

ABSTRACT
Myxovirus resistance (Mx) proteins are dynamin-like GTPases that are inducible by interferons (IFNs) following virus infections. Most studies investigating Mx proteins have focused on their activity against influenza A viruses (IAV), although emerging evidence suggests that some Mx proteins may exhibit broader antiviral activity. Herein, we demonstrate that in addition to IAV, overexpression of mouse Mx1 (mMx1), but not mMx2, resulted in potent inhibition of growth of the human alphaherpesviruses herpes simplex virus 1 (HSV-1) and HSV-2, whereas neither inhibited the mouse betaherpesvirus murine cytomegalovirus (MCMV) in vitro. IFN induction of a functional endogenous mMx1 in primary mouse fibroblasts ex vivo was also associated with inhibition of HSV-1 growth. Using an in vitro overexpression approach, we demonstrate that mutations that result in redistribution of mMx1 from the nucleus to the cytoplasm or in loss of its combined GTP binding and GTPase activity also abrogated its ability to inhibit HSV-1 growth. Overexpressed mMx1 did not inhibit early HSV-1 gene expression but was shown to inhibit both replication of the HSV-1 genome as well as subsequent late gene expression. In a mouse model of cutaneous HSV-1 infection, mice expressing a functional endogenous mMx1 showed significant reductions in the severity of skin lesions as well as reduced HSV-1 titers in both the skin and dorsal root ganglia (DRG). Together, these data demonstrate that mMx1 mediates potent antiviral activity against human alphaherpesviruses by blocking replication of the viral genome and subsequent steps in virus replication. Moreover, endogenous mMx1 potently inhibited pathogenesis in the zosteriform mouse model of HSV-1 infection.
IMPORTANCE While a number of studies have demonstrated that human Mx proteins can inhibit particular herpesviruses in vitro, we are the first to report the antiviral activity of mouse Mx1 (mMx1) against alphaherpesviruses both in vitro and in vivo. We demonstrate that both overexpressed mMx1 and endogenous mMx1 potently restrict HSV-1 growth in vitro. mMx1-mediated inhibition of HSV-1 was not associated with inhibition of virus entry and/or import of the viral genome into the nucleus, but rather with inhibition of HSV-1 genomic replication as well as subsequent late gene expression. Therefore, inhibition of human alphaherpesviruses by mMx1 occurs by a mechanism that is distinct from that reported for human Mx proteins against herpesviruses. Importantly, we also provide evidence that expression of a functional endogenous mMx1 can limit HSV-1 pathogenesis in a mouse model of infection.
KEYWORDS: Mx protein, herpesviruses, host-cell interactions, innate immunity, interferons, restriction factor
INTRODUCTION
Following virus infection, host cells rapidly produce type I interferons (IFNs), which in turn activate hundreds of interferon-stimulated genes (ISGs) in both infected and neighboring cells. Dynamin-like GTPase proteins, including myxovirus resistance (Mx) proteins, are among the many ISG proteins induced following virus infections. Mx genes tend to be highly conserved across all vertebrates and can range from one to nine genes in different species, although most mammals express two (1, 2). In humans and mice, these genes each encode two Mx proteins that localize to different intracellular compartments (i.e., nuclear Mx1 versus cytoplasmic Mx2 in mice and nuclear MxB versus cytoplasmic MxA in humans), suggesting that they may have evolved to control replication of particular viruses that replicate in distinct intracellular compartments. While murine Mx1 (mMx1) is an ortholog of human MxA (hMxA), mMx2 is a paralog of mMx1 rather than an ortholog of hMxB (2). It is well established that mMx1 inhibits a number of negative-strand RNA viruses, including influenza A virus (IAV), Thogoto virus, Dhori virus, and Batken virus (reviewed in reference 2), whereas mMx2 inhibits distinct RNA viruses, including vesicular stomatitis virus (VSV) and hantavirus (3–5).
Of the different Mx proteins described, there is increasing evidence that some also mediate antiviral activity against particular DNA viruses, including members of the herpesvirus family. For example, human glioblastoma U87MG cells stably expressing hMxB were shown to restrict human alphaherpesviruses (herpes simplex virus 1 [HSV-1] and HSV-2), mouse betaherpesviruses (mouse cytomegalovirus [MCMV]), and mouse gammaherpesviruses (MHV-68), whereas overexpression of hMxA did not (6). Consistent with this, early studies showed that transfection of mouse 3T3 cells to express hMxA did not result in inhibition of HSV-1 replication; however, subsequent studies in human primary fibroblasts reported that a 76-kDa isoform of hMxA induced by IFN treatment was associated with inhibition of HSV-1 growth, whereas a 56-kDa isoform induced by HSV-1 infection actually enhanced HSV-1 replication (7). To date, the mechanisms by which different isoforms of hMxA may inhibit or augment HSV-1 infection are yet to be defined. Currently, little is known regarding the ability of mMx1 and mMx2 to inhibit different herpesviruses, although neither inhibited MCMV growth following overexpression in 293T cells (8).
The first studies addressing the antiviral activity of mouse Mx proteins were described in the 1960s, where an inbred mouse strain (A2G) was shown to be resistant to IAV infection (9), and this trait was inherited as a single dominant allele, which was named Mx (10). Subsequent studies showed resistance to be associated with a single gene (Mx1) located on chromosome 16 and that the Mx locus contained a second Mx gene, Mx2, also on chromosome 16 (11). While the Mx1 and Mx2 genes contain mutations and/or deletions and are therefore nonfunctional in most inbred laboratory strains (e.g., C57BL/6, BALB/c, and CBA/J), both genes are intact in wild mice such as Mus musculus musculus and Mus spretus (12–14). mMx1 mediates antiviral activity against IAV by targeting the RNA-dependent RNA polymerase, thereby prevent primary transcription (15). Endogenous mMx1 has also been shown to inhibit IAV in intranasal mouse models of infection (9, 16).
While the anti-IAV activity of mMx1 has been intensively studied, less is known regarding the ability of mMx1 and mMx2 to inhibit members of other virus families. Moreover, recent reports indicate that human MxA and/or MxB can inhibit one or more herpesviruses in vitro. Herein, we used overexpression studies to demonstrate that mMx1, but not mMx2, potently inhibits replication of the human alphaherpesviruses HSV-1 and HSV-2, but not the murine betaherpesvirus MCMV, in vitro. We confirm critical residues of mMx1 required for inhibition of HSV-1 and demonstrate that mMx1 does not inhibit HSV-1 entry and early gene expression, but can block HSV-1 genomic replication and subsequent late gene expression. Complementary in vitro studies and in vivo studies confirm that expression of functional endogenous mMx1 is also associated with potent antiviral activity against HSV-1.
RESULTS
Generation of LA-4 cell lines overexpressing nuclear mMx1 and cytoplasmic mMx2.
We generated LA-4 cells with doxycycline (DOX)-inducible overexpression of either mMx1 or mMx2, each with an N-terminal FLAG tag, as well as control (CTRL) LA-4 cells with inducible overexpression of cytoplasmic hen egg ovalbumin (cOVA) lacking the sequence for cell surface trafficking (17) and without a FLAG tag. LA-4 cells were utilized as they were a mouse cell line and therefore would contain any species-specific accessory proteins that might be required for Mx1/Mx2 to mediate antiviral activity. Moreover, our preliminary experiments indicated that they supported growth of IAV, HSV-1, and other viruses of interest to our studies, namely, Sendai virus (SeV [strain Cantrell]), lymphocytic choriomeningitis virus (LCMV [strain Armstrong]), and encephalomyocarditis virus (EMCV).
To examine the kinetics of mMx protein induction, LA-4 cells cultured in the presence or absence (no DOX) of 1 μg/mL DOX were analyzed by flow cytometry using an anti-FLAG monoclonal antibody (Mab). FLAG-tagged mMx1/2 proteins were detected after 4 h of culture in DOX and increased markedly by 12 h, but further culture (24 h) did not substantially increase protein expression (Fig. 1ai, protein induction, upper panels). To gain insight regarding inducible mMx protein levels at various times after removal of DOX, cells were cultured in DOX-containing medium for 24 h and then washed and cultured in DOX-free medium for various times. mMx1/2 protein expression was very stable following DOX induction, with only a modest reduction noted in the levels of expression of each protein 48 h after removal of DOX-containing medium (Fig. 1aii, protein stability, lower panels). As expected, LA-4 CTRL cells did not express FLAG-tagged protein in the presence or absence of DOX. These data confirm that mMx1 and mMx2 are rapidly induced by DOX in each LA-4 cell line and that high levels of Mx1 and Mx2 could still be detected 48 h following removal of DOX.
FIG 1.

DOX-inducible overexpression of mMx1 and mMx2 in lentivirus-transduced LA-4 cells. LA-4 cells with stable DOX-inducible overexpression of intracellular ovalbumin (CTRL) or FLAG-tagged mMx1 or mMx2 were seeded at 105 cells/well into 12-well tissue culture plates and incubated overnight at 37°C in 5% CO2. (ai) Cells were cultured in medium supplemented with 1 μg/mL DOX for the indicated times or for 24 h in the absence of DOX (no DOX) and then detached, fixed, permeabilized, and stained for intracellular FLAG expression. (aii) Cells were cultured in medium supplemented with 1 μg/mL DOX for 24 h, washed, and then cultured in DOX-free medium for the indicated times. Cells were then fixed and stained as described above. Histograms show FLAG expression at (ai) different times after DOX induction and (aii) different times after DOX withdrawal following 24 h of induction in DOX. A no-DOX control was also included (filled histograms). (b) After incubation with or without 1 μg/mL DOX for 24 h, cell lysates were subjected to SDS-PAGE, transferred to a polyvinylidene difluoride (PVDF) membrane, and probed with anti-FLAG MAb (upper panel) or with anti-β-actin MAb (lower panel). Molecular weight (MW) markers are shown in kilodaltons. (c) Cells cultured in 8-well tissue culture plates with 1 μg/mL DOX were washed, stained for cell surface lectin MAL II (lectin), and then fixed, permeabilized, and stained for intracellular FLAG, as well as with DAPI. Representative images from confocal microscopy show cell surface MAL II (green [Lectin]), cell nucleus (blue [DAPI]), and expression of FLAG (red [FLAG]), as well as a merged panel (Merged). White arrowheads in merged images highlight localization of mMx1 to the nucleus and mMx2 to the cytoplasm. The scale bar represents 50 μm.
Western blot analysis with anti-FLAG antibody confirmed expression of proteins of the expected size for mMx1 (72 kDa) and mMx2 (74 kDa) (5) in the presence of DOX, which were not present in the absence of DOX or in LA-4 CTRL cells (Fig. 1b). Finally, immunofluorescent staining in conjunction with confocal microscopy confirmed nuclear expression of FLAG-tagged mMx1 and cytoplasmic expression of FLAG-tagged mMx2 (Fig. 1c), consistent with previous studies (3, 4, 18). Together, these data confirm that DOX-inducible overexpression of mMx1 and mMx2 results in expression of proteins of the expected size and with appropriate cellular localization patterns in LA-4 cells.
DOX-inducible overexpression of mMx1, but not mMx2, inhibits growth of HSV-1 and HSV-2 in vitro.
Human MxA and/or MxB proteins can inhibit particular human or mouse herpesviruses (6–8), whereas the ability of mouse Mx proteins to inhibit herpesviruses is less clear. This allows us to examine the ability of Mx proteins from different species to inhibit herpesviruses, including murine herpesviruses. Moreover, studies of mouse Mx proteins allow us to assess their antiviral activities against herpesviruses in vivo for the first time. Therefore, LA-4 cell lines overexpressing mMx1 or mMx2 were cultured in the presence or absence of DOX and then infected with IAV (a virus known to be potently inhibited by mMx1) or with different strains of HSV-1 (the KOS, SC-16, or F strain) or with HSV-2 (strain 186). Overexpression of mMx1, but not mMx2, resulted in reduced titers of infectious IAV, HSV-1, and HSV-2 from LA-4 cells at 24 (IAV) or 48 (HSV-1/2) h postinfection (hpi) (Fig. 2a). Given that HSV-1 and -2 are human alphaherpesviruses, we also assessed the ability of mMx1/2 to inhibit murine cytomegalovirus (MCMV), a betaherpesvirus and a natural pathogen of mice with cytoplasmic replication. While titers of infectious MCMV increased over time between 24 and 96 hpi, no significant differences were observed in virus titers recovered under DOX versus no-DOX conditions for any cell line tested (Fig. 2b). Of interest, additional mouse viruses that also replicate in the cytoplasm, namely, Sendai virus (SeV strain Cantrell), lymphocytic choriomeningitis virus (LCMV strain Armstrong), and encephalomyocarditis virus (EMCV), were not inhibited by inducible overexpression of mMx1 or mMx2 in LA-4 cells (data not shown). The strength of our conclusions regarding the inability of inducible Mx2 to inhibit herpesvirus replication was limited by our inability to include an appropriate control virus sensitive to Mx2-mediated inhibition, namely, hantavirus and/or VSV (3–5), in our studies. Despite this, our studies clearly demonstrate that overexpressed mMx1 displays potent antiviral activity against human alphaherpesviruses in vitro.
FIG 2.

DOX-inducible mMx1, but not mMx2, overexpression inhibits growth of IAV and HSV-1, but not MCMV, in LA-4 cells. LA-4 cells with DOX-inducible overexpression of mMx1 or mMx2, as well as CTRL cells, were seeded and incubated overnight before medium was supplemented with or without 1 μg/mL DOX for 24 h. After DOX induction, cells were incubated with IAV (HKx31; MOI = 1), different strains of HSV-1 (KOS, SC-16, strain F,), HSV-2 (strain 186), and MCMV (strain K181-Perth) at the MOI indicated for 60 min at 37°C and washed and cultured at 37°C. At 24 (IAV and MCMV), 48 (HSV-1), or 96 (MCMV) hpi, supernatants were clarified, and titers of infectious virus were determined using ViroSpot (VS) assays on MDCK cells (IAV) or plaque assays on Vero cells (HSV-1) or M210B4 cells (MCMV). IAV supernatants were activated with 2 μg/mL tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin prior to VS assay. Graphs show the mean (±SD) virus titers from triplicate samples in the presence (black bars) or absence (white bars) of DOX. Where a significant result was obtained, the percentage of reduction in virus titer after DOX induction is also shown. Data show the mean (±SD) of (a) representative results from at least 2 independent experiments or (b) six samples pooled from 2 independent experiments. Dashed lines indicate the detection limit of each assay. Statistical significance was determined by Student's t test. ***, P < 0.001; ns, not significant.
GTPase activity and the nuclear localization signal of mMx1 are required for its ability to inhibit HSV-1 infection.
To determine particular residues of mMx1 required for antiviral activity against HSV-1, we generated LA-4 cells overexpressing mMx1 with specific mutations known to abolish its anti-IAV activity. The mMx1(K49A) mutant lacks GTP binding activity (15, 19), while the mMx1(T69A) mutant lacks both GTP binding capacity and GTPase activity (15). The mMx1(R614E) mutant is reported to facilitate redistribution of mMx1 from the nucleus to the cytoplasm (20). First, we investigated expression and cellular localization of the different mMx1 mutants following overexpression in LA-4 cells. Flow cytometry confirmed expression of parental mMx1 and all mMx1 mutants in LA-4 cells following DOX induction (Fig. 3a). While ~72% of parental mMx1 cells expressed FLAG-tagged mMx1 protein following DOX induction, the mMx1(K49A), mMx1(T69A), and mMx1(R614E) mutants showed ~28%, 78%, and 78% FLAG expression, respectively (Fig. 3a). Immunofluorescent staining in conjunction with confocal microscopy demonstrated that the cellular localizations of the mMx1 mutants are consistent with previous studies assessing their activity against IAV (15, 21). We confirmed that (i) mMx1(K49A) was expressed diffusely throughout the nucleus, similar to parental mMx1, (ii) mMx1(T69A) was predominantly expressed in nuclear aggregates, and (iii) mMx1(R614E) was expressed diffusely throughout the cytoplasm (data not shown). LA-4 cells overexpressing different mMx1 mutants were incubated in the presence or absence of DOX and then infected with HSV-1 or with IAV since each mutant has been shown to abolish the ability of mMx1 to inhibit this virus (15, 19, 20). While mMx1 overexpression (DOX) resulted in significant inhibition of growth of both HSV-1 and IAV, overexpression of each mMx1 mutant did not inhibit growth of either virus (Fig. 3b). Thus, as previously reported for IAV (15, 19), the GTP binding/GTPase activity [mMx1(T69A)] and nuclear localization [mMx1(R614E)] of mMx1 were also critical for antiviral activity toward HSV-1 in LA-4 cells.
FIG 3.

Single-amino-acid substitutions in mMx1 that disrupt GTPase/GTP binding and nuclear localization abrogate antiviral activity against HSV-1. (a and b) LA-4 CTRL or cells with DOX-inducible overexpression of parental mMx1 or an mMx1(K49A), mMx1(T69A), or mMx1(R614E) mutant were seeded, cultured overnight, and then incubated in medium supplemented with or without 1 μg/mL DOX for 24 h. (a) After DOX induction, cells were detached, fixed, permeabilized, and stained for intracellular FLAG expression. Histograms show FLAG expression after incubation in the presence (open) or absence (shaded) of DOX. (b) After DOX induction, cells were incubated with (i) IAV strain HKx31 (MOI = 1) or (ii) HSV-1 KOS (MOI = 0.1) for 60 min and washed and cultured at 37°C. (c) 293T cells constitutively overexpressing CTRL or parental mMx1, an mMx1(K49A), mMx1(T69A), or mMx1(R614E) mutant, or mMx2 were stained for intracellular FLAG staining as described above. Representative histograms are shown. (d) 293T cells (5 × 105) constitutively overexpressing CTRL or parental mMx1, an mMx1(K49A), mMx1(T69A), or mMx1(R614E) mutant, or mMx2 were infected with (i) IAV HKx31 (MOI = 1) or (ii) HSV-1 KOS (MOI = 0.1) for 60 min and washed and cultured at 37°C. (b and d) At 24 (IAV) or 48 (HSV-1) hpi, supernatants were removed and clarified. IAV supernatants were activated with 2 μg/mL TPCK-treated trypsin and then titrated on MDCK cells by ViroSpot assay. HSV-1 supernatants were titrated on Vero cells by plaque assay. Data show the mean (±SD) from triplicate samples and are representative of two independent experiments. The dashed line indicates the detection limit of plaque and VS assays. Statistical significance was determined by Student's t test. ***, P < 0.001. ns, not significant.
An important caveat to these studies relates to the reduced expression levels of the mMx1(K49A) mutant in LA-4 cells compared to parental mMx1, as also reported in a previous study (15). To circumvent the issue that the lack of expression may be associated with reduced antiviral activity, we generated 293T cell lines with stable constitutive overexpression of FLAG-tagged parental mMx1, Mx2, and mMx1 mutants, confirming ~97% of parental mMx1 293T cells were FLAG+, and high levels of FLAG staining were also noted for 293T cells overexpressing the mMx1(K49A), mMx1(T69A), and mMx1(R614E) mutants, where the percentages of FLAG-positive cells were ~85%, 88%, and 72%, respectively (Fig. 3c). Of interest, the mMx1(K49A) and mMx1(T69A) mutants were constitutively overexpressed at higher levels in 293T cells (based on the percentage of cells stained positive for FLAG) compared to DOX-inducible overexpression in LA-4 cells (Fig. 3a), and the fluorescence intensity of mMx1(K49A) and other mutants was equivalent to or higher than that of parental mMx1. The high expression levels of the mMx1(K49A) mutant in 293T cells enabled us to assess of the importance of the GTP binding activity to HSV-1 restriction.
As expected, 293T cells expressing parental mMx1 (mMx1) inhibited the growth of both IAV and HSV-1 compared to 293T cells expressing control (CTRL) protein, whereas expression of Mx2 in 293T cells did not inhibit either virus (Fig. 3d). Inhibition of growth following constitutive overexpression of mMx1 in 293T cells was >99% for IAV and HSV-1 titers, respectively, which is more potent than DOX-inducible overexpression in LA-4 cells, where percentages of inhibition were ~88% and ~95% for IAV and HSV-1, respectively (Fig. 3b). Of note, quantitative real-time PCR (qRT-PCR) confirmed that the fold induction of mMx1 between 293T-CTRL versus 293T-mMx1 (67.31 ± 11.22-fold) was greater than in LA-4 mMx1 cells when comparing cells cultured for 24 h in the presence or absence of DOX (15.43 ± 3.94-fold). In contrast, culture of parental LA-4 cells for 24 h in type I IFN resulted in a 370 ± 105-fold induction of mMx1 compared to untreated cells.
As expected, overexpression of each of the three mMx1 mutant proteins in 293T cells did not result in significant inhibition of IAV growth, indicating that GTP binding, GTPase activity, and nuclear localization of mMx1 were critical for anti-IAV activity (Fig. 3d). When examining HSV-1 growth, overexpression of mMx1(T69A) (GTPase activity/GTP binding) and mMx1(R614E) (nuclear localization) did not inhibit HSV-1, whereas overexpression of mMx1(K49A) (GTP binding) resulted in a modest, but significant, reduction in HSV-1 titers compared to CTRL 293T cells (Fig. 3d). Assessment of HSV-1 genome copy number in cell lysates at 18 hpi (expressed as log10 genome copy number per 100 ng input DNA) confirmed that compared to 293T CTRL cells (mean log10 genome copy number = 5.89 ± 0.02), cells overexpressing mMx1 and the mMx1(K49A) mutant showed significant reductions (mean log10 genome copy numbers = 3.57 ± 0.09 and 5.22 ± 0.06, respectively; P < 0.001), whereas cells overexpressing mMx1(T69A) and mMx1(R614E) mutants did not (mean log10 genome copy numbers = 5.71 ± 0.09 and 5.77 ± 0.21, respectively; P > 0.05). Thus, when mMx1(K49A) was expressed to equivalent levels to parental mMx1 in 293T cells, we observed modest but significant antiviral activity against HSV-1, but not IAV. This indicates that the GTP binding activity of mMx1 (as determined by residue K49) is required for complete antiviral activity toward IAV, but not HSV-1.
mMx1 does not inhibit HSV-1 immediate early gene expression during infection.
mMx1 inhibits IAV by targeting the RNA-dependent RNA polymerase, thereby prevent primary transcription (15). While both IAV and HSV-1 replicate in the nucleus, IAV has a negative-sense single-stranded RNA (ssRNA) genome and replication and transcription are dependent on the viral RNA (vRNA)-dependent RNA polymerase, comprising the PA, PB1, and PB2 viral proteins (22). In contrast, the double-stranded DNA (dsDNA) genome of HSV-1 requires a viral DNA polymerase for replication (23), and transcription requires temporally regulated expression of immediate early (IE), early (E), and late (L) genes (24, 25). To examine the impact of mMx1 on early events in HSV-1 replication, LA-4 CTRL or LA-4 mMx1 cells were infected with HSV-1 KOS and stained for ICP4 protein (an IE gene product) at 3, 5, and 8 hpi. As seen in Fig. 4a, no significant differences in ICP4 expression were observed between cells cultured in the presence of DOX and those in its absence. As a second approach to investigate the impacts of mMx1 on virus entry and/or import of viral genome into the nucleus, we used a recombinant HSV-1 strain with a disruption in the E gene for thymidine kinase (TK) that expresses green fluorescent protein (GFP) under the HCMV IE promoter and therefore independently of HSV-1 genomic replication (i.e., KOS-TK-GFP). Following infection of LA-4 CTRL and LA-4 mMx1 cells with KOS-TK-GFP, we observed no significant differences in the percentage of GFP+ cells in each cell line at 8 hpi following culture in the presence or absence of DOX (Fig. 4bi). In contrast, LA-4 mMx1 cells cultured in DOX showed a significant reduction in the percentage of IAV-infected cells at 8 hpi as detected by flow cytometry (Fig. 4bii). Together, these data indicate that inducible mMx1 overexpression inhibits synthesis of IAV nucleoprotein (NP) in infected cells, consistent with its well-established ability to inhibit IAV primary transcription (15). In contrast, overexpressed mMx1 does not interfere with HSV-1 entry and import of the viral genome into the nucleus.
FIG 4.

DOX-inducible mMx1 overexpression does not inhibit virus entry and early HSV-1 gene expression. LA-4 CTRL cells or LA-4 cells with DOX-inducible overexpression of mMx1 were seeded (105 cells/well), cultured overnight, and then incubated with medium supplemented with or without 1 μg/mL DOX for 24 h. After DOX induction, cells were incubated with (a) HSV-1 KOS (MOI = 2 or 10), (bi) HSV-1 KOS-TK-GFP (MOI = 1 or 10), or (bii) IAV strain HKx31 (H3N2; MOI = 10) for 60 min at 37°C and washed and cultured at 37°C. (a) At 3, 5, and 8 hpi, cells were detached, fixed, permeabilized, and stained with a MAb to the immediate early (IE) HSV-1 protein ICP4, followed by Alexa Fluor 488-conjugated chicken anti-mouse Ig. (b) At 8 hpi, cells were detached and (i) fixed or (ii) fixed, permeabilized, and stained with a fluorescein isothiocyanate (FITC)-conjugated anti-IAV NP MAb. For panels a and b, cells were then analyzed by flow cytometry to quantitate the percentage of ICP4+, GFP+, or NP+ cells. Data are from at least two independent experiments and show the mean (±SD) of results from triplicate samples. Statistical significance was determined by Student's t test. ***, P < 0.001; ns, not significant.
mMx1 inhibits HSV-1 late gene expression during infection.
To investigate the impact of mMx1 on HSV-1 late gene expression, we used a recombinant virus in which enhanced green fluorescent protein (EGFP) and red fluorescent protein (RFP) are expressed from the HSV-1 glycoprotein B (gB) and gC promoters, respectively (HSV-1 RE-pgB-EGFP/pgC-RFP virus, here referred to as double-fluorescent (DF) HSV-1 (26). The HSV-1 gB gene is classified as a leaky late (γ1) gene as it can be expressed early (prior to DNA replication) and late (after DNA replication) during infection (27), whereas the gC gene is a true late (γ2) gene that requires DNA replication for transcription (26, 28). Consistent with this, our flow cytometric analysis confirmed that EGFP, but not RFP, is expressed at 8 hpi, whereas both EGFP and RFP are expressed at 18 hpi (Fig. 5a). Moreover, 300 μg/mL phosphonoacetic acid (PAA), a known inhibitor of HSV-1 DNA replication (29), inhibited RFP expression at 18 hpi after infection with DF HSV-1 (Fig. 5a) but did not affect cell viability (data not shown).
FIG 5.

mMx1 overexpression inhibits viral late gene expression during HSV-1 infection. (a) Parental LA-4 cells seeded (2 × 105 cells/well) into 12-well tissue culture plates were incubated overnight and then infected with HSV-1 DF at the MOI indicated for 60 min at 37°C, washed, and then cultured with or without 300 μg/mL of PAA. At 8 hpi (left panel) or 18 hpi (right panel), cells were detached, stained with fixable viability dye eFluor 780, and fixed before EGFP and RFP expression was analyzed by flow cytometry. (b, c, and d) LA-4 CTRL or LA-4 mMx1 cells were seeded, incubated overnight, and then cultured in medium supplemented with or without 1 μg/mL DOX for an additional 24 h. After DOX induction, cells were infected with (b and c) HSV-1 DF or (d) HSV-1 KOS (MOI = 1) and washed and cultured at 37°C (b) in the presence of 300 μg/mL of PAA to inhibit genomic replication and late gene expression or (c and d) in the absence of PAA. At 18 hpi, (c) cells were fixed and analyzed for EGFP and RFP expression or (d) fixed, permeabilized, and stained for intracellular expression of VP5. Data are representative of at least two independent experiments and show the mean (±SD) of results from triplicate samples. Statistical significance was determined by Student's t test. ***, P < 0.001; ns, not significant.
Next, LA-4 CTRL and LA-4 mMx1 cells cultured in the presence or absence of DOX were infected with DF HSV-1 and then cultured in the presence or absence of PAA before analysis by flow cytometry at 18 hpi. In the presence of PAA, we would still expect some EGFP expression in the absence of genomic replication (i.e., leaky late gene expression), but RFP expression should be suppressed. No major differences in EGFP were observed between DOX-induced and uninduced LA-4 CTRL or LA-4 mMx1 cells at 18 hpi, and RFP expression was suppressed in all cells (Fig. 5b). In the absence of PAA, EGFP and RFP were expressed at similar levels in CTRL cells in the presence or absence of DOX, whereas DOX-inducible mMx1 overexpression was associated with significantly reduced EGFP and RFP, and this was particularly potent for gC-RFP (Fig. 5c). To confirm mMx1-mediated inhibition of HSV-1 late gene expression, LA-4 cells overexpressing mMx1 were infected with HSV-1 KOS and stained for the L gene product, capsid protein VP5, at 18 hpi. While no differences were noted in the percentage of VP5+ LA-4 CTRL cells in the presence and absence of DOX (no DOX = 35.2% ± 3.9% versus DOX = 32.3% ± 1.8%; P > 0.05 by Student's t test), DOX-induced overexpression of mMx1 significantly reduced the percentage of VP5+ LA-4 mMx1 cells at 18 hpi (no DOX = 15.1% ± 1.3% versus DOX = 5.7% ± 0.5%; P < 0.001 by Student's t test) (Fig. 5d). Overall, these data indicate that mMx1 does not affect HSV-1 entry or expression of IE gene product ICP4, and the expression of HSV-1 L genes is strongly inhibited by mMx1.
mMx1 inhibits replication of the HSV-1 viral genome during infection.
Next, we assessed the impact of mMx1 on genomic replication by quantitating HSV-1 viral DNA during infection using qPCR. In these studies, we determined HSV-1 genome copy numbers in LA-4 CTRL and LA-4 mMx1 at 2 and 18 hpi. As seen in Fig. 6a, genome copy number increased markedly between 2 and 18 hpi in all cell lines tested. Moreover, while no differences in copy number were detected in LA-4 CTRL cells in the presence or absence of DOX, a significant (~70%) reduction in genome copy number was observed in LA-4 in the presence of mMx1 (DOX) compared to the absence of mMx1 (no DOX) overexpression. To substantiate these findings, we also assessed genome copy number in 293T cells constitutively overexpressing mMx1 or CTRL proteins. Given that HSV-1 growth was so potently inhibited in 293T-mMx1 cells (Fig. 3dii), we first infected cells with KOS-TK-GFP and examined GFP expression at 8 hpi to confirm there was no major disruption in virus entry and/or import of genome into the nucleus in 293T-mMx1 cells (Fig. 6b). Despite no differences in virus entry, we observed an ~99% reduction in the copy number of genomic HSV-1 DNA at 18 hpi when comparing 293T-CTRL to 293T-mMx1 cells (Fig. 6c). While we did not normalize copy numbers against genomic DNA, we note that the inhibitory effect of Mx1 was observed across biological replicates, in different cell lines, and at multiple multiplicities of infection (MOI).
FIG 6.
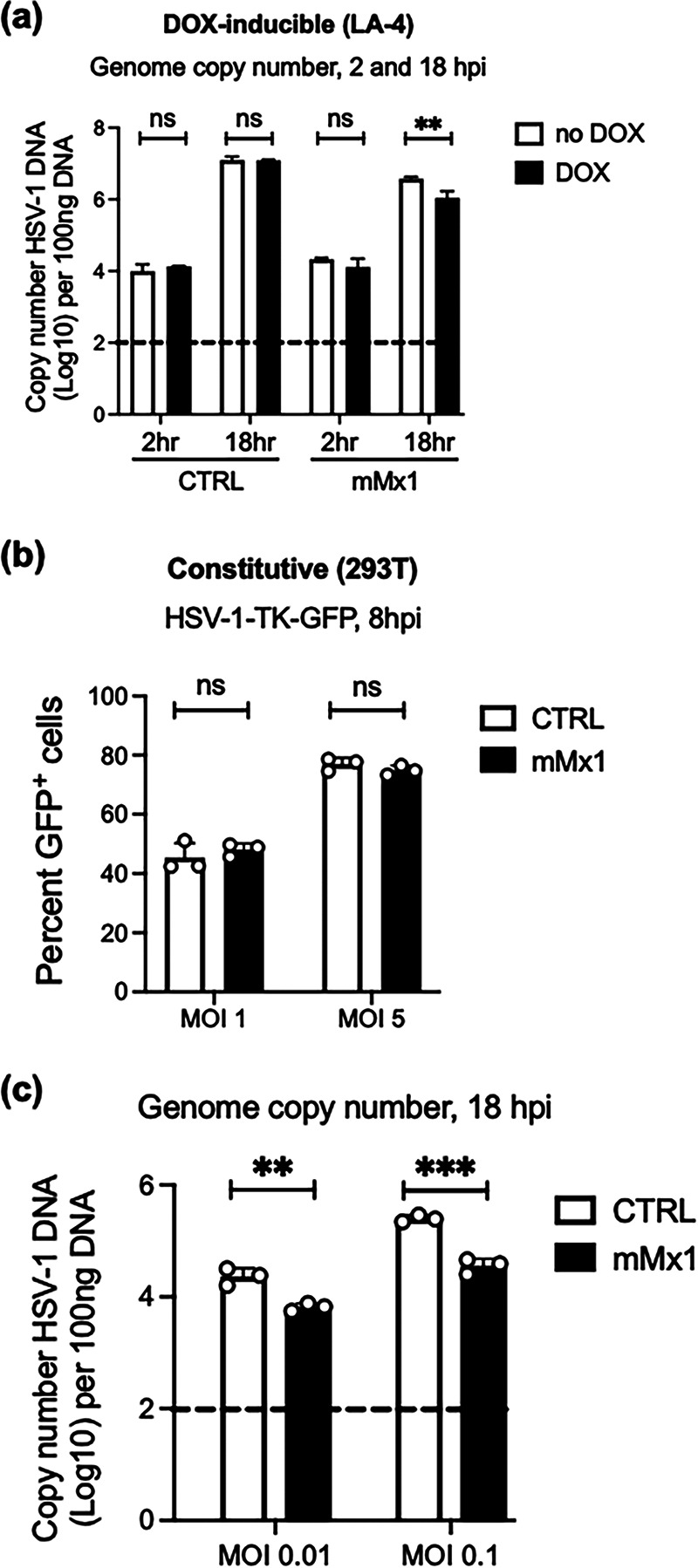
Inducible or constitutive overexpression of mMx1 results in reduced levels of HSV-1 genomic DNA in virus-infected cells. (a) LA-4 CTRL or mMx1 cells were seeded, cultured overnight, and then incubated with medium supplemented with or without 1 μg/mL DOX for 24 h. After DOX induction, cells were incubated with HSV-1 KOS (MOI = 1) for 60 min and washed and cultured at 37°C. At 2 and 18 hpi, total cellular DNA was extracted for real-time qPCR. (b) 293T cells constitutively expressing CTRL or mMx1 proteins were seeded, cultured overnight, and then incubated with HSV-1-TK-GFP (MOI = 2) and analyzed by flow cytometry at 8 hpi. (c) 293T cells constitutively expressing CTRL or mMx1 proteins were seeded, cultured overnight, and then incubated with HSV-1 KOS (MOI = 2) for 60 min at 37°C and washed and cultured at 37°C. At 18 hpi, total cellular DNA was extracted for real-time qPCR. For panels a and c, HSV-1 DNA copy number was determined relative to a standard curve generated using a plasmid expressing the HSV-1 TK gene. Data are from one of two independent experiments and show the mean (±SD) of results from triplicate samples. The dashed line indicates the detection limit of the assay. Statistical significance was determined by Student's t test. **, P < 0.01; ***, P < 0.001; ns, not significant.
Induction and antiviral activity of endogenous mMx1 against HSV-1.
While functional Mx1 and Mx2 alleles are found in wild mice, most inbred strains of laboratory mice (e.g., C57BL/6, BALB/c, and others) do not express functional mMx1 or Mx2 proteins (3, 12). The gene encoding functional mMx1 was discovered in the 1960s based on the resistance of the A2G mouse strain to IAV infection (9, 30), and congenic C57BL/6 mice expressing A2G Mx1 (B6.A2G-Mx1) are highly resistant to IAV infection (16). We have used C57BL/6 mice expressing a nonfunctional (B6.WT) or functional (B6.A2G-Mx1) mMx1 protein to assess induction and antiviral activity of mMx1 against HSV-1, using IAV as a control virus known to be potently inhibited by endogenous mMx1 (9). The B6.WT and B6.A2G-Mx1 mice used in these experiments were derived from breeding of heterozygote founder animals to ensure consistency in the C57BL/6 background. First, primary lung fibroblasts isolated from B6.WT or B6.A2G-Mx1 mice were treated ex vivo with recombinant mouse IFN-α (type I IFN) or infected with IAV or HSV-1 prior to qRT-PCR to quantitate mMx1 gene expression, expressed as fold change relative to mock-treated cells using the threshold cycle (2−ΔΔCT) method (31). As expected, mouse IFN-α potently upregulated mMx1 in fibroblasts from both mouse strains (Fig. 7ai). While infection with both IAV and HSV-1 also upregulated mMx1 (Fig. 7aii and iii), this was generally delayed and peaked at lower levels than in response to mouse IFN-α, which may relate to the time required for IFN production in response to the IAV and HSV-1 infections, which would subsequently amplify expression of ISGs, including that coding for mMx1 (32).
FIG 7.

Induction and antiviral activity of endogenous mMx1 gene expression in primary lung fibroblasts following IAV and HSV-1 infection. Lung fibroblasts isolated from B6.WT and B6.A2G-Mx1 mice were seeded in triplicate into 12-well tissue culture plates (2 × 105 cells/well) and cultured overnight. (a) Cells were then treated with 1,000 U/mL of recombinant mouse IFN-α or infected with IAV strain IAV (HKx31; MOI = 10) or HSV-1 (KOS; MOI = 1) for 1 h at 37°C and washed and cultured at 37°C. At the indicated times, cells were lysed, and total RNA was extracted to determine mMx1 gene expression by qRT-PCR. Shown is induction of mMx1 over time in response to IFN, IAV, or HSV-1. Expression was normalized to the mean of three housekeeping genes and expressed as fold induction relative to untreated cells. (b) Untreated cells (no IFN) or cells pretreated with mouse IFN-α were then infected with either HSV-1-TK-GFP (at MOI = 2, as indicated) and analyzed by flow cytometry at 8 hpi. (c) Cells were (i) left untreated (no IFN) or treated with 1,000 U/mL of mouse IFN-α (IFN) or (ii) transfected with RIG-I agonist RNA (IVT4) or with control RNA (GAC) and then cultured for an additional 24 h. After washing, cells were infected with HSV-1 (MOI = 10) for 1 h and then washed and cultured further. At 2 and 48 hpi, cell supernatants were removed and clarified, and virus titers were determined by plaque assay on Vero cells. Virus titers are shown, and where significant, the percentage of reduction in virus titer between B6.WT and B6.A2G-Mx1 cells is indicated for each condition on the graph. The dashed line indicates the detection limit of the plaque assay. Statistical significance was determined by Student's t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Next, we assessed the impact of endogenous mMx1 on HSV-1 entry and infection. In these experiments, lung fibroblasts were untreated (no IFN) or pretreated with mouse IFN-α (IFN) prior to infection with HSV-1-TK-GFP or IAV, and the percentage of infected cells was determined at 8 hpi. As seen in Fig. 7bi, B6.WT and B6.A2G-Mx1 cells showed a similar susceptibility to infection by HSV-1-TK-GFP, and while this was reduced by IFN pretreatment, no differences were noted between cells from B6.WT and B6.A2G-Mx1 mice. In contrast, B6.A2G-Mx1 cells showed a significant reduction in the percentage of IAV-infected cells at 8 hpi compared to B6.WT cells, and differences were more pronounced by pretreatment with IFN (Fig. 7bii). To assess the impact of endogenous mMx1 on HSV-1 growth, cells were (i) untreated (no IFN) or pretreated with IFN or (ii) treated with control RNA (GAC) or RIG-I agonist (IVT4) prior to HSV-1 infection. As expected, virus titers increased markedly between 2 and 48 hpi following infection of untreated cells, and only negligible differences were observed between titers obtained from B6.WT and B6.A2G-Mx1 cells (Fig. 7c). However, pretreatment of fibroblasts from both mice strains with recombinant mouse IFN-α markedly reduced virus titers observed at 48 hpi, and this was even further reduced in lung fibroblasts isolated from B6.A2G-Mx1 mice expressing functional Mx1 (Fig. 7c). Similar effects were observed in fibroblasts isolated from B6.A2G-Mx1 mice and transfected with RIG-I agonist IVT4 (Fig. 7c). Together, these data indicate that endogenous mMx1 does not inhibit HSV-1 entry and/or import of the viral genome to the nucleus but does act as a potent inhibitor of HSV-1 growth.
Mice expressing a functional mMx1 protein show reduced lesion severity and virus replication in the HSV-1 zosteriform model of infection.
The antiviral activity of functional mMx1 protein has been clearly established in the mouse model of IAV infection (9, 16). In preliminary studies, we infected B6.WT and B6.A2G-Mx1 mice via the intranasal route with 104 PFU of IAV strain HKx31 (H3N2). While IAV infection of B6.WT mice resulted in significant weight loss 3 to 10 days postinfection (dpi), B6.A2G-Mx1 did not lose any weight after infection (data not shown). Moreover, if IAV-infected mice were killed at 5 dpi, virus titers recovered from the lungs of B6.A2G-Mx1 mice (3.0 ± 0.3 log10 PFU/lung; n = 5/group) were significantly lower than those recovered from B6.WT mice (6.3 ± 0.3 log10 PFU/lung; n = 5/group; P < 0.001). These are expected data, and confirmed previous studies using these mice.
Mice can be infected with HSV-1 by a number of different routes, including flank skin zosteriform infection (33–36). The zosteriform model is characterized by virus replication in epithelial cells of the skin at the inoculation site, followed by infection of sensory neurons innervating the skin and subsequent retrograde transport to infect the dorsal root ganglia (DRG). Following lytic replication in the DRG, the virus can quickly reemerge from the innervating DRG to appear at secondary sites along the dermatome, giving rise to a zosteriform band of vesiculating lesions. Skin lesions first appear as transient vesicular lesions in the epidermal layer and generally heal without scarring within 2 weeks of inoculation (33–36). Ultimately, HSV-1 also establishes latent infection in the DRG, although it may periodically reemerge to form recurrent lesions at or near the initial site of infection in response to different stimuli.
We performed flank skin zosteriform infections of B6.WT and B6.A2G-Mx1 mice with HSV-1. Lesion development was monitored and photographed every second day over an 11-day observation period. Ulceration at the primary site of infection was observed in both mouse strains by 3 dpi, before B6.WT mice developed zosteriform lesions involving ulceration at secondary sites by 5 dpi, peaking at 7 to 11 dpi. Formation of zosteriform lesions was delayed, and lesions were less severe in B6.A2G-Mx1 mice. Figure 8a shows representative images of lesions from B6.WT and B6.A2G-Mx1 mice at 7 dpi. Using a lesion scoring system previously published by our group (34), three readers each blindly scored the lesion severity of infected animals over time. Using this approach, we found that the lesion score of B6.A2G-Mx1 mice expressing a functional mMx1 protein was significantly reduced compared to that of B6.WT animals at 5 to 11 dpi (Fig. 8b). B6.WT mice developed lesions characterized by local vesicles, ulcerated vesicles, and severe ulcerations throughout the dermatome and/or contralateral spread, peaking at 7 to 11 dpi, whereas lesions observed in B6.A2G-Mx1 mice were much milder, with little evidence of ulceration along the dermatome and therefore with significantly lower lesion scores over time. Next, mice were killed at 5 dpi to allow for assessment of virus titers in the skin at the primary site (i.e., the site of inoculation) and the secondary site (i.e., the site of zosteriform lesions along the entire dermatome), as well as the DRG. This time point was chosen based on previous studies from our group that showed this to be the time of peak virus replication at each of the three sites (34). As seen in Fig. 8c, virus was recovered from the primary inoculation site and secondary skin lesions, as well as the DRG, of infected B6.WT and B6.A2G-Mx1 mice. Consistent with the reduced lesion severity recorded, titers of infectious virus recovered from each site of B6.A2G-Mx1 mice were significantly reduced compared to titers recovered from B6.WT mice.
FIG 8.

Expression of a functional endogenous mMx1 protein is associated with reduced lesion severity and HSV-1 virus titers in the zosteriform model of HSV-1 infection. Groups of B6.WT and B6.A2G-Mx1 mice (n = 5/group) were infected via flank scarification with 106 PFU of HSV-1 KOS. Mice were monitored daily, and lesions were photographed every second day. (a) Images of representative skin lesions from the group (left panels) or an individual (right panel) B6.WT and B6.A2G-Mx1 mouse at 7 days postinfection are shown. Regions corresponding to the primary (P) and secondary (S) sites are indicated. (b) Lesion severity was determined using the scoring system described in Materials and Methods. Lesion images were photographed every second day and then scored blind by three assessors. Average scores from individual mice across the three assessors are shown as circles, and bars represent the mean lesion scores (±SD). (c) At 5 dpi, animals were killed, skin samples (corresponding to the primary and secondary sites) and the DRG were removed, and titers of infectious virus were determined in clarified homogenates by plaque assay on Vero cells. (d) To assess reactivation of latent virus, DRG collected at day 35 postinfection were cultured in vitro for 5 days before the plaque assay was performed on clarified supernatants in Vero cells. Data were pooled from two experiments (n = 7/group). For panels c and d, the dashed line indicates the detection limit of the plaque assay. Data were from one of two independent experiments for panel a and pooled from two independent experiments for panels b to d. Statistical significance was determined by Student's t test. **, P < 0.01; ***, P < 0.001; ns, not significant.
Following lytic replication in the skin and the DRG, the HSV-1 zosteriform model is characterized by latent infection of the DRG, whereby the virus enters a quiescent state and is transcriptionally inactive. Given the marked reduction in titers of infectious virus in the skin and DRG of B6.A2G-Mx1 mice at 5 dpi, we investigated if expression of a functional mMx1 protein might also impact the development of latency and/or reactivation of lytic virus from latently infected DRG. Previous studies have demonstrated that following removal of latently infected DRG, in vitro culture for 3 to 5 days is sufficient to reactivate HSV-1, resulting in lytic virus replication (37). Therefore, mice flank infected with HSV-1 were killed at 35 dpi, and DRG were removed and cultured in serum-free medium at 37°C for 5 days. The DRG were then homogenized and titrated to determine titers of infectious virus. As shown in Fig. 8d, virus could be reactivated from DRG recovered from both B6.WT and B6.A2G-Mx1 mice, and no major differences were noted in recovery or titers of virus from mice which did or did not express a functional endogenous mMx1. It is important to note that multiple rounds of lytic virus replication will occur during this 5-day culture period, and a reactivation time course could be performed in future studies to investigate if any early differences in reactivation kinetics and/or virus titers are observed in DRG from B6.A2G-Mx1 mice.
DISCUSSION
While several studies have demonstrated antiviral activities of human Mx proteins against different herpesviruses in vitro, we are the first to report the antiviral activity of mouse Mx1 against alphaherpesviruses both in vitro and in vivo. Our study adds to a growing body of evidence that particular Mx proteins inhibit certain members of the herpesvirus family. From studies to date, hMxB has been described as a “pan-herpesvirus restriction factor” due to its ability to inhibit members of the alphaherpesvirus (e.g., HSV-1, HSV-2), betaherpesvirus (e.g., MCMV), and gammaherpesvirus (e.g., Kaposi’s sarcoma-associated herpesvirus [KSHV] and MHV-68) subfamilies (6, 8). Of note, hMxB is active against a range of both human (HSV-1/2, KSHV) and mouse (MCMV, MHV-68) viruses. While early studies indicated that transfection of mouse 3T3 cells with hMxA resulted in inhibition of IAV and VSV, but not HSV-1 (38), subsequent studies indicated that IFN treatment of human fibroblasts resulted in induction of a 76-kDa cytoplasmic form of hMxA that could inhibit HSV-1 replication (7). Surprisingly, in the absence of IFN, HSV-1 infection resulted in the induction of a 56-kDa isoform of hMxA expressed in the nucleus, which actually enhanced virus replication (7). This led the authors to speculate that expression of the alternatively spliced 56-kDa variant of hMxA represented a novel immune evasion mechanism. In another study, 293T cells stably transduced to overexpress hMxB were shown to inhibit MCMV replication, whereas cell lines expressing hMxA, mMx1, or mMx2 did not (8). Of interest, while the authors studied the antiviral effects of mMx1/2 expression on MCMV using 293T cells, they reported that they could not generate mouse (3T3) lines with stable overexpression of either mouse Mx protein. In our study, we generated human 293T cells with constitutive mMx overexpression, as well as mouse LA-4 cells with DOX-inducible mMx overexpression. In either overexpression system, only mMx1 inhibited HSV-1 replication, noting that this restriction of viral replication was particularly potent in 293T cells. Despite this, the DOX-inducible overexpression system offered several advantages, including tightly regulated overexpression in a mouse cell line at levels likely to be more physiologically relevant. Of note, our results contrast with those of an earlier study reporting that overexpression of mMx1 in 3T3 cells did not inhibit HSV-1 growth (38), which likely reflect differences in virus strains and other experimental parameters.
mMx-mediated inhibition of HSV-1 occurs via a mechanism that is distinct from that by which hMxB has been reported to inhibit herpesviruses. Crameri et al. reported that hMxB blocks HSV-1 infection after virus entry and tegument dissociation from the capsid, but prior to uncoating of viral DNA from the capsid and nuclear import of viral DNA (39). Similarly, hMxB also inhibited delivery of viral genomes to the host cell nucleus during MCMV infection (8). While hMxB localizes to the nuclear envelope, mMx1 is expressed within the nucleus, and our studies clearly demonstrate that early HSV-1 gene expression was not impacted by mMx1 overexpression or by IFN-induced endogenous mMx1. Thus, mMx1 does not appear to mediate the same “gatekeeper” function proposed for hMxB in restricting nuclear entry of herpesvirus DNA (39). Instead, while HSV-1 expression of IE gene product ICP4 was not inhibited by mMx1, restriction of HSV-1 was associated with reduced levels of HSV-1 genomic DNA. This alone would be sufficient to account for the impacts on subsequent late gene expression and HSV-1 growth, although we cannot rule out the possibility of additional direct effects of mMx1 on later steps in the virus replication cycle which further enhance inhibition of late gene expression and/or virus assembly and egress. Moreover, a more comprehensive analysis of IE, E, and L gene expression in the presence or absence of functional mMx1 may provide further insights regarding the specific step or steps in HSV-1 that are blocked.
In our studies, an intact GTPase domain and the nuclear localization of mMx1 were required for inhibition of HSV-1 replication. hMxB-mediated restriction of herpesviruses was also shown to be dependent on its GTPase function (8, 39), although the GTPase activity of hMxB was not required for inhibition of lentivirus replication (40). For mMx1, GTP binding and/or GTPase activity are also required for its antiviral activity against IAV (15, 19). We used an mMx1(K49A) mutant lacking GTP binding activity (15, 19), as well as an mMx1(T69A) mutant lacking both GTP binding capacity and GTPase activity (12), to determine how these mutations affected the ability of mMx1 to restrict HSV-1 growth. While overexpression of either mutant did not inhibit HSV-1 growth in LA-4 cells, interpretation of these results was confounded by the very low levels of mMx1(K49A) protein expressed in these cells. Of interest, previous studies used the same mutants to investigate the impact of mMx1 on assembly of the viral ribonucleoprotein (vRNP) complex during IAV infection and also reported low levels of mMx1(K49A) protein expression following transient transfection of 293T cells (15). While the authors increased the amount of plasmid used for transient transfection to obtain higher expression of the mMx1(K49A) mutant in the IAV study, we used an alternative approach and generated 293T cells with stable constitutive overexpression of parental and mMx1 mutants. Using this approach, we confirmed robust expression of all mMx1 proteins by flow cytometry and found that expression of mMx1(K49A) markedly reduced, but did not abolish, its ability to restrict HSV-1 growth. This contrasts with the complete loss of anti-IAV activity of mMx1(K49A) in a minireplicon system containing a firefly luciferase reporter gene and the viral proteins PB1, PB2, PA, and NP (15) or in impacting IAV growth in 293T cells in our study.
mMx1 localizes to the nucleus, and mutations in mMx1 that result in its redistribution to the cytoplasm are known to abrogate its ability to inhibit IAV (20). We confirmed that the mMx1(R614E) mutant of mMx1 localized to the cytoplasm instead of the nucleus, and this mutant no longer inhibited HSV-1 growth. mMx1 inhibits primary transcription of IAV in the nucleus, and this is likely to occur by interfering with the assembly of the RNA-dependent RNA polymerase (15, 41). Of note, this is distinct from the anti-IAV activity of cytoplasmic hMxA, which appears to act early to prevent transport of incoming vRNPs into the nucleus (42), as well blocking newly translated viral proteins from entering the nucleus to participate in secondary transcription (41). Like IAV, HSV-1 also replicates within the nucleus; however, it does not express an RNA-dependent RNA polymerase but instead utilizes viral replication machinery consisting of a DNA polymerase (UL30), processivity factor (UL42), helicase-primase complex (UL5/UL8/UL52), a single-stranded DNA binding protein (UL29 or ICP8), and an origin binding protein (UL9), along with several host proteins that contribute to the process (23). While it is clear from our studies that genomic replication and therefore subsequent steps such as late gene expression are inhibited by mMx1, further studies are required to identify the specific viral and/or host proteins recognized by mMx1 to inhibit replication of the HSV-1 genome in infected cells.
Human and mouse Mx proteins are dynamin-like GTPases originally identified as potent inhibitors of influenza A virus (IAV), but subsequently shown to inhibit a number of other viruses. While the ability of Mx1 to inhibit IAV was described over 50 years ago, the molecular mechanisms by which it inhibits IAV replication are still not fully elucidated. Human Mx proteins localize to different cellular compartments, and this impacts how they interact with HSV-1. For example, human MxB localizes to the nuclear envelope and blocks herpesviruses by inhibiting entry of the herpesvirus genome into the nucleus (6, 39). Our studies provide an important contrast to these findings, as Mx1 is expressed within the nucleus and does not stop entry nuclear entry of the HSV-1 genome, but rather blocks subsequent genomic replication within the nucleus. Of interest, GTP binding and hydrolysis are required for human MxB (6, 39) and mouse Mx1 to inhibit HSV-1, as well as for inhibition of IAV by Mx1 (15, 19), although the specific mechanisms by which these features determine viral inhibition are yet to be determined. In future studies, it will be of interest to determine if relocation of human MxB from the nuclear membrane to within the nucleus (as for mouse Mx1) also results in inhibition of HSV-1 genomic replication. Additional studies to define interactions between Mx1/MxB with specific viral proteins and/or the HSV-1 genome itself will be of major interest in elucidating differences in inhibition of HSV-1 by different Mx proteins.
Our studies with constitutive or DOX-induced overexpression of mMx1 were complemented by studies using mice that do or do not express a functional endogenous mMx1 protein. It is well known that most inbred strains of laboratory mice do not express functional mMx1 protein due to deletions or mutations within the mMx1 gene (12), whereas the feral A2G mouse strain is known to express a functional protein (9, 30). Previously published in vivo studies using mice expressing a functional mMx1 (A2G mice) demonstrated that mMx1 protected mice against infections with IAV (9, 30, 43, 44) and Thogoto virus, both belonging to the Orthomyxoviridae family (45). Herein, we have used the zosteriform model of flank infection to confirm the impact of a functional endogenous mMx1 during HSV-1 infection. Strikingly, the severity of skin lesions was markedly reduced in B6.A2G-Mx1 mice, with little evidence of secondary lesion development and ulceration, as was seen in the B6.WT mice. Reduced lesion severity in B6.A2G-Mx1 mice correlated with reduced virus titers at the primary inoculation site, as well as at the sites of secondary lesion formation. Of note, virus titers were also reduced at 5 dpi in the DRG, noting that despite lower titers, infectious virus was recovered from the DRG of all HSV-1-infected B6.A2G-Mx1 mice. In future studies, it would be of interest to determine if viral components (e.g., viral capsid and/or tegument) and Mx1 protein colocalize in skin lesions and DRG, as well as to determine levels of Mx1 mRNA expressed at these sites over the course of infection.
Expression of a functional mMx1 might inhibit HSV-1 replication in vivo in a number of ways. Following skin inoculation, local HSV-1 replication occurs in the skin dermis and epidermis, from where HSV-1 spreads to infect innervating sensory neurons and travels retrograde to the DRG. Then the virus establishes a latent infection in the DRG or reactivates to enter a lytic stage and migrates anterograde toward flank skin, resulting in the formation of secondary zosteriform lesions that appear along sensory neurons of the dermis (33–36). We observed that B6.A2G-Mx1 mice had a lower HSV-1 titer at the primary inoculation site, as well as in secondary lesions and in the DRG. The presence of reduced titers at the primary inoculation site likely reflects direct inhibition of HSV-1 replication by mMx1 in epithelial cells, and virus-induced IFN production is likely to amplify this early effect over time. It is also possible that mMx1-mediated inhibition at the primary site results in reduced infection of innervating sensory neurons, leading to reduced virus replication in the DRG and secondary sites in the skin. Alternatively, mMx1 may be induced in the DRG and the secondary sites early after infection, and this therefore limits HSV-1 replication once the virus arrives. The difference in virus titers between B6.WT and B6.A2G-Mx1 mice is very pronounced in the secondary lesions, which could be the cumulative effect of mMx1 inhibition at each site as mMx1 is broadly expressed with little tissue specificity. However, the presence of higher virus titers in secondary lesions compared to the primary site and the DRG suggests that the mMx1 has a profound impact at this site, which might relate to local production of IFNs in the skin (as a result of replication at the primary site) by the time the virus arrives to the secondary lesions from the DRG. Our in vitro data using lung fibroblasts confirmed that the antiviral effect of a functional mMx1 against HSV-1 is much more potent following IFN prestimulation. Given the potent reduction in HSV-1 titers in the skin and DRG, it would be of interest to use RT-qPCR to measure both IFN and mMx1 induction at these sites over time during HSV-1 infections. In a previous study, IAV infection and IFN treatment of A2G mice resulted in systemic induction of mMx1 in several organs, with the highest levels found in the target organ for virus replication (i.e., the lungs) (46). Future studies could investigate mMx1 induction following HSV-1 infection in the skin and DRG over time using qPCR.
Overall, our studies examined the effects of overexpressed and endogenously expressed mMx1 protein on the lytic replication of HSV-1 in cell lines and primary cells, as well as in skin and DRG from HSV-1-infected mice. It is also worth noting that we performed initial experiments examining reactivation of HSV-1 from latently infected DRG in mice with or without a functional mMx1. While endogenous expression of functional mMx1 protein effectively reduces lytic HSV-1 replication, latent virus could be reactivated from the DRG of all B6.WT and B6.A2G-Mx1 mice tested, suggesting that a functional mMx1 did not prevent establishment of latency, nor did it prevent reactivation of latent virus from within infected DRG. A previous study showed that a tripartite protein (TRIM19), also known as promyelocytic leukemia (PML) protein, is an IFN-inducible protein involved in control of HSV-1 latency by negatively regulating the expression of the latency-associated transcript (LAT) genes (47). Thus, while some ISG proteins are involved in regulating HSV-1 latency in neurons, our studies suggest that mMx1 is not.
Finally, our findings that Mx1 inhibited HSV-1/2, but not MCMV, suggest that certain mouse viruses may have evolved to avoid detection by Mx1, thereby promoting their persistence. This appears particularly relevant given that human MxB does inhibit MCMV (8), but mouse Mx1 does not. While MCMV replicates in the nucleus, where Mx1 is expressed, we also found that DOX-inducible cytoplasmic Mx2 did not inhibit mouse viruses that replicate in the cytoplasm (Sendai virus, lymphocytic choriomeningitis virus, and encephalomyocarditis virus) Thus, it could be speculated that evasion of Mx1 and/or Mx2 might be an important feature of at least some viruses that naturally infect mice, noting that this may not be true for all as hantaviruses were reported to be sensitive to Mx2 (5).
MATERIALS AND METHODS
Cell lines and viruses.
Mouse airway epithelial LA-4 cells (American Type Culture Collection [ATCC] CCL-196) were maintained and passaged in Ham’s F-12K (Kaighn's) medium (Gibco) containing 10% (vol/vol) fetal calf serum (FCS) (Sigma-Aldrich, USA) and supplemented with 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco-BRL, NY) (F-12K10). Madin-Darby canine kidney (MDCK) cells (ATCC CCL-34) were maintained and passaged in RPMI 1640 medium supplemented with 10% (vol/vol) FCS and supplements as described above. 293T cells (ATCC CRL-3216) and Vero cells (CSL, Parkville, Australia) were maintained and passaged in Dulbecco's modified Eagle medium (DMEM) (Gibco) containing 10% (vol/vol) FCS and supplements (DMEM10).
The HSV-1 KOS, SC-16, and F strains and HSV-2 strain 186, as well as a recombinant HSV-1 KOS virus with green fluorescent protein (GFP) inserted to disrupt the TK locus and expressed under the control of a HCMV promoter as described previously (48), were kind gifts from Francis Carbone, The University of Melbourne. Recombinant HSV-1 viruses on the RE strain background expressing GFP and/or red fluorescent protein (RFP) under the control of the HSV-1 gB or gC promoters, respectively, were a kind gift from Paul (Kip) Kinchington, University of Pittsburgh, USA, and have been described previously (26). All HSV-1 stocks were propagated in Vero cells and titrated by standard plaque assay on Vero cells (36). MCMV (K181-Perth strain) was propagated and viral titers determined by plaque assay using M210B4 fibroblasts as described previously (49). The IAV strain used in this study was HKx31, which is a high-yield reassortant of A/Philippines/2/82 (H3N2) with PR8, which was obtained from the WHO Collaborating Centre for Reference and Research on Influenza (WHO CCRRI), Melbourne, Australia, and was grown in embryonated hen’s eggs by standard procedures (50). Titers of virus stock were determined by standard plaque assay or ViroSpot (VS) immunoassay on MDCK cells as described below and expressed in PFU per milliliter or VS per milliliter, respectively.
Generating LA-4 cell lines with doxycycline-inducible expression of intracellular proteins.
To produce lentiviruses for the generation of cell lines with doxycycline (DOX)-inducible expression of mouse Mx1 (mMx1) and mMx2, a two-step cloning strategy was required. mMx1 (GenBank accession no. NM_010846.1) and mMx2 (GenBank accession no. NM_013606.1) engineered to express an N-terminal FLAG tag were cloned into the pTRE-tight plasmid vector and then subcloned into the pFUV1-mCherry lentivirus transfer plasmid (51, 52) (kindly provided by Marco Herold, The Walter and Eliza Hall Institute, Parkville, Australia). A cell line expressing cytoplasmic hen egg ovalbumin (cOVA) lacking the sequence for cell surface trafficking (17, 53) was prepared as a control (CTRL). Lentivirus stocks to be used for subsequent LA-4 cell transduction were generated in 293T cells following transfection of (i) pMDL (gag and pol), (ii) pRSV-REV packaging plasmids, (iii) pMD2G.VSVg envelope plasmid, and (iv) pFUV1-mCherry transfer plasmid as described previously (52, 54). Lentivirus-transduced LA-4 cells were sorted based on mCherry-positive cells using a FACSAria III instrument (BD Biosciences, NJ, USA).
Site-directed mutagenesis to generate mMx1 mutants.
Site-directed mutagenesis was performed using the pTREtight-mMx1 plasmid and Pfu DNA polymerase (Agilent Technologies) according to the manufacturer’s instructions. Three different mMx1 mutants were generated using degenerate primers for mMx1(K49A) (forward, 5′-CCAGAGTTCAGGGGCGAGCTCTGTTCTG-3′; reverse, 5′-CAGAACAGAGCTCGCCCCTGAACTCTGG-3′), mMx1(T69A) (forward, 5′-GTGGTATTGTCGCCAGATGCCCTC-3′; reverse, 5′-GAGGGCATCTGGCGACAATACCACTG-3′), and mMx1(R614E) (forward, 5′-GAAGTTCCTGAAAGAGCGGCTTTTAAGGC-3′; reverse, 5′-GCCTTAAAAGCCGCTCTTTCAGGAACTTC-3′). After confirmation of mutagenesis by sequencing (AGRF), inserts were subcloned into pFUV1-mCherry to generate lentivirus stocks.
Generation of 293T cell lines with stable constitutive expression of Mx proteins.
Genes encoding mMx1, mMx2, or mutants of mMx1 with an N-terminal FLAG tag were cloned into pcDNA3.1 expression plasmids (Invitrogen). cOVA lacking an N-terminal FLAG tag was also cloned into the same vector to generate a cell line expressing an irrelevant cytoplasmic protein as a control (CTRL) (53). 293T cells were transfected with 1 μg of pcDNA3.1 containing FLAG-tagged Mx (or CTRL) using Lipofectamine 2000 (Life Technologies, CA, USA), cultured at 37°C for 2 days, before culture in medium supplemented with 100 μg/mL hygromycin B (InvivoGen). After 12 to 14 days, mCherry-positive cells were sorted twice to enrich for mCherry-positive cells before use in subsequent experiments.
Detection of FLAG-tagged proteins in LA-4 cell lines. (i) Flow cytometry.
FLAG-tagged mMx1 and mMx2 proteins were detected in LA-4 cells (cultured for 24 h in the presence or absence of 1 μg/mL DOX) by intracellular staining with allophycocyanin (APC)-conjugated anti-FLAG MAb (clone L5; Biolegend) in conjunction with fixable viability dye eFluor 780 (eBioscience) before flow cytometric analysis.
(ii) Western blot.
FLAG-tagged mMx1 and mMx2 proteins were detected in LA-4 cells (cultured for 24 h in the presence or absence of 1 μg/mL DOX) by Western blot as described previously (55), using anti-FLAG-APC monoclonal antibody (clone L5; Bioloegend). Cellular β-actin was monitored to ensure equivalent protein loading of all samples using a mouse monoclonal antibody (clone sc-47778; Santa Cruz, CA, USA) followed by chicken anti-mouse IgG conjugated to Alexa Fluor 488 (Invitrogen). Bound antibodies were detected after scanning for fluorescence using Molecular Imager PharosFX Plus system (Bio-Rad) and analyzed using Fiji ImageJ software.
(iii) Confocal microscopy.
FLAG-tagged mMx1 and mMx2 proteins were visualized in LA-4 cells by confocal microscopy. Cells were seeded into 8-well chamber slides (Lab-Tek, Nunc, USA) (cultured for 24 h in the presence or absence of 1 μg/mL DOX), and cell surface staining was performed prior to fixation using biotinylated Maackia amurensis lectin II (MAL II) (Vector Laboratories, CA, USA), followed by Alexa Fluor 488-streptavidin (Molecular Probes) in cold 0.05 M Tris-HCl–0.15 M NaCl buffer (pH 7.2) containing 10 mM CaCl2 and 1% bovine serum albumin (BSA). Cells were then fixed in phosphate-buffered saline (PBS) containing 4% (vol/vol) paraformaldehyde (PFA) before permeabilization in PBS containing 5% (wt/vol) bovine serum albumin, 5% (vol/vol) FCS, and 0.1% (vol/vol) Triton X-100. FLAG was detected by using anti-FLAG antibody conjugated to Alexa Fluor 647 (L5; BioLegend). Prior to visualization, cells were counterstained with Vectashield antifade mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector, Burlingame, CA, USA). Images were acquired with a Zeiss LSM780 microscope and were processed using Fiji ImageJ software.
Quantitation of endogenous mMx1 gene expression in murine cells.
Primary lung fibroblasts were generated from mice as previously described (56). Induction of endogenous mMx1 gene expression in primary mouse lung fibroblasts was determined by qRT-PCR. Lung fibroblasts (2 × 105 cells per well) seeded into 12-well tissue culture plates (Corning, NY) were cultured overnight and then treated with 1,000 U/mL of recombinant mouse IFN-α (Miltenyi Biotec, Germany) or incubated with IAV (HKx31) or HSV-1 (KOS) at the MOI indicated. Mock-treated cells were included for comparison. Cellular RNA was extracted 2, 6, 12, and 24 h after type I IFN treatment/virus infection using the RNeasy minikit (Qiagen, Germany) according to the manufacturer’s instructions. RNA purity and quantity were determined by Nanodrop (Thermo Scientific, USA), and extracted RNA was treated with DNase I (Sigma-Aldrich). Next, a total of 80 ng RNA was reverse transcribed into cDNA using the SensiFAST cDNA synthesis kit (Bioline, United Kingdom). cDNA (10 ng) was used for qPCR with the SensiFAST SYBR Lo-ROX kit (Bioline) and primers for mMx (forward, 5′-TGTACCCCAGCAAAACATCA-3′; reverse, 5′-TTGGAAGCGCTAAAGTGGAA-3′) and housekeeping genes, including those coding for mouse GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (forward, 5′-CCAGGTTGTCTCCTGCGACTT-3′; reverse, 5′-CCTGTTGCTGTAGCCGTATTCA-3′), mouse TBP (TATA box binding protein) (forward, 5′-CCCCACAACTCTTCCATTCT-3′; reverse, 5′-GCAGGAGTGATAGGGGTCAT-3′), and mouse HPRT1 (hypoxanthine guanine phosphoribosyl transferase 1) (forward, 5′-CCTAAGATGAGCGCAAGTTGAA-3′; reverse, 5′-CCACAGGACTAGAACACCTGCTAA-3′). Data were acquired using a QuantStudio 7 Flex RT-PCR system (Applied Biosystems) with the following cycling conditions: denaturation at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 5 s, annealing at 60°C for 10 s, and extension at 72°C for 20 s. The relative gene expression of mMx1 was determined compared to those of the three housekeeping genes. The fold change in mMx1 gene induction relative to mock conditions was calculated by averaging the fold change obtained relative to each of the housekeeping genes. Relative expression levels for each sample were determined using the 2−ΔΔCT method (31).
Virus infection of cells.
For infection of different cell types with viruses of interest, cells were seeded into 12-well tissue culture plates (Corning, NY) and cultured overnight at 37°C with 5% (vol/vol) CO2 in a humidified incubator (unless stated otherwise). Duplicate wells were washed and detached for viable cell counts to allow for calculation of the multiplicity of infection (MOI) of each virus. For infections, cell monolayers were washed once with serum-free medium and incubated with 1 mL per well of virus diluted in serum-free medium. After 60 min at 37°C, cells were washed three times and cultured at 37°C for (i) 0 to 18 h postinfection (hpi) prior to determining the percentage of virus-infected cells by flow cytometry (see below) or (ii) 24 to 48 hpi to measure release of infectious virus (see below) and levels of soluble mediators in cell-free supernatants (3.5.3). For virus growth experiments, cell-free supernatants harvested 24 to 48 hpi were clarified by centrifugation (2,000 × g for 5 min) and stored at –80°C prior to titration by plaque assay or VS assay, as described below.
Plaque assays and ViroSpot assays to determine titers of infectious virus.
Titers of infectious IAV were determined by standard plaque assay (57) or by ViroSpot (VS) immunostain assay (52) on MDCK cells. Titers of infectious HSV-1 were determined by standard plaque assay on Vero cells (36). For MCMV, viral titers were determined using a plaque assay on M210B4 cells as described previously (49). For plaque assays, wells containing 10 to 100 plaques were counted 3 to 4 days after the assay was performed, and titers of infectious virus were expressed as PFU/mL of original sample. For VS assays, wells containing 10 to 100 spots were counted at titers of infectious virus expressed as VS per milliliter of original sample. The lower limit of detection (LLOD) was set to 10 plaques/well and 10 spots/well of original sample for each assay.
Detection of virus-infected cells by flow cytometry.
Virus infection of cells was assessed using flow cytometry by staining intracellularly expressed viral proteins. After virus infection (at the indicated times), cells were detached, washed, and resuspended in 200 μL fluorescence-activated cell sorter (FACS) buffer before transfer to a U-bottom 96-well plate (Corning, NY). Cells were then stained with fixable viability dye eFluor 780 (eBioscience) and fixed in 2 to 4% (vol/vol) PFA, permeabilized with 0.5% (vol/vol) Triton X-100 in PBS for 10 min at room temperature, and then washed in permeabilization buffer (PBS containing 0.25% [vol/vol] Triton X-100, 1% [vol/vol] FCS, and 1 mM EDTA) prior to staining for intracellular expression of viral proteins. IAV-infected cells were stained with fluorescein isothiocyanate (FITC)-conjugated MAb specific for IAV nucleoprotein (Thermo Fisher Scientific) diluted in permeabilization buffer. Cells infected with HSV-1 were stained with mouse MAbs specific for HSV-1 ICP4 and ICP5 (VP5) (Abcam) followed by chicken anti-mouse IgG conjugated to Alexa Fluor 488 (Invitrogen). Cells infected with fluorescent HSV-1 were stained with fixable viability dye eFluor 780 (eBioscience) and then fixed in 2 to 4% (vol/vol) PFA. Cells were then washed and resuspended in FACS buffer for subsequent analysis by flow cytometry. Samples were acquired on a FACSCanto II (BD Biosciences) or a LSRFortessa flow cytometer (BD Bioscience) and analyzed using FlowJo analysis software version 10.7.1.
qPCR to quantitate HSV-1 genomic DNA.
To determine HSV-1 DNA levels, virus-infected cells were detached using trypsin versine. Total DNA was extracted using DNeasy Blood & Tissue kits (Qiagen, Germany) according to the manufacturer’s instructions. Total DNA (100 ng) was then amplified by real-time PCR using the QuantStudio 7 Flex real-time PCR system (Applied Biosystems, Foster City, CA, USA). PCRs were performed in a total volume of 10 μL containing TaqMan Fast Advanced master mix (Thermo Fisher Scientific, MA, USA), 100 ng DNA, primers (forward, 5′-TTGTCTCCTTCCGTGTTTCAGTTAGCC-3′; reverse, 5′-GGCTCCATACCGACGATCTGCG-3′ specific for the HSV-1 TK gene, and probe (5′-6-carboxyfluorescein [FAM]-CCATCTCCCGGGCAAACGTGC-MGBNFQ-3′). Amplification conditions were 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. To calculate viral copy number, a 10-point standard curve was generated using serial 10-fold dilutions of pGEMT-Easy TK qPCR plasmid (a kind gift from David Tscharke, Australian National University, Canberra), such that 4 μL contained 1010 to 100 copies of TK gene plasmid DNA. The copy number of HSV-1 DNA from each sample was then calculated using the standard curve. The LLOD for the assay was set to 2.
Flank infection of mice with HSV-1.
HSV-1 flank infection of mice was performed using a flank scarification method described previously (34–36, 58). All research complied with the University of Melbourne’s Animal Experimentation Ethics guidelines and policies in accordance with the NHMRC Australian code of practice for the care and use of animals for scientific purposes. Congenic C57BL/6 mice expressing a nonfunctional (B6.WT) or functional (B6.A2G-Mx1) mMx1 protein were derived from breeding of heterozygote littermates. Briefly, 6- to 10-week-old B6.WT and B6.A2G-Mx1 mice were anesthetized by intraperitoneal injection with a 1:1 ketamine–Xylazil-20 solution in PBS (10 μL/g body weight). The left flank was shaved and depilated, and a small area of skin (2 to 4 mm2) situated above the dorsal tip of the spleen was scarified using a MultiPro Dremel drill (Dremel, Racine, WI, USA) with a grindstone tip attachment (3.2 mm) held on the skin for 20 s. A 10-μL drop containing 106 PFU of HSV-1 (KOS) in PBS was applied to the abraded inoculation site, which was then rubbed in with a water-soaked cotton-tipped applicator, and mice were bandaged for 48 h after virus application. Mice were examined daily for lesion development, and photos were taken on 3, 5, 7, 9, and 11 dpi. Lesion severity was scored blind from photographs by three independent scorers using a scoring system based on that described by Wojtasiak et al. (34). Lesion severity (0 to 5) of each mouse was recorded according to the following scale: 0, no sign of vesicles; 1, ulcerated local vesicles, erythema, and/or inflammation; 2, ulcerated vesicles throughout the dermatome; 3, regions of severe ulceration and presence of large lesions; 4, severe ulcerations throughout the dermatome; 5, continuous ulceration over entire dermatome, appearance of vesicles in adjacent dermatomes, and/or contralateral spread.
At 5 dpi, mice were killed and skin and dorsal root ganglia (DRG) were removed as described previously (36). A 1- by 1-cm piece of skin was removed from the primary site of inoculation into 1 mL serum-free medium, and a 1- by 2-cm piece of skin was taken from the secondary site (lower flank between the excised primary site and the anterior midline) into 1 mL serum-free medium, with a 0.5-cm section of skin left intact between the two excised sites. Mice were then perfused with PBS through the left ventricle, and DRG innervating the infected dermatome (thoracic 8 [T8] to -12) were removed with the aid of a dissecting microscope. All DRG from one mouse were pooled into 1 mL of serum-free medium. Skin and DRG samples were then homogenized, and titers of infectious virus in clarified tissue samples were determined using standard plaque assays on Vero cells. In some experiments, mice were killed at 35 dpi after all skin lesions had resolved. Mice were perfused with PBS, and DRG (T8 to -12) were removed from each mouse, pooled into 1 mL of serum-free medium, and cultured at 37°C with 5% CO2 for 5 days to allow for reactivation of latent virus in infected DRG (37). After culture, DRG were homogenized, and titers of infectious HSV-1 in clarified supernatants were determined by plaque assays on Vero cells.
Data analysis.
Flow cytometry data were analyzed using FlowJo software version 10.7.1 (Becton Dickinson and Company, NJ, USA). Fluorescence microscopy and Western blot images were analyzed using ImageJ software version 2.1.0/1.53c. Graphs and statistical analysis (as indicated in the figure legends) were performed using GraphPad Prism version 9.2.0 (GraphPad Software, CA, USA). Statistical significance was determined by Student's t test or one-way analysis of variance (ANOVA) as indicated. Data are shown as means ± standard deviation (SD).
ACKNOWLEDGMENTS
We acknowledge the Melbourne Cytometry Platform (Peter Doherty Institute node) for providing flow cytometry services. Fluorescent images were taken by confocal microscopes located at the Biological Optical Microscopy Platform, The University of Melbourne. We also thank Marco Herold at the Walter and Eliza Hall Institute for providing the lentiviral constructs.
This study was supported by Project Grant APP1143154 from The National Health and Medical Research Council (NHMRC) of Australia. The Melbourne WHO Collaborating Centre for Reference and Research on Influenza is supported by the Australian Government Department of Health and Ageing.
Contributor Information
Patrick C. Reading, Email: preading@unimelb.edu.au.
Felicia Goodrum, University of Arizona.
REFERENCES
- 1.Busnadiego I, Kane M, Rihn SJ, Preugschas HF, Hughes J, Blanco-Melo D, Strouvelle VP, Zang TM, Willett BJ, Boutell C, Bieniasz PD, Wilson SJ. 2014. Host and viral determinants of Mx2 antiretroviral activity. J Virol 88:7738–7752. 10.1128/JVI.00214-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Verhelst J, Hulpiau P, Saelens X. 2013. Mx proteins: antiviral gatekeepers that restrain the uninvited. Microbiol Mol Biol Rev 77:551–566. 10.1128/MMBR.00024-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin HK, Takada A, Kon Y, Haller O, Watanabe T. 1999. Identification of the murine Mx2 gene: interferon-induced expression of the Mx2 protein from the feral mouse gene confers resistance to vesicular stomatitis virus. J Virol 73:4925–4930. 10.1128/JVI.73.6.4925-4930.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zürcher T, Pavlovic J, Staeheli P. 1992. Mouse Mx2 protein inhibits vesicular stomatitis virus but not influenza virus. Virology 187:796–800. 10.1016/0042-6822(92)90481-4. [DOI] [PubMed] [Google Scholar]
- 5.Jin HK, Yoshimatsu K, Takada A, Ogino M, Asano A, Arikawa J, Watanabe T. 2001. Mouse Mx2 protein inhibits hantavirus but not influenza virus replication. Arch Virol 146:41–49. 10.1007/s007050170189. [DOI] [PubMed] [Google Scholar]
- 6.Schilling M, Bulli L, Weigang S, Graf L, Naumann S, Patzina C, Wagner V, Bauersfeld L, Goujon C, Hengel H, Halenius A, Ruzsics Z, Schaller T, Kochs G. 2018. Human MxB protein is a pan-herpesvirus restriction factor. J Virol 92:e01056-18. 10.1128/JVI.01056-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ku CC, Che XB, Reichelt M, Rajamani J, Schaap-Nutt A, Huang KJ, Sommer MH, Chen YS, Chen YY, Arvin AM. 2011. Herpes simplex virus-1 induces expression of a novel MxA isoform that enhances viral replication. Immunol Cell Biol 89:173–182. 10.1038/icb.2010.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaguva Vasudevan AA, Bähr A, Grothmann R, Singer A, Häussinger D, Zimmermann A, Münk C. 2018. MxB inhibits murine cytomegalovirus. Virology 522:158–167. 10.1016/j.virol.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 9.Lindenmann J, Lane CA, Hobson D. 1963. The resistance of A2G mice to myxoviruses. J Immunol 90:942–951. [PubMed] [Google Scholar]
- 10.Lindenmann J. 1964. Inheritance of resistance to influenza virus in mice. Proc Soc Exp Biol Med 116:506–509. 10.3181/00379727-116-29292. [DOI] [PubMed] [Google Scholar]
- 11.Staeheli P, Pravtcheva D, Lundin L, Acklin M, Ruddle F, Lindenmann J, Haller O. 1986. Interferon-regulated influenza virus resistance gene Mx is localized on mouse chromosome 16. J Virol 58:967–969. 10.1128/JVI.58.3.967-969.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Staeheli P, Grob R, Meier E, Sutcliffe JG, Haller O. 1988. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol Cell Biol 8:4518–4523. 10.1128/mcb.8.10.4518-4523.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Staeheli P, Pitossi F, Pavlovic J. 1993. Mx proteins: GTPases with antiviral activity. Trends Cell Biol 3:268–272. 10.1016/0962-8924(93)90055-6. [DOI] [PubMed] [Google Scholar]
- 14.Pavlovic J, Schröder A, Blank A, Pitossi F, Staeheli P. 1993. Mx proteins: GTPases involved in the interferon-induced antiviral state. Ciba Found Symp 176:233–247. 10.1002/9780470514450.ch15. [DOI] [PubMed] [Google Scholar]
- 15.Verhelst J, Parthoens E, Schepens B, Fiers W, Saelens X. 2012. Interferon-inducible protein Mx1 inhibits influenza virus by interfering with functional viral ribonucleoprotein complex assembly. J Virol 86:13445–13455. 10.1128/JVI.01682-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shin DL, Hatesuer B, Bergmann S, Nedelko T, Schughart K. 2015. Protection from severe influenza virus infections in mice carrying the Mx1 influenza virus resistance gene strongly depends on genetic background. J Virol 89:9998–10009. 10.1128/JVI.01305-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Londrigan SL, Turville SG, Tate MD, Deng Y-M, Brooks AG, Reading PC. 2011. N-linked glycosylation facilitates sialic acid-independent attachment and entry of influenza A viruses into cells expressing DC-SIGN or L-SIGN. J Virol 85:2990–3000. 10.1128/JVI.01705-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dreiding P, Staeheli P, Haller O. 1985. Interferon-induced protein Mx accumulates in nuclei of mouse cells expressing resistance to influenza viruses. Virology 140:192–196. 10.1016/0042-6822(85)90460-x. [DOI] [PubMed] [Google Scholar]
- 19.Pitossi F, Blank A, Schroder A, Schwarz A, Hussi P, Schwemmle M, Pavlovic J, Staeheli P. 1993. A functional GTP-binding motif is necessary for antiviral activity of Mx proteins. J Virol 67:6726–6732. 10.1128/JVI.67.11.6726-6732.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zurcher T, Pavlovic J, Staeheli P. 1992. Nuclear localization of mouse Mx1 protein is necessary for inhibition of influenza virus. J Virol 66:5059–5066. 10.1128/JVI.66.8.5059-5066.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, Wu Y, Wu XD, Zhou J, Liang XD, Baloch AS, Qiu YF, Gao S, Zhou B. 2020. The R614E mutation of mouse Mx1 protein contributes to the novel antiviral activity against classical swine fever virus. Vet Microbiol 243:108621. 10.1016/j.vetmic.2020.108621. [DOI] [PubMed] [Google Scholar]
- 22.Dou D, Revol R, Östbye H, Wang H, Daniels R. 2018. Influenza A virus cell entry, replication, virion assembly and movement. Front Microbiol 9:1581. 10.3389/fimmu.2018.01581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weller SK, Coen DM. 2012. Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb Perspect Biol 4:a013011. 10.1101/cshperspect.a013011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Honess RW, Roizman B. 1974. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J Virol 14:8–19. 10.1128/JVI.14.1.8-19.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Honess RW, Roizman B. 1975. Regulation of herpesvirus macromolecular synthesis: sequential transition of polypeptide synthesis requires functional viral polypeptides. Proc Natl Acad Sci USA 72:1276–1280. 10.1073/pnas.72.4.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramachandran S, Knickelbein JE, Ferko C, Hendricks RL, Kinchington PR. 2008. Development and pathogenic evaluation of recombinant herpes simplex virus type 1 expressing two fluorescent reporter genes from different lytic promoters. Virology 378:254–264. 10.1016/j.virol.2008.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mueller SN, Jones CM, Chen W, Kawaoka Y, Castrucci MR, Heath WR, Carbone FR. 2003. The early expression of glycoprotein B from herpes simplex virus can be detected by antigen-specific CD8+ T cells. J Virol 77:2445–2451. 10.1128/jvi.77.4.2445-2451.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shapira M, Homa FL, Glorioso JC, Levine M. 1987. Regulation of the herpes simplex virus type 1 late (gamma 2) glycoprotein C gene: sequences between base pairs −34 to +29 control transient expression and responsiveness to transactivation by the products of the immediate early (alpha) 4 and 0 genes. Nucleic Acids Res 15:3097–3111. 10.1093/nar/15.7.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Overby LR, Robishaw EE, Schleicher JB, Rueter A, Shipkowitz NL, Mao JC-H. 1974. Inhibition of herpes simplex virus replication by phosphonoacetic acid. Antimicrob Agents Chemother 6:360–365. 10.1128/AAC.6.3.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hug H, Costas M, Staeheli P, Aebi M, Weissmann C. 1988. Organization of the murine Mx gene and characterization of its interferon- and virus-inducible promoter. Mol Cell Biol 8:3065–3079. 10.1128/mcb.8.8.3065-3079.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Haller O, Staeheli P, Kochs G. 2007. Interferon-induced Mx proteins in antiviral host defense. Biochimie 89:812–818. 10.1016/j.biochi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 33.Simmons A, Nash AA. 1984. Zosteriform spread of herpes simplex virus as a model of recrudescence and its use to investigate the role of immune cells in prevention of recurrent disease. J Virol 52:816–821. 10.1128/JVI.52.3.816-821.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wojtasiak M, Pickett DL, Tate MD, Bedoui S, Job ER, Whitney PG, Brooks AG, Reading PC. 2010. Gr-1+ cells, but not neutrophils, limit virus replication and lesion development following flank infection of mice with herpes simplex virus type-1. Virology 407:143–151. 10.1016/j.virol.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 35.Wakim LM, Jones CM, Gebhardt T, Preston CM, Carbone FR. 2008. CD8(+) T-cell attenuation of cutaneous herpes simplex virus infection reduces the average viral copy number of the ensuing latent infection. Immunol Cell Biol 86:666–675. 10.1038/icb.2008.47. [DOI] [PubMed] [Google Scholar]
- 36.van Lint A, Ayers M, Brooks AG, Coles RM, Heath WR, Carbone FR. 2004. Herpes simplex virus-specific CD8+ T cells can clear established lytic infections from skin and nerves and can partially limit the early spread of virus after cutaneous inoculation. J Immunol 172:392–397. 10.4049/jimmunol.172.1.392. [DOI] [PubMed] [Google Scholar]
- 37.Li L, Li Z, Li X, Wang E, Lang F, Xia Y, Fraser NW, Gao F, Zhou J. 2016. Reactivation of HSV-1 following explant of tree shrew brain. J Neurovirol 22:293–306. 10.1007/s13365-015-0393-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pavlovic J, Zurcher T, Haller O, Staeheli P. 1990. Resistance to influenza virus and vesicular stomatitis virus conferred by expression of human MxA protein. J Virol 64:3370–3375. 10.1128/JVI.64.7.3370-3375.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crameri M, Bauer M, Caduff N, Walker R, Steiner F, Franzoso FD, Gujer C, Boucke K, Kucera T, Zbinden A, Münz C, Fraefel C, Greber UF, Pavlovic J. 2018. MxB is an interferon-induced restriction factor of human herpesviruses. Nat Commun 9:1980. 10.1038/s41467-018-04379-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goujon C, Moncorgé O, Bauby H, Doyle T, Ward CC, Schaller T, Hué S, Barclay WS, Schulz R, Malim MH. 2013. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502:559–562. 10.1038/nature12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pavlovic J, Haller O, Staeheli P. 1992. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J Virol 66:2564–2569. 10.1128/JVI.66.4.2564-2569.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiao H, Killip MJ, Staeheli P, Randall RE, Jackson D. 2013. The human interferon-induced MxA protein inhibits early stages of influenza A virus infection by retaining the incoming viral genome in the cytoplasm. J Virol 87:13053–13058. 10.1128/JVI.02220-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Salomon R, Staeheli P, Kochs G, Yen HL, Franks J, Rehg JE, Webster RG, Hoffmann E. 2007. Mx1 gene protects mice against the highly lethal human H5N1 influenza virus. Cell Cycle 6:2417–2421. 10.4161/cc.6.19.4779. [DOI] [PubMed] [Google Scholar]
- 44.Tumpey TM, Szretter KJ, Van Hoeven N, Katz JM, Kochs G, Haller O, García-Sastre A, Staeheli P. 2007. The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. J Virol 81:10818–10821. 10.1128/JVI.01116-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spitaels J, Van Hoecke L, Roose K, Kochs G, Saelens X. 2019. Mx1 in hematopoietic cells protects against Thogoto virus infection. J Virol 93:e00193-19. 10.1128/JVI.00193-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horisberger MA, De Staritzky K. 1989. Expression and stability of the Mx protein in different tissues of mice, in response to interferon inducers or to influenza virus infection. J Interferon Res 9:583–590. 10.1089/jir.1989.9.583. [DOI] [PubMed] [Google Scholar]
- 47.Catez F, Picard C, Held K, Gross S, Rousseau A, Theil D, Sawtell N, Labetoulle M, Lomonte P. 2012. HSV-1 genome subnuclear positioning and associations with host-cell PML-NBs and centromeres regulate LAT locus transcription during latency in neurons. PLoS Pathog 8:e1002852. 10.1371/journal.ppat.1002852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rinaldi A, Marshall KR, Preston CM. 1999. A non-cytotoxic herpes simplex virus vector which expresses Cre recombinase directs efficient site specific recombination. Virus Res 65:11–20. 10.1016/s0168-1702(99)00102-1. [DOI] [PubMed] [Google Scholar]
- 49.Schuster IS, Wikstrom ME, Brizard G, Coudert JD, Estcourt MJ, Manzur M, O'Reilly LA, Smyth MJ, Trapani JA, Hill GR, Andoniou CE, Degli-Esposti MA. 2014. TRAIL+ NK cells control CD4+ T cell responses during chronic viral infection to limit autoimmunity. Immunity 41:646–656. 10.1016/j.immuni.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 50.Anders EM, Hartley CA, Jackson DC. 1990. Bovine and mouse serum beta inhibitors of influenza A viruses are mannose-binding lectins. Proc Natl Acad Sci USA 87:4485–4489. 10.1073/pnas.87.12.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herold MJ, van den Brandt J, Seibler J, Reichardt HM. 2008. Inducible and reversible gene silencing by stable integration of an shRNA-encoding lentivirus in transgenic rats. Proc Natl Acad Sci USA 105:18507–18512. 10.1073/pnas.0806213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Villalón-Letelier F, Brooks AG, Londrigan SL, Reading PC. 2021. MARCH8 restricts influenza A virus infectivity but does not downregulate viral glycoprotein expression at the surface of infected cells. mBio 12:e01484-21. 10.1128/mBio.01484-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boyle JS, Koniaras C, Lew AM. 1997. Influence of cellular location of expressed antigen on the efficacy of DNA vaccination: cytotoxic T lymphocyte and antibody responses are suboptimal when antigen is cytoplasmic after intramuscular DNA immunization. Int Immunol 9:1897–1906. 10.1093/intimm/9.12.1897. [DOI] [PubMed] [Google Scholar]
- 54.Meischel T, Fritzlar S, Villalon-Letelier F, Tessema MB, Brooks AG, Reading PC, Londrigan SL. 2021. IFITM proteins that restrict the early stages of respiratory virus infection do not influence late-stage replication. J Virol 95:e00837-21. 10.1128/JVI.00837-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Londrigan SL, Short KR, Ma J, Gillespie L, Rockman SP, Brooks AG, Reading PC. 2015. Infection of mouse macrophages by seasonal influenza viruses can be restricted at the level of virus entry and at a late stage in the virus life cycle. J Virol 89:12319–12329. 10.1128/JVI.01455-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Edelman BL, Redente EF. 2018. Isolation and characterization of mouse fibroblasts. Methods Mol Biol 1809:59–67. 10.1007/978-1-4939-8570-8_5. [DOI] [PubMed] [Google Scholar]
- 57.Anders EM, Hartley CA, Reading PC, Ezekowitz RA. 1994. Complement-dependent neutralization of influenza virus by a serum mannose-binding lectin. J Gen Virol 75:615–622. 10.1099/0022-1317-75-3-615. [DOI] [PubMed] [Google Scholar]
- 58.Whitney PG, Makhlouf C, MacLeod B, Ma JZ, Gressier E, Greyer M, Hochheiser K, Bachem A, Zaid A, Voehringer D, Heath WR, Wagle MV, Parish I, Russell TA, Smith SA, Tscharke DC, Gebhardt T, Bedoui S, Jung JU. 2018. Effective priming of herpes simplex virus-specific CD8+ T cells in vivo does not require infected dendritic cells. J Virol 92:e01508-17. 10.1128/JVI.01508-17. [DOI] [PMC free article] [PubMed] [Google Scholar]