

ABSTRACT
Herpes simplex virus 1 (HSV-1) maintains a lifelong latent infection in neurons and periodically reactivates, resulting in the production of infectious virus. The exact cellular pathways that induce reactivation are not understood. In primary neuronal models of HSV latency, the cellular protein dual leucine zipper kinase (DLK) has been found to initiate a wave of viral gene expression known as phase I. Phase I occurs independently of both viral DNA replication and the activities of histone demethylase enzymes required to remove repressive heterochromatin modifications associated with the viral genome. In this study, we investigated whether phase I-like gene expression occurs in ganglia reactivated from infected mice. Using the combined trigger of explant-induced axotomy and inhibition of phosphatidylinositide 3-kinase (PI3K) signaling, we found that HSV lytic gene expression was induced rapidly from both sensory and sympathetic neurons. Ex vivo reactivation involved a wave of viral late gene expression that occurred independently of viral genome synthesis and histone demethylase activity and preceded the detection of infectious virus. Importantly, we found that DLK was required for the initial induction of lytic gene expression. These data confirm the essential role of DLK in inducing HSV-1 gene expression from the heterochromatin-associated genome and further demonstrate that HSV-1 gene expression during reactivation occurs via mechanisms that are distinct from lytic replication.
IMPORTANCE Reactivation of herpes simplex virus from a latent infection is associated with clinical disease. To develop new therapeutics that prevent reactivation, it is important to understand how viral gene expression initiates following a reactivation stimulus. Dual leucine zipper kinase (DLK) is a cellular protein that has previously been found to be required for HSV reactivation from sympathetic neurons in vitro. Here, we show that DLK is essential for reactivation from sensory ganglia isolated from infected mice. Furthermore, we show that DLK-dependent gene expression ex vivo occurs via mechanisms that are distinct from production replication, namely, lytic gene expression that is independent of viral DNA replication and histone demethylase activity. The identification of a DLK-dependent wave of lytic gene expression from sensory ganglia will ultimately permit the development of novel therapeutics that target lytic gene expression and prevent the earliest stage of reactivation.
KEYWORDS: herpes simplex virus, latent infection
INTRODUCTION
The ubiquitous human pathogen herpes simples virus (HSV) persists for life in the form of a latent infection in neurons. In response to a variety of different stimuli, the virus can reactivate from the latent state, resulting in the release of infectious virus and subsequent replication in the surrounding tissue. Clinically, reactivation of the virus can manifest as a variety of disease states, including lesions at the body surface, keratitis, and encephalitis. In addition, there is growing evidence of a link between HSV infection and the development of late onset Alzheimer’s disease, particularly in individuals with the ApoE4 variant (1–9). There are potentially different stimuli that can induce HSV to reactivate from latency. These stimuli may converge on single cellular pathways or operate via distinct mechanisms to induce reactivation (10). Identifying the cellular pathways important in reactivation is required to understand how viral gene expression initiates from the latent genome and to ultimately develop therapeutics to prevent reactivation.
During a latent infection of neurons, the HSV genome is assembled into repressive heterochromatin. This has been characterized by the enrichment of posttranslational modifications on histone H3, namely, di- and trimethyl lysine 9 (H3K9me2/3) and trimethyl lysine 27 (H3K27me3) on lytic promoters (11–17). By assembling into heterochromatin, viral lytic transcripts are maintained in a silent state. In addition, both host and viral miRNAs target lytic mRNAs (18–22). Therefore, the action of transcriptional and translational silencing results in limited synthesis of viral lytic proteins. This lack of lytic proteins suggests that HSV is reliant upon host signaling to initiate gene expression and reactivation. To understand the mechanism of HSV reactivation, it is important to determine how activated host cell pathways ultimately converge on the repressed viral genome to induce lytic gene expression.
There is evidence that the initial induction of HSV-1 lytic gene expression following reactivation occurs in a manner that is distinct from the mechanisms of viral gene expression during lytic replication. HSV lytic genes can be divided into groups characterized by their requirements for viral protein synthesis and viral DNA replication during lytic replication (23). Immediate early (IE) genes are expressed independently of viral protein synthesis during lytic infection and instead require the viral tegument protein VP16 for maximal expression. Early (E) genes are expressed following the production of IE proteins. Certain IE proteins (including ICP4, ICP0, and ICP27) stimulate viral E protein synthesis. Late (L) genes require viral DNA synthesis and are subdivided into genes that are expressed at low levels even prior to DNA replication but increased with genome synthesis (leaky L) and those that are fully dependent on viral DNA replication (true L). The dependence on DNA replication for L gene expression is not fully understood but likely involves a shift in genome accessibility and increased binding of host transcriptional machinery (RNA polymerase [Pol] II, TATA-binding protein [TBP], and TATA-Box binding protein associated factor 1 [TAF1]) (24). In contrast to this regulated cascade, in models of HSV reactivation, an initial burst of lytic gene expression, named phase I of reactivation, has been observed in which inhibition of protein synthesis prior to the accumulation of IE transcripts does not prevent E gene expression (25). In addition, L gene expression is unaffected by inhibition of viral DNA replication (25). Together, these data indicate that initial expression of lytic transcripts during the early stages of reactivation does not resemble the early stages of de novo infection in nonneuronal cells.
Phase I of HSV reactivation has largely been identified in primary neuronal models of HSV latency in sympathetic neurons (26). In these experimental models, a variety of stimuli have been found to induce HSV to reactivate from a latent infection, including loss of neurotrophic factor support (25, 27–29), increased neuronal excitation (30), modulation of DNA damage/repair (31), and exposure to corticosteroids (27). Precisely how activation of these pathways permits expression of the viral gene transcripts for reactivation to occur is not fully understood. Previously, we have identified a role for the cell stress protein, dual leucine zipper kinase (DLK), in inducing phase I of HSV reactivation (27). DLK is activated by the loss of neurotrophic factor support, which can be mimicked by inhibition of phosphoinositide 3-kinase (PI3K) activity (27), and during heightened neuronal excitation and interleukin-1 (IL-1) treatment (30). DLK is a master regulator of axonal responses to stress and can mediate a variety of responses, including Wallerian degeneration, axon regeneration, apoptosis, and axon pruning (32). Upon activation, DLK is known to redirect the cell stress protein c-Jun N-terminal kinase (JNK) from its physiological role in neurons, maintaining synaptic arborization, to its cell stress function (33). Accordingly, we and others have identified a role for JNK in reactivation of HSV from latency (27, 30, 31). JNK is also important in reactivation of the related alphaherpesvirus varicella-zoster virus from a latent infection (34), highlighting its potential central role in reactivation of human alphaherpesviruses.
A role for DLK and JNK in HSV reactivation has been mostly widely studied in primary neuronal models of latency in murine sympathetic neurons (27, 30). In these neurons, latency is established in the presence of the HSV DNA replication inhibitor acyclovir (ACV). After the removal of acyclovir, reactivation can be induced by PI3K inhibition or forskolin (25, 27, 30, 35, 36). In these systems, full reactivation occurs around 48h poststimulus, which requires the activities of histone demethylase enzymes, indicating that reactivation requires removal of repressive heterochromatin (27, 30). However, the DLK-dependent phase I peaks around 18 h poststimulus and (27, 30, 37), importantly, phase I occurs independently of lysine 9 and lysine 27 histone demethylase activity (27, 30). Instead, JNK activation induces histone phosphorylation on H3 serine 10 (H3S10) on histones that maintain the H3K9me3 modification (27); this is known as a histone methyl/phospho switch and presumably permits transcription by overriding the repressive H3K9me3 modification. However, although demonstrated in vitro, the possibility of a DLK/JNK-dependent wave of gene expression, characteristic of phase I, following in vivo infections has not been explored.
A robust mode of HSV reactivation from infected mice is explanation of the sensory trigeminal ganglia (TG). The action of severing the axon (axotomy) is thought to be the trigger that induces HSV to reactivate. In this model, whether a phase I-like wave of gene expression occurs has not been fully explored. Data from a previous study suggests that a H3 lysine 9 demethylase functions to promote lytic gene expression at an early time point (6 h postexplant) (38). This indicates that the wave of gene expression that is independent of histone demethylase activity occurs at an earlier time point or that explant-induced reactivation does not involve a phase-I like wave of viral gene expression. There are multiple differences between the in vivo experiments and in vitro latency models, including the presence of a host immune system in vivo. In addition, explant-induced reactivation has been investigated mostly in sensory neurons, whereas models for in vitro infection often use sympathetic neurons; both neuronal types are targets of HSV latent infection in humans (2, 39–41). In vitro models also use acyclovir to promote the establishment of latency. Finally, potential differences resulting from viral strains used cannot be ruled out. In experiments investigating the role for histone demethylases ex vivo, HSV strain F was used (16, 38), whereas the in vitro models have been performed with the KOS and Patton strains (25, 27).
To determine whether previous observations of a DLK/JNK-triggered phase I reactivation in vitro were recapitulated ex vivo and in sensory neurons, we dissected trigeminal ganglia from latently infected mice and determined whether there was lytic gene expression in response to PI3K inhibition. We found that after only 5 h postexcision, in treated ganglia, there was robust expression of IE, E, and L viral genes and this gene expression was independent of viral DNA replication. Supporting previous findings, we also found that this initial burst of lytic gene expression was not dependent upon LSD1 (H3K9 demethylase) or JMJD3 and UTX (H3K27 demethylases). Therefore, phase I of reactivation was observed ex vivo and was not reliant upon the removal of repressive heterochromatic marks. We found that this phase I was dependent upon DLK, thus indicating that neuronal stress pathways can trigger biphasic reactivation of HSV-1 in ganglia ex vivo.
RESULTS
Explant combined with PI3K inhibition triggers robust HSV-1 lytic gene expression.
We first set out to determine whether a wave of lytic gene expression occurs following reactivation of explanted sensory trigeminal ganglia (TG), similar to what has been observed in primary sympathetic neuronal cultures. Female mice were infected via the ocular route and reactivation studies were carried out at least 28 days postinfection. Previously, studies from our lab and others have shown that addition of a reactivation stimulus to sympathetic neurons infected in vitro yields induction of lytic viral gene expression around 15 to 20 h poststimulus (25, 27). In addition, a previous study examining explant-induced reactivation of TG combined with deprivation of nerve growth factor (NGF) also resulted in lytic gene induction around 12 to 15 h postexplant (42). Therefore, we initially examined the effect of PI3K inhibition (using LY294002 at 40 μM) at 20 h postreactivation to determine whether loss of the PI3K/AKT branch of the NGF signaling pathway also promotes reactivation from TG ex vivo. Acyclovir (ACV; 100 μM) was also introduced alongside LY294002 because late gene expression during the initial activation of viral gene expression in in vitro models has been reported to proceed independently of DNA replication (25).
In ganglia that were explanted and maintained in NGF to provide continued neurotrophin support, very little induction of lytic gene expression was observed at 20 h postexplant, especially for representative IE and E transcripts (Fig. 1B and C). A slight increase in gC mRNA was observed (Fig. 1D), although this increase was not statistically significant (P = 0.1255, Mann-Whitney U test). However, the addition of the PI3K inhibitor (LY294002) increased viral gene expression 100- to 1,000-fold. Reverse transcription-quantitative PCR (RT-qPCR) was carried out using primers against IE (ICP27 [Fig. 1B]), E (ICP8 [Fig. 1C]), and L (gC [Fig. 1D]) genes. All three genes were significantly induced at 20 h postexcision. The addition of ACV to inhibit viral DNA replication did not inhibit the induction of IE or E gene expression, as expected. However, expression of the gC mRNA was significantly reduced in the presence of ACV, indicating that its maximal expression was dependent on viral genome synthesis. However, gC mRNA levels with ACV were still significantly increased compared to the latent samples. At 20 h postreactivation, an increase in viral genomes was also observed (Fig. 1A). Together, these data show that at 20 h postreactivation, viral gene expression does not resemble the phase I observed in primary neuronal cultures, as genome synthesis had occurred, and full late gene expression was dependent on viral DNA replication. However, this did not rule out the possibility of a phase I-like wave of gene expression occurring prior to 20 h, especially as gC mRNA was still induced compared to the case with the 0 h (latent) samples.
FIG 1.

Explant combined with PI3K inhibition triggers robust HSV-1 lytic gene expression. Mice were infected via corneal scarification and at least 28 days postinfection, trigeminal ganglia (TG) were excised. Ganglia were either snap-frozen at 0 h or reactivated for 20 h in neuronal media alone containing nerve growth factor (NGF), with LY294002 (LY; 40 μM) or LY294002 with acyclovir (ACV; 100 μM). (A) Viral genome copy number was quantified by qPCR. (B to D) Viral gene expression was quantified by RT-qPCR for immediate early (ICP27) (B), early (ICP8) (C), and late (gC) (D) genes. Transcript copy number was normalized to cellular control (GAPDH). Six biological replicates from 1 representative infection group were used. The Mann-Whitney U test was used to determine statistical significance of differences. Individual biological replicates along with the means and SEMs are represented. *, P < 0.05; **, P < 0.01. ns, not significant.
PI3K inhibition/explant of trigeminal ganglia results in rapid late gene expression in the absence of detectable genome synthesis.
To determine whether viral lytic gene expression occurred prior to 20 h poststimulus in explanted TG with features characteristic of phase I gene expression, we decided to infect both male and female mice and determine changes in lytic gene expression following PI3K inhibition of explanted ganglia. Because we infected both male and female mice, we assessed if sex impacted clinical manifestations of infection. Both male and female mice had minimal mortality, with 92 and 88% survival, respectively (Fig. 2A). To further analyze clinical symptoms, mice were evaluated by scoring lesions, neurological symptoms, and eye health based on a previously described scoring metric (43). Infection of female mice resulted in more severe clinical manifestations at days 7 and 9 postinfection (Fig. 2B). Despite these differences, there was no significant differences in the percent weight loss following infection between male and female mice (Fig. 2C). Based on these criteria, we concluded there were only minor sex-dependent differences upon HSV infection.
FIG 2.
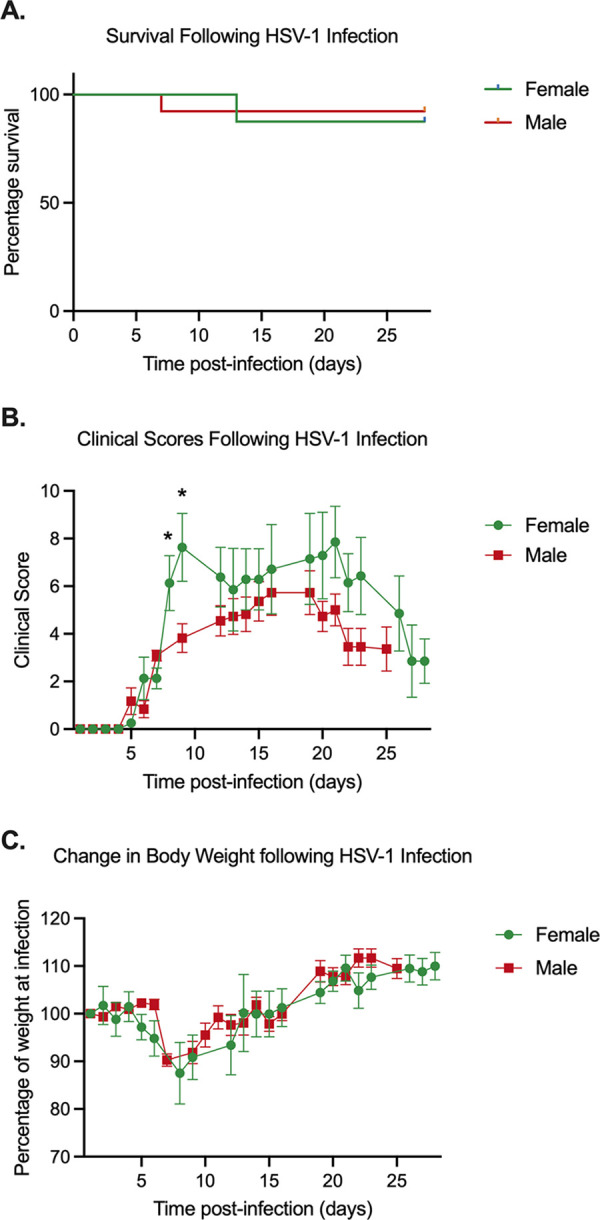
Sex-dependent phenotypes during HSV-1 in vivo infection. Mice were infected via corneal scarification and monitored postinfection. (A) Survival over time was observed, and the significance of the percent survival between female and male mice was analyzed using Kaplan-Meier survival analysis. (B) Clinicals scores were calculated by scoring lesion, neurological, and eye phenotypes as outlined in Table 1. (C) Percent weight compared to preinfection weights. Student’s t test was used to determine statistical significance of differences (B and C). The mean and SEM are represented. Thirteen females and 8 males from 2 independent infection groups were tested. *, P < 0.05.
After at least 28 days postinfection, TG were explanted and incubated in the presence of LY294002, with and without ACV. Values from male and female mice were combined (Fig. 3A to D). Quantification of viral DNA loads showed that the copy number of viral genomes stayed constant up to 15 h poststimulus and were not affected by the presence of ACV, indicating that detectable viral genome synthesis did not occur in this time period. In contrast, a robust induction of viral lytic gene expression occurred by 5 h poststimulus, as indicated by an increase in IE (ICP27), E (ICP8), and late (gC) mRNA copy number. For all gene classes examined, the increases in copy number were 100-fold for IE mRNA (Fig. 3B), 20-fold for E mRNA (Fig. 3C), and 10-fold for L mRNA (Fig. 3D) at 5 h postexplant. The robust increase in late gene expression in the absence of detectable viral genome synthesis between 0 and 15 h postexplant shows that late gene expression could occur even prior to DNA replication. To further support this conclusion, late gene induction occurred to equivalent levels even in the presence of ACV. The inclusion of ACV did prevent genome synthesis at 20 h poststimulus (Fig. 1A), indicating that the ACV was capable of acting on the explanted ganglia. Therefore, these data indicate that the induction of lytic gene expression following PI3K inhibition in explanted sensory neurons resembles at least one feature of phase I gene expression, as viral late gene expression occurred independently of viral DNA replication.
FIG 3.

PI3K inhibition of explanted trigeminal ganglia induces rapid lytic gene expression in the absence of detectable genome synthesis. Latently infected TG from male and female mice were reactivated for 5, 10, or 15 h with LY294002 in the presence and absence of acyclovir. (A) Viral genome copy number was quantified by qPCR. (B to D) Viral gene expression also was quantified by RT-qPCR for immediate early (ICP27) (B), early (ICP8) (C), and late (gC) (D) genes. The average genome copy number (E) and ICP27 mRNA transcripts (F) for male and female mice were calculated. Transcript copy numbers were normalized to cellular control (18S rRNA). The limit of detection is indicated by a black dashed line. At least 19 biological replicates (A to D) from 6 independent infection groups were used. The Mann-Whitney U test was used to determine statistical significance of differences. The means and SEMs are represented. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 compared to the 0-h time point.
To confirm that reactivation had no sex-dependent effects, we performed quantification of viral lytic gene expression in both male and female mice. We found no difference in viral genome copy number (Fig. 3E) or ICP27 lytic gene expression (Fig. 3F) at any time point. Therefore, we see no obvious differences in the efficiency of lytic gene expression following reactivation in male and female mice.
PI3K inhibition in combination with axotomy triggers rapid lytic gene expression in sympathetic neurons ex vivo.
Previous studies investigating phase I gene expression in vitro have largely used sympathetic neurons, in which the induction of lytic gene expression has been found to occur 15 to 20 h poststimulus (25–27). However, we found that lytic gene expression induced ex vivo from sensory neurons was robustly induced by 5 h poststimulus (Fig. 2). To determine whether the enhanced kinetics of reactivation observed ex vivo from sensory neurons in the TG could result from the use of different neuronal types, we investigated reactivation ex vivo from the sympathetic superior cervical ganglia (SCG). Latently infected SCG were explanted and incubated in the presence of the PI3K inhibitor LY294002. Quantification of viral DNA loads showed that the copy number of viral genomes remained constant at 5 h after stimulation, indicating that no detectable viral genome synthesis occurred during this time (Fig. 4A). In concordance with our sensory ganglia data (Fig. 1), there was a significant (10-fold) increase in viral genome copy number at 20 h postexcision (Fig. 4A). By 5 h poststimulus, a robust stimulation of viral lytic gene expression had occurred, as evidenced by a 10-fold increase in IE (ICP27 [Fig. 4B]) mRNA and a 20-fold increase in late (gC [Fig. 4C]) mRNA copy numbers. This indicates that the faster kinetics observed ex vivo in sensory neurons was not due to different neuronal subtypes and instead likely resulted from either the combination of triggers or in vivo infection.
FIG 4.

PI3K inhibition of explanted superior cervical ganglia induces rapid lytic gene expression in the absence of detectable genome synthesis. Latently infected SCG from female mice were reactivated for 5 or 20 h with LY294002. (A) Viral genome copy number was quantified by PCR. (B and C) Viral gene expression was quantified by RT-qPCR for immediate early (ICP27) (B) and late (gC) (C) genes. Transcript copy numbers were normalized to the cellular control (18S rRNA). The limit of detection is indicated by a black dashed line. At least 12 biological replicates from 3 independent infection groups were used. The Mann-Whitney U test was used to determine statistical significance of differences. The means and SEMs are represented. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Analysis of preformed virus production in sensory neurons ex vivo.
Based on our data that viral late gene expression was detectable by 5 h and robustly expressed by 10 h postexplant, we investigated when preformed infectious virus could be detected in the explanted ganglia. The rationale for this was that phase I lytic gene expression occurs in cultured neurons before the production of de novo virus (25). To ensure that we detected only preformed virus and not virus produced from remaining intact cells, the ganglia were homogenized and subjected to three rounds of sonication and two cycles of freezing and thawing. Using a viral stock of known titer, we confirmed that that this procedure did not result in a detectable loss in viral titer (data not shown). Infectious virus was robustly detected at 20 h postexplant in 7 out of the 8 ganglia tested (Fig. 5). At 15 h, virus was detected in only 5 out of 8 ganglia tested, and at 10 h, only 2 ganglia had detectable virus. No infectious virus was detected at 7 h postexplant. These findings suggest that a detectable increase in late gene expression occurs 10 to 15 h before infectious virus production, observed at 20 h, in a population of reactivating ganglia.
FIG 5.

Robust detection of preformed virus occurs 20 h postexcision Latently infected TG from female mice were explanted and reactivated for 0, 7, 10, 15, or 20 h with LY294002 and titers of preformed virus were quantified on Vero cells. The number of ganglia with detectable virus is displayed beneath the x axis. At least 14 biological replicates from 5 independent infection groups were used. The median titer is represented.
DLK activity is required for reactivation ex vivo.
Previously, we found that the neuronal cell stress protein dual leucine zipper kinase (DLK) is required for HSV reactivation and acts to induce phase I lytic gene expression (27, 30). To determine whether DLK was also required for the induction of lytic gene expression in sensory neurons reactivated ex vivo by PI3K inhibition/axotomy, the DLK inhibitor GNE-3511 (44) was added to the explanted ganglia and viral RNA was quantified at 5 h postreactivation. Inclusion of the DLK inhibitor resulted in ICP27, ICP8, and gC mRNA (Fig. 6) levels that were equivalent to those in the unreactivated samples and significantly decreased compared to those in the ganglia treated with LY204002 only. These data indicate that DLK is required for reactivation from sensory neurons induced by the combined trigger of explant and PI3K inhibition. This is the first demonstration of a role for DLK in reactivation from sensory neurons and in response to ex vivo axotomy.
FIG 6.

DLK activity is required for the induction of lytic gene expression following explant/PI3K inhibition of latently infected TG. Latently infected TG were explanted and incubated with LY294002 and the DLK inhibitor GNE-3511 (8 μM) for 5 h postexcision. Viral gene expression was quantified by RT-qPCR for immediate early (ICP27) (A), early (ICP8) (B), and late (gC) (C) genes. The limit of detection is indicated by a dashed line. Twelve biological replicates from 2 independent infection groups were used. The Mann-Whitney U test was used to determine statistical significance of differences. Individual biological replicates along with the means and SEMs are represented. *, P < 0.05; **, P < 0.01.
Lytic gene induction upon axotomy/PI3K inhibition is independent of histone H3 histone lysine 9 and lysine 27 demethylase inhibitors.
HSV promoters are known to be enriched with histone H3 di- and trimethyl at lysine 9 (H3K9me2/3) and histone H3 trimethyl at lysine 27 (H3K27me3) during latency. The removal of restrictive histone modifications, specifically the aforementioned methylation marks, was previously shown to be essential for full reactivation of HSV in vitro, yet it was not required for initial lytic gene expression during phase I of reactivation (27, 30). To determine if initial lytic gene expression was independent of histone demethylation ex vivo, we used OG-L002 (60 μM), a drug that inhibits the histone lysine 9 demethylase LSD1 (45), and GSK-J4 (20 μM), which inhibits the histone lysine 27 demethylases UTX and JMJD3 (46, 47). HSV reactivation has been demonstrated to be inhibited by both of these inhibitors (27, 30, 45, 48, 49). Five hours postexcision, the addition of OG-L002 and GSK-J4 had no effect on the induction of ICP27, ICP8, or gC mRNA (Fig. 7). These results indicate that histone demethylase activity is not required for the initial induction of lytic gene expression induced by axotomy and PI3K inhibition ex vivo.
FIG 7.

Lytic gene induction following axotomy/PI3K inhibition is unaffected by histone demethylase inhibitors. Latently infected TG were explanted and incubated with LY294002 along with the LSD1 inhibitor OG-L1002 (50 μM) or GSK-J4 (20 μM). Viral gene expression was quantified by RT-qPCR for immediate early (ICP27) (A), early (ICP8) (B), and late (gC) (C) genes. At least 8 biological replicates from 2 independent infection groups. The Mann-Whitney U test was used to determine statistical significance of differences. Individual biological replicates along with the means and SEMs are represented. **, P < 0.01; ***, P < 0.001.
Together, our results demonstrate that features of phase I gene expression, namely, late gene expression in the absence of viral DNA replication and lytic gene expression in the presence of histone demethylase inhibitors, can occur from ganglia isolated from infected mice. In addition, ex vivo reactivation was dependent on DLK activity.
DISCUSSION
In vitro models of HSV latency are incredibly powerful for the study of molecular mechanisms of HSV latency and reactivation as well as the contribution of host factors and viral factors during specific stages of infection. By including components of the host immune response, such as interferon at different stages of infection, in vitro models can be used to investigate how the host immune response can modulate latent infection and reactivation (36, 50). Using these models, unique aspects regarding entry into lytic gene expression during reactivation have been uncovered, including the dependence on DLK/JNK for reactivation, the ability of late gene expression to occur in the absence of DNA replication, and transcription despite the presence of histone demethylase inhibitors. Therefore, it was highly important to validate these patterns of gene expression from ganglia infected in vivo. Here, we have confirmed that the features of gene expression observed during phase I reactivation in vitro also occur following reactivation ex vivo when PI3K inhibition and axotomy are used as a trigger. These data therefore demonstrate the validity of using in vitro model systems to investigate the mechanisms of initiation of lytic gene expression during reactivation. We also demonstrate for the first time that phase I gene expression occurs from sensory neurons.
There are certain caveats to our study, especially relating to the ability of inhibitors to act on intact ganglia. However, we show that the DLK inhibitor, PI3K inhibitor, and acyclovir can all act on intact ganglia. Therefore, we think it unlikely that the histone demethylase inhibitors used were unable to penetrate the tissue and have an effect on viral lytic gene induction. The concentrations of histone demethylase inhibitors were higher than previously used to inhibit full HSV reactivation in primary neuronal models (27, 30). We have shown that OG-L002 can inhibit forskolin-mediated reactivation at 30 μM (30); in this study, we used 60 μM. Similarly, we have shown that GSK-J4 can inhibit both forskolin- and LY294002-induced reactivation at 3 μM (27, 30); in this study, we used it at 20 μM. Therefore, we think it unlikely that the concentrations used were too low to inhibit the histone demethylases. Experiments using higher concentrations of both GSK-J4 and OG-L002 were performed (at 60 μM and 150 μM, respectively). However, these higher concentrations resulted in decreased viral genome copy number at 5 h compared to that at latency. Because there was no detectable viral DNA replication at this time point, this indicates neuronal loss and therefore neuronal toxicity at higher concentrations.
This study demonstrates that phase I of reactivation occurs from ganglia explanted from latently infected mice. Unlike in previous in vitro studies, we found that phase I of reactivation in intact neurons in vitro and ex vivo had enhanced kinetics of lytic gene induction in response to axotomy and PI3K stimulation. We observed this enhanced phenotype in both sensory and sympathetic ganglia. Therefore, we posit that the most likely explanation of the difference in reactivation kinetics between in vitro and ex vivo studies is the combined trigger of axotomy and PI3K inhibition. It has previously been demonstrated that when nerve growth factor deprivation is paired with axotomy, treated neurons undergo cell death more rapidly than untreated neurons. (51). In addition, both PI3K inhibition and axotomy result in activation of DLK (32, 52). However, there are some differences in how DLK is activated and the resulting downstream response following the two stimuli. Upon axotomy, DLK is rapidly activated at the proximal segment of the severed axon in response to loss of cytoplasmic integrity. A resulting calcium influx from cytoplasmic membrane rupture leads to DLK-dependent transcription of genes involved in axon regeneration (53). The exact mechanism of DLK activation upon loss of NGF signaling that mediates neuronal cell death or axon pruning is not fully understood. However, it is known that in response to NGF deprivation, DLK protein levels are stabilized, DLK is phosphorylated (54), and this phosphorylation results in the downstream activation of JNK, c-Jun, and other transcription factors. The activation of these transcription factors promotes the expression of proapoptotic genes (55), although in mature neurons there are multiple brakes downstream that prevent apoptosis (56). It is therefore conceivable that these two independent pathways to DLK activation may converge following the dual trigger of axotomy/PI3K inhibition to result in more rapid and robust DLK activation and HSV reactivation.
Consistent with cellular pathways converging the enhance DLK activity, we observed a robust induction of lytic gene expression at 5 h postreactivation. For the IE gene ICP27, this was an approximate 100-fold induction, compared to the 5- to 20-fold increase often observed in an in vitro mouse model with PI3K inhibition (27). The use of an ex vivo model system will therefore be incredibly powerful for studying the mechanisms of gene expression that occur during phase I. We have previously observed a histone methyl/phospho switch on lytic promoters during phase I reactivation (27), which permits lytic gene expression independently of recruitment of histone demethylase enzymes. There is evidence that the methyl/phospho switch permits gene expression because repressive histone readers (for example, HP1) are no longer capable of interacting with the methylated residue on histone H3K9 because phosphorylation at serine 10 occludes binding (57, 58). Because of the robust lytic gene induction ex vivo, and because we have now shown that the gene expression both in vitro and ex vivo is DLK dependent and histone demethylase independent, the complementary models can now be used answer key questions on the mechanism of viral lytic gene induction during phase I reactivation.
MATERIALS AND METHODS
Cells and viruses.
Stocks of HSV-1 KOS were grown and titrated on Vero cells obtained from the American Type Culture Collection (Manassas, VA) as described previously (11). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% FetalPlex (Gemini Bio-Products) and 2 mM l-glutamine. KOS was kindly provided by David Knipe, Harvard Medical School.
Reagents.
Compounds used in the study are as follows: acycloguanosine (acyclovir [ACV]), GSK-J4 and GNE-3511 (Millipore Sigma), SP600125 (Thermo Fisher Scientific), OG-L002 (Tocris), and nerve growth factor 2.5S (Alomone Labs).
Mouse infections.
Six-week-old male and female CD-1 mice (Charles River Laboratories) were anesthetized by intraperitoneal injection of ketamine hydrochloride (80 mg/kg of body weight) and xylazine hydrochloride (10 mg/kg) and inoculated with 1.5 × 106 PFU/eye of virus (in a 5-μL volume) onto scarified corneas, as described previously (11). Mice were housed in accordance with institutional and National Institutes of Health guidelines on the care and use of animals in research, and all procedures were approved by the Institutional Animal Care and Use Committee of the University of Virginia. Criteria used for clinical scoring based on the formation of lesions and neurological and eye symptoms are shown in Table 1 and were based on a previously establishing scoring scale (43). Mice were randomly assigned to groups, and all experiments included biological replicates from independent litters.
TABLE 1.
Clinical scoring scale for HSV-infected mice
| Score | Description of score by type |
||
|---|---|---|---|
| Lesion | Eye | Neurological | |
| 0 | Lesion free | Symptom free | Symptom free |
| 1 | Small area of broken skin <0.5 cm | Pus around edges | Hunched posture, normal movement |
| 2 | Area of broken skin 0.5–1 cm | Pus and squint | Hunched posture, slow movement |
| 3 | Bleeding, scabbing, or pustules | Closed | Hunched posture, labored breathing and/or hind-leg paralysis |
| 4 | Broken skin >1 cm with multiple pustules or scabbing | Scab formation | Hunched, ruffled fur, little to no movement |
| 5 | Severe scabbing or bleeding with pustules | Severe scabbing | Moribund or dead |
Explant-induced reactivation.
Trigeminal ganglia (TG) and superior cervical ganglia (SCG) were removed at least 28 days postinfection. The ganglia were maintained intact and immediately placed in reactivation medium containing DMEM/F-12 (Gibco) supplemented with 10% fetal bovine serum and mouse NGF 2.5S (50 ng/mL). Compounds were added at the following concentrations: LY294002, 40 μM; GNE-3511, 8 μM; ACV, 100 μM; GSK-J4, 10 μM; and OG-L002, 50 μM. For treatments with GNE-3511, ACV, GSK-J4, and OG-L002, ganglia were treated with the compounds 3 to 60 min prior to the addition of LY294002. Ganglia were placed on a shaking platform at 50 rpm and 37°C.
Quantification of viral transcripts and genome copy number.
At the required time point, the reactivation medium was removed and ganglia were snap-frozen in liquid nitrogen. Lysis was carried out by addition of the excised ganglia to BeadBug homogenization microtubules then homogenized for 60 s using the BeadBug microtube homogenizer. DNA and RNA were isolated from the homogenized mixture using the Quick DNA/RNA miniprep kit. Following RNA isolation, the TURBO DNA-free kit (Invitrogen) was used to remove any contaminant DNA. mRNA was reverse transcribed into cDNA using MaximaRT (Thermo Fisher) using random hexamers for first-strand synthesis. Equal amounts of mRNA were used for each reverse transcriptase experiment (20 to 30 ng/reaction). Power SYBR green PCR master mix (Applied Biosystems) was used for qPCR. Standard curves were used to calculate the relative mRNA or DNA copy number per genome and were standardized to cellular glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or 18S rRNA. All RNA samples were run in triplicate and all DNA samples were run in duplicate on an Applied Biosystems QuantStudio 6 Flex real-time PCR system. The primers that were used are described in Table 2.
TABLE 2.
Oligonucleotides used in this study
| Primer | Sequence 5′–3′ |
|---|---|
| GAPDH F | CAT GGC CTT CCG TGT TCC TA |
| GAPDH R | GCG GCA CGT CAG ATC CA |
| 18S F | CAC GGA CAG GAT TGA CAG ATT |
| 18S R | GCC AGA GTC TCG TTC GTT ATC |
| ICP27 F | GCA TCC TTC GTG TTT GTC ATT CTG |
| ICP27 R | GCA TCT TCT CTC CGA CCC CG |
| ICP8 F | GGA GGT GCA CCG CAT ACC |
| ICP8 R | GGC TAA AAT CCG GCA TGA AC |
| gC F | GAG TTT GTC TGG TTC GAG GAC |
| gC R | ACG GTA GAG ACT GTG GTG AA |
Titration of preformed virus.
Sterile milk was added to media containing reactivated ganglia and samples were snap-frozen. Ganglia were thawed at 37°C and then homogenized using a BeadBug microtube homogenizer. The homogenized ganglia were then sonicated at 25% for 30 s three times on ice in a Fisherbrand model 120 sonic dismembrator. After sonication, homogenates of ganglia were frozen on dry ice and thawed at 37°C. Homogenates from all time points were simultaneously titrated on Vero cells.
ACKNOWLEDGMENTS
We thank David Knipe (Harvard Medical School) for the KOS virus.
This work was supported by grant NIH/NINDS R01NS105630 (A.R.C.), The Owens Family Foundation (ARC), NIH/NIAID grant T32AI007046 (A.L.W. and J.B.S.), NIH/NEI grant F30EY030397 (J.B.S.), and NIH/NIGMS grant T32GM008136 (S.A.D.).
Contributor Information
Anna R. Cliffe, Email: cliffe@virginia.edu.
Felicia Goodrum, University of Arizona.
REFERENCES
- 1.Jamieson GA, Maitland NJ, Wilcock GK, Craske J, Itzhaki RF. 1991. Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J Med Virol 33:224–227. 10.1002/jmv.1890330403. [DOI] [PubMed] [Google Scholar]
- 2.Baringer JR, Pisani P. 1994. Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann Neurol 36:823–829. 10.1002/ana.410360605. [DOI] [PubMed] [Google Scholar]
- 3.Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. 1997. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 349:241–244. 10.1016/S0140-6736(96)10149-5. [DOI] [PubMed] [Google Scholar]
- 4.Mori I, Kimura Y, Naiki H, Matsubara R, Takeuchi T, Yokochi T, Nishiyama Y. 2004. Reactivation of HSV-1 in the brain of patients with familial Alzheimer’s disease. J Med Virol 73:605–611. 10.1002/jmv.20133. [DOI] [PubMed] [Google Scholar]
- 5.Piacentini R, De Chiara G, Li Puma DD, Ripoli C, Marcocci ME, Garaci E, Palamara AT, Grassi C. 2014. HSV-1 and Alzheimer’s disease: more than a hypothesis. Front Pharmacol 5:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itzhaki RF. 2018. Corroboration of a major role for herpes simplex virus type 1 in Alzheimer’s disease. Front Aging Neurosci 10:324. 10.3389/fnagi.2018.00324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Chiara G, Piacentini R, Fabiani M, Mastrodonato A, Marcocci ME, Limongi D, Napoletani G, Protto V, Coluccio P, Celestino I, Li Puma DD, Grassi C, Palamara AT. 2019. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog 15:e1007617. 10.1371/journal.ppat.1007617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tzeng NS, Chung CH, Lin FH, Chiang CP, Yeh CB, Huang SY, Lu RB, Chang HA, Kao YC, Yeh HW, Chiang WS, Chou YC, Tsao CH, Wu YF, Chien WC. 2018. Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections—a nationwide, population-based cohort study in Taiwan. Neurotherapeutics 15:417–429. 10.1007/s13311-018-0611-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P, Haroutunian V, Sano M, Liang WS, Beckmann ND, Price ND, Reiman EM, Schadt EE, Ehrlich ME, Gandy S, Dudley JT. 2018. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99:64–82.e7. 10.1016/j.neuron.2018.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzich JB, Cliffe AR. 2018. Strength in diversity: understanding the pathways to herpes simplex virus reactivation. Virology 522:81–91. 10.1016/j.virol.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83:8182–8190. 10.1128/JVI.00712-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6:211–221. 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 13.Wang QY, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM. 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci USA 102:16055–16059. 10.1073/pnas.0505850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwiatkowski DL, Thompson HW, Bloom DC. 2009. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol 83:8173–8181. 10.1128/JVI.00686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicoll MP, Hann W, Shivkumar M, Harman LE, Connor V, Coleman HM, Proenca JT, Efstathiou S. 2016. The HSV-1 latency-associated transcript functions to repress latent phase lytic gene expression and suppress virus reactivation from latently infected neurons. PLoS Pathog 12:e1005539. 10.1371/journal.ppat.1005539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. 2009. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med 15:1312–1317. 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dochnal SA, Francois AK, Cliffe AR. 2021. De novo polycomb recruitment: lessons from latent herpesviruses. Viruses 13:1470. 10.3390/v13081470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454:780–783. 10.1038/nature07103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jurak I, Kramer MF, Mellor JC, van Lint AL, Roth FP, Knipe DM, Coen DM. 2010. Numerous conserved and divergent microRNAs expressed by herpes simplex viruses 1 and 2. J Virol 84:4659–4672. 10.1128/JVI.02725-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kramer MF, Jurak I, Pesola JM, Boissel S, Knipe DM, Coen DM. 2011. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology 417:239–247. 10.1016/j.virol.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jurak I, Hackenberg M, Kim JY, Pesola JM, Everett RD, Preston CM, Wilson AC, Coen DM. 2014. Expression of herpes simplex virus 1 microRNAs in cell culture models of quiescent and latent infection. J Virol 88:2337–2339. 10.1128/JVI.03486-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan D, Flores O, Umbach JL, Pesola JM, Bentley P, Rosato PC, Leib DA, Cullen BR, Coen DM. 2014. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe 15:446–456. 10.1016/j.chom.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roizman B, Knipe DM, Whitley R. 2013. Herpes simplex viruses, pp 1823–1897. In Knipe DM, Howley P, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B, (eds) Fields’ Virology. Wolters Kluwer/Lippincott-Williams and Wilkins, New York, New York. [Google Scholar]
- 24.Dremel SE, DeLuca NA. 2019. Genome replication affects transcription factor binding mediating the cascade of herpes simplex virus transcription. Proc Natl Acad Sci USA 116:3734–3739. 10.1073/pnas.1818463116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. 2012. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog 8:e1002540. 10.1371/journal.ppat.1002540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cliffe AR, Wilson AC. 2017. Restarting lytic gene transcription at the onset of herpes simplex virus reactivation. J Virol 91:e01419-16. 10.1128/JVI.01419-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M. 2015. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 18:649–658. 10.1016/j.chom.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilcox CL, Johnson EM, Jr.. 1987. Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J Virol 61:2311–2315. 10.1128/JVI.61.7.2311-2315.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yanez AA, Harrell T, Sriranganathan HJ, Ives AM, Bertke AS. 2017. Neurotrophic factors NGF, GDNF and NTN selectively modulate HSV1 and HSV2 lytic infection and reactivation in primary adult sensory and autonomic neurons. Pathogens 6:5–13. 10.3390/pathogens6010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuddy SR, Schinlever AR, Dochnal S, Seegren PV, Suzich J, Kundu P, Downs TK, Farah M, Desai BN, Boutell C, Cliffe AR. 2020. Neuronal hyperexcitability is a DLK-dependent trigger of herpes simplex virus reactivation that can be induced by IL-1. Elife 9:e58037. 10.7554/eLife.58037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu HL, Shiflett LA, Kobayashi M, Chao MV, Wilson AC, Mohr I, Huang TT. 2019. TOP2β-dependent nuclear DNA damage shapes extracellular growth factor responses via dynamic AKT phosphorylation to control virus latency. Mol Cell 74:466–480.e4. 10.1016/j.molcel.2019.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tedeschi A, Bradke F. 2013. The DLK signalling pathway—a double-edged sword in neural development and regeneration. EMBO Rep 14:605–614. 10.1038/embor.2013.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sengupta Ghosh A, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. 2011. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol 194:751–764. 10.1083/jcb.201103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurapati S, Sadaoka T, Rajbhandari L, Jagdish B, Shukla P, Ali MA, Kim YJ, Lee G, Cohen JI, Venkatesan A. 2017. Role of the JNK pathway in varicella-zoster virus lytic infection and reactivation. J Virol 91:e00640-17. 10.1128/JVI.00640-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV. 2010. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell Host Microbe 8:320–330. 10.1016/j.chom.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzich JB, Cuddy SR, Baidas H, Dochnal S, Ke E, Schinlever AR, Babnis A, Boutell C, Cliffe AR. 2021. PML-NB-dependent type I interferon memory results in a restricted form of HSV latency. EMBO Rep 22:e52547. 10.15252/embr.202152547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu HL, Srinivas KP, Wang S, Chao MV, Lionnet T, Mohr I, Wilson AC, Depledge DP, Huang TT. 2022. Single-cell transcriptomics identifies Gadd45b as a regulator of herpesvirus-reactivating neurons. EMBO Rep 23:e53543. 10.15252/embr.202153543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang Y, Vogel JL, Arbuckle JH, Rai G, Jadhav A, Simeonov A, Maloney DJ, Kristie TM. 2013. Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency. Sci Transl Med 5:167ra5. 10.1126/scitranslmed.3005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baringer JR, Swoveland P. 1973. Recovery of herpes-simplex virus from human trigeminal ganglions. N Engl J Med 288:648–650. 10.1056/NEJM197303292881303. [DOI] [PubMed] [Google Scholar]
- 40.Richter ER, Dias JK, Gilbert JE, II, Atherton SS. 2009. Distribution of herpes simplex virus type 1 and varicella zoster virus in ganglia of the human head and neck. J Infect Dis 200:1901–1906. 10.1086/648474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Warren KG, Brown SM, Wroblewska Z, Gilden D, Koprowski H, Subak-Sharpe J. 1978. Isolation of latent herpes simplex virus from the superior cervical and vagus ganglions of human beings. N Engl J Med 298:1068–1069. 10.1056/NEJM197805112981907. [DOI] [PubMed] [Google Scholar]
- 42.Du T, Zhou G, Roizman B. 2011. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc Natl Acad Sci USA 108:18820–18824. 10.1073/pnas.1117203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riccio RE, Park SJ, Longnecker R, Kopp SJ. 2019. Characterization of sex differences in ocular herpes simplex virus 1 infection and herpes stromal keratitis pathogenesis of wild-type and herpesvirus entry mediator knockout mice. mSphere 4:e00322-19. 10.1128/mSphere.00322-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel S, Cohen F, Dean BJ, De La Torre K, Deshmukh G, Estrada AA, Ghosh AS, Gibbons P, Gustafson A, Huestis MP, Le Pichon CE, Lin H, Liu W, Liu X, Liu Y, Ly CQ, Lyssikatos JP, Ma C, Scearce-Levie K, Shin YG, Solanoy H, Stark KL, Wang J, Wang B, Zhao X, Lewcock JW, Siu M. 2015. Discovery of dual leucine zipper kinase (DLK, MAP3K12) inhibitors with activity in neurodegeneration models. J Med Chem 58:401–418. 10.1021/jm5013984. [DOI] [PubMed] [Google Scholar]
- 45.Liang Y, Quenelle D, Vogel JL, Mascaro C, Ortega A, Kristie TM. 2013. A novel selective LSD1/KDM1A inhibitor epigenetically blocks herpes simplex virus lytic replication and reactivation from latency. mBio 4:e00558-12. 10.1128/mBio.00558-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, Katso R, Leveridge M, Mander PK, Mosley J, Ramirez-Molina C, Rowland P, Schofield CJ, Sheppard RJ, Smith JE, Swales C, Tanner R, Thomas P, Tumber A, Drewes G, Oppermann U, Patel DJ, Lee K, Wilson DM. 2012. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488:404–408. 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heinemann B, Nielsen JM, Hudlebusch HR, Lees MJ, Larsen DV, Boesen T, Labelle M, Gerlach LO, Birk P, Helin K. 2014. Inhibition of demethylases by GSK-J1/J4. Nature 514:E1–E2. 10.1038/nature13688. [DOI] [PubMed] [Google Scholar]
- 48.Messer HG, Jacobs D, Dhummakupt A, Bloom DC. 2015. Inhibition of H3K27me3-specific histone demethylases JMJD3 and UTX blocks reactivation of herpes simplex virus 1 in trigeminal ganglion neurons. J Virol 89:3417–3420. 10.1128/JVI.03052-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dochnal SA, Merchant H, Schinlever A, Babnis A, Depledge DP, Wilson A, Cliffe AR. 26 March 2022. DLK-dependent biphasic reactivation of herpes simplex virus latency established in the absence of antivirals. bioRxiv 10.1101/2022.02.25.482019. [DOI] [PMC free article] [PubMed]
- 50.Linderman JA, Kobayashi M, Rayannavar V, Fak JJ, Darnell RB, Chao MV, Wilson AC, Mohr I. 2017. Immune escape via a transient gene expression program enables productive replication of a latent pathogen. Cell Rep 18:1312–1323. 10.1016/j.celrep.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fletcher GC, Xue L, Passingham SK, Tolkovsky AM. 2000. Death commitment point is advanced by axotomy in sympathetic neurons. J Cell Biol 150:741–754. 10.1083/jcb.150.4.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu CC, Wu HJ, Wang CH, Lin CH, Hsu SC, Chen YR, Hsiao M, Schuyler SC, Lu FL, Ma N, Lu J. 2015. Akt suppresses DLK for maintaining self-renewal of mouse embryonic stem cells. Cell Cycle 14:1207–1217. 10.1080/15384101.2015.1014144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Asghari Adib E, Smithson LJ, Collins CA. 2018. An axonal stress response pathway: degenerative and regenerative signaling by DLK. Curr Opin Neurobiol 53:110–119. 10.1016/j.conb.2018.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huntwork-Rodriguez S, Wang B, Watkins T, Ghosh AS, Pozniak CD, Bustos D, Newton K, Kirkpatrick DS, Lewcock JW. 2013. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J Cell Biol 202:747–763. 10.1083/jcb.201303066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW. 2013. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci USA 110:4039–4044. 10.1073/pnas.1211074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kole AJ, Annis RP, Deshmukh M. 2013. Mature neurons: equipped for survival. Cell Death Dis 4:e689. 10.1038/cddis.2013.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fischle W, Wang Y, Allis CD. 2003. Binary switches and modification cassettes in histone biology and beyond. Nature 425:475–479. 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 58.Fischle W, Tseng BS, Dormann HL, Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H, Allis CD. 2005. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature 438:1116–1122. 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]