

Abstract
Background
The prevalence of epilepsy among people with intellectual disabilities is much higher than in the general population. Seizures in this population are often complex and refractory to treatment and antiepileptic medication may have a profound effect upon behaviour (Kerr 1997).
This is an updated version of a Cochrane Review first published in Issue 3, 2007.
Objectives
To assess the data available from randomised controlled trials (RCTs) of the efficacy of antiepileptic drug (AED) interventions in people with epilepsy and intellectual disabilities.
Search methods
For the latest update of this review, we searched the Cochrane Epilepsy Group Specialised Register (2 September 2014), the Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online (CRSO) (2 September 2014), MEDLINE (Ovid, 1946 to 3 September 2014) and PsycINFO (EBSCOhost, 1887 to 3 September 2014).
Selection criteria
Randomised and quasi‐randomised controlled trials (RCTs) of pharmacological interventions for people with epilepsy and a learning disability.
Data collection and analysis
Two review authors independently assessed trial quality and extracted data. We contacted study authors for additional information. We assessed epilepsy/seizure, behavioural and cognitive outcomes, as well as quality of life and adverse effects.
Main results
We included 14 RCTs (1116 participants) in the present review. Data were heterogenous and a descriptive analysis is presented. In the majority of cases where antiepileptic drugs (AEDs) were trialled in this population, we found moderate reductions in seizure frequency in that there was a significantly higher rate of responders (reduction of 50% or more) in the treatment group compared with the placebo group, with some studies reporting a higher incidence of seizure freedom in the treatment group. In general, AEDs that are proven to be effective in the general epilepsy population are also effective for refractory epilepsy in people with intellectual disability. It is not possible to comment on the relative efficacy of medications, making clinical decisions difficult.
In trial settings patients continued on treatment in the majority of cases. Placebo groups often experienced fewer adverse events. Where adverse events were experienced they appeared similar to those in the general population. The methods by which adverse events were recorded and reported appeared to be inconsistent, resulting in very large variation between studies. This is problematic as clinically relevant interpretation of these findings is limited.
The quality of evidence provided in the present review is low to moderate. Additionally the majority of studies lacked or used non‐reliable measures of behavioural exacerbation. However, where measured, little obvious impact on behaviour was seen in terms of behaviour disorder.
Authors' conclusions
This review broadly supports the use of AEDs to reduce seizure frequency in people with refractory epilepsy and intellectual disability. The evidence suggests that adverse events are similar to those in the general population and that behavioural adverse events leading to discontinuation are rare; however, other adverse effects are under‐researched.
Plain language summary
Pharmacological interventions for epilepsy in people with intellectual disability
Background
Epilepsy is a common condition. Approximately 3% of the general population are diagnosed with epilepsy at some point in their life (Rugg‐Gunn 2012). However, epilepsy is significantly more common in people with intellectual disabilities where estimates range from 14% to 44% (Bowley 2000). People with intellectual disability and epilepsy often do not respond as well to antiepileptic drugs (AEDs) as the general population and behavioural disturbances are frequent. We review the use of AEDs in this population.
Study characteristics
The evidence is current to 2 September 2014. The included studies assessed the effectiveness of pharmacological interventions in people with epilepsy and intellectual disability. In total we included 14 studies and data from 1116 participants in the present review update. Five studies were funded by pharmaceutical companies, seven studies had insufficient information regarding how the trial was funded and one study was free from other bias.
Key results
This updated review confirms that in the majority of cases where intellectually disabled populations participated in trials of AEDs, moderate reductions in seizure frequency and occasional seizure freedom were obtained. The present update found that participants in the AED groups were more likely to report a 50% or greater reduction in seizure frequency compared with the control groups. The results also suggested that participants in the AED group were more likely to become seizure‐free and report a 50% or greater reduction in seizure frequency compared with participants in the placebo group, however these results were not significant. Additionally, the results of the present update suggest that more participants in the AED groups withdrew from treatment and reported adverse effects compared with the placebo group, however these results were also not significant. Where adverse events were reported they appeared to be similar to those seen in people without intellectual disability and thus they were not specific to this population.
In summary, this review broadly supports the use of AEDs to reduce seizure frequency in patients with refractory epilepsy and intellectual disability. Side effects seem to be the same as in people with epilepsy without intellectual disabilities and behavioural adverse events leading to withdrawal from the treatment are rare.
Quality of the evidence
The quality of evidence provided by this review is low to moderate. There is a large amount of variation across the studies in that different studies used different AEDs and reported different outcomes. Due to the inconsistencies between studies, not all studies could be included in the meta‐analyses. Therefore the statistical analysis in the present update is limited.
Overall there appear to be very few high quality studies assessing pharmacological interventions for people with epilepsy and intellectual disability. We hope that further research will be conducted in this area to provide a more thorough understanding.
Summary of findings
for the main comparison.
| Antiepileptic drug (AED) compared with placebo for people with epilepsy and intellectual disability | ||||
|
Patient or population: people with epilepsy and intellectual disability Settings: UK, USA Intervention: AEDs Comparison: placebo | ||||
| Outcomes | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments |
|
Retention on treatment Duration of retention on treatment was not reported in these studies. The retention on treatment in this analysis is assessed by the number of withdrawals in the intervention and placebo groups The results show a trend in favour of the placebo groups |
RR 1.43 (0.76 to 2.69) | 310 (3) |
⊕⊕⊕⊝ moderate | The included studies assessed different AEDs (topiramate and rufinamide). 1 study did not include participants under 12 years of age, 1 study did not include participants under 4 years of age and 1 study did not include participants under 1 year of age. The duration of intervention was similar across the studies (84 days, 11 weeks and 3 months). 2 studies had low risk of bias and 1 study was unclear |
|
Seizure freedom 1 study reported the incidence of seizure frequency in the intervention and placebo group The results show a trend in favour of the AED group |
RR 2.92 (0.32 to 26.77) | 73 (1) |
⊕⊕⊝⊝ low | The included study had good methodological quality and low risk of bias. However, given that only 1 study was included in this analysis, the overall quality of evidence is low |
|
Responder rate (≥ 50% reduction in overall seizure frequency) 3 studies included data for responder rate for overall seizure reduction in the AED and placebo groups The results show a significant trend in favour of the AED groups |
RR 2.58 (1.60 to 4.14) | 382 (3) |
⊕⊕⊝⊝ low | The included studies assessed different AEDs (lamotrigine, rufinamide, topiramate). Similar age ranges were included in the studies and they had a similar duration of treatment. 2 studies were rated as having unclear risk of bias and 1 study had low risk of bias |
|
Responder rate (≥ 50% reduction in rate of drop seizures) 2 studies reported the frequency of responder rate in the reduction of drop seizures only in the AED and placebo groups The results show a trend in favour of the AED groups |
RR 6.04 (0.27 to 133.89) | 313 (2) |
⊕⊕⊝⊝ low | The 2 studies assessed different AEDs (clobazam, topiramate). They had a similar duration of treatment period (11 weeks, 12 weeks). 1 study was rated as low risk of bias and 1 study was rated as unclear risk of bias |
|
Adverse events 4 studies reported the frequency of self reported adverse events in the AED and placebo groups The results show a trend in favour of the placebo groups |
RR 1.13 (0.95 to 1.35) | 527 (4) |
⊕⊕⊕⊝ moderate | The included studies assessed different AEDs (topiramate, clobazam, rufinamide). They all had similar duration treatment periods (around 3 months). 1 study did not include participants under 12 years of age, 1 study did not include participants under 4 years of age, 1 study did not include participants under 2 years of age and 1 study did not include participants under 1 year of age. 2 studies were rated as having low risk of bias and 2 studies were rated as having unclear risk of bias |
| CI: confidence interval; RR: risk ratio; AED: antiepileptic drugs | ||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||
Background
This review is an update of a review first published in The Cochrane Library in 2007, Issue 3.
Description of the condition
Epilepsy is a common neurological disorder with approximately 3% of people being diagnosed at some point in their life (Rugg‐Gunn 2012). The development of epilepsy in a person with intellectual disability is significantly more common, with an estimated overall prevalence rate of 14% to 44% and prevalence increases with the severity of the disability (Bowley 2000). A community‐based study of epilepsy in intellectually disabled people showed a prevalence of 16%, significantly higher than in the general population (Morgan 2003). In those with additional disabilities, such as cerebral palsy or postnatal brain injury, the prevalence of epilepsy can be as high as 75% (Goulden 1991; Shepherd 1989). The management of epilepsy in people with an intellectual disability provides additional challenges due to a number of factors, including the aetiology and severity of the epilepsy, a limited evidence base for interventions and difficulties in investigation and communication.
People with an intellectual disability who also have epilepsy exhibit different types and frequency of seizures; they have a higher frequency of certain epilepsy syndromes, in particular the Lennox‐Gastaut syndrome (Mariani 1993). Furthermore, the underlying cause of the intellectual disability may have an impact on seizure type and outcome, for example tuberous sclerosis is associated with a particular seizure disorder (Webb 1991), as is Down syndrome (Stafstrom 1993). Authors suggest that generalised seizures are the most commonly encountered seizure type in people with intellectual disabilities. According to two population‐based cohort studies, 68% and 83% of the study population had generalised seizures (Deb 1999; Forsgren 1990).
Rates of behavioural disturbance and psychiatric disorder have been shown to be significantly higher in people with epilepsy compared with the general population (Hoare 1984), and in people with an intellectual disability (Bouras 1992; Deb 1997; Eaton 1982; Mansell 1993). Currently, there is no consensus on whether people with both epilepsy and intellectual disability have higher rates of psychological or psychiatric morbidity compared with their peers with either epilepsy or an intellectual disability alone. A recent prospective controlled study has suggested that there is an increase in psychopathology in people with intellectual disability who have active epilepsy as compared with their peers with no seizures (Turky 2011). Espie 1989, Espie 1990 and Gillies 1989 reported that people with both epilepsy and intellectual disability have poorer life skills, together with behavioural disturbances such as aggression and self injury.
There are a number of causes of behavioural disturbances in people with epilepsy and intellectual disability; antiepileptic medication is one of those causes. According to Besag 2001, much of the published literature (unreferenced in this article) regarding behavioural disturbance associated with new antiepileptic drugs consists of unblinded studies or anecdotal case reports in people without an intellectual disability. Some of this anecdotal evidence suggests that adverse behavioural disturbances as a result of antiepileptic drug treatment are more likely to occur in people (adults and children) with an intellectual disability and epilepsy than in people with epilepsy alone (Beran 1998; Dam 1990; Dulac 1991; Ettinger 1998; Khurana 1996; Lee 1996; Mikati 1998; Wallace 1996; Wolf 1995). Conversely, beneficial behavioural effects in response to antiepileptic drugs have also been reported in people with epilepsy and intellectual disability (Gay 1995; Mullens 1996).
Description of the intervention
This review will focus only on pharmacological interventions for epilepsy in people with intellectual disability. Pharmacological interventions in epilepsy aim to reduce or stop seizures. Antiepileptic medication is the standard treatment modality for epilepsy. Medication is given daily. Ideally the target is for monotherapy to control seizures but when cases do not respond to monotherapy then polytherapy is often used.
How the intervention might work
Antiepileptic drugs (AEDs) have numerous mechanisms of action; some AEDs may be more effective for certain seizure types whereas others are broad spectrum and can be used to treat a range of seizure types. These therapies aim to target neuronal receptors in order to either reduce neuronal excitation or increase inhibition, thus decreasing the likelihood of seizure discharges and therefore reducing seizure frequency.
Why it is important to do this review
Seizure activity has a profound impact on both life expectancy and quality of life in people with an intellectual disability. A clinical decision regarding the introduction of antiepileptic medication in people with epilepsy and an intellectual disability involves an assessment of the impact of seizures on an individual, the effect that antiepileptic medication might have on an individual's seizures and an assessment of medication adverse effects.
In spite of the high prevalence of epilepsy in people with intellectual disability, interventional studies for the treatment of epilepsy in this group are relatively rare. In view of the fact that seizures in intellectually disabled people are often complex and refractory to treatment, and that antiepileptic medication can have a profound effect upon behaviour in this patient group, it is evident that good quality randomised controlled trials (RCTs) are needed in this population.
The aim of our study was to assess the data available from RCTs of AED interventions in people with epilepsy and intellectual disability and to identify the reasons for success or failure of the intervention in these studies. We assessed both beneficial and harmful outcomes.
Objectives
To assess the data available from randomised controlled trials (RCTs) of the efficacy of antiepileptic drug (AED) interventions in people with epilepsy and intellectual disabilities.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs; individual or cluster‐randomised) using random allocation sequences (e.g. using computer‐generated sequences) and using an adequate method of allocation concealment (for example, sealed, opaque envelopes, central randomisation by telephone or interactive voice response systems).
Quasi‐RCTs in which an inadequate method of allocation concealment is used (e.g. patients allocated by day of week).
Cross‐over and parallel designs.
Blinded or unblinded trials.
Types of participants
Individuals who were 12 years and over, with epilepsy and an intellectual disability. We excluded studies specifically recruiting children under 12 years of age and those restricted to patients with infantile spasms or West syndrome.
-
We considered participants to have an intellectual disability if they met any of the following criteria:
population defined as having an intellectual disability according to IQ, clinical description or if the population is sampled from an environment likely to have an intellectually disabled population, for example a special needs school or learning disabled day service;
population known to have a high percentage of intellectual disability, such as people with Fragile X, Down syndrome or Lennox‐Gastaut syndrome. To ensure a remit that is as inclusive as possible, we excluded studies reporting results for a population with less than 50% of participants with intellectual disability. We included studies including these populations that did not report the percentage of participants with intellectual disabilities but the conclusions drawn from these studies were more conservative
Participants may have any type of epilepsy, classified either by the investigator and where possible according to the International League Against Epilepsy guidelines (Commission on Classification ILAE 1981; ILAE 1989), or by other recognised classifications or using clinical descriptions.
Types of interventions
Pharmacological intervention licensed for the treatment of epileptic seizures, including but not limited to: carbamazepine; gabapentin; phenytoin; clobazam; clonazepam; ethosuximide; lamotrigine; methylphenobarbitone; paraldehyde; phenobarbitone; primidone; topiramate; sodium valproate; vigabatrin; acetazolamide; levetiracetam; oxcarbazepine; tiagabine; felbamate; zonisamide and pregabalin.
Types of outcome measures
Primary outcomes
Epilepsy/seizure outcomes
Retention on treatment (days, weeks or months).
Seizure freedom.
-
Reduction in seizure frequency in the treatment period compared with the pre‐randomisation baseline period:
overall reduction (median/mean);
responder rates (50% or greater reduction in seizure frequency).
Seizure severity scales.
Secondary outcomes
Behavioural outcomes
Changes from baseline in validated behavioural measures.
Cognitive outcomes
Changes from baseline in validated cognitive measures.
Adverse effects
Any reported adverse effect.
Adverse effects requiring treatment withdrawal.
Quality of life
Changes from baseline on a validated quality of life scale.
Global rating scale
This is often a non‐validated, clinician‐generated view on overall outcome without focusing on a specific change. These outcomes are usually reported as 'better', 'the same' or 'worse'.
Search methods for identification of studies
Electronic searches
The search for the original review was run in 2007 and subsequent searches were run in February 2010, January 2011, September 2012 and September 2014. For the latest update, we searched the following electronic databases. We did not apply any language restrictions.
The Cochrane Epilepsy Group Specialised Register (2 September 2014) using the strategy outlined in Appendix 1.
The Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online (CRSO, 2 September 2014) using the strategy outlined in Appendix 2. CENTRAL includes the Specialised Registers of the Cochrane Schizophrenia Group and the Cochrane Developmental, Psychosocial and Learning Problems Group.
MEDLINE (Ovid, 1946 to 3 September 2014) using the strategy outlined in Appendix 3.
PsycINFO (EBSCOhost, 1887 to 3 September 2014) using the strategy outlined in Appendix 4.
The National Research Register Archive was searched on 7 February 2010 using 'epilepsy' as a keyword and 'trial' in the methodology field, but this no longer needs to be searched because it is a closed archive to which no new items are being added.
The search terms used for the first version of this review (The Cochrane Library 2007, Issue 3) are set out in Appendix 5. We originally searched EMBASE (1980 to April 2005) but we no longer have direct access to that database. However, a project to identify reports of trials in EMBASE is being carried out by the UK Cochrane Centre. This search is updated annually and these records are published in CENTRAL. These records are therefore available to us via our searches of CENTRAL.
Searching other resources
We handsearched any journals for which there were hits in our electronic searches, if these journals had not already been handsearched by members of The Cochrane Collaboration.
We contacted other researchers in the field in an attempt to identify unpublished studies.
We also contacted pharmaceutical companies in an attempt to locate unpublished studies.
Data collection and analysis
Selection of studies
For the update, two review authors (CJ, SMM) independently assessed trials identified from the search strategies for inclusion. We discussed and resolved any disagreements. The same two review authors (CJ, SMM) completed data extraction and 'Risk of bias' assessments on each of the included studies. Again, we discussed and resolved any disagreements.
Data extraction and management
Two review authors (CJ, SMM) extracted the following information from the included studies. We discussed and resolved any disagreements.
Methodological design
Study design.
Method of randomisation and concealment.
Method of double‐blinding.
Whether any participants had been excluded from the reported analysis and reasons why they were excluded.
Confounding variables considered and controlled for.
Duration of baseline period.
Duration of treatment period.
Type and dose of medication.
Participants
Total number of participants allocated to each group.
Setting.
Inclusion criteria.
Exclusion criteria.
Age.
Gender.
Nature and severity of intellectual disability.
Nature and severity of seizures.
Seizure frequency during baseline period.
Number of background drugs.
Outcomes
Definition of outcome.
Units of measurement.
Results
Number of participants allocated to each intervention and control group that completed the study.
Sample size for each outcome.
Missing data.
Summary data for intervention and control groups (for example, means and standard deviations for all outcomes); see Types of outcome measures.
Assessment of risk of bias in included studies
Two review authors (CJ, SMM) independently assessed the risk of bias for each trial in accordance with the Cochrane 'Risk of bias' tool as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We discussed and resolved any disagreements. We rated studies as having high, low or unclear risk of bias for six domains applicable to RCTs: randomisation method, allocation concealment, blinding methods, incomplete data outcome, selective outcome reporting and other sources of bias.
Measures of treatment effect
Where data were available, we reported dichotomous outcomes, such as responder rate, seizure freedom and adverse effects as risk ratios with 95% confidence intervals. In the case of zero events where a risk ratio cannot be calculated, we also reported the risk difference (RD). For continuous outcomes, such as seizure severity, retention, behavioural outcomes, cognitive outcomes and quality of life outcomes, we reported mean differences or standardised mean differences if there was significant heterogeneity.
Where there were insufficient data or significant heterogeneity and meta‐analysis was deemed inappropriate, we discussed outcomes narratively.
Unit of analysis issues
In the event of unit of analysis issues being present across the included studies (for example, cross‐over studies, cluster‐randomised or repeated measures), we planned to:
determine whether the methods used in such studies were appropriate;
combine extracted effect sizes from such studies through a generic inverse variance meta‐analysis.
For studies assessing more than one treatment arm, such as different doses, we planned to pool the data from the intervention groups and compare these with the placebo group. We also planned to discuss the effect of different doses narratively. For studies comparing two or more treatment groups, such as different AEDs and a placebo, we planned to combine intervention groups in a general AED versus placebo meta‐analysis. If there were sufficient data, we also planned to carry out subgroup analysis stratifying by AED. Therefore we would complete separate, specific AED versus placebo meta‐analyses.
Dealing with missing data
In the event of missing data, we sought the reasons for this by contacting study authors in order to conclude whether data were missing at random or not.
Assessment of heterogeneity
As this review evaluates various AEDs, we expected to see differences in control groups, measures, interventions and timescales that may not necessarily relate to one another. In this instance, we would not combine data in meta‐analysis.
Two authors planned to assess visually the clinical and methodological heterogeneity of the included studies, and we planned to use the I2 statistic and Chi2 tests where applicable to assess statistical heterogeneity. We judged a Chi2 P value of less than 0.1 and an I2 value greater than 50% to indicate statistical heterogeneity.
Assessment of reporting biases
We planned to request protocols from study authors and investigate outcome reporting bias using the ORBIT matrix system (Kirkham 2010).
To examine publication bias, we searched for unpublished data by carrying out a comprehensive search of multiple sources and requesting any unpublished data from study authors. We also planned to look for small study effects to establish the likelihood of publication bias.
Data synthesis
Trials are summarised in the text and in tables. Where two or more studies investigated the same intervention in similar populations and using the same outcomes, we considered combining the results in a fixed‐effect meta‐analysis. However, in the event of statistical heterogeneity being identified, we planned to complete a random‐effects meta‐analysis.
Subgroup analysis and investigation of heterogeneity
We planned to stratify subgroup analysis by type of intervention and duration of AED. If there was statistical heterogeneity across studies, we planned to carry out a random‐effects meta‐analysis.
Sensitivity analysis
In the event of any inconsistencies or peculiarities being identified, we planned to carry out a sensitivity analysis.
Results
Description of studies
For the original review, we carried out literature searches in the following databases: Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2006, Issue 4), MEDLINE (Ovid; 1966 to present), PsycINFO (Ovid 1966 to present), EMBASE (Ovid; 1980 to April 2005) and the National Research Register Archive. For more information about this please see Electronic searches.
The original search in October 2006 found 18 studies relevant studies. We excluded six of these studies as they did not satisfy the inclusion criteria (Bielmann 1978; Espe‐Lillo 1992; Luna 1989; Schlumberger 1994; Smith 1968; Turner 1970). Therefore we included 12 studies in the original review: five cross‐over trials (Battaglia 1991; Eriksson 1998; Gigli 1988; Kaski 1991; Siegel 1999), six parallel studies (Crumrine 1989; Kerr 2005; Motte 1997; Ritter 1993; Sachdeo 1999; Yamatogi 1997), and one parallel, open‐label study (Crawford 2001).
The updated searches in Septembere 2012 found four relevant studies; we added one study to the included studies (Glauser 2008), we excluded one study (Donati 2006), we added one as an ongoing study (Bjurulf 2008), and we added one study as awaiting classification (Hellings 2009).
Results of the search
We carried out updated searches to include all studies published from the date of the previous search (September 2012) to 2 September 2014. We identified 68 studies in total. After initial screening, we excluded 60 studies as they were irrelevant. We carried out full‐text review on the remaining eight studies. At this stage, we excluded seven papers (Bjornaes 2013; Conry 2014; Gidal 2013; Hagebeuk 2011; Hodoba 2013; Santos 2013; Tolbert 2014). Additionally, we excluded the previously identified study awaiting classification as it did not satisfy the inclusion criteria (Hellings 2009). We located no published data for the previously identified study Bjurulf 2008. We contacted the study authors and discovered that a paper had been submitted for publication; therefore this study remains in the ongoing studies section (see Characteristics of ongoing studies). We have therefore added one study to the included studies in this review (Ng 2011). Details of the search results can be found in Figure 1.
1.

Study flow diagram.
In total, the present review includes 14 studies and data from 1116 participants. Nine AEDs were included in the studies: lamotrigine, carbamazepine, gabapentin, topiramate, felbamate, clobazam, cinromide, flunarizine and rufinamide.
Included studies
Battaglia 1991 This was a randomised, placebo‐controlled, double‐blind, cross‐over trial studying the effect of flunarizine add‐on therapy in refractory childhood epilepsy. Inclusion criteria included being aged 18 years or less, having clearly and reliably documented epileptic seizures, a seizure incidence for absences and myoclonic attacks of not less than one per day and for all other seizure types of not less than four per month. Antiepileptic drug regimen was optimised as far as possible by adjustment of serum levels. No evidence of non‐compliance and informed consent was obtained. Exclusion criteria included any serious illness other than epilepsy, the regular taking of drugs other than antiepileptic drugs, pregnancy or risk of conception in females of reproductive age. Twenty patients (10 male, 10 female) aged six to 18 years (mean age 12 years seven months) were entered into the study; however, seven were withdrawn (three male, four female). The reasons for withdrawal included: one non‐compliance, two withdrawal of consent (not caused by deterioration of seizures or flunarizine adverse events), one drowsiness (probably drug‐related), one due to intercurrent illness unrelated to therapy, one patient missed several review appointments, and one missed the final examination because of non‐medical problems. All data therefore related to 13 patients (two patients were aged below 12 years). Twelve patients had 'mental retardation'; one mild, one moderate and 10 with severe 'retardation'. Epilepsy types included: symptomatic generalised epilepsy (10), cryptogenic generalised epilepsy (two), symptomatic partial epilepsy (one), myoclonic absences (one) and temporal lobe epilepsy (one). The dosage regime of flunarizine was 5 mg per day (for patients less than 10 years of age) or 10 mg per day (for patients older than 10 years of age). Evaluation of the activity of flunarizine was based on the total number of seizures observed during the treatment phase (four months for each cross‐over leg). No adverse effects were seen in those patients completing the trial.
Crawford 2001 This was an unblinded, parallel, add‐on RCT study comparing the efficacy and safety of gabapentin and lamotrigine in adults with learning disabilities. Inclusion criteria included: aged 12 years and over, of either sex and with localisation‐related epilepsy that was not satisfactorily controlled by existing antiepileptic drugs. Participants had to be taking one, two or three standard antiepileptic drugs (not including gabapentin or lamotrigine) but still not achieving satisfactory control; and have a minimum of four seizures in each 28‐day period and no seizure‐free 28‐day period in the preceding three months. Patients had to have a degree of learning disability and meet any level of the Diagnostic and Statistical Manual of Mental Disorders IV (DSM‐IV) criteria for mental retardation.
Exclusion criteria included: individuals who had had primary generalised seizures, symptomatic generalised epilepsy or a history of non‐epileptic seizures; concurrent therapy with antacids or a recent participation in any clinical trial; pregnant women and women of childbearing age not using adequate contraception; and a known hypersensitivity to gabapentin or lamotrigine or significant renal or hepatic dysfunction. Eighty‐three patients were randomised (53 male, 30 female), age range 15 to 59 years; the mean age for the gabapentin‐treated group was 38 years. Thirty‐four patients who were treated with gabapentin completed the study and 35 patients treated with lamotrigine completed the study, indicating that five gabapentin‐treated patients and nine lamotrigine‐treated patients either withdrew or dropped out. Results were analysed by intention‐to‐treat. The reasons for withdrawal or dropout in the gabapentin‐treated group included: adverse events (n = 3), other (n = 1) and protocol violation (n = 1). In the lamotrigine‐treated group the reasons included: carer withdrawal of consent (n = 1), adverse events (n = 4) and other (n = 4). All patients had a degree of intellectual disability and had to meet any level of the DSM‐IV criteria for mental retardation. The seizure types present in the gabapentin group included: simple partial (9%), complex partial (49%), secondary generalised (34%), absence (3%), tonic (0%) and other (5%) and in the lamotrigine group they included simple partial (13%), complex partial (43%), secondary generalised (37%), absence (2%), tonic (2%) and other (3%). The dose of gabapentin was increased during the 14‐week titration period at the investigators' discretion, to a maximum of 3600 mg. The mean dose during the 10‐week evaluation period was 1749 mg per day (minimum = 400 mg and maximum = 3600 mg). The primary efficacy outcome measure was the reduction in seizure frequency between the baseline period and the last eight weeks of the treatment period, assessed using the R‐ratio= (T ‐ B)/(T + B) where T and B were the seizure frequencies per 28 days during treatment and baseline respectively. Secondary outcome measures included the number of responders (seizure frequency reduced by 50% or more) and an assessment of mood, behaviour and dependency. The overall incidence of adverse events was similar in both treatment groups (62% with gabapentin and 50% with lamotrigine). Approximately 10% reported serious adverse events on gabapentin and 11% on lamotrigine.
Crumrine 1989 This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel trial evaluating the effects of cinromide in patients with Lennox‐Gastaut syndrome. Inclusion criteria included: only patients with Lennox‐Gastaut syndrome; patients aged two to 18 years; patients must have had seizures for at least six months and could be receiving no more than three concomitant antiepileptic drugs; parents/guardians had to demonstrate their ability to administer medications as instructed and to maintain satisfactory seizure counts according to the study protocol. Exclusion criteria consisted of: patients with a history of tonic‐clonic status epilepticus within the previous six months; an anoxic episode requiring resuscitation during the previous year; a progressive central nervous system (CNS) lesion or progressive hepatic; cardiovascular, haematopoetic, renal, pulmonary, gastrointestinal, ophthalmologic or endocrine disease; a predisposing condition which might interfere with absorption, distribution or excretion of drugs; severe adverse effects from pre‐study medications; changes to medication regimen within the previous four weeks; if they had been maintained on a ketogenic diet within the previous two weeks or treated with adrenocorticotropic hormone (ACTH) or corticosteroids within the previous four weeks; previous exposure to cinromide; severe learning disability or other impairment that safety and efficacy evaluations would have been unreliable or difficult to perform. Seventy‐three patients were randomised but sufficient data for analysis were available for only 56 patients (26 treated with cinromide and 30 on placebo). No information was provided regarding withdrawals or dropouts. The age range for the 56 evaluable patients was two to 18 years; the mean for the cinromide group was 7.38 (SD 3.65) and the mean for the placebo group was 7.93 (SD 4.87). Thirty‐four patients were male and 22 female. The level of intellectual disability was stated and patients with severe retardation were excluded from the study. Epilepsy was defined according to a modification of the International Classification of Epileptic Seizures. Seizure types included: absence seizures (39), myoclonic seizures (36), tonic seizures (25), atonic seizures (30) and tonic clonic seizures (20). Cinromide was initiated at doses of 20 mg/kg/day to 40 mg/kg/day and further increases (to a total daily maximum of 83 mg/kg to 109 mg/kg) were prescribed at weekly visits according to a fixed dosing schedule. The objective was to achieve an optimal dose during the first six treatment weeks and to maintain this dose during the last 12 treatment weeks. Concomitant AEDs could be changed during the treatment period if plasma drug concentrations exceeded specified limits above or below baseline means. The primary statistical analysis of seizure frequency compared the changes from the baseline period (six weeks) to the treatment period (12 weeks for most patients) in the cinromide and placebo groups. Global rating scale evaluations at weeks 12, 18 and 24 were also compared. No information was given on adverse events. This study was terminated early when it became clear that cinromide was not effective.
Eriksson 1998 This was a randomised, double‐blind, placebo‐controlled, cross‐over trial evaluating the efficacy of lamotrigine in children and adolescents with refractory generalised epilepsy. Inclusion criteria were all children older than two years and adolescents with refractory or intractable generalised epilepsy with more than two seizures per month. Exclusion criteria were the presence of liver, renal or progressive neurologic disease or the diagnosis of focal disease. Thirty consecutive patients (15 male, 15 female) were entered into the study; the age range was 2.5 to 22 years, median age 9.9 years. Patients entered into an open‐phase study, with a duration of two to 12 months, during which time the optimal lamotrigine dose was found for each child. Those who were categorised as responders (more than a 50% reduction in seizure frequency, severity or both; or improvements in behaviour or motor skills, or both) were then randomised into the double‐blind phase. Seventeen patients were randomised to receive either lamotrigine or placebo. Two patients were withdrawn and data from 15 patients were used in the analyses. Of the randomised patients, all had mental retardation (IQ less than 70). Seizure types included tonic‐clonic (nine), tonic/atonic (15), myoclonic (16), atypical absences (14) and other seizure types (two). The dosing regime was individualised and determined during the open‐phase period. Patients were taking one to three other antiepileptic drugs. The primary outcome measure was the percentage reduction in mean monthly seizure frequency. Secondary outcome measures included severity of seizures, functional status of patient and frequency of adverse events. During the cross‐over phase no adverse events were seen in the lamotrigine group; however, in the placebo group 10 had fatigue and four had more intense seizures.
Gigli 1988 (Italian article) This was a randomised, double‐blind, placebo‐controlled, cross‐over study evaluating the effect of flunarizine in an intellectually disabled population with epilepsy. Inclusion criteria were not stated. Twenty‐six patients were randomised to one of two groups: group I would receive flunarizine in the first arm and a placebo in the second whereas group II would receive the placebo in the first arm and flunarizine in the second. The mean age of group I (10 males, five females) was 11.7 years and of group II (one male, 10 females) was 18.3 years (the groups differed in respect of the proportion of patients with secondary generalised epilepsy and partial epilepsy). Intellectual disability was described in 25 patients as slight (three), moderate (seven) and severe (15). Seizure types included: six patients with secondary generalised epilepsy and nine patients with partial epilepsy with or without secondary generalisation (Group I); and seven patients with secondary generalised epilepsy and four with partial epilepsy with or without secondary generalisation (Group II). There was a baseline period of three months followed by a three‐month treatment period, a three‐month washout period and a three‐month treatment period. The dosing regime was not described, however the dose of flunarizine was 10 mg/day; patients were maintained on the medication they were receiving prior to the start of the study. A primary efficacy variable was not stated but alterations in seizure frequency were recorded together with behavioural observations. In group I additional data were obtained in patients with the ability to respond on antiepileptic drugs (AEDs): electroencephalography (EEG) together with an attention test (Toulouse modified). No adverse effects or withdrawals were reported. For the update in 2014, this paper was unavailable. We contacted the study authors but access to the full‐text paper was unavailable.
Glauser 2008 This was randomised, double‐blind, placebo‐controlled study of antiepileptic drug rufinamide in patients with Lennox‐Gastaut syndrome. The eligible patients were between four and 30 years of age (median 12), and had multiple types of seizures (including tonic‐atonic and atypical absences) with a minimum of 90 seizures in the month before baseline and a recent history of a slow spike and wave pattern on EEG. Patients who were receiving more than three AEDs, pregnant or not using adequate contraception, had correctable aetiology of their seizures, had history of generalised tonic‐clonic status epilepticus within 30 days of baseline or had a history of any clinically significant non‐neurological medical condition were excluded. One hundred and thirty‐eight patients were randomised, receiving either rufinamide (n = 74) or placebo (n = 64). The two treatment groups had a compatible distribution of concomitant AEDs with valproic acid, lamotrigine and topiramate being the most frequently used. This study consisted of a 28‐day baseline period followed by a 84‐day, double‐blind, placebo‐controlled, parallel‐group treatment period. This phase consisted of a 14‐day titration period followed by a 70‐day maintenance period. The target dosage was approximately 45 mg/kg of rufinamide per day. The primary efficacy was defined as reduction in total seizure frequency, tonic‐atonic ("drop attack") seizure frequency and seizure severity rating. The secondary efficacy measures were response to treatment (% of patients with at least 50% reduction in seizure frequency during the double‐blind phase), % change in seizure frequency per 28 days relative to baseline (for each seizure subtype other than tonic‐atonic) and parental global evaluation (alertness, interaction with environment, daily activity performance, responsiveness to verbal request and seizure severity). Ten patients in the rufinamide group discontinued therapy (six because of adverse effects, three for unsatisfactory therapeutic effect and one for withdrawal of consent). Five patients in the placebo group did not complete the study (two for protocol violations, one for unsatisfactory therapeutic effect, one due to an administrative problem and one following withdrawal of consent).
Kaski 1991 This was a randomised, double‐blind, comparator‐controlled, cross‐over study evaluating the pharmacokinetic properties and efficacy of a conventional carbamazepine preparation (three daily doses) and a slow‐release carbamazepine preparation (two daily doses) in intellectually disabled patients. Inclusion criteria were: only people previously treated with carbamazepine and whose carbamazepine concentrations had been on a therapeutic level for at least two months, and in spite of treatment, were suffering four seizures per month or more. All patients were intellectually disabled. Twenty‐one patients (nine males, 11 females) were randomised but one was withdrawn due to appendicitis. The mean age of the 20 evaluable patients was 24.9 years (range six to 38 years). Seizure types/syndromes included: partial seizures (four), secondary generalised (eight), primary generalised (two) and Lennox‐Gastaut syndrome (six). There was a baseline period of two months followed by two 10‐week treatment periods. During the two study periods, the daily dose of carbamazepine was the same as during the baseline period. Conventional carbamazepine was given three times daily and the slow‐release carbamazepine twice daily. Placebo tablets were used to make the daily number of tablets taken identical; the double‐dummy technique was used. Primary efficacy variables were not stated, however, in the aims section the authors stated that the aims were: (a) to carry out a 24‐hour pharmacokinetic trial comparing slow‐release carbamazepine and conventional carbamazepine when given at different dosing frequencies, and (b) to look at what effect a reduction in dosing frequency of carbamazepine had on seizure control, that is the difference between slow‐release and conventional carbamazepine. No information was given regarding adverse events.
Kerr 2005 This was a randomised, double‐blind, placebo‐controlled, add‐on trial evaluating the efficacy and safety of topiramate in patients with epilepsy and an intellectual disability. Patients were included if they were aged 12 years and above; weighed at least 45 kg; had a diagnosis of epilepsy with a documented six‐month history of at least four seizures per month; and an intellectual disability, defined as significantly below average general intellectual functioning and an IQ of less than 70. Patients had to be on treatment with one to three other antiepileptic drugs and to have an identified carer. Patients were excluded if they had absence seizures only, or non‐epileptic seizures; a history of psychosis or psychiatric problems in the last year; nephrolithiasis or renal impairment; previous treatment with topiramate; or treatment in the last three months with acetazolamide, zonisamide, triamterene, more than 2 g/day of vitamin C or chronic use of antacids or calcium supplements; or women who were pregnant, lactating or without adequate contraception. Seventy‐four patients were randomised in equal numbers to the topiramate and placebo groups. Seventeen patients withdrew and an intention‐to‐treat analysis was performed both for the efficacy data (n = 72) and the safety data (n = 74). The actual numbers completing the study were 28 in the topiramate group and 29 in the placebo group. The age of the patients was 12 years and above, with 39 males and 34 females. All patients had an intellectual disability, defined as a significant below‐average general intellectual functioning and an IQ of 70 or less. Seizures were classified in accordance with the International Classification of Seizures. Seizure types included generalised tonic‐clonic (n = 36), partial (n = 54), partial with generalisation (n = 34) and other types (n = 33). The drug dose regime consisted of an 18‐week titration phase to achieve the optimum dose of study drug, to a maximum of 400 mg/day topiramate (for adults) or 9 mg/kg/day (children) and a 12‐week maintenance period where the dose of study medication and concomitant medications remained constant. Patients were taking up to three concomitant antiepileptic drugs. The primary efficacy variables were the mean change in total seizure frequency and the mean change in seizure severity during the treatment phase. Adverse events were reported by 92% of patients in the topiramate group and 84% in the placebo group. Sixteen per cent of the topiramate group reported serious adverse events, possibly drug‐related in four cases and 11% in the placebo group. The number of patients reporting at least one adverse event leading to a permanent stop in study medication was 18.9% (n = 7) in the topiramate group and 16.2% (n = 6) in the placebo group.
Motte 1997 This was a randomised, double‐blind, placebo‐controlled, parallel study examining the effect of lamotrigine in patients with Lennox‐Gastaut syndrome. Inclusion criteria included patients having more than one type of predominantly generalised seizure for at least one year, under 11 years of age at the onset of epilepsy, seizures at least every other day, intellectual disability based on IQ or developmental assessments and recent EEGs showing an abnormal background and a pattern of slow spike‐and‐wave complexes. Exclusion criteria included if patients had a progressive neurodegenerative disorder, were receiving more than three antiepileptic drugs, weighed less than 15 kg or were taking valproate. A total of 169 patients were randomised, 79 to lamotrigine (68% male) and 90 to placebo (50% male). For safety data analyses 169 patients were evaluable and for efficacy data 167 patients were evaluable; two patients were excluded due to lack of data. Seven patients in the lamotrigine group were withdrawn early, four due to protocol violations and three due to adverse events. Fourteen patients in the placebo group did not complete the study; three due to protocol violations, seven due to adverse events and two due to deterioration of seizure control. These 21 patients were included in the analyses using an 'as treated analysis' rather than a true intention‐to‐treat analysis. Age range was three to 25 years (mean = 9.6 years). All patients had intellectual impairment: 92% (n = 73) had moderate to severe intellectual disability in the lamotrigine group and 91% (n = 82) in the placebo group. Patients were assigned to one of four dosing regimens according to concomitant valproate use and body weight. The 16‐week treatment period comprised a six‐week titration period, two weeks in which the dose was fixed and an additional eight weeks during which the fixed dose could be increased during week eight or 12 to no more than the maximal allowable daily dose. Primary efficacy variables included: (a) percentage change from baseline in the frequency of major motor seizures, and (b) median changes from baseline in the frequency of drop attacks, tonic clonic seizures and atypical absences. The primary safety variable was the frequency of reported adverse events. The adverse event most frequently responsible for withdrawal was clinical deterioration of seizure control (one patient in the lamotrigine group and six in the placebo group).
Ng 2011 This was a multi‐centre randomised, double‐blind, placebo‐controlled, parallel‐group trial examining the effect of clobazam in patients with Lennox‐Gastaut syndrome. It was conducted at 51 sites in the United States, India, Europe and Australia between August 2007 and December 2009. Inclusion criteria included: aged two to 60 years old, weigh 12.5 kg or over, onset of Lennox‐Gastaut syndrome before the age of 11, one or more generalised seizures (including drop seizures) for six months or longer and a previous EEG report showing generalised, slow spike‐and‐wave patterns (< 2.5 Hz). A total of 238 patients were randomised to one of four groups: 59 participants in the placebo group, 58 participants in the low‐dose group (clobazam 0.25 mg/kg/day; maximum 10 mg/day), 62 participants in the medium‐dose group (clobazam 0.5 mg/kg/day; maximum 20 mg/day) and 59 participants in the high‐dose group (clobazam 1.0 mg/kg/day; maximum 40 mg/day). The study included a four‐week baseline period, a three‐week titration period and a 12‐week maintenance period, followed by either continuation in an open‐label study or a two to three‐week taper period. A total of 177 participants completed the study: 41/59 participants in the placebo group, 50/58 in the low‐dose group, 45/62 in the medium‐dose group and 41/59 in the high‐dose group. Intention‐to‐treat analysis was carried out on all participants who did not have one or more daily seizure measurement during the maintenance period, which was 217 participants. The percentage of patients who reported experiencing one or more adverse effects was 67.8% for placebo, and 72.4%, 88.7% and 76.3% for the low‐, medium‐ and high‐dosage groups, respectively. Adverse effects that had more than a 10% difference in reports from participants in the placebo group and any clobazam groups were somnolence, pyrexia, lethargy, drooling and constipation. Sedation was reported in 4.5% (8/177) clobazam‐treated patients (one in the low‐, two in the medium‐ and five in the high‐dosage group). Of the adverse effects, somnolence and drooling increased with increasing clobazam dosages. Twenty‐nine participants (one in the placebo group, and four in the low‐, nine in the medium‐ and 15 in the high‐dosage group) had their dosage reduced during the study because of an adverse effect. A dose‐related trend was observed for overall adverse effects, which led to discontinuation from the study. In total 27 participants (two in the placebo group, and four in the low‐, eight in the medium‐ and 13 in the high‐dosage group) discontinued because of adverse effects. Sixteen participants reported serious adverse effects (two in the placebo group, and three in the low‐, six in the medium‐ and five in the high‐dosage group). Serious adverse effects included lobar pneumonia and pneumonia.
Ritter 1993 This was a randomised, double‐blind, placebo‐controlled, add‐on, parallel study aimed at evaluating the efficacy and safety of felbamate in patients with Lennox‐Gastaut syndrome. Inclusion criteria included: males and females with Lennox‐Gastaut syndrome who had a history of multiple types of seizures and a minimum of 90 atonic or atypical absence seizures per month during an eight‐week pre‐study screening phase; were taking no more than two antiepileptic drugs; had no evidence of progressive central nervous system lesions on magnetic resonance imaging or computed tomography; weighed at least 11.3 kg and had a slow spike‐wave complex on EEG. Exclusion criteria were: females who were pregnant or not using adequate contraception; a history of identifiable progressive neurologic disorders; anoxic episodes within the past year; poor compliance with past antiepileptic therapy; recent drug or alcohol abuse; a major medical illness or previous suicide attempts; recently received corticotropin; following a ketogenic diet; or inadequate supervision. Seventy‐three patients were randomised, 37 to the felbamate treatment group and 36 to the placebo group. All patients were included in the statistical analyses for efficacy and safety. Two patients were withdrawn from the study due to adverse events, one from each group. The age range was four to 36 years; 51 were male and 22 female. The authors give no specific details on the level of intellectual disability in the randomised population, however the introduction indicates that Lennox‐Gastaut syndrome is "characterised by mental retardation". Patients had multiple types of seizures as characterised by Lennox‐Gastaut syndrome. Seizures were classified according to the ILAE 1981 classification. Felbamate was titrated during the first 14 days of the treatment phase to a maximum of 45 mg/kg/day or 3600 mg/day, whichever was less, followed by a 56‐day maintenance period. Felbamate was given in addition to the patients' current antiepileptic drugs, but patients could not be on more than two antiepileptic drugs before study initiation. The primary efficacy variables were the per cent change in average seizure frequency between baseline and the treatment phase for the two groups, parents'/guardians' global evaluations of the patients' quality of life and the total number of atonic seizures. Efficacy was also assessed by two secondary variables: parental counts of total seizures and parental counts of generalised tonic‐clonic seizures. The evaluation of safety included monitoring adverse events, vital signs and body weight, general physical and neurologic examinations, measurements of plasma concentrations of felbamate and standard antiepileptic drugs and clinical laboratory evaluations. Adverse events were similar in the two groups although anorexia, vomiting and somnolence occurred more frequently in the felbamate group and diarrhoea occurred more frequently in the placebo group. Severe effects were reported by eight patients in the felbamate group and three in the placebo group. One patient in the felbamate group withdrew due to somnolence and ataxia and one patient in the placebo group withdrew due to pancreatitis.
Sachdeo 1999 This study was a randomised, double‐blind, placebo‐controlled, add‐on trial evaluating the efficacy and safety of topiramate in patients with Lennox‐Gastaut syndrome. Patients had to be aged between one and 30 years, weigh at least 11.5 kg, have an EEG showing a slow spike‐and‐wave pattern, seizure types including tonic‐atonic and either a history of or active atypical absences. Other types could include tonic‐clonic, myoclonic and partial onset seizures. Patients had to have at least 60 seizures during the month prior to entering the baseline phase while being maintained on one or two standard antiepileptic drugs. Patients were excluded if they had a history of recent cardiovascular, respiratory, hepatic, renal, gastrointestinal or haematologic illness or malignancy; seizures due to progressive disease, documented status within three months of baseline; drug or alcohol abuse; a psychiatric or mood disorder requiring medication or electroconvulsant therapy, within six months of baseline; poor compliance with therapy; anoxic episodes requiring resuscitation within one year before the study or nephrolithiasis; treatment with, or use of, an experimental drug or device within 60 days of baseline, use of acetazolamide or zonisamide within 60 days of baseline; treatment with a ketogenic diet or adrenocorticotropic hormone within six months before the study; use of benzodiazepines on more than an occasional basis; presence of clinically significant electrocardiogram (ECG) abnormalities; and history of inability to take medication or maintain a seizure calendar, independently or with assistance. Ninety‐eight patients were randomised to the study, 48 patients to receive topiramate and 50 to receive placebo. One patient was considered to be a premature withdrawal and seizure counts were estimated from data provided at the final visit. Age ranges for the topiramate group were two to 29 years (mean 11.2 years) and were two to 42 years (mean 11.2 years) for the placebo group. Fifty‐three patients were male and 45 were female. The authors give no specific details on the level of intellectual disability in the randomised population, however they state that Lennox‐Gastaut syndrome is "characterised by mental retardation". Seizures were classified according to the ILAE 1989 classification. Seizure types included tonic‐atonic, atypical absences, tonic‐clonic, myoclonic and partial seizures. The most common seizure types recorded during the baseline period were; atonic (90 patients), atypical absence (70), tonic (51), myoclonic (46), tonic‐clonic (38), complex partial (16), absence (nine), partial evolving to secondary generalised (four), clonic (two) and unspecified (six). The dosing schedule involved a titration period divided into three one‐week intervals. In the first week the drug dosage was approximately 1 mg/kg/day administered twice daily in equal doses. Dosage was then increased to approximately 3 mg/kg/day during the second week and to the target dose of 6 mg/kg/day during the third week. Patients were then followed for an eight‐week maintenance period on 6 mg/kg/day or their maximum tolerated dose. At study entry all patients were taking concomitant antiepileptic drugs. The primary determinant of efficacy was based on a statistically significant between‐group difference with respect to either a reduction in average monthly seizure rate for all seizure types combined or each component of a compound variable consisting of a per cent reduction in drop attacks (tonic and atonic) and the parental global evaluation of seizure severity. Secondary efficacy variables were a reduction in the average monthly rate of major seizures (drop attacks and tonic‐clonic) and the percentage of patients considered to be treatment responders, defined as those with a 50% or more, 75% or more or 100% reduction from baseline for drop attacks, major seizures and all seizures. Twenty‐three per cent of topiramate‐treated patients had severe adverse events compared with 10% of the placebo‐treated patients. Three patients in the placebo group and nine in the topiramate‐treated group experienced at least one adverse event that required either a dosage reduction or temporary discontinuation of treatment. However, no patient discontinued the study due to an adverse event.
Siegel 1999 This was a randomised, placebo‐controlled, double‐blind, cross‐over trial evaluating the efficacy of felbamate as add‐on therapy to valproic acid in Lennox‐Gastaut syndrome. Patients were included if they had a clinical diagnosis of Lennox‐Gastaut syndrome and were able to tolerate valproic acid monotherapy, had discontinued all benzodiazepines for at least four weeks and carbamazepine or phenytoin for at least two weeks prior to study entry. Patients were excluded based on the following criteria: history of generalised tonic‐clonic status epilepticus within six months prior to entrance; the presence of serious medical or psychiatric disorders requiring medication; clinical imaging or electrographic evidence for a partial seizure disorder or structural abnormality; presence of a metabolic disorder; presence of a known, treatable seizure aetiology; alcohol or drug abuse or progressive neurological disorders; a history of poor compliance with antiepileptic drug therapy; if routine laboratory screening yielded any results of a white blood cell count of less than 3000/mm³ and less than 50% granulocytes, a platelet count of less than 100,000/mm³, a haemoglobin count of less than 11 or an SGOT (serum glutamate oxaloacetic transaminase), SGPT (serum glutamate pyruvate transaminase) or alkaline phosphatase level greater than twice the upper limit of normal. Fourteen patients were randomised to the study; there was one withdrawal (reason not stated) and therefore, 13 patients were used in the data analysis. The age range was 4.2 to 15.7 years and there were seven males and seven females. The authors do not indicate any specific levels of learning disability except that all patients had Lennox‐Gastaut syndrome. The authors also do not state which classification system was used to diagnose seizures; however, seizure types included generalised tonic‐clonic, tonic, atonic or myoclonic seizures. Patients needed to be on sodium valproate, the concomitant drug, as monotherapy (at steady state) prior to adding in the felbamate or placebo. Felbamate/placebo titration began on day one at 15 mg/kg/day in four daily doses, and was then increased by 15 mg/kg/day every three days up to a dose of 45 mg/kg/day on day seven, with a maximum dose of 3600 mg/day. Observations started on day 10 and lasted seven weeks. On days 59 to 71, cross‐over to the alternate regime and washout occurred at the same rate as for the initial titration. Observations for the second cross‐over period lasted from days 72 to 121. The only concomitant therapy was valproic acid. A primary efficacy variable was not stated, however the outcomes measured were: the percentage change in total seizure frequency after felbamate/placebo add‐on to valproic acid, compared with baseline (valproic acid monotherapy); and seizures, both clinical and electrographic, counted from video‐EEG tapes. Patients' parents reported no adverse events from felbamate; however, there was a significant decrease in the mean white blood counts, which was related to the felbamate levels and mean platelet counts associated with increased valproic acid levels.
Yamatogi 1997 (Japanese article) This was a randomised, single‐blind, comparator‐controlled, parallel trial evaluating the effect of clobazam in intractable childhood epilepsies. Patients were included if they had intractable symptomatic/cryptogenic generalised childhood epilepsy, experienced one or more seizures per week, were aged 17 years or less and the number of concomitant antiepileptic drugs was three or less. Patients were excluded if they were taking benzodiazepines at the time of recruitment. Eighty patients were randomised, 38 to clobazam and 42 to the comparator drug clonazepam. Sixty‐six patients (34 clobazam, 32 clonazepam) were analysed for efficacy data and 76 (36 clobazam, 40 clonazepam) were analysed for safety data. The age range of the patients was 10 months to 17 years, with 39 males and 27 females. Ninety‐two per cent (61/66) of the patients were classed as intellectually disabled. Seizure types included simple partial (two), complex partial (five), secondary generalised (three), atypical absence (eight), myoclonic (15), tonic (29), tonic spasm (33), generalised tonic clonic (14), atonic (seven) and other (three). Fifty per cent of patients had Lennox‐Gastaut syndrome. Patients were started on clobazam at 0.2 mg/kg/day and increased stepwise 0.2 mg/kg/day over two to four weeks, up to 0.8 mg/kg/day. All patients except for two were taking concomitant antiepileptic drugs. The primary efficacy variable was the number of patients with a 50% or greater reduction in seizure frequency. Seizure frequency at the end of the treatment phase (12 weeks) was compared with the frequency at the end of the baseline period of four weeks. A total of 41.7% of patients treated with clobazam experienced an adverse event compared with 57.5% of patients treated with clonazepam. Fourteen patients were withdrawn from the study, only two due to drug adverse events.
Excluded studies
In the original review, we excluded six studies: two articles were not randomised trials (Luna 1989; Schlumberger 1994); one article was in abstract form and appeared to be a report of an open‐label follow‐up study from a double‐blind trial for which we are awaiting further details from the authors (Espe‐Lillo 1992); one study included participants with an IQ that was higher than our inclusion criteria (Smith 1968); one study had only one participant who was aged 12 or over and only six out of 18 participants had Lennox‐Gastaut syndrome (Turner 1970); and one study where the data were complicated by the use of antipsychotics (Bielmann 1978).
In the 2011 update, one identified study was excluded as it did not include participants with intellectual disability (Donati 2006). This update also identified one study awaiting classification (Hellings 2009), and one study as ongoing (Bjurulf 2008).
In the present update, we excluded seven identified studies: three studies were not randomised controlled trials (Conry 2014; Hodoba 2013; Tolbert 2014), three studies did not include people with intellectual disability (Bjornaes 2013; Gidal 2013; Santos 2013), and one study did not include any participants over 12 years of age (Hagebeuk 2011).
We contacted the authors of the previously identified ongoing study to enquire about published work (Bjurulf 2008). We were informed that this study is still ongoing and they hope to publish some data later this year. Therefore this study has remained as ongoing in the present review.
Hellings 2009 compared two different methods of transitioning between AEDs; this is not relevant to this review and therefore we have excluded this study.
Risk of bias in included studies
For an overview of the 'Risk of bias' assessments across the domains, please see Figure 2. For more detail of the 'Risk of bias' assessment for each study, please see Characteristics of included studies.
2.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
We rated two studies as having low risk of bias for both sequence generation and allocation concealment (Ng 2011; Sachdeo 1999). Five studies did not describe the method of randomisation but we rated them as having low risk of bias for allocation concealment (Eriksson 1998; Glauser 2008; Kaski 1991; Kerr 2005; Yamatogi 1997). We rated one study as having high risk of bias for randomisation sequence generation but low risk of bias for allocation concealment (Kaski 1991). We rated one study as having high risk of bias for sequence generation but it did not describe how allocation concealment was achieved (Crawford 2001). The remaining five studies did not give sufficient information about how sequence generation or allocation concealment was achieved (Battaglia 1991; Crumrine 1989; Gigli 1988; Motte 1997; Siegel 1999).
Blinding
Five studies were double‐blinded trials and it was clear that the method of blinding was sufficient (Eriksson 1998; Glauser 2008; Kaski 1991; Kerr 2005; Sachdeo 1999). One study was described as a double‐blinded trial but there was insufficient information about how participants were blinded (Ritter 1993). Six studies were described as double‐blinded trials but there was insufficient information about how blinding was achieved and therefore we rated these studies as having unclear risk of bias (Battaglia 1991; Crumrine 1989; Gigli 1988; Motte 1997; Ng 2011; Siegel 1999). One study was a single‐blinded trial and we rated it as having low risk of performance bias as participants were blinded but high risk of detection bias as outcome assessors and study personnel were unblinded (Yamatogi 1997). One study was an open‐label trial and therefore participants, study personnel and outcome assessors were unblinded (Crawford 2001).
Incomplete outcome data
Five studies completed an intention‐to‐treat analysis (Crawford 2001; Glauser 2008; Motte 1997; Ng 2011; Ritter 1993), two studies reported reasons for withdrawals that were unlikely to affect outcomes (Eriksson 1998; Kaski 1991), one study reported comparable rates of withdrawals between the groups (Kerr 2005), and one study stated that there were no withdrawals. We rated one study as having high risk of attrition bias as the reasons for withdrawals were likely to be associated with the interventions and no intention‐to‐treat analysis was carried out (Battaglia 1991). Four studies did not provide reasons for withdrawals (Crumrine 1989; Sachdeo 1999; Siegel 1999; Yamatogi 1997).
Selective reporting
We rated six studies as having low risk of reporting bias (Battaglia 1991; Crawford 2001; Kaski 1991; Ritter 1993; Sachdeo 1999; Siegel 1999). One study did not report many P values for outcomes (Crumrine 1989), and one study did not appear to report outcomes from the neurological evaluation and therefore we rated it as high risk of bias (Motte 1997). There was insufficient information to establish the risk of reporting bias for six studies (Eriksson 1998; Gigli 1988; Glauser 2008; Kerr 2005; Ng 2011; Yamatogi 1997).
Other potential sources of bias
We rated three studies as having high risk of other bias (Battaglia 1991; Crumrine 1989; Ng 2011), one study as having low risk of other bias (Kerr 2005), and 10 studies as having unclear risk of other bias (Crawford 2001; Eriksson 1998; Gigli 1988; Glauser 2008; Kaski 1991; Motte 1997; Ritter 1993; Sachdeo 1999; Siegel 1999; Yamatogi 1997).
Effects of interventions
See: Table 1
Epilepsy/seizure outcomes
Retention on treatment
Retention time could only be established from one study: Ritter 1993 reported that one patient (2.7%) in the felbamate group withdrew from the trial on study day 47 due to somnolence and ataxia and one patient (2.8% of those randomised to placebo) from the placebo group withdrew on study day 42 due to pancreatitis. All other studies did not provide information on when patients were withdrawn from the study. The number of dropouts at the endpoint was described in a number of the studies. We obtained no additional retention time information for the present review update. Total reported withdrawal rates were 35% (7/20; Battaglia 1991), 25.6% (61/238; Ng 2011), 23.3% (17/73; Crumrine 1989), 23.0% (Kerr 2005), 17.5% (Yamatogi 1997), 16.9% (Crawford 2001), 12.4% (Motte 1997), 11.8% (Eriksson 1998), 10.8% (Glauser 2008), 7.1% (Siegel 1999), 4.8% (Kaski 1991), and 2% (Sachdeo 1999). Gigli 1988 did not report any withdrawals.
Of the studies that provided information for withdrawals specific to each group: in the clobazam group (Ng 2011), 24.0% (43/179) of participants withdrew from the study: 13.8% from the low‐dosage group, 27.4% from the medium‐dosage group and 10.5% from the high‐dosage group; in the gabapentin group (Crawford 2001), 12.8% of participants withdrew from the study and in the lamotrigine group (Crawford 2001), 20.5% of participants withdrew; in the rufinamide group (Glauser 2008), 13.5% of participants withdrew from the study; in one topiramate group no withdrawals were reported (Sachdeo 1999), whereas in another topiramate group (Kerr 2005), 24.3% of patients withdrew.
Seizure freedom
Four studies recorded seizure freedom rates:
Eriksson 1998 showed one patient (6.7%) to be seizure‐free during the lamotrigine‐treatment phase.
Ritter 1993 recorded three (8.1%) patients to be seizure‐free during a 10‐week treatment phase on felbamate. This compared with one patient (2.8%) seizure‐free whilst on placebo.
Ng 2011 reported that two participants (3.5%) in the placebo group were drop seizure‐free during the 12‐week maintenance period, compared with four (7.5%), seven (12.1%) and 12 (24.5%) participants in the low‐, medium‐ and high‐dosage groups.
Crawford 2001 recorded three (7.7%) patients on gabapentin and five (11.4%) patients on lamotrigine as seizure‐free during a 10‐week minimum evaluation phase. One patient in the lamotrigine group was seizure‐free during the titration phase (a period of up to 14 weeks).
Statistical adjustments were not possible for the Eriksson 1998 study due to the cross‐over design and the zero events within the placebo group. Ng 2011 only reported seizure freedom for drop seizures. Therefore only one study compared the rates of seizure freedom between antiepileptic drug (AED) and placebo groups (Ritter 1993). This analysis indicated that there was no significant difference in the number of participants becoming seizure‐free between the treatment and placebo groups (risk ratio (RR) 2.92, 95% confidence interval (CI) 0.32 to 26.77).
Reduction in seizure frequency
Eight studies presented data on the reduction in seizure frequency observed during the treatment period compared with the baseline period: Crumrine 1989 reported no significant reduction in seizure frequency in patients treated with cinromide as compared with the placebo‐treated patients. This study was terminated early resulting in some patients having less than the 12 weeks of observation in the treatment period. In the study by Glauser 2008 there was a highly significant reduction in the median percentage of total seizure frequency between the rufinamide therapy group compared with the placebo group (32.7% versus 11.7%, P value = 0.0015). There was also a highly significant reduction in tonic‐atonic ("drop attack") seizure frequency between the rufinamide group (42.5% median percentage reduction) and placebo (1.4% increase) (P value < 0.0001). Ritter 1993 found an 11% decrease in seizure frequency in the felbamate‐treated group as compared with a 1% increase in the placebo‐treated group (P value = 0.32). Motte 1997 reported that the median reduction from baseline for all major seizures in the lamotrigine‐treated group was 32% and in the placebo‐treated group was 9% (P value = 0.002) during a 16‐week treatment period. The results were similar when drop attacks and tonic‐clonic attacks were examined separately.
Crawford 2001 reported a mean percentage reduction in seizure frequency of 50.6% in the gabapentin group and 50.8% reduction in the lamotrigine group. Sachdeo 1999 reported that the median percentage reduction from baseline in the average monthly seizure rate for generalised and partial seizures was 20.6% for the topiramate group and 8.8% for the placebo group, which was not significant. However, the median percentage reduction from baseline in the average monthly seizure rate of drop attacks was significantly greater for the topiramate group (14.8%) compared with the placebo‐treated group, a 5.1% increase (P value = 0.041) during a treatment period of 11 weeks. The median percentage reduction from baseline in the average monthly rate of major seizures (drop attacks and tonic‐clonic seizures) in the topiramate group was 25.8% as compared with a 5.2% increase in the placebo group (P value = 0.015).
Kerr 2005 observed no significant difference (P value = 0.099) in the reduction in mean total seizure frequency between the topiramate‐treated group (32.43 ± 46.59% reduction) and the placebo group (1.08 ± 80.63% reduction) during a 12‐week treatment period.
Ng 2011 reported that the mean percentage decrease in the average weekly rate of drop seizures from baseline to maintenance period was 12.1% for drop seizures compared with 41.2% (P value = 0.0120), 49.4% (P value = 0.0015) and 68.3% (P value < 0.0001) for low, medium and high dosages of clobazam. Additionally, Yamatogi 1997 reported significantly higher seizure reduction in the treatment group (clobazam) compared with the control group (clonazepam) (P value = 0.001).
Five studies were cross‐over studies and recorded seizure change as compared with placebo or comparator drug during the cross‐over leg:
Kaski 1991 looked at the effects of conventional carbamazepine as compared with slow‐release carbamazepine and found the mean total number of seizures during the treatment phases was nearly equal: 42.7 (range 4 to 107) during conventional carbamazepine and 44.0 (range 9 to 133) during slow‐release carbamazepine treatment. Each cross‐over leg was a period of 10 weeks each.
Siegel 1999 looked at the effects of felbamate plus valproic acid compared with placebo plus valproic acid. Video EEG counts showed that total seizure frequency was 46% lower (P value < 0.014) and drop attacks 39% lower (P value < 0.027) on felbamate plus valproic acid compared with placebo plus valproic acid. There was no significant difference between parental seizure counts and the EEG seizure count recordings.
Battaglia 1991 showed there was a lower total seizure incidence during the flunarizine period in comparison to the placebo period in five out of 13 patients. The difference between the flunarizine period and the placebo period was not statistically significant. The median total number of seizures during the flunarizine period was 74 (range 13 to 6000) and during the placebo period was 78 (range 22 to 6000). Three of the responders were on a flunarizine‐placebo sequence and the other two were on a placebo‐flunarizine sequence.
Gigli 1988 demonstrated no significant differences in seizure frequency between the flunarizine and placebo groups (although no p values were given).
Eriksson 1998 showed one patient to have a greater than 75% decrease in seizure frequency and nine out of 15 patients showed a greater than 50% seizure reduction during the lamotrigine phase compared with the placebo phase. Five of the remaining children showed some reduction in seizures during the lamotrigine phase and only one patient did not show any difference between the two periods.
Due to insufficient information and the varied methods of reporting seizure frequency, meta‐analysis was not possible.
Responder rates (50% or greater reduction in seizure frequency)
Eight studies recorded responder rates:
Crawford 2001 found the percentage of patients achieving a greater than or equal to 50% reduction in seizure frequency on gabapentin was 50% and on lamotrigine was 48.6%, showing no significant difference.
Glauser 2008 indicated a higher 50% responder rate in the rufinamide group compared with placebo for total seizures (P value = 0.0045) and tonic‐atonic seizures (P value = 0.002).
Motte 1997 found that 33% of lamotrigine‐treated patients and 16% of placebo‐treated patients had a reduction of at least 50% in the frequency of all types of major seizures (P value = 0.01). Significantly more lamotrigine‐treated patients than placebo‐treated patients had a reduction of at least 50% in the frequency of drop attacks and tonic‐clonic seizures (P value = 0.04 and P value = 0.007).
Eriksson 1998 found that nine patients (60%) showed a greater than 50% seizure reduction during the lamotrigine phase; one patient showed a greater than 75% decrease in seizure frequency and one patient was seizure‐free. No patients showed any improvement during the placebo phase.
Sachdeo 1999 showed that the percentage of patients with a 50% or greater reduction from baseline in major seizures (drop attacks and tonic‐clonic seizures) during the double‐blind phase was significantly greater in the topiramate group compared with placebo (33% versus 8%, P value = 0.002). However, there was no significant difference between the treatment group and the placebo group with respect to all seizures in the 50% or greater responder group. Eight topiramate‐treated (17%) versus two (4%) placebo‐treated patients had a 75% or greater reduction in major seizure rates and one patient in the topiramate group was free of major seizures during the double‐blind phase. During the double‐blind phase, 28% (13/46) of the topiramate‐treated patients achieved a 50% or greater reduction in drop attacks compared with 14% (7/49) of the placebo‐treated patients. A 75% or greater reduction in the frequency of drop attacks was achieved in 17% (8/46) of the topiramate group and 6% (3/49) of the placebo group. One patient (2%) from the topiramate group had no drop attacks during the double‐blind phase. However, Kerr 2005 found there was no statistical difference between the topiramate and placebo‐treated groups in the number of responders: 29.7% and 25.7%, respectively.
Ng 2011 reported percentages of participants who had a greater than or equal to 50% reduction in average weekly drop seizures: 31.6% of participants in the placebo group compared with 43.3%, 58.3% (P value = 0.0159) and 77.6% (P value < 0.0001) for low, medium and high clobazam dosage groups, respectively. Yamatogi 1997 found that 61.8% of clobazam‐treated patients and 21.9% of clonazepam‐treated patients had a 50% or greater reduction in seizure frequency.
We included four studies in a meta‐analysis of responder rates for overall seizure frequency (50% or greater reduction in overall seizure frequency) (Eriksson 1998; Glauser 2008; Motte 1997; Sachdeo 1999). We identified no statistical heterogeneity (I2 = 17%). The results of the analysis showed that significantly more people in the AED groups had a 50% or greater reduction in seizure frequency compared with people in the placebo groups (RR 2.58, 95% CI 1.60 to 4.14). As there were zero events in one trial, we also calculated the risk difference (RD 0.16, 95% CI 0.09 to 0.24), see Analysis 1.1. However, it is unclear why Sachdeo 1999 reported this result out of 46 people in the intervention group as it was previously stated that an intention‐to‐treat analysis was carried out and therefore it would be expected that the results would be out of 48 participants.
1.1. Analysis.

Comparison 1 AED vs placebo, Outcome 1 Responder rate (≥ 50% reduction in overall seizure frequency).
We combined data from two studies in a meta‐analysis of responder rates for drop seizure frequency (50% or greater reduction in drop seizure frequency) (Ng 2011; Sachdeo 1999). As Ng 2011 included multiple‐dose arms, we pooled the intervention groups in a meta‐analysis. We identified significant statistical heterogeneity (I2 = 80%) therefore we adopted a random‐effects model. The results of the analysis showed a trend in favour of the AED groups in that more people in the intervention groups reported a 50% or greater reduction in drop seizure frequency, however, this result was not significant (RR 6.04, 95% CI 0.27 to 133.89). However, there was a very large, imprecise effect in Sachdeo 1999 due to zero events in the placebo group, therefore we calculated the risk difference and this indicated that participants in the treatment group showed a significant decrease in the frequency of drop seizures (RD 0.30, 95% CI 0.20 to 0.40), see Analysis 1.2.
1.2. Analysis.

Comparison 1 AED vs placebo, Outcome 2 Responder rate (≥ 50% reduction in rate of drop seizures).
Seizure severity scales
Five studies in the original review reported data on seizure severity:
Crumrine 1989 reported that global evaluation data, which included seizure severity ratings, failed to show any differences between the cinromide and placebo‐treated patients.
Crawford 2001 reported that the key carer rating scale showed significant improvement in seizure severity in the gabapentin‐treated patients and also in the lamotrigine comparator‐treated patients. The physicians' rating scale showed significant improvement in seizure severity in the gabapentin‐treated group.
In the study by Glauser 2008, the rufinamide group had a greater improvement in seizure severity compared with the placebo group (P value = 0.0041).
Sachdeo 1999 found that patients treated with topiramate were nearly twice as likely to show an improvement in seizure severity based on parental global evaluation (P value = 0.037), however Kerr 2005 showed that there was no significant difference between the topiramate and placebo groups in the mean seizure severity scores for the different seizures as assessed by the National Hospital Seizure Severity Scale (NHS3).
The different measures used to calculate seizure severity suggest that there is considerable clinical heterogeneity across the studies and therefore combining data in meta‐analysis is not feasible.
Behavioural outcomes
Three studies reported behavioural outcomes:
Crawford 2001 reported data using the Crichton Royal Behaviour Rating Scale, which showed significant improvement, compared with baseline, in co‐operation and restlessness in the gabapentin‐treated patients. Significant differences between the gabapentin and lamotrigine groups were seen in communication, co‐operation and restlessness on the Crichton Royal Behaviour Rating Scale. The total score also improved significantly (P value = 0.01) in the gabapentin group but not in lamotrigine‐treated patients. The Whelan and Speake Rating Scale measuring challenging behaviour showed that gabapentin was similar to lamotrigine, with both drugs reducing the level of challenging behaviour as a total score over the duration of the trial.
Kerr 2005 showed that there was no significant difference between the topiramate and placebo‐treated groups in the mean total EOS (Epilepsy Outcome Scale) scores, mean total ABC (Aberrant Behaviour Checklist) or mean total ELDQOL (Epilepsy and Learning Disabilities Quality of Life) scores. There was, however, a trend toward significance for improvement of the mean ELDQOL behaviour subscale score for patients treated with topiramate.
Eriksson 1998 noted in their study a clear difference in behaviour and alertness between the lamotrigine‐treated patients and the placebo‐treated patients, even without concomitant seizure reduction. No new data are available for this update.
Due to the different measures used to calculate behavioural outcomes across the studies for behavioural outcomes, meta‐analysis was not feasible.
Cognitive outcomes
No studies reported cognitive outcomes.
Adverse effects
Three studies did not report adverse effects data (Crumrine 1989; Gigli 1988; Kaski 1991).
Glauser 2008 indicated similar rates of adverse effects between the group treated with rufinamide and placebo (81.1% and 81.3%, respectively). However, somnolence, (24.3%), vomiting (21.6%) and pyrexia (13.5%) were the only adverse events reported in more than 10% of patients in the rufinamide treatment arm. The adverse event most often resulting in withdrawal from the study was vomiting.
Ritter 1993 reported that adverse events were similar in patients treated with felbamate or placebo. Only adverse events that occurred at least five times were listed. Anorexia, vomiting and somnolence occurred more frequently in the felbamate group, whereas diarrhoea occurred more frequently in the placebo group. Most adverse events were mild or moderate in severity and self limited. Severe adverse events were reported in eight patients in the felbamate group and three in the placebo group. Somnolence and ataxia resulted in the withdrawal of one patient in the felbamate group and pancreatitis resulted in the withdrawal of one patient in the placebo group.
Siegel 1999 found that patients' parents reported no adverse effects from felbamate, however laboratory results showed that felbamate significantly decreased the mean white blood cell count and valproic acid decreased the platelet counts.
Battaglia 1991 reported no significant adverse events in the 13 patients completing the trial; however, seven patients were withdrawn during the trial, one due to drowsiness, probably related to the drug. The authors do not state whether the patients were taking the drug or placebo when the withdrawals occurred.
Crawford 2001 found that the incidence of all adverse events was 62% (24) in the gabapentin group and 50% (22) in the lamotrigine group. Drug‐related adverse events were 33% (13) in the gabapentin group and 25% (11) in the lamotrigine group. Ten per cent of patients (four) in the gabapentin group and 11% (five) in the lamotrigine group experienced serious adverse events and 8% of patients (three) and 9% (four) were withdrawn due to adverse events in the gabapentin and lamotrigine groups, respectively. One patient on gabapentin was withdrawn due to vomiting (considered to be probably drug‐related); three patients on lamotrigine were withdrawn due to rash and peripheral oedema. One patient treated with gabapentin died due to myocardial infarction but this was not considered to be study drug‐related.
Motte 1997 found that there were no significant differences between the groups in the incidence of adverse events except for colds or viral illnesses, which were more common in the lamotrigine group (p = 0.05) compared with the placebo group. Only adverse events reported by four or more patients in either treatment group were listed. Three patients in the lamotrigine group and seven in the placebo group withdrew from the study due to adverse events. The adverse event most frequently responsible for withdrawal was clinical deterioration of seizure control (one in the lamotrigine group, six in the placebo group). Seven patients in the lamotrigine group (9%) and six in the placebo group (7%) reported a rash that led to the withdrawal of two lamotrigine‐treated patients and one placebo‐treated patient, all of whom were also receiving valproate.
Eriksson 1998 found that no adverse effects were reported during the lamotrigine phase in the double‐blind period. When receiving placebo during the double‐blind phase 10 patients complained of fatigue and four had more severe seizures. No withdrawals were reported due to adverse events.
Sachdeo 1999 showed that 23% of topiramate‐treated patients had severe adverse events, compared with 10% of the placebo‐treated patients. Three patients in the placebo group and nine in the topiramate‐treated group experienced at least one adverse event that required either a dosage reduction or temporary discontinuation of treatment. These adverse events included nervousness, personality disorder, nausea, arthrosis, rash and urinary incontinence in the placebo group and gait abnormality, fatigue, pallor, aggression, personality disorder, somnolence and agitation in the topiramate group. However, no patient discontinued the study due to an adverse event.
Kerr 2005 found that 92% in the topiramate group reported adverse events and 84% in the placebo group. Adverse events were recorded if they were reported in 5% or more of all patients. Serious adverse events were reported by 16% (six) in the topiramate group, possibly drug‐related in four cases. Eleven per cent (four) in the placebo group reported serious adverse events, considered to be drug‐related in one individual. The number of patients reporting at least one adverse event leading to a permanent stop in study medication was 18.9% (seven) in the topiramate group and 16.2% (six) in the placebo group. One patient died in the active group following hospitalisation with increasing drowsiness and confusion. This was reported as likely to be associated with the drug by the investigator.
Ng 2011 reported the percentages of participants who experienced one or more adverse effect: 67.8% in the placebo group compared with 72.4%, 88.7% and 76.3% for the low, medium and high clobazam dosage groups respectively. Adverse effects that were more common in clobazam groups than placebo groups (10% or more difference) were somnolence, pyrexia, lethargy, drooling and constipation. They reported that somnolence and drooling increased in frequency with clobazam dosage. Yamatogi 1997 reported drowsiness, dizziness and vomiting in 41.7% of patients in the clobazam group and in 57.5% in the clonazepam group. Fourteen patients were withdrawn from the study, only two due to drug adverse events of clonazepam.
Quality of life
Only one study assessed the quality of life of patients (Kerr 2005). The Epilepsy Outcome Scale (EOS, a measure of quality of life specific to epilepsy and intellectual disability) showed no significant difference (P value = 0.856) between the topiramate‐treated group and the placebo‐treated group in the mean total EOS score or in any of the subscales. The ELDQOL (Epilepsy and Learning Disabilities Quality of Life) scale also showed no significant difference in the total mean score, however a trend towards significance for improvement of the mean ELDQOL behaviour subscale score was found for topiramate‐treated patients (P value = 0.08). No additional information is available for this review update.
Global rating scale
Crumrine 1989 stated that global evaluation data, which included seizure frequency, seizure severity, signs and symptoms and general functional ability, failed to show any differences between the cinromide and placebo‐treated patients. Ritter 1993 showed that global rating scale outcomes provided by parents or guardians were significantly improved in the felbamate group compared with the placebo group. Kerr 2005 found that there was no significant difference between the groups in the global rating scale outcomes provided by patients, their carers or investigators. This review update does not provide any new information. Ng 2011 reported the percentages of participants who were at least minimally improved on global rating scale outcomes provided by physicians and caregivers: 47.3% (physicians' assessments) and 45.5% (caregivers' assessments) in the placebo group compared with 71.2% to 80.7% (physicians' assessments) and 79.2% to 81.6% (caregivers' assessments) across the clobazam groups. They also reported the percentage of participants who were much improved or very much improved: 23.6% (physicians' assessments) and 25.5% (caregivers' assessments) for the placebo group compared with 46.2% to 64.9% (physicians' assessments) and 41.5% to 59.2% across the clobazam groups. The different measures used and the different way in which the authors identified, interpreted and reported results from global rating scales meant that the combination of data available was not possible and therefore we did not attempt meta‐analysis.
Discussion
Summary of main results
The aim of this study was to assess the data available from randomised controlled trials (RCTs) of antiepileptic drug interventions in people with epilepsy and intellectual disability. This review includes a total of 14 RCTs, which comprised data from 1116 participants.
In terms of retention on treatment, only one study reported the time at which dropouts occurred. The majority of studies, however, did report the number of dropouts. The frequency of dropouts varied considerably across the included studies with dropouts in the intervention groups ranging from 0% to 24.3%.
Three studies assessed the efficacy of lamotrigine: two studies used a placebo group as a comparator (Eriksson 1998; Motte 1997), and one study compared lamotrigine against gabapentin (Crawford 2001). Eriksson 1998 and Motte 1997 concluded that the use of lamotrigine as an adjunctive intervention to existing drug regimes is both well tolerated and produces a positive clinical response in people with epilepsy and intellectual disability. Similarly, Crawford 2001 concluded that the use of lamotrigine in this population is an effective antiepileptic medication comparable to gabapentin. One study found that topiramate used as an adjunctive intervention is well tolerated and can have a significant positive clinical response in people with epilepsy and intellectual disability (Sachdeo 1999). Additionally, Kerr 2005 found a similar trend for adjunctive topiramate intervention in seizure reduction, however this result did not reach significance. Two studies found that clobazam was an effective adjunctive intervention for people with epilepsy and intellectual disability compared with a placebo (Ng 2011), and compared with clonazepam (Yamatogi 1997). Although one study suggested that flunarizine, used as an adjunctive therapy, showed a tendency to produce lower seizure incidence, neither study assessing the effectiveness of flunarizine as an adjunctive intervention found any significant beneficial effect in reducing seizure frequency (Battaglia 1991; Gigli 1988). One study found no significant effect of cinromide compared with a placebo in people with epilepsy and intellectual disability (Crumrine 1989). The results from one study showed that rufinamide, used as an adjunctive intervention, was well tolerated and efficacious in people with epilepsy and Lennox‐Gastaut syndrome (Glauser 2008). One study compared the efficacy of carbamazepine against slow‐release carbamazepine and concluded that reducing the fluctuation in serum concentrations may also reduce seizure frequency (Kaski 1991). Ritter 1993 concluded that felbamate can be beneficial to patients with epilepsy and Lennox‐Gastaut syndrome. Furthermore, Siegel 1999 found that felbamate combined with valproic acid may be synergistic in reducing seizure frequency, although they did acknowledge that the reduction in seizure is in part due to the increased doses of valproic acid.
With respect to the effect of antiepileptic drugs (AEDs) in the population of people with learning disability, overall the studies indicate relatively good efficacy. Seizure freedom was achieved in some patients. We combined incidence data for seizure frequency in AED groups compared with placebo groups from two studies and the results suggested a trend in favour of the AED intervention (Eriksson 1998; Ritter 1993). However, this result was not significant. We combined data for responder rates for overall seizure frequency (≥ 50% reduction in overall seizure frequency) from four studies in a meta‐analysis and the results suggested that AEDs produced significantly higher rates of responders than placebo (Eriksson 1998; Glauser 2008; Motte 1997; Sachdeo 1999). We combined data for responder rates for drop seizures (≥ 50% reduction in drop seizure frequency) from two studies (Ng 2011; Sachdeo 1999). However, although there was a trend in favour of AEDs over placebo, this result was not significant. Six out of ten studies recording seizure reduction in comparison to baseline showed a benefit of the drug compared with placebo; Motte 1997, Glauser 2008 and Ng 2011 showed a significant decrease in seizures in the treatment group compared with placebo; Ritter 1993, Sachdeo 1999 and Battaglia 1991 showed a decrease but this was not statistically significant. The study by Glauser 2008 also showed a significant reduction in the frequency of tonic‐clonic seizures between the rufinamide and placebo‐treated groups. Of the remaining three studies, Crawford 2001 showed equivalence between the active drug and comparator, Yamatogi 1997 showed a significantly higher seizure reduction in the clobazam‐treated group compared with the comparator (clonazepam), whilst Crumrine 1989 and Kerr 2005 showed no significant difference between drug (cinromide and topiramate, respectively) and placebo groups.
Of the four cross‐over studies comparing the active drug to placebo, three showed positive improvements; Eriksson 1998 found lamotrigine to be significantly more effective than placebo, Siegel 1999 also showed total seizure frequency and drop attacks to be significantly reduced in the felbamate group compared with placebo and Battaglia 1991 showed that there was a lower total seizure incidence during the flunarizine period in comparison to placebo in five out of 13 patients but that this difference was not statistically significant. However, Gigli 1988 demonstrated no significant differences in seizure frequency between flunarizine and placebo groups (although no p values were reported). The cross‐over study carried out by Kaski 1991 was an active comparator study and showed equivalence in seizure frequency between carbamazepine and slow‐release carbamazepine during the early phase of the study but during the last two weeks the seizure frequency was significantly lower during the comparator treatment phase.
The effect of AEDs on seizure severity showed that of the five studies reporting this outcome, Crumrine 1989 failed to show a difference between the treatment and placebo group and Kerr 2005 showed that there was no significant difference between the drug and placebo groups. However, both Crawford 2001 and Sachdeo 1999 showed significant improvement in seizure severity in the treatment groups. Following the recent review, we identified that the study by Glauser 2008 showed an improvement in seizure severity and a higher 50% responder rate for total and tonic‐atonic seizures in the rufinamide‐treated arm compared with placebo.
Crawford 2001 reported significant improvements in behavioural outcomes for both the active drug and comparator drug, however the validity of the rating scales used cannot be verified in the literature. Kerr 2005, using validated rating scales, showed that there was no significant difference between the treatment and placebo groups.
Quality of life measures were only recorded in one study (Kerr 2005) and showed no significant differences between the drug and placebo‐treated groups.
The incidence of adverse events reported in participants taking rufinamide was 81.1%. This was similar to the rate of adverse events reported in the placebo group in the same study. Three studies reported the rate of adverse events in felbamate groups. One study reported a similar rate of adverse events in both the felbamate and placebo groups. One of three studies involving felbamate indicated a possible increase in serious adverse events in the treatment group compared with placebo, although the total number of adverse events was comparable between the groups. In two studies, no adverse events were reported in participants taking felbamate, however the laboratory outcomes of one study indicated that it may be associated with a decrease in the white blood cell count. The incidence of adverse events in the gabapentin group was reported by one study and indicated a rate of 33% of drug‐related adverse events, 10% of which were considered serious, compared with an incidence of 25% in the lamotrigine group, 11% of which were considered serious. A separate study involving lamotrigine reported greater withdrawals due to adverse events in the placebo group compared with patients taking lamotrigine. Both studies involving topiramate recorded adverse events although the rates were variable, ranging from 23% to 92% for the topiramate groups and from 10% to 84% for the placebo groups. Both studies indicate a slightly higher incidence of adverse events in the treatment groups. The higher rates were reported within the same study, which may reflect the criteria they used for reporting adverse events. Both studies involving clobazam reported adverse events. One study reported rates ranging from 72.4% to 88.7% in the treatment group and 67.8% in the placebo group. A separate study reported the incidence rate of adverse events in the clobazam group to be 41.7%, compared with 57.5% in the clonazepam group. Therefore, the rates of adverse events in the clobazam group were elevated compared with placebo but lower compared with clonazepam. No adverse events were recorded in the studies involving flunarizine, cinromide or carbamazepine. Included studies adopted various methods to record adverse events, which may explain some variability between the rates of adverse events across the studies. Without the use of a standardised means to record adverse events, there is poor consistency between studies in categorising and recording adverse events.
Overall completeness and applicability of evidence
In the present review we identified only 14 RCTs from our literature search, which illustrates how under‐investigated this patient population is with regards to pharmacological interventions for epilepsy. The reasons for this paucity may be multifactorial, including the difficulty in obtaining informed consent, co‐medication and adherence to the study protocol. This is a difficult area of research but the production of these 14 studies does demonstrate that relevant evaluative research is feasible.
The main focus of this review was to assess the effectiveness and tolerability of pharmacological interventions in people with epilepsy and intellectual disabilities. To assess this with the limited number of studies, we pooled and compared all pharmacological interventions with placebo groups. Whilst this provides some evidence of the benefits of pharmacological interventions compared to placebo groups, it fails to address the relative effectiveness of each AED separately and therefore offers no information regarding which pharmacological intervention is most beneficial within this population.
As a result of the variable designs, small sample sizes and high dropout rates in some studies, analysis and therefore the reliability of the results is limited and few clinically relevant conclusions can be drawn from this review.
Quality of the evidence
Of the 14 studies identified in this review, we rated seven studies as having a low risk of bias (Eriksson 1998; Glauser 2008; Kaski 1991; Kerr 2005; Ng 2011; Ritter 1993; Sachdeo 1999), six studies as having an unclear risk of bias (Battaglia 1991; Crumrine 1989; Gigli 1988; Motte 1997; Siegel 1999; Yamatogi 1997), and one study as high risk of bias (Crawford 2001).
Potential biases in the review process
None identified.
Agreements and disagreements with other studies or reviews
Despite not being able to draw robust conclusions from the present review, the results suggest that pharmacological interventions are effective in reducing seizure frequency in people with epilepsy and intellectual disability. As discussed, refractory epilepsy is common in people with intellectual disability. This is consistent with other Cochrane reviews, which have found that lamotrigine (Ramaratnam 2001), clobazam (Michael 2008), gabapentin (Al‐Bachari 2013), and topiramate (Pulman 2014) are effective interventions for refractory epilepsy. However, each of the previous reviews also identified a paucity of high quality RCTs and therefore meta‐analysis was not possible.
Contrary to the findings of the present review, a previous Cochrane review identified three RCTs assessing the effectiveness of felbamate in refractory epilepsy, two of which found no significant benefit (Shi 2014).
Authors' conclusions
Implications for practice.
Since the initial version of this review we identified one additional study relevant to this topic but the previous conclusions remain the same. Professionals in the field of epilepsy see numerous people with epilepsy and intellectual disability in need of treatment change. This review shows that in the majority of cases where antiepileptic drugs (AEDs) were trialled in this population, moderate reduction in seizure frequency and occasional seizure freedom were obtained. It seems reasonable to say, in general, that AEDs with proven effectiveness in the general epilepsy population are also effective in refractory epilepsy in people with intellectual disability.
It is not possible to comment on the relative efficacy of medications, making clinical choice decisions difficult.
Clinical decisions are often guided by concern over adverse events. This is particularly pertinent for people with complex co‐morbidity who are often in supported care environments. It is difficult to draw reliable conclusions from this review due to clinical and methodological heterogeneity. However, from the limited evidence provided by this review, it appears that the adverse events experienced by people with epilepsy and intellectual disability are similar to those reported within the general population.
One key area of concern is that of behavioural exacerbation. The majority of studies are unhelpful due to their lack of or unreliable measures in this area. However, where measured, little obvious impact on behaviour was seen in terms of behaviour disorder.
In summary, the review offers tentative support for the use of AEDs to reduce seizure frequency in people with epilepsy and intellectual disability. Additionally, the evidence presented in this review suggests that the adverse events are similar to those known to affect the general population. In this review we pooled all AEDs together, therefore it is difficult to differentiate the effectiveness of each AED individually.
Implications for research.
Key areas for research have been identified by this review.
There is a lack of data on many interventions. Neither the efficacy of newer AEDs such as levetiracetam, pregabalin, tiagabine and zonisamide, or older AEDs such as sodium valproate, have been investigated in this population in terms of seizure reduction or safety.
The issue of behavioural and cognitive safety is of concern to patients, carers and professionals, but has been inadequately investigated for the majority of AEDs. Investigators often used unreliable or no measures for assessment. Specific studies to investigate behavioural and cognitive safety are needed for this population.
Access to research is also an issue. The vast majority of studies recruit small numbers and this is compounded by the paucity of studies in general. The research community and its funders need to redress this imbalance positively through recognition of recruitment and access difficulties in this population.
What's new
| Date | Event | Description |
|---|---|---|
| 2 September 2014 | New search has been performed | Searches updated 2 September 2014; one new study has been included. |
| 2 September 2014 | New citation required but conclusions have not changed | Conclusions remain unchanged. |
History
Protocol first published: Issue 3, 2005 Review first published: Issue 3, 2007
| Date | Event | Description |
|---|---|---|
| 17 October 2012 | New search has been performed | Searches updated on 13 September 2012 but no new studies found. |
| 16 December 2010 | New search has been performed | Searches updated January 2011; we have included one new study and added one as an ongoing study. |
| 20 August 2008 | Amended | Converted to new review format. |
Acknowledgements
We would like to thank Janine Beavis and Ivana Dojcinov for contributions to previous versions of this review.
Appendices
Appendix 1. Cochrane Epilepsy Group's Specialised Register search strategy
#1 MeSH DESCRIPTOR Learning Disorders Explode All WITH BL CF CI CL CO DI DH DT EC EN EP EH ET GE HI IM ME MI MO NU PS PA PP PC PX RA RI RH SU TH US UR VI
#2 MeSH DESCRIPTOR Intellectual Disability Explode All WITH BL CF CI CL CO DI DH DT EC EM EN EP EH ET GE HI IM ME MI MO NU PS PA PP PC PX RA RI RT RH SU TH US UR VE VI
#3 MeSH DESCRIPTOR Mentally Disabled Persons Explode All WITH CL HI LJ PX RH SN
#4 MeSH DESCRIPTOR Mental Disorders Explode All WITH BL CF CI CL CO CN DI DH DT EC EM EN EP EH ET GE HI IM ME MI MO NU PS PA PP PC PX RA RI RT RH SU TH US UR VE VI
#5 MeSH DESCRIPTOR Developmental Disabilities Explode All WITH BL CF CI CL CO DI DH DT EC EN EP EH ET GE HI IM ME MI MO NU PS PA PP PC PX RA RI RH SU TH US UR VI
#6 MeSH DESCRIPTOR Fragile X Syndrome Explode All WITH BL CF CI CL CO DI DH DT EC EM EN EP EH ET GE HI IM ME MI MO NU PS PA PP PC PX RA RI RT RH SU TH US UR VE VI
#7 MeSH DESCRIPTOR Rett Syndrome Explode All WITH BL CF CI CL CO DI DH DT EC EM EN EP EH ET GE HI IM ME MI MO NU PS PA PP PC PX RA RI RT RH SU TH US UR VE VI
#8 MeSH DESCRIPTOR Down Syndrome Explode All WITH BL CF CI CL CO DI DH DT EC EM EN EP EH ET GE HI IM ME MI MO NU PS PA PP PC PX RA RI RT RH SU TH US UR VE VI
#9 MeSH DESCRIPTOR Autistic Disorder Explode All WITH BL CF CI CL CO DI DH DT EC EN EP EH ET GE HI IM ME MI MO NU PS PA PP PC PX RA RI RH SU TH US UR VI
#10 MeSH DESCRIPTOR Angelman Syndrome Explode All WITH BL CF CI CL CO DI DH DT EC EM EN EP EH ET GE HI IM ME MI MO NU PS PA PP PC PX RA RI RT RH SU TH US UR VE VI
#11 MeSH DESCRIPTOR Cerebral Palsy Explode All WITH BL CF CI CL CO CN DI DH DT EC EM EN EP EH ET GE HI IM ME MI MO NU PS PA PP PC PX RA RI RT RH SU TH US UR VE VI
#12 MeSH DESCRIPTOR Tuberous Sclerosis Explode All WITH BL CF CI CL CO CN DI DH DT EC EM EN EP EH ET GE HI IM ME MI MO NU PS PA PP PC PX RA RI RT RH SU TH US UR VE VI
#13 learning NEXT (disab* or difficult* or problem* or disorder* or handicap*)
#14 mental* NEXT (retard* or disab* or deficien* or handicap* or incapacity or disorder*)
#15 intellect* NEXT (disab* or impair* or handicap*)
#16 cognitive NEXT impairment
#17 development* NEXT disab*
#18 multipl* NEXT handicap*
#19 Down* NEAR2 syndrome
#20 "Fragile X" NEXT syndrome
#21 Rett* NEAR2 syndrome
#22 Lennox NEXT Gastaut NEXT syndrome
#23 tuberous NEXT sclerosis
#24 autism OR autistic
#25 Angelman* NEAR2 syndrome
#26 West* NEAR2 syndrome
#27 cerebral NEXT palsy
#28 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27
#29 MeSH DESCRIPTOR Epilepsy Explode All WITH DT
#30 MeSH DESCRIPTOR Anticonvulsants Explode All WITH AD AE AG AN AI BL CF CS CH CL CT DU EC HI IM IP ME PK PD PO RE ST SD TU TO UR
#31 antiepilep* OR anti‐epilep* OR anticonvuls* OR anti‐convuls* OR aed OR aeds OR acetazolamide OR alodorm OR antilepsin OR arem OR ativan OR barbexaclone OR beclamide OR brivaracetam OR carbagen OR carbamazepine OR celontin OR cerebyx OR chlonazepam OR chloracon OR cloazepam OR clobazam OR clonazepamum OR clonex OR clonopin OR clorazepate OR convulex OR depacon OR depak* OR depamide OR desitin OR diacomit OR diamox OR diastat OR diazepam OR dilantin OR diphenin* OR diphenylhydantoin OR divalpr* OR dormicum OR ecovia OR emeside OR epanutin OR epiject OR epilim OR episenta OR epival OR eptoin OR ergenyl OR erimin OR eslicarbazepine OR ethadione OR ethosuximide OR ethotoin OR ethylphenacemide OR exalief OR excegran OR ezogabine OR fanatrex OR felbamate OR felbatol OR fosphenytoin OR frisium OR fycompa OR gabapentin OR gabarone OR gabitril OR gabrene OR ganaxolone OR garene OR gralise OR halogabide OR halogenide OR hibicon OR hypnovel OR iktorivil OR inovelon OR insoma OR intensl OR keppra OR klonopin OR kriadex OR lacosamide OR lamict* OR lamitor OR lamitrin OR lamogine OR lamotrigine OR lamotrine OR landsen OR levetiracetam OR liskantin OR loraz OR lorazepam OR losigamone OR luminal OR lyrica OR mebaral OR mephenytoin OR mephobarbit* OR mephyltaletten OR mesantoin OR mesuximide OR methazolamide OR methsuximide OR methylphenobarbit* OR midazolam OR mogadon OR mylepsinum OR mysoline OR neogab OR neptazane OR neurontin OR nimetazepam OR nitrados OR nitrazadon OR nitrazepam OR normison OR novo‐clopate OR nupentin OR nydrane OR onfi OR orfiril OR orlept OR ormodon OR ospolot OR oxcarbazepine OR pacisyn OR paraldehyde OR paramethadione OR paxadorm OR paxam OR peganone OR perampanel OR petinutin OR petril OR phemiton OR phenacemide OR pheneturide OR phenobarbit* OR phensuximide OR phenytek OR phenytoin OR posedrine OR potiga OR pregabalin OR primidone OR prodilantin OR progabide OR prominal OR prysoline OR ravotril OR remacemide OR remnos OR resimatil OR restoril OR retigabine OR riv?tril OR rufinamide OR sabril OR seclar OR selenica OR seletracetam OR sertan OR somnite OR stavzor OR stedesa OR stiripentol OR sulthiam* OR sultiam* OR talampanel OR tegretol OR temazepam OR temesta OR teril OR tiagabine OR timonil OR topamax OR topiramate OR tranxene OR tridione OR trileptal OR trimethadione OR trobalt OR urbanol OR valance OR valcote OR valium OR valnoctamide OR valparin OR valpro* OR versed OR vigabatrin OR vimpat OR zalkote OR zarontin OR zebinix OR zonegran OR zonisamide
#32 #29 OR #30 OR #31
#33 #28 AND #32
#34 >2010:YR AND INREGISTER
#35 #33 AND #34
Appendix 2. CENTRAL (CRSO) search strategy
#1 MESH DESCRIPTOR Learning Disorders EXPLODE ALL TREES
#2 MESH DESCRIPTOR Intellectual Disability EXPLODE ALL TREES
#3 MESH DESCRIPTOR Mentally Disabled Persons EXPLODE ALL TREES
#4 MESH DESCRIPTOR Mental Disorders EXPLODE ALL TREES
#5 MESH DESCRIPTOR Developmental Disabilities EXPLODE ALL TREES
#6 MESH DESCRIPTOR Fragile X Syndrome EXPLODE ALL TREES
#7 MESH DESCRIPTOR Rett Syndrome EXPLODE ALL TREES
#8 MESH DESCRIPTOR Down Syndrome EXPLODE ALL TREES
#9 MESH DESCRIPTOR Autistic Disorder EXPLODE ALL TREES
#10 MESH DESCRIPTOR Angelman Syndrome EXPLODE ALL TREES
#11 MESH DESCRIPTOR Cerebral Palsy EXPLODE ALL TREES
#12 MESH DESCRIPTOR Tuberous Sclerosis EXPLODE ALL TREES
#13 learning NEXT (disab* or difficult* or problem* or disorder* or handicap*)
#14 mental* NEXT (retard* or disab* or deficien* or handicap* or incapacity or disorder*)
#15 intellect* NEXT (disab* or impair* or handicap*)
#16 cognitive NEXT impairment
#17 development* NEXT disab*
#18 multipl* NEXT handicap*
#19 Down* NEAR2 syndrome
#20 "Fragile X" NEXT syndrome
#21 Rett* NEAR2 syndrome
#22 Lennox NEXT Gastaut NEXT syndrome
#23 tuberous NEXT sclerosis
#24 autism OR autistic
#25 Angelman* NEAR2 syndrome
#26 West* NEAR2 syndrome
#27 cerebral NEXT palsy
#28 #1 OR #2 OR #3 OR #4 OR #5 OR #6 OR #7 OR #8 OR #9 OR #10 OR #11 OR #12 OR #13 OR #14 OR #15 OR #16 OR #17 OR #18 OR #19 OR #20 OR #21 OR #22 OR #23 OR #24 OR #25 OR #26 OR #27
#29 (epilep* OR seizure* OR convuls*):TI,AB,KY
#30 MESH DESCRIPTOR Epilepsy EXPLODE ALL TREES
#31 MESH DESCRIPTOR Seizures EXPLODE ALL TREES
#32 #29 OR #30 OR #31
#33 #28 AND #32
#34 MESH DESCRIPTOR Epilepsy EXPLODE ALL TREES WITH QUALIFIERS DT
#35 MESH DESCRIPTOR Anticonvulsants EXPLODE ALL TREES
#36 (antiepilep* or anti‐epilep* or anticonvuls* or anti‐convuls* or AED or AEDs):TI,AB,KY
#37 MESH DESCRIPTOR Acetazolamide EXPLODE ALL TREES
#38 MESH DESCRIPTOR Carbamazepine EXPLODE ALL TREES
#39 MESH DESCRIPTOR Clonazepam EXPLODE ALL TREES
#40 MESH DESCRIPTOR Clorazepate Dipotassium EXPLODE ALL TREES
#41 MESH DESCRIPTOR Diazepam EXPLODE ALL TREES
#42 MESH DESCRIPTOR Ethosuximide EXPLODE ALL TREES
#43 MESH DESCRIPTOR Lorazepam EXPLODE ALL TREES
#44 MESH DESCRIPTOR Mephenytoin EXPLODE ALL TREES
#45 MESH DESCRIPTOR Mephobarbital EXPLODE ALL TREES
#46 MESH DESCRIPTOR Midazolam EXPLODE ALL TREES
#47 MESH DESCRIPTOR Methazolamide EXPLODE ALL TREES
#48 MESH DESCRIPTOR Nitrazepam EXPLODE ALL TREES
#49 MESH DESCRIPTOR Paraldehyde EXPLODE ALL TREES
#50 MESH DESCRIPTOR Phenobarbital EXPLODE ALL TREES
#51 MESH DESCRIPTOR Phenytoin EXPLODE ALL TREES
#52 MESH DESCRIPTOR Primidone EXPLODE ALL TREES
#53 MESH DESCRIPTOR Temazepam EXPLODE ALL TREES
#54 MESH DESCRIPTOR Trimethadione EXPLODE ALL TREES
#55 MESH DESCRIPTOR Valproic Acid EXPLODE ALL TREES
#56 MESH DESCRIPTOR Vigabatrin EXPLODE ALL TREES
#57 (acetamidothiadiazolesulfonamide OR acetamox OR acetazolam OR acetazolamid OR acetazolamide OR acetazolamine OR acetazoleamide OR acetozalamide OR ak zol atenezol OR carbonic anhydrase inhibitor 6063 OR cidamex OR dazamide OR defiltran OR dehydratin OR diacarb OR diakarb OR diamox OR didoc OR diluran OR diuramid OR diureticum holzinger OR diuriwas OR diutazol OR donmox OR duiramid OR edemox OR eumicton OR fonurit OR glaupax OR glupax OR natrionex OR nephramid OR nephramide OR phonurit OR storzolamide OR vetamox):TI,AB,KY
#58 (barbexaclone):TI,AB,KY
#59 (beclamide):TI,AB,KY
#60 (brivaracetam):TI,AB,KY
#61 (atretol OR biston OR calepsin OR carbamazepen OR carbamazepine OR carbamezepine OR carbatrol OR carbazepine OR carbelan OR cbz OR epitol OR equetro OR finlepsin OR karbamazepin OR lexin OR neurotol OR novo carbamaz OR sirtal OR spd417 OR stazepin OR stazepine OR tegretal OR tegretol OR telesmin OR teril OR timonil):TI,AB,KY
#62 (aedon OR anxirloc OR castilium OR chlorepin OR clarmyl OR clobam OR clobamax OR clobator OR clobazam OR clofritis OR clopax OR clorepin OR frisium OR grifoclobam OR karidium OR lucium OR mystan OR noiafren OR onfi OR sederlona OR sentil OR urbadan OR urbanil OR urbanol OR urbanyl):TI,AB,KY
#63 (antelepsin OR antilepsin OR chlonazepam OR cloazepam OR clonazepam OR clonazepamum OR clonopin OR iktorivil OR klonopin OR landsen OR rivotril):TI,AB,KY
#64 (chlorazepate OR chlorazepic acid OR clorazepate OR clorazepic acid OR gen xene OR novo clopate OR tranxene):TI,AB,KY
#65 (alboral OR aliseum OR alupram OR amiprol OR an ding OR ansiolin OR ansiolisina OR apaurin OR apozepam OR armonil OR assival OR atensine OR atilen OR bensedin OR bialzepam OR calmocitene OR calmpose OR cercine OR ceregulart OR diacepan OR dialag OR dialar OR diapam OR diastat OR diazemuls OR diazemulus OR diazepam OR diazepan OR diazetard OR dienpax OR dipam OR dipezona OR dizac OR domalium OR duksen OR duxen OR e pam OR eridan OR eurosan OR evacalm OR faustan OR faustan OR freudal OR frustan OR gewacalm OR gihitan OR kabivitrum OR kiatrium OR la iii OR lamra OR lembrol OR levium OR liberetas OR mandrozep OR methyldiazepinone OR morosan OR neurolytril OR noan OR novazam OR novo dipam OR paceum OR paranten OR paxate OR paxel OR plidan OR pro pam OR q pam OR quetinil OR quiatril OR quievita OR relaminal OR relanium OR renborin OR ruhsitus OR saromet OR sedapam OR sedipam OR seduksen OR seduxen OR serenack OR serenamin OR serenzin OR servizepam OR setonil OR sibazon OR sibazone OR solis OR sonacon OR stesolid OR stesolin OR tensopam OR tranimul OR tranqdyn OR tranquase OR tranquirit OR tranquo OR umbrium OR unisedil OR usempax OR valaxona OR valeo OR valiquid OR valitran OR valium OR valrelease OR vatran OR velium OR vival OR vivol OR zetran OR zipan):TI,AB,KY
#66 (divalproex):TI,AB,KY
#67 (eslicarbazepine):TI,AB,KY
#68 (aethosuximide OR asamid OR atysmal OR capitus OR emeside OR epileo petit mal OR ethosuccimide OR ethosuccinimide OR ethosuxide OR ethosuximide OR ethymal OR etomal OR etosuximida OR mesentol OR pemal OR pemalin OR pentinimid OR peptinimid OR petinimid OR petnidan OR piknolepsin OR pyknolepsinum OR ronton OR simatin OR succimal OR succimitin OR suxilep OR suximal OR suxin OR suxinutin OR thetamid OR thilopemal OR zaraondan OR zarodan OR zarondan OR zarontin OR zartalin):TI,AB,KY
#69 Ethadione:TI,AB,KY
#70 (accenon OR ethotoin OR ethotoine OR ethotoinum OR etotoina OR peganone OR pegoanone):TI,AB,KY
#71 (felbamate OR felbamyl OR felbatol OR taloxa):TI,AB,KY
#72 (phenytoin OR cerebyx OR fosfenitoina OR fosphenytoin OR fosphenytoine OR fosphenytoinum OR prodilantin):TI,AB,KY
#73 (aclonium OR gabapentin OR gabapentine OR gabapentino OR gabapentinum OR gabapetin OR neurontin):TI,AB,KY
#74 (ganaxolone):TI,AB,KY
#75 (erlosamide OR harkoseride OR lacosamide OR spm 927 OR vimpat):TI,AB,KY
#76 (gw 273293 OR lamictal OR lamictin OR lamotrigina OR lamotrigine OR lamotriginum):TI,AB,KY
#77 (keppra OR levetiracetam OR levetiracetamum OR levitiracetam):TI,AB,KY
#78 (almazine OR alzapam OR anxiedin OR aplacassee OR ativan OR bonatranquan OR delormetazepam OR emotival OR idalprem OR lorabenz OR lorax OR loraz OR lorazepam OR lorsilan OR chlorooxazepam OR chloroxazepam OR pro dorm OR psicopax OR punktyl OR quait OR securit OR sedatival OR sedazin OR somagerol OR tavor OR temesta OR wypax):TI,AB,KY
#79 (losigamone):TI,AB,KY
#80 (epiazin OR gerot epilan OR insulton OR mephentoin OR mephenytoin OR mesdontoin OR mesontoin OR methoin OR methyl hydantoin OR methylphenetoin OR metydan OR phenantoin OR sacerno OR sedantional OR sedantoin OR sedantoinal OR triantoin):TI,AB,KY
#81 Mesuximide:TI,AB,KY
#82 (enfenemal OR enphenemal OR enphenemalum OR mebaral OR meberal OR mebroin OR menta bal OR mephobarbital OR mephobarbitone OR mephytal OR methyl phenobarbitone OR methyl calminal OR methylphenobarbital OR methylphenobarbitalum OR methylphenobarbitonum OR methylphenolbarbital OR methylphenylbarbituric acid OR metilfenobarbital OR metilfenobarbitale OR metylfenemal OR metyna OR morbusan OR ethylmethylphenylbarbituric acid OR methylethylphenylbarbituric acid OR methylphenolbarbitol OR phemetone OR phemiton OR phemitone OR phenmiton OR prominal):TI,AB,KY
#83 (dea no 2884 OR dormicum OR midazolam OR midazolamum OR versed):TI,AB,KY
#84 (methazolamide OR methenamide OR mzm OR naptazane OR neptazane OR neptazaneat):TI,AB,KY
#85 (nimetazepam):TI,AB,KY
#86 (benzodiazepin OR alodorm OR apo nitrazepam OR apodorm OR benzalin OR calsamin OR calsmin OR cerson OR dormin OR dormo puren OR dumolid OR eatan OR epibenzalin OR epinelbon OR eunoctin OR eunoktin OR gerson OR hipnax OR hipsal OR ibrovek OR imeson OR imesont OR insomin OR ipersed OR magadon OR megadon OR mitidin OR mogadan OR mogadon OR mogadone OR desmethylnimetazepam OR nelbon OR nelmat OR neozepam OR neuchlonic OR nitrados OR nitravet OR nitrazadon OR nitrazepam OR nitrazepamum OR nitrempax OR nitrenpax OR noctesed OR pacisyn OR paxisyn OR pelson OR persopit OR radedorm OR relact OR remnos OR somitran OR somnased OR somnibel OR somnite OR sonebon OR sonnolin OR surem OR trazenin OR unisomnia):TI,AB,KY
#87 (actinium OR barzepin OR carbox OR deprectal OR gp 47680 OR lonazet OR ocbz OR oxalepsy OR oxcarbamazepine OR oxcarbazepine OR oxetol OR oxpin OR oxrate OR oxtellar OR oxypine OR pharozepine OR prolepsi OR timox OR trexapin OR trileptal OR trileptin):TI,AB,KY
#88 (paraldehyde):TI,AB,KY
#89 Paramethadione:TI,AB,KY
#90 (folsaeure OR deoxypteroylglutamate OR amino pga OR aminofolic acid OR aminopteroylglutamic acid OR a 771726 OR ninopterin OR aminopteridine OR aminopterine OR apga OR bi 1356 OR braf v600e kinase inhibitor ro5185426 OR e 2007 OR er 155055 90 OR hmr1726 OR minopterin OR diamino 6 pteridinyl methyl amino benzoyl glutamic acid OR perampanel OR plx4032 OR pteramina OR rg7204 OR ro5185426 OR trajenta):TI,AB,KY
#91 Phenacemide:TI,AB,KY
#92 (pheneturide):TI,AB,KY
#93 (bartol OR cardenal OR chinoin OR fenemal OR fenobarbital OR gardenal OR lumen OR luminal OR phenemal OR phenobarbital OR phenobarbitol OR phenobarbitone OR phob):TI,AB,KY
#94 Phensuximide:TI,AB,KY
#95 (diphenylhydantoin OR dwufenylohydantoina OR aleviatin OR antisacer OR auranile OR causoin OR citrullamon OR citrulliamon OR comital OR comitoina OR convul OR danten OR dantinal OR dantoinal OR dantoine OR denyl OR di hydan OR di lan OR di phetine OR didan OR difenilhidantoina OR difenin OR difetoin OR difhydan OR dihycon OR dihydantoin OR dilabid OR dilantin OR dilantine OR dillantin OR dintoin OR dintoina OR diphantoin OR diphedal OR diphedan OR diphenat OR diphenin OR diphenine OR diphentoin OR diphentyn OR diphenylan OR diphenylhydantoine OR diphenylhydatanoin OR ditoinate OR ekko OR elepsindon OR enkelfel OR epamin OR epanutin OR epasmir OR epdantoin OR epdantoine OR epelin OR epifenyl OR epihydan OR epilan OR epilantin OR epinat OR epised OR eptal OR eptoin OR fenantoin OR fenidantoin OR fenitoina OR fentoin OR fenylepsin OR fenytoin OR fenytoine OR gerot epilan OR hidan OR hidantal OR hidantilo OR hidantina OR hidantomin OR hindatal OR hydantal OR hydantin OR hydantoin OR hydantoinal OR hydantol OR ictalis OR idantoil OR idantoin OR iphenylhydantoin OR kessodanten OR labopal OR lehydan OR lepitoin OR lepsin OR mesantoin OR minetoin OR neos hidantoina OR neosidantoina OR novantoina OR novophenytoin OR om hidantoina OR om hydantoine OR oxylan OR phanantin OR phanatine OR phenatine OR phenatoine OR phenhydan OR phenhydanin OR phenitoin OR phentoin OR phentytoin OR phenytex OR phenytoin OR phenytoine OR phenytoinum OR ritmenal OR saceril OR sanepil OR silantin OR sinergina OR sodanthon OR sodantoin OR sodanton OR solantin OR solantoin OR solantyl OR sylantoic OR tacosal OR thilophenyl OR toin OR zentronal OR zentropil):TI,AB,KY
#96 (ci 1008 OR lyrica OR pregabalin OR isobutylgaba):TI,AB,KY
#97 (cyral OR desoxyphenobarbitone OR hexadiona OR hexamidine OR lepimidin OR lepsiral OR liskantin OR majsolin OR medi pets OR midone OR milepsin OR misodine OR misolyne OR mizodin OR mizolin OR myidone OR mylepsin OR mylepsinum OR mysedon OR mysoline OR neurosyn OR prilepsin OR primacione OR primaclone OR primacone OR primakton OR primidon OR primidone OR primoline OR prysoline OR pyrimidone OR resimatil OR sertan):TI,AB,KY
#98 (gabren OR gabrene OR halogabide OR progabida OR progabide OR progabidum):TI,AB,KY
#99 (remacemide):TI,AB,KY
#100 (d 23129 OR ezg OR ezogabine OR potiga OR retigabine OR rtg):TI,AB,KY
#101 (banzel OR cgp 33101 OR e 2080 OR inovelon OR ruf 331 OR rufinamide OR xilep):TI,AB,KY
#102 Seletracetam:TI,AB,KY
#103 (stiripentol):TI,AB,KY
#104 (Sultiame OR sulthiame):TI,AB,KY
#105 (talampanel):TI,AB,KY
#106 (cerepax OR crisonar OR euhypnos OR euipnos OR gelthix OR hydroxydiazepam OR levanxene OR levanxol OR levanzene OR mabertin OR methyloxazepam OR normison OR oxydiazepam OR perdorm OR planum OR remestan OR restoril OR signopam OR temaz OR temazepam):TI,AB,KY
#107 (gabitril OR tiagabina OR tiagabine OR tiagabinum):TI,AB,KY
#108 (tipiramate OR topamax OR topiramate OR topiramatum OR topiramic acid):TI,AB,KY
#109 (absentol OR absetil OR convenixa OR convexina OR edion OR epidione OR epidone OR epixal OR etydion OR mino aleviatin OR minoaleuiatin OR minoaleviatin OR petidion OR petidon OR petilep OR petimalin OR pitmal OR ptimal OR tioxanona OR tredione OR tricione OR tridilona OR tridion OR tridione OR tridone OR trilidona OR trimedal OR trimedone OR trimetadione OR trimethadion OR trimethadione OR trimethdione OR trimethin OR trimetin OR trioxanona OR triozanona OR tromedone OR troxidone):TI,AB,KY
#110 (valnoctamide):TI,AB,KY
#111 (propylpentanoic acid OR propylvaleric acid OR avugane OR baceca OR convulex OR delepsine OR depacon OR depakene OR depakine OR depakote OR deproic OR di propylacetic acid OR propylessigsaure OR dipropylacetic acid OR dpa OR epilex OR epilim OR epival OR ergenyl OR mylproin OR sprinkle OR stavzor OR valcote OR valparin):TI,AB,KY
#112 (valproate OR valproic):TI,AB,KY
#113 (valpromide):TI,AB,KY
#114 (gamma vinyl gaba OR gvg OR sabril OR sabrilan OR sabrilex OR vigabatrin OR vigabatrina OR vigabatrine OR vigabatrinum):TI,AB,KY
#115 (exceglan OR excegram OR excegran OR zonegran OR zonisamida OR zonisamide OR zonisamidum):TI,AB,KY
#116 #34 OR #35 OR #36 OR #37 OR #38 OR #39 OR #40 OR #41 OR #42 OR #43 OR #44 OR #45 OR #46 OR #47 OR #48 OR #49 OR #50 OR #51 OR #52 OR #53 OR #54 OR #55 OR #56 OR #57 OR #58 OR #59 OR #60 OR #61 OR #62 OR #63 OR #64 OR #65 OR #66 OR #67 OR #68 OR #69 OR #70 OR #71 OR #72 OR #73 OR #74 OR #75 OR #76 OR #77 OR #78 OR #79 OR #80 OR #81 OR #82 OR #83 OR #84 OR #85 OR #86 OR #87 OR #88 OR #89 OR #90 OR #91 OR #92 OR #93 OR #94 OR #95 OR #96 OR #97 OR #98 OR #99 OR #100 OR #101 OR #102 OR #103 OR #104 OR #105 OR #106 OR #107 OR #108 OR #109 OR #110 OR #111 OR #112 OR #113 OR #114 OR #115
#117 #33 AND #116
#118 * NOT INMEDLINE AND 17/10/2012 TO 30/09/2014:DL
#119 #117 AND #118
Appendix 3. MEDLINE search strategy
This strategy is based on the Cochrane Highly Sensitive Search Strategy for identifying randomised trials published in Lefebvre 2011.
1. exp Learning Disorders/
2. exp Intellectual Disability/
3. exp Mentally Disabled Persons/
4. exp Mental Disorders/
5. exp Developmental Disabilities/
6. exp Fragile X Syndrome/
7. exp Rett Syndrome/
8. exp Down Syndrome/
9. exp Autistic Disorder/
10. exp Angelman Syndrome/
11. exp Cerebral Palsy/
12. exp Tuberous Sclerosis/
13. (learning adj1 (disab$ or difficult$ or problem$ or disorder$ or handicap$)).tw.
14. (mental$ adj1 (retard$ or disab$ or deficien$ or handicap$ or incapacity or disorder$)).tw.
15. (intellect$ adj1 (disab$ or impair$ or handicap$)).tw.
16. cognitive impairment.tw.
17. (development$ adj1 disab$).tw.
18. (multipl$ adj1 handicap$).tw.
19. fragile x syndrome.tw.
20. Rett$ syndrome.tw.
21. Lennox Gastaut syndrome.tw.
22. Down$ syndrome.tw.
23. tuberous sclerosis.tw.
24. (autism or autistic).tw.
25. Angelman$ syndrome.tw.
26. West$ syndrome.tw.
27. cerebral palsy.tw.
28. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27
29. exp Epilepsy/
30. exp Seizures/
31. (epilep$ or seizure$ or convuls$).tw.
32. 29 or 30 or 31
33. exp *Pre‐Eclampsia/ or exp *Eclampsia/
34. 32 not 33
35. (randomized controlled trial or controlled clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
36. clinical trials as topic.sh.
37. trial.ti.
38. 35 or 36 or 37
39. exp animals/ not humans.sh.
40. 38 not 39
41. 28 and 34 and 40
42. exp *Epilepsy/dt [Drug Therapy]
43. exp Anticonvulsants/
44. exp Acetazolamide/
45. exp Carbamazepine/
46. exp Clonazepam/
47. exp Clorazepate Dipotassium/
48. exp Diazepam/
49. exp Ethosuximide/
50. exp Lorazepam/
51. exp Mephenytoin/
52. exp Mephobarbital/
53. exp Midazolam/
54. exp Methazolamide/
55. exp Nitrazepam/
56. exp Paraldehyde/
57. exp Phenobarbital/
58. exp Phenytoin/
59. exp Primidone/
60. exp Temazepam/
61. exp Trimethadione/
62. exp Valproic Acid/
63. exp Vigabatrin/
64. (antiepilep$ or anticonvulsant$ or AED$1 or Acetazolamide or Alodorm or Antilepsin or Arem or Ativan or Barbexaclone or Beclamide or Brivaracetam or Carbagen or Carbamazepine or Celontin or Cerebyx or Chlonazepam or Chloracon or Cloazepam or Clobazam or Clonazepamum or Clonex or Clonopin or Clorazepate or Convulex or Depacon or Depak$ or Depamide or Desitin or Diacomit or Diamox or Diastat or Diazepam or Dilantin or Diphenin$ or Diphenylhydantoin or Divalpr$ or Dormicum or Ecovia or Emeside or Epanutin or Epiject or Epilim or Episenta or Epival or Eptoin or Ergenyl or Erimin or Eslicarbazepine or Ethadione or Ethosuximide or Ethotoin or Ethylphenacemide or Exalief or Excegran or Ezogabine or Fanatrex or Felbamate or Felbatol or Fosphenytoin or Frisium or Fycompa or Gabapentin or Gabarone or Gabitril or Gabrene or Ganaxolone or Garene or Gralise or Halogabide or Halogenide or Hibicon or Hypnovel or Iktorivil or Inovelon or Insoma or Intensl or Keppra or Klonopin or Kriadex or Lacosamide or Lamict$ or Lamitor or Lamitrin or Lamogine or Lamotrigine or Lamotrine or Landsen or Levetiracetam or Liskantin or Loraz or Lorazepam or Losigamone or Luminal or Lyrica or Mebaral or Mephenytoin or Mephobarbit$ or Mephyltaletten or Mesantoin or Mesuximide or Methazolamide or Methsuximide or Methylphenobarbit$ or Midazolam or Mogadon or Mylepsinum or Mysoline).tw.
65. (Neogab or Neptazane or Neurontin or Nimetazepam or Nitrados or Nitrazadon or Nitrazepam or Normison or Novo‐Clopate or Nupentin or Nydrane or Onfi or Orfiril or Orlept or Ormodon or Ospolot or Oxcarbazepine or Pacisyn or Paraldehyde or Paramethadione or Paxadorm or Paxam or Peganone or Perampanel or Petinutin or Petril or Phemiton or Phenacemide or Pheneturide or Phenobarbit$ or Phensuximide or Phenytek or Phenytoin or Posedrine or Potiga or Pregabalin or Primidone or Prodilantin or Progabide or Prominal or Prysoline or Ravotril or Remacemide or Remnos or Resimatil or Restoril or Retigabine or Riv?tril or Rufinamide or Sabril or Seclar or Selenica or Seletracetam or Sertan or Somnite or Stavzor or Stedesa or Stiripentol or Sulthiam$ or Sultiam$ or Talampanel or Tegretol or Temazepam or Temesta or Teril or Tiagabine or Timonil or Topamax or Topiramate or Tranxene or Tridione or Trileptal or Trimethadione or Trobalt or Urbanol or Valance or Valcote or Valium or Valnoctamide or Valparin or Valpro$ or Versed or Vigabatrin or Vimpat or Zalkote or Zarontin or Zebinix or Zonegran or Zonisamide).tw.
66. 42 or 43 or 44 or 45 or 46 or 47 or 48 or 49 or 50 or 51 or 52 or 53 or 54 or 55 or 56 or 57 or 58 or 59 or 60 or 61 or 62 or 63 or 64 or 65
67. 41 and 66
68. limit 67 to ed=20120901‐20140903
Appendix 4. PsycINFO search strategy
| S20 | S15 AND S18 Published 20110901‐ |
| S19 | S15 AND S18 |
| S18 | S16 OR S17 |
| S17 | TI antiepilep* or anti‐epilep* or anticonvuls* or anti‐convuls* or AED or AEDs or Acetazolamide or Alodorm or Antilepsin or Arem or Ativan or Barbexaclone or Beclamide or Brivaracetam or Carbagen or Carbamazepine or Celontin or Cerebyx or Chlonazepam or Chloracon or Cloazepam or Clobazam or Clonazepamum or Clonex or Clonopin or Clorazepate or Convulex or Depacon or Depak* or Depamide or Desitin or Diacomit or Diamox or Diastat or Diazepam or Dilantin or Diphenin* or Diphenylhydantoin or Divalpr* or Dormicum or Ecovia or Emeside or Epanutin or Epiject or Epilim or Episenta or Epival or Eptoin or Ergenyl or Erimin or Eslicarbazepine or Ethadione or Ethosuximide or Ethotoin or Ethylphenacemide or Exalief or Excegran or Ezogabine or Fanatrex or Felbamate or Felbatol or Fosphenytoin or Frisium or Fycompa or Gabapentin or Gabarone or Gabitril or Gabrene or Ganaxolone or Garene or Gralise or Halogabide or Halogenide or Hibicon or Hypnovel or Iktorivil or Inovelon or Insoma or Intensl or Keppra or Klonopin or Kriadex or Lacosamide or Lamict* or Lamitor or Lamitrin or Lamogine or Lamotrigine or Lamotrine or Landsen or Levetiracetam or Liskantin or Loraz or Lorazepam or Losigamone or Luminal or Lyrica or Mebaral or Mephenytoin or Mephobarbit* or Mephyltaletten or Mesantoin or Mesuximide or Methazolamide or Methsuximide or Methylphenobarbit* or Midazolam or Mogadon or Mylepsinum or Mysoline or Neogab or Neptazane or Neurontin or Nimetazepam or Nitrados or Nitrazadon or Nitrazepam or Normison or Novo‐Clopate or Nupentin or Nydrane or Onfi or Orfiril or Orlept or Ormodon or Ospolot or Oxcarbazepine or Pacisyn or Paraldehyde or Paramethadione or Paxadorm or Paxam or Peganone or Perampanel or Petinutin or Petril or Phemiton or Phenacemide or Pheneturide or Phenobarbit* or Phensuximide or Phenytek or Phenytoin or Posedrine or Potiga or Pregabalin or Primidone or Prodilantin or Progabide or Prominal or Prysoline or Ravotril or Remacemide or Remnos or Resimatil or Restoril or Retigabine or Riv?tril or Rufinamide or Sabril or Seclar or Selenica or Seletracetam or Sertan or Somnite or Stavzor or Stedesa or Stiripentol or Sulthiam* or Sultiam* or Talampanel or Tegretol or Temazepam or Temesta or Teril or Tiagabine or Timonil or Topamax or Topiramate or Tranxene or Tridione or Trileptal or Trimethadione or Trobalt or Urbanol or Valance or Valcote or Valium or Valnoctamide or Valparin or Valpro* or Versed or Vigabatrin or Vimpat or Zalkote or Zarontin or Zebinix or Zonegran or Zonisamide |
| S16 | MM "Anticonvulsive Drugs" OR MM "Carbamazepine" OR MM "Chloral Hydrate" OR MM "Clonazepam" OR MM "Diphenylhydantoin" OR MM "Nitrazepam" OR MM "Oxazepam" OR MM "Pentobarbital" OR MM "Phenobarbital" OR MM "Pregabalin" OR MM "Primidone" OR MM "Valproic Acid" |
| S15 | S10 AND S13 AND S14 |
| S14 | TI ( (randomiz* OR randomis* OR controlled OR placebo OR blind* OR unblind* OR "parallel group" OR crossover OR "cross over" OR cluster OR "head to head") N2 (trial OR method OR procedure OR study) ) OR AB ( (randomiz* OR randomis* OR controlled OR placebo OR blind* OR unblind* OR "parallel group" OR crossover OR "cross over" OR cluster OR "head to head") N2 (trial OR method OR procedure OR study) ) |
| S13 | S11 OR S12 |
| S12 | epilep* OR seizure* OR convuls* |
| S11 | MM "Epilepsy" OR MM "Epileptic Seizures" OR MM "Experimental Epilepsy" OR MM "Grand Mal Seizures" OR MM "Petit Mal Seizures" OR MM "Status Epilepticus" |
| S10 | S1 OR S2 OR S3 OR S4 OR S5 OR S6 OR S7 OR S8 OR S9 |
| S9 | Rett* syndrome or Lennox Gastaut syndrome or Down* syndrome or Angelman* syndrome or West* syndrome |
| S8 | autistic or autism or cerebral palsy or fragile X syndrome |
| S7 | development* disab* or multipl* handicap* or cognitive impairment or tuberous sclerosis |
| S6 | intellect* disab* or intellect* impair* or intellect* handicap* |
| S5 | mental* disab* or mental* retard* or mental* deficien* or mental* handicap* or mental* incapacity or mental* disorder* |
| S4 | learning disab* or learning difficult* or learning problem* or learning disorder* or learning handicap* |
| S3 | DE "autism" or "cerebral palsy" or "downs syndrome" |
| S2 | DE "learning disabilities" or "learning disorders" or "mental disorders" or "rett syndrome" |
| S1 | DE "cognitive impairment" or "mental retardation" or "developmental disabilities" or "fragile x syndrome" |
Appendix 5. Original search terms
The following search terms were used (free text and as index terms, as appropriate, for each database) when searching for the first version of this review (The Cochrane Library, Issue 3, 2007)
‐ Learning difficulties ‐ Learning problems ‐ Mental retardation ‐ Mentally disabled ‐ Mental handicap ‐ Mental deficiency ‐ Mental incapacity ‐ Mental illness ‐ Mental disorders ‐ Intellectual disability ‐ Intellectual impairment ‐ Intellectual handicap ‐ Cognitive impairment ‐ Developmental disabilities ‐ Subnormal ‐ Fragile X syndrome ‐ Rett syndrome ‐ Lennox‐Gastaut syndrome ‐ Down's syndrome ‐ Tuberous sclerosis ‐ Autism ‐ West syndrome ‐ Angelman's syndrome ‐ Cerebral palsy ‐ Multiple handicap ‐ Epilepsy ‐ Seizures ‐ Convulsion ‐ Anticonvulsant ‐ Antiepileptic
To identify randomised controlled trials in MEDLINE, we combined the search terms with phases 1 and 2 of the Cochrane highly sensitive search strategy as set out in Appendix 5b of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Data and analyses
Comparison 1. AED vs placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Responder rate (≥ 50% reduction in overall seizure frequency) | 3 | 382 | Risk Difference (M‐H, Fixed, 95% CI) | 0.16 [0.09, 0.24] |
| 2 Responder rate (≥ 50% reduction in rate of drop seizures) | 2 | 313 | Risk Difference (M‐H, Random, 95% CI) | 0.30 [0.20, 0.40] |
| 3 Seizure freedom | 1 | 73 | Risk Difference (M‐H, Fixed, 95% CI) | 0.05 [‐0.05, 0.16] |
1.3. Analysis.
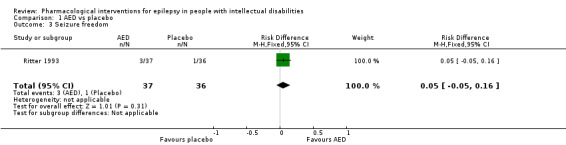
Comparison 1 AED vs placebo, Outcome 3 Seizure freedom.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Battaglia 1991.
| Methods | RCT, cross‐over | |
| Participants | Total randomised: 20 Number of patients over the age of 12 = 11/13. Age range 6 to 18 years (mean age 12 years 7 months). Number of patients with intellectual disability = 12/13 Most patients on 2 to 3 other AEDs Baseline = 16 weeks No titration period Treatment phase 1 and 2 = 16 weeks each | |
| Interventions | Flunarizine versus placebo | |
| Outcomes | Primary efficacy = total number of seizures | |
| Notes | Numbers used in data analysis = 13/20 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | States "randomised sequence" but does not say how this was achieved |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Although the trial is described as "double blind", no information is given to describe how blinding was achieved |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Although the trial is described as "double blind", no information is given to describe how blinding was achieved |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition may be due to adverse effects and no intention‐to treat‐analysis appears to have been carried out |
| Selective reporting (reporting bias) | Low risk | All outcomes discussed in the methods section were reported in the results and all P values were reported |
| Other bias | High risk | Lack of protocol and insufficient information regarding the methodology. Funded by a pharmaceutical company |
Crawford 2001.
| Methods | Open‐label, parallel RCT | |
| Participants | Total randomised: 83 All patients were 12 years and over. Age range = 15 to 59. All patients had intellectual disability All patients were taking 1 to 3 other AEDs Baseline period = 8 weeks. Titration period up to 14 weeks. Treatment period = minimum of 10 weeks | |
| Interventions | Gabapentin versus lamotrigine | |
| Outcomes | Primary efficacy = reduction in seizure frequency between baseline period and the last 8 weeks of the treatment period assessed using the R‐ratio=(T‐B)/(T+B) where T and B are the seizure frequencies per 28 days during treatment and baseline, respectively Secondary = responders (seizure frequency reduced by 50% or more. Non‐responders = seizure frequency reduced by less than 50% and those withdrawing Mood, behaviour and dependency were assessed |
|
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Participants were sequentially randomised in blocks |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants were unblinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Outcome assessment unblinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Although missing data and study attrition are unclear, an intention‐to‐treat analysis was carried out |
| Selective reporting (reporting bias) | Low risk | All outcomes discussed were reported. There is no evidence to suggest outcomes were selectively reported |
| Other bias | Unclear risk | Funded by a pharmaceutical company but no other evidence for other risk of bias |
Crumrine 1989.
| Methods | RCT, parallel | |
| Participants | Total randomised: 73 Age range of patients = 2 to 18 years. Mean age for cinromide group = 7.38 (± 3.65), mean age for placebo group = 7.93 (± 4.87). All patients had Lennox‐Gastaut syndrome, although level of intellectual disability was not stated Patients could be on no more than 3 other AEDs Baseline period = 6 weeks. Treatment period = 18 weeks | |
| Interventions | Cinromide versus placebo | |
| Outcomes | The primary statistical analysis of seizure frequency compared the changes from the baseline period (6 weeks) to the treatment period (12 weeks for most patients) in the cinromide and placebo groups Global evaluations at weeks 12, 18 and 24 were also compared |
|
| Notes | Numbers used in data analysis = 56 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Although it states that participants were randomly assigned, information is not given about how randomisation was achieved |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Although it describes the trial as "double‐blind", there is insufficient information as to how blinding was achieved. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Although it describes the trial as "double‐blind", there is insufficient information as to how blinding was achieved. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Study attrition reported but reasons unclear |
| Selective reporting (reporting bias) | High risk | Very few P values reported |
| Other bias | High risk | Study was terminated early when it became clear to the funder that cinromide was not effective |
Eriksson 1998.
| Methods | RCT, cross‐over | |
| Participants | Total randomised: 17 Age range of patients = 4.6 to 20.7 years, mean = 10.1 years (5/17 patients over 12 years) All randomised patients had intellectual disability All patients were taking 1 to 3 other AEDs Baseline period = 8 weeks. Open phase = 2 to 12 months. Washout phase = 3 weeks. Each cross‐over leg = 12 weeks | |
| Interventions | Lamotrigine versus placebo | |
| Outcomes | Primary = % reduction in mean monthly seizure frequency Secondary = severity of seizures, functional status of patient and frequency of adverse events | |
| Notes | Numbers used in data analysis = 15 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | LMG and placebo administered in random order ‐ does not say how random order was determined |
| Allocation concealment (selection bias) | Low risk | Randomisation carried out by pharmacological department |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and study personnel blinded through use of identical placebo and LMG |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Reasons for dropouts unlikely to effect outcomes |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information |
| Other bias | Unclear risk | No information about how trial was funded and no other evidence to suggest other risk of bias |
Gigli 1988.
| Methods | RCT, cross‐over | |
| Participants | Total randomised: 26 Age range of patients = 11.7 to 18.3 Patients were divided into 2 groups according to seizure type and severity Group I: 6 patients had secondary generalised epilepsy and 9 patients had partial epilepsy with or without secondary generalisation Group II: 7 patients had secondary generalised epilepsy and 4 had partial epilepsy with or without secondary generalisation 3 patients had slight mental retardation, 7 had moderate mental retardation, 15 patients had serious mental retardation Number of concomitant AEDs were not specified, however, patients were maintained on the drugs they were taking prior to the start of the study Baseline period = 3 months, each cross‐over leg = 3 months, washout period = 3 months | |
| Interventions | Flunarizine versus placebo | |
| Outcomes | Primary efficacy variable not stated. Alterations in seizure frequency were recorded together with behavioural observations In group I additional data were obtained, in patients with the ability to respond, on AEDs, EEG, attention test (Toulouse modified) | |
| Notes | Numbers used in data analysis = 26 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Does not say how patients were randomised |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Although it states that the trial was blinded, there is insufficient information regarding how blinding was achieved |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Although it states that the trial was blinded, there is insufficient information regarding how blinding was achieved |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participants withdrew from the study |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information |
| Other bias | Unclear risk | Insufficient information |
Glauser 2008.
| Methods | Double‐blind, randomised, placebo‐controlled trial | |
| Participants | Total randomised: 138 Age range of patients = 4 to 30 The patients were all diagnosed with Lennox‐Gastaut syndrome. No specific mention of the level of intellectual disability was made. The patients had a minimum of 90 seizures in the month. They were on 1 to 3 fixed‐dose regimens of 1 to 3 concomitant AEDs. Baseline period = 28 days. Treatment period = 84 days (14 days titration + 17 days treatment) |
|
| Interventions | Rufinamide versus placebo | |
| Outcomes | Primary = reduction in total seizure frequency, tonic‐atonic ("drop attack") seizure frequency and seizure severity rating Secondary = response to treatment (% of patient with at least 50% reduction in seizure frequency during the double‐blind phase); % change in seizure frequency per 28 days relative to baseline (for each seizure subtype other than tonic‐atonic); parental global evaluation (alertness, interaction with environment, daily activity performance, responsiveness to verbal request and seizure severity) |
|
| Notes | Number used in data analysis 138 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about how randomisation was achieved |
| Allocation concealment (selection bias) | Low risk | Numbers assigned to identical drugs |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and study personnel blinded by use of identical drugs |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comparable dropout rates across the groups. Intention‐to‐treat analysis carried out |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable |
| Other bias | Unclear risk | No information about how trial was funded but no other evidence to suggest unclear risk of bias |
Kaski 1991.
| Methods | RCT, cross‐over | |
| Participants | Total randomised: 21 Age range = 6 to 38. Mean = 24.9. All patients had intellectual disabilities Most patients on 1 to 3 other AEDs, except 2 patients who were not taking concomitant AEDs Baseline period = 8 weeks. Each cross‐over leg = 10 weeks | |
| Interventions | Carbamazepine versus slow‐release carbamazepine | |
| Outcomes | Primary efficacy variables not stated. The study aims state (a) to carry out a 24‐hour pharmacokinetic trial comparing slow‐release CBZ and conventional CBZ at different dosing frequencies; (b) to look at the effect the reduction in dosing frequency of CBZ has on seizure control, that is the difference between slow‐release and conventional CBZ | |
| Notes | Numbers used in data analysis = 20 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Study states that randomisation was achieved according to order participants were recruited to study |
| Allocation concealment (selection bias) | Low risk | Pharmacist‐controlled allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and study personnel blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study personnel blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only 1 participant dropped out and reasons for dropout unlikely to affect study outcome |
| Selective reporting (reporting bias) | Low risk | All outcomes discussed were reported. There is no evidence to suggest outcomes were selectively reported |
| Other bias | Unclear risk | No information about how trial was funded and no other evidence to suggest unclear risk of bias |
Kerr 2005.
| Methods | RCT, parallel | |
| Participants | Total randomised: 74 Age range = 12 years and above. Mean for topiramate group = 29.9 years and mean for placebo group = 31.9 years All patients had intellectual disabilities All patients taking 1 to 3 other AEDs Baseline period = 4 weeks. Titration period = 18 weeks. Maintenance period = 12 weeks | |
| Interventions | Topiramate versus placebo | |
| Outcomes | Efficacy variables: (1) Change in total seizure frequency between baseline and end of on‐drug phase (12 weeks) (2) Change in seizure severity between baseline and end of on‐drug phase (12 weeks) (3) Number of responders (response defined as a 50% reduction in seizure frequency) (4) Changes from baseline to the end of the on‐drug phase were compared for the ABC total score, EOS total score and subscale scores and ELDQOL subscale scores (5) Global assessments | |
| Notes | Numbers used in data analysis: efficacy data = 72, safety data = 74 (intention‐to‐treat analysis) Actual numbers completing the study: topiramate = 28, placebo = 29 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about how randomisation was achieved |
| Allocation concealment (selection bias) | Low risk | Pharmacist‐controlled allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and study personnel were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comparable dropout rates across groups |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable |
| Other bias | Low risk | No other sources of bias identified |
Motte 1997.
| Methods | RCT, parallel | |
| Participants | Total randomised: 169 Age range = 3 to 25 years. Mean = 9.6 years. All had intellectual impairment but 92% (n = 73) had moderate/severe intellectual disabilities in LMG group and 91% (82) in placebo group All patients were on at least 1 other AED Baseline period = 4 weeks. Treatment period = 16 weeks | |
| Interventions | Lamotrigine versus placebo | |
| Outcomes | Primary efficacy = % change from baseline in frequency of major motor seizures. In addition, the median changes from baseline in frequency of drop attacks, tonic‐clonic seizures and atypical absences Primary safety = frequency of reported adverse events | |
| Notes | Numbers used in data analysis: 169 = safety data, 167 = efficacy data | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information how randomisation was achieved |
| Allocation concealment (selection bias) | Unclear risk | "a double blinded fashion" but no details as to how they achieved this |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | "a double blinded fashion" but no details as to how they achieved this |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | "a double blinded fashion" but no details as to how they achieved this |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Similar dropout rates across the 2 groups and a modified intention‐to‐treat analysis completed |
| Selective reporting (reporting bias) | High risk | Does not appear to have reported on neurological examination outcomes |
| Other bias | Unclear risk | Trial funded by a pharmaceutical company but no other evidence to suggest unclear risk of bias |
Ng 2011.
| Methods | Double‐blind, placebo‐controlled RCT | |
| Participants | Total randomised: 238 patients with Lennox‐Gastaut syndrome; 177 participants completed the study Age range: 2 to 54 years; mean age: 12.4 years Approximately half of all participants were receiving concomitant valproic acid, valproate semisodium or valproate sodium | |
| Interventions | Placebo Low‐dose clobazam (0.25 mg/kg/day; maximum 10 mg/day) Medium‐dose clobazam (0.5 mg/kg/day; maximum 20 mg/day) High‐dose clobazam (1.0 mg/kg/day; maximum 40 mg/day) |
|
| Outcomes | Baseline period: 4 weeks, treatment period 12 weeks. Outcomes assessed at week 4 and week 16 8‐item quality of life (QOL) questionnaire % decrease in weekly seizure frequency; overall seizure frequency Wechsler Adult Intelligence Scale (WAIS); Wechsler Intelligence Scale for Children (WISC) |
|
| Notes | 217 participants were included in the analysis. Modified intention‐to‐treat analysis excluded 21 patients who did not have one or more daily seizures measurement in the 12‐week maintenance period | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Central randomisation via interactive voice |
| Allocation concealment (selection bias) | Low risk | Central randomisation via interactive voice |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Although it states the trial was double‐blind there is insufficient information about how blinding was achieved |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Although it states the trial was double‐blind there is insufficient information about how blinding was achieved |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | High attrition rate. Modified intention‐to‐treat analysis carried out |
| Selective reporting (reporting bias) | Unclear risk | Protocol unavailable |
| Other bias | High risk | Researchers were employed by pharmaceutical companies |
Ritter 1993.
| Methods | RCT, parallel | |
| Participants | Total randomised: 73 Age range = 4 to 36 years. All patients had Lennox‐Gastaut syndrome, although the article does not make specific mention of the level of intellectual disability in this population Patients could not be on more than 2 AEDs before study initiation Baseline period = 28 days. Felbamate or placebo was administered for 70 days. Felbamate was titrated during the 1st 14 days of the treatment phase to a maximum of 45 mg/kg/day or 3600 mg/day, whichever was less. Followed by a 56‐day maintenance period | |
| Interventions | Felbamate versus placebo | |
| Outcomes | Primary efficacy = (a) frequency of seizures during a 4‐hour period of video recording performed at same time of day; (b) a compound variable consisting of parents' or guardians' global evaluations and their counts of atonic seizures Secondary efficacy = (a) parental counts of total seizures; (b) parental counts of generalised tonic‐clonic seizures Evaluation of safety = monitoring adverse events, vital signs and body weight General physical and neurologic examinations, measurements of plasma concentrations of felbamate and standard antiepileptic drugs and clinical laboratory evaluations | |
| Notes | Numbers used in data analysis = 71 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was computer‐generated |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Although it states that participants and study personnel were blinded, there is no information about how this blinding was achieved |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Study personnel blinded. Independent team completed data analysis |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 dropout in each group; all participants included in the analysis |
| Selective reporting (reporting bias) | Low risk | All outcomes discussed were reported. There is no evidence to suggest outcomes were selectively reported |
| Other bias | Unclear risk | Information on funding unavailable and no other evidence to suggest unclear risk of bias |
Sachdeo 1999.
| Methods | RCT, parallel | |
| Participants | Total randomised: 98 Age range: topiramate group = 2 to 29 years (mean 11.2 years), placebo group = 2 to 42 years (mean 11.2 years). All patients had Lennox‐Gastaut syndrome, although the article does not make specific mention of the level of intellectual disability in this population All patients were receiving 1 to 3 concomitant AEDs Baseline period = 4 weeks. Followed by a 3‐week titration phase and an 8‐week maintenance phase | |
| Interventions | Topiramate versus placebo | |
| Outcomes | Primary efficacy determinants = (a) reduction in average monthly seizure rate for all seizure types combined; or (b) each component of a compound variable consisting of % reduction in drop attacks (tonic + atonic) and the parental global evaluation of seizure severity Secondary efficacy = (a) reduction in average monthly rate of major seizures (drop attacks + TC); (b) the % of patients considered to be treatment responders defined as those with a >= 50%, >= 75% or 100% reduction from baseline for drop attacks, major seizures and all seizures | |
| Notes | Numbers used in data analysis = 98 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was computer‐generated |
| Allocation concealment (selection bias) | Low risk | Coded, opaque containers |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants and study personnel "remained blinded to codes" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Participants and study personnel "remained blinded to codes" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unequal dropout rates across groups; reasons for dropouts not given |
| Selective reporting (reporting bias) | Low risk | All outcomes discussed were reported. There is no evidence to suggest outcomes were selectively reported |
| Other bias | Unclear risk | Funded by pharmaceutical company but no other evidence to suggest unclear risk of bias |
Siegel 1999.
| Methods | RCT, cross‐over | |
| Participants | Total randomised: 14 Age range = 4.2 to 15.7 years. All patients had Lennox‐Gastaut syndrome, although the article does not make specific mention of the level of intellectual disability in this population | |
| Interventions | Felbamate versus placebo in presence of valproic acid | |
| Outcomes | Primary variable not stated. However, the measures include: (1) the % change in total seizure frequency after FBM/placebo add‐on compared with baseline (baseline = VPA monotherapy) Seizures were determined by either (a) video‐EEG counts; (b) family seizure records; (c) electrographic seizures without a clinical correlate viewed on the video‐EEG tapes | |
| Notes | Numbers used in data analysis = 13 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about how randomisation was achieved |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unclear how blinding was achieved |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear how blinding was achieved |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 1 participant withdrew but reasons not given |
| Selective reporting (reporting bias) | Low risk | All outcomes discussed were reported. There is no evidence to suggest outcomes were selectively reported |
| Other bias | Unclear risk | No information about how trial was funded and no other evidence to suggest unclear risk of bias |
Yamatogi 1997.
| Methods | RCT, parallel | |
| Participants | Total randomised: 80 Age range = 10 months to 17 years. Number of patients with intellectual disabilities = 61/66 Most patients were receiving 1 to 3 concomitant AEDs Baseline period = 4 weeks. Treatment period = 12 weeks | |
| Interventions | Clobazam versus clonazepam as comparator | |
| Outcomes | Primary efficacy = number of patients with a 50% or greater reduction in seizure frequency. Seizure frequency at the end of the treatment phase (12 weeks) was compared with the frequency at the end of the baseline period | |
| Notes | Numbers used in data analysis: 66 (CLB = 34, CZP = 32) used for efficacy data and 76 (CLB = 36, CZP = 40) used for safety data | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about how randomisation achieved |
| Allocation concealment (selection bias) | Low risk | Sealed, opaque envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Participants were blinded |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Single‐blinded study |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 80 patients were randomised but only 66 and 76 were included in the analyses. Reasons for dropouts unavailable |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information |
| Other bias | Unclear risk | Insufficient information |
ABC: Aberrant Behaviour Checklist AED: antiepileptic drug CLB: clobazam CZP: clonazepam EEG: electroencephalography ELDQOL: Epilepsy and Learning Disabilities Quality of Life EOS: Epilepsy Outcome Scale FBM: felbamate LMG: lamotrigine RCT: randomised controlled trial VPA: valproic acid
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bielmann 1978 | This was a complex study that looked at the effect of clonazepam as an adjunct to phenytoin and phenobarbital in patients with mental retardation. It was complicated, however, by the stratification of patients into those receiving and those not receiving an antipsychotic. Age ranges were also not provided |
| Bjornaes 2013 | Not people with intellectual disability |
| Conry 2014 | Not a RCT |
| Donati 2006 | The participants did not have an intellectual disability |
| Espe‐Lillo 1992 | This conference proceedings abstract appears to be an open‐label, follow‐up study from a double‐blind trial. Requested further information from the authors but no reply received |
| Gidal 2013 | Not people with intellectual disability |
| Hagebeuk 2011 | Participants were all under 12 years old |
| Hellings 2009 | Comparing methods of transitioning between 2 types of AED |
| Hodoba 2013 | Not a RCT |
| Luna 1989 | This study was not randomised |
| Santos 2013 | Not people with intellectual disability |
| Schlumberger 1994 | This study was not randomised |
| Smith 1968 | IQ level of participants is above our inclusion criteria |
| Tolbert 2014 | Not a RCT |
| Turner 1970 | Only 1 patient was aged 12 or above. In addition, only 6/18 patients had Lennox‐Gastaut syndrome |
AED: antiepileptic drug RCT: randomised controlled trial
Characteristics of ongoing studies [ordered by study ID]
Bjurulf 2008.
| Trial name or title | Comparing ketogenic diet with the most appropriate antiepileptic drug ‐ a randomised study of children with mental retardation and refractory epilepsy |
| Methods | RCT open, parallel assignment |
| Participants | Estimated enrolment 60 |
| Interventions | Ketogenic diet versus antiepileptic drugs |
| Outcomes | Primary: reduction in number of seizures Secondary: quality of life and cognitive function between groups; adverse events; slow activity and epileptic activity with 24‐hour EEG between groups; adverse events, number of seizures, change in EEG, quality of life and cognitive function after 13 months on ketogenic diet; effect of ketogenic diet on children with severe and less severe mental retardation |
| Starting date | November 2007 |
| Contact information | SSE, Neurological Department, Rikshospitalet University Hospital, Oslo, Norway |
| Notes | — |
EEG: electroencephalography RCT: randomised controlled trial
Contributions of authors
Cerian F Jackson was responsible for assessing studies for eligibility, data extraction, assessing risk of bias and writing up the review update.
Selina M Makin contributed to assessing studies for eligibility, data extraction, assessing risk of bias and supervising the write up of this review update.
Michael Kerr has supervised this review update and provided expert opinion and feedback.
Anthony G Marson has supervised this review update and provided expert opinion and feedback.
Sources of support
Internal sources
No sources of support supplied
External sources
Baily Thomas Foundation Charitable Fund, UK.
-
National Institute of Health Research (NIHR), UK.
This review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Epilepsy Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Declarations of interest
MK was an author on two of the papers reviewed in this article.
CFJ;SMM;AGM have no known declarations of interest.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Battaglia 1991 {published data only}
- Battaglia A, Ferrari AR, Guerrini R. Double‐blind placebo‐controlled trial of flunarizine as add‐on therapy in refractory childhood epilepsy. Brain & Development 1991;13(4):217‐22. [DOI] [PubMed] [Google Scholar]
Crawford 2001 {published data only}
- Crawford P, Brown S, Kerr M, Parke Davis Clinical Trials Group. A randomized open‐label study of gabapentin and lamotrigine in adults with learning disability and resistant epilepsy. Seizure 2001;10(2):107‐15. [DOI] [PubMed] [Google Scholar]
Crumrine 1989 {published data only}
- Crumrine P, Dreifuss FE, Corwin H, et al. Double‐blind, placebo‐controlled evaluation of cinromide in patients with the Lennox‐Gastaut Syndrome. Epilepsia 1989;30(4):422‐9. [DOI] [PubMed] [Google Scholar]
Eriksson 1998 {published data only}
- Eriksson AS, Nergardh A, Hoppu K. The efficacy of lamotrigine in children and adolescents with refractory generalized epilepsy: a randomized, double‐blind, crossover study. Epilepsia 1998;39(5):495‐501. [DOI] [PubMed] [Google Scholar]
Gigli 1988 {published data only}
- Gigli GL, Mazza S, Valente RM, Colognola RM, Musumeci R. Ineffectiveness of add‐on treatment with flunarizine in patients with epilepsy and mental retardation [Inefficacia dell'aggiunta di flunarizina al trattamento di pazienti con epilessia e ritardo mental: risultati di uno studio cross‐over in doppio cieco]. Il Bolletino della Lega Italiana contro l'Epilessia 1988;62(63):417‐8. [Google Scholar]
Glauser 2008 {published data only}
- Glauser T, Kluger G, Sachdeo R, Krauss G, Perdomo C, Arroyo S. Rufinamide for generalized seizures associated with Lennox‐Gastaut syndrome. Neurology 2008;70(21):1950‐8. [DOI] [PubMed] [Google Scholar]
Kaski 1991 {published data only}
- Kaski M, Heinonen E, Sivenius J, Tuominen J, Anttila M. Treatment of epilepsy in mentally retarded patients with a slow‐release carbamazepine preparation. Journal of Mental Deficiency Research 1991;35(3):231‐9. [DOI] [PubMed] [Google Scholar]
Kerr 2005 {published data only}
- Kerr MP, Baker GA, Brodie MJ. A randomized, double‐blind, placebo‐controlled trial of topiramate in adults with epilepsy and intellectual disability: impact on seizures, severity, and quality of life. Epilepsy & Behavior 2005;7(3):472‐80. [DOI] [PubMed] [Google Scholar]
Motte 1997 {published data only}
- Billard C, Motte J, Arvidsson D, Trevathan E, Talladai M, Campos J, et al. Double‐blind, placebo‐controlled evaluation of the safety and efficacy of lamotrigine (Lamictal) for the treatment of patients with a clinical diagnosis of Lennox‐Gastaut syndrome. Epilepsia 1996;37(Suppl 4):92. [Google Scholar]
- Motte J, Trevathan E, Arvidsson JFV, Barrera MN, Mullens EL, Manasco P. Lamotrigine for generalized seizures associated with the Lennox‐Gastaut syndrome. New England Journal of Medicine 1997;337(25):1807‐12. [DOI] [PubMed] [Google Scholar]
- Mullens L, Gallagher J, Manasco P. Improved neurological function accompanies effective control of the Lennox‐Gastaut syndrome with Lamictal: results of a multinational, placebo‐controlled trial. Epilepsia 1996;37(Suppl 5):163. [Google Scholar]
Ng 2011 {published data only}
- Ng YT, Conry JA, Drummond R, Stolle J, Weinberg MA. Randomized, phase III study of clobazam in Lennox‐Gastaut syndrome. Neurology 2011;77(15):1473‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ritter 1993 {published data only}
- Jensen PK. Felbamate in the treatment of Lennox‐Gastaut syndrome. Epilepsia 1994;35(Suppl 5):S54‐7. [DOI] [PubMed] [Google Scholar]
- Ritter F, Dreifuss FE, Sackellares JC, Shields D, French J, Dodson WE. A double‐blind trial of felbamate in Lennox‐Gastaut syndrome. Epilepsia 1991;32(Suppl 3):13. [Google Scholar]
- Ritter FJ, Leppik IE, Dreifuss FE, et al. Efficacy of felbamate in childhood epileptic encephalopathy (Lennox‐Gastaut syndrome). New England Journal of Medicine 1993;328(1):29‐33. [DOI] [PubMed] [Google Scholar]
- Vigevano F, Cilio MR, Antonini L. Clinical experience with felbamate in paediatric age [Esperienze con Felbamato in eta pediatrica]. Lega Italiana conto l'Epilessia 1994;86‐7:63‐6. [Google Scholar]
Sachdeo 1999 {published data only}
- Glauser TA, Sachdeo RC, Ritter FJ, Reife R, Lim P. A double‐blind trial of topiramate in Lennox‐Gastaut syndrome (LGS). Epilepsia 1997;38(Suppl 3):131. [DOI] [PubMed] [Google Scholar]
- Sachdeo RC, Glauser TA, Ritter F, Reife R, Lim P, Pledger G. A double‐blind, randomized trial of topiramate in Lennox‐Gastaut syndrome. Topiramate YL Study Group. Neurology 1999;52(9):1882‐7. [DOI] [PubMed] [Google Scholar]
Siegel 1999 {published data only}
- Oletsky H, Kelley K, Stertz B, Reeves‐Tyer P, Flamini R, Malow B, et al. The efficacy of felbamate as add‐on therapy to valproic acid in the Lennox‐Gastaut syndrome (LGS). Epilepsia 1996;37(Suppl 5):155. [DOI] [PubMed] [Google Scholar]
- Siegel H, Kelley K, Stertz B, Reeves‐Tyer P, Flamini R, Malow B, et al. The efficacy of felbamate as add‐on therapy to valproic acid in the Lennox‐Gastaut syndrome. Epilepsy Research 1999;34(2‐3):91‐7. [DOI] [PubMed] [Google Scholar]
Yamatogi 1997 {published data only}
- Yamatogi Y, Ohtahara S, Shigematsu H, et al. Single blind comparative study of clobazam (NH‐15) with clonazepam in intractable childhood epilepsies. Journal of the Japan Epilepsy Society 1997;15(2):110‐21. [Google Scholar]
References to studies excluded from this review
Bielmann 1978 {published data only}
- Bielmann P, Levac T, Gagnon MA. Clonazepam: its efficacy in association with phenytoin and phenobarbital in mental patients with generalized major motor seizures. International Journal of Clinical Pharmacology and Biopharmacy 1978;16(6):268‐73. [PubMed] [Google Scholar]
Bjornaes 2013 {published data only}
- Bjornaes H, Bakke KA, Larsson PG, Heminghyt E, Rytter E, Brager‐Larsen LM. Subclinical epileptiform activity in children with electrical status epilepticus during sleep: effects on cognition and behaviour before and after treatment with levetiracetam. Epilepsy & Behavior 2013;27(1):40‐8. [DOI] [PubMed] [Google Scholar]
Conry 2014 {published data only}
- Conry JA, Ng T‐T, Kernitsky L, Mitchell WG, Veidemanis R, Drummond R, et al. Stable dosages of clobazam for Lennox‐Gastaut syndrome are associated with sustained drop‐seizure and total seizure improvements over 3 years. Epilepsia 2014;55(4):558‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Donati 2006 {published data only}
- Donati F, Gobbi G, Campistol J, Rapatz G, Daehler M, Sturm Y, et al. Effects of oxcarbazepine on cognitive function in children and adolescents with partial seizures. Neurology 2006;67(4):679‐82. [DOI] [PubMed] [Google Scholar]
Espe‐Lillo 1992 {published data only}
- Espe‐Lillo J, Ritter FJ. Long‐term follow‐up of felbamate treatment in children with Lennox‐Gastaut Syndrome. Epilepsia 1992;33(Suppl 3):118. [Google Scholar]
Gidal 2013 {published data only}
- Gidal BE, Ferry J, Majid O, Hussein Z. Concentration‐effect relationships with perampanel in patients with pharmacoresistant partial‐onset seizures. Epilepsia 2013;54(8):1490‐7. [DOI] [PubMed] [Google Scholar]
Hagebeuk 2011 {published data only}
- Hagebeuk EEO, Koelman J, Duran M, Abeling NG, Vyth A, Poll‐The BT. Clinical and electroencephalographic effects of folinic acid treatment in Rett syndrome patients. Journal of Child Neurology 2011;26(6):718‐23. [DOI] [PubMed] [Google Scholar]
Hellings 2009 {published data only}
- Hellings JA, Barth FX, Logan M, Cook‐Wiens G, Osorio I, Reed RC. Overnight versus progressive conversion of multiple daily‐dose divalproex to once‐daily divalproex extended release: which strategy is better tolerated by adults with intellectual disabilities?. Journal of Clinical Psychopharmacology 2009;29(5):492‐5. [DOI] [PubMed] [Google Scholar]
Hodoba 2013 {published data only}
- Hodoba S, Santic AM, Schmidt D. Adjunctive Madopar for ultrarefractory epilepsy? Preliminary observations. Epilepsy & Behavior 2013;28(2):201‐2. [DOI] [PubMed] [Google Scholar]
Luna 1989 {published data only}
- Luna D, Dulac O, Pajot N, Beaumont D. Vigabatrin in the treatment of childhood epilepsies: a single‐blind placebo‐controlled study. Epilepsia 1989;30(4):430‐7. [DOI] [PubMed] [Google Scholar]
Santos 2013 {published data only}
- Santos K, Palmini A, Radziuk AL, Robert R, Bastos F, Booij L, et al. The impact of methylphenidate on seizure frequency and severity in children with attention‐deficit‐hyperactivity disorder and difficult‐to‐treat epilepsies. Developmental Medicine & Child Neurology 2013;55(7):654‐60. [DOI] [PubMed] [Google Scholar]
Schlumberger 1994 {published data only}
- Schlumberger E, Dulac O. A simple, effective and well‐tolerated treatment regime for West syndrome. Developmental Medicine and Child Neurology 1994;36(10):863‐72. [DOI] [PubMed] [Google Scholar]
Smith 1968 {published data only}
- Smith WL, Philippus MJ, Guard HL. Psychometric study of children with learning problems and 14‐6 positive spike EEG patterns, treated with ethosuximide (Zarontin) and placebo. Archives of Disease in Childhood 1968;43(231):616‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tolbert 2014 {published data only}
- Tolbert D, Harris SI, Bekersky I, Lee D, Isojarvi J. Withdrawal‐related adverse events from clinical trials of clobazam in Lennox‐Gastaut syndrome. Epilepsy & Behavior 2014;37:11‐5. [DOI] [PubMed] [Google Scholar]
Turner 1970 {published data only}
- Turner M, Cordero Funes JR, Aspinwall R, Cantlon B, Fejerman N, Loñ JC. Clinico‐electroencephalographic evaluation of a new benzodiazepine (Ro 05‐4023) by oral administration in epileptic patients by double blind technique. Acta Neurologica Latinoamericana 1970;16(1):158‐69. [PubMed] [Google Scholar]
References to ongoing studies
Bjurulf 2008 {published data only}
- Bjurulf B. Comparing ketogenic diet with the most appropriate antiepileptic drug ‐ a randomized study of children with mental retardation and drug resistant epilepsy. www.clinicaltrials.gov/ct/show/NCT00552526 (accessed 7 August 2008).
Additional references
Al‐Bachari 2013
- Al‐Bachari S, Pulman J, Hutton JL, Marson AG. Gabapentin add‐on for drug‐resistant partial epilepsy. Cochrane Database of Systematic Reviews 2013, Issue 7. [DOI: 10.1002/14651858.CD001415.pub2] [DOI] [PubMed] [Google Scholar]
Beran 1998
- Beran RG, Gibson RJ. Aggressive behaviour in intellectually challenged patients with epilepsy treated with lamotrigine. Epilepsia 1998;39(3):280‐2. [DOI] [PubMed] [Google Scholar]
Besag 2001
- Besag FM. Behavioural effects of the new anticonvulsants. Drug Safety 2001;24(7):513‐36. [DOI] [PubMed] [Google Scholar]
Bouras 1992
- Bouras N, Drummond C. Behaviour and psychiatric disorders of people with mental handicaps living in the community. Journal of Intellectual Disability Research 1992;36(4):349‐57. [DOI] [PubMed] [Google Scholar]
Bowley 2000
- Bowley C, Kerr M. Epilepsy and intellectual disability. Journal of Intellectual Disability Research 2000;44(5):529‐43. [DOI] [PubMed] [Google Scholar]
Dam 1990
- Dam M. Vigabatrin and behaviour disturbances (Letter). Lancet 1990;335:8689. [PubMed] [Google Scholar]
Deb 1997
- Deb S. Mental disorder in adults with mental retardation and epilepsy. Comprehensive Psychiatry 1997;38(3):179‐84. [DOI] [PubMed] [Google Scholar]
Deb 1999
- Deb S, Joyce J. Characteristics of epilepsy in a population based cohort of adults with learning disability. Irish Journal of Psychological Medicine 1999;16(1):5‐9. [Google Scholar]
Dulac 1991
- Dulac O, Chiron C, Luna D, Cusmai R, Pajot N, Beaumont D, et al. Vigabatrin in childhood epilepsy. Journal of Child Neurology 1991;6(Suppl 2):S30‐7. [PubMed] [Google Scholar]
Eaton 1982
- Eaton LF, Menolascino FJ. Psychiatric disorders in the mentally retarded: types, problems, and challenges. American Journal of Psychiatry 1982;139(10):1297‐303. [DOI] [PubMed] [Google Scholar]
Espie 1989
- Espie CA, Pashley AS, Bonham KG, Sourindhrin I, O'Donovan M. The mentally handicapped person with epilepsy: a comparative study investigating psychosocial functioning. Journal of Mental Deficiency Research 1989;33(2):123‐35. [DOI] [PubMed] [Google Scholar]
Espie 1990
- Espie CA, Gillies JB, Montgomery JM. Antiepileptic polypharmacy, psychosocial behaviour and locus of control orientation among mentally handicapped adults living in the community. Journal of Mental Deficiency Research 1990;34(4):351‐60. [DOI] [PubMed] [Google Scholar]
Ettinger 1998
- Ettinger AB, Weisbrot DM, Saracco J, Dhoon A, Kanner A, Devinsky O. Positive and negative psychotropic effects of lamotrigine in patients with epilepsy and mental retardation. Epilepsia 1998;39(8):874‐7. [DOI] [PubMed] [Google Scholar]
Forsgren 1990
- Forsgren L, Edvinsson SO, Blomquist HK, Heijbel J, Sidenvall R. Epilepsy in a population of mentally retarded children and adults. Epilepsy Research 1990;6(3):234‐48. [DOI] [PubMed] [Google Scholar]
Gay 1995
- Gay PE, Mecham GF, Coskey JS, Sadler T, Thompson JA. Behavioural effects of felbamate in childhood epileptic encephalopathy (Lennox‐Gastaut syndrome). Psychological Reports 1995;77(3 Pt 2):1208‐10. [DOI] [PubMed] [Google Scholar]
Gillies 1989
- Gillies JB, Espie CA, Montgomery JM. The social and behavioural functioning of people with mental handicaps attending adult training centres: a comparison with those with and without epilepsy. Mental Handicap Research 1989;2:129‐36. [Google Scholar]
Goulden 1991
- Goulden K, Shinnar S, Koller H, Katz M, Richardson S. Epilepsy in children with mental retardation: a cohort study. Epilepsia 1991;34:690‐7. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hoare 1984
- Hoare P. The development of psychiatric disorder among school children with epilepsy. Developmental Medicine and Child Neurology 1984;26(1):3‐13. [DOI] [PubMed] [Google Scholar]
ILAE 1981
- Commission on Classification and Terminology of the ILAE: proposal for revised clinical and EEG classification of epileptic seizures. Epilepsia 1981;22:489‐501. [DOI] [PubMed] [Google Scholar]
ILAE 1989
- Commission on Classification and Terminology of the ILAE: proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30:389‐99. [DOI] [PubMed] [Google Scholar]
Kerr 1997
- Kerr MP, Espie CA. Learning disability and epilepsy. Seizure 1997;6(5):331‐6. [DOI] [PubMed] [Google Scholar]
Khurana 1996
- Khurana DS, Riviello J, Helmers S, Holmes G, Anderson J, Mikati MA. Efficacy of gabapentin therapy in children with refractory partial seizures. Journal of Pediatrics 1996;128(6):829‐33. [DOI] [PubMed] [Google Scholar]
Kirkham 2010
- Kirkham JJ, Swan KM, Altman DG, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:365. [DOI] [PubMed] [Google Scholar]
Lee 1996
- Lee DO, Steingard RJ, Cesena M, Helmers SL, Riviello JJ, Mikati MA. Behavioral side effects of gabapentin in children. Epilepsia 1996;37(1):87‐90. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Mansell 1993
- Mansell JL. Services for People with Learning Disabilities and Challenging Behaviour or Mental Health Needs. London: HMSO, 1993. [Google Scholar]
Mariani 1993
- Mariani E, Ferini‐Strambi L, Sala M, Erminio C, Smirne S. Epilepsy in institutionalized patients with encephalopathy: clinical aspects and nosological considerations. American Journal of Mental Retardation 1993;98:27‐33. [PubMed] [Google Scholar]
Michael 2008
- Michael B, Marson AG. Clobazam as an add‐on in the management of refractory epilepsy. Cochrane Database of Systematic Reviews 2008, Issue 2. [DOI: 10.1002/14651858.CD004154.pub4] [DOI] [PubMed] [Google Scholar]
Mikati 1998
- Mikati MA, Choueri R, Khurana DS, Riviello J, Helmers S, Holmes G. Gabapentin in the treatment of refractory partial epilepsy in children with intellectual disability. Journal of Intellectual Disability Research 1998;1:57‐62. [PubMed] [Google Scholar]
Morgan 2003
- Morgan CL, Baxter H, Kerr MP. Prevalence of epilepsy and associated health service utilization and mortality among patients with intellectual disability. American Journal of Mental Retardation 2003;108(5):293‐300. [DOI] [PubMed] [Google Scholar]
Mullens 1996
- Mullens L, Gallagher J, Manasco P. Improved neurological function accompanies effective control of the Lennox‐Gastaut syndrome with Lamictal: results of a multinational, placebo‐controlled trial. Epilepsia 1996;37(Suppl 5):163. [Google Scholar]
Pulman 2014
- Pulman J, Jette N, Dykeman J, Hemming K, Hutton JL, Marson AG. Topiramate add‐on for drug‐resistant partial epilepsy. Cochrane Database of Systematic Reviews 2014, Issue 2. [DOI: 10.1002/14651858.CD001417.pub3] [DOI] [PubMed] [Google Scholar]
Ramaratnam 2001
- Ramaratnam S, Marson AG, Baker G. Lamotrigine add‐on for drug‐resistant partial epilepsy. Cochrane Database of Systematic Reviews 2001, Issue 3. [DOI: 10.1002/14651858.CD001909] [DOI] [PubMed] [Google Scholar]
Rugg‐Gunn 2012
- Rugg‐Gunn FJ, Sander JW. Management of chronic epilepsy. BMJ (Clinical Research Ed.) 2012;345:4576. [DOI] [PubMed] [Google Scholar]
Shepherd 1989
- Shepherd C, Hosking G. Epilepsy in school children with intellectual impairments in Sheffield: the size and nature of the problem and its implications in service provision. Journal of Mental Deficiency Research 1989;33:511‐4. [DOI] [PubMed] [Google Scholar]
Shi 2014
- Shi LL, Dong J, Ni H, Geng J, Wu T. Felbamate as an add‐on therapy for refractory epilepsy. Cochrane Database of Systematic Reviews 2014, Issue 7. [DOI: 10.1002/14651858.CD008295.pub3] [DOI] [PubMed] [Google Scholar]
Stafstrom 1993
- Stafstrom C. Epilepsy in Down's syndrome: clinical aspects and possible mechanisms. American Journal of Mental Retardation 1993;98:12‐26. [PubMed] [Google Scholar]
Turky 2011
- Turky A, Felce D, Jones G, Kerr M. A prospective case control study of psychiatric disorders in adults with epilepsy and intellectual disability. Epilepsia 2011;52:1223‐30. [DOI] [PubMed] [Google Scholar]
Wallace 1996
- Wallace SJ. A comparative review of the adverse effects of anticonvulsants in children with epilepsy. Drug Safety 1996;15(6):378‐93. [DOI] [PubMed] [Google Scholar]
Webb 1991
- Webb D, Fryer A, Osborne J. On the incidence of fits and mental retardation in tuberous sclerosis. Journal of Medical Genetics 1991;6:395‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wolf 1995
- Wolf SM, Shinnar S, Kang H, Gil KB, Moshe SL. Gabapentin toxicity in children manifesting as behavioural changes. Epilepsia 1995;36(12):1203‐5. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Beavis 2007a
- Beavis J, Kerr M, Marson AG. Pharmacological interventions for epilepsy in people with intellectual disabilities. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD005399.pub2] [DOI] [PubMed] [Google Scholar]
Beavis 2007b
- Beavis J, Kerr M, Marson AG, Dojcinov I. Pharmacological interventions for epilepsy in people with intellectual disabilities. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD005399.pub2] [DOI] [PubMed] [Google Scholar]