

Abstract
Development of targeted anticancer modalities has prompted a new era in cancer treatment that is notably different from the age of radical surgery and highly toxic chemotherapy. Behind each effective compound is a rich and complex history from first identification of chemical matter, detailed optimization, and mechanistic investigations, ultimately leading to exciting molecules for drug development. Herein we review the history and on-going journey of one such anticancer scaffold, the 3-(4-hydroxyphenyl)indoline-2-ones. With humble beginnings in 19th century Bavaria, we review this scaffold's synthetic history and anticancer optimization, including its recent demonstration of tumor eradication of drug-resistant, estrogen receptor-positive breast cancer. Compounds containing the 3-(4-hydroxyphenyl)indoline-2-one pharmacophore are emerging as intriguing candidates for the treatment of cancer.
Some compounds possessing the 3-(4-hydroxyphenyl)indoline-2-one pharmacophore (ErSO, ErSO-DFP, and others) have significant antitumor activity in preclinical models and induce cancer cell death via an intriguing mode-of-action.
1. Introduction
While biologics (e.g. antibodies, CAR T cells) are beginning to populate the anticancer arsenal with impressive clinical outcomes against traditionally recalcitrant disease, small molecule drugs continue to be the cornerstone of anticancer regimens.1 Beyond their translational potential, molecular mechanisms of small molecules continue to expand the spectrum of possibilities,2 exemplified by the rise of molecular glues,3 monovalent/divalent degraders,4,5 and other unanticipated modes of action.6 Iterative synthesis and biological evaluation of diverse compounds enables discovery of intriguing leads for further development.
Targeted anticancer therapy often exploits the ‘addiction’ of cancer cells to oncogenic pathways or other specific vulnerabilities (e.g. mutant-selective or synthetic lethality strategies).7 Often times, interesting anticancer compounds that warrant further exploration have strong differential activity between different cancer cell lines, i.e. potent activity against only a subset of cell lines when screened across a large panel. Typically this differential activity is the result of a drug's exploitation of a specific translocation,8,9 mutation,10 or protein overexpression.11 In contrast, drugs that have minimal selectivity between cancer cell lines in culture typically have narrow therapeutic indices in patients, for example traditional chemotherapeutics (paclitaxel, doxorubicin, gemcitabine and others) as well as more modern drugs that target essential process in all cells (MEK1/2 inhibitors, CDK4/6 inhibitors). Truly selective compounds typically have superior therapeutic indices and thus are better positioned for translation to the clinic.7
Similar pharmacophores – the core scaffold necessary for the biological effect of a small molecule – can be found across a variety of biologically active compounds. Regardless of the exact reason, these ‘privileged’ scaffolds are highly pervasive within chemical libraries and drug leads.12 One such privileged scaffold in drug discovery is the 3,3-disubstituted oxindole core (Scheme 1), a subfamily of the indoline heterocycle that is seen in a wide range of natural products and drugs.13 Within the general 3,3-disubstituted oxindole family, 3-(4-hydroxyphenyl)indoline-2-ones (Scheme 1) specifically denote oxindoles substituted in the 3-postion with a phenol (4-hydroxyphenyl). Herein, we focus solely on the 3-(4-hydroxyphenyl)indoline-2-ones as a pharmacophore found in anticancer small molecules. We review the emergence of this core scaffold, from foundational discoveries, to provocative anticancer activity, through competing mechanistic hypotheses, and finally to synthetic advances that ultimately allow access to a wide-variety of 3-(4-hydroxyphenyl)indoline-2-ones, leading to compounds with impressive antitumor activity that are advancing for selective treatment of cancer.
Scheme 1. Generalized chemical structure for 3,3-disubstituted oxindoles and 3-(4-hydroxyphenyl)indoline-2-ones.

2. Oxyphenisatin: origins and anticancer activity
The founding member of the 3-(4-hydroxyphenyl)indoline-2-one compound class is the drug oxyphenisatin (Scheme 2; also called oxyphenisatine, phenolisatin; CAS#: 125-13-3). Oxyphenisatin and its acetylated prodrug form, oxyphenisatin acetate (Scheme 2; CAS#: 115-33-3), have potent cathartic properties and thus were active ingredients in a variety of marketed laxative drugs (e.g. Herbex, Isacen).14 Until its withdrawal from the clinic in 1971 due to rare liver toxicities, oxyphenisatin was a mainstay drug given to numerous patients worldwide.14 In this section, we briefly review the initial synthesis of oxyphenisatin, its use as a laxative, and highlight early signs of its anticancer efficacy.
Scheme 2. Chemical structure of oxyphenisatin and its pro-drug, oxyphenisatin acetate.

2.1. Adolf von Baeyer and oxyphenisatin
Well-known for his many remarkable achievements in organic chemistry, Adolf von Baeyer was awarded the Nobel Prize in Chemistry in 1905 for his work on organic dyes and hydroaromatic compounds. The vast portfolio of von Baeyer's work has been comprehensively covered elsewhere,15,16 and his pioneering contributions to the discipline of organic chemistry cannot be overstated. Of specific merit, Fruton17 describes von Baeyer's legacy beyond that of an excellent experimentalist, but as a mentor to many budding organic chemists who we now know as pioneers in their own right: Emil Fischer (Nobel Prize in Chemistry 1902), Heinrich Otto Wieland (Nobel Prize in Chemistry 1927), Walter Dieckmann (Dieckmann condensation), Victor Villiger (Baeyer–Villiger oxidation), Kurt Meyer (Meyer–Schuster rearrangement), and many other notables.15,16
In 1885 during his time at the University of Munich, von Baeyer described the synthesis of a variety of oxindoles (oxindol in older nomenclature).18 Through the reaction of isatin and excess phenol in the presence of sulfuric acid, ‘phenolisatin’ was isolated and its structure (correctly) proposed (Scheme 3).18 This is the first report of oxyphenisatin's synthesis and this facile approach of accessing 3,3-bis(4-hydroxyphenyl)indoline-2-ones via acid-promoted Friedel–Crafts reaction remains a major synthetic route for accessing such core scaffolds today.
Scheme 3. Synthesis of oxyphenisatin as reported by von Baeyer (structure on the right is reproduced from original report in 1885).18.

2.2. Phenolphthalein and oxyphenisatin: clinically used laxatives
Even though it is widely reported as a laxative, the exact historical discovery of oxyphenisatin's cathartic activity is challenging to precisely track down. The story, however, appears to begin at the turn of the 20th century at the University of Budapest, where Vamossy reported attempts to use structurally-related phenolphthalein (Scheme 4) as a pH indicator to identify poor quality, imported white wine.19 Of note, von Baeyer is also credited with the first synthesis and use of phenolphthalein in 1871.20,21 Astonishingly, Vamossy was so convinced by the excellent oral tolerability of phenolphthalein in rabbits that he and a colleague decided to test phenolphthalein on themselves.19 They tested doses as high as 1.5 g of phenolphthalein, but 150–200 mg were sufficient for “reichliche” (translation: copious) laxative effects.19 Ultimately, Vamossy concluded that this cathartic effect only occurred with compound concentrations well-above those needed for use in wines and likely a person would suffer from the effects of excess alcohol prior to any laxative effects.19
Scheme 4. Chemical structure of phenolphthalein.

Interestingly, this unusual report of phenolphthalein spurred sufficient interest in the United States, where Hungarian pharmacist Max Kiss saw an opportunity to establish a new laxative alternative to castor oil.22 In 1906, Kiss marketed a chocolate containing phenolphthalein called Ex-Lax.22 Phenolphthalein was later shown to have carcinogenic activity.23 Accordingly, in 1997 Ex-Lax was reformulated without phenolphthalein (replaced by sennoside),24 followed by the FDA officially banning phenolphthalein in 1999.25
Scientists at F. Hoffman-La Roche in the early 20th century were interested in further building on the discovery of phenolphthalein as a marketed laxative.26 In 1925, M. Guggenheim disclosed his work with oxyphenisatin (noting its similar chemical structure to phenolphthalein) and its pro-drug, oxyphenisatin acetate (he called “Isacen”).27 The exact molecular mechanism for oxyphenisatin's laxative activity is still unknown. However, it is clearly distinct from cAMP-modulating laxatives28 and oxyphenisatin leads to rapid epithelial permeability in vivo.29,30
Remarkably (in what seems to be a common thread in this history), Guggenheim dosed himself with 5 mg tablets of oxyphenisatin acetate two to four times daily to explore its laxative activity and observed no “untoward results”.31 More controlled human clinical testing of oxyphenisatin acetate was conducted in Geneva with 300 patients taking a variety of doses (5–40 mg).32 An additional unknown number of patients were given oxyphenisatin acetate after the nursing staff “employé couramment” (translation: widely used) oxyphenisatin acetate because they were observing promising results during the trial.32 Further clinical testing of oxyphenisatin acetate was conducted with private individuals and patients at the Home of the Daughters of Jacob (nursing home) in New York in 1926.31 The ultimate conclusion was that oxyphenisatin acetate was an excellent alternative to phenolphthalein and recommended for clinical use.26,33 With a modern-day lens, these reports paint an eye-opening picture of therapeutic development and clinical testing of the past.
In the late 1960's and early 1970's, sporadic reports of jaundice and chronic liver disease resulting from the use of oxyphenisatin and oxyphenisatin acetate-containing laxatives began amassing; colloquially referred to as “laxative hepatitis”.14,34–40 With this growing body of evidence coupled with the minimal tolerance for laxatives with major chronic side effects, oxyphenisatin and oxyphenisatin acetate were withdrawn from the clinic.41,42 However, given the sheer number of patients treated with these drugs, this liver toxicity is likely a rare hypersensitivity response to the drug, so uncommon that oxyphenisatin (and its acetylated derivative) was in wide use for over four decades prior to the discovery of this hepatic toxicity.14
2.3. Prunes and oxyphenisatin
Oxyphenisatin is often referred to as a compound of synthetic origin. However, there is a report that it may be a natural product. In 1951, Baum et al. identified a ‘diphenyl isatin’ in chloroform extracts from California prunes using a qualitative read out.43 These authors then showed that this isolated ‘diphenyl isatin’ had similar cathartic properties to oxyphenisatin acetate in an animal model.43 This report provided initial evidence for a possible causative relationship between oxyphenisatin and the known laxative effects of prunes.44 However, this claim has not been independently validated and thus there is a need for more concrete, quantitative chemical studies before this connection can truly be made.45
2.4. Oxyphenisatin: anticancer activity
Although overlooked by many subsequent publications, the first report of oxyphenisatin's antiproliferative effects on transformed cells was published in Arzneimittelforschung in 1981.46 Halperin and coworkers subsequently reported rapid Ca2+ homeostasis changes, increased phosphorylation of eIF2α, and growth inhibition of NIH 3T3 cells when treated with 3,3-diaryl-indoline-2-ones (Scheme 5) and structurally related compounds in 2004.47 In 2007 using standard methods for determining anticancer activity in culture, Uddin et al. reported the antiproliferative effects of oxyphenisatin, showing potent activity against MDA-MB-468 breast cancer cells, with minimal activity against MDA-MB-231 breast cancer cells.48
Scheme 5. Generalized chemical structure of 3,3-diaryl-indoline-2-ones.

Newton and coworkers further evaluated oxyphenisatin anticancer activity with studies in 2013 using its pro-drug, oxyphenisatin acetate.49 In this study, oxyphenisatin acetate was shown to have potent activity against estrogen receptor alpha (ERα) positive cancer cell lines (MCF-7, T47D) with modest activity against MDA-MB-468 and Hs578t breast cancer cells (classically ERα-negative cell lines), and again no activity against MDA-MB-231 cells (ERα-negative cell line).49 Newton et al. pursued a variety of mechanistic experiments including showing profound phosphorylation of eIF2α and other endoplasmic reticulum stress markers.49 While an exact molecular target was not elucidated nor suggested, the effects of oxyphenisatin acetate on mitochondrial function, endoplasmic reticulum stress induction, and the fundamental anticancer phenotype were clear. Important for discussions below, a possible ERα-dependence for oxyphenisatin's anticancer activity was suggested in 2013 by these authors.49
3. Anticancer activity of 3,3-bis(4-hydroxy phenyl)indoline-2-ones
3.1. Synthesis of 3,3-bis(4-hydroxyphenyl)indoline-2-ones
For the purpose of moving beyond oxyphenisatin or simple derivatives thereof, isatin starting materials are well positioned to enable rapid construction 3,3-diaryl-indoline-2-ones by simple addition of the desired Friedel–Crafts reaction partner (e.g. phenol, toluene) and strong acid as evidenced by von Baeyer's earlier work (Scheme 3).18 Isatins are excellent starting points for complex molecule synthesis and have been reviewed extensively.50–54 Accessing a collection of isatins was well-established as early as the 1920s with synthetic methods like the Sandmeyer isatin synthesis (Scheme 6).55 A comprehensive Organic Synthesis procedure published by Carl Shipp “Speed” Marvel in 1925 further demonstrates the early ease associated with accessing initial isatin building blocks.56 There are a variety of modifications and variations to the Sandmeyer isatin synthesis that all use anilines as a shared starting point.50–54 With the importance of isatins as starting materials and final products for a variety of applications,53 more recent methods have been developed which utilize less harsh conditions than those employed by classical methods, which often use strong acids and elevated temperatures.57 One emerging method is the reaction of commercially available indoles with an iodine source and oxidant to rapidly access isatins (Scheme 6).58 There are numerous other methods for synthesizing isatins with a wide range of substrate scopes and starting materials (Scheme 6).50–54,57–62 Of course, in modern times many isatins are commercially available from a wide variety of vendors.
Scheme 6. Retrosynthetic disconnection to isatin is an ideal starting point for the synthesis of 3,3-diaryl-indoline-2-ones.

As noted by von Baeyer in 1885,18 some Friedel–Crafts nucleophiles, like benzene and others, are not competent substrates in forming 3,3-diaryl-indoline-2-one products when utilizing sulfuric acid as a strong acid, even in combination with other forcing conditions (e.g. elevated temperatures and reaction times). As such, a major advance in expanding the synthesis of 3,3-diaryl-indoline-2-ones was the Olah laboratory's use of triflic acid to generate ‘superelectrophilic’ species that rapidly react with desired Friedel–Crafts nucleophiles (Scheme 7).63 With this advance in synthetic methodology and others, there are now robust methods to generate a range of 3,3-diaryl-indoline-2-one derivatives that can be applied to exploring the anticancer activity of 3-(4-hydroxyphenyl)indoline-2-ones.
Scheme 7. Summary of reaction scope reported by Olah and coworkers.63 Ph: phenyl, TfOH: triflic acid (CF3SO3H), r.t.: room temperature.

3.2. Derivatives of oxyphenisatin
In 2007, Uddin and coworkers were exploring derivatives of oxyphenisatin, establishing key structure activity relationships (SAR) and foraging for variants with more potent anticancer activity.48 In a campaign to find anticancer small molecules, the authors first identified 6-chloro-3,3-bis(4-hydroxyphenyl)-7-methylindolin-2-one (later renamed TOP001,64Table 1) as an intriguing initial ‘hit’ molecule. Utilizing isatin synthesis followed by acid-catalyzed Friedel–Crafts reaction, a variety of derivatives were synthesized and evaluated for antiproliferative activity against MDA-MB-468/231 cells. Interesting compounds only exhibited activity against MDA-MB-468 (IC50 values < 0.150 μM) and were not active against MDA-MB-231 cells (IC50 > 3 μM); some examples are shown in Table 1, including potent compound 3f with a 7-trifluoromethyl substitution on the oxindole core.48
Selected examples of compounds reported by Uddin et al.48.

|

|

|
|
|---|---|---|---|
| TOP001 | Compound 3f | TOP216 | |
| MDA-MB-468 | 0.020 μM | 0.004 μM | 0.003 μM |
| MDA-MB-231 | >3 μM | >3 μM | >3 μM |
This study also demonstrated that the phenol was necessary for the anticancer activity of 3-(4-hydroxyphenyl)indoline-2-ones, and simple phenol bioisosteres (e.g. –NH2, –NHSO2CH3) were not productive substitutions;48 indeed, any derivatives lacking the 4-hydroxyphenyl (4-phenol) did not have potent activity.48 Complicating interpretation of this report is the lack of detailed biological experimental protocols, for example drug incubation times and assay read-outs were not well described. No suggestion of the underlying mechanism was discussed for these molecules, but authors noted that these molecules had a spectrum of activity similar to mTOR inhibitors.48 An important distinction for this review, all compounds described by Uddin et al. were ‘symmetric’ meaning that the 3,3-disubstitutions were the same, resulting in an achiral compound.
Later work further investigated the biological activities of TOP001 and TOP216 (named by scientists at TopoTarget).64 These authors once again showed that 3,3-bis(4-hydroxyphenyl)indoline-2-ones have potent, selective anticancer activity against a variety of cancer cell lines with a stark contrast between sensitive cancer cell lines (single to double digit nanomolar, nM, potencies) and insensitive cell lines (>1000 nM).64 These authors demonstrated that this cell culture data translated to antitumor activity in a variety of in vivo tumor models (e.g. MCF-7, PC-3, MDA-MB-468, MiaPaca, and A2780) in which TOP216 treatment (both oral and intravenously administered) induced robust antitumor effects.64
Again, a specific biological target was not discussed nor discovered in this follow-up report on TOP001/216.64 Rapid global protein synthesis inhibition, corresponding increases in phosphorylated eIF2α (p-eIF2α), and a variety of other mechanistic components were shown as key markers for their biological activity,64 similar to reported effects of oxyphenisatin acetate.49 A new observation was noted, namely that cellular amino acids levels decreased upon TOP216 treatment and this lower amino acid level was associated with an increase in phosphorylated AMP-activated protein kinase (AMPK). This new observation led the authors to argue that these compounds were dissimilar to traditional mTOR inhibitors (as proposed by Uddin48) in many regards and may simply “target amino acid uptake”;64 this speculation was not supported with comprehensive mechanistic experiments and the authors stated the need for more in-depth exploration of the causative mechanism and target for TOP216 and other compounds.
Interestingly, this report does not discuss the ERα correlation with TOP216's observed anticancer activity. Using modern information about these cell lines, TOP001/216 does have activity against MCF-7, T47D, and other ERα-positive cancer cell lines, and is inactive in some classically defined ERα-negative cell lines (e.g. MDA-MB-231) but is active in some other classically defined ERα-negative cell lines (e.g. MDA-MB-468, MDA-MB-453).64 This discordance in relation to reported ERα status and activity blurs the interpretation of the spectrum of activity of these 3,3-bis(4-hydroxyphenyl)indoline-2-ones. However, we now know that many of these reported ERα-negative cell lines actually do express low/extremely low levels of ERα (e.g. MDA-MB-468/453,65,66 SK-BR-3,67 U87MG,68,69 OVCAR-3 (ref. 70 and 71)). When looking at the in vivo tumor models reported in the work, TOP216 shows efficacy in models that all express ERα (MCF-7,72 PC-3,73,74 MDA-MB-468,65,66 MiaPaca,75 and A2780 (ref. 71 and 76)). While the correlation is not perfect and it is still an open area of research on what the exact causative molecular target is for TOP216 and related compounds, there is a possible ERα-dependence in the anticancer activity of these molecules that was not noted.
4. Anticipatory unfolded protein response, breast cancer, and the discovery of BHPI
The unfolded protein response (UPR) has been the subject of wide exploration and interest. In short, the classical UPR is a ‘reactive’ pathway by which cells respond to endoplasmic reticulum (EnR) stress, often initiated by the buildup of unfolded protein.77 The importance of UPR for cancer cells is well-known,78,79 and inhibiting the UPR in cancer has been an attempted anticancer strategy for some time. It is clear from these failed campaigns of trying to inhibit classical UPR machinery that a major problem is the lack of cancer-specific activity as many of these therapies suffer from poor toxicity profiles.80 In the opposite mode, pharmacological activation of the classical UPR yields similar results, with molecules like thapsigargin that do not selectively kill cancer cells and are generally toxic to all cells.81 To date, a cancer-selective approach to the UPR has not been accomplished clinically.
However, other studies (discussed below) have shown another mode in which UPR signaling is activated, not by unfolded protein, but rather upon mitogenic signals and importantly in the absence of underlying EnR stress. This non-classical UPR activation has been coined the anticipatory UPR (a-UPR) due to its apparent nature where cells activate UPR pathways in ‘anticipation’ of necessary future needs in protein folding associated with increased cellular growth. This non-classical mode was first suggested by Walter et al. in his landmark review on the UPR,79 and was initially observed in B cell immunoglobulin production.82–84 a-UPR's role in the context of cancer was first suggested by Shapiro and coworkers.85–87 In this section, we will discuss the current evidence for the a-UPR pathway in cancer cells, and how small molecules (hormones and others) activate this pathway leading to anticancer activity.
4.1. Estrogen, mitogenic hormones, and the a-UPR
There has been considerable exploration of the function and downstream signaling of mitogenic hormones, including estrogen (E2), androgen (DHT), epidermal growth factor (EGF), vascular endothelial growth factor (VEGF).87 Specifically, E2 binds estrogen receptors (most importantly estrogen receptor alpha, ERα) leading to a turn-on of an estrogen-associated transcriptional program.88 This classical description of E2-ERα nuclear activity has been the focus of most ERα-related research since Elwood Jensen's89 and Jack Gorski's90,91 initial discovery of ERα in the 1960's.88,92 However, there are numerous functions for ERα outside this canonical nuclear activity93–95 and there is a significant amount of ERα in the cytoplasm.96 Amazingly, new functions for ERα continue to be discovered, including the recently elucidated role of ERα as an RNA-binding protein.97,98
Another emerging non-nuclear role of ERα is its function in activating the a-UPR.80,87 In this manifold, E2 binding ERα exerts canonical nuclear actions, as well as non-canonical activation of a E2-ERα-Src-phospholipase C γ (PLCγ) leading to inositol triphosphate (IP3) production which binds inositol triphosphate receptors of the EnR, liberating calcium ions (Ca2+) from the EnR into the cytoplasm (Fig. 1).80,87,99 The downstream consequences of this rapid Ca2+ flux are still being investigated, but what has been observed in concurrence with this Ca2+ flux is the rapid turn-on of all three arms of the UPR and activation of the ATP-dependent sarcoplasmic/EnR calcium ATPase (SERCA) pump which depletes cellular ATP levels, increasing phosphorylation of AMPK. This activating hormone–hormone receptor–Src-PLCγ complex leading to a-UPR turn-on is likely conserved across multiple mitogenic hormones (E2, DHT, EGF and others).80,87
Fig. 1. Summary of a-UPR signalling pathway. For a review of a-UPR see: Shapiro, D. J.; et al. 2016.87 Red circles are calcium (Ca2+) ions. E2: estrogen/estradiol.

What are the ultimate cellular consequences of this complex pathway? Like many biology processes it depends on the degree of activation, meaning that the a-UPR can be both cytoprotective or cytotoxic depending on the duration and level of activation.80,87 The ability of E2 to activate the a-UPR is transitory and ultimately cytoprotective for breast cancers.85,100 In concordance with this idea, an a-UPR gene signature (consisting of genes encoding known components of the UPR pathway) is correlated with poorer breast cancer patient outcomes.85 Since the a-UPR is hormone receptor-dependent pathway and many cancer cells overexpress these receptors, Shapiro et al. has hypothesized that the a-UPR (unlike activation of traditional UPR) may be an excellent way of selectively inducing death of cancer cells.87
While the annotation and description of the ERα-dependent a-UPR is relatively new (first reported in 2015),85 the ability of high doses of E2 or other ‘super estrogens’ to activate cell death via a UPR-associated activity (as defined by an increase in a variety of genes known to impart UPR function) has been a well-explored breast cancer therapeutic approach. Interestingly in 1935, Alexander Haddow showed that carcinogenic levels of estrogens could induce tumor regression.101 This antitumor effect (termed the Haddow effect) was comprehensively shown to be an effective treatment of late-stage breast cancer patients in 1944.102 In line with this, diethylstilbestrol (DES) (a non-steroidal estrogen) was used to treat breast cancer until it was supplanted in the late 1970's by inhibition of ERα with tamoxifen.103 The move away from using estrogens to treat breast cancer was likely due to the superior tolerability profile of tamoxifen, and indeed inhibiting ERα signaling has been the flagship therapy for hormone-positive breast cancer ever since.104 However there is some historical data and more recent data that suggests an activation approach may be highly efficacious, albeit with significant estrogen-related toxicity.105,106 New-age estrogens, termed selective human estrogen receptor partial agonists (ShERPAs), are being developed to leverage this antitumor effect.107,108
While the high dose E2 therapeutic strategy was known from clinical reports, the underlying mechanism was poorly understood until 2011 when V. Craig Jordan's laboratory showed that E2 treatment leads to a toxic turn-on of a UPR gene signature.103 This link between E2 treatment and UPR turn-on could be the result of now understood a-UPR activity and/or related to IRE1α role as an ERα-target gene; this distinction has not been explored with differentiating assays such as Ca2+ flux or PLCγ activity assay. The combination of the a-UPR pathway annotation and the potent antitumor effects seen with activating the UPR with estrogens (or ShERPAs) implies that modulation of the a-UPR with a selective agonist may be able to finally produce a cancer cell specific-induction of the UPR and bring about a clinically relevant UPR activator.
4.2. BHPI: mechanistic implications for other 3,3-bis(4-hydroxyphenyl)indoline-2-ones
In 2015, Shapiro and coworkers reported the discovery of a small molecule, BHPI (Scheme 8, 3,3,-bis(4-hydroxyphenyl)-7-methylindoline-2-one), that led to hyperactivation of the a-UPR.86 In ERα-positive cancer cell lines, BHPI treatment led to upstream activation of PLCγ, increases in [IP3], increasing intracellular [Ca2+], followed by activation of all three arms of the UPR, global protein synthesis inhibition, as well as activation of SERCA pumps in the EnR that rapidly deplete cellular [ATP]. BHPI does not affect protein synthesis of ERα-negative MCF10A cells, but does inhibit protein synthesis in isogenic MCF10A cells stably expressing ERα, demonstrating BHPI's ERα-dependent a-UPR activation.86 This a-UPR activation cascade inhibits the growth of cancer cells, leading to the antiproliferative activity of BHPI.87
Scheme 8. Chemical structure of BHPI.
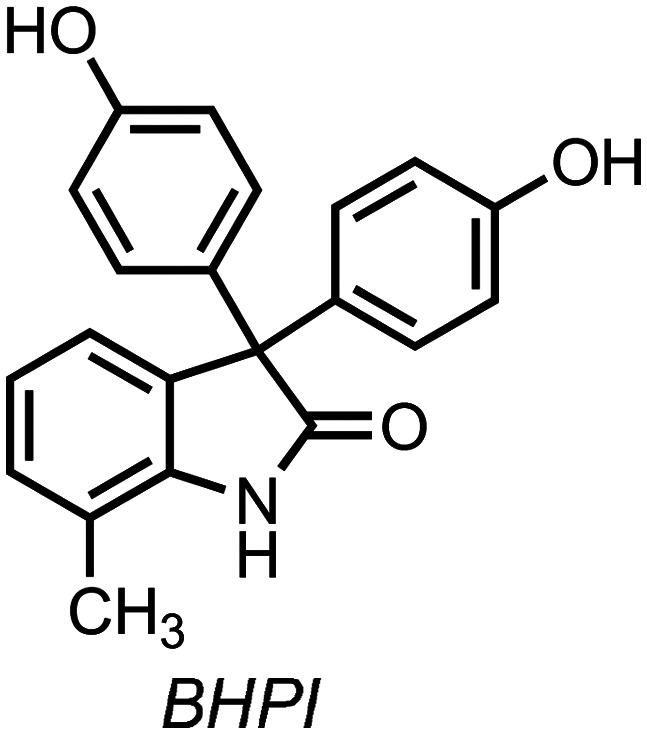
Similar to this result with BHPI, treatment with other 3,3-bis(4-hydroxyphenyl)indoline-2-ones also leads to increases in a-UPR markers (although they were not referred to as such), as indicated by significant increases in p-EIF2α, protein synthesis inhibition, increases in phosphorylated AMPK, and others.49,64 Together with the mechanistic studies using BHPI and given their structural similarity, these results suggest that other 3,3-bis(4-hydroxyphenyl)indoline-2-ones, like TOP001, TOP216, and oxyphenisatin, exert their published antiproliferative activity, at least partially, through activation of the a-UPR.
5. Development of chiral 3-(4-hydroxyphenyl)indoline-2-ones
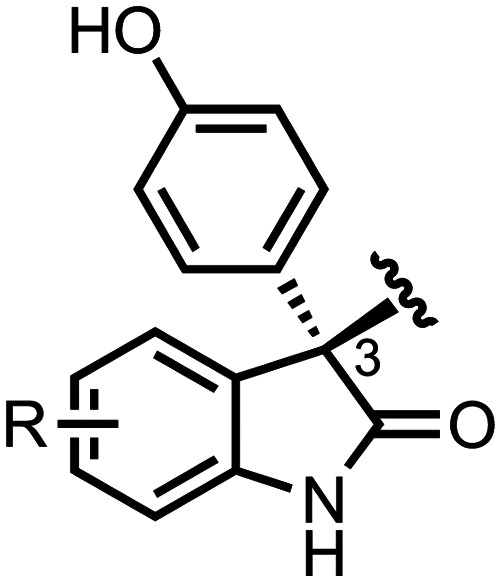
The above sections have focused on an achiral, subclass of 3-(4-hydroxyphenyl)indoline-2-ones, the 3,3-bis(4-hydroxyphenyl)indoline-2-ones, in which the 3-position of the indoline-2-one is disubstituted with 4-hydroxylphenyl groups. Examples of these achiral 3,3-bis(4-hydroxyphenyl)indoline-2-ones are oxyphenisatin, BHPI, TOP001, and others.49,64,86 However, a 3-(4-hydroxyphenyl)indoline-2-one can be optionally substituted with a different chemical moiety, yielding a chiral 3-(4-hydroxyphenyl)indoline-2-one (Scheme 9). These chiral molecules are of interest for their potent, single enantiomer anticancer activity. Below, we review the recent explorations of chiral 3-(4-hydroxyphenyl)indoline-2-ones that exhibit potent anticancer activity.
Scheme 9. Generalized chemical structure of a chiral 3-(4-hydroxyphenyl)indoline-2-one.
5.1. Compound (S)-38
In a follow-up on TOP001/TOP216 work,48 Christensen et al. reported their exploration of the 3-(4-hydroxyphenyl)indoline-2-one pharmacophore109 by assessing antiproliferative activity against ERα-positive cancer cell lines, MCF-7 (breast cancer)110 and PC-3 (prostate cancer).73,74 In the course of their medicinal chemistry campaign, the authors reported consistent SAR for the class,48 for example the necessity for at least one 4-hydroxylphenyl (4-phenol) for biological activity.109 However, replacement of one of the 4-hydroxyphenyl with unsubstituted cycloalkanes yielded a chiral class of compounds with potent anticancer activity.109 These molecules were synthesized via reaction of isatin starting materials with Grignard reagents, followed by Friedel–Crafts reaction with phenol (Scheme 10). This two-step reaction sequence enabled access to a range of racemic 3-(4-hydroxyphenyl)indoline-2-ones, albeit with limited diversity beyond simple unsubstituted cycloalkanes.
Scheme 10. Synthesis of racemic 3-(4-hydroxyphenyl)indoline-2-ones as reported by Christensen et al.109 R = H; 7-CF3; 5,7-di-CH3; 5-F,7-CH3; 6,7-di-CH3; 6-F,7-CH3; 6-Cl,7-CH3; 6-CH3,7-Cl; 6-OCH3,7-CH3. n = 1 (cyclopentyl), 2 (cyclohexyl), 3 (cycloheptyl), 4 (cyclooctyl). X = Cl or Br.

Upon optimization and chiral separation, the authors reported compound (S)-38 with potent antiproliferative activity while its opposite enantiomer ((R)-38) was completely devoid of that potent activity (Table 2).109 Further mechanistic work demonstrated that (S)-38 led to increases in p-EIF2α. No direct molecular target nor mechanism of action for (S)-38 or similar compounds reported therein were suggested (no mention of ERα expression and correlation to cellular activity).109 Given the structural similarity of (S)-38 to the general 3-(4-hydroxyphenyl)indoline-2-one class and the profound increase in p-EIF2α observed, a-UPR activation may be a mechanism of action for (S)-38 and similar compounds.
Chemical structures and the reported biological activity (IC50) of (S)-38 and its enantiomer (R)-38 (ref. 109).
 (S)-38
(S)-38 |
 (R)-38
(R)-38 |
|
|---|---|---|
| MCF-7 | 0.0005 μM | >1 μM |
| PC-3 | 0.002 μM | >1 μM |
For racemic compounds of interest, in vivo tolerability and antitumor activity were then investigated. In broad strokes, all active compounds in cell culture had profound antitumor (either tumor static and/or tumor regressions) activity against PC-3 xenografts in mice with sufficient therapeutic indices in mice (all compounds administered once-a-week for three total doses intravenously).109 In studies with tumor-bearing rats, again the compounds were potent antitumor agents, however some tolerability problems arose.109 Achiral and racemic 3,3-diaryl-indoline-2-ones clearly exhibited low maximum tolerated doses (MTD) in rats ranging from 1–15 mg kg−1 (intravenously administered).109 However, compounds substituted with a cycloheptyl (like (S)-38) had better tolerability in rats while maintaining antitumor activity. For example, (S)-38 showed tumor regressions in rat models (PC-3 xenograft) at 10 mg kg−1 (IV), with an MTD of 70 mg kg−1 (IV), and thus an estimated therapeutic index of 7.109 The differential toxicity observed between rats and mice was noted, although the excellent tolerability in mice and much worse tolerability in rats is not “easily explained” and the exact underlying mechanism is unknown.109
5.2. Cytotoxic a-UPR activator: ErSO
In 2021, we reported the discovery of ErSO (Fig. 2A), a member of the chiral 3-(4-hydroxyphenyl)indoline-2-one class that activates the a-UPR and has profound antitumor activity.111 Of note, most assays employed for annotating the anticancer effects of 3-(4-hydroxyphenyl)indoline-2-ones measure a combination of antiproliferative and cell death effects. This can complicate interpretation of results as authors report cytotoxic (killing) or antiproliferative (cytostatic) activity sometimes interchangeably.112 Shapiro and coworkers have shown previously that in some instances BHPI can be cytotoxic, but in most cell death assays, BHPI (even at 1 μM) cell death is <20%,111,113 even though its antiproliferative activity (4-day IC50) is 15 nM.86,114 With this initial clue that a cytotoxic anticancer phenotype could be optimized, we sought to identify highly cytotoxic a-UPR activating molecules and discovered that ErSO quantitatively kills ERα positive cancer cell lines in culture (Fig. 2B).111 This cytotoxic phenotype starkly contrasts other ERα targeted therapies (e.g. 4-hydroxytamoxifen (OHT) and fulvestrant (Fulv.) as shown in Fig. 2B), BHPI, and other 3-(4-hydroxyphenyl)indoline-2-ones.111,115
Fig. 2. ErSO is a potent killer of ERα-positive cancer in both cell culture and in vivo models. A, Chemical Structure of ErSO. B, Crystal violet staining of T47D cells (ERα-positive breast cancer) 24 hours post-treatment with indicated compounds. (±)-1 is the racemic mixture of ErSO (active enantiomer) and (S)-1 (inactive enantiomer). OHT: 4-hydroxytamoxifen, Fulv.: fulvestrant. C, ErSO IC50 values in indicated ERα+/− (ESR1 status) cancer cell lines after 24 hours compound incubation. D, ErSO treatment of ST941/HI patient derived xenograft (PDX). po: per os (oral administration). E and F, ErSO's antitumor effect is ERα-dependent. More comprehensive details of these models (D–F) are reported elsewhere.111 Panels B–F were reproduced from Boudreau, et al.111 Reprinted with permission from American Association for the Advancement of Science (AAAS), copyright 2021.

ErSO, like other chiral 3-(4-hydroxyphenyl)indoline-2-ones (as mentioned above),109 has single enantiomer activity, with its opposite enantiomer lacking anticancer activity ((S)-1, Fig. 2B).111ErSO displays an ERα-dependent activity as evidenced by dramatic sensitization of ERα-negative MDA-MB-231 cells after knock-in of ERα expression, including in a mouse model.111 To determine the robustness of this ERα-dependent effect, the activity of ErSO was assessed in a broader panel of breast cancer and other cancer types with variable amounts of ERα expression. Similar to other 3-(4-hydroxyphenyl)indoline-2-ones,48,86,109ErSO has potent activity against ERα-positive cancer cells (average 34 nM IC50) while being inactive in an array of other cancer cell lines (average IC50: 12 400 nM) (Fig. 2C).111 Of note and as discussed further in the next section, there is a time-dependence for this wide ERα-dependent window.111
However, similar to TOP001/216,64ErSO does have potent activity against some classically defined ERα-negative cell lines, namely Hs578t, MDA-MB-453, MDA-MB-468, and BT-20. The exact molecular understanding of this activity remains to be fully understood; however, we showed by western blot analysis111 that BT-20 does express low levels of ERα (in line with other reports116,117). Indeed, in a variety of other reports, MDA-MB-453 and MDA-MB-468 are shown to express low levels of ERα.65,66 Ultimately, a relationship between reported ESR1 (gene encoding ERα) promoter methylation (which squelches ERα expression) and ErSO activity was proposed as a useful correlation (Fig. 2C).111 Even in Hs578t cells where ERα expression could not be detected, siRNA-induced knockdown of ESR1 and fulvestrant-induced ERα degradation largely reversed ErSO's activity, suggesting that extremely low levels of ERα may be sufficient for cancer cell killing.111 Absolute level of ERα expression was not correlated with potency, but rapid cytotoxic activity seemed to track with relative ERα expression.111 As discussed later (Conclusions), future work is warranted to fully understand the molecular mechanism behind ErSO's and related 3-(4-hydroxyphenyl)indoline-2-ones' spectrum of activity, especially its potent activity in breast cancer cells that express very low levels of ERα.
ErSO's rapid cell killing phenotype in vitro translates into profound in vivo efficacy against ERα-positive tumors in murine models, including multiple examples of quantitative tumor regressions. In orthotopic xenograft models in mice with MCF-7 (ERα WT, ERαY537S, ERαD538G) and T47D (ERαY537S, ERαD538G), ErSO treatment (including both oral or injected administrations daily or once-a-week) was sufficient to eradicate tumors, with some quantitatively regressing after just three doses (40 mg kg−1).111 These profound tumor regressions were also observed when assessed against the patient derived xenograft model ST941/HI (Fig. 2D),111 which is a highly drug-resistant PDX model for low expressing ERαY537S breast cancer.118 These remarkable in vivo results were also recapitulated in a variety of metastatic mouse models, with ErSO treatment leading to full to near-quantitative regression of bone, lung, liver, and brain metastases.111 In agreement with in vitro work, ErSO's antitumor activity appears to also be ERα-dependent in vivo, most powerfully suggested by eradication of orthotopic xenografts of tumors formed from MDA-MB-231 cells where the gene ERα had been knocked in (Fig. 2E) and no effect on xenografts formed from parental ERα-negative MDA-MB-231 cells (Fig. 2F).111
ErSO's eradication of many ERα-positive breast cancer tumors – even in once-a-week dosing a low mg kg−1 levels – in preclinical models is remarkable and starkly contrasts the often cytostatic antitumor activity seen with other ERα-targeted breast cancer drugs and drug combinations.111,118–125 This initial preclinical body of work provides sufficient support for future investigations of ErSO to establish its translational potential.
5.3. Reducing lipophilicity: discovery of ErSO-DFP and ErSO-TFPy
As noted above, the ERα-dependency of ErSO, in some cell lines, erodes over time111 and this ERα-independent activity is not well-understood. As such, while preclinical evidence suggests ErSO holds tremendous translational promise, we undertook a campaign to discover derivatives that have: (1) enhanced selectivity for ERα+ cancer cells in culture, (2) maintain ErSO's remarkable ability to eradicate tumors in vivo, and (3) have wider therapeutic windows in vivo.126 Notably, these ‘second generation’ compounds would also be better positioned as chemical probes for studying the underlying biological mechanism (e.g. a-UPR) that is leading to the profound antitumor responses.
An optimization strategy needed to be established in the absence of any direct target engagement information. When analyzing ErSO, one key metric that is clear is the relatively high lipophilic nature of ErSO (clog D7.4 = 6.4) and other 3-(4-hydroxyphenyl)indoline-2-ones. Lipophilicity is an important consideration for drug development;127 while hydrophobic interactions can help enhance potency, they can also lead to poor ‘drug-like’ properties and increases the likelihood of pleotropic effects and promiscuity.127,128 In the absence of an exact molecular target, we sought to decrease the general lipophilic nature of ErSO and assess these derivatives in a battery of ERα-positive and ERα-negative breast cancer cell lines.
This pursuit of decreasing lipophilicity led to the design and synthesis of nitrogen heterocycle-containing derivatives with clog D7.4 values ranging from 2.6 to 4.9 (Scheme 11).126 One of the most intriguing of this set of compounds was a 4,4-difluoropiperidine containing derivative, ErSO-DFP (Scheme 11), with a clog D7.4 of 4.4. The importance of this difluorination on anticancer efficacy is clear, as variants lacking these fluorine atoms were devoid of activity.126
Scheme 11. Summary of chemical derivatization that arrived at ErSO-DFP. Further details of exact derivatives synthesized and displayed clog D7.4 values are reported by Boudreau et al.126.

ErSO-DFP has potent anticancer activity against MCF-7 and other ERα-positive cancer cell lines. ErSO-DFP's lipophilic efficiency (LipE) is 3.4, a >2 unit increase when compared to progenitor ErSO (LipE: 1.3).126 Importantly, ErSO-DFP maintains an ERα-dependent activity with an often >2000 fold difference between activity seen against ERα-positive and ERα-negative cancer cell lines (Fig. 3). This increase in selectivity in cell culture also translated to an increase in the tolerability seen with in vivo preclinical models, namely significant increases in the maximum tolerated doses (MTD) of ErSO-DFP in mice and rats with >3 fold increases in tolerability in both species. As mentioned previously, ErSO shows a similar speciation toxicity in rats111,126 to that of compound other 3-(4-hydroxyphenyl)indoline-2-ones.109 In contrast, ErSO-DFP is well-tolerated in rats up to its solubility limit.126ErSO-DFP also maintains ErSO-like tumor regressions as shown in an initial MCF-7 xenograft model (intravenous dosing 5 mg kg−1 once-a-week for 3 total doses).126
Fig. 3. ErSO-DFP and ErSO-TFPy have enhanced selectivity for ERα-positive cancer cells in cell culture. Chemical structures, lipophilicity, and biological activity values (IC50 and LipE) plotted as reported by Boudreau et al.126 ERα+: ERα-positive; ERα−: ERα-negative; LipE (lipophilic efficiency) was calculated clog D7.4 − pIC50.

Further exploration of this novel difluorinated nitrogen heterocycle chemical space revealed that difluorinated and tetrafluorinated pyrrolidines were also active derivatives with corresponding changes in lipophilicity. The most potent derivative, ErSO-TFPy (Fig. 3), has single enantiomer activity, a LipE of 3.8, and ≥3500 fold changes between ERα-positive and ERα-negative cancer cell lines, which is even larger than ErSO-DFP.126 The exact reason for why difluorination/tetrafluorination is necessary for these small molecules is unknown, but fluorination has well-reported effects on compound pKa, conformation, and target engagement.126 Future work is focused on more in-depth studies of ErSO-DFP, ErSO-TFPy, and related derivatives to fully explore their antitumor activity and therapeutic potential.
Conclusions
Herein, we outline the journey of the 3-(4-hydroxyphenyl)indoline-2-one pharmacophore through time and how the initial work with oxyphenisatin spurred an exciting area of anticancer research in the modern era. Even with the considerable body of work on 3-(4-hydroxyphenyl)indoline-2-ones, including outstanding candidates for translational development, there are still significant open questions surrounding this class of molecules with multiple avenues for future investigation. The remarkable antitumor activity of these molecules is clear (e.g.ErSO) and this phenotype is driving the interest in these molecules as drugs. However (as discussed above and elsewhere111,126), the exact causative direct molecular target of 3-(4-hydroxyphenyl)indoline-2-ones remains elusive. Establishment of the direct molecular target of ErSO (and derivatives) will likely lead to an explanation of some of the more puzzling observations (e.g. potent activity in cancer cells lines with very low ERα expression), and may also facilitate and inform translational studies. The a-UPR elucidation and its role in anticancer activity of 3-(4-hydroxyphenyl)indoline-2-ones has provided an initial step in understanding the profound antitumor activity of these compounds. The identification of the direct cellular binding partner will no doubt lead to a new understanding of this cell death pathway.
“The dose makes the poison” is an adage often discussed in drug discovery and it is of course true with our development of the ErSO series of compounds. The antitumor efficacy in preclinical models is compelling. However, ErSO's idiosyncratic toxicity observed in rats, time-dependent effect on some ERα-negative cancer cell lines, and ‘drug-like’ character leave room for optimization. Indeed, ErSO-DFP and ErSO-TFPy address these concerns and as such represent true second-generation versions. In-depth studies must be conducted to determine the full translational potential of these compounds; most critically, rigorous determination of therapeutic indices prior to explorations in human cancer patients. Our preclinical assessments are encouraging with the ErSO series,111,126 but in no way comprehensive. The importance of exhaustive safety annotation cannot be overstated, with the preclinical safety bar rightly very high prior to any investigational new drug application.
Inherent in the story of 3-(4-hydroxyphenyl)indoline-2-ones is the role of synthetic chemistry to access a wide-variety of compounds to investigate hypotheses and enhance the anticancer activity of these molecules. Of course, further optimization of this scaffold will continue to be a part of the future with in-depth exploration of an even wider scope of the indoline-2-one core, other fluorinated nitrogen heterocycles, phenol substitutions and/or replacements, prodrug forms, and many other chemical modifications. These medicinal chemistry efforts may yield even more potent and more tolerated derivatives in the future. Ideally, these optimizations will be informed by some sort of ligand–target relationship (e.g. crystal structure with biological target).
The application of 3-(4-hydroxyphenyl)indoline-2-ones for the treatment of ERα-positive breast cancer has been the main focus of this class of compounds. However, ERα overexpression is not solely found in breast cancer and the use of these molecules for the treatment of other cancers (especially those that are ERα expressing but ERα-independent and thus resistant to traditional inhibitory modes of endocrine therapy) is an ongoing pursuit. Again, as the molecular target and mechanistic underpinnings thereof are better understood, the ability to leverage 3-(4-hydroxyphenyl)indoline-2-ones for the treatment of even more different forms of cancer will become evident. For example, discovery of the exact molecular target could shine light on the current unclear trends seen with 3-(4-hydroxyphenyl)indoline-2-ones' spectrum of cancer cell line selectivity (e.g. extremely low ERα-expressing cell lines). Arguably even more exciting, elucidation of ErSO's molecular mechanism may inform other strategies that promote whole new opportunities that cannot even be imagined with today's ignorance. There are many examples where comprehensive understanding of a molecule's mechanism of action creates whole new fields and technologies.2 Therefore while clinical translation research with 3-(4-hydroxyphenyl)indoline-2-ones is ongoing, there is an equally rigorous ongoing investigation of the basic science underlying their anticancer activity, ultimately with the goal of developing highly efficacious drugs that contain this exciting pharmacophore.
Author contributions
M. W. B. conducted the literature review, and wrote the manuscript with input from P. J. H.
Conflicts of interest
The University of Illinois has filed patents on some compounds described herein on which M. W. B. and P. J. H. are inventors. This intellectual property has been licensed to Systems Oncology, LLC, and P. J. H. is a consultant and a member of the Scientific Advisory Board of Systems Oncology.
Supplementary Material
Acknowledgments
We would like to thank the University of Illinois Library services for their help in finding literature from the late 18th and early 19th century. For funding this work, we would like to thank the NIH (CA258746), and we extend our gratitude to the University of Illinois and the Cancer Center at Illinois (CCIL). M. W. B. is a member of the NIH Chemistry-Biology Interface Training Program (T32-GM136629), was an ACS Medicinal Chemistry Predoctoral Fellow, and he is supported by an NCI F99/K00 predoctoral fellowship (F99-CA253731; K00-CA253731).
Biographies
Biography
Matthew W. Boudreau.

Matthew W. Boudreau received his B.S. in Chemistry at North Carolina State University in 2016. He completed his Ph.D. in Chemistry at the University of Illinois at Urbana-Champaign in 2022 under the direction of Prof. Paul J. Hergenrother. Matthew develops and studies a variety of anticancer strategies. Matthew is currently an NCI F99/K00 fellow at Dana-Farber Cancer Institute as a member of Prof. William G. Kaelin Jr.'s laboratory.
Biography
Paul J. Hergenrother.

Prof. Paul J. Hergenrother is the Kenneth L. Rinehart Endowed Chair in Natural Products Chemistry at the University of Illinois at Urbana-Champaign. His laboratory is focused on molecular solutions to problems at the interface of organic chemistry and chemical biology, including the discovery and development of novel anticancer and antibacterial strategies and compounds.
Notes and references
- Zhong L. Li Y. Xiong L. Wang W. Wu M. Yuan T. Yang W. Tian C. Miao Z. Wang T. Yang S. Signal Transduction Targeted Ther. 2021;6:201. doi: 10.1038/s41392-021-00572-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedy A. M. Liau B. B. Nat. Chem. Biol. 2021;17:1219–1229. doi: 10.1038/s41589-021-00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S. L. Cell. 2021;184:3–9. doi: 10.1016/j.cell.2020.12.020. [DOI] [PubMed] [Google Scholar]
- Hanan E. J. Liang J. Wang X. Blake R. A. Blaquiere N. Staben S. T. J. Med. Chem. 2020;63:11330–11361. doi: 10.1021/acs.jmedchem.0c00093. [DOI] [PubMed] [Google Scholar]
- Bekes M. Langley D. R. Crews C. M. Nat. Rev. Drug Discovery. 2022;21:181–200. doi: 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerry C. J. Schreiber S. L. Nat. Chem. Biol. 2020;16:369–378. doi: 10.1038/s41589-020-0469-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L. Ruiz P. Ito T. Sellers W. R. Cancer Cell. 2021;39:466–479. doi: 10.1016/j.ccell.2020.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker B. J. Lydon N. B. J. Clin. Invest. 2000;105:3–7. doi: 10.1172/JCI9083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardini E. Menichincheri M. Banfi P. Bosotti R. De Ponti C. Pulci R. Ballinari D. Ciomei M. Texido G. Degrassi A. Avanzi N. Amboldi N. Saccardo M. B. Casero D. Orsini P. Bandiera T. Mologni L. Anderson D. Wei G. Harris J. Vernier J. M. Li G. Felder E. Donati D. Isacchi A. Pesenti E. Magnaghi P. Galvani A. Mol. Cancer Ther. 2016;15:628–639. doi: 10.1158/1535-7163.MCT-15-0758. [DOI] [PubMed] [Google Scholar]
- Joseph E. W. Pratilas C. A. Poulikakos P. I. Tadi M. Wang W. Taylor B. S. Halilovic E. Persaud Y. Xing F. Viale A. Tsai J. Chapman P. B. Bollag G. Solit D. B. Rosen N. Proc. Natl. Acad. Sci. U. S. A. 2010;107:14903–14908. doi: 10.1073/pnas.1008990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson E. I. Hergenrother P. J. Acc. Chem. Res. 2015;48:2715–2723. doi: 10.1021/acs.accounts.5b00365. [DOI] [PubMed] [Google Scholar]
- Welsch M. E. Snyder S. A. Stockwell B. R. Curr. Opin. Chem. Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khetmalis Y. M. Shivani M. Murugesan S. Chandra Sekhar K. V. G. Biomed. Pharmacother. 2021;141:111842. doi: 10.1016/j.biopha.2021.111842. [DOI] [PubMed] [Google Scholar]
- Reynolds T. B. Peters R. L. Yamada S. N. Engl. J. Med. 1971;285:813–820. doi: 10.1056/NEJM197110072851501. [DOI] [PubMed] [Google Scholar]
- Huisgen R. Angew. Chem., Int. Ed. Engl. 1986;98:297–311. doi: 10.1002/ange.19860980404. [DOI] [Google Scholar]
- de Meijere A. Angew. Chem., Int. Ed. 2005;44:7836–7840. doi: 10.1002/anie.200503351. [DOI] [PubMed] [Google Scholar]
- Fruton J. S., Methods and Styles in the Development of Chemistry, American Philosophical Society, 2002 [Google Scholar]
- Baeyer A. Lazarus M. J. Ber. Dtsch. Chem. Ges. 1885;18:2637–2643. doi: 10.1002/cber.188501802170. [DOI] [Google Scholar]
- Vamossy Z. V. Chem.-Ztg. 1900;24:679–680. [Google Scholar]
- Baeyer A. Ber. Dtsch. Chem. Ges. 1871;4:555–558. doi: 10.1002/cber.18710040209. [DOI] [Google Scholar]
- Baeyer A. Ber. Dtsch. Chem. Ges. 1871;4:658–665. doi: 10.1002/cber.18710040247. [DOI] [Google Scholar]
- Radl S. Chem. Listy. 2020;114:791–803. [Google Scholar]
- Dunnick J. K. Hailey J. R. Cancer Res. 1996;56:4922–4926. [PubMed] [Google Scholar]
- Murphy J. JAMA, J. Am. Med. Assoc. 2009;301:1770. doi: 10.1001/jama.2009.585. [DOI] [PubMed] [Google Scholar]; , author reply 1770
- Fed. Regist., 1999, 64, 4535–4540 [PubMed] [Google Scholar]
- Christensen E. V. Arch. Pharm. Chemi. 1931;88(47–56):69–78. [Google Scholar]
- Guggenheim M. Die spezifische Kolonwirkung der Abführmittel und ihre Abhängigkeit von der chemischen Konstitution. Schweiz. Med. Wochenschr. 1925;55(16) [Google Scholar]
- Farack U. M. Nell G. Digestion. 1984;30:191–194. doi: 10.1159/000199105. [DOI] [PubMed] [Google Scholar]
- Nell G. Forth W. Rummel W. Wanitschke R. Naunyn-Schmiedeberg's Arch. Pharmacol. 1976;293:31–37. doi: 10.1007/BF00498868. [DOI] [PubMed] [Google Scholar]
- Wanitschke R. Klin. Wochenschr. 1980;58:267–278. doi: 10.1007/BF01476568. [DOI] [PubMed] [Google Scholar]
- Einhorn M. Rafsky H. A. JAMA, J. Am. Med. Assoc. 1926;86:1754–1755. doi: 10.1001/jama.1926.02670490016005. [DOI] [Google Scholar]
- Katzenelbogen S. Güder R. Note thérapeutique sur un nouveau purgatif. Schweiz. Med. Wochenschr. 1925;55(18) [Google Scholar]
- Hirschberg A. Klin. Wochenschr. 1927;16:767. doi: 10.1007/BF01748021. [DOI] [Google Scholar]
- Keeley A. F. Trey C. Gottlieb C. B. Gottlieb L. S. Gastroenterology. 1971;60:195. [Google Scholar]
- McHardy G. Balart L. A. JAMA, J. Am. Med. Assoc. 1970;211:83–85. doi: 10.1001/jama.1970.03170010037006. [DOI] [PubMed] [Google Scholar]
- Pearson A. J. G. Scheuer P. J. Grainger J. M. Mcintyre N. Lancet. 1971;1:994–996. doi: 10.1016/S0140-6736(71)91388-2. [DOI] [PubMed] [Google Scholar]
- Reynolds T. B. Redeker A. G. JAMA, J. Am. Med. Assoc. 1970;213:2273. doi: 10.1001/jama.1970.03170390063027. [DOI] [PubMed] [Google Scholar]
- Naess K. JAMA, J. Am. Med. Assoc. 1970;212:1961. doi: 10.1001/jama.1970.03170240163029. [DOI] [PubMed] [Google Scholar]
- Reynolds T. B. Lapin A. C. Peters R. L. Yamahiro H. S. JAMA, J. Am. Med. Assoc. 1970;211:86–90. doi: 10.1001/jama.1970.03170010040007. [DOI] [PubMed] [Google Scholar]
- Reynolds T. B. Gastroenterology. 1969;56:418. doi: 10.1016/S0016-5085(69)80077-6. [DOI] [PubMed] [Google Scholar]
- Hubbard W. K. Fed. Regist. 1999;64:10944–10947. [PubMed] [Google Scholar]
- Med. J. Aust. 1972. 1 1051-1053 [Google Scholar]
- Baum H. M. Sanders R. G. Straub G. J. J. Am. Pharm. Assoc. 1951;40:348–349. doi: 10.1002/jps.3030400713. [DOI] [PubMed] [Google Scholar]
- Emerson G. A. Proc. Soc. Exp. Biol. Med. 1933;31:278–281. doi: 10.3181/00379727-31-7091C. [DOI] [Google Scholar]
- Stacewicz-Sapuntzakis M. Bowen P. E. Hussain E. A. Damayanti-Wood B. I. Farnsworth N. R. Crit. Rev. Food Sci. Nutr. 2001;41:251–286. doi: 10.1080/20014091091814. [DOI] [PubMed] [Google Scholar]
- Nishikawa J. Arzneimittelforschung. 1981;31:1872–1875. [PubMed] [Google Scholar]
- Natarajan A. Fan Y. H. Chen H. Guo Y. Iyasere J. Harbinski F. Christ W. J. Aktas H. Halperin J. A. J. Med. Chem. 2004;47:1882–1885. doi: 10.1021/jm0499716. [DOI] [PubMed] [Google Scholar]
- Uddin M. K. Reignier S. G. Coulter T. Montalbetti C. Granas C. Butcher S. Krog-Jensen C. Felding J. Bioorg. Med. Chem. Lett. 2007;17:2854–2857. doi: 10.1016/j.bmcl.2007.02.060. [DOI] [PubMed] [Google Scholar]
- Morrison B. L. Mullendore M. E. Stockwin L. H. Borgel S. Hollingshead M. G. Newton D. L. Cancer Med. 2013;2:687–700. doi: 10.1002/cam4.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumpter W. C. Chem. Rev. 1944;34:393–434. doi: 10.1021/cr60109a003. [DOI] [Google Scholar]
- da Silva J. F. M. Garden S. J. Pinto A. C. J. Braz. Chem. Soc. 2001;12:273–324. doi: 10.1590/S0103-50532001000300002. [DOI] [Google Scholar]
- Wang Z., Sandmeyer Isatin Synthesis, in Comprehensive organic name reactions and reagents, Wiley, Hoboken, N.J, 2010 [Google Scholar]
- Singh G. S. Desta Z. Y. Chem. Rev. 2012;112:6104–6155. doi: 10.1021/cr300135y. [DOI] [PubMed] [Google Scholar]
- Varun Sonam Kakkar R. MedChemComm. 2019;10:351–368. doi: 10.1039/C8MD00585K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmeyer T. Helv. Chim. Acta. 1919;2:234–242. doi: 10.1002/hlca.19190020125. [DOI] [Google Scholar]
- Marvel C. S. Hiers G. S. Org. Synth. 1925;5:71–74. doi: 10.15227/orgsyn.005.0071. [DOI] [Google Scholar]
- Satish G. Polu A. Ramar T. Ilangovan A. J. Org. Chem. 2015;80:5167–5175. doi: 10.1021/acs.joc.5b00581. [DOI] [PubMed] [Google Scholar]
- Zi Y. Cai Z. J. Wang S. Y. Ji S. J. Org. Lett. 2014;16:3094–3097. doi: 10.1021/ol501203q. [DOI] [PubMed] [Google Scholar]
- Kurihara T. Nasu K. Mizuhara Y. Hayashi K. Chem. Pharm. Bull. 1982;30:2742–2746. doi: 10.1248/cpb.30.2742. [DOI] [Google Scholar]
- Ilangovan A. Satish G. Org. Lett. 2013;15:5726–5729. doi: 10.1021/ol402750r. [DOI] [PubMed] [Google Scholar]
- Wei W. T. Ying W. W. Zhu W. M. Wu Y. Huang Y. L. Cao Y. Q. Wang Y. N. Liang H. Z. Synlett. 2017;28:2307–2310. doi: 10.1055/s-0036-1590965. [DOI] [Google Scholar]
- Chandra A. Yadav N. R. Moorthy J. N. Tetrahedron. 2019;75:2169–2174. doi: 10.1016/j.tet.2019.02.033. [DOI] [Google Scholar]
- Klumpp D. A. Yeung K. Y. Prakash G. K. S. Olah G. A. J. Org. Chem. 1998;63:4481–4484. doi: 10.1021/jo980588g. [DOI] [Google Scholar]
- Trojel-Hansen C. Erichsen K. D. Christensen M. K. Jensen P. B. Sehested M. Nielsen S. J. Cancer Chemother. Pharmacol. 2011;68:127–138. doi: 10.1007/s00280-010-1453-3. [DOI] [PubMed] [Google Scholar]
- Wang W. Smith, 3rd R. Burghardt R. Safe S. H. Mol. Cell. Endocrinol. 1997;133:49–62. doi: 10.1016/S0303-7207(97)00142-1. [DOI] [PubMed] [Google Scholar]
- Bhatt S. Xiao Z. Meng Z. Katzenellenbogen B. S. Mol. Cell. Biol. 2012;32:1928–1943. doi: 10.1128/MCB.06561-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan W. S. Vazhappilly C. G. Saleh E. M. Menon V. AlAzawi A. M. El-Serafi A. T. Mansour W. El-Awady R. Cancers. 2018;11:13. doi: 10.3390/cancers11010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Vega A. M. Del Moral-Morales A. Zamora-Sanchez C. J. Pina-Medina A. G. Gonzalez-Arenas A. Camacho-Arroyo I. Cell. 2020;9:1930. doi: 10.3390/cells9091930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Vega A. M. Camacho-Arroyo I. Brain Sci. 2021;11:564. doi: 10.3390/brainsci11050564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton T. C. Young R. C. McKoy W. M. Grotzinger K. R. Green J. A. Chu E. W. Whang-Peng J. Rogan A. M. Green W. R. Ozols R. F. Cancer Res. 1983;43:5379–5389. [PubMed] [Google Scholar]
- Matsumura S. Ohta T. Yamanouchi K. Liu Z. Sudo T. Kojimahara T. Seino M. Narumi M. Tsutsumi S. Takahashi T. Takahashi K. Kurachi H. Nagase S. Cancer Biol. Ther. 2017;18:730–739. doi: 10.1080/15384047.2016.1235656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comsa S. Cimpean A. M. Raica M. Anticancer Res. 2015;35:3147–3154. [PubMed] [Google Scholar]
- Pisolato R. Lombardi A. P. Vicente C. M. Lucas T. F. Lazari M. F. Porto C. S. Steroids. 2016;107:74–86. doi: 10.1016/j.steroids.2015.12.021. [DOI] [PubMed] [Google Scholar]
- Lau K. M. LaSpina M. Long J. Ho S. M. Cancer Res. 2000;60:3175–3182. [PubMed] [Google Scholar]
- Guo J. M. Xiao B. X. Dai D. J. Liu Q. Ma H. H. World J. Gastroenterol. 2004;10:860–863. doi: 10.3748/wjg.v10.i6.860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. H. Lee K. T. Leung P. C. Carcinogenesis. 2011;32:589–596. doi: 10.1093/carcin/bgq276. [DOI] [PubMed] [Google Scholar]
- Ma Y. Hendershot L. M. Nat. Rev. Cancer. 2004;4:966–977. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- Ron D. Walter P. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Walter P. Ron D. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Livezey M. Kim J. E. Shapiro D. J. Front. Endocrinol. 2018;9:325. doi: 10.3389/fendo.2018.00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaskulska A. Janecka A. E. Gach-Janczak K. Int. J. Mol. Sci. 2020;22:4. doi: 10.3390/ijms22010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimold A. M. Iwakoshi N. N. Manis J. Vallabhajosyula P. Szomolanyi-Tsuda E. Gravallese E. M. Friend D. Grusby M. J. Alt F. Glimcher L. H. Nature. 2001;412:300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- van Anken E. Romijn E. P. Maggioni C. Mezghrani A. Sitia R. Braakman I. Heck A. J. Immunity. 2003;18:243–253. doi: 10.1016/S1074-7613(03)00024-4. [DOI] [PubMed] [Google Scholar]
- Hu C. C. Dougan S. K. McGehee A. M. Love J. C. Ploegh H. L. EMBO J. 2009;28:1624–1636. doi: 10.1038/emboj.2009.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andruska N. Zheng X. Yang X. Helferich W. G. Shapiro D. J. Oncogene. 2015;34:3760–3769. doi: 10.1038/onc.2014.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andruska N. D. Zheng X. Yang X. Mao C. Cherian M. M. Mahapatra L. Helferich W. G. Shapiro D. J. Proc. Natl. Acad. Sci. U. S. A. 2015;112:4737–4742. doi: 10.1073/pnas.1403685112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro D. J. Livezey M. Yu L. Zheng X. Andruska N. Trends Endocrinol. Metab. 2016;27:731–741. doi: 10.1016/j.tem.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen E. V. Jordan V. C. Clin. Cancer Res. 2003;9:1980–1989. [PubMed] [Google Scholar]
- Flesher J. W. Gupta G. N. Jacobson H. I. Jensen E. V. Fed. Proc. 1960;19:170–170. [Google Scholar]
- Toft D. Shyamala G. Gorski J. Proc. Natl. Acad. Sci. U. S. A. 1967;57:1740–1743. doi: 10.1073/pnas.57.6.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toft D. Gorski J. Proc. Natl. Acad. Sci. U. S. A. 1966;55:1574–1581. doi: 10.1073/pnas.55.6.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenellenbogen B. S. Mol. Endocrinol. 2006;20:2611–2612. doi: 10.1210/mend.20.11.5432. [DOI] [PubMed] [Google Scholar]
- Hammes S. R. Levin E. R. Endocr. Rev. 2007;28:726–741. doi: 10.1210/er.2007-0022. [DOI] [PubMed] [Google Scholar]
- Levin E. R. Am. J. Physiol. 2014;307:E133–E140. doi: 10.1152/ajpendo.00626.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acconcia F. Kumar R. Cancer Lett. 2006;238:1–14. doi: 10.1016/j.canlet.2005.06.018. [DOI] [PubMed] [Google Scholar]
- Welsh A. W. Lannin D. R. Young G. S. Sherman M. E. Figueroa J. D. Henry N. L. Ryden L. Kim C. Love R. R. Schiff R. Rimm D. L. Clin. Cancer Res. 2012;18:118–126. doi: 10.1158/1078-0432.CCR-11-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. Huangyang P. Wang Y. Xue L. Devericks E. Nguyen H. G. Yu X. Oses-Prieto J. A. Burlingame A. L. Miglani S. Goodarzi H. Ruggero D. Cell. 2021;184:5215–5229.e17. doi: 10.1016/j.cell.2021.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzenellenbogen B. S. Cell. 2021;184:5086–5088. doi: 10.1016/j.cell.2021.09.012. [DOI] [PubMed] [Google Scholar]
- Yu L. Wang L. Kim J. E. Mao C. Shapiro D. J. Biochim. Biophys. Acta, Mol. Cell Res. 2020;1867:118765. doi: 10.1016/j.bbamcr.2020.118765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P. Griffith O. L. Agboke F. A. Anur P. Zou X. McDaniel R. E. Creswell K. Kim S. H. Katzenellenbogen J. A. Gray J. W. Jordan V. C. Cancer Res. 2013;73:4510–4520. doi: 10.1158/0008-5472.CAN-12-4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddow A. Nature. 1935;136:868–869. doi: 10.1038/136868a0. [DOI] [Google Scholar]
- Haddow A. Watkinson J. M. Paterson E. Koller P. C. Br. Med. J. 1944;2:393–398. doi: 10.1136/bmj.2.4368.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariazi E. A. Cunliffe H. E. Lewis-Wambi J. S. Slifker M. J. Willis A. L. Ramos P. Tapia C. Kim H. R. Yerrum S. Sharma C. G. Nicolas E. Balagurunathan Y. Ross E. A. Jordan V. C. Proc. Natl. Acad. Sci. U. S. A. 2011;108:18879–18886. doi: 10.1073/pnas.1115188108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanker A. B. Sudhan D. R. Arteaga C. L. Cancer Cell. 2020;37:496–513. doi: 10.1016/j.ccell.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingle J. N. Ahmann D. L. Green S. J. Edmonson J. H. Bisel H. F. Kvols L. K. Nichols W. C. Creagan E. T. Hahn R. G. Rubin J. Frytak S. N. Engl. J. Med. 1981;304:16–21. doi: 10.1056/NEJM198101013040104. [DOI] [PubMed] [Google Scholar]
- Ellis M. J. Gao F. Dehdashti F. Jeffe D. B. Marcom P. K. Carey L. A. Dickler M. N. Silverman P. Fleming G. F. Kommareddy A. Jamalabadi-Majidi S. Crowder R. Siegel B. A. JAMA, J. Am. Med. Assoc. 2009;302:774–780. doi: 10.1001/jama.2009.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong R. Patel H. K. Gutgesell L. M. Zhao J. Delgado-Rivera L. Pham T. N. D. Zhao H. Carlson K. Martin T. Katzenellenbogen J. A. Moore T. W. Tonetti D. A. Thatcher G. R. J. J. Med. Chem. 2016;59:219–237. doi: 10.1021/acs.jmedchem.5b01276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abderrahman B. Maximov P. Y. Curpan R. F. Fanning S. W. Hanspal J. S. Fan P. Foulds C. E. Chen Y. Malovannaya A. Jain A. Xiong R. Greene G. L. Tonetti D. A. Thatcher G. R. J. Jordan V. C. Mol. Cancer Ther. 2021;20:11–25. doi: 10.1158/1535-7163.MCT-20-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen M. K. Erichsen K. D. Trojel-Hansen C. Tjornelund J. Nielsen S. J. Frydenvang K. Johansen T. N. Nielsen B. Sehested M. Jensen P. B. Ikaunieks M. Zaichenko A. Loza E. Kalvinsh I. Bjorkling F. J. Med. Chem. 2010;53:7140–7150. doi: 10.1021/jm100763j. [DOI] [PubMed] [Google Scholar]
- Dai X. Cheng H. Bai Z. Li J. J. Cancer. 2017;8:3131–3141. doi: 10.7150/jca.18457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau M. W. Duraki D. Wang L. Mao C. Kim J. E. Henn M. A. Tang B. Fanning S. W. Kiefer J. Tarasow T. M. Bruckheimer E. M. Moreno R. Mousses S. Greene G. L. Roy E. J. Park B. H. Fan T. M. Nelson E. R. Hergenrother P. J. Shapiro D. J. Sci. Transl. Med. 2021;13:eabf1383. doi: 10.1126/scitranslmed.abf1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz H. R. Richards R. Fontana R. E. Joyce A. J. Honeywell M. E. Lee M. J. Cell Rep. 2020;31:107800. doi: 10.1016/j.celrep.2020.107800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livezey M. Huang R. Hergenrother P. J. Shapiro D. J. Cell Death Differ. 2018;25:1796–1807. doi: 10.1038/s41418-018-0143-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C. Livezey M. Kim J. E. Shapiro D. J. Sci. Rep. 2016;6:34753. doi: 10.1038/srep34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakas B. Aka Y. Giray A. Temel S. G. Acikbas U. Basaga H. Gul O. Kutuk O. Cell Death Discovery. 2021;7:189. doi: 10.1038/s41420-021-00573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu I. Arnaout A. Loiseau S. Sun J. Seth A. McMahon C. Chun K. Hennessy B. Mills G. B. Nawaz Z. Slingerland J. M. J. Clin. Invest. 2007;117:2205–2215. doi: 10.1172/JCI21739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castles C. G. Fuqua S. A. Klotz D. M. Hill S. M. Cancer Res. 1993;53:5934–5939. [PubMed] [Google Scholar]
- Bihani T. Patel H. K. Arlt H. Tao N. Jiang H. Brown J. L. Purandare D. M. Hattersley G. Garner F. Clin. Cancer Res. 2017;23:4793–4804. doi: 10.1158/1078-0432.CCR-16-2561. [DOI] [PubMed] [Google Scholar]
- Liang J. Zbieg J. R. Blake R. A. Chang J. H. Daly S. DiPasquale A. G. Friedman L. S. Gelzleichter T. Gill M. Giltnane J. M. Goodacre S. Guan J. Hartman S. J. Ingalla E. R. Kategaya L. Kiefer J. R. Kleinheinz T. Labadie S. S. Lai T. Li J. Liao J. Liu Z. Mody V. McLean N. Metcalfe C. Nannini M. A. Oeh J. O'Rourke M. G. Ortwine D. F. Ran Y. Ray N. C. Roussel F. Sambrone A. Sampath D. Schutt L. K. Vinogradova M. Wai J. Wang T. Wertz I. E. White J. R. Yeap S. K. Young A. Zhang B. Zheng X. Zhou W. Zhong Y. Wang X. J. Med. Chem. 2021;64:11841–11856. doi: 10.1021/acs.jmedchem.1c00847. [DOI] [PubMed] [Google Scholar]
- Osborne C. K. Coronado E. B. Robinson J. P. Eur. J. Cancer Clin. Oncol. 1987;23:1189–1196. doi: 10.1016/0277-5379(87)90154-4. [DOI] [PubMed] [Google Scholar]
- Fanning S. W. Hodges-Gallagher L. Myles D. C. Sun R. Fowler C. E. Plant I. N. Green B. D. Harmon C. L. Greene G. L. Kushner P. J. Nat. Commun. 2018;9:2368. doi: 10.1038/s41467-018-04413-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y. Laws M. J. Guillen V. S. Ziegler Y. Min J. Sharma A. Kim S. H. Chu D. Park B. H. Oesterreich S. Mao C. Shapiro D. J. Nettles K. W. Katzenellenbogen J. A. Katzenellenbogen B. S. Cancer Res. 2017;77:5602–5613. doi: 10.1158/0008-5472.CAN-17-1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puyang X. Furman C. Zheng G. Z. Wu Z. J. Banka D. Aithal K. Agoulnik S. Bolduc D. M. Buonamici S. Caleb B. Das S. Eckley S. Fekkes P. Hao M. H. Hart A. Houtman R. Irwin S. Joshi J. J. Karr C. Kim A. Kumar N. Kumar P. Kuznetsov G. Lai W. G. Larsen N. Mackenzie C. Martin L. A. Melchers D. Moriarty A. Nguyen T. V. Norris J. O'Shea M. Pancholi S. Prajapati S. Rajagopalan S. Reynolds D. J. Rimkunas V. Rioux N. Ribas R. Siu A. Sivakumar S. Subramanian V. Thomas M. Vaillancourt F. H. Wang J. Wardell S. Wick M. J. Yao S. Yu L. Warmuth M. Smith P. G. Zhu P. Korpal M. Cancer Discovery. 2018;8:1176–1193. doi: 10.1158/2159-8290.CD-17-1229. [DOI] [PubMed] [Google Scholar]
- O'Brien N. Conklin D. Beckmann R. Luo T. Chau K. Thomas J. Mc Nulty A. Marchal C. Kalous O. von Euw E. Hurvitz S. Mockbee C. Slamon D. J. Mol. Cancer Ther. 2018;17:897–907. doi: 10.1158/1535-7163.MCT-17-0290. [DOI] [PubMed] [Google Scholar]
- Nardone A. Weir H. Delpuech O. Brown H. De Angelis C. Cataldo M. L. Fu X. Shea M. J. Mitchell T. Veeraraghavan J. Nagi C. Pilling M. Rimawi M. F. Trivedi M. Hilsenbeck S. G. Chamness G. C. Jeselsohn R. Osborne C. K. Schiff R. Br. J. Cancer. 2019;120:331–339. doi: 10.1038/s41416-018-0354-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau M. W. Mulligan M. P. Shapiro D. J. Fan T. M. Hergenrother P. J. J. Med. Chem. 2022;65:3894–3912. doi: 10.1021/acs.jmedchem.1c01730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. W. Gallego R. A. Edwards M. P. J. Med. Chem. 2018;61:6401–6420. doi: 10.1021/acs.jmedchem.8b00077. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Expert Opin. Drug Discovery. 2010;5:235–248. doi: 10.1517/17460441003605098. [DOI] [PubMed] [Google Scholar]