

ABSTRACT
Infectious bronchitis virus (IBV), a γ-coronavirus, causes the economically important poultry disease infectious bronchitis. Cellular stress response is an effective antiviral strategy that leads to stress granule (SG) formation. Previous studies suggested that SGs were involved in the antiviral activity of host cells to limit viral propagation. Here, we aimed to delineate the molecular mechanisms regulating the SG response to pathogenic IBV strain infection. We found that most chicken embryo kidney (CEK) cells formed no SGs during IBV infection and IBV replication inhibited arsenite-induced SG formation. This inhibition was not caused by changes in the integrity or abundance of SG proteins during infection. IBV nonstructural protein 15 (Nsp15) endoribonuclease activity suppressed SG formation. Regardless of whether Nsp15 was expressed alone, with recombinant viral infection with Newcastle disease virus as a vector, or with EndoU-deficient IBV, the Nsp15 endoribonuclease activity was the main factor inhibiting SG formation. Importantly, uridine-specific endoribonuclease (EndoU)-deficient IBV infection induced colocalization of IBV N protein/dsRNA and SG-associated protein TIA1 in infected cells. Additionally, overexpressing TIA1 in CEK cells suppressed IBV replication and may be a potential antiviral factor for impairing viral replication. These data provide a novel foundation for future investigations of the mechanisms by which coronavirus endoribonuclease activity affects viral replication.
IMPORTANCE Endoribonuclease is conserved in coronaviruses and affects viral replication and pathogenicity. Infectious bronchitis virus (IBV), a γ-coronavirus, infects respiratory, renal, and reproductive systems, causing millions of dollars in lost revenue to the poultry industry worldwide annually. Mutating the viral endoribonuclease poly(U) resulted in SG formation, and TIA1 protein colocalized with the viral N protein and dsRNA, thus damaging IBV replication. These results suggest a new antiviral target design strategy for coronaviruses.
KEYWORDS: γ-coronavirus, infectious bronchitis virus, nonstructural protein 15 (Nsp15), uridylate-specific endoribonuclease (EndoU), stress granule, TIA1 cytotoxic granule-associated RNA-binding protein
INTRODUCTION
Coronaviruses are enveloped viruses carrying a single-stranded positive-sense RNA genome of ~30 kb (1–3) and are classified into four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus. Replication of infectious bronchitis virus (IBV), a Gammacoronavirus, starts when the virus binds to a specific viral receptor and then fuses with host cell membranes. The incoming genomic RNA is translated into two large polyproteins via 1 ribosomal frameshift from open reading frame (ORF) 1a and 1b, pp1a and pp1ab. The polyproteins are processed by Papain-like and 3C-like protease to yield 15 mature Nsps (Nsp2–16), most of which are essential for viral RNA synthesis (4–7). Nsp15 protein has a uridine-specific endoribonuclease (EndoU) domain, which is highly conserved among coronaviruses family and attributed to the formation of the replicase-transcriptase complex (RTC) (7–9). Our previous study indicated that IBV Nsp15 also encodes viral EndoU and that residues His223, His238, Lys278, and Tyr314 located in the C terminus are four key EndoU active sites (10).
Viruses regulate various cellular processes to form an environment that is suitable for generation of progeny virus. The activation of some pathways in favor of virus replication and antagonizing innate immune system is an important process during virus infection. The cellular stress granule (SG) is a macromolecular center for mRNAs that contains translation stalled messenger ribonucleoproteins (mRNPs), 40S ribosomal subunits, and RNA-binding proteins. It can regulate host-cell pathways and play a key role in activating antiviral responses. Thus, SG is often targeted by viruses during replication (11–14).
RNA viruses will generate double-stranded RNA (dsRNA) intermediates during genome replication. The dsRNA can activate protein kinase R (PKR), an important molecule of antiviral defense (15). PKR is activated after binding to dsRNA and then activated PKR phosphorylates the eukaryotic translation initiation factor 2α (eIF2α) on Ser51 (15, 16). The eIF2α phosphorylation binding to eIF2B prevents recycling of ternary complex tRNA-Met-GTP-eIF2 that is essential to cap-dependent translation initiation, causing host translation inhibition (17). Viruses utilize host translation system to produce their own proteins so the global translation inhibition of the host cell will severely impair viral replication. Inhibition of translation initiation causes elongating ribosomes to “run off” their transcripts, leading to decomposition of polysome. The stalled initiation, polysome disassembly, and mRNP aggregation trigger formation of discrete foci, termed SGs in a series of reversible stages.
Because viral protein synthesis relies on host translation machinery, translational arrest mediated by SG formation is an efficient strategy for inhibiting viral replication, and consequently, many viruses have evolved mechanisms to overcome this (18, 19). Common viral strategies for disrupting SG formation include degrading or sequestering key SG nucleation proteins, such as G3BP1 and TIA1 (20–23), and suppressing PKR activation (24, 25). The impact of viral infection on SG formation varies. Most viruses can inhibit SGs formation via different mechanisms to promote virus replication; for example, Influenza A virus NS1 protein inhibits SG formation by sequestering dsRNA from PKR (26). Herpes simplex virus 1 host shutoff RNase, U(S)11 protein, and viral glycoprotein B could block SG formation (27–29). The 4a accessory protein of Middle East Respiratory Syndrome-coronavirus (MERS-CoV) inhibits SG formation via binding to PKR (30). Mosquito-borne alphaviruses nsP3 macrodomain hydrolase activity disassembles virus-induced SG and suppresses SG formation (31). There are some viruses, such as Newcastle disease virus (NDV) and vesicular stomatitis virus (VSV), that induce the formation of SG or SG-like structures to promote viral replication (32–34). Hepatitis C virus induces eIF2-dependent SG assembly and disassembly, and some SG components play pivotal roles in several steps of the viral life cycle (35, 36). A previous study reported that IBV strain Beau-R induced SG formation in 10% to 25% of infected Vero cells (37, 38). However, these studies were based on data from avirulent strains in nontarget animal cells and may not reflect the true intracellular signaling and antiviral responses of prevalent and virulent strains infecting host cells. However, whether virulent IBV has evolved strategic mechanisms to modulate SGs and how SG formation affects viral replication are unclear.
Here, we show that a pathogenic IBV strain failed to trigger SG formation at any time postinfection and virulent IBV replication inhibited SG formation. The endoribonuclease activity of Nsp15 suppressed SG formation. Importantly, EndoU-deficient IBV infection induced colocalization of viral N protein/dsRNA and SG-associated protein TIA1 in infected cells, and TIA1 suppressed IBV replication.
RESULTS
The stress response pathway was suppressed during IBV infection.
To determine whether epidemic pathogenic IBV strain infection induces SG formation, particularly at earlier time points in host cells, we analyzed the distribution of the SG marker protein G3BP1 in IBV-infected chicken embryo kidney (CEK) cells. The viral infection efficiency was monitored by visualizing Nsp2. In IBV-infected cells, despite efficient viral infection and replication indicated by accumulation of considerable amounts of Nsp2 in the cytosol, no SGs were observed at 6, 18, or 24 h postinfection (hpi). However, SGs were detected at 12 hpi, but only in <5% of infected cells (Fig. 1). The lack of SGs was not caused by an intrinsic defect in the stress response pathway of CEK cells, as clear SGs were formed upon arsenic acid treatment (Fig. 2A). Examining the distribution of another canonical SG protein, TIA1, confirmed that no SGs were present in IBV-infected cells at any time point examined (data not shown). Hence, IBV failed to effectively trigger SG formation in cells during infection.
FIG 1.

Infectious bronchitis virus (IBV) replication did not trigger stress granules (SGs) in infected cells. SG formation in chicken embryo kidney (CEK) cells infected with IBV rSD were analyzed by immunofluorescent staining at indicated times postinfection (hpi). Scale bars = 20 μm. Immunofluorescent staining for viral nonstructural protein (Nsp2; red) and cellular SG marker G3B1 (green). Small arrows indicate SG formation in the cells.
FIG 2.

IBV replication abrogated eukaryotic translation initiation factor 2α (eIF2α)-dependent SG formation. (A) CEK cells were treated with 1 mM sodium arsenite for 30 min and then immunostained. (B) CEK cells were infected with IBV SD at 18 hpi and then with 1 mM sodium arsenite for 30 min and then immunostained. Cells were detected with anti-Nsp2 antibody (red), SGs with anti-G3BP1 (green), and cell nuclei with DAPI (blue). Representative images of three independent experiments are shown. Scale bars = 10 μm. (C) CEK cells were mock infected (control [C]) or infected with 106.0 50% egg infective dose of IBV rSD. At 6, 12, 24, and 36 hpi, cell lysates (10 μg per lane) were analyzed via Western blot to detect G3BP1, IBV N, TIA1, and tubulin. Small asterisks indicate SG formation in the cells.
To investigate whether IBV inhibits SG formation, we treated cells with 1 mM sodium arsenite for 30 min. In mock-infected cells treated with sodium arsenite, 80% to 100% of the cells contained G3BP1 SGs in the cytoplasm. Conversely, G3BP1-containing SGs were observed in only 24% of IBV-infected cells when treated with sodium arsenite (Fig. 2B). Most IBV-infected cells (Nsp2-positive cells) produced no SGs, indicating that IBV replication significantly inhibited sodium arsenite-induced SG formation.
To investigate whether IBV inhibits SG assembly by promoting proteolytic cleavage of and/or degrading of SG-nucleating proteins, we infected CEK cells with IBV and investigated the abundance and integrity of select SG proteins at 6, 12, 24, and 36 hpi. Western blot analysis revealed that the SG protein abundance remained the same between the mock- and IBV-infected samples, and no cleavage products were identified (Fig. 2C). Thus, changes in G3BP1 abundance or proteolytic cleavage did not cause IBV to block SG assembly. These findings indicate that IBV encodes one or more antagonists to suppress activation of the stress response pathway.
IBV Nsp15 endoribonuclease activity suppressed SG formation.
Our previous studies revealed that IBV Nsp15 endoribonuclease activity prevented activation of PKR, which phosphorylates the alpha subunit of eIF2 (10). This inhibits translation and is intended to block viral replication. A downstream effect of translational shutoff involves SG formation. We suspect that IBV Nsp15 might be an antagonist to suppress activation of the stress response pathway.
To prove whether IBV Nsp15 can suppress the SG formation, we transfected different plasmids into 293T cells to observe SG formation referenced previous study (39). The results showed that plasmids encoding Nsp15 and IAV NS1 (an PKR antagonist served as a positive control) inhibit SG formation at 36 h posttransfection, whereas IBV Nsp8 protein (an unrelated protein control) and EndoU-deficient proteins (Nsp15-H238A and Nsp15-Y334A) can induce 30% to 40% cells to form SGs (Fig. 3A and B). We further tested the ability of these IBV Nsp15s to suppress the sodium arsenite-induced SG formation. We transfected enhanced green fluorescent protein (EGFP) fusion proteins into Vero and baby hamster kidney (BHK) cells. After treating cells with 1 mM sodium arsenite for 30 min, IBV Nsp15 suppressed SG formation, which was detected with the SG marker G3BP1. This inhibition was more pronounced in BHK cells. We repeated the experiment in Vero cells by replacing the labeled antibody and achieved consistent results (data not shown). The inhibitory effect of Nsp15 was less pronounced in this assay, possibly because the relatively large amounts of sodium arsenite may have exceeded the maximum capacity of the PKR antagonists. These data suggest that IBV Nsp15 inhibits cellular stress response pathways, which is associated with endoribonuclease activity of Nsp15.
FIG 3.
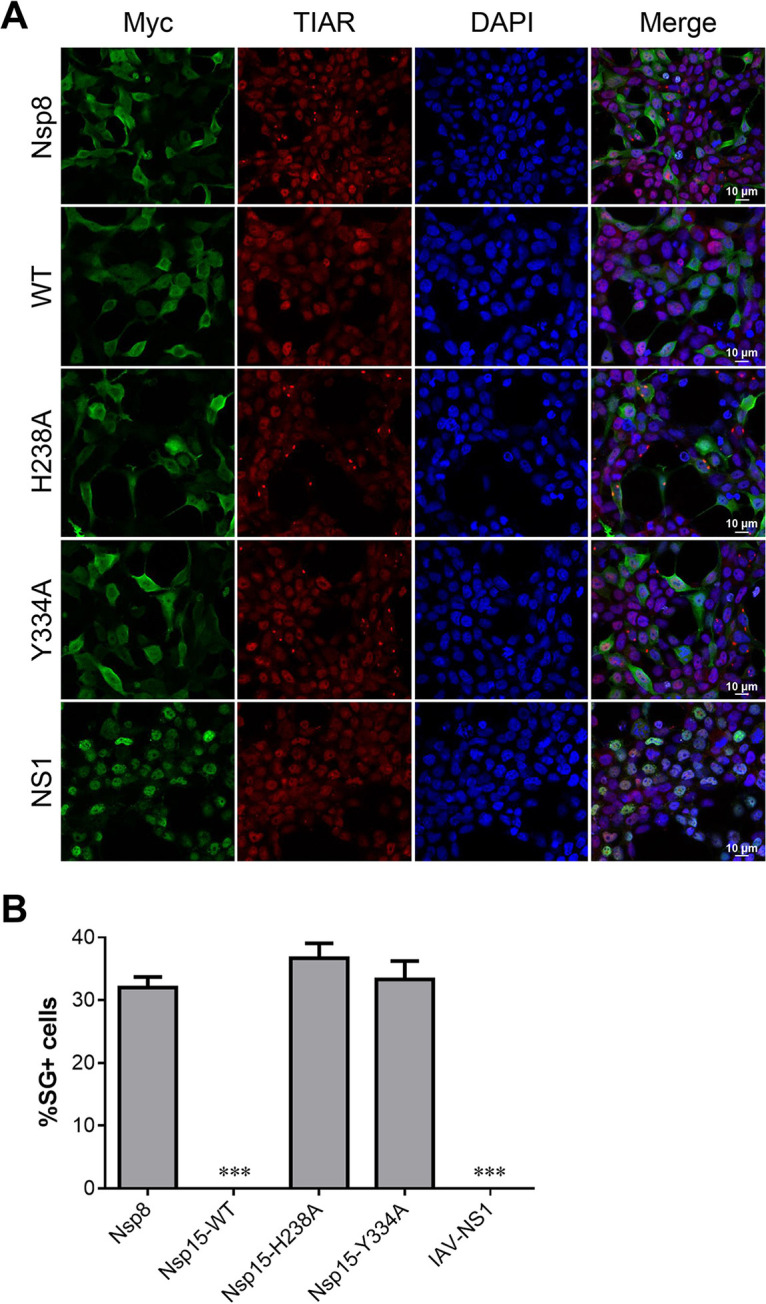
IBV Nsp15 endoribonuclease activity suppressed SG formation. (A) 293T cells were transfected with plasmids encoding Myc-tagged IBV-wild-type (WT) Nsp15, uridine-specific endoribonuclease (EndoU)-deficient mutants (IBV Nsp15-H238A and IBV Nsp15-Y334A), IAV NS1 protein, or pCMV-Myc vector. At 36 h posttransfection (hpt), cells were immunostained. Proteins were stained with anti-Myc antibody (green); SGs were detected with anti-TIAR (red). Cell nuclei were stained with DAPI (blue). Representative images of three independent experiments are shown. (B) SG formation was quantified by counting cells with and without SGs in 3–5 randomly selected fields of view at 36 hpt (>100 cells). Error bars = SD for three independent experiments. ***, P < 0.001.
IBV Nsp15 endoribonuclease activity partially inhibited NDV-induced SG production.
NDV triggered stable SG formation by activating the PKR/eIF2a pathway (32). We used the properties of NDV to induce SG production to explore whether Nsp15 inhibits virus-induced SG production. We constructed recombinant NDV that stably expressed Nsp15 and its mutant protein to infect CEK cells (Fig. 4A). We used Flag tag to indicate foreign protein expression and G3BP1 as an SG marker. Infecting CEK cells with NDV induced SG production in the infected cells as well as in neighboring cells. Therefore, we used the total number of cells that produced SGs in our data. NDV-EGFP, NDV-Nsp15-H238A, and NDV-Nsp15-Y334A induced 83% to 90% of cells to produce SGs, whereas NDV-Nsp15 induced only 29% of cells to produce SGs. Thus, NDV expressing Nsp15 inhibited SG formation postinfection (Fig. 4B and C). More interestingly, IAV NS1 strongly inhibited NDV-induced SGs, and no obvious SGs were produced in either infected or uninfected cells. Hence, IBV Nsp15 endoribonuclease activity partially inhibited NDV-induced SG production activated via the PKR/eIF2a pathway.
FIG 4.
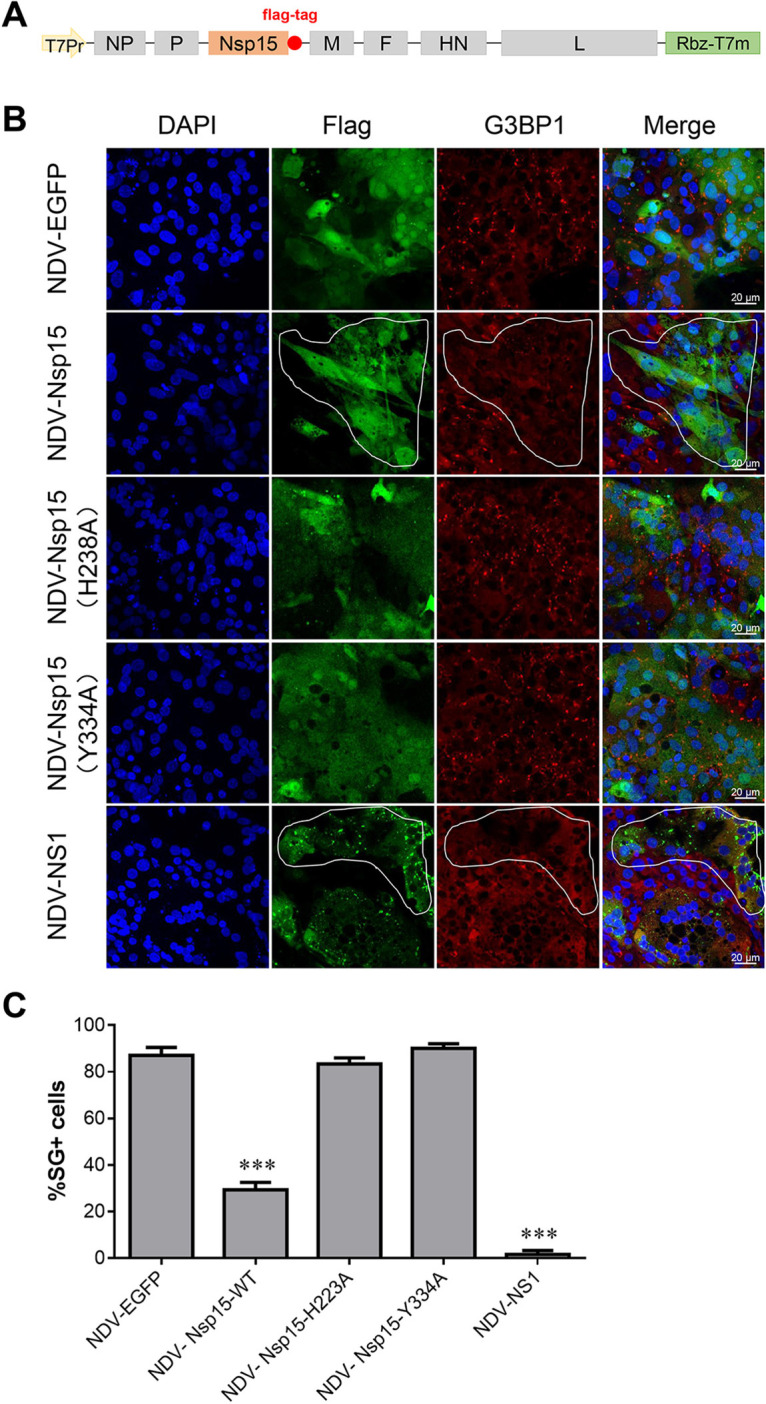
IBV Nsp15 endoribonuclease activity partially inhibited Newcastle disease virus (NDV)-induced SG production. (A) Cloning strategy to incorporate the Nsp15, Nsp15-H238A, Nsp15-Y334A, and IAV-NS1 genes into the full-length NDV. (B) CEK cells were infected with recombinant NDV virus and stained for immunofluorescence at 18 hpi. Proteins were stained with anti-Flag antibody (green); SGs were detected with anti-G3BP1 (red). Cell nuclei were stained with DAPI (blue). Representative images of three independent experiments are shown. The selected outlined fields represent cells that are infected by NDV and have no SGs formation. (C) SG formation was quantified by counting infected cells with and without SGs in 3–5 randomly selected fields of view at 18 hpi (>100 cells). Error bars = SD for three independent experiments. ***, P < 0.001.
Absence of endoribonuclease activity induced more SG formation and less viral replication during IBV infection.
Our former studies showed that EndoU-deficient IBVs exhibit impaired replication capacities in CEK cells and attenuated virulence in chickens (10). To explore whether Nsp15 inhibits SG formation during IBV infection and whether SG formation in cells is related to viral replication and pathogenicity, we examined SG formation in CEK cells infected with wild-type IBV (IBV-WT) or EndoU-deficient IBV (IBV-H238A or IBV-Y334A), which were rescued via a reverse genetics system (40). Immunofluorescence analysis using antibodies specific for the SG marker G3BP1, together with IBV Nsp2, showed SG accumulation in EndoU-deficient IBV-infected cells. IBV-WT replication did not induce SG formation in CEK cells, but SGs formed in the EndoU-deficient IBV-infected cells (Fig. 5A). At 18 hpi, only 5% of IBV-WT-infected CEK cells contained SGs, whereas approximately 75% of the IBV-Nsp15-H238A-infected CEK cells and 82% of the IBV-Nsp15-Y334A-infected CEK cells contained SGs (Fig. 5B). Thus, absence of endoribonuclease activity induced more SG formation and less viral replication during IBV infection.
FIG 5.
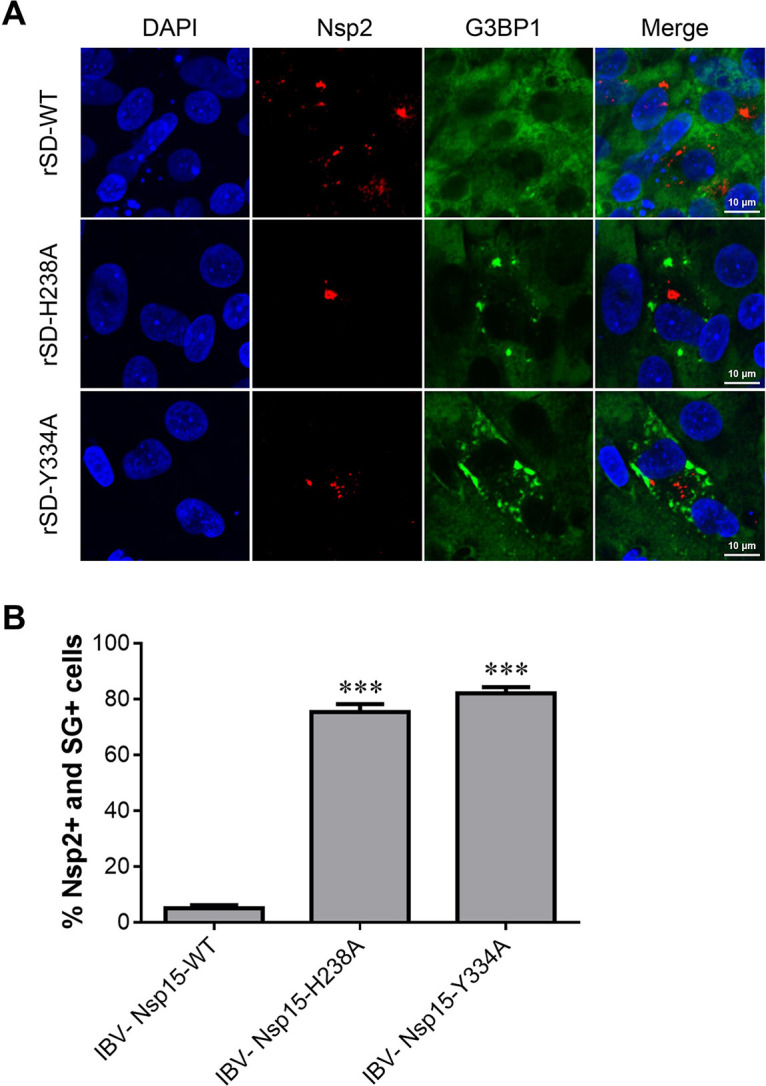
Absence of endoribonuclease activity induced more SG formation and less viral replication during IBV infection. (A) CEK cells were infected with parent IBV rSD strain or EndoU-deficient virus (rSD-H238A or rSD-Y334A) for 18 hpi and then immunostained. Infected cells were identified with anti-IBN-Nsp2 (red); SGs were detected with anti-G3BP1 (green). Cell nuclei were stained with DAPI (blue). (B) SG formation was quantified; the bar graphs show the percentage of SG-positive cells to total infected cells (Nsp2-positive), which were calculated over 20 random fields, presented as the mean ± SD. ***, P < 0.001.
EndoU-deficient IBV infection induced colocalization of N protein/dsRNA and TIA1 in infected cells.
To further explore the relationship between changes in SG-related proteins and viral replication after endoribonuclease deletion, we used TIA1 for verification. Immunofluorescence analysis using TIA1-specific antibodies together with the IBV N protein showed SG accumulation in EndoU-deficient IBV-infected cells. Interestingly, TIA1 lacked the typical SG morphology but formed larger aggregates, showing a strong positive signal in EndoU-deficient IBV-infected cells. Viral N protein partially colocalized with TIA1 during EndoU-deficient IBV infection. This phenomenon was not seen in IBV-WT-infected CEK cells, in which TIA1 was evenly distributed in the infected CEK cells as it was in the control group (Fig. 6A). The coronavirus N protein is a structural protein that forms complexes with genomic RNA and is critical in enhancing viral transcription and assembly efficiency. N protein is often localized to coronavirus RTCs, and we observed dsRNA, which is produced by positive-strand RNA viruses during replication. As with the N protein, viral dsRNA localized to TIA1 aggregation in EndoU-deficient IBV-infected cells (Fig. 6B). These results indicate that infection with EndoU-deficient mutants changed the TIA1 distribution and induced viral N protein and dsRNA partly localized with TIA1 in the infected CEK cells.
FIG 6.
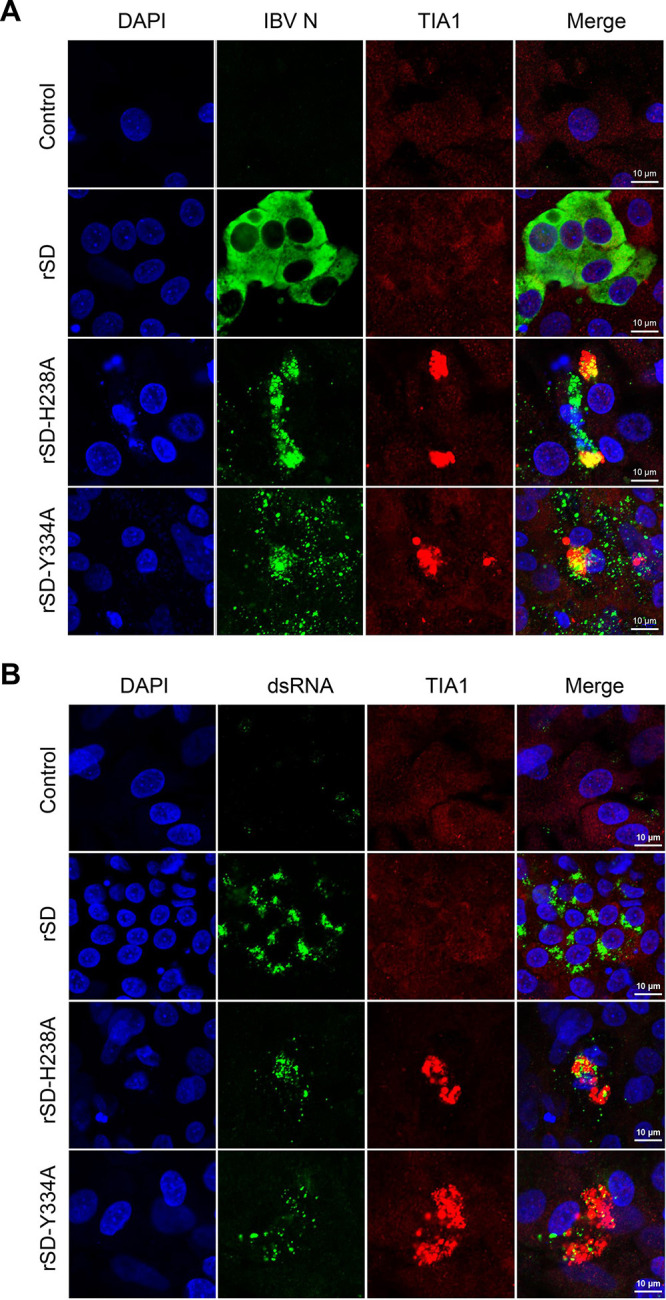
EndoU-deficient IBV infection induced colocalization of N protein/double-strand (ds)RNA and TIA1 in infected cells. CEK cells were infected with parent IBV rSD strain or EndoU-deficient virus (rSD-H238A or rSD-Y334A) for 18 hpi and then immunostained. (A) Infected cells were immunostained; IBV N was detected with anti-N (green); TIA1 was detected with anti-TIA1 (red). (B) Infected cells were immunostained; IBV dsRNA was detected with J2 antibody (green); and TIA1 was detected with anti-TIA1 (red).
TIA1 overexpression inhibited IBV replication.
EndoU-deficient viral propagation was severely impaired in CEK cells, which was evidenced by reduced viral dsRNA accumulation and protein synthesis (10). This induced TIA1 to aggregate into larger particles and localize to the viral N protein and dsRNA in infected cells. We suspected that TIA1 may help reduce viral replication during EndoU-deficient IBV infection. We constructed a PRK-EGFP-TIA1 plasmid electroporated with CEK cells and used the PRK-EGFP plasmid as a control. At 36 h posttransfection, the CEK cells were infected with IBV virus, and viral N protein was detected. N protein synthesis showed no differences at 12 hpi, while the N protein was not significantly increased in the TIA1-overexpressing cells at 24 hpi compared with that in the control cells (Fig. 7A). Furthermore, we compared the IBV replication kinetics in EGFP-expressing cells and TIA1-expressing cells over 48 h. The amount of virus in the supernatant of the TIA1-expressing cells was less than that of the EGFP-expressing cells from 24 hpi, and this phenomenon was even more obvious at 48 hpi (Fig. 7B). Thus, TIA1 inhibited IBV replication. TIA1 may be a potential antiviral protein that interferes with normal viral replication in cells by interacting with the N protein and the genomic RNA of IBV.
FIG 7.

TIA1 overexpression inhibited IBV replication. (A) TIA1 was constructed into PRK-EGFP, the plasmid was electroporated in CEK cells, and PRK-EGFP plasmid was used as a control. At 36 hpt, CEK cells were infected with IBV and the viral protein synthesis was observed via Western blot. TIA1 overexpression was evident at 48 and 60 hpt. Relative levels of the density ratio were quantified. The N protein/tubulin density ratio of the PRK-EGFP control group was set as 1 and compared with the other experimental groups. (B) Viral replication kinetics were compared in EGFP-expressing cells and TIA1-expressing cells. Data are shown as the mean ± SD of at least three independent experiments. P values were calculated using t tests. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns = not significant.
Transient Nsp15 expression did not affect cellular gene expression.
SG usually forms after translation inhibition owing to the aggregation of stalled mRNPs, translation initiation factors, and RNA-binding proteins. A previous study indicated that translation was inhibited during IBV replication from 12 hpi, and the degree of inhibition increased as the infection progressed (41). Using Renilla luciferase (Rluc) assays, we assessed IBV Nsps for their ability to interfere with host-cell gene expression. Human embryonic kidney (HEK-293T) cells were cotransfected with plasmids expressing Nsps and pRL-TK plasmids expressing TK promoter-driven Rluc. As a positive control, we used Nsp1 of severe acute respiratory syndrome coronavirus (SARS-CoV) because previous studies have shown that it inhibits host gene expression (42). IBV Nsp6 and Nsp15 significantly reduced luciferase reporter gene expression (Fig. 8A). The inhibitory effect of Nsp15 was dose dependent, and greater transfection doses more strongly inhibited reporter gene expression (Fig. 8B). This inhibitory effect is related to the Nsp15 endoribonuclease; destroying this endoribonuclease removes its inhibitory effect (Fig. 8C). To further explore whether IBV Nsp15 could broadly inhibit protein synthesis in HEK-293T cells, we examined host proteins using a ribopuromycylation assay. Interestingly, IBV Nsp6 and Nsp15 exerted no broad-spectrum inhibitory activity on host protein synthesis in this experiment (Fig. 8D). These results indicate that the viral Nsp15 only inhibits reporter gene expression but not host gene expression, which differs from the function of Nsp1. EndoU-deficient viruses induced defective replication but not by inhibiting host protein expression.
FIG 8.

Transient Nsp15 expression did not affect cellular gene expression. (A) IBV Nsp6 and Nsp15 reduced luciferase reporter gene expression. HEK-293T cells were cotransfected with plasmids expressing the IBV Nsp2-16 protein in vector pCMV-Myc and pRL-TK plasmids expressing TK promoter-driven Rluc. SARS-CoV Nsp1 was used as a positive control. At 24 hpt, cells were used for luciferase activity assays. (B) The inhibitory effect of Nsp15 was dose dependent. Different doses of Nsp15 protein (0.5–2.0 μg) and pRL-TK plasmids were cotransfected into 293T cells, and luciferase activity was detected. (C) Endoribonuclease activity affected inhibition of reporter genes. Nsp15 and EndoU-mutants (Nsp15-H223A, Nsp15-H238A, Nsp15-K278A, and Nsp15-Y334A) were transfected with pRL-TK plasmids. (D) Cells were transfected with IBV Nsp6 and Nsp15 plasmid for 24 h. The cells were pulsed with 3 μm puromycin for 1 h at 37°C and then subjected to Western blot analysis (left). The grayscale values of the protein bands were analyzed using ImageJ (right). ***, P < 0.001.
DISCUSSION
The increasing number of viruses that disrupt or evade SG formation during infection has led to recognition of cellular stress responses, which are important antiviral mechanisms. We found that IBV infection induced no SG formation in most CEK cells at any time point. Several other viruses, including the Ebola, Zika, West Nile, and Junin viruses, can inhibit SG signaling by regulating the eIF2α-dependent pathway (43–46). These viruses achieve this by inhibiting PKR activation, thereby preventing eIF2α phosphorylation, or by dephosphorylating eIF2α. SG formation or inhibition has been reported for various coronaviruses; for example, mouse hepatitis virus and transmissible gastroenteritis virus induce SGs or SG-like granules, whereas Middle East respiratory syndrome virus does not (47–49). IBV was reported to induce SG formation but only in 20% of infected Vero cells (50). These results are based on IBV strain Beau-R, an apathogenic strain that differs completely from the current epidemic and pathogenic strains. However, the biological significance of SGs in epidemic IBV replication is unclear. Here, we identified epidemic IBV-induced SGs in approximately 5% of CEK cells, which is less than that of the apathogenic virus, and IBV infection inhibited arsenite-induced eIF2α-dependent SG formation. Thus, SGs likely have antiviral functions during CoV replication.
Our previous studies showed that IBV Nsp15 alone can inhibit phosphorylation of PKR, which phosphorylates the alpha subunit of eIF2 (10). This inhibits translation and leads to SG formation. Thus, we suspect that Nsp15 is a specific stress-response antagonist of coronaviruses. We found that Nsp15 inhibited SG production in CEK cells and depended on endoribonuclease activity. Immunofluorescence analysis of fixed cells infected with recombinant EndoU-deficient IBV revealed SGs in a substantial fraction of the infected cells. Previous studies indicated that coronaviral Nsp15 is an EndoU RNase and the main interferon antagonist for coronaviruses to evade host innate immune responses (9, 50). EndoU mediates evasion of viral dsRNA recognition by host sensors in macrophages, and the RNase is a unique strategy for viruses to subvert SGs. Several reports have shown that SGs serve as platforms for viral dsRNA sensing by retinoic acid-inducible gene I-like receptors and subsequent activation of viral immune responses (51, 52). Recent studies reported that several other interferon-regulatory molecules, such as MEX3C, Riplet, DHX36, and Pumilios, also localize to SGs (53). EndoU-deficient IBV infection revealed that deficient replication may be related to the antiviral responses of SGs, which requires further study.
G3BP1 and TIA1 are two classic SG markers, which we used to show SG formation. Immunofluorescence analysis showed that TIA1 aggregated into larger granules that were morphologically distinct from classic SGs and the viral N protein localized with TIA1 in EndoU-deficient IBV-infected cells, while viral dsRNA simultaneously localized with TIA1. Coronavirus Nsp15 has uridine-specific endoribonuclease activity (8). EndoU-deficient mutant infection induced more AU-rich RNA. TIA1 is a robust SG marker, and the N-terminal RNA recognition motifs domain preferentially binds to RNA transcripts with AU-rich or C-rich elements in the 5′- or 3′-untranslated regions (54–56). Colocalization of N protein/dsRNA and TIA1 in EndoU-deficient virus-infected cells may be related to these factors. Hackbart et al. (57) reported that EndoU cleaved the 5′-polyuridines from negative-sense viral RNA, termed PUN RNA, which is the product of polyA-templated RNA synthesis, and the EndoU-deficient coronavirus-infected cytoplasm contained more PUN RNA than did WT-infected cells. In our study, viral dsRNA localized with TIA1 in EndoU-deficient IBV-infected CEK cells. Many PUN RNAs were present in EndoU-deficient IBV-infected CEK cells. These PUN RNAs are often produced in viral (v)RNA synthesis sites and recruit cellular TIA1 aggregates with them because TIA1 preferentially binds to AU-rich RNAs. Coronavirus N protein, an RNA-binding protein, and dsRNA are located in vRNA synthesis sites (58); therefore, colocalization of N protein/dsRNA and TIA1 can be visualized in EndoU-deficient IBV-infected cells. Coronavirus vRNA replication occurs in a special double-membrane vesicle (DMV) structure, which can effectively prevent the innate immune system from recognizing vRNA (59). Whether TIA1 can interact with PUN RNA, how TIA1 crosses the DMV structure into the interior, and whether the DMV structure is destroyed in the absence of viral endoribonuclease activity are interesting questions that can be explored in future studies.
TIA1 may be a factor in reducing viral replication during EndoU-deficient IBV infection. Infecting cells with IBV after overexpressing TIA1 restricted IBV replication, meaning that TIA1 may be a potential antiviral molecule. To replicate IBV genome, the replicase gene ORFs 1a and 1b are first translated into large polyproteins in host cells, which is further cleaved by Papain-Like and 3C-like protease to yield 15 mature Nsps (5). Multiple Nsps compose viral RTCs to initiate the positive-strand genomic RNA, subgenomic and negative-strand RNAs synthesis. We suspect that TIA1 preferentially binds to AU-rich RNA, which is abundant in EndoU-deficient IBV-infected cells, and it colocalizes with the viral N protein and dsRNA, which are RTC components, resulting in aggregation of larger particles. Such aggregation further interferes with viral replication.
We further explored whether the endoribonuclease activity of Nsp15 affected viral replication by inhibiting host protein synthesis. Many viruses disrupt host gene expression by degrading host mRNA and/or manipulating translation activities to create a cellular environment favorable for viral replication. Various α- and β-coronaviruses have been shown to induce host shutoff via viral Nsp1 (42, 60), whereas γ- and δ-coronaviruses lack Nsp1. Additionally, dsRNA generated during viral replication activates PKR, which inhibits protein synthesis by phosphorylating the eIF2α on Ser51. Phosphorylation of eIF2α blocks GTP/GDP exchange; thus, eIF2 must be recycled between rounds of translation initiation. Nsp15 inhibited PKR activation; therefore, we speculated whether it affects protein synthesis after PKR activation. IBV Nsp15 did not affect host protein expression, indicating that the endoribonuclease activity of Nsp15 affected viral replication but not by inhibiting host protein synthesis.
Insufficient endoribonuclease activity reduced the viral replication levels, thus attenuating the viral pathogenicity to chickens. This may have occurred for the following reasons. First, SG formation in infected cells recruited PKR to promote multiple innate antiviral responses when CEK cells were infected with EndoU-deficient virus. Second, coronavirus Nsp15 is a uridine-specific endoribonuclease, and PUN RNA was increased in infected cells, which activated the host’s innate immune response. Third, TIA1 preferentially binds to AU-rich RNA, which is abundant in EndoU-deficient coronavirus-infected cells and interferes with viral replication. Fourth, RTCs are critical replication factories in coronaviruses, and certain cellular stress proteins, such as TIA1, enter these factories and interfere with normal viral replication progression.
How the conserved EndoU activity of coronaviruses regulates viral replication remains unclear. Meaningful discoveries of the role of EndoU were revealed in recent studies: EndoU mediates the evasion of viral double-stranded RNA recognition by host sensors in macrophages (61), suppresses type I and type III interferon responses (9, 62), attenuates pathogenesis (10), and targets viral polyuridine sequences to evade activating host sensors (57). In this study, we provide another way that coronavirus EndoU promote virus replication via inhibiting SGs formation and regulated SG-associated protein TIA1 distribution, which is a potential antiviral factor that impairs viral replication. These results provide a solid foundation for future investigations of the mechanisms by which coronavirus endoribonuclease activity affects viral replication.
MATERIALS AND METHODS
Cells and viruses.
Cell lines used in this study included African green monkey kidney cells (Vero and Vero E6; CRL-1586, ATCC, Manassas, VA, USA), human embryonic kidney cells (HEK293T; ATCC CRL-11268), and hamster kidney cells (BHK; ATCC CCL-10). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (50 U/mL), and streptomycin (50 mg/mL). The recombinant viruses rSD (GenBank accession number MW351623), rSD-H238A (GenBank accession number MW351624), and rSD-Y334A (GenBank accession number MW351628) were constructed in our laboratory.
Plasmid construction and transfection.
IBV Nsp15, Nsp15-H238A, and Nsp15-Y334A carrying a C-terminal EGFP were cloned into PRK5 vectors, and a linker sequence (GGGGS) was inserted between the target proteins (Nsp15, Nsp15-H238A, and Nsp15-Y334A) and EGFP, thus yielding PRK5-IBV-Nsp15, PRK5-IBV-Nsp15-H238A, and PRK5-IBV-Nsp15-Y334A. The plasmids encoding IBV Nsp2, Nsp3, Nsp4, Nsp5, Nsp6, Nsp7, Nsp8, Nsp9, Nsp12, Nsp15, Nsp16, and IAV NS1 were generated by amplification of cDNA from IBV rSD or IAV using corresponding primers and cloned into pCMV-myc or pRK5-EGFP. The SARS-CoV Nsp1 gene was synthesized and purchased from Sangon Biotech. The catalytic mutant plasmids of Nsp15 were cloned using the Fast MultiSite Mutagenesis System (TransGen Biotech, Beijing, China). The chicken-derived TIA1 gene was amplified from chicken embryo primary kidney cells.
Cells were seeded on glass coverslips in a 24-well cluster (25,000 cells/well). The indicated plasmids were transfected into cells using Lipofectamine 2000 (Invitrogen) per the manufacturer’s handbook. Briefly, 0.5 μg plasmid and 1.5 μL Lipofectamine 2000 (m/vol = 1:3) were diluted and incubated in 0.10 mL Opti-MEM (Gibco). After 5 min, the plasmid and Lipofectamine 2000 were mixed and incubated at room temperature for 20 min, allowing formation of the lipid-plasmid complex, which was added to the cultured cells and incubated for 24 h.
Antibodies and chemicals.
The following antibodies were used in the experiments: anti-IBV-N (mouse; 3BN1; HyTest, Turku, Finland), anti-G3BP1 (mouse; Ab32506; Abcam, Cambridge, UK), anti-TIAR (rabbit; Ab32036; Abcam), anti-TIA1 (rabbit; 12133-2-AP; Proteintech, Wuhan, China), anti-β-actin (mouse; 3700S; Cell Signaling Technology, Danvers, MA, USA), anti-α/β-tubulin (rabbit; 2148; Cell Signaling Technology), anti-Myc-tag (mouse; 2276; Cell Signaling Technology), and J2 (mouse, anti-dsRNA; English and Scientific Consulting Kft, Hungary). Goat anti-rabbit IgG (H+L) (AS014) and goat anti-mouse IgG (H+L) (AS003) conjugated with horseradish peroxidase (HRP) were from Abcam. Alexa Fluor goat anti-rabbit-488 (4412), Alexa Fluor goat anti-rabbit-555 (4413), Alexa Fluor goat anti-mouse-488 (4408), and Alexa Fluor goat anti-mouse-555 (4409) were obtained from Cell Signaling Technology. Sodium arsenite (S7400) was purchased from Merck.
Indirect immunofluorescence and confocal microscopy.
Cells seeded onto glass coverslips in 12-well plates were transfected with plasmids by Lipofectamine 2000 or infected with IBV (106.0 50% egg infective dose [EID50]). At the indicated time points, cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min at room temperature (RT), permeabilized in 0.5% Triton X-100 for 15 min, and incubated in blocking buffer (5% bovine serum albumin in PBS) for 30 min. Permeabilized cells were stained with the following primary antibody overnight at 4°C: mouse monoclonal anti-Myc antibody (Cell Signaling Technology); mouse monoclonal anti-G3BP1 antibody (Abcam); rabbit polyclonal anti-TIAR antibody (Abcam); rabbit polyclonal anti-TIA1 antibody (Proteintech); mouse monoclonal anti-IBV-N antibody (HyTest); rabbit polyclonal anti-IBV-Nsp2 antibody; and mouse monoclonal anti-dsRNA J2 (English and Scientific Consulting). Next, cells were incubated with Alexa Fluor conjugated secondary antibody (Cell Signaling Technology; dilution 1:1,000) for 1 h at RT. DAPI (4′,6-diamidino-2-phenylindole) (Sigma-Aldrich, St. Louis, MO, USA) was used to stain the nuclei for 5 min at RT.
Western blotting.
CEK cells were infected with IBV (106.0 EID50), and cells were harvested at indicated time points. Whole-cell lysates were extracted from the infected CEK cells using the ProteinExt mammalian total protein extraction kit (TransGen). The concentration of total protein was detected with a bicinchoninic acid protein assay kit (CWBIO, Beijing, China). Protein samples were separated on 10% SDS-PAGE and then transferred to Immun-Blot PVDF membrane (Amersham Biosciences, Freiburg, Germany). Membranes were blocked with 5% skim milk in 0.1% Tween 20 in Tris-buffered saline for 2 h at RT and then incubated with indicated primary antibody overnight at 4°C and subsequently incubated with the HRP-conjugated anti-mouse or anti-rabbit antibody for 1 h at RT. Primary antibodies used in this experiment were anti-G3BP1 antibody (Abcam); anti-TIA1 antibody (Proteintech); anti-IBV-N antibody (HyTest); anti-Myc antibody (Cell Signaling Technology); anti-actin antibody (Abcam); anti-tubulin antibody (Abcam); and anti-puromycin (Sigma-Aldrich). Results were observed with BeyoECL Moon (Beyotime).
Construction of recombinant virus NDV-Nsp15/H238A/Y334A/NS1.
The full-length aSG10 cDNA clone pOK-aSG10 was used as the backbone to construct a recombinant cDNA clone containing the IBV Nsp15, IBV Nsp15-H238A, IBV Nsp15-Y334A, and IAV-NS1 genes between the P and M genes in the aSG10 genome as an additional transcription unit (Fig. 4A). Briefly, the unique restriction enzyme site PmeI was introduced into the region between the P and M genes of the infectious pOK-aSG10 clone using overlap extension PCR. The target gene, amplified with the appropriate primer pair, was engineered to contain the NDV gene-start and gene-end signal sequences and then inserted into the PmeI restriction sites in pOK-aSG10 according to the “rule of six.” To rescue the infectious NDV, BSR T7/5 cells stably expressing the T7 phage RNA polymerase were grown to 90% confluence in six-well plates and then cotransfected with 10 μg DNA consisting of a mixture of pOK-rSG10-target gene, pCI-NP, pCI-P, and pCI-L at a 4:2:1:1 ratio using Lipofectamine 2000 per the manufacturer’s instructions. At 6 hpi, the cells were washed once with PBS and maintained in DMEM containing 2% (vol/vol) FBS. Three days later, the culture supernatants and cell monolayers were harvested by freeze-thawing the infected cells three times. The allantoic cavities of 10-day-old embryonated specific-pathogen-free chicken eggs were then inoculated with 200 μL of cells to amplify the recovered viruses. After incubating for 4 days, the allantoic fluid was harvested, and the rescued virus was detected via the hemagglutination test. The target genes were confirmed by nucleotide sequencing.
Growth kinetics and titration.
In a 12-well plate, CEK cells were electroporated with PRK-EGFP or PRK-EGFP-TIA1 plasmids. After 36 h posttransfection, cells were infected with IBV at 106.0 EID50 per well. The inoculum was removed and replaced with cell maintenance medium after 1.5 h of incubation. Cell culture supernatant was collected after infection and titrated using viral copies based on standard curves (63).
Reporter and ribopuromycylation assays.
To measure the effect of Nsp15 on the amount of luciferase (Rluc) production, a Rluc reporter assay was performed. Briefly, the functional plasmid and Rluc reporter plasmid pRL-TK were cotransfected into HEK-293T cells with Lipofectamine 2000. Twenty-four hours later, cells were subjected to Rluc activity assay according to the manufacturer’s instructions (Promega). A ribopuromycylation assay was performed as described previously (64). Briefly, cultured HEK-293T cells were transfected with plasmids expressing IBV-WT Nsp15 or mutants. At 36 h posttransfection, the cells were pulse labeled with 3 μM puromycin and cultured for 1 h at 37°C with 5% CO2. After three washes with PBS, the cells protein lysates were extracted and subjected to Western-blot analysis. Total protein expression was analyzed using an anti-puromycin antibody (Sigma-Aldrich) and quantified by ImageJ.
Statistical analysis.
The data were analyzed with GraphPad Prism8 software (GraphPad Software, Inc., La Jolla, CA, USA). The data are presented as the mean ± standard deviation of three independent experiments. Significance was determined using t test analysis of variance. ImageJ (NIH, Bethesda, MD, USA) was used to quantify the intensities of the corresponding bands on the Western blots.
ACKNOWLEDGMENTS
This study was supported by the National Key Research and Development Program of China Grant 2021YFD1801103 and the 2115 Talent Development Program of China Agricultural University.
We thank Traci Raley from Liwen Bianji (Edanz) (www.liwenbianji.cn/) for editing a draft of this manuscript.
Contributor Information
Guozhong Zhang, Email: zhanggz@cau.edu.cn.
Rebecca Ellis Dutch, University of Kentucky College of Medicine.
REFERENCES
- 1.Lee HJ, Shieh CK, Gorbalenya AE, Koonin EV, La Monica N, Tuler J, Bagdzhadzhyan A, Lai MM. 1991. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology 180:567–582. 10.1016/0042-6822(91)90071-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lomniczi B. 1977. Biological properties of avian coronavirus RNA. J Gen Virol 36:531–533. 10.1099/0022-1317-36-3-531. [DOI] [PubMed] [Google Scholar]
- 3.Lomniczi B, Kennedy I. 1977. Genome of infectious bronchitis virus. J Virol 24:99–107. 10.1128/JVI.24.1.99-107.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hurst-Hess KR, Kuo L, Masters PS. 2015. Dissection of amino-terminal functional domains of murine coronavirus nonstructural protein 3. J Virol 89:6033–6047. 10.1128/JVI.00197-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fang S, Shen H, Wang J, Tay FP, Liu DX. 2010. Functional and genetic studies of the substrate specificity of coronavirus infectious bronchitis virus 3C-Like proteinase. J Virol 84:7325–7336. 10.1128/JVI.02490-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neuman BW, Chamberlain P, Bowden F, Joseph J. 2014. Atlas of coronavirus replicase structure. Virus Res 194:49–66. 10.1016/j.virusres.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prajapat M, Sarma P, Shekhar N, Prakash A, Avti P, Bhattacharyya A, Kaur H, Kumar S, Bansal S, Sharma AR, Medhi B. 2020. Update on the target structures of SARS-CoV-2: a systematic review. Indian J Pharmacol 52:142–149. 10.4103/ijp.IJP_338_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krishnan DA, Sangeetha G, Vajravijayan S, Nandhagopal N, Gunasekaran K. 2020. Structure-based drug designing towards the identification of potential anti-viral for COVID-19 by targeting endoribonuclease NSP15. Inform Med Unlocked 20:100392. 10.1016/j.imu.2020.100392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Volk A, Hackbart M, Deng X, Cruz-Pulido Y, O’Brien A, Baker SC. 2020. Coronavirus endoribonuclease and deubiquitinating interferon antagonists differentially modulate the host response during replication in macrophages. J Virol 94:e00178-20. 10.1128/JVI.00178-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao J, Sun L, Zhao Y, Feng D, Cheng J, Zhang G. 2021. Coronavirus endoribonuclease ensures efficient viral replication and prevents protein kinase R activation. J Virol 95:e02103-20. 10.1128/JVI.02103-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCormick C, Khaperskyy DA. 2017. Translation inhibition and stress granules in the antiviral immune response. Nat Rev Immunol 17:647–660. 10.1038/nri.2017.63. [DOI] [PubMed] [Google Scholar]
- 12.Walsh D, Mathews MB, Mohr I. 2013. Tinkering with translation: protein synthesis in virus-infected cells. Cold Spring Harb Perspect Biol 5:a12351. 10.1101/cshperspect.a012351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, Anderson P. 2002. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell 13:195–210. 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kedersha N, Stoecklin G, Ayodele M, Yacono P, Lykke-Andersen J, Fritzler MJ, Scheuner D, Kaufman RJ, Golan DE, Anderson P. 2005. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J Cell Biol 169:871–884. 10.1083/jcb.200502088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia MA, Meurs EF, Esteban M. 2007. The dsRNA protein kinase PKR: virus and cell control. Biochimie 89:799–811. 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Vattem KM, Staschke KA, Zhu S, Wek RC. 2001. Inhibitory sequences in the N-terminus of the double-stranded-RNA-dependent protein kinase, PKR, are important for regulating phosphorylation of eukaryotic initiation factor 2alpha (eIF2alpha). Eur J Biochem 268:1143–1153. 10.1046/j.1432-1327.2001.01979.x. [DOI] [PubMed] [Google Scholar]
- 17.Jackson RJ, Hellen CU, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11:113–127. 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.White JP, Lloyd RE. 2012. Regulation of stress granules in virus systems. Trends Microbiol 20:175–183. 10.1016/j.tim.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beckham CJ, Parker R. 2008. P bodies, stress granules, and viral life cycles. Cell Host Microbe 3:206–212. 10.1016/j.chom.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthews JD, Frey TK. 2012. Analysis of subcellular G3BP redistribution during rubella virus infection. J Gen Virol 93:267–274. 10.1099/vir.0.036780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson HG, McInerney GM. 2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol Biol Cell 23:4701–4712. 10.1091/mbc.E12-08-0619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White JP, Lloyd RE. 2011. Poliovirus unlinks TIA1 aggregation and mRNA stress granule formation. J Virol 85:12442–12454. 10.1128/JVI.05888-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scholte FE, Tas A, Albulescu IC, Zusinaite E, Merits A, Snijder EJ, van Hemert MJ. 2015. Stress granule components G3BP1 and G3BP2 play a proviral role early in Chikungunya virus replication. J Virol 89:4457–4469. 10.1128/JVI.03612-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elde NC, Child SJ, Geballe AP, Malik HS. 2009. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 457:485–489. 10.1038/nature07529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dauber B, Wolff T. 2009. Activation of the antiviral kinase PKR and viral countermeasures. Viruses 1:523–544. 10.3390/v1030523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khaperskyy DA, Hatchette TF, McCormick C. 2012. Influenza A virus inhibits cytoplasmic stress granule formation. FASEB J 26:1629–1639. 10.1096/fj.11-196915. [DOI] [PubMed] [Google Scholar]
- 27.Sciortino MT, Parisi T, Siracusano G, Mastino A, Taddeo B, Roizman B. 2013. The virion host shutoff RNase plays a key role in blocking the activation of protein kinase R in cells infected with herpes simplex virus 1. J Virol 87:3271–3276. 10.1128/JVI.03049-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cassady KA, Gross M. 2002. The herpes simplex virus type 1 U(S)11 protein interacts with protein kinase R in infected cells and requires a 30-amino-acid sequence adjacent to a kinase substrate domain. J Virol 76:2029–2035. 10.1128/jvi.76.5.2029-2035.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mulvey M, Arias C, Mohr I. 2007. Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J Virol 81:3377–3390. 10.1128/JVI.02191-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakagawa K, Narayanan K, Wada M, Makino S. 2018. Inhibition of stress granule formation by Middle East Respiratory Syndrome Coronavirus 4a accessory protein facilitates viral translation, leading to efficient virus replication. J Virol 92:e00902-18. 10.1128/JVI.00902-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jayabalan AK, Adivarahan S, Koppula A, Abraham R, Batish M, Zenklusen D, Griffin DE, Leung A. 2021. Stress granule formation, disassembly, and composition are regulated by alphavirus ADP-ribosylhydrolase activity. Proc Natl Acad Sci USA 118:e2021719118. 10.1073/pnas.2021719118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Y, Dong L, Yu S, Wang X, Zheng H, Zhang P, Meng C, Zhan Y, Tan L, Song C, Qiu X, Wang G, Liao Y, Ding C. 2017. Newcastle disease virus induces stable formation of bona fide stress granules to facilitate viral replication through manipulating host protein translation. FASEB J 31:1482–1493. 10.1096/fj.201600980R. [DOI] [PubMed] [Google Scholar]
- 33.Heinrich BS, Cureton DK, Rahmeh AA, Whelan SP. 2010. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog 6:e1000958. 10.1371/journal.ppat.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dinh PX, Beura LK, Das PB, Panda D, Das A, Pattnaik AK. 2013. Induction of stress granule-like structures in vesicular stomatitis virus-infected cells. J Virol 87:372–383. 10.1128/JVI.02305-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garaigorta U, Heim MH, Boyd B, Wieland S, Chisari FV. 2012. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J Virol 86:11043–11056. 10.1128/JVI.07101-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruggieri A, Dazert E, Metz P, Hofmann S, Bergeest JP, Mazur J, Bankhead P, Hiet MS, Kallis S, Alvisi G, Samuel CE, Lohmann V, Kaderali L, Rohr K, Frese M, Stoecklin G, Bartenschlager R. 2012. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 12:71–85. 10.1016/j.chom.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brownsword MJ, Doyle N, Brocard M, Locker N, Maier HJ. 2020. Infectious bronchitis virus regulates cellular stress granule signaling. Viruses 12:536. 10.3390/v12050536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao B, Gong X, Fang S, Weng W, Wang H, Chu H, Sun Y, Meng C, Tan L, Song C, Qiu X, Liu W, Forlenza M, Ding C, Liao Y. 2021. Inhibition of anti-viral stress granule formation by coronavirus endoribonuclease nsp15 ensures efficient virus replication. PLoS Pathog 17:e1008690. 10.1371/journal.ppat.1008690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nejepinska J, Malik R, Moravec M, Svoboda P. 2012. Deep sequencing reveals complex spurious transcription from transiently transfected plasmids. PLoS One 7:e43283. 10.1371/journal.pone.0043283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Y, Cheng J, Xu G, Thiel V, Zhang G. 2019. Successful establishment of a reverse genetic system for QX-type infectious bronchitis virus and technical improvement of the rescue procedure. Virus Res 272:197726. 10.1016/j.virusres.2019.197726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kint J, Langereis MA, Maier HJ, Britton P, van Kuppeveld FJ, Koumans J, Wiegertjes GF, Forlenza M. 2016. Infectious bronchitis coronavirus limits interferon production by inducing a host shutoff that requires accessory protein 5b. J Virol 90:7519–7528. 10.1128/JVI.00627-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanaka T, Kamitani W, DeDiego ML, Enjuanes L, Matsuura Y. 2012. Severe acute respiratory syndrome coronavirus nsp1 facilitates efficient propagation in cells through a specific translational shutoff of host mRNA. J Virol 86:11128–11137. 10.1128/JVI.01700-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amorim R, Temzi A, Griffin BD, Mouland AJ. 2017. Zika virus inhibits eIF2alpha-dependent stress granule assembly. PLoS Negl Trop Dis 11:e5775. 10.1371/journal.pntd.0005775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basu M, Courtney SC, Brinton MA. 2017. Arsenite-induced stress granule formation is inhibited by elevated levels of reduced glutathione in West Nile virus-infected cells. PLoS Pathog 13:e1006240. 10.1371/journal.ppat.1006240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linero FN, Thomas MG, Boccaccio GL, Scolaro LA. 2011. Junin virus infection impairs stress-granule formation in Vero cells treated with arsenite via inhibition of eIF2alpha phosphorylation. J Gen Virol 92:2889–2899. 10.1099/vir.0.033407-0. [DOI] [PubMed] [Google Scholar]
- 46.Nelson EV, Schmidt KM, Deflubé LR, Doğanay S, Banadyga L, Olejnik J, Hume AJ, Ryabchikova E, Ebihara H, Kedersha N, Ha T, Mühlberger E. 2016. Ebola virus does not induce stress granule formation during infection and sequesters stress granule proteins within viral inclusions. J Virol 90:7268–7284. 10.1128/JVI.00459-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raaben M, Groot KM, Rottier PJ, de Haan CA. 2007. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell Microbiol 9:2218–2229. 10.1111/j.1462-5822.2007.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sola I, Galan C, Mateos-Gomez PA, Palacio L, Zuniga S, Cruz JL, Almazan F, Enjuanes L. 2011. The polypyrimidine tract-binding protein affects coronavirus RNA accumulation levels and relocalizes viral RNAs to novel cytoplasmic domains different from replication-transcription sites. J Virol 85:5136–5149. 10.1128/JVI.00195-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rabouw HH, Langereis MA, Knaap RC, Dalebout TJ, Canton J, Sola I, Enjuanes L, Bredenbeek PJ, Kikkert M, de Groot RJ, van Kuppeveld FJ. 2016. Middle east respiratory coronavirus accessory protein 4a inhibits PKR-mediated antiviral stress responses. PLoS Pathog 12:e1005982. 10.1371/journal.ppat.1005982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng X, Baker SC. 2018. An “Old” protein with a new story: coronavirus endoribonuclease is important for evading host antiviral defenses. Virology 517:157–163. 10.1016/j.virol.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoneyama M, Jogi M, Onomoto K. 2016. Regulation of antiviral innate immune signaling by stress-induced RNA granules. J Biochem 159:279–286. 10.1093/jb/mvv122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oh SW, Onomoto K, Wakimoto M, Onoguchi K, Ishidate F, Fujiwara T, Yoneyama M, Kato H, Fujita T. 2016. Leader-containing uncapped viral transcript activates RIG-I in antiviral stress granules. PLoS Pathog 12:e1005444. 10.1371/journal.ppat.1005444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Onomoto K, Yoneyama M, Fung G, Kato H, Fujita T. 2014. Antiviral innate immunity and stress granule responses. Trends Immunol 35:420–428. 10.1016/j.it.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Damgaard CK, Lykke-Andersen J. 2011. Translational coregulation of 5'TOP mRNAs by TIA-1 and TIAR. Genes Dev 25:2057–2068. 10.1101/gad.17355911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bauer WJ, Heath J, Jenkins JL, Kielkopf CL. 2012. Three RNA recognition motifs participate in RNA recognition and structural organization by the pro-apoptotic factor TIA-1. J Mol Biol 415:727–740. 10.1016/j.jmb.2011.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim HS, Kuwano Y, Zhan M, Pullmann RJ, Mazan-Mamczarz K, Li H, Kedersha N, Anderson P, Wilce MC, Gorospe M, Wilce JA. 2007. Elucidation of a C-rich signature motif in target mRNAs of RNA-binding protein TIAR. Mol Cell Biol 27:6806–6817. 10.1128/MCB.01036-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hackbart M, Deng X, Baker SC. 2020. Coronavirus endoribonuclease targets viral polyuridine sequences to evade activating host sensors. Proc Natl Acad Sci USA 117:8094–8103. 10.1073/pnas.1921485117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McBride R, van Zyl M, Fielding BC. 2014. The coronavirus nucleocapsid is a multifunctional protein. Viruses 6:2991–3018. 10.3390/v6082991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wolff G, Melia CE, Snijder EJ, Barcena M. 2020. Double-membrane vesicles as platforms for viral replication. Trends Microbiol 28:1022–1033. 10.1016/j.tim.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakagawa K, Makino S. 2021. Mechanisms of coronavirus nsp1-mediated control of host and viral gene expression. Cells 10:300. 10.3390/cells10020300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deng X, Hackbart M, Mettelman RC, O'Brien A, Mielech AM, Yi G, Kao CC, Baker SC. 2017. Coronavirus nonstructural protein 15 mediates evasion of dsRNA sensors and limits apoptosis in macrophages. Proc Natl Acad Sci USA 114:E4251–E4260. 10.1073/pnas.1618310114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deng X, van Geelen A, Buckley AC, O’Brien A, Pillatzki A, Lager KM, Faaberg KS, Baker SC. 2019. Coronavirus endoribonuclease activity in porcine epidemic diarrhea virus suppresses type I and type III interferon responses. J Virol 93:e02000-18. 10.1128/JVI.02000-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu G, Cheng J, Ma S, Jia W, Yan S, Zhang G. 2018. Pathogenicity differences between a newly emerged TW-like strain and a prevalent QX-like strain of infectious bronchitis virus. Vet Microbiol 227:20–28. 10.1016/j.vetmic.2018.10.019. [DOI] [PubMed] [Google Scholar]
- 64.Shen Z, Ye G, Deng F, Wang G, Cui M, Fang L, Xiao S, Fu ZF, Peng G. 2018. Structural basis for the inhibition of host gene expression by porcine epidemic diarrhea virus nsp1. J Virol 92:e01896-17. 10.1128/JVI.01896-17. [DOI] [PMC free article] [PubMed] [Google Scholar]