;17:353–377. doi: 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- Cohen P. Cross D. Jänne P. A. Nat. Rev. Drug Discovery. 2021;20:551–569. doi: 10.1038/s41573-021-00195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J. Zhou H. Zhu S. Huang J. Zhao X. Ding H. Pan Y. Cancer Manage. Res. 2018;10:2289–2301. doi: 10.2147/CMAR.S170105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant D. E. Morrison D. K. Br. J. Cancer. 2018;118:3–8. doi: 10.1038/bjc.2017.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman P. B. Hauschild A. Robert C. Haanen J. B. Ascierto P. Larkin J. Dummer R. Garbe C. Testori A. Maio M. Hogg D. Lorigan P. Lebbe C. Jouary T. Schadendorf D. Ribas A. O'Day S. J. Sosman J. A. Kirkwood J. M. Eggermont A. M. M. Dreno B. Nolop K. Li J. Nelson B. Hou J. Lee R. J. Flaherty K. T. McArthur G. A. N. Engl. J. Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchook G. S. Long G. V. Kurzrock R. Kim K. B. Arkenau T. H. Brown M. P. Hamid O. Infante J. R. Millward M. Pavlick A. C. O'Day S. J. Blackman S. C. Curtis C. M. Lebowitz P. Ma B. Ouellet D. Kefford R. F. Lancet. 2012;379:1893–1901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. Spevak W. Zhang Y. Burton E. A. Ma Y. Habets G. Zhang J. Lin J. Ewing T. Matusow B. Tsang G. Marimuthu A. Cho H. Wu G. Wang W. Fong D. Nguyen H. Shi S. Womack P. Nespi M. Shellooe R. Carias H. Powell B. Light E. Sanftner L. Walters J. Tsai J. West B. L. Visor G. Rezaei H. Lin P. S. Nolop K. Ibrahim P. N. Hirth P. Bollag G. Nature. 2015;526:583–586. doi: 10.1038/nature14982. [DOI] [PubMed] [Google Scholar]
- Agianian B. Gavathiotis E. J. Med. Chem. 2018;61:5775–5793. doi: 10.1021/acs.jmedchem.7b01306. [DOI] [PubMed] [Google Scholar]
- Poulikakos P. I. Zhang C. Bollag G. Shokat K. M. Rosen N. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoulia Z. Wu Y. Ahmed T. A. Xin Q. Bollard J. Krepler C. Wu X. Zhang C. Bollag G. Herlyn M. Fagin J. A. Lujambio A. Gavathiotis E. Poulikakos P. I. Cancer Cell. 2016;30:485–498. doi: 10.1016/j.ccell.2016.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigglestone C. E. Yeung K.-S. ACS Med. Chem. Lett. 2021;12:1629–1632. doi: 10.1021/acsmedchemlett.1c00543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J. J. Zhang C. Nat. Chem. Biol. 2020;16:1154–1155. doi: 10.1038/s41589-020-0647-1. [DOI] [PubMed] [Google Scholar]
- Yu F. Cai M. Shao L. Zhang J. Front. Chem. 2021;9:679120. doi: 10.3389/fchem.2021.679120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paiva S.-L. Crews C. M. Curr. Opin. Chem. Biol. 2019;50:111–119. doi: 10.1016/j.cbpa.2019.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z. Crews C. M. ChemBioChem. 2022;23:e202100270. doi: 10.1002/cbic.202100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida T. Ciulli A. SLAS Discovery. 2020;26:484–502. doi: 10.1177/2472555220965528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. Chen F. Pei S. Gou S. Bioorg. Chem. 2019;87:191–199. doi: 10.1016/j.bioorg.2019.03.035. [DOI] [PubMed] [Google Scholar]
- Posternak G. Tang X. Maisonneuve P. Jin T. Lavoie H. Daou S. Orlicky S. Goullet de Rugy T. Caldwell L. Chan K. Aman A. Prakesch M. Poda G. Mader P. Wong C. Maier S. Kitaygorodsky J. Larsen B. Colwill K. Yin Z. Ceccarelli D. F. Batey R. A. Taipale M. Kurinov I. Uehling D. Wrana J. Durocher D. Gingras A.-C. Al-Awar R. Therrien M. Sicheri F. Nat. Chem. Biol. 2020;16:1170–1178. doi: 10.1038/s41589-020-0609-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.-R. Chen L. Wei Y. Yu W. Chen Y. Zhang C. Jiao B. Shi T. Sun L. Zhang C. Xu Y. Lee M. R. Luo Y. Plewe M. B. Wang J. J. Med. Chem. 2020;63:4069–4080. doi: 10.1021/acs.jmedchem.9b02083. [DOI] [PubMed] [Google Scholar]
- Alabi S. Jaime-Figueroa S. Yao Z. Gao Y. Hines J. Samarasinghe K. T. G. Vogt L. Rosen N. Crews C. M. Nat. Commun. 2021;12:920. doi: 10.1038/s41467-021-21159-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J. Meng F. Park K.-S. Yim H. Velez J. Kumar P. Wang L. Xie L. Chen H. Shen Y. Teichman E. Li D. Wang G. G. Chen X. Kaniskan H. Ü. Jin J. J. Am. Chem. Soc. 2021;143:15073–15083. doi: 10.1021/jacs.1c04841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau K. Coen M. Zhang A. X. Pachl F. Castaldi M. P. Dahl G. Boyd H. Scott C. Newham P. Br. J. Pharmacol. 2020;177:1709–1718. doi: 10.1111/bph.15014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinebach C. Ng Y. L. D. Sosic I. Lee C.-S. Chen S. Lindner S. Vu L. P. Bricelj A. Haschemi R. Monschke M. Steinwarz E. Wagner K. G. Bendas G. Luo J. Gütschow M. Krönke J. Chem. Sci. 2020;11:3474–3486. doi: 10.1039/D0SC00167H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gockel L. M. Pfeifer V. Baltes F. Bachmaier R. D. Wagner K. G. Bendas G. Gütschow M. Sosič I. Steinebach C. Arch. Pharm. 2022;355(5):2100467. doi: 10.1002/ardp.202100467. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T. Deacon S. W. Devarajan K. Ma H. Peterson J. R. Nat. Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso M. Estrada M. A. Ventocilla C. Samanta M. Maksimoska J. Villanueva J. Winkler J. D. Marmorstein R. ACS Chem. Biol. 2016;11:2876–2888. doi: 10.1021/acschembio.6b00529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G. Hirth P. Tsai J. Zhang J. Ibrahim P. N. Cho H. Spevak W. Zhang C. Zhang Y. Habets G. Burton E. A. Wong B. Tsang G. West B. L. Powell B. Shellooe R. Marimuthu A. Nguyen H. Zhang K. Y. J. Artis D. R. Schlessinger J. Su F. Higgins B. Iyer R. D'Andrea K. Koehler A. Stumm M. Lin P. S. Lee R. J. Grippo J. Puzanov I. Kim K. B. Ribas A. McArthur G. A. Sosman J. A. Chapman P. B. Flaherty K. T. Xu X. Nathanson K. L. Nolop K. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The X-ray crystallographic data collection of 3 was performed on a Bruker X8 –Kappa Apex-II diffractometer at 100(2) K. The diffractometer was equipped with a low-temperature device (Bruker Kryoflex I, Bruker AXS) and used Mo-Kα Radiation (λ=0.71073 Å). intensities were measured by fine-slicing φ- and ω-scans and corrected for background, polarization and lorentz effects. A semi-empirical absorption correction was applied for the data sets by using Bruker's SADABS program including a semi-empirical absorption correction according to Blessing's method. The structures were solved by intrinsic phasing methods and refined anisotropically by the least-squares procedure implemented in the ShelX-2014/7 program system. The hydrogen atoms were included isotropically using a riding model on the bound carbon atoms. CCDC 2143596 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the Cambridge Crystallographic Data Centre.
- Steinebach C. Voell S. A. Vu L. P. Bricelj A. Sosič I. Schnakenburg G. Gütschow M. Synthesis. 2020;52:2521–2527. doi: 10.1055/s-0040-1707400. [DOI] [Google Scholar]
- Steinebach C. Kehm H. Lindner S. Vu L. P. Köpff S. López Mármol Á. Weiler C. Wagner K. G. Reichenzeller M. Krönke J. Gütschow M. Chem. Commun. 2019;55:1821–1824. doi: 10.1039/C8CC09541H. [DOI] [PubMed] [Google Scholar]
- Steinebach C. Sosič I. Lindner S. Bricelj A. Kohl F. Ng Y. L. D. Monschke M. Wagner K. G. Krönke J. Gütschow M. Med. Chem. Commun. 2019;10:1037–1041. doi: 10.1039/C9MD00185A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey K. Cory M. Davis R. Frye S. V. Harris P. A. Hunter R. N. Jung D. K. McDonald O. B. McNutt R. W. Peel M. R. Rutkowske R. D. Veal J. M. Wood E. R. Bioorg. Med. Chem. Lett. 2000;10:223–226. doi: 10.1016/S0960-894X(99)00668-X. [DOI] [PubMed] [Google Scholar]
- Gadd M. S. Testa A. Lucas X. Chan K.-H. Chen W. Lamont D. J. Zengerle M. Ciulli A. Nat. Chem. Biol. 2017;13:514–521. doi: 10.1038/nchembio.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X. Wang C. Qin C. Xiang W. Fernandez-Salas E. Yang C.-Y. Wang M. Zhao L. Xu T. Chinnaswamy K. Delproposto J. Stuckey J. Wang S. J. Med. Chem. 2019;62:941–964. doi: 10.1021/acs.jmedchem.8b01631. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.