

Abstract
Mal de Meleda (MDM) is a rare autosomal recessive type of palmoplantar keratoderma that is characterized by transgradient keratoderma with scleroatrophy, pseudoainhum around the fingers, and perioral erythema. Its features may also include lichenoid lesions, brachydactyly, and nail dystrophy. The disease has high morbidity and significantly impairs quality of life. Here, we describe two sisters with typical clinical presentations of MDM and a history of consanguinity between the parents.
Keywords: Acroerythrokeratoderma, consanguinity, genodermatosis
Introduction
Mal de Meleda (MDM), which takes its name from the Croatian island of Meleda (Mljet), is a rare autosomal recessive skin disease. It is also known by a variety of other names: keratoderma palmoplantaris transgrediens, acroerythrokeratoderma, and keratosis extremitatum hereditaria progrediens.[1]
Clinically, the disease occurs after birth. Its estimated prevalence in the general population is 1 in 100,000. Consanguinity is a high-risk factor for the disease.[2]
The hands (palms) and feet (soles and inner parts) are primarily affected. The lesions are characterized by hyperkeratosis with wide margins and erythema, starting from the center of the body of palms and soles and gradually expanding to other areas.[3] The involvement of the lips has also been reported.[4]
Due to the chronic nature of the disease, MDM can lead to serious flexion contractures and long-term disability, which significantly limits patients’ daily activities and increases the burden of this disease on them and their families.[5] Here, we present the cases of two sisters with the familial occurrence of MDM who are the offspring of consanguineous parents. The aim of this article is to describe this rare genodermatosis and discuss these cases in relation to the existing literature.
Case Report
A 32-year-old female patient born from a second-degree consanguineous marriage presented with complaints of intense sweating, desquamation, and thickening of the skin on the palms of her hands and soles of her feet since she was 3 years old. The clinical picture was worsened by the involvement of the dorsal parts of the hands and feet. She also complained about flexion contractures of her hands and feet, which had a significant negative impact on her daily life. The patient reported that her parents had four children, two of whom were healthy and one of whom suffered from the disease. Dermatological examination revealed intense transgressive whitish-yellow palmoplantar hyperkeratosis with maceration and erythema extending to the dorsal of the hands and feet covered with a yellowish and waxy substance accompanied by loss of dermatoglyphics. The nails were normal [Figure 1 a and b]. A biopsy was taken from the lesion on the right foot. Histopathology revealed hyperkeratosis and acanthosis without epidermolysis in the epidermis, accompanied by perivascular lymphocytic infiltrate in the dermis [Figure 2 a and b]. A clinical diagnosis of MDM was made. Laboratory tests were unremarkable. Oral acitretin at 25 mg/day and topical emollients were prescribed. After she took these treatments for nearly 2 months, physical examinations indicated that the hyperkeratosis was in remission and joint movement in the hands began to improve. However, due to the significant side effects (dry eyes and lips), the patient stopped taking oral acitretin.
Figure 1.

Clinical features of the hands and foot in patient 1. (a) Transgradient palmoplantar keratoderma with sharp margins. (b) The palms and toes are covered by yellowish, waxy, and thick hyperkeratosis with an obvious erythematosus border
Figure 2.

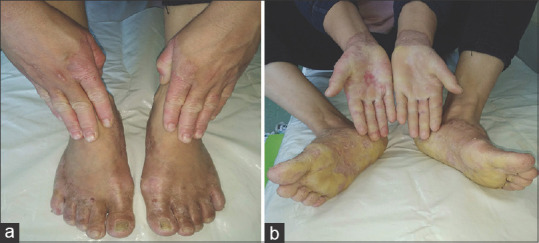
Clinical features of the hands and foot in patient 2. (a) Diffuse palmoplantar keratoderma involving bilateral palms and soles and extending proximally up to the wrists and the dorsal of hands and feet with well-defined margins. (b) Appearance of palms and soles thickness, waxy and yellow with loss of dermatoglyphics
Case 2
A 41-year-old female, the sister of the patient described in case 1, presented with a complaint of desquamation on her hands and feet since birth and the development of painful fissures. She also complained of excessive sweating but denied that she had any lesions on other parts of her body. Her lesions were less severe than those of her sibling, and there was no associated flexion contracture. On examination, the patient was found to have palmoplantar hyperkeratosis on the feet and hands that extended proximally up to the wrist and dorsum with well-defined margins and in a transgrediens pattern. Hyperkeratosis of the nails on all fingers and toes was also present [Figure 3 a and b]. Laboratory tests were normal. A diagnosis of MDM was made based on the abovementioned features and clinical history. However, the patient refused to take systemic retinoids and chose treatment with emollients instead. To identify the genetic defect, genetic analysis for SLURP-1 could not be performed in both cases because of lack of access to the proper facilities.
Figure 3.
(a and b) Hyperkeratosis and acanthosis without epidermolysis in the epidermis, accompanied by perivascular lymphocytic infiltrate in the dermis. (H and E ×100, ×40)
Discussion
MDM was first observed by Luca Stulli in 1826 on the island of Meleda (Mljet) in Dalmatia, which is on a Mediterranean trade route. Almost all the described cases belong to descendants of individuals from this region, but the disease has also been seen in America and Africa. For over 50 years, it was thought to be a form of Hansen’s disease. However, Hovorka and Ehlers realized that this was not a contagious disease and used the term “MDM.”[6]
MDM is characterized by autosomal recessive inheritance of symmetric palmoplantar hyperkeratosis that progressively extends to the dorsal surfaces of the hands and feet and ichthyotic changes elsewhere that have a material basis in homozygous mutation of the SLURP-1 gene located on chromosome 8q24. SLURP-1 has been found to be an epidermal secreted neuromodulator that affects the inhibition of tumor necrosis factor-alpha release by macrophages during both epidermal homeostasis and the wound healing process. Missense mutation of SLURP-1 in MDM may result in changes to protein’s 3-dimensional structure and eventually cause nonfunctional protein, which can lead to severe hyperkeratosis in MDM.[7] Although previously published studies have investigated the molecular mechanism underlying MDM, it remains uncertain and more research is needed.
Clinically, disease onset typically occurs within the 1st months of life. Bilateral symmetrical diffuse erythema of the palms and soles is followed by the development of palmoplantar hyperkeratosis with a distinct yellow hue and well-defined margins. The severity of the disease increases with time. Hyperkeratotic plaques extend to the dorsal surface of the hands and feet (transgredient pattern), but they are even more prominent on the joints and may involve the lower legs. Psoriasiform or lichenoid plaques are frequently seen on the knees and elbows and may be accompanied by perioral erythema.[4] In both of our cases, keratoderma appeared after birth and showed clinical features such as glove-and-stocking distribution with a sharp margin, gradually extending to the dorsum of the hands and feet. Other findings included nail dystrophy affecting all the toes and fingers, palmoplantar hyperhidrosis, brachydactyly, lichenoid eruption, and contracture of the fingers with functional loss.
Due to the wide clinical spectrum of MDM, it should be differentiated from other syndromes associated with palmoplantar keratoderma (PPK).
Greither syndrome may present with symptoms similar to those of MDM, but it has an autosomal dominant inheritance pattern, displays progressive evolution, and is manifested by epidermolysis. Richner‒Hanhart syndrome presents with more prominent keratoderma, intellectual disabilities, and high tyrosine metabolite levels.[6] In our cases, there were no intellectual disabilities, and tyrosine metabolites were normal.
Vohwinkel syndrome, an autosomal dominant condition characterized by mutilating PPK, can progress to spontaneous amputation, alopecia, ichthyosis, and deafness.[8] The absence of honeycomb keratoderma and sensorineural hearing loss in our two cases led to the exclusion of this diagnosis.
Papillon‒Lefèvre syndrome, which is an autosomal recessive condition, develops due to a mutation in the cathepsin C gene located on chromosome 11q14. It is characterized by diffuse keratoderma and associated with gingivitis, premature tooth loss, periosteal changes, and intracranial calcifications.[9] We ruled out this syndrome in our cases due to the presence of normal teeth and the absence of periodontitis.
Unna‒Thost syndrome is a nontransgressive PPK that develops as a result of a mutation in the keratin 9 gene and shows autosomal dominant inheritance. Unna‒Thost PPK is characterized by symmetrical keratosis circumscribed to the palms and soles.[10] Transgressive lesions at the palmoplantar regions and consanguineous marriage in their family and also acanthosis without epidermolysis in the epidermis as histopathological finding showed the possibility of MDM in our cases.
Huriez syndrome, which is also an autosomal dominant disease, develops due to a mutation in locus on chromosome 4q28-q31. It is manifested by diffuse PPK accompanied by sclerodactyly, atrophy, and koilonychia.[6] Scleroatrophy and nail changes were not observed in our patients to support this disease.
MDM treatment is difficult because it is a rare genetic condition. Before retinoids became available, the main regimen was emollients and keratolytics. The most common treatment used today is acitretin, which is effective in curing hyperkeratosis but has little effect on erythema. However, to relieve their symptoms, patients may need long-term oral acitretin, which has side effects that most often include drug-induced dry eyes, lips, and skin.[11] Therefore, patients must be provided with appropriate health education and moisturizers to ensure they adapt well to this therapy.
Painful cracks and secondary infection with malodorous interdigital maceration are the most common complications of MDM. Very few cases of melanoma have been reported to have developed on MDM-affected skin.[12] To date, no evidence of malignancies has been found in our patients; however, they still require long-term follow-up and observation.
Conclusion
MDM is a rare autosomal recessive skin disorder characterized by PPK with glove-and-stocking distribution, keratotic skin lesions, perioral erythema, brachydactyly, and nail abnormalities. In the cases presented here, treatment with retinoids led to partial improvement, and they are recommended unless there are contraindications. Their cases detail a rare genodermatosis that should be included in the differential diagnosis of PPK.
Statement of Ethics
The study is exempt from ethical approval.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Judge MR, McLean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, Griffiths C, editors. Rook's Textbook of Dermatology. 8th ed. Singapore: Wiley-Blackwell; 2010. pp. 19.93–19.119. [Google Scholar]
- 2.Bchetnia M, Laroussi N, Youssef M, Charfeddine C, Ben Brick AS, Boubaker MS, et al. Particular Mal de Meleda phenotypes in Tunisia and mutations founder effect in the Mediterranean region. Biomed Res Int. 2013;2013:206803. doi: 10.1155/2013/206803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thappa DM. Mal de Meleda type of keratoderma. Indian J Dermatol. 2002;47:196. [Google Scholar]
- 4.Nath AK, Chaudhuri S, Thappa DM. Mal de meleda with lip involvement:A report of two cases. Indian J Dermatol. 2012;57:390–3. doi: 10.4103/0019-5154.100497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan Y, Zhao H, Chen A, Huang X. A Mal de Meleda patient with severe flexion contractures of hands and feet:A case report in West China. Medicine (Baltimore) 2017;96:e7972. doi: 10.1097/MD.0000000000007972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silva FA, Cunha TV, Boeno Edos S, Steiner D. Mal de Meleda:A report of two cases of familial occurrence. An Bras Dermatol. 2011;86:S100–3. doi: 10.1590/s0365-05962011000700026. [DOI] [PubMed] [Google Scholar]
- 7.Adeyo O, Oberer M, Ploug M, Fong LG, Young SG, Beigneux AP. Heterogeneity in the properties of mutant secreted lymphocyte antigen 6/urokinase receptor-related protein 1 (SLURP1) in Mal de Meleda. Br J Dermatol. 2015;173:1066–9. doi: 10.1111/bjd.13868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cavalcante LI, Holanda EM, Almeida TL, Accioly-Filho JW. Vohwinkel´s mutilating keratoderma:Report of three familial cases. An Bras Dermatol. 2003;78:311–8. [Google Scholar]
- 9.Proença N, Rotberg A, Todescan JH. Palmoplantar keratosis with periodontal disease (Papillon-Lèfevre) An Bras Dermatol. 1970;45:249–54. [PubMed] [Google Scholar]
- 10.Hinterberger L, Pföhler C, Vogt T, Müller CS. Diffuse epidermolytic palmoplantar keratoderma (Unna-Thost-) BMJ Case Rep. 2012;2012:bcr2012006443. doi: 10.1136/bcr-2012-006443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van de Kerkhof PC, van Dooren-Greebe RJ, Steijlen PM. Acitretin in the treatment of mal de Meleda. Br J Dermatol. 1992;127:191–2. doi: 10.1111/j.1365-2133.1992.tb08060.x. [DOI] [PubMed] [Google Scholar]
- 12.Mozzillo N, Nunziata CA, Caracò C, Fazioli F, Botti G. Melanoma Cooperative Group. Malignant melanoma developing in an area of hereditary palmoplantar keratoderma (Mal de Meleda) J Surg Oncol. 2003;84:229–33. doi: 10.1002/jso.10317. [DOI] [PubMed] [Google Scholar]