

Abstract
The diagnosis of a neurofibroma or a malignant peripheral nerve sheath tumor (MPNST) often raises the question of whether the patient has genetic disorder neurofibromatosis type 1 (NF1) as well as how this will impact the patient’s outcome, what their risk is for developing additional neoplasms and whether treatment options differ for NF1-associated and sporadic peripheral nerve sheath tumors. Establishing a diagnosis of NF1 is challenging as this disorder has numerous neoplastic and non-neoplastic manifestations which are variably present in individual patients. Further, other genetic diseases affecting the Ras signaling cascade (RASopathies) mimic many of the clinical features of NF1. Here, we review the clinical manifestations of NF1 and compare and contrast them with those of the RASopathies. We also consider current approaches to genetic testing for germline NF1 mutations. We will then focus on NF1-associated neurofibromas, considering first the complicated clinical behavior and pathology of these neoplasms and then discussing our current understanding of the genomic abnormalities that drive their pathogenesis, including the mutations encountered in atypical neurofibromas. As several neurofibroma subtypes are capable of undergoing malignant transformation to become malignant peripheral nerve sheath tumors (MPNSTs), we compare and contrast patient outcomes in sporadic, NF1-associated and radiation-induced MPNSTs and review the challenging pathology of these lesions. The mutations involved in neurofibroma-MPNST progression, including the recent identification of mutations affecting epigenetic regulators,are then considered. Finally, we explore how our current understanding of neurofibroma and MPSNT pathogenesis is inform the design of new therapies for these neoplasms.
Keywords: Neurofibroma, malignant peripheral nerve sheath tumor, RASopathies, tumor genomics, epigenetic regulation
When diagnosing a neurofibroma or a malignant peripheral nerve sheath tumor (MPNST), pathologists often have to consider whether the patient might also have the genetic disorder neurofibromatosis type 1 (NF1). If NF1 is likely, the clinician caring for the patient may need to consider how this will impact the patient’s outcome, what their risk is for developing additional neoplasms and whether treatment options differ for NF1-associated and sporadic peripheral nerve sheath tumors. These questions are reasonable but answering them can be problematic. NF1 has numerous neoplastic and non-neoplastic clinical features, which are variably present in different patients; this variable presentation can make it difficult to diagnose NF1 with confidence. To complicate matters further, several genetic diseases that dysregulate the Ras pathway (RASopathies) have clinical manifestations that overlap with those of NF1. Predicting outcomes in patients with neurofibromas and MPNSTs continues to be complicated, in part because of ongoing controversies regarding the grading of these neoplasms. Distinguishing sporadic and NF1-associated neurofibromas and MPNSTs on the basis of genomic abnormalities is also not possible because we have not yet identified abnormalities that are unique to sporadic or NF1-associated tumors. Indeed, there is a surprising degree of overlap in the driver mutations found in sporadic and NF1-associated neurofibromas and MPNSTs; there are differences, though, which suggests that there is more than one pathway to neoplasia in the peripheral nervous system (PNS). These genomic studies are also providing hope, as they are generating intriguing new ideas as to how to treat neurofibromas and MPNSTs.
In this review, we will discuss our current understanding of NF1 and the pathology of NF1-associated peripheral nerve sheath tumors. We will first consider the clinical and genetic features of NF1, comparing these features to those seen in NF1 mimics. We will then focus on NF1-associated neurofibromas and MPNSTs, considering first the complicated clinical behavior and pathology of these neoplasms and then discussing our current understanding of the genomic abnormalities that drive their pathogenesis. Finally, we will explore how recent studies have informed the design of new, potentially effective therapies for neurofibromas and MPNSTs.
Presentation and Diagnosis of NF1
Clinical Features of NF1
NF1 (OMIM #162200) is the most common genetic disease affecting the human nervous system, occurring in every one in 2500–3500 newborn infants1–4. This condition, which is also known as peripheral neurofibromatosis or von Recklinghausen disease, is caused by the mutation of the NF1 tumor suppressor gene located on chromosome 17 (17q11.2). In families with a history of NF1, the disease typically manifests as a fully penetrant autosomal dominant disorder. The absence of a family history does not eliminate NF1 as a diagnostic possibility, however, as 50% of individuals with NF1 are born into families with no previous history of the disease. This reflects the fact that the NF1 gene has one of the highest rates of de novo mutation known for a single gene disorder. Intriguingly, these new NF1 mutations occur in the paternally derived chromosome and so are thought to arise during spermatogenesis5.
The lifespan of individuals with NF1 is typically about 15 years shorter than that of the general population6. In large part, this shortened lifespan reflects the fact that NF1 patients are at increased risk for the development of several types of benign and malignant neoplasms (Fig. 1). Although many of these tumors arise in the central nervous system (CNS) and the PNS, several types of NF1-associated neoplasms occur elsewhere including skin, the gastrointestinal tract, bone marrow, breast and soft tissues (Figure 1). In the central nervous system, NF1 patients develop astrocytic neoplasms, most commonly optic gliomas (pilocytic astrocytomas) that arise during childhood or adolescence. NF1-associated optic gliomas can occur anywhere along the optic nerves, the optic chiasm or the optic tracts and are found in approximately 15% of children with NF14, 7. As WHO grade I neoplasms, optic gliomas typically follow a benign clinical course. Nonetheless, they can be clinically problematic as nearly 50% of NF1 patients with optic gliomas develop moderate to severe visual impairment8, 9. Precocious puberty also occurs in a small fraction of NF1 patients whose optic gliomas involve the optic chiasm10, 11. Although less common than optic gliomas, NF1 patients are also at increased risk for the development of higher grade diffuse astrocytomas (WHO grade II-III) and glioblastomas (WHO grade IV), many of which occur in the brainstem12. Strikingly, The Cancer Genome Atlas Research Network has found that 18% of sporadic glioblastomas have homozygous deletion or mutation of the NF1 gene13, underscoring the important role that NF1 loss plays in glioblastoma pathogenesis. In the PNS, NF1 patients primarily develop neurofibromas and malignant peripheral nerve sheath tumors (MPNSTs; see below for a detailed discussion of these lesions).
FIGURE 1.
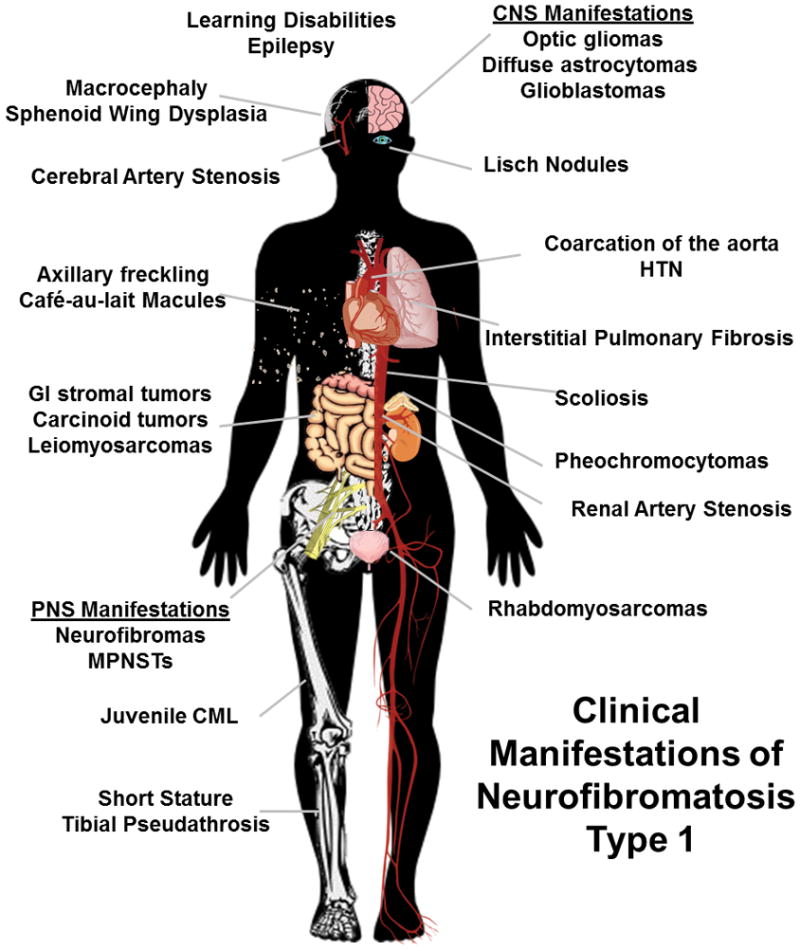
Diagram indicating the neoplastic and non-neoplastic clinical findings potentially encountered in NF1 patients.
Although tumors of the nervous system are most commonly encountered in NF1, individuals with this disorder also develop tumors at other anatomic sites. Several series have estimated that 0.1–5.7% of NF1 patients will develop pheochromocytomas, typically in the fifth decade of life14–18. Gastric carcinoids are also associated with NF1, although this is a rare manifestation of the disorder19. NF1 patients are at increased risk (45-fold higher than that of the general population) for the development of gastrointestinal stromal tumors (GISTs)20. Curiously, 60% of NF1-associated GISTs occur in the small intestine, whereas sporadic GISTs are most commonly gastric21. Young children with NF1 are prone to develop juvenile myelomonocytic leukemia, with boys being particularly susceptible to this malignancy22. Glomus tumors, which are small benign but exquisitely painful tumors that develop at the end of digits, have also been suggested to be a feature of NF123. Compared to the general population, NF1 patients are also at increased risk for the development of embryonal rhabdomyosarcomas24–26; this is, however, one of the less common manifestations of NF1, occurring in less than 1% of individuals with this disorder. Likewise, leiomyosarcomas and osteosarcomas occur rarely in NF1, but still at a rate higher than that of the general population27. Finally, it was recently found that the lifetime risk of developing breast cancer is doubled in women with NF1 and that the survival of NF1 patients with breast cancer is poorer than that of other breast cancer patients28, 29.
In addition to these neoplastic manifestations, there are a number of non-neoplastic findings that are commonly encountered in NF1 patients (Fig. 1). Macrocephaly is consistently present in NF1 patients; many individuals with NF1 also have a short stature, although this finding is more variable30–32. Melanocytic lesions of the skin (café au lait macules, axillary freckling) and iris (Lisch nodules) are very common findings in NF1. Skeletal abnormalities such as scoliosis, vertebral scalloping, unilateral sphenoid wing dysplasia and tibial pseudoarthrosis, are present in nearly half of NF1 patients33. Decreased bone mineral density is also frequent in NF1 patients. This is potentially related to the fact that serum 25-hydroxy-vitamin D concentrations are reduced in individuals with a large numbers of dermal neurofibromas34—in fact, 25-hydroxy-vitamin D levels are inversely proportion to the burden of these tumors. Cardiovascular abnormalities ranging from renal, coronary and cerebral artery stenosis to pulmonary stenosis, valvular malformations and coarctation of the abdominal aorta occur in nearly 10% of NF1 patients6, 35–38 and are another factor contributing to mortality in these patients, particularly those who die before 30 years of age6, 39. Although their IQ scores are normal or slightly below normal, learning disabilities are reported in up to 80% of children with NF140. Further, nearly 40% of these children have attention deficit/hyperactivity disorder, 30% have autism spectrum disorder41 and many have visual spatial deficits42. T2-weighted magnetic resonance imaging often identifies “unidentified bright objects” (UBOs) in the basal ganglia, thalamus, brainstem, cerebellum or subcortical white matter of these children43–45. These UBOs, which have variously been interpreted as hamartomas46, regions of abnormal myelination47, heterotopias48 or vacuolated myelin49, are potentially related to the learning disabilities seen in children with NF150. UBOs can also be confused with the radiologic abnormalities associated with a glioma.
It is thus apparent that a wide variety of clinical abnormalities can be encountered in NF1 patients. NF1 is completely penetrant and affects males and females equally. However, the constellation of findings that are present in individual patients is highly variable, even among closely related family members who have NF1. As a result of this variability, clinicians attempting to establish a diagnosis of NF1 rely on key clinical findings that are most commonly present in patients with this disorder. At least two of the following must be present to diagnose NF1: 1) six café-au-lait macules (>1.5 cm in diameter post puberty, >0.5 cm pre-puberty); 2) armpit or inguinal freckling; 3) one plexiform neurofibroma or two or more cutaneous neurofibromas; 4) an optic glioma; 5) two or more Lisch nodules; 6) bony dysplasia; or 7) a first-degree relative with NF1. In most instances, these criteria, which were developed by the 1988 National Institutes of Health Consensus Development Conference on Neurofibromatosis, work well. However, in patients where the diagnosis has relied primarily on pigmentary abnormalities rather than the occurrence of key neoplasms, NF1 can be confused with other NF1 mimics.
Distinguishing NF1 from NF1 Mimics Clinically
To understand why some RASopathies have clinical findings that overlap with NF1, one must first understand the signaling pathways affected by NF1 loss. Neurofibromin, the protein encoded by the NF1 gene, includes up to 2,818 amino acids and, as the result of alternative mRNA splicing, occurs as 220 kDa and 250 kDa protein species. Shortly after the NF1 gene was cloned, it was noted that a domain in the neurofibromin protein was highly homologous to the yeast Ras GTPase-activating proteins (Ras GAPs) IRA1 and IRA251. Functional analyses subsequently confirmed that the IRA-homologous domain of neurofibromin (amino acids 1203–1549 of GenBank sequence NP_000258.1), like the yeast IRA proteins, functions as a Ras GAP52. Ras proteins exist in two forms: an active GTP-bound form and an inactive GDP-bound form. The conversion of active Ras to the inactive state is dependent on a GTPase activity that is intrinsic to Ras. However, this intrinsic GTPase activity is very inefficient and Ras thus requires a “helper” protein to stimulate its own GTPase activity. The neurofibromin Ras GAP performs this function; neurofibromin interacts with Ras to stimulate its intrinsic GTPase activity, resulting in the cleavage of the terminal triphosphate of GTP, thus converting GTP (active) to GDP (inactive). In light of this neurofibromin function, it can be readily appreciated that loss-of-function mutations of the NF1 gene and the accompanying loss of neurofibromin expression result in unopposed activation of Ras and Ras-regulated signaling pathways. The regulation of Ras signaling, however, is not the sole function of neurofibromin. Indeed, several studies have shown that neurofibromin interacts with the cytoskeleton53 and focal adhesion kinase (FAK)54, regulates calcium55, 56 and cAMP-modulated57–59 signaling pathways and binds to glycerophospholipids60–62; all of these neurofibromin functions are independent of neurofibromin’s Ras GAP domain. However, neurofibromin’s ability to inhibit Ras signaling is its best understood function and it is clearly biologically relevant. Consequently, most attempts to design therapies targeting NF1-associated neoplasms have focused on Ras-associated signaling cascades (see below).
Ras proteins regulate a wide variety of cellular functions such as proliferation, survival, migration and differentiation63. Given the central role of Ras signaling in the life of the cell and the need to precisely regulate this signaling, it is not surprising that a number of proteins other than neurofibromin enhance or inhibit Ras-modulated signaling pathways (Fig. 2). Mutations in several of the genes encoding these proteins also dysregulate Ras-associated signaling cascades, thereby producing the genetic syndromes that are collectively known as the RASopathies64, 65. In addition to NF1, at least nine RASopathies have been defined to date: Legius syndrome, Noonan syndrome, LEOPARD syndrome (also known as Noonan syndrome with multiple lentigines), Noonan-like syndrome (recently renamed CBL syndrome), Noonan-like syndrome with loose anagen hair, Costello syndrome, cardiofaciocutaneous (CFC) syndrome, hereditary gingival fibromatosis and capillary malformation-arteriovenous malformation syndrome. The clinical manifestations of many of these RASopathies overlap with one another and with NF1. Intriguingly, however, although the mutations causing these syndromes all dysregulate the Ras signaling cascade, their clinical manifestations are not identical (Table 1). Of note, the findings encountered in non-NF1 RASopathies most commonly overlap with the non-neoplastic manifestations of NF1. For example, Legius syndrome, which is also known as NF1-like syndrome, is characterized by the occurrence of multiple café-au-lait macules, skin fold freckling, macrocephaly and learning disabilities, much as is seen in NF1. However, Legius syndrome patients, unlike NF1 patients, do not typically develop large numbers of dermal neurofibromas (we have, however, seen doubts raised by the chance co-occurrence of a sporadic neurofibroma in a Legius patient) or the spectrum of malignancies commonly encountered in NF1. Noonan syndrome patients also demonstrate a number of the non-neoplastic findings such as macrocephaly, short stature and mild cognitive impairment often seen in NF1; individuals with Noonan syndrome are also prone to develop myeloproliferative disorders similar to juvenile myelomonocytic leukemia. However, again, classic Noonan syndrome patients do not develop numerous neurofibromas or the spectrum of other neoplasms characteristic of NF1. The clinical features of the other RASopathies indicated in Table 1 show less overlap with NF1 than either Legius or Noonan syndrome. Consequently, a careful assessment of the non-neoplastic features of these patients and an appreciation of the fact that they either don’t develop malignancies or develop a spectrum of malignancies quite different from that seen in NF1 will usually keep the clinician from falling into the trap of misdiagnosing one of these RASopathies as NF1.
FIGURE 2.

The Ras-associated signaling cascades and the inherited causative genes responsible for the clinical manifestations of RASopathies. RASopathies include neurofibromatosis type 1 (NF1), Legius syndrome, Noonan syndrome (NS), CBL syndrome (Noonan-syndrome like with or without juvenile myelomonocytic leukemia), Noonan syndrome with multiple lentigines (NSML or LEOPARD), Noonan syndrome-like disorder with loose anagen hair (NLSAH), cardio-facio-cutaneous syndrome (CFC), Costello syndrome, and capillary malformation-ateriovenous malformation syndrome (CM-AVM). The most common mutated genes and the resultant proteins associated with each effected disease are color matched as indicated in the key. Note KRAS and BRAF are found mutated in both NS and CFC. Not depicted above are potential causal genes with germline mutations requiring further validation such as SOS2, A2ML1, RASA2, and LZTR1 (Noonan syndrome), RRAS (CBL syndrome) and BRAF (LEOPARD). RASopathy related germline gain-of-function mutations are designated with a “*”, loss-of-function mutations are designated with a “x”.
Abbreviations: A2ML1, alpha-2-macroglobulin like 1; BRAF, B-Raf proto-oncogene serine/threonine-protien kinase, CBL, the Cbl proto-oncogene; GRB2, growth factor receptor bound protein 2; LZTR1, leucine zipper like transcription regulator 1; MEK1/MEK2, mitogen-activated protein kinase kinase 1 and 2; neurofibromin, NF1; polo-like-kinase 1 (PLK1); protein phosphatase 1 (PP1), protein phosphatase 1 catalytic subunit beta (PPP1CB); PTPN11, protein tyrosine phosphatase non-receptor typ3 11; RAF1, Raf-1 proto-oncogene; RASA1, Ras p21 protein activator 1; RASA2, Ras p21 protein activator 2; RIT1, Ras-Like without CAAX 1; SHOC2, SHOC2 leucine rich repeat scaffold protein; SOS1, SOS Ras/Rac guanine nucleotide exchange factor 1; SOS2, SOS Ras/Rho guanine nucleotide exchange factor 2; SPRED1, sprout related EVH1 domain containing 1.
Table 1.
Clinical Features of RASopathies
| Syndrome | Non-Neoplastic Features | Neoplastic Features |
|---|---|---|
| Legius Syndrome/NF1-Like Syndrome | Multiple café-au-lait macules; macrocephaly; skin fold freckling; learning disability; Noonan syndrome-like facies; cardiac valvular abnormalities | Lipomas |
| Noonan Syndrome | Macrocephaly; distinctive facies; short stature; mild cognitive impairment; webbed neck; pectus excavatum; cryptorchidism | Juvenile myelomonocytic leukemia-like myeloproliferative disorders |
| LEOPARD Syndrome/ Noonan Syndrome with Multiple Lentigines | Multiple lentigines; electrocardiographic conduction abnormalities; ocular hypertelorism; pulmonary stenosis; abnormal genitalia; retardation of growth; sensorineural deafness | None |
| Noonan-Like Syndrome | Noonan syndrome-like facies; hyperpigmented skin lesions; microcephaly; cardiomyopathy; arrthymias; valvular abnormalities; developmental delays | None |
| Noonan-Like Syndrome with Loose Anagen Hair | Cardiovascular abnormalities; macrocephaly; short stature; mild cognitive impairment; fine sparse and easily pluckable hair | None |
| Costello syndrome | Cardiovascular abnormalities; coarse facies with facial warts; short stature; failure to thrive; severe feeding difficulties; curly hair; palmar keratosis. Variant exists presenting with a congenital myopathy with excess of muscle spindles. | Increased risk of malignant tumors including transitional cell carcinomas of bladder, rhabdomyosarcomas and ganglioneuroblastomas |
| Cardiofaciocutaneous (CFC) Syndrome | Cardiovascular abnormalities; distinct facies; failure to thrive; nevi, lentigines, palmar-plantar keratosis; curly hair; seizures; severe intellectual disability | None |
| Hereditary Gingival Fibromatosis | None | Gingival fibromatosis |
| Capillary Malformation-Arteriovenous Malformation Syndrome | Capillary malformations; arteriovenous malformations | None |
While the guidelines outlined above are typically sufficient for distinguishing NF1 from other RASopathies, we would note that there are some patients where diagnosis will still be problematic. This is because a small subset of NF1 patients exist who present with clinical features that more extensively overlap with other RASopathies. As an illustration of this, we would point to the subset of patients with the NF1 variant known as neurofibromatosis-Noonan syndrome (OMIM #601321). Individuals with neurofibromatosis-Noonan syndrome have café-au-lait macules, Lisch nodules and axillary freckling in combination with features of Noonan syndrome such as hypertelorism, epicanthal folds, a webbed neck, low-set ears and pulmonic stenosis; they do not, however, have neurofibromas66, 67. Genomic analyses have shown that neurofibromatosis-Noonan syndrome patients carry heterozygous mutations of the NF1 gene, with in-frame deletions affecting exons 24 and 25, which encode part of the Ras GAP sequence, being particularly common68. On occasion, co-existing mutations of the PTPN11 gene, which is linked to Noonan syndrome, are present in neurofibromatosis-Noonan syndrome patients69. However, this appears be to the exception rather than the rule.
NF1 Mutations and Genotype-Phenotype Correlations
In families in which NF1 has not been previously encountered or properly diagnosed, genomic testing is often performed to establish the diagnosis and to facilitate genetic counseling for family members. However, this testing is complicated, both because of the large size of the NF1 gene and because of the wide spectrum of mutations that are encountered in NF1 patients. The human NF1 gene spans approximately 283,000 base pairs and contains 60 exons, some of which are alternatively spliced in the mRNAs transcribed from this locus70, 71. In light of the high frequency of new mutations affecting the NF1 locus, it is not surprising that the types of mutations encountered in NF1 patients are diverse. The overwhelming majority of germline NF1 mutations are point mutations and include nonsense mutations, missense mutations, frameshift mutations and mutations affecting mRNA splicing72, 73. Only 5% of NF1 patients have whole gene deletion74, which apparently results from non-allelic homologous recombination between sequences flanking the NF1 locus. These homologous recombination events, which are also referred to as NF1 microdeletions, are classified as type I or type II recombination events. Type I deletions span 1.4 MB and result from recombination between the NF1-REP A and NF1-REP C clusters of paralogous loci75–78. Type II deletions result in the loss of 1.2 MB of sequence and arise by recombination between the SUZ12 gene and the SUZ12P1 pseudogene79, 80. Interestingly, three genes known as OMGP, EVI2B and EVI2A are encoded in an intron of the NF1 gene (although running in the opposite direction). These genes are also deleted in type I and type II recombination events, raising the question of whether their loss might also contribute to the phenotype of patients with NF1 microdeletions.
Germline mutations in the NF1 gene tend to cluster in particular locations, with mutations affecting exons 10a-c and 37 being particularly common72. However, given the frequency with which new mutations occur in the NF1 gene and the variety of NF1 mutation types described above, it is not surprising that germline NF1 mutations are also encountered at numerous other sites in this locus. To address this complexity, a comprehensive approach was developed several years ago which combined deep sequencing of all 60 NF1 exons, copy number analyses and screening for intronic splicing site mutations. Although complicated, this testing methodology was effective, identifying NF1 mutations in approximately 95% of clinically suspicious cases72; the 5% of candidate cases in which an NF1 diagnosis could not be established were likely NF1 cases in which the mutation occurred in gene regions that were not examined (e.g., promoter sequences of the gene), cases in which the patient was mosaic for the NF1 mutation or an NF1 mimic such as those described above. With the advent of massively parallel sequencing methodologies, an increasing number of diagnostic laboratories have now turned to sequencing the entire sequence of the NF1 gene to identify possible causative mutations. Curiously, however, many reference laboratories are reporting diagnostic success in only 75–85% of candidate cases. It is not clear whether this difference reflects a more careful selection of cases with the older diagnostic methodology or some other factor.
The question often arises as to whether the NF1 genotype predicts the phenotype in patients. The answer is “sometimes”. There is good evidence that patients with NF1 microdeletion (i.e., the 5% of NF1 patients with whole gene deletion) show a strong correlation between genotype and phenotype. NF1 microdeletion syndrome patients characteristically have large numbers of dermal neurofibromas (in the thousands), dysmorphic features and substantial cognitive impairment and are at increased risk for the development of plexiform neurofibromas and MPNSTs81. Another notable example is those pedigrees in which family members carry a three base pair in-frame deletion in exon 17 of the NF1 gene. While these patients do not develop dermal neurofibromas, they do manifest the other features characteristic of NF182.
On the other hand, the manifestations of NF1 can be highly variable in patients with other mutations of the NF1 gene. This is true even when comparing the clinical findings in family members who carry the exact same NF1 mutation. These clinical observations have led to the suggestion that modifier genes exist that interact with the NF1 gene and influence what manifestations of NF1 will be evident in an individual patient. As an initial assessment of this possibility, eight traits commonly encountered in NF1 (the number of café-au-lait macules, number of dermal neurofibromas and head circumference as well as the occurrence of plexiform neurofibromas, optic gliomas, scoliosis, epilepsy and referral for remedial education) were examined in 48 pedigrees (a total of 175 patients) that included 6 pairs of monozygotic twins19. This study showed that the number of café-au-lait macules and dermal neurofibromas had the highest correlation between monozygotic twins, with a lower degree of correlation between first-degree relatives and an even lower correlation among more distant relatives. The presence or absence of plexiform neurofibromas, optic gliomas, scoliosis, epilepsy and referral for remedial education also showed significant familial clustering, in a pattern most consistent with polygenic effects. There is also evidence that aberrant Ras activation must cross a threshold to drive tumorigenesis and that some “milder” NF1 mutations cannot accomplish this on their own83; in this circumstance, other genes must interact with NF1 mutations to drive tumorigenesis.
There is thus good evidence that modifier genes exist that interact with NF1 to modify the phenotype of NF1 patients. However, the identity of these modifier genes has not yet been established. Thus far, the most promising approaches to identifying the relevant modifier genes have come from studies in rodent models. It is clear that the pathogenesis of MPNSTs in mice carrying cis-linked Nf1 and p53 null mutations (cis-NP mice) is influenced by strain background84. Two unlinked polymorphic loci known as nerve sheath tumor resistance 1 (Nstr1) and 2 (Nstr2) have been mapped and shown to epistatically influence MPNST pathogenesis in cis-NP mice. Genome-wide association studies in BDIV (tumor resistant) and BDIX (tumor susceptible) rats exposed to the chemical mutagen ethylnitrosourea have also identified two genomic regions known as Mss1 and Mss7 that lower the risk of developing an MPNST85, 86. It remains to be determined whether the human counterparts of these rodent loci contribute to phenotypic variability in NF1 patients.
Neurofibromas
Pathology of Neurofibromas
It is generally accepted that multiple neurofibroma subtypes exist, that these subtypes have distinct natural histories and that they differ in their potential for undergoing malignant transformation and becoming an MPNST. However, how these neurofibroma subtypes are classified depends on the specialists’ perspective—at present, pathologists, dermatologists, geneticists and basic scientists all use different classification schemes. In an attempt to resolve this confusion, a summit that included experts from all of these specialities recently convened and devised a consensus classification scheme for neurofibromas (Ortonne et al, submitted). Hopefully, this consensus classification scheme will be widely accepted in the near future. However, at present, pathologists most commonly use the World Health Organization (WHO) criteria for defining neurofibroma subtypes. Consequently, we will use the WHO classification scheme here as a framework for discussing the biology of different neurofibroma subtypes.
The WHO classification scheme defines five neurofibroma subtypes (Table 2), two of which are found exclusively found in skin and related tissues (cutaneous or dermal neurofibromas). Localized cutaneous neurofibromas, as indicated by their designation, are well circumscribed non-encapsulated lesions that are found in the dermis and subcutaneous tissues of the skin. Sporadic localized cutaneous neurofibromas are very commonly encountered in the general population as solitary lesions—indeed localized cutaneous neurofibromas are the most common subtype of neurofibroma encountered clinically. Only about 10% of localized cutaneous neurofibromas are NF1-associated. NF1 patients also tend to have multiple localized cutaneous neurofibromas, which are often overlain by café-au-lait macules. Curiously, localized cutaneous neurofibromas are typically not present at birth in NF1 patients. These lesions instead begin to appear when the patient enters puberty and accumulate thereafter. In addition, localized cutaneous neurofibromas increase in number and enlarge in women with NF1 who get pregnant, suggesting that these lesions are hormonally regulated. Despite their at times rapid growth, localized cutaneous neurofibromas are benign tumors that have essentially no malignant potential. The second type of skin-associated neurofibromas, which are known as diffuse cutaneous neurofibromas, are poorly defined plaque-like lesions that, like localized cutaneous neurofibromas, are localized in the dermis and subcutaneous region of skin in children and young adults. As 90% of diffuse cutaneous neurofibromas are sporadic, the presence of these uncommon lesions is not necessarily indicative of NF1. Unlike localized cutaneous neurofibromas, however, diffuse cutaneous neurofibromas do have a very low, but nonetheless real, malignant potential. Although dermal neurofibromas have long been held to originate from peripheral nerves, it typically has been difficult to find a nerve of origin in either localized or diffuse cutaneous neurofibromas; even if a small cutaneous nerve can be identified, it is difficult to rule out the possibility that the nerve was simply entrapped by the growing neurofibroma. These observations, as well as observations in genetically engineered mouse models, have led to the suggestion that dermal neurofibromas instead arise from skin-derived precursors, which are a population of neural crest-derived multipotent progenitor cells found at the base of hair follicles87.
Table 2.
Characteristics of Neurofibroma Subtypes Defined by the WHO
| Subtype | Location | Relation to NF1 | Malignant Potential | Comments |
|---|---|---|---|---|
| Localized cutaneous | Skin (dermis and subcutaneous) | 10% NF1-associated, 90% sporadic | None | Most common subtype. In NF1, may be overlain by café-au-lait macules |
| Diffuse cutaneous | Skin (dermis and subcutaneous) | 10% NF1-associated, 90% sporadic | Very low, but does occur | Uncommon lesions that present as plaque-like thickenings of skin |
| Localized intraneural | Cranial, spinal or autonomic nerves | Can be either sporadic or NF1-associated | Intermediate | Second most common subtype |
| Plexiform | Cranial, spinal or autonomic nerve plexuses | Exclusively NF1-associated | Highest | Key diagnostic for NF1 |
| Massive soft tissue | Extremities, extensive soft tissue expansion with underlying large nerve | Exclusively NF1-associated | Intermediate, but low clinical impact due to rarity | Least common subtype, also known as elephantiasis neuromatosa |
The other three neurofibroma subtypes are more clearly associated with large nerves and are thought to originate from these structures. Localized intraneural neurofibromas, which are the second most common type of neurofibroma, occur at any point along cranial, spinal or autonomic nerves. Localized intraneural neurofibromas may be either sporadic or NF1-associated. Grossly, localized intraneural neurofibromas are evident as a segmental fusiform enlargement of the affected nerve rather than as a diffuse enlargement of multiple nerve fascicles as is seen in a plexiform neurofibroma. Localized intraneural neurofibromas can transform into a MPNST but do so less frequently than plexiform neurofibromas. Plexiform neurofibromas occur in cranial, spinal or autonomic nerve plexuses and are often thought to be exclusively NF1-associated. In contrast to localized intraneural neurofibromas, these produce diffuse enlargement of a nerve plexus rather than segmental enlargement of the nerve. Grossly, plexiform neurofibromas are often described as having a “bag of worms” appearance. Of the five neurofibroma subtypes, plexiform neurofibromas have the highest potential for undergoing transformation into a MPNST. Massive soft tissue neurofibromas are the rarest subtype of neurofibroma. These neoplasms occur in the extremities, where they trigger massive hypertrophy of soft tissue, resulting in the condition sometimes referred to as elephantiasis neurofibromatosa. An underlying plexiform neurofibroma is often found in massive soft tissue neurofibromas, which can lead to confusion as to how these neurofibromas should be subtyped. Like plexiform neurofibromas, massive soft tissue neurofibromas are solely NF1-associated neoplasms and often undergo malignant transformation. However, their clinical impact is lessened by the fact that massive soft tissue neurofibromas are by far the least common neurofibroma subtype and so are only rarely identified as the source of an MPNST.
Despite their differences in location and malignant potential, all five neurofibroma subtypes have an identical microscopic appearance. Examination of a hematoxylin and eosin stained section of a neurofibroma (Fig. 3A) shows a bland, moderately cellular neoplasm composed predominantly of spindled cells set again a richly collagenous or focally mucinous background. Despite this bland appearance, however, neurofibromas are actually composed of a very complex mixture of cell types. Between 40–80% of the cells within a neurofibroma are Schwann cell-like elements that are immunoreactive for the calcium-binding protein S100β, the low affinity neurotrophin receptor (p75LNTR) and the transcription factor Sox10 (Fig. 3B). Mast cells, which are recognizable by their immunoreactivity for c-Kit (CD117; Fig. 3C), are scattered throughout neurofibromas and are thought to be responsible for the pruritus that NF1 patients often experience in conjunction with their dermal neurofibromas. Neurofibromas also contain a peculiar population of CD34-immunoreactive dendritic cells (Fig. 3D). The identity of these cells remains controversial, with some investigators believing that these CD34-positive cells are a type of macrophage88 and others suggesting that they may be another, as yet undescribed, cell type89. A population of cells with the ultrastructural characteristics of perineurial cells (long thin cytoplasmic processes, frequent pinocytotic vesicles and a discontinuous basal lamina) are also present in neurofibromas; although these cells lack the epithelial membrane antigen immunoreactivity seen in normal perineurial cells, they do mark for the perineurial markers Glut1 and claudin. At least some of the CD34 immunoreactivity in neurofibromas is in small blood vessels, however, as demonstrated by the fact that these structures also label for CD31 (Fig. 3E). Fibroblasts are also numerous in neurofibromas and can be readily identified by their immunoreactivity for the transcription factor TCF4 (Fig. 3F). In addition, it was recently pointed out that neurofibromas contain large numbers of macrophages90 that label for the pan-macrophage marker Iba1 (Fig. 3G). These cells include both the anti-inflammatory (M2 macrophages, identifiable by CD163 staining; Fig. 3H) and pro-inflammatory (M1 macrophages, identifiable by immunoreactivity for CD86; Fig. 3I) subsets of macrophages.
FIGURE 3.

Cellular composition of neurofibromas. A, hematoxylin and eosin stained section of a dermal neurofibroma (40x). All subsequent images are taken from this same tumor at a 63x magnification. B, Immunoreactivity for the transcription factor Sox10 (green, representative cell indicated by an arrow) in the Schwann cell component of this dermal neurofibroma. The section has been counterstained with bisbenzamide to highlight the nuclei of all cells in the tumor. C, Immunoreactivity for the mast cell marker CD117 (also known as c-Kit; red, representative cell indicated by arrow) in the dermal neurofibroma. D, Immunoreactivity for CD34 (red) is apparent in vasculature (arrow) and in small dendritic cells in the tumor. E, Immunoreactivity (red) for the fibroblast nuclear marker TCF4 (transcription factor 4; representative cell indicated by arrow) in the tumor. F, Immunoreactivity for the pan-macrophage marker Iba1 (red, representative cell indicated by arrow) in the tumor. G, Immunoreactivity for CD163 (red), a marker of the M2 (anti-inflammatory) subclass of macrophages, in the tumor (representative cell indicated by arrow). H, Immunoreactivity for CD86 (red), a marker of the M1 (pro-inflammatory) subclass of macrophages, in the tumor (representative cell indicated by arrow).
Given the complex composition of neurofibromas, it is not surprising that for many decades pathologists argued about: 1) whether neurofibromas were hamartomas or were true neoplasms and 2) if they were neoplasms, which cell type within the neurofibroma was the neoplastic cell type. The identification of the NF1 gene in the early 1990s provided the tools that made it possible to settle these questions. The germline of NF1 patients is heterozygous for the NF1 mutation, carrying both a mutant allele and a wild-type allele. As clinical evidence suggested that NF1 functions as a tumor suppressor, it was anticipated that a “second hit” deleting the remaining wild-type (functional) NF1 gene would be required for tumorigenesis. In keeping with this, NF1 loss of heterozygosity (LOH) was identified in cells isolated from neurofibromas. However, NF1 LOH was evident in Schwann cells, but not fibroblasts, isolated from these tumors91, 92. These observations both confirmed the identification of neurofibromas as true neoplasms and argued that Schwann cells were the neoplastic component within neurofibromas. The subsequent generation of genetically engineered mouse models in which the ablation of Nf1 in Schwann cells triggered neurofibroma formation provided direct experimental proof that Schwann cells are the neoplastic element in neurofibromas93, 94.
Although it is now clear that Schwann cells are the neoplastic component of neurofibromas, a growing body of evidence indicates that the other cell types present in these neoplasms also contribute to tumorigenesis. Loss of the remaining functional NF1 allele in Schwann cells results in changes in the biology of Schwann cells (e.g., increased proliferation) that promote tumor formation. However, NF1 loss in Schwann cells also induces hyper-reactive paracrine interactions that act in concert to drive neurofibroma pathogenesis. At present, the role that paracrine interactions between NF1−/− Schwann cells and NF1+/− mast cells play in neurofibroma pathogenesis is best understood. Kit ligand, which promotes the proliferation, survival, migration and activation of mast cells is secreted by the Schwann cells within neurofibromas95, 96. A comparison of Kit ligand secretion by Schwann cells isolated from mice with ablation of the Nf1 gene showed that secretion of this growth factor was dependent on Nf1 gene dosage, with Nf1−/− Schwann cells secreting 6 times more Kit ligand than either Nf1+/− or wild-type Schwann cells97. Intriguingly, the proliferation, survival and migration of Nf1+/− mast cells challenged with Kit ligand is exaggerated compared to wild-type mast cells97–99, which facilitates the recruitment of mast cells into a developing neurofibroma. These recruited, hyperactive Nf1+/− mast cells are required for tumorigenesis, as mice with Schwann cell-specific Nf1 ablation that are grafted with Nf1+/− bone marrow develop neurofibromas, whereas those grafted with wild-type bone marrow do not100.
Follow Kit ligand stimulation, these recruited mast cells in turn demonstrate enhanced secretion of transforming factor-β (TGFβ)101, which acts on fibroblasts to stimulate collagen production. As Nf1+/− fibroblasts are themselves hyper-responsive to TGFβ compared to their wild-type counterparts, this results in collagen production that is in excess of what is produced by similarly stimulated wild-type fibroblasts. The existence of a paracrine network that connects Schwann cells, mast cells and fibroblasts in neurofibroma pathogenesis suggests that other, as yet undiscovered, paracrine factors promote additional interactions between other cell types intrinsic to these neoplasms. Investigations directed towards further defining these paracrine networks and identifying key signaling molecules within them will likely be a fruitful avenue of future study that has great potential for therapeutic development.
Genomic abnormalities in neurofibromas
As noted above, Schwann cells derived from NF1-associated neurofibromas have both a germline NF1 mutation and a somatic NF1 mutation91, 92, 102, which is often evident as NF1 LOH. The essential role of NF1 loss in neurofibroma pathogenesis is supported by the development of neurofibromas in mice following ablation of the Nf1 gene in Schwann cells93, 94. Consequently, it is clear that NF1 loss is necessary for the pathogenesis of neurofibromas in NF1 patients. However, there is also reason to think that NF1 loss alone is not sufficient for the development of a neurofibroma and that additional genomic or other abnormalities are required for tumorigenesis. This reasoning is supported, in part, by our current understanding of the regulation of the Ras signaling pathway. Because of the central role it plays in a number of key cellular functions, surveillance mechanisms exist that reign in excessive Ras activation by triggering a process known as oncogene-induced senescence. This process is clearly relevant to the pathogenesis of NF1-associated neoplasms as experimental ablation of NF1 expression in fibroblasts results in an initial activation of Ras and Ras-regulated signaling pathways which is then followed by inactivation of these pathways, growth arrest and cellular senescence103. As senescent neurofibromin-negative Schwann cells are present in neurofibromas, oncogene-induced senescence is likely operant in NF1-null Schwann cells and must be overcome for tumor growth. However, the mechanism by which NF1-null Schwann cells overcome oncogene-induced senescence is not yet understood. In principal, this mechanism could involve the occurrence of mutations in other genes, alterations in the epigenome or biochemical alterations that oppose oncogene-induced senescence.
Several laboratories have examined the genomes of NF1-associated neurofibromas, both to search for other mutations that interact with NF1 loss to drive tumor growth and to determine whether the multiple neurofibromas that occur in these patients arise independently or are instead clones derived from a single progenitor event. At present, there is little evidence that implicates mutations in genes other than NF1 in neurofibroma pathogenesis. Emmerich et al104 performed whole exome sequencing on seven spatially distinct dermal neurofibromas and two reference tissues from a single NF1 patient. Although these tumors shared a common germline NF1 mutation, the “second hit” NF1 mutation was distinct in each tumor, indicating these neoplasms arose independently. These authors reported somatic mutations in other genes in only two of their seven neoplasms, which led them to suggest that mutations in genes other than NF1 are not necessary for the pathogenesis of NF1-associated dermal neurofibromas. In contrast, Faden et al also performed whole exome sequencing on six dermal neurofibromas from a single individual and found 15–26 somatic mutations per sample105. Although NF1 was the only recurrently mutated gene in their tumors, pathway analyses indicated that mutations affecting Hippo pathway components were over-represented in their tumors. Findings similar to those of Emmerich et al have been reported in plexiform neurofibromas. Whole exome sequencing, RNA-Seq and genome-wide copy number assays performed on 23 Schwann cell cultures established from plexiform neurofibromas consistently identified NF1 mutations in these tumors, but found no evidence of other recurrent somatic inactivating mutations or copy number changes106. Consequently, there little evidence at present to indicate that specific genes other than NF1 are consistently mutated in dermal or plexiform neurofibromas. These observations suggest that it is more likely that other types of abnormalities (e.g., epigenetic changes, biochemical alterations) instead interact with NF1 loss to promote neurofibroma growth.
In contrast, there is good evidence that the accumulation of additional genomic abnormalities occurs when a benign neurofibroma transitions to become an atypical neurofibroma. In keeping with the studies cited above, array comparative genomic hybridization (aCGH) analyses of 16 atypical neurofibromas, 15 benign neurofibromas (11 plexiform, 4 dermal) and 34 MPNSTs [including (WHO grade II), intermediate (WHO grade III) and high grade (WHO grade IV) lesions] showed no evidence of copy number changes in the benign neurofibromas107. However, 15 of the 16 atypical neurofibromas showed deletions of CDKN2A/CDKN2B, a locus encoding two key inhibitors of the cell cycle (p16INK4A and p19ARF). This same abnormality was present in the MPNSTs, which led these investigators to suggest that CDKN2A/CDKN2B loss is an early step in neurofibroma-MPNST progression and that this loss can be used as a diagnostic marker for the premalignant state in neurofibromas.
Malignant Peripheral Nerve Sheath Tumors (MPNSTs)
Clinical Features and Diagnosis of MPNSTs
Malignant peripheral nerve sheath tumors (MPNSTs) are aggressive spindle-cell neoplasms derived from the Schwann cell lineage2, 108. As a group, MPNSTs represent approximately 2–5% of all soft tissue sarcomas109, 110. However, MPNSTs are encountered in three very different clinical settings, which raises the question of whether there are distinct MPNST subtypes that arise via different pathogenic mechanisms. About 40–50% of MPNSTs arise in NF1 patients2. Indeed, MPNSTs are the most common malignancy encountered in NF1 patients—these individuals’ lifetime risk of developing an MPNST has been estimated at 8–13%111 and 5.9–10.3%112. Another 40–47% of MPNSTs are sporadic, with the remaining 10–13% occurring at sites of previous radiation therapy111, 113–115. Although there is evidence that NF1-associated, sporadic and radiation-induced MPNSTs have different prognoses, this evidence is controversial and there is an ongoing debate as to which of these groups have the worst prognosis. Some investigators have found that NF1-associated MPNSTs have a poorer outcome than sporadic or radiation-induced MPNSTs115–118. In contrast, other groups have reported that radiation-induced MPNSTs have the poorest outcome114, 118. This controversy aside, there is general agreement that MPNSTs have a poor prognosis, with multiple centers reporting that the five year disease-free survival rates of patients with these tumors are between 34–60%113, 115, 117–122.
Despite their different prognoses, there are no histologic features that distinguish sporadic, NF1-associated and radiation-induced MPNSTs. Irrespective of their origin, MPNSTs are poorly differentiated spindle cell neoplasms in which mitotic figures are usually readily identified (Fig. 4). Unfortunately, MPNSTs are histologically similar to several other types of spindle cell malignancies. Further, the immunohistochemical markers that are commonly used to diagnose an MPNST and eliminate other diagnostic possibilities have significant limitations. To begin with, S100β, the most widely used marker for MPNSTs, is only positive in 30 to 65% of MPNSTs. Glial fibrillary acidic protein (GFAP), an intermediate filament normally expressed in non-myelinating Schwann cells, is positive in an even smaller fraction of MPNSTs (20 to 30% of cases). Leu-7 (CD57) shows variable staining in MPNSTs, with the amount of Leu-7 immunoreactivity present being dependent on the degree of Schwannian differentiation. In our experience, basement membrane markers such as collagen type IV and laminin are patchy and tend to be more prominent only in low-grade MPNSTs. It has been reported that strong immunoreactivity for the intermediate filament nestin (despite the misconception that this is a stem cell marker, nestin is strongly expressed in non-neoplastic Schwann cells) is common in MPNSTs123. Again, however, other tumor types that are in the differential for MPNSTs also stain for nestin. A recent study suggested using loss of neurofibromin immunoreactivity as a means of distinguishing MPNSTs from other spindle cell neoplasms124. This approach likely will be useful for identifying NF1-associated MPNSTs and a subset of sporadic and radiation-induced MPNSTs, as at least some of these latter tumors also have NF1 loss (see Genomic Abnormalities in MPNSTs, below). However, NF1 is maintained in other sporadic and radiation-induced MPNSTs, so this approach likely won’t distinguish all MPNSTs from their histologic mimics. Due to the often poor differentiation encountered in MPNSTS and the lack of specific ultrastructural markers, electron microscopy also has limited utility. MPNSTs typically show rudimentary to poorly formed cell junctions, long-spacing fibrous collagen (Luse bodies) and partial investment of tumor cells by basement membranes; these features may be more obvious in low-grade MPNSTs.
FIGURE 4.

Pathology of a MPNST that arose from a plexiform neurofibroma involving the brachial plexus of a 32 year old Caucasian man with NF1. A, section of the MPNST, which is evident as a markedly hypercellular spindle cell neoplasm. The arrow indicates one of the numerous mitotic figures that were detected in this malignancy. B, regions of the original plexiform neurofibroma were readily detected in this resection specimen.
Grading of MPNSTs also continues to be problematic. The WHO classification scheme currently assigns three grades to MPNSTs (WHO grades II-IV). Per their diagnostic criteria, WHO grade II (low grade) MPNSTs are hypercellular lesions that contain cells with nuclei that are greater than 3-fold larger than typical neurofibroma nuclei and show nuclear hyperchromasia. Although the presence of mitotic figures supports the diagnosis, these features are assessed independent of mitotic activity. This is a diagnostically challenging grade, as it is quite difficult to use these findings to distinguish between an atypical neurofibroma and a low grade MPNST. A WHO grade III (intermediate grade) MPNST is characterized by prominent hypercellularity, clear cytologic atypia and brisk mitotic activity (4 or more mitoses per 10 high power fields). WHO grade IV (high grade) MPNSTs show tumor necrosis in addition to the other features present in a WHO grade III lesion. In addition to the difficulties noted for low grade MPNSTs, this grading system has been criticized because it does not clearly predict patient outcomes. Consequently, many pathologists prefer to simply classify MPNSTs as either low grade or high grade tumors.
Genomic Abnormalities in MPNSTs
The difficulties inherent to diagnosing and grading MPNSTs and the need for effective new therapeutic regimens has stimulated interest in understanding the genomic abnormalities in these neoplasms. In contrast to neurofibromas, the genomes of MPNSTs are highly complex. Classic karyotyping indicates that MPNSTs tend to be hypodiploid or near triploid, with multiple regions of chromosomal gain and loss, both cytogenetically and with array CGH125–133. Initial reports were that gains are commonly seen on chromosome 7, 8p and 19q, whereas losses are more widespread (chromosomes 1p, 9p, 11,12p, 141, 17q, 18, 22q, X and Y). The genomic changes in MPNSTs are likely more complex than was indicated by these earlier studies. We recently examined a series of human MPNST cell lines and surgically resected MPNSTs using high density SNP arrays capable of identifying both balanced and unbalanced regions of chromosomal gain or loss (Huenerberg et al, in preparation). We found a characteristic pattern in the genomes of these tumors, with unusually high levels of loss of heterozygosity and frequent regions of chromosomal gain. Overall, LOH occurred in up to 20 chromosomes (median, 14) with gains or high copy number gains in up to 23 chromosomes (median, 18). Analysis of the genomic segments in these tumors indicated that a massive loss of genomic material occurs early in MPNST pathogenesis that is followed by whole or partial genome reduplication. In keeping with these high density SNP array analyses, our whole exome sequencing and RNA-Seq analyses of these same specimens also shows extensive genomic rearrangement, which results in the production of multiple fusion genes (Longo et al, in preparation).
At present, our understanding of the specific mutations that drive MPNST pathogenesis is rudimentary. Like neurofibromas, MPNSTs show biallelic inactivation of the NF1 gene134. Since this observation makes it clear that NF1 loss alone is not sufficient for MPNST pathogenesis, the progression of a plexiform neurofibroma to become a MPNST must involve the mutation of additional driver genes. The first mutations other than NF1 that were identified in MPNSTs were in genes encoding proteins in the p19ARF-MDM2-p53 and p16INK4A-cyclin D-Rb signaling cascades, both of which regulate the progression of the cell cycle. Mutations of TP53 are quite common in MPNSTs, with several series reporting mutations of this tumor suppressor gene in up to 75% of MPNSTs135–138. Intriguingly, complete p53 loss is not required for MPNSTZ pathogenesis as loss or mutation of only one copy of TP53 is common in MPNSTs139. Loss of the CDKN2A gene, which simultaneously dysregulates the p19ARF-Mdm2-p53 and p16INK4A-cyclin D-Rb pathways, is evident in about half of all MPNSTs140, 141. Further emphasizing the importance of these cell cycle regulatory pathways in MPNSTs, RB1 loss is observed in approximately 25% of MPNSTs142, 143. In addition to the mutations of genes encoding proteins in the p19ARF-MDM2-p53 and p16INK4A-cyclin D-Rb signaling cascades, mutations of some genes previously shown to be important in other tumor types are encountered in MPNSTs. Decreased expression of PTEN, a key inhibitor of the PI3 kinase-Akt signaling pathway, has been reported in MPNSTs144, 145; PTEN deletions and epigenetic silencing of the PTEN locus have both been identified in MPNSTs146, 147 and likely account for this decreased expression. Amplification of the EGFR148, 149 gene, which encodes a key upstream activator of the Ras signaling cascade also occurs in MPNSTs. In addition to these genes, several other genes have been shown to be mutated, amplified or deleted in MPNSTs. However, many of these mutations occur in small subsets of MPNSTs and their functional impact has not yet been thoroughly assessed.
With the advent of massively parallel sequencing methodologies, we are now getting our first comprehensive look at the genomic abnormalities present in MPNSTs. Two groups recently reported the results of using whole exome/genome sequencing, copy number analyses and whole transcriptome sequencing on 15150 and 4151 MPNSTs; these tumors included NF1-associated, sporadic and radiation-induced MPNSTs. These investigators found that NF1 loss was uniformly present in the NF1-associated MPNSTs and in 72% of the sporadic and radiation-induced MPNSTs150. Both groups also found loss-of-function mutations in genes encoding proteins in Polycomb repressive complex 2 (PRCs), an epigenetic regulator that modulates histone methylation, in up to 80% of MPNSTs. The affected genes included SUZ12, EED, EPC1 and CHD4; intriguingly, SUZ12 is the same gene that is involved in the type II recombination events that produce NF1 microdeletions (see NF1 Mutations and Genotype-Phenotype Correlations, above). SUZ12 and EED mutations are associated with a loss of di- and trimethylation of lysine 27 in histone H3 (H3K27me3 marks) and a transcriptome signature that is distinct from the signature of MPNSTs with intact SUZ12 and EED genes. Restoring SUZ12 expression in SUZ12-null MPNST cells restores H3K27me3 marks and inhibits tumor growth, indicating that this gene acts as a tumor suppressor in MPNST cells. SUZ12 and EED are not lost in neurofibromas, indicating that these mutations occur either during neurofibroma-MPNST progression or in MPNSTs as they progress from low grade to high grade. These observations have led to the suggestion that a loss of H3K27me3 immunoreactivity can be useful for distinguishing MPNSTs from neurofibromas and for distinguishing MPNSTs from their histologic mimics152–155. Undoubtedly, these findings are the first of many observations that will further clarify the role that specific genomic and epigenetic abnormalities play in MPNST pathogenesis.
New Approaches to the Treatment of NF1-Associated Peripheral Nerve Sheath Tumors
The poor survival of patients with MPNSTs and the morbidity associated with neurofibromas reflects the fact that the currently employed radio- and chemotherapeutic regimens are ineffective against these tumors. This has led several laboratories to ask whether neurofibromas and MPNSTs could be treated by targeting the signaling abnormalities that result from NF1 loss, since this mutation is present in all neurofibromas and MPNSTs arising in NF1 patients2 as well as in the majority of sporadic and radiation-induced MPNSTs150. Given neurofibromin’s role as a Ras GTPase-activating protein, most attempts to develop new therapies for neurofibromas and MPNSTs initially focused on Ras and Ras-regulated signaling cascades. In an initial clinical trial, the farnesyltransferase inhibitor tipifarnib, which prevents an essential modification of H-Ras, was used to treat children with plexiform neurofibromas. Although well tolerated in Phase I trials156, tipifarnib ultimately proved ineffective for the treatment of these tumors. This failure likely reflects the fact that neurofibromin acts as a Ras GAP for multiple members of the classic Ras (H-Ras, K-Ras, N-Ras) and R-Ras (R-Ras, R-Ras2/TC21 and M-Ras/R-Ras3) subfamilies of small GTP-binding proteins52, 157–159, most of which are not sensitive to tipifarnib. We have found that all six of these Ras proteins are expressed and activated in MPNST cells; knockdown of one Ras protein simply results in increased expression and activation of other Ras family members160. Since targeting these Ras proteins by other means has proven difficult63, subsequent efforts instead focused on inhibiting downstream mediators of Ras signaling such as the Raf/MEK/ERK and PI3 kinase/AKT/mTOR effector pathways. Unfortunately, this approach has also had limited success. Although MEK inhibitors are effective against neurofibromas, MPNSTs are only moderately and variably responsive to these agents161, 162. The Raf inhibitor sorafenib also showed no objective responses in plexiform neurofibromas163 or MPNSTs164. The mTORC1 inhibitor rapamycin showed promising effects in culture165, but had only transient effects on MPNST xenograft growth in vivo166. While these efforts were logical and merit additional investigation, it is clear that therapeutic agents targeting key signaling pathways downstream of Ras have not yet been shown to be effective against MPNSTs.
An alternative approach to treating neurofibromas and MPNSTs would be to inhibit key molecules upstream of Ras proteins that are essential for Ras activation. As noted above, MPNST pathogenesis is driven by NF1 mutation-induced Ras hyperactivation and other incompletely understood mutations that accumulate during neurofibroma-MPNST progression2. In some other sporadic cancer types that are Ras-dependent, Ras hyperactivation is promoted via multiple mechanisms that activate receptor tyrosine kinases (RTKs). This suggests that Ras-mediated increases in proliferation, invasion and survival in MPNSTs could be inhibited by therapeutically targeting one or more RTK(s). Initiating experiments to test the effects of inhibiting the key RTK(s) in MPNSTs, however, have been confounded by varying reports in the literature. Several laboratories have presented evidence that one or another RTK drives MPNST growth. However, these reports often contradict one another and the strength of the evidence supporting each candidate RTK is variable. For instance, an amplicon involving chromosomal segment 4q12 occurs in 19% of MPNSTs; this amplicon contains genes encoding three RTKs (PDGFRA, KIT and KDR)167. Platelet-derived growth factor (PDGF) and its two receptors (PDGFRA and PDGFRB) are co-expressed in neurofibromas168 and MPNSTs169 and these receptors are constitutively activated in at least some MPNSTs170. VEGF and its KDR (VEGFR2) receptor are similarly co-expressed in MPNST cells and have been suggested to form an autocrine signaling loop171. Some, but not all, MPNST lines express c-Kit and the baseline proliferation of MPNST cells has reportedly been inhibited with agents targeting this RTK172. Co-expression of hepatocyte growth factor (HGF) and its c-Met receptor have been detected in neurofibromas173–175 and MPNSTs173, 175, raising the possibility that these molecules also form an autocrine signaling loop in these tumors. The EGF receptor is inappropriately expressed in neurofibromas and MPNSTs148 and the EGFR gene is amplified in a subset of MPNSTs149. Introduction of an Egfr hypomorphic mutation into cis-Nf1+/−;Trp53+/− mice (a widely used model of NF1-associated MPNSTs) impedes tumorigenesis176, suggesting that EGF signaling is important for MPNST growth. Finally, we have shown that the neuregulin-1 (NRG1) receptors erbB2, erbB3 and erbB4 are present and activated in MPNSTs, where they promote proliferation177 and migration178. When we overexpressed the NRG1 isoform GGFβ3 in the Schwann cells of transgenic mice, these animals developed plexiform neurofibromas that progressed to become MPNSTs at a high frequency179; our genetic complementation experiments indicated that GGFβ3 promotes tumorigenesis via its effects on Nf1-regulated signaling pathways180.
In light of these conflicting reports, it is clear that a comprehensive analysis needs to be performed that will look simultaneously at the role that all 58 of the RTKs encoded in the human genome play in individual MPNSTs. This is needed because at present we cannot rule out the possibility that RTKs other than those noted above also contribute to MPNST pathogenesis; indeed, the role of many of the 58 RTKs found in humans has not yet been explored in MPNSTs. Based on experience in other tumor types (e.g., glioblastomas), we also have to consider the possibility that multiple RTKs are co-activated in MPNSTs and simultaneously contribute to tumorigenesis181, 182. Finally, it is conceivable that there are distinct MPNST subtypes that depend on different RTKs for growth. Nonetheless, considering the large number of drugs currently available that effectively inhibit RTKs, this is a promising line of investigation that is being actively pursued by multiple laboratories. Identifying an RTK inhibitor, or more likely a combination of RTK inhibitors with or without inhibitors of key Ras-regulated pathways, would finally give us an effective means of treating MPNSTs or preventing the malignant transformation of neurofibromas.
In summary, there have been great advances in our understanding of the clinical abnormalities associated with NF1 and the features that distinguish NF1 from other RASopathies. Although distinguishing these conditions remains challenging, we now have genetic testing available that can facilitate this process. Recent advances in understanding the genomic abnormalities found in neurofibromas, atypical neurofibromas and MPNSTs are also providing new tools that can be used better classify these lesions pathologically. As we build upon these advances in coming years, it is our hope that our growing understanding of the genomic abnormalities in NF1-associated peripheral nerve sheath tumors will allow the rational design of effective new therapies for these clinically challenging neoplasms.
Acknowledgments
Sources of Funding: This work was supported by the National Institute of Neurological Diseases and Stroke (R01 NS048353), the National Cancer Institute (R01 CA122804), the Department of Defense (X81XWH-09-1-0086, W81XWH-12-1-0164, W81XWH-14-1-0073 and W81XWH-15-1-0193) and the Children’s Tumor Foundation (2015-05-007 and 2016-04-001).
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest.
References
- 1.Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590–605. doi: 10.1002/glia.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carroll SL. Molecular mechanisms promoting the pathogenesis of Schwann cell neoplasms. Acta Neuropathol. 2012;123:321–48. doi: 10.1007/s00401-011-0928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen MH, Harper PS, Upadhyaya M. Molecular genetics of neurofibromatosis type 1 (NF1) J Med Genet. 1996;33:2–17. doi: 10.1136/jmg.33.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics. 2009;123:124–33. doi: 10.1542/peds.2007-3204. [DOI] [PubMed] [Google Scholar]
- 5.Jadayel D, Fain P, Upadhyaya M, Ponder MA, Huson SM, Carey J, Fryer A, Mathew CG, Barker DF, Ponder BA. Paternal origin of new mutations in von Recklinghausen neurofibromatosis. Nature. 1990;343:558–9. doi: 10.1038/343558a0. [DOI] [PubMed] [Google Scholar]
- 6.Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. 2001;68:1110–8. doi: 10.1086/320121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44:81–8. doi: 10.1136/jmg.2006.045906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balcer LJ, Liu GT, Heller G, Bilaniuk L, Volpe NJ, Galetta SL, Molloy PT, Phillips PC, Janss AJ, Vaughn S, Maguire MG. Visual loss in children with neurofibromatosis type 1 and optic pathway gliomas: relation to tumor location by magnetic resonance imaging. Am J Ophthalmol. 2001;131:442–5. doi: 10.1016/s0002-9394(00)00852-7. [DOI] [PubMed] [Google Scholar]
- 9.Thiagalingam S, Flaherty M, Billson F, North K. Neurofibromatosis type 1 and optic pathway gliomas: follow-up of 54 patients. Ophthalmology. 2004;111:568–77. doi: 10.1016/j.ophtha.2003.06.008. [DOI] [PubMed] [Google Scholar]
- 10.Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125:63–6. doi: 10.1016/s0022-3476(94)70122-9. [DOI] [PubMed] [Google Scholar]
- 11.Listernick R, Darling C, Greenwald M, Strauss L, Charrow J. Optic pathway tumors in children: the effect of neurofibromatosis type 1 on clinical manifestations and natural history. J Pediatr. 1995;127:718–22. doi: 10.1016/s0022-3476(95)70159-1. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez FJ, Perry A, Gutmann DH, O’Neill BP, Leonard J, Bryant S, Giannini C. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol. 2008;67:240–9. doi: 10.1097/NEN.0b013e318165eb75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riccardi VM. Neurofibromatosis: past, present, and future. N Engl J Med. 1991;324:1283–5. doi: 10.1056/NEJM199105023241812. [DOI] [PubMed] [Google Scholar]
- 15.Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen’s disease and pheochromocytomas. J Urol. 1999;162:1582–6. [PubMed] [Google Scholar]
- 16.Bausch B, Koschker AC, Fassnacht M, Stoevesandt J, Hoffmann MM, Eng C, Allolio B, Neumann HP. Comprehensive mutation scanning of NF1 in apparently sporadic cases of pheochromocytoma. J Clin Endocrinol Metab. 2006;91:3478–81. doi: 10.1210/jc.2006-0780. [DOI] [PubMed] [Google Scholar]
- 17.Mannelli M, Castellano M, Schiavi F, Filetti S, Giacche M, Mori L, Pignataro V, Bernini G, Giache V, Bacca A, Biondi B, Corona G, Di Trapani G, Grossrubatscher E, Reimondo G, Arnaldi G, Giacchetti G, Veglio F, Loli P, Colao A, Ambrosio MR, Terzolo M, Letizia C, Ercolino T, Opocher G Italian Pheochromocytoma/Paraganglioma N. Clinically guided genetic screening in a large cohort of italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab. 2009;94:1541–7. doi: 10.1210/jc.2008-2419. [DOI] [PubMed] [Google Scholar]
- 18.Eisenhofer G, Lenders JW, Timmers H, Mannelli M, Grebe SK, Hofbauer LC, Bornstein SR, Tiebel O, Adams K, Bratslavsky G, Linehan WM, Pacak K. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem. 2011;57:411–20. doi: 10.1373/clinchem.2010.153320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Easton DF, Ponder MA, Huson SM, Ponder BA. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. Am J Hum Genet. 1993;53:305–13. [PMC free article] [PubMed] [Google Scholar]
- 20.Barahona-Garrido J, Aguirre-Gutierrez R, Gutierrez-Manjarrez JI, Tellez-Avila FI, Lopez-Arce G, Fomperoza-Torres A, Criales S, Sanchez-Cortes E, Sarti HM, Yamamoto-Furusho JK. Association of GIST and somatostatinoma in a patient with type-1 neurofibromatosis: is there a common pathway? Am J Gastroenterol. 2009;104:797–9. doi: 10.1038/ajg.2008.133. [DOI] [PubMed] [Google Scholar]
- 21.Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30:90–6. doi: 10.1097/01.pas.0000176433.81079.bd. [DOI] [PubMed] [Google Scholar]
- 22.Stiller CA, Chessells JM, Fitchett M. Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer. 1994;70:969–72. doi: 10.1038/bjc.1994.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brems H, Park C, Maertens O, Pemov A, Messiaen L, Upadhyaya M, Claes K, Beert E, Peeters K, Mautner V, Sloan JL, Yao L, Lee CC, Sciot R, De Smet L, Legius E, Stewart DR. Glomus tumors in neurofibromatosis type 1: genetic, functional, and clinical evidence of a novel association. Cancer Res. 2009;69:7393–401. doi: 10.1158/0008-5472.CAN-09-1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGaughran JM, Harris DI, Donnai D, Teare D, MacLeod R, Westerbeek R, Kingston H, Super M, Harris R, Evans DG. A clinical study of type 1 neurofibromatosis in north west England. J Med Genet. 1999;36:197–203. [PMC free article] [PubMed] [Google Scholar]
- 25.Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. 2012;17:101–16. doi: 10.1634/theoncologist.2010-0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crucis A, Richer W, Brugieres L, Bergeron C, Marie-Cardine A, Stephan JL, Girard P, Corradini N, Munzer M, Lacour B, Minard-Colin V, Sarnacki S, Ranchere-Vince D, Orbach D, Bourdeaut F. Rhabdomyosarcomas in children with neurofibromatosis type I: A national historical cohort. Pediatr Blood Cancer. 2015;62:1733–8. doi: 10.1002/pbc.25556. [DOI] [PubMed] [Google Scholar]
- 27.Afsar CU, Kara IO, Kozat BK, Demiryurek H, Duman BB, Doran F. Neurofibromatosis type 1, gastrointestinal stromal tumor, leiomyosarcoma and osteosarcoma: four cases of rare tumors and a review of the literature. Crit Rev Oncol Hematol. 2013;86:191–9. doi: 10.1016/j.critrevonc.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Uusitalo E, Kallionpaa RA, Kurki S, Rantanen M, Pitkaniemi J, Kronqvist P, Harkonen P, Huovinen R, Carpen O, Poyhonen M, Peltonen S, Peltonen J. Breast cancer in neurofibromatosis type 1: overrepresentation of unfavourable prognostic factors. Br J Cancer. 2017;116:211–7. doi: 10.1038/bjc.2016.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Teer JK, Tousignant RN, Levin AM, Boulware D, Chitale DA, Shaw BM, Chen Z, Zhang Y, Blakeley JO, Acosta MT, Messiaen LM, Korf BR, Tainsky MA. Breast cancer risk and germline genomic profiling of women with neurofibromatosis type 1 who developed breast cancer. Genes Chromosomes Cancer. 2018;57:19–27. doi: 10.1002/gcc.22503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clementi M, Milani S, Mammi I, Boni S, Monciotti C, Tenconi R. Neurofibromatosis type 1 growth charts. Am J Med Genet. 1999;87:317–23. doi: 10.1002/(sici)1096-8628(19991203)87:4<317::aid-ajmg7>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 31.Szudek J, Birch P, Friedman JM. Growth charts for young children with neurofibromatosis 1 (NF1) Am J Med Genet. 2000;92:224–8. [PubMed] [Google Scholar]
- 32.Szudek J, Birch P, Friedman JM. Growth in North American white children with neurofibromatosis 1 (NF1) J Med Genet. 2000;37:933–8. doi: 10.1136/jmg.37.12.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elefteriou F, Kolanczyk M, Schindeler A, Viskochil DH, Hock JM, Schorry EK, Crawford AH, Friedman JM, Little D, Peltonen J, Carey JC, Feldman D, Yu X, Armstrong L, Birch P, Kendler DL, Mundlos S, Yang FC, Agiostratidou G, Hunter-Schaedle K, Stevenson DA. Skeletal abnormalities in neurofibromatosis type 1: approaches to therapeutic options. Am J Med Genet A. 2009;149A:2327–38. doi: 10.1002/ajmg.a.33045. [DOI] [PubMed] [Google Scholar]
- 34.Lammert M, Friedman JM, Roth HJ, Friedrich RE, Kluwe L, Atkins D, Schooler T, Mautner VF. Vitamin D deficiency associated with number of neurofibromas in neurofibromatosis 1. J Med Genet. 2006;43:810–3. doi: 10.1136/jmg.2006.041095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin AE, Birch PH, Korf BR, Tenconi R, Niimura M, Poyhonen M, Armfield Uhas K, Sigorini M, Virdis R, Romano C, Bonioli E, Wolkenstein P, Pivnick EK, Lawrence M, Friedman JM. Cardiovascular malformations and other cardiovascular abnormalities in neurofibromatosis 1. Am J Med Genet. 2000;95:108–17. doi: 10.1002/1096-8628(20001113)95:2<108::aid-ajmg4>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 36.Oderich GS, Sullivan TM, Bower TC, Gloviczki P, Miller DV, Babovic-Vuksanovic D, Macedo TA, Stanson A. Vascular abnormalities in patients with neurofibromatosis syndrome type I: clinical spectrum, management, and results. J Vasc Surg. 2007;46:475–84. doi: 10.1016/j.jvs.2007.03.055. [DOI] [PubMed] [Google Scholar]
- 37.Rea D, Brandsema JF, Armstrong D, Parkin PC, deVeber G, MacGregor D, Logan WJ, Askalan R. Cerebral arteriopathy in children with neurofibromatosis type 1. Pediatrics. 2009;124:e476–83. doi: 10.1542/peds.2009-0152. [DOI] [PubMed] [Google Scholar]
- 38.D’Arco F, D’Amico A, Caranci F, Di Paolo N, Melis D, Brunetti A. Cerebrovascular stenosis in neurofibromatosis type 1 and utility of magnetic resonance angiography: our experience and literature review. Radiol Med. 2014;119:415–21. doi: 10.1007/s11547-013-0358-8. [DOI] [PubMed] [Google Scholar]
- 39.Friedman JM, Arbiser J, Epstein JA, Gutmann DH, Huot SJ, Lin AE, McManus B, Korf BR. Cardiovascular disease in neurofibromatosis 1: report of the NF1 Cardiovascular Task Force. Genet Med. 2002;4:105–11. doi: 10.1097/00125817-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 40.Schwetye KE, Gutmann DH. Cognitive and behavioral problems in children with neurofibromatosis type 1: challenges and future directions. Expert Rev Neurother. 2014;14:1139–52. doi: 10.1586/14737175.2014.953931. [DOI] [PubMed] [Google Scholar]
- 41.Garg S, Green J, Leadbitter K, Emsley R, Lehtonen A, Evans DG, Huson SM. Neurofibromatosis type 1 and autism spectrum disorder. Pediatrics. 2013;132:e1642–8. doi: 10.1542/peds.2013-1868. [DOI] [PubMed] [Google Scholar]
- 42.Cutting LE, Levine TM. Cognitive profile of children with neurofibromatosis and reading disabilities. Child Neuropsychol. 2010;16:417–32. doi: 10.1080/09297041003761985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Itoh T, Magnaldi S, White RM, Denckla MB, Hofman K, Naidu S, Bryan RN. Neurofibromatosis type 1: the evolution of deep gray and white matter MR abnormalities. AJNR Am J Neuroradiol. 1994;15:1513–9. [PMC free article] [PubMed] [Google Scholar]
- 44.Van Es S, North KN, McHugh K, De Silva M. MRI findings in children with neurofibromatosis type 1: a prospective study. Pediatr Radiol. 1996;26:478–87. doi: 10.1007/BF01377205. [DOI] [PubMed] [Google Scholar]
- 45.Griffiths PD, Blaser S, Mukonoweshuro W, Armstrong D, Milo-Mason G, Cheung S. Neurofibromatosis bright objects in children with neurofibromatosis type 1: a proliferative potential? Pediatrics. 1999;104:e49. doi: 10.1542/peds.104.4.e49. [DOI] [PubMed] [Google Scholar]
- 46.Braffman BH, Bilaniuk LT, Zimmerman RA. The central nervous system manifestations of the phakomatoses on MR. Radiol Clin North Am. 1988;26:773–800. [PubMed] [Google Scholar]
- 47.Smirniotopoulos JG, Murphy FM. The phakomatoses. AJNR Am J Neuroradiol. 1992;13:725–46. [PMC free article] [PubMed] [Google Scholar]
- 48.Bognanno JR, Edwards MK, Lee TA, Dunn DW, Roos KL, Klatte EC. Cranial MR imaging in neurofibromatosis. AJR Am J Roentgenol. 1988;151:381–8. doi: 10.2214/ajr.151.2.381. [DOI] [PubMed] [Google Scholar]
- 49.DiPaolo DP, Zimmerman RA, Rorke LB, Zackai EH, Bilaniuk LT, Yachnis AT. Neurofibromatosis type 1: pathologic substrate of high-signal-intensity foci in the brain. Radiology. 1995;195:721–4. doi: 10.1148/radiology.195.3.7754001. [DOI] [PubMed] [Google Scholar]
- 50.Torres Nupan MM, Velez Van Meerbeke A, Lopez Cabra CA, Herrera Gomez PM. Cognitive and Behavioral Disorders in Children with Neurofibromatosis Type 1. Front Pediatr. 2017;5:227. doi: 10.3389/fped.2017.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu GF, O’Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62:599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- 52.Xu GF, Lin B, Tanaka K, Dunn D, Wood D, Gesteland R, White R, Weiss R, Tamanoi F. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae Cell. 1990;63:835–41. doi: 10.1016/0092-8674(90)90149-9. [DOI] [PubMed] [Google Scholar]
- 53.Bollag G, McCormick F, Clark R. Characterization of full-length neurofibromin: tubulin inhibits Ras GAP activity. EMBO J. 1993;12:1923–7. doi: 10.1002/j.1460-2075.1993.tb05841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kweh F, Zheng M, Kurenova E, Wallace M, Golubovskaya V, Cance WG. Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol Carcinog. 2009;48:1005–17. doi: 10.1002/mc.20552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korkiamaki T, Yla-Outinen H, Koivunen J, Karvonen SL, Peltonen J. Altered calcium-mediated cell signaling in keratinocytes cultured from patients with neurofibromatosis type 1. Am J Pathol. 2002;160:1981–90. doi: 10.1016/S0002-9440(10)61148-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dang I, DeVries GH. Schwann cell lines derived from malignant peripheral nerve sheath tumors respond abnormally to platelet-derived growth factor-BB. J Neurosci Res. 2005;79:318–28. doi: 10.1002/jnr.20334. [DOI] [PubMed] [Google Scholar]
- 57.Dasgupta B, Dugan LL, Gutmann DH. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci. 2003;23:8949–54. doi: 10.1523/JNEUROSCI.23-26-08949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dang I, De Vries GH. Aberrant cAMP metabolism in NF1 malignant peripheral nerve sheath tumor cells. Neurochem Res. 2011;36:1697–705. doi: 10.1007/s11064-011-0433-2. [DOI] [PubMed] [Google Scholar]
- 59.Kim HA, Ratner N, Roberts TM, Stiles CD. Schwann cell proliferative responses to cAMP and Nf1 are mediated by cyclin D1. J Neurosci. 2001;21:1110–6. doi: 10.1523/JNEUROSCI.21-04-01110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.D’Angelo I, Welti S, Bonneau F, Scheffzek K. A novel bipartite phospholipid-binding module in the neurofibromatosis type 1 protein. EMBO Rep. 2006;7:174–9. doi: 10.1038/sj.embor.7400602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Welti S, Fraterman S, D’Angelo I, Wilm M, Scheffzek K. The sec14 homology module of neurofibromin binds cellular glycerophospholipids: mass spectrometry and structure of a lipid complex. J Mol Biol. 2007;366:551–62. doi: 10.1016/j.jmb.2006.11.055. [DOI] [PubMed] [Google Scholar]
- 62.Welti S, Kuhn S, D’Angelo I, Brugger B, Kaufmann D, Scheffzek K. Structural and biochemical consequences of NF1 associated nontruncating mutations in the Sec14-PH module of neurofibromin. Hum Mutat. 2011;32:191–7. doi: 10.1002/humu.21405. [DOI] [PubMed] [Google Scholar]
- 63.Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2–27. doi: 10.4161/sgtp.1.1.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aoki Y, Niihori T, Inoue S, Matsubara Y. Recent advances in RASopathies. J Hum Genet. 2016;61:33–9. doi: 10.1038/jhg.2015.114. [DOI] [PubMed] [Google Scholar]
- 65.Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell. 2017;170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nystrom AM, Ekvall S, Allanson J, Edeby C, Elinder M, Holmstrom G, Bondeson ML, Anneren G. Noonan syndrome and neurofibromatosis type I in a family with a novel mutation in NF1. Clin Genet. 2009;76:524–34. doi: 10.1111/j.1399-0004.2009.01233.x. [DOI] [PubMed] [Google Scholar]
- 67.Stevenson DA, Viskochil DH, Rope AF, Carey JC. Clinical and molecular aspects of an informative family with neurofibromatosis type 1 and Noonan phenotype. Clin Genet. 2006;69:246–53. doi: 10.1111/j.1399-0004.2006.00576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.De Luca A, Bottillo I, Sarkozy A, Carta C, Neri C, Bellacchio E, Schirinzi A, Conti E, Zampino G, Battaglia A, Majore S, Rinaldi MM, Carella M, Marino B, Pizzuti A, Digilio MC, Tartaglia M, Dallapiccola B. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet. 2005;77:1092–101. doi: 10.1086/498454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bertola DR, Pereira AC, Passetti F, de Oliveira PS, Messiaen L, Gelb BD, Kim CA, Krieger JE. Neurofibromatosis-Noonan syndrome: molecular evidence of the concurrence of both disorders in a patient. Am J Med Genet A. 2005;136:242–5. doi: 10.1002/ajmg.a.30813. [DOI] [PubMed] [Google Scholar]
- 70.Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, Culver M, Carey JC, Copeland NG, Jenkins NA, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62:187–92. doi: 10.1016/0092-8674(90)90252-a. [DOI] [PubMed] [Google Scholar]
- 71.Wallace MR, Marchuk DA, Andersen LB, Letcher R, Odeh HM, Saulino AM, Fountain JW, Brereton A, Nicholson J, Mitchell AL, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–6. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 72.Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, Speleman F, Paepe AD. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000;15:541–55. doi: 10.1002/1098-1004(200006)15:6<541::AID-HUMU6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 73.Wimmer K, Roca X, Beiglbock H, Callens T, Etzler J, Rao AR, Krainer AR, Fonatsch C, Messiaen L. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5’ splice-site disruption. Hum Mutat. 2007;28:599–612. doi: 10.1002/humu.20493. [DOI] [PubMed] [Google Scholar]
- 74.Wimmer K, Yao S, Claes K, Kehrer-Sawatzki H, Tinschert S, De Raedt T, Legius E, Callens T, Beiglbock H, Maertens O, Messiaen L. Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer. 2006;45:265–76. doi: 10.1002/gcc.20289. [DOI] [PubMed] [Google Scholar]
- 75.Dorschner MO, Sybert VP, Weaver M, Pletcher BA, Stephens K. NF1 microdeletion breakpoints are clustered at flanking repetitive sequences. Hum Mol Genet. 2000;9:35–46. doi: 10.1093/hmg/9.1.35. [DOI] [PubMed] [Google Scholar]
- 76.Lopez-Correa C, Dorschner M, Brems H, Lazaro C, Clementi M, Upadhyaya M, Dooijes D, Moog U, Kehrer-Sawatzki H, Rutkowski JL, Fryns JP, Marynen P, Stephens K, Legius E. Recombination hotspot in NF1 microdeletion patients. Hum Mol Genet. 2001;10:1387–92. doi: 10.1093/hmg/10.13.1387. [DOI] [PubMed] [Google Scholar]
- 77.Jenne DE, Tinschert S, Reimann H, Lasinger W, Thiel G, Hameister H, Kehrer-Sawatzki H. Molecular characterization and gene content of breakpoint boundaries in patients with neurofibromatosis type 1 with 17q11. 2 microdeletions. Am J Hum Genet. 2001;69:516–27. doi: 10.1086/323043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raedt TD, Stephens M, Heyns I, Brems H, Thijs D, Messiaen L, Stephens K, Lazaro C, Wimmer K, Kehrer-Sawatzki H, Vidaud D, Kluwe L, Marynen P, Legius E. Conservation of hotspots for recombination in low-copy repeats associated with the NF1 microdeletion. Nat Genet. 2006;38:1419–23. doi: 10.1038/ng1920. [DOI] [PubMed] [Google Scholar]
- 79.Kehrer-Sawatzki H, Kluwe L, Sandig C, Kohn M, Wimmer K, Krammer U, Peyrl A, Jenne DE, Hansmann I, Mautner VF. High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 gene. Am J Hum Genet. 2004;75:410–23. doi: 10.1086/423624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Steinmann K, Cooper DN, Kluwe L, Chuzhanova NA, Senger C, Serra E, Lazaro C, Gilaberte M, Wimmer K, Mautner VF, Kehrer-Sawatzki H. Type 2 NF1 deletions are highly unusual by virtue of the absence of nonallelic homologous recombination hotspots and an apparent preference for female mitotic recombination. Am J Hum Genet. 2007;81:1201–20. doi: 10.1086/522089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pasmant E, Sabbagh A, Spurlock G, Laurendeau I, Grillo E, Hamel MJ, Martin L, Barbarot S, Leheup B, Rodriguez D, Lacombe D, Dollfus H, Pasquier L, Isidor B, Ferkal S, Soulier J, Sanson M, Dieux-Coeslier A, Bieche I, Parfait B, Vidaud M, Wolkenstein P, Upadhyaya M, Vidaud D members of the NFFN. NF1 microdeletions in neurofibromatosis type 1: from genotype to phenotype. Hum Mutat. 2010;31:E1506–18. doi: 10.1002/humu.21271. [DOI] [PubMed] [Google Scholar]
- 82.Upadhyaya M, Huson SM, Davies M, Thomas N, Chuzhanova N, Giovannini S, Evans DG, Howard E, Kerr B, Griffiths S, Consoli C, Side L, Adams D, Pierpont M, Hachen R, Barnicoat A, Li H, Wallace P, Van Biervliet JP, Stevenson D, Viskochil D, Baralle D, Haan E, Riccardi V, Turnpenny P, Lazaro C, Messiaen L. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c. 2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007;80:140–51. doi: 10.1086/510781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brohl AS, Kahen E, Yoder SJ, Teer JK, Reed DR. The genomic landscape of malignant peripheral nerve sheath tumors: diverse drivers of Ras pathway activation. Sci Rep. 2017;7:14992. doi: 10.1038/s41598-017-15183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hawes JJ, Tuskan RG, Reilly KM. Nf1 expression is dependent on strain background: implications for tumor suppressor haploinsufficiency studies. Neurogenetics. 2007;8:121–30. doi: 10.1007/s10048-006-0078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koelsch B, van den Berg L, Fischer C, Winzen-Reichert B, Kutritz A, Kindler-Rohrborn A. Chemically Induced Oncogenesis in the Peripheral Nervous System Is Suppressed in Congenic BDIX. BDIV-Mss1 and -Mss7 Rats. G3 (Bethesda) 2015;6:59–65. doi: 10.1534/g3.115.021170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van den Berg L, Koelsch BU, Winzen-Reichert B, Fischer C, Kutritz A, Kindler-Rohrborn A. Genetic dissection of the Mss4 locus mediating sex-biased cancer resistance in the rat peripheral nervous system. Int J Cancer. 2015;136:2099–108. doi: 10.1002/ijc.29256. [DOI] [PubMed] [Google Scholar]
- 87.Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009;4:453–63. doi: 10.1016/j.stem.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cohen PR, Rapini RP, Farhood AI. Expression of the human hematopoietic progenitor cell antigen CD34 in vascular and spindle cell tumors. J Cutan Pathol. 1993;20:15–20. doi: 10.1111/j.1600-0560.1993.tb01243.x. [DOI] [PubMed] [Google Scholar]
- 89.Weiss SW, Nickoloff BJ. CD-34 is expressed by a distinctive cell population in peripheral nerve, nerve sheath tumors, and related lesions. Am J Surg Pathol. 1993;17:1039–45. doi: 10.1097/00000478-199310000-00009. [DOI] [PubMed] [Google Scholar]
- 90.Prada CE, Jousma E, Rizvi TA, Wu J, Dunn RS, Mayes DA, Cancelas JA, Dombi E, Kim MO, West BL, Bollag G, Ratner N. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathol. 2013;125:159–68. doi: 10.1007/s00401-012-1056-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kluwe L, Friedrich R, Mautner VF. Loss of NF1 allele in Schwann cells but not in fibroblasts derived from an NF1-associated neurofibroma. Genes Chromosomes Cancer. 1999;24:283–5. doi: 10.1002/(sici)1098-2264(199903)24:3<283::aid-gcc15>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 92.Rutkowski JL, Wu K, Gutmann DH, Boyer PJ, Legius E. Genetic and cellular defects contributing to benign tumor formation in neurofibromatosis type 1. Hum Mol Genet. 2000;9:1059–66. doi: 10.1093/hmg/9.7.1059. [DOI] [PubMed] [Google Scholar]
- 93.Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–2. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, Stemmer-Rachamimov AO, Cancelas JA, Ratner N. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105–16. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hirota S, Nomura S, Asada H, Ito A, Morii E, Kitamura Y. Possible involvement of c-kit receptor and its ligand in increase of mast cells in neurofibroma tissues. Arch Pathol Lab Med. 1993;117:996–9. [PubMed] [Google Scholar]
- 96.Ryan JJ, Klein KA, Neuberger TJ, Leftwich JA, Westin EH, Kauma S, Fletcher JA, DeVries GH, Huff TF. Role for the stem cell factor/KIT complex in Schwann cell neoplasia and mast cell proliferation associated with neurofibromatosis. J Neurosci Res. 1994;37:415–32. doi: 10.1002/jnr.490370314. [DOI] [PubMed] [Google Scholar]
- 97.Yang FC, Ingram DA, Chen S, Hingtgen CM, Ratner N, Monk KR, Clegg T, White H, Mead L, Wenning MJ, Williams DA, Kapur R, Atkinson SJ, Clapp DW. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/− mast cells. J Clin Invest. 2003;112:1851–61. doi: 10.1172/JCI19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ingram DA, Yang FC, Travers JB, Wenning MJ, Hiatt K, New S, Hood A, Shannon K, Williams DA, Clapp DW. Genetic and biochemical evidence that haploinsufficiency of the Nf1 tumor suppressor gene modulates melanocyte and mast cell fates in vivo. J Exp Med. 2000;191:181–8. doi: 10.1084/jem.191.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ingram DA, Hiatt K, King AJ, Fisher L, Shivakumar R, Derstine C, Wenning MJ, Diaz B, Travers JB, Hood A, Marshall M, Williams DA, Clapp DW. Hyperactivation of p21(ras) and the hematopoietic-specific Rho GTPase, Rac2, cooperate to alter the proliferation of neurofibromin-deficient mast cells in vivo and in vitro. J Exp Med. 2001;194:57–69. doi: 10.1084/jem.194.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, Yang X, Knowles S, Horn W, Li Y, Zhang S, Yang Y, Vakili ST, Yu M, Burns D, Robertson K, Hutchins G, Parada LF, Clapp DW. Nf1-dependent tumors require a microenvironment containing Nf1+/−- and c-kit-dependent bone marrow. Cell. 2008;135:437–48. doi: 10.1016/j.cell.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yang FC, Chen S, Clegg T, Li X, Morgan T, Estwick SA, Yuan J, Khalaf W, Burgin S, Travers J, Parada LF, Ingram DA, Clapp DW. Nf1+/− mast cells induce neurofibroma like phenotypes through secreted TGF-beta signaling. Hum Mol Genet. 2006;15:2421–37. doi: 10.1093/hmg/ddl165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Serra E, Rosenbaum T, Winner U, Aledo R, Ars E, Estivill X, Lenard HG, Lazaro C. Schwann cells harbor the somatic NF1 mutation in neurofibromas: evidence of two different Schwann cell subpopulations. Hum Mol Genet. 2000;9:3055–64. doi: 10.1093/hmg/9.20.3055. [DOI] [PubMed] [Google Scholar]
- 103.Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein PE, MacCollin M, Cichowski K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10:459–72. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Emmerich D, Zemojtel T, Hecht J, Krawitz P, Spielmann M, Kuhnisch J, Kobus K, Osswald M, Heinrich V, Berlien P, Muller U, Mautner VF, Wimmer K, Robinson PN, Vingron M, Tinschert S, Mundlos S, Kolanczyk M. Somatic neurofibromatosis type 1 (NF1) inactivation events in cutaneous neurofibromas of a single NF1 patient. Eur J Hum Genet. 2015;23:870–3. doi: 10.1038/ejhg.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Faden DL, Asthana S, Tihan T, DeRisi J, Kliot M. Whole Exome Sequencing of Growing and Non-Growing Cutaneous Neurofibromas from a Single Patient with Neurofibromatosis Type 1. PLoS One. 2017;12:e0170348. doi: 10.1371/journal.pone.0170348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pemov A, Li H, Patidar R, Hansen NF, Sindiri S, Hartley SW, Wei JS, Elkahloun A, Chandrasekharappa SC, Program NCS, Boland JF, Bass S, Mullikin JC, Khan J, Widemann BC, Wallace MR, Stewart DR Laboratory NDCGR. The primacy of NF1 loss as the driver of tumorigenesis in neurofibromatosis type 1-associated plexiform neurofibromas. Oncogene. 2017;36:3168–77. doi: 10.1038/onc.2016.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Beert E, Brems H, Daniels B, De Wever I, Van Calenbergh F, Schoenaers J, Debiec-Rychter M, Gevaert O, De Raedt T, Van Den Bruel A, de Ravel T, Cichowski K, Kluwe L, Mautner V, Sciot R, Legius E. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011;50:1021–32. doi: 10.1002/gcc.20921. [DOI] [PubMed] [Google Scholar]
- 108.Brossier NM, Carroll SL. Genetically engineered mouse models shed new light on the pathogenesis of neurofibromatosis type I-related neoplasms of the peripheral nervous system. Brain Res Bull. 2012;88:58–71. doi: 10.1016/j.brainresbull.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brennan MF, Antonescu CR, Moraco N, Singer S. Lessons learned from the study of 10,000 patients with soft tissue sarcoma. Ann Surg. 2014;260:416–21. doi: 10.1097/SLA.0000000000000869. discussion 21–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ng VY, Scharschmidt TJ, Mayerson JL, Fisher JL. Incidence and survival in sarcoma in the United States: a focus on musculoskeletal lesions. Anticancer Res. 2013;33:2597–604. [PubMed] [Google Scholar]
- 111.Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–4. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McCaughan JA, Holloway SM, Davidson R, Lam WW. Further evidence of the increased risk for malignant peripheral nerve sheath tumour from a Scottish cohort of patients with neurofibromatosis type 1. J Med Genet. 2007;44:463–6. doi: 10.1136/jmg.2006.048140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zou C, Smith KD, Liu J, Lahat G, Myers S, Wang WL, Zhang W, McCutcheon IE, Slopis JM, Lazar AJ, Pollock RE, Lev D. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann Surg. 2009;249:1014–22. doi: 10.1097/SLA.0b013e3181a77e9a. [DOI] [PubMed] [Google Scholar]
- 114.LaFemina J, Qin LX, Moraco NH, Antonescu CR, Fields RC, Crago AM, Brennan MF, Singer S. Oncologic outcomes of sporadic, neurofibromatosis-associated, and radiation-induced malignant peripheral nerve sheath tumors. Ann Surg Oncol. 2013;20:66–72. doi: 10.1245/s10434-012-2573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stucky CC, Johnson KN, Gray RJ, Pockaj BA, Ocal IT, Rose PS, Wasif N. Malignant peripheral nerve sheath tumors (MPNST): the Mayo Clinic experience. Ann Surg Oncol. 2012;19:878–85. doi: 10.1245/s10434-011-1978-7. [DOI] [PubMed] [Google Scholar]
- 116.Kolberg M, Holand M, Agesen TH, Brekke HR, Liestol K, Hall KS, Mertens F, Picci P, Smeland S, Lothe RA. Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro Oncol. 2013;15:135–47. doi: 10.1093/neuonc/nos287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Porter DE, Prasad V, Foster L, Dall GF, Birch R, Grimer RJ. Survival in Malignant Peripheral Nerve Sheath Tumours: A Comparison between Sporadic and Neurofibromatosis Type 1-Associated Tumours. Sarcoma. 2009;2009:756395. doi: 10.1155/2009/756395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Watson KL, Al Sannaa GA, Kivlin CM, Ingram DR, Landers SM, Roland CL, Cormier JN, Hunt KK, Feig BW, Ashleigh Guadagnolo B, Bishop AJ, Wang WL, Slopis JM, McCutcheon IE, Lazar AJ, Torres KE. Patterns of recurrence and survival in sporadic, neurofibromatosis Type 1-associated, and radiation-associated malignant peripheral nerve sheath tumors. J Neurosurg. 2017;126:319–29. doi: 10.3171/2015.12.JNS152443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57:2006–21. doi: 10.1002/1097-0142(19860515)57:10<2006::aid-cncr2820571022>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 120.Anghileri M, Miceli R, Fiore M, Mariani L, Ferrari A, Mussi C, Lozza L, Collini P, Olmi P, Casali PG, Pilotti S, Gronchi A. Malignant peripheral nerve sheath tumors: prognostic factors and survival in a series of patients treated at a single institution. Cancer. 2006;107:1065–74. doi: 10.1002/cncr.22098. [DOI] [PubMed] [Google Scholar]
- 121.Fan Q, Yang J, Wang G. Clinical and molecular prognostic predictors of malignant peripheral nerve sheath tumor. Clin Transl Oncol. 2014;16:191–9. doi: 10.1007/s12094-013-1061-x. [DOI] [PubMed] [Google Scholar]
- 122.Valentin T, Le Cesne A, Ray-Coquard I, Italiano A, Decanter G, Bompas E, Isambert N, Thariat J, Linassier C, Bertucci F, Bay JO, Bellesoeur A, Penel N, Le Guellec S, Filleron T, Chevreau C. Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO) Eur J Cancer. 2016;56:77–84. doi: 10.1016/j.ejca.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 123.Shimada S, Tsuzuki T, Kuroda M, Nagasaka T, Hara K, Takahashi E, Hayakawa S, Ono K, Maeda N, Mori N, Illei PB. Nestin expression as a new marker in malignant peripheral nerve sheath tumors. Pathol Int. 2007;57:60–7. doi: 10.1111/j.1440-1827.2006.02059.x. [DOI] [PubMed] [Google Scholar]
- 124.Reuss DE, Habel A, Hagenlocher C, Mucha J, Ackermann U, Tessmer C, Meyer J, Capper D, Moldenhauer G, Mautner V, Frappart PO, Schittenhelm J, Hartmann C, Hagel C, Katenkamp K, Petersen I, Mechtersheimer G, von Deimling A. Neurofibromin specific antibody differentiates malignant peripheral nerve sheath tumors (MPNST) from other spindle cell neoplasms. Acta Neuropathol. 2014;127:565–72. doi: 10.1007/s00401-014-1246-6. [DOI] [PubMed] [Google Scholar]
- 125.Mertens F, Rydholm A, Bauer HF, Limon J, Nedoszytko B, Szadowska A, Willen H, Heim S, Mitelman F, Mandahl N. Cytogenetic findings in malignant peripheral nerve sheath tumors. Int J Cancer. 1995;61:793–8. doi: 10.1002/ijc.2910610609. [DOI] [PubMed] [Google Scholar]
- 126.Plaat BE, Molenaar WM, Mastik MF, Hoekstra HJ, te Meerman GJ, van den Berg E. Computer-assisted cytogenetic analysis of 51 malignant peripheral-nerve-sheath tumors: sporadic vs. neurofibromatosis-type-1-associated malignant schwannomas. Int J Cancer. 1999;83:171–8. doi: 10.1002/(sici)1097-0215(19991008)83:2<171::aid-ijc5>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 127.Forus A, Weghuis DO, Smeets D, Fodstad O, Myklebost O, van Kessel AG. Comparative genomic hybridization analysis of human sarcomas: I. Occurrence of genomic imbalances and identification of a novel major amplicon at 1q21-q22 in soft tissue sarcomas. Genes Chromosomes Cancer. 1995;14:8–14. doi: 10.1002/gcc.2870140103. [DOI] [PubMed] [Google Scholar]
- 128.Lothe RA, Karhu R, Mandahl N, Mertens F, Saeter G, Heim S, Borresen-Dale AL, Kallioniemi OP. Gain of 17q24-qter detected by comparative genomic hybridization in malignant tumors from patients with von Recklinghausen’s neurofibromatosis. Cancer Res. 1996;56:4778–81. [PubMed] [Google Scholar]
- 129.Mechtersheimer G, Otano-Joos M, Ohl S, Benner A, Lehnert T, Willeke F, Moller P, Otto HF, Lichter P, Joos S. Analysis of chromosomal imbalances in sporadic and NF1-associated peripheral nerve sheath tumors by comparative genomic hybridization. Genes Chromosomes Cancer. 1999;25:362–9. [PubMed] [Google Scholar]
- 130.Schmidt H, Wurl P, Taubert H, Meye A, Bache M, Holzhausen HJ, Hinze R. Genomic imbalances of 7p and 17q in malignant peripheral nerve sheath tumors are clinically relevant. Genes Chromosomes Cancer. 1999;25:205–11. [PubMed] [Google Scholar]
- 131.Schmidt H, Taubert H, Wurl P, Bache M, Bartel F, Holzhausen HJ, Hinze R. Cytogenetic characterization of six malignant peripheral nerve sheath tumors: comparison of karyotyping and comparative genomic hybridization. Cancer Genet Cytogenet. 2001;128:14–23. doi: 10.1016/s0165-4608(01)00393-4. [DOI] [PubMed] [Google Scholar]
- 132.Schmidt H, Taubert H, Meye A, Wurl P, Bache M, Bartel F, Holzhausen HJ, Hinze R. Gains in chromosomes 7, 8q, 15q and 17q are characteristic changes in malignant but not in benign peripheral nerve sheath tumors from patients with Recklinghausen’s disease. Cancer Lett. 2000;155:181–90. doi: 10.1016/s0304-3835(00)00426-2. [DOI] [PubMed] [Google Scholar]
- 133.Mertens F, Dal Cin P, De Wever I, Fletcher CD, Mandahl N, Mitelman F, Rosai J, Rydholm A, Sciot R, Tallini G, van Den Berghe H, Vanni R, Willen H. Cytogenetic characterization of peripheral nerve sheath tumours: a report of the CHAMP study group. J Pathol. 2000;190:31–8. doi: 10.1002/(SICI)1096-9896(200001)190:1<31::AID-PATH505>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 134.Legius E, Marchuk DA, Collins FS, Glover TW. Somatic deletion of the neurofibromatosis type 1 gene in a neurofibrosarcoma supports a tumour suppressor gene hypothesis. Nat Genet. 1993;3:122–6. doi: 10.1038/ng0293-122. [DOI] [PubMed] [Google Scholar]
- 135.Birindelli S, Perrone F, Oggionni M, Lavarino C, Pasini B, Vergani B, Ranzani GN, Pierotti MA, Pilotti S. Rb and TP53 pathway alterations in sporadic and NF1-related malignant peripheral nerve sheath tumors. Lab Invest. 2001;81:833–44. doi: 10.1038/labinvest.3780293. [DOI] [PubMed] [Google Scholar]
- 136.Holtkamp N, Atallah I, Okuducu AF, Mucha J, Hartmann C, Mautner VF, Friedrich RE, Mawrin C, von Deimling A. MMP-13 and p53 in the progression of malignant peripheral nerve sheath tumors. Neoplasia. 2007;9:671–7. doi: 10.1593/neo.07304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Legius E, Dierick H, Wu R, Hall BK, Marynen P, Cassiman JJ, Glover TW. TP53 mutations are frequent in malignant NF1 tumors. Genes Chromosomes Cancer. 1994;10:250–5. doi: 10.1002/gcc.2870100405. [DOI] [PubMed] [Google Scholar]
- 138.Menon AG, Anderson KM, Riccardi VM, Chung RY, Whaley JM, Yandell DW, Farmer GE, Freiman RN, Lee JK, Li FP, et al. Chromosome 17p deletions and p53 gene mutations associated with the formation of malignant neurofibrosarcomas in von Recklinghausen neurofibromatosis. Proc Natl Acad Sci U S A. 1990;87:5435–9. doi: 10.1073/pnas.87.14.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Upadhyaya M, Kluwe L, Spurlock G, Monem B, Majounie E, Mantripragada K, Ruggieri M, Chuzhanova N, Evans DG, Ferner R, Thomas N, Guha A, Mautner V. Germline and somatic NF1 gene mutation spectrum in NF1-associated malignant peripheral nerve sheath tumors (MPNSTs) Hum Mutat. 2008;29:74–82. doi: 10.1002/humu.20601. [DOI] [PubMed] [Google Scholar]
- 140.Kourea HP, Orlow I, Scheithauer BW, Cordon-Cardo C, Woodruff JM. Deletions of the INK4A gene occur in malignant peripheral nerve sheath tumors but not in neurofibromas. Am J Pathol. 1999;155:1855–60. doi: 10.1016/S0002-9440(10)65504-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nielsen GP, Stemmer-Rachamimov AO, Ino Y, Moller MB, Rosenberg AE, Louis DN. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am J Pathol. 1999;155:1879–84. doi: 10.1016/S0002-9440(10)65507-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mantripragada KK, Spurlock G, Kluwe L, Chuzhanova N, Ferner RE, Frayling IM, Dumanski JP, Guha A, Mautner V, Upadhyaya M. High-resolution DNA copy number profiling of malignant peripheral nerve sheath tumors using targeted microarray-based comparative genomic hybridization. Clin Cancer Res. 2008;14:1015–24. doi: 10.1158/1078-0432.CCR-07-1305. [DOI] [PubMed] [Google Scholar]
- 143.Mawrin C, Kirches E, Boltze C, Dietzmann K, Roessner A, Schneider-Stock R. Immunohistochemical and molecular analysis of p53, RB, and PTEN in malignant peripheral nerve sheath tumors. Virchows Arch. 2002;440:610–5. doi: 10.1007/s00428-001-0550-4. [DOI] [PubMed] [Google Scholar]
- 144.Bradtmoller M, Hartmann C, Zietsch J, Jaschke S, Mautner VF, Kurtz A, Park SJ, Baier M, Harder A, Reuss D, von Deimling A, Heppner FL, Holtkamp N. Impaired Pten expression in human malignant peripheral nerve sheath tumours. PLoS One. 2012;7:e47595. doi: 10.1371/journal.pone.0047595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gregorian C, Nakashima J, Dry SM, Nghiemphu PL, Smith KB, Ao Y, Dang J, Lawson G, Mellinghoff IK, Mischel PS, Phelps M, Parada LF, Liu X, Sofroniew MV, Eilber FC, Wu H. PTEN dosage is essential for neurofibroma development and malignant transformation. Proc Natl Acad Sci U S A. 2009;106:19479–84. doi: 10.1073/pnas.0910398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kawaguchi K, Oda Y, Saito T, Takahira T, Yamamoto H, Tamiya S, Iwamoto Y, Tsuneyoshi M. Genetic and epigenetic alterations of the PTEN gene in soft tissue sarcomas. Hum Pathol. 2005;36:357–63. doi: 10.1016/j.humpath.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 147.Holtkamp N, Malzer E, Zietsch J, Okuducu AF, Mucha J, Mawrin C, Mautner VF, Schildhaus HU, von Deimling A. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro Oncol. 2008;10:946–57. doi: 10.1215/15228517-2008-053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.DeClue JE, Heffelfinger S, Benvenuto G, Ling B, Li S, Rui W, Vass WC, Viskochil D, Ratner N. Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumors and NF1 animal models. J Clin Invest. 2000;105:1233–41. doi: 10.1172/JCI7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Perry A, Kunz SN, Fuller CE, Banerjee R, Marley EF, Liapis H, Watson MA, Gutmann DH. Differential NF1, p16, and EGFR patterns by interphase cytogenetics (FISH) in malignant peripheral nerve sheath tumor (MPNST) and morphologically similar spindle cell neoplasms. J Neuropathol Exp Neurol. 2002;61:702–9. doi: 10.1093/jnen/61.8.702. [DOI] [PubMed] [Google Scholar]
- 150.Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A, Tap WD, Fletcher JA, Huberman KH, Qin LX, Viale A, Singer S, Zheng D, Berger MF, Chen Y, Antonescu CR, Chi P. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1227–32. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R, Wang Q, Belzberg AJ, Chaichana K, Gallia GL, Gokaslan ZL, Riggins GJ, Wolinksy JP, Wood LD, Montgomery EA, Hruban RH, Kinzler KW, Papadopoulos N, Vogelstein B, Bettegowda C. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1170–2. doi: 10.1038/ng.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Schaefer IM, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol. 2016;29:4–13. doi: 10.1038/modpathol.2015.134. [DOI] [PubMed] [Google Scholar]
- 153.Cleven AH, Sannaa GA, Briaire-de Bruijn I, Ingram DR, van de Rijn M, Rubin BP, de Vries MW, Watson KL, Torres KE, Wang WL, van Duinen SG, Hogendoorn PC, Lazar AJ, Bovee JV. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol. 2016;29:582–90. doi: 10.1038/modpathol.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Asano N, Yoshida A, Ichikawa H, Mori T, Nakamura M, Kawai A, Hiraoka N. Immunohistochemistry for trimethylated H3K27 in the diagnosis of malignant peripheral nerve sheath tumours. Histopathology. 2017;70:385–93. doi: 10.1111/his.13072. [DOI] [PubMed] [Google Scholar]
- 155.Mito JK, Qian X, Doyle LA, Hornick JL, Jo VY. Role of Histone H3K27 Trimethylation Loss as a Marker for Malignant Peripheral Nerve Sheath Tumor in Fine-Needle Aspiration and Small Biopsy Specimens. Am J Clin Pathol. 2017;148:179–89. doi: 10.1093/ajcp/aqx060. [DOI] [PubMed] [Google Scholar]
- 156.Widemann BC, Salzer WL, Arceci RJ, Blaney SM, Fox E, End D, Gillespie A, Whitcomb P, Palumbo JS, Pitney A, Jayaprakash N, Zannikos P, Balis FM. Phase I trial and pharmacokinetic study of the farnesyltransferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatosis type I and plexiform neurofibromas. J Clin Oncol. 2006;24:507–16. doi: 10.1200/JCO.2005.03.8638. [DOI] [PubMed] [Google Scholar]
- 157.Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O’Connell P, Cawthon RM, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell. 1990;63:843–9. doi: 10.1016/0092-8674(90)90150-d. [DOI] [PubMed] [Google Scholar]
- 158.Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, Collins F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851–9. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 159.Ohba Y, Mochizuki N, Yamashita S, Chan AM, Schrader JW, Hattori S, Nagashima K, Matsuda M. Regulatory proteins of R-Ras, TC21/R-Ras2, and M-Ras/R-Ras3. J Biol Chem. 2000;275:20020–6. doi: 10.1074/jbc.M000981200. [DOI] [PubMed] [Google Scholar]
- 160.Brossier NM, Prechtl AM, Longo JF, Barnes S, Wilson LS, Byer SJ, Brosius SN, Carroll SL. Classic Ras Proteins Promote Proliferation and Survival via Distinct Phosphoproteome Alterations in Neurofibromin-Null Malignant Peripheral Nerve Sheath Tumor Cells. J Neuropathol Exp Neurol. 2015;74:568–86. doi: 10.1097/NEN.0000000000000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mattingly RR, Kraniak JM, Dilworth JT, Mathieu P, Bealmear B, Nowak JE, Benjamins JA, Tainsky MA, Reiners JJ., Jr The mitogen-activated protein kinase/extracellular signal-regulated kinase kinase inhibitor PD184352 (CI-1040) selectively induces apoptosis in malignant schwannoma cell lines. J Pharmacol Exp Ther. 2006;316:456–65. doi: 10.1124/jpet.105.091454. [DOI] [PubMed] [Google Scholar]
- 162.Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, Eaves D, Widemann B, Kim MO, Dombi E, Sabo J, Hardiman Dudley A, Niwa-Kawakita M, Page GP, Giovannini M, Aronow BJ, Cripe TP, Ratner N. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123:340–7. doi: 10.1172/JCI60578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kim A, Dombi E, Tepas K, Fox E, Martin S, Wolters P, Balis FM, Jayaprakash N, Turkbey B, Muradyan N, Choyke PL, Reddy A, Korf B, Widemann BC. Phase I trial and pharmacokinetic study of sorafenib in children with neurofibromatosis type I and plexiform neurofibromas. Pediatr Blood Cancer. 2013;60:396–401. doi: 10.1002/pbc.24281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Maki RG, D’Adamo DR, Keohan ML, Saulle M, Schuetze SM, Undevia SD, Livingston MB, Cooney MM, Hensley ML, Mita MM, Takimoto CH, Kraft AS, Elias AD, Brockstein B, Blachere NE, Edgar MA, Schwartz LH, Qin LX, Antonescu CR, Schwartz GK. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27:3133–40. doi: 10.1200/JCO.2008.20.4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005;102:8573–8. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Johansson G, Mahller YY, Collins MH, Kim MO, Nobukuni T, Perentesis J, Cripe TP, Lane HA, Kozma SC, Thomas G, Ratner N. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol Cancer Ther. 2008;7:1237–45. doi: 10.1158/1535-7163.MCT-07-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Holtkamp N, Okuducu AF, Mucha J, Afanasieva A, Hartmann C, Atallah I, Estevez-Schwarz L, Mawrin C, Friedrich RE, Mautner VF, von Deimling A. Mutation and expression of PDGFRA and KIT in malignant peripheral nerve sheath tumors, and its implications for imatinib sensitivity. Carcinogenesis. 2006;27:664–71. doi: 10.1093/carcin/bgi273. [DOI] [PubMed] [Google Scholar]
- 168.Kadono T, Soma Y, Takehara K, Nakagawa H, Ishibashi Y, Kikuchi K. The growth regulation of neurofibroma cells in neurofibromatosis type-1: increased responses to PDGF-BB and TGF-beta 1. Biochem Biophys Res Commun. 1994;198:827–34. doi: 10.1006/bbrc.1994.1118. [DOI] [PubMed] [Google Scholar]
- 169.Badache A, De Vries GH. Neurofibrosarcoma-derived Schwann cells overexpress platelet-derived growth factor (PDGF) receptors and are induced to proliferate by PDGF BB. J Cell Physiol. 1998;177:334–42. doi: 10.1002/(SICI)1097-4652(199811)177:2<334::AID-JCP15>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 170.Perrone F, Da Riva L, Orsenigo M, Losa M, Jocolle G, Millefanti C, Pastore E, Gronchi A, Pierotti MA, Pilotti S. PDGFRA, PDGFRB, EGFR, and downstream signaling activation in malignant peripheral nerve sheath tumor. Neuro Oncol. 2009;11:725–36. doi: 10.1215/15228517-2009-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zietsch J, Ziegenhagen N, Heppner FL, Reuss D, von Deimling A, Holtkamp N. The 4q12 amplicon in malignant peripheral nerve sheath tumors: consequences on gene expression and implications for sunitinib treatment. PLoS One. 2010;5:e11858. doi: 10.1371/journal.pone.0011858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Badache A, Muja N, De Vries GH. Expression of Kit in neurofibromin-deficient human Schwann cells: role in Schwann cell hyperplasia associated with type 1 neurofibromatosis. Oncogene. 1998;17:795–800. doi: 10.1038/sj.onc.1201978. [DOI] [PubMed] [Google Scholar]
- 173.Rao UN, Sonmez-Alpan E, Michalopoulos GK. Hepatocyte growth factor and c-MET in benign and malignant peripheral nerve sheath tumors. Hum Pathol. 1997;28:1066–70. doi: 10.1016/s0046-8177(97)90060-5. [DOI] [PubMed] [Google Scholar]
- 174.Fukuda T, Ichimura E, Shinozaki T, Sano T, Kashiwabara K, Oyama T, Nakajima T, Nakamura T. Coexpression of HGF and c-Met/HGF receptor in human bone and soft tissue tumors. Pathol Int. 1998;48:757–62. doi: 10.1111/j.1440-1827.1998.tb03834.x. [DOI] [PubMed] [Google Scholar]
- 175.Su W, Gutmann DH, Perry A, Abounader R, Laterra J, Sherman LS. CD44-independent hepatocyte growth factor/c-Met autocrine loop promotes malignant peripheral nerve sheath tumor cell invasion in vitro. Glia. 2004;45:297–306. doi: 10.1002/glia.10340. [DOI] [PubMed] [Google Scholar]
- 176.Ling BC, Wu J, Miller SJ, Monk KR, Shamekh R, Rizvi TA, Decourten-Myers G, Vogel KS, DeClue JE, Ratner N. Role for the epidermal growth factor receptor in neurofibromatosis-related peripheral nerve tumorigenesis. Cancer Cell. 2005;7:65–75. doi: 10.1016/j.ccr.2004.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Stonecypher MS, Byer SJ, Grizzle WE, Carroll SL. Activation of the neuregulin-1/ErbB signaling pathway promotes the proliferation of neoplastic Schwann cells in human malignant peripheral nerve sheath tumors. Oncogene. 2005;24:5589–605. doi: 10.1038/sj.onc.1208730. [DOI] [PubMed] [Google Scholar]
- 178.Eckert JM, Byer SJ, Clodfelder-Miller BJ, Carroll SL. Neuregulin-1 beta and neuregulin-1 alpha differentially affect the migration and invasion of malignant peripheral nerve sheath tumor cells. Glia. 2009;57:1501–20. doi: 10.1002/glia.20866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Huijbregts RP, Roth KA, Schmidt RE, Carroll SL. Hypertrophic neuropathies and malignant peripheral nerve sheath tumors in transgenic mice overexpressing glial growth factor beta3 in myelinating Schwann cells. J Neurosci. 2003;23:7269–80. doi: 10.1523/JNEUROSCI.23-19-07269.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Brosius SN, Turk AN, Byer SJ, Brossier NM, Kohli L, Whitmire A, Mikhail FM, Roth KA, Carroll SL. Neuregulin-1 overexpression and Trp53 haploinsufficiency cooperatively promote de novo malignant peripheral nerve sheath tumor pathogenesis. Acta Neuropathol. 2014;127:573–91. doi: 10.1007/s00401-013-1209-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pillay V, Allaf L, Wilding AL, Donoghue JF, Court NW, Greenall SA, Scott AM, Johns TG. The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia. 2009;11:448–58. 2–58. doi: 10.1593/neo.09230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, Stegh AH, Bradner JE, Ligon KL, Brennan C, Chin L, DePinho RA. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318:287–90. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]