

Abstract
Decavanadate (V10O286− or V10) is a paradigmatic member of the polyoxidometalate (POM) family, which has been attracting much attention within both materials/inorganic and biomedical communities due to its unique structural and electrochemical properties. In this work we explored the utility of high-resolution electrospray ionization (ESI) mass spectrometry (MS) and ion exclusion chromatography LC/MS for structural analysis of V10 species in aqueous solutions. While ESI generates abundant molecular ions representing the intact V10 species, their isotopic distributions show significant deviations from the theoretical ones. A combination of high-resolution MS measurements and hydrogen/deuterium exchange allows these deviations to be investigated and interpreted as a result of partial reduction of V10. While the redox processes are known to occur in the ESI interface and influence the oxidation state of redox-active analytes, the LC/MS measurements using ion exclusion chromatography provide unequivocal evidence that the mixed-valence V10 species exist in solution, as extracted ion chromatograms representing V10 molecular ions at different oxidation states exhibit distinct elution profiles. The spontaneous reduction of V10 in solution is seen even in the presence of hydrogen peroxide and has not been previously observed. The susceptibility to reduction of V10 is likely to be shared by other redox active POMs. In addition to the molecular V10 ions, a high-abundance ionic signal for a V10O262− anion was displayed in the negative-ion ESI mass spectra. None of the V10O26 cations were detected in ESI MS, and only a low-abundance signal was observed for V10O26 anions with a single negative charge, indicating that the presence of abundant V10O262− anions in ESI MS reflects gas-phase instability of V10O28 anions carrying two charges. The gas-phase origin of the V10O262− anion was confirmed in tandem MS measurements, where mild collisional activation was applied to V10 molecular ions with an even number of hydrogen atoms (H4V10O282−), resulting in a facile loss of H2O molecules and giving rise to V10O262− as the lowest-mass fragment ion. Water loss was also observed for V10O28 anions carrying an odd number of hydrogen atoms (e.g., H5V10O28−), followed by a less efficient and incomplete removal of an OH• radical, giving rise to both HV10O26− and V10O25− fragment ions. Importantly, at least one hydrogen atom was required for ion fragmentation in the gas phase, as no further dissociation was observed for any hydrogen-free V10 ionic species. The presented workflow allows a distinction to be readily made between the spectral features revealing the presence of non-canonical POM species in the bulk solution from those that arise due to physical and chemical processes occurring in the ESI interface and/or the gas phase.
Graphical Abstract

Polyoxidometalates (POMs) are a unique class of polynuclear inorganic compounds, which are mostly composed of transition metal and oxygen atoms, although other atoms (such as phosphorus and silicon) may also be involved.1 POMs are frequently viewed as the “missing link” between the single-molecule and the bulk material scales.2 Apart from their obvious importance for the field of mesoscopics and helping understand how the physical properties of materials are determined by the chemical properties of constituent molecules, POMs continue to enjoy considerable attention in fields as diverse as catalysis,3 electronics4,5 and medicine.6–8 Decavanadate (V10O286−, or V10) is one particularly interesting member of the POM family, which had been shown to interact with a variety of biomolecules in vivo, eliciting a range of biological effects that are distinct from those typically associated with smaller oxidovanadate species.9 Despite being a focus of extensive research efforts in the past two decades, several aspects of V10 structural and chemical properties remain incompletely understood. As is the case with many other POMs, V10 can interconvert between multiple species in solution, but the mechanisms of such processes (which likely include an intricate interplay of acid-base and redox chemistry) remain elusive. For example, the existence of a partially reduced V10 species (V10O264−) has been reported based on spectroscopic measurements,10 but never confirmed independently. Likewise, the proton association with V10 species in solution and the hydrogen bonding phenomena have been shown to play important roles in dissociation/association of this paradigmatic member of the POM family.11 Lastly, the notion of the low-energy metastable configurations of V10 (distinct from its crystal structure12) has been invoked to explain several phenomena related to its interaction with the solvent.13,14
The majority of work on V10 (as well as other POMs) characterization has been traditionally carried out using X-ray crystallography in the solid state and spectroscopic methods in solution. While the former provides atomic-level structural data for stable POM molecules, it is insensitive to those species that are present in solution in minute quantities or become populated only transiently. In contrast, NMR can be used to speciate heterogeneous populations of POMs. The natural abundance of the 17O isotope and the NMR activity of many metal centers allow for facile application of the technique to a wide array of POMs. However, the atomic-level resolution afforded by the NMR is frequently insufficient for complete characterization of distinct ensembles of POMs that may co-exist in solution. Indeed, while the state of a single atom and its unique chemical environment can be monitored with unprecedented precision using NMR, grouping such states together to yield distinct molecular states is not straightforward.
In comparison to X-ray crystallography and NMR, mass spectrometry (MS) has played a less prominent role in characterization of POMs, even though the initial application of MS in this field dates back nearly four decades ago.15 Introduction of electrospray ionization (ESI) and, to a lesser extent, matrix-assisted laser desorption/ionization (MALDI) provided further impetus to developing robust MS-based methods to characterize a variety of POMs,16–19 including polyoxidovanadates.20 In many ways, MS offers an attractive alternative for the analysis of POMs compared to the traditional approaches, as it enables POMs speciation in solution and allows oxidation states to be distinguished from one another. At the same time, the possibility of ion fragmentation occurring in the gas phase raises the concern of introducing artifacts in MS-based POM speciation measurements. Likewise, the frequent occurrence of electrochemical processes within the liquids being electrosprayed21 may cast doubt on the validity of oxidation state profiling obtained with ESI MS. In fact, the complete elimination of interfering redox processes may require the use of complicated setups and extensive front-end modification of commercial MS equipment.22 The main goal of this work was to evaluate and illustrate the utility of high-resolution MS for characterization of both structure and chemistry of decavanadate species in aqueous solutions with a particular emphasis on exploring both similarities and differences between V10 behavior in the bulk solution and in the solvent-free environment.
In addition to its obvious importance via-a-vis validation of the ESI MS-based methods of POM structural analysis, the juxtaposition of the solution- and gas-phase behavior for this class of analytes may provide important insights into the POMs/solvent interactions (and, by extension, the phenomena occurring at the insoluble metal oxide/water interface). For example, previous computational studies have indicated that the presence of hydrogen at an oxometallate surface facilitates the abstraction of oxygen from the surface.23 Oxygen abstraction from polyoxidovanadate clusters has also been shown to be triggered by partial reduction of the latter,24 although no distinct partially reduced species could be detected; instead, their existence was inferred from the spectroscopic data. MS is unique in that it allows such species to be detected even if they account only for a minor fraction of the entire ensemble of molecules present in solution. In this work we use high resolution MS as an on-line detection tool for ion exclusion chromatography to provide conclusive evidence that multiple partially reduced V10 species exist in solution alongside the canonical (fully oxidized) V10 prior to the MS measurement. These reduced species accommodate a larger number of protons during the ESI process, leading to formation of ions that readily dissociate via a facile loss of water molecules and, to a lesser extent, OH· radicals, thus mirroring processes that were previously hypothesized to take place in solution.
EXPERIMENTAL SECTION
Materials.
The decavanadate samples in the form of (NH4)6V10O28, Na6V10O28 and K4Na2V10O28 salts were prepared using a procedure described elsewhere25,26 and the speciation was characterized using 51V NMR and absorbance spectroscopy.27 Hydrogen peroxide (H2O2) and heavy water (2H2O) were purchased from Millipore-Sigma (St. Louis, MO). All other solvents and buffers used in this work were of analytical grade or higher.
Methods.
All MS measurements were carried out using a SolariX 7 (Bruker Daltonics, Billerica, MA) hybrid quadrupole-Fourier transform ion cyclotron resonance mass spectrometer equipped with a standard electrospray ionization source. V10 samples were dissolved in a 5 mM aqueous solution of ammonium acetate whose pH was adjusted with formic acid to 4.85±0.15. Final decavanadate concentrations were 1 mM (negative ion mode) or 0.1 mM (positive ion mode). Decavanadate was typically kept in solution for 30 min prior to MS analysis. The ESI source conditions and ESI MS interface ion optics parameters were optimized to produce abundant signal in the 400–1100 m/z; the typical settings in the negative ion mode were as follows: ESI capillary, 3800 V; end plate offset, 500 V; nebulizer, 1.6 bar; dry gas, 4.0 L/min; dry temperature, 150 °C; capillary exit, −180 V; deflector plate, −180 V; funnel 1, −150 V, skimmer 1, −10 V; funnel RF amplitude, 200 Vp-p). External calibration was used to determine ionic masses in mass spectra acquired in the broad-band mode for both positive and negative ions. Post-calibration mass accuracy was determined to be at least ±3 ppm throughout the entire 200–1200 m/z region. Internal calibration was used for mass measurements in the narrow-band mode, providing mass accuracy of at least 0.3 ppm. Theoretical isotopic distributions of ionic species and accurate masses of relevant isotopologues were calculated using a web-based tool at https://www.sisweb.com/mstools/isotope.htm. Fragment ion spectra were obtained by mass-selecting precursor ions of interest in the front-end quadrupole (mass selection window 3.0 m/z units unless specified otherwise) followed by their activation in the collision cell using argon as a collision gas. Ion exclusion LC and LC-MS measurements were carried out with an Agilent 1100 (Agilent Technologies, Santa Clara, CA) HPLC system using a 4.1 mm × 250 mm PRP-X300 (Hamilton Co., Reno, NV) ion exclusion column. The mobile phase consisted of 30 mM triethylamine, 100 mM ammonium acetate and 0.8% formic Acid. The UV detection of V10 was based on the absorbance at 254 nm. On-line LC-MS measurements were carried out by directing the eluate flow off the column directly to the ESI source of the mass spectrometer without splitting.
RESULTS AND DISCUSSION
V10 (in a variety of protonation states) is the predominant vanadium species in moderately concentrated (> 1 mM) and mildly acidic (3 ≤ pH ≤ 6) aqueous solutions, but it readily converts to other oxo-forms (e.g., VO43ˉ/HVO42ˉ at basic pH, V4O124ˉ/H2VO4ˉ at neutral pH, or VO3−/HVO3 upon dilution).28,29 Although these and other smaller vanadates are expected to be present in lower quantities at mildly acidic pH as well, the negative ion mass spectrum of Na6V10O28 acquired at pH 4.7 (Figure 1) shows only two oxidovanadate species in the low-m/z region of the mass spectrum, V6O162− and (H/Na)V4O11ˉ. Outside of this region, the mass spectrum is dominated by two clusters of abundant ions with distinct charge states: a group of singly charged ions (m/z region 950–1100) and the doubly charged ones (m/z 450–550). The nominal masses of the most abundant peaks in each cluster indicate that these two species (H5V10O28ˉ and H4V10O282ˉ) are produced by partial balancing of the net charge of the presumed most abundant V10 species in solution (V10O286ˉ, HV10O285ˉ and H2V10O284ˉ) with “extra” protons or ubiquitous alkali metal cations upon electrospraying. This behavior is similar to the ESI production of the moderately-charged species representing high charge-density organic polyelectrolytes, such as heparin oligomers.30
Figure 1.
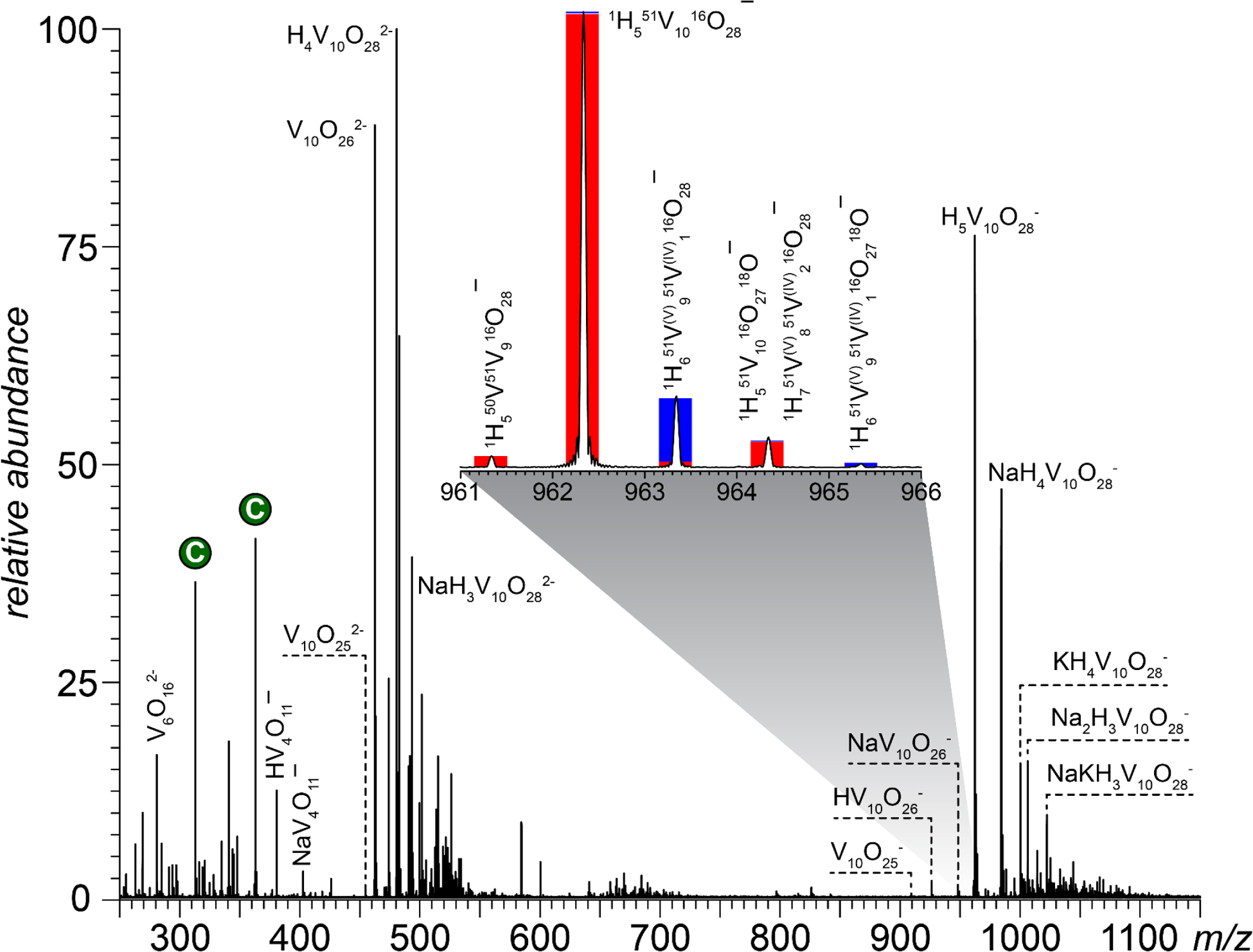
ESI mass spectrum of a 1 mM aqueous solution of sodium V10 in 5 mM ammonium acetate (pH adjusted to 4.7). The inset shows the isotopic distribution of the most abundant mono-anionic species (black trace) and the calculated isotopic distributions for the fully oxidized species (H5V10O28ˉ, red bars) and a partially reduced one (H6V10O28ˉ, blue).
It is now commonly accepted that the extent of protonation (and, more broadly, multiple charging in general) of polyprotic acids in ESI MS does not reflect the acid-base equilibria in solution, but is instead dictated by the physical size of the analyte.31 Consistent with this notion, V10 generates abundant ionic signal in the positive-ion ESI MS as well, where it is represented by H(7-n-k)NanKkV10O28+ (n, k = 0 – 4) ions (see Supplementary Material for more detail).
While the extent of protonation of the dominant V10 ionic species in ESI MS cannot be related to the acid/base equilibria in solution, the total charge accommodated by these ions in both negative- and positive ion modes is consistent with the notion of all metal ions being fully oxidized (i.e., charge state +5). Indeed, the most abundant peak in the isotopic distribution of the mono-anionic V10 species m/z region 961–966 can be confidently assigned as the most abundant isotopologue of H5V10O28ˉ (the calculated mass for 1H551V1016O28ˉ is 962.3369, a value that is within 0.2 ppm of the measured mass). The lowest-mass detectable monoisotopic peak of the H5V10O28ˉ species (m/z 961.3373) can also be readily assigned as 1H550V51V916O28ˉ (calculated mass 961.34031, and the intensity ratio of these two isotopologues (which we will term M and M-1, respectively) is in excellent agreement with the known 51V : 50V natural abundance ratio (the theoretical isotopic distribution of the H5V10O28ˉ is represented with red bars in the inset in Figure 1). However, the abundance of the third isotopologue in this distribution (M+1, measured m/z 963.3443) appears anomalously high, exceeding the expected contribution of 1H551V1017O16O27ˉ by more than an order of magnitude. Furthermore, the spacing between the M+1 and M species (1.0071) shows a significant deviation from the mass difference between the 16O and 17O isotopes (1.0042), but is reasonably close to the hydrogen atom mass (1.0078). Therefore, this species was tentatively assigned as 1H651V(V)951V(IV)116O28ˉ, which represents a partially reduced decavanadate, where an extra proton is needed in order to keep the overall single net charge of the anion (compensating for the lost positive charge on one of the metals). Such satellite species are observed for all other reasonably abundant anions containing the decavanadium core, and their mass shifts in 2H2O solutions (vide infra) confirm the presence of an “extra” hydrogen atom, lending strong support to the notion of partial V10 reduction.
It is worth noting that the partially reduced V10 species (see the inset in Figure 1) are present in all V10 samples examined in this work, regardless of the nature of the counterion (Na+, K+ or NH4+, see Supplementary Material for more detail). The relative abundance of the partially reduced species shows some variations among different V10 samples with distinct counter-ions, and even within the same sample temporal variations in the intensity ratios of the M and M+1 species are apparent (data not shown). In many instances, low-intensity ionic signal was also observed for V10 species where more than a single reduction event takes place (e.g., H7V(V)8V(IV)2O28ˉ and H8V(V)7V(IV)3O28ˉ incorporating two and three vanadium atoms in oxidation state IV, respectively, to ensure the charge balance). Such seemingly random variations naturally lead to a suggestion that the partial reduction occurs during the ESI process, a phenomenon that has been documented for a range of redox-active species in the past.32 Furthermore, the observation of the partially reduced V10 species in the mass spectra of V10 acquired in the presence of hydrogen peroxide in solution appears to lend further support to the notion of the ESI-driven redox processes being responsible for the partial reduction of these POM assemblies. Should the partial reduction take place in the ESI source, it would be reversed by simply switching the polarity of the ion source. However, examination of the V10 mass spectra acquired in the positive-ion mode also reveals the presence of the partially reduced V10 species, such as NaH7V10O28+ (see Supplementary Material), contradicting the notion of V10 partial reduction being an ESI artifact.
To obtain definitive evidence with regards to the origin of the partially reduced V10 species, ion exclusion chromatography (IEC) with on-line ESI MS detection was used. IEC is a mixed-mode chromatographic modality, in which differential exclusion of weak electrolytes from the anionic resin pores is used as a physical principle enabling analyte separation.33 Although in the past IEC has been applied almost exclusively to separation of weak organic acids (based on their sizes and pKa values34), it can be used for separation of inorganic anions as well.35,36 Since V10, as well as many smaller vanadates, are weak acids, we hypothesized that it may be possible to achieve their retention on IEC. Furthermore, partial reduction of V10 in solution should have an effect on the pKa values, and unless the redox equilibria are too fast on the chromatographic time scale (e.g., the H5V(V)10O28ˉ ⇌ H6V(IV)V (V)9O28ˉ ⇌ H7(IV)V2(V)V8O28ˉ interconversion occurs within < 1 min), it should be possible to achieve chromatographic separation of V10 species at different oxidation states. In contrast, if the partial reduction of V10 is triggered by electrochemical processes in the ESI source, no separation of the V10 species at different oxidation states would be possible with IEC. Unfortunately, achieving efficient retention of V10 with IEC is not straightforward, as the principal species for the canonical, fully oxidized form of this POM in the pH range of interest are H2V10O284ˉ and HV10O285ˉ, while the pKa value of the fully protonated (electrically neutral) V10 species is significantly below 1 (and possibly below 0).37 This means that V10 retention on anionic resins is very ineffective, unless an appropriate ion pairing reagent is identified. Indeed, our initial attempts to achieve V10 retention with an IEC column failed until trimethylamine (TEA) was selected as an ion pairing agent. The elution time of V10 off the IEC column in the presence of 30 mM TEA was equivalent to application of 3 column volumes of the eluent.
Importantly, the extracted ion chromatograms (XICs) recorded for the individual monoisotopic peaks representing H5+nV10O28ˉ for n = 0 – 3 clearly show incongruent elution profiles (Figure 2). These chromatograms provide clear and conclusive evidence of the solution phase origin of the partially reduced species. Furthermore, this observation also indicates that the interconversion rate among the V10 species at different oxidation states in solution is relatively low, allowing at least partial chromatographic separation to be achieved on a time scale exceeding 10 min. Indeed, the elution profiles of chemically reactive species that are at dynamic equilibrium during the elution process are very sensitive to the kinetics of the interconversion reactions, yielding a single unresolved peak in the case of fast equilibria and allowing separation of the reactants only for systems with slower kinetics.38,39 The significant overlap of the elution profiles of different V10 species shown in Figure 2 would yield an unresolved asymmetric peak should UV absorption be used as the only means of the analyte detection. However, the unique ability of MS to distinguish the species of interest by examining the corresponding XICs allows kinetic analysis to be carried out in situations when the normal methods of detection (such as UV absorbance) fail, as was recently demonstrated for on-column association/dissociation equilibria of large protein complexes.40 The elution order of the four ionic species representing different oxidation states of V10 correlate with the extent of oxidation (with the fully oxidized V10 exhibiting the most efficient retention), in line with the expectation that the higher electron density (the result of partial reduction) disfavors the POM interaction with the anionic resin. Another intriguing feature revealed by the XICs shown in Figure 2 is the apparent correlation between the oxidation state of V10 and the peak shape. Both peak widths (measured at half-maximum) and the extent of tailing of the four XICs follow the order H5V(V)10O28ˉ >> H6V(IV)V (V)9O28ˉ >> H7V(IV)2V (V)8O28ˉ ≈ H8V(IV)3V (V)7O28ˉ. The XIC shapes of reactive analytes in dynamic systems can be used to extract the rates of relevant reactions,40 and even though such a task is beyond the scope of this work, these calculations can be performed in the future to determine the rates of V10 partial reduction and re-oxidation.
Figure 2.
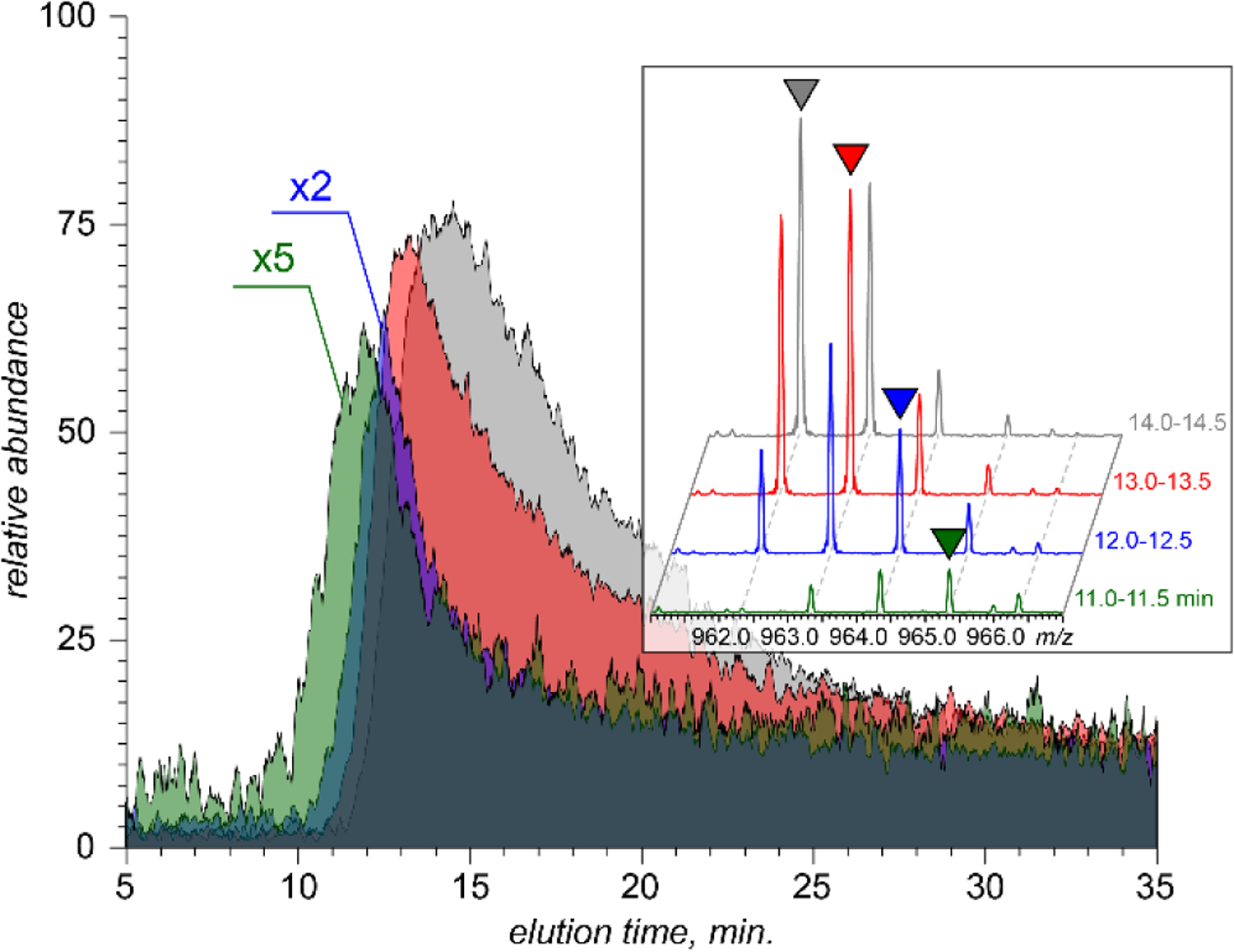
Extracted ion chromatograms representing the fully oxidized V10 (gray) and the partially reduced species incorporating one (red), two (blue) and three (olive) metal atoms at oxidation state IV obtained using ion exclusion LC with on-line MS detection. The inset shows the evolution of the ionic signal within the m/z window 961–967 as a function of elution time.
In addition to the partial reduction of the molecular ions, another unexpected feature of the V10 mass spectra acquired in the negative ion mode is the presence of prominent ions representing neutral H2O (and, to a significantly lesser extent, OH) loss from the molecular ions (Figure 1). Intriguingly, while the V10O262ˉ ion consistently appears nearly equi-abundant to the doubly charged molecular V10 ion, its singly-charged counterpart (HV10O26ˉ) is only a minor feature in the mass spectrum of V10. Furthermore, it is the loss of two water molecules that gives rise to these oxygen-deficient V10 anions. Oxygen loss from smaller oxidovanadates (such as V6) has been reported in the past,22 although the proposed scenario invoked a removal of a single oxygen atom from the metal oxide cluster upon addition of V(Mes)3THF in dichloromethane, leaving behind a reactive (III)V within the molecule (inconsistent with the results of this work, where the most abundant signal in the isotopic clusters of V10O262ˉ and HV10O26ˉ represents the fully oxidized species). Earlier molecular modeling studies carried out with large oxidovanadate clusters (V20) concluded that it was the protonation of the surface oxygen atoms that weakened their interaction with the neighboring metal atoms and facilitated their removal from the cluster.23
Although it might be tempting to conclude that the H2O/OH loss from V10 clusters observed in our work takes place in solution, one must remember that the extent of protonation of V10 molecular species generated by ESI is significantly higher compared to that in solution (vide supra). Therefore, it is possible that the water abstraction process occurs in the gas phase, rather than in solution. In order to investigate the origins of oxygen-deficient V10 species, tandem MS measurements were carried out on several molecular species of V10 using low-energy collisional excitation (Figure 3). All parent ions displayed a remarkable degree of instability, with fragmentation ensuing immediately upon the precursor ion isolation even before any additional collisional activation is applied. Low-energy collisional activation results in robust dissociation of all precursor ions, giving rise to the oxygen-deficient species, all of which had been observed in the mass spectrum of V10 prior to fragmentation. Interestingly, the isotopic distributions of the surviving precursor ions no longer show the presence of the partially reduced V10 species carrying “extra” hydrogen atoms (e.g., see the right-hand-side inset in Figure 3), which appear to be particularly vulnerable to collisional activation. In fact, it is the gas-phase dissociation of such partially reduced species that give rise to the “terminal” fragmentation products in Figure 3, e.g. V10O252ˉ, which appears to be generated as follows:
Figure 3.
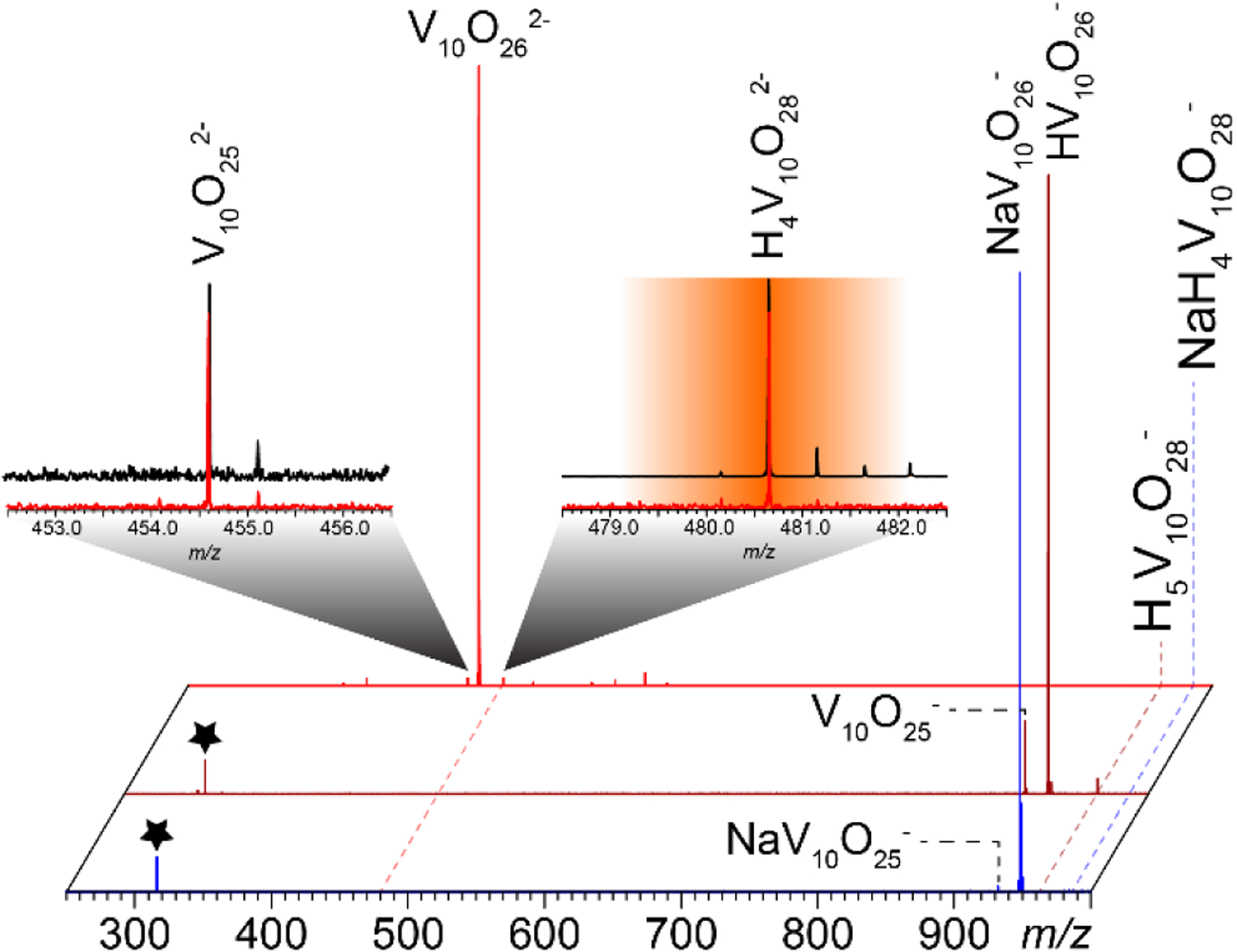
Low-energy CAD mass spectra of H4V10O282ˉ (red trace), H5V10O28ˉ (brown) and NaH4V10O28ˉ (blue) acquired by placing the most abundant isotopologue of each species in the center of a 3-m/z unit wide mass selection window and applying low (5 V) collisional activation. The dotted lines show positions of the three precursor ions, and the stars represent the FT harmonics. The insets show isotopic distributions of the surviving precursor ions (right) and the “terminal” fragment ion, V10O25ˉ (left). The precursor ion isolation window highlighted in orange and the black traces represent isotopic distributions of these ions in MS1.
The ease of generating the oxygen-deficient V10 ions (V10O262ˉ in particular) provides strong evidence that they are MS artifacts. Further increase of collisional activation energy results in a dramatic expansion of the fragment ion repertoire by breaking the metal framework of the V10 cluster and giving rise to abundant V4 and V5 and less abundant V3 and V6 species (see Supplementary Material for more detail), only one of which is observed in the original mass spectrum of V10 (vide infra).
Although the mass spectrum of V10 is dominated by decavanadate anions, two smaller (and lower-abundance) oxidovanadate species, V6O162ˉ and (H/Na)V4O11ˉ are detected as well (Figure 1). While one of them (HV4O11ˉ) can be produced upon collisional activation of H4V10O282ˉ, its generation requires considerable collisional energy (see Supplementary Material for more detail). V6O162ˉ was never observed in our tandem MS experiments (although its singly charged counterpart, HV6O16ˉ has been detected at elevated collisional energy - see Supplementary Material for more detail). The absence of these ions among the fragments produced upon low-energy collisional activation of V10 anions suggests that they represent solution-phase oxidovanadates. Furthermore, should they arise as a result of dissociation of metastable V10 species, one would expect a similar share of partially reduced species among them (e.g., V6O162ˉ should have an anomalous satellite peak M+1 corresponding to HV(IV)V(V)5O162ˉ). However, the recorded isotopic distribution appears consistent with that calculated using the natural isotopic abundance, and acquiring the mass spectrum in 2H2O does not result in a shift of the M+1 peak to the M+3 position, as is the case for V10-derived fragments, such as V10O262ˉ (see Figure 4). Therefore, we conclude that V6O162ˉ and (H/Na)V4O11ˉ represent smaller oxidovanadate species that are present in solution alongside V10 (rather than being the products of its dissociation in the gas phase), and are not as susceptible to the partial reduction in solution under ambient conditions as V10 is.
Figure 4.
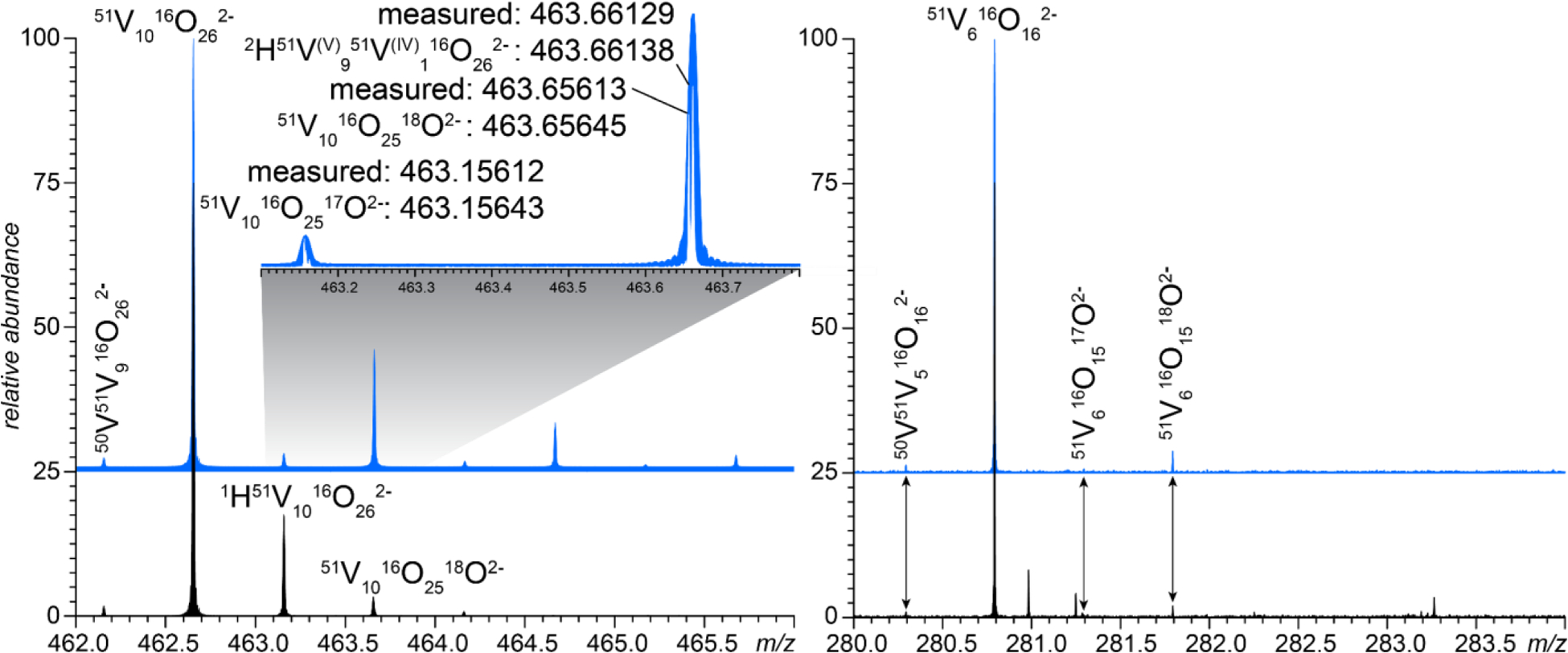
Zoomed views of mass spectra of V10 (sodium salt) acquired in H2O and 2H2O (black and blue traces, respectively) showing isotopic clusters corresponding to V10O262ˉ (left) and V6O162ˉ (right). The inset on the left panel shows the results of high-resolution measurements acquired in the narrow-band mode.
A careful examination of V10 behavior using an array of MS-based methods presented in this work indicates that interpretation of straightforward MS measurements, particularly those carried out at lower resolution offered by conventional instruments, should be approached with extreme caution. Indeed, the additional protonation of V10 polyanions in the course of ESI process gave rise to metastable molecular ions. Water is readily abstracted from these metastable ions in the gas phase generating oxygen deficient V10 species. Furthermore, the apparent susceptibility of V10 to partial reduction in solution generates anions whose extent of protonation is even higher than that observed for fully oxidized V10 species, facilitating more extensive oxygen abstraction (via H2O/OH loss) in the gas phase. Although V10 dissociation gives rise to oxygen-deficient anions in the gas phase, rather than in solution, the underlying processes and the critical role of the surface-bound hydrogen atoms bear remarkable similarity to generation of “oxygen vacancies” within large oxidovanadate clusters and indeed the bulk material surface that had been predicted in earlier computational studies.23 Oxygen vacancies are believed to be key to a range of catalytic properties of oxidovanadates,41 but their experimental investigation has been notoriously difficult, particularly on a larger scale. In this respect, high-resolution MS and MS/MS may lend themselves as potent tools capable of unveiling the most intimate details of oxygen vacancy formation within large vanadate clusters unaffected by solvent.
CONCLUSIONS
Decavanadate is a versatile member of the POM family displaying a variety of unique properties that can be exploited for a range of diverse applications. Our own interest in this molecule stems from its polyanionic nature that lends V10 as a viable candidate for applications in medicine as a disruptor of electrostatically-driven biopolymer interactions such as the platelet factor 4/heparin interactions42 underlying the etiology of heparin-induced thrombocytopenia. The unique chemical properties of V10 are also enjoying considerable attention in areas ranging from chemical catalysis to electronics. Last, but not the least, POMs in general and V10 in particular offer unique opportunities for fundamental studies in material sciences by serving as an atomically defined bridge between micro- and macro-scales.
Mass spectrometry, particularly ESI MS, has had long and illustrious history in inorganic structural analysis and related fields, although complex physical processes occurring within the ESI interface frequently require that the conclusions derived from such measurements be taken cum grano salis. For example, high-resolution measurements of ionic isotopic distributions allow the oxidation states to be determined at both single43,44 and multi-metal level,45 but the measurement outcomes can be affected by the redox processes in the ESI interface. Likewise, mass measurements afforded by high-resolution MS provide a unique opportunity to unequivocally identify a wide range of metallo-compounds, but can be complicated by the metastable nature of molecular ions giving rise to their dissociation in the gas-phase.
In this work we demonstrate that the origin of the redox processes affecting the oxidation state of POMs can be reliably traced to the bulk solution when incorporating ion exclusion LC as an on-line front-end separation tool in ESI MS measurements. We also use two orthogonal MS-based methods, tandem MS and hydrogen/deuterium exchange, to determine the origin of V10 species whose structures deviate from that of the “canonical” species. While this work is focused entirely on V10, the experimental strategies reported in this manuscript will undoubtedly advance the on-going efforts to elucidate structure and reactivity of other members of the POM family.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the grants R01 GM112666 from the National Institutes of Health and CHE-1709552 from the National Science Foundation. The FT ICR mass spectrometer used in this work was acquired through the grant CHE-0923329 from the National Science Foundation (Major Research Instrumentation program) and is now a part of the Mass Spectrometry Core facility at UMass-Amherst (RRID:SCR_019063).
Footnotes
Supporting Information
PDF files containing (i) a positive-ion ESI mass spectrum of V10, (ii) zoomed views of the negative ion ESI mass spectrum of (NH4)6V10O28 (full spectrum shown in Figure 1), (iii) negative-ion ESI mass spectra of ammonium- and potassium salts of V10, and (iv) mass spectra of fragment ions derived from the ESI-generated V10O262ˉ ion over a range of collisional energies. The Supporting Information is available free of charge on the RCS Publications website.
REFERENCES
- (1).Müller A; Peters F; Pope MT; Gatteschi D Polyoxometalates: Very Large ClustersNanoscale Magnets. Chem. Rev 1998, 98, 239–272. [DOI] [PubMed] [Google Scholar]
- (2).Gouzerh P; Che M From Scheele and Berzelius to Muller - Polyoxometalates (POMs) revisited and the “missing link” between the bottom up and top down approaches. Actual Chim. 2006, 9–22. [Google Scholar]
- (3).Hill CL Progress and challenges in polyoxometalate-based catalysis and catalytic materials chemistry. J. Mol. Catal. A-Chem 2007, 262, 2–6. [Google Scholar]
- (4).Vila-Nadal L; Mitchell SG; Markov S; Busche C; Georgiev V; Asenov A; Cronin L Towards Polyoxometalate-Cluster-Based Nano-Electronics. Chem.-Eur. J 2013, 19, 16502–16511. [DOI] [PubMed] [Google Scholar]
- (5).Wang SM; Hwang J; Kim E Polyoxometalates as promising materials for electrochromic devices. J. Mater. Chem. C 2019, 7, 7828–7850. [Google Scholar]
- (6).Van Rompuy LS; Parac-Vogt TN Interactions between polyoxometalates and biological systems: from drug design to artificial enzymes. Curr. Opin. Biotechnol 2019, 58, 92–99. [DOI] [PubMed] [Google Scholar]
- (7).Čolović MB; Lacković M; Lalatović J; Mougharbel AS; Kortz U; Krstić DZ Polyoxometalates in Biomedicine: Update and Overview. Curr. Med. Chem 2020, 27, 362–379. [DOI] [PubMed] [Google Scholar]
- (8).Aureliano M; Gumerova NI; Sciortino G; Garribba E; Rompel A; Crans DC Polyoxovanadates with emerging biomedical activities. Coord. Chem. Rev 2021, 447, 214143. [Google Scholar]
- (9).Aureliano M; Crans DC Decavanadate (V10O286-) and oxovanadates: Oxometalates with many biological activities. J. Inorg. Biochem 2009, 103, 536–546. [DOI] [PubMed] [Google Scholar]
- (10).Forster J; Rosner B; Khusniyarov MM; Streb C Tuning the light absorption of a molecular vanadium oxide system for enhanced photooxidation performance. Chem. Commun 2011, 47, 3114–3116. [DOI] [PubMed] [Google Scholar]
- (11).Kojima T; Antonio MR; Ozeki T Solvent-Driven Association and Dissociation of the Hydrogen-Bonded Protonated Decavanadates. J. Am. Chem. Soc 2011, 133, 7248–7251. [DOI] [PubMed] [Google Scholar]
- (12).Evans HT; Swallow AG; Barnes WH Structure of decavanadate ion. J. Am. Chem. Soc 1964, 86, 4209-&. [Google Scholar]
- (13).Comba P; Helm L The solution structure and reactivity of decavanadate. Helvetica Chim. Acta 1988, 71, 1406–1420. [Google Scholar]
- (14).Rustad JR; Casey WH Metastable structures and isotope exchange reactions in polyoxometalate ions provide a molecular view of oxide dissolution. Nat. Mater 2012, 11, 223–226. [DOI] [PubMed] [Google Scholar]
- (15).Finke RG; Droege MW; Cook JC; Suslick KS Fast atom bombardment mass spectroscopy (FABMS) of polyoxoanions. J. Am. Chem. Soc 1984, 106, 5750–5751. [Google Scholar]
- (16).Vilà-Nadal L; Wilson EF; Miras HN; Rodríguez-Fortea A; Cronin L; Poblet JM Combined Theoretical and Mass Spectrometry Study of the Formation-Fragmentation of Small Polyoxomolybdates. Inorg. Chem 2011, 50, 7811–7819. [DOI] [PubMed] [Google Scholar]
- (17).Boulicault JE; Alves S; Cole RB Negative Ion MALDI Mass Spectrometry of Polyoxometalates (POMs): Mechanism of Singly Charged Anion Formation and Chemical Properties Evaluation. J. Am. Soc. Mass Spectrom 2016, 27, 1301–1313. [DOI] [PubMed] [Google Scholar]
- (18).Surman AJ; Robbins PJ; Ujma J; Zheng Q; Barran PE; Cronin L Sizing and Discovery of Nanosized Polyoxometalate Clusters by Mass Spectrometry. Journal of the American Chemical Society 2016, 138, 3824–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Izzet G; Macdonell A; Rinfray C; Piot M; Renaudineau S; Derat E; Abécassis B; Afonso C; Proust A Metal-Directed Self-Assembly of a Polyoxometalate-Based Molecular Triangle: Using Powerful Analytical Tools to Probe the Chemical Structure of Complex Supramolecular Assemblies. Chem. Eur. J 2015, 21, 19010–19015. [DOI] [PubMed] [Google Scholar]
- (20).Warzok U; Mahnke LK; Bensch W Soluble Hetero-Polyoxovanadates and Their Solution Chemistry Analyzed by Electrospray Ionization Mass Spectrometry. Chem. Eur. J 2019, 25, 1405–1419. [DOI] [PubMed] [Google Scholar]
- (21).Pozniak BP; Cole RB Perspective on Electrospray Ionization and Its Relation to Electrochemistry. J. Am. Soc. Mass Spectrom 2015, 26, 369–385. [DOI] [PubMed] [Google Scholar]
- (22).Lubbert C; Peukert W How to avoid interfering electrochemical reactions in ESI-MS analysis. J. Mass Spectrom 2019, 54, 301–310. [DOI] [PubMed] [Google Scholar]
- (23).Hermann K; Witko M; Druzinic R; Tokarz RJAPA Oxygen vacancies at oxide surfaces: ab initio density functional theory studies on vanadium pentoxide. Appl. Phys. A 2001, 72, 429–442. [Google Scholar]
- (24).Petel BE; Brennessel WW; Matson EM Oxygen-Atom Vacancy Formation at Polyoxovanadate Clusters: Homogeneous Models for Reducible Metal Oxides. J. Am. Chem. Soc 2018, 140, 8424–8428. [DOI] [PubMed] [Google Scholar]
- (25).Johnson GK; Murmann RK In Inorganic Syntheses, Shriver DF, Ed.; John Wiley and Sons: New York, 1979, pp 140–145. [Google Scholar]
- (26).Lee U; Joo HC Potassium-sodium double salt of decavanadate, K4Na2 V10O28 center dot 10H(2)O. Acta Crystallogr. E 2003, 59, I122–I124. [Google Scholar]
- (27).Crans DC; Peters BJ; Wu X; McLauchlan CC Does anion-cation organization in Na+-containing X-ray crystal structures relate to solution interactions in inhomogeneous nanoscale environments: Sodium-decavanadate in solid state materials, minerals, and microemulsions. Coord. Chem. Rev 2017, 344, 115–130. [Google Scholar]
- (28).Heath E; Howarth OW Vanadium-51 and oxygen-17 nuclear magnetic resonance study of vanadate(V) equilibria and kinetics. J. Chem. Soc. Dalton Trans 1981, 1981, 1105–1110. [Google Scholar]
- (29).Gorzsás A; Andersson I; Pettersson L Speciation in aqueous vanadate-ligand and peroxovanadate-ligand systems. J. Inorg. Biochem 2009, 103, 517–526. [DOI] [PubMed] [Google Scholar]
- (30).Abzalimov RR; Dubin PL; Kaltashov IA Glycosaminoglycans as naturally occurring combinatorial libraries: Developing a mass spectrometry-based strategy for characterization of anti-thrombin interaction with low molecular weight heparin and heparin oligomers. Anal. Chem 2007, 79, 6055–6063. [DOI] [PubMed] [Google Scholar]
- (31).Kaltashov IA; Abzalimov RR Do ionic charges in ESI MS provide useful information on macromolecular structure? J. Am. Soc. Mass Spectrom 2008, 19, 1239–1246. [DOI] [PubMed] [Google Scholar]
- (32).Indelicato S; Bongiorno D; Ceraulo L Recent Approaches for Chemical Speciation and Analysis by Electrospray Ionization (ESI) Mass Spectrometry. Front. Chem 2020, 8, 625945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lodi G; Storti G; Pellegrini LA; Morbidelli M Ion Exclusion Chromatography: Model Development and Experimental Evaluation. Ind. Eng. Chem. Res 2017, 56, 1621–1632. [Google Scholar]
- (34).Glód BK Ion exclusion chromatography: parameters influencing retention. Neurochem. Res 1997, 22, 1237–1248. [DOI] [PubMed] [Google Scholar]
- (35).Ye M; Nesterenko PN; Yan Z; Xie P; Chen M Determination of inorganic anions in weak acids by using ion exclusion chromatography - Capillary ion chromatography switching column technique. J. Chromatogr. A 2019, 1588, 169–173. [DOI] [PubMed] [Google Scholar]
- (36).Tanaka K; Mori M Milestone Studies on Ion-exclusion Chromatography of Ionic and Nonionic Substances Utilizing Multifunctional Separation Mechanism of Ion-exchange Resins. Anal. Sci 2021, 37, 93–105. [DOI] [PubMed] [Google Scholar]
- (37).Rehder D Bioinorganic Vanadium Chemistry; John Wiley & Sons, Ltd.: Chichester, West Sussex, UK, 2008, p 1–224. [Google Scholar]
- (38).Moriyasu M; Hashimoto Y; Endo M Kinetic Studies of Fast Equilibrium by Means of High-performance Liquid Chromatography. IV. Separation of Rotamers of Palladium(II) Dithiocarbamates. Bull. Chem. Soc. Jpn 1983, 56, 1972–1977. [Google Scholar]
- (39).Melander WR; Lin HJ; Jacobson J; Horvath C Dynamic effect of secondary equilibria in reversed-phase chromatography. J. Phys. Chem 1984, 88, 4527–4536. [Google Scholar]
- (40).Kaltashov IA; Pawlowski JW; Yang W; Muneeruddin K; Yao H; Bobst CE; Lipatnikov AN LC/MS at the whole protein level: studies of biomolecular structure and interactionsusing native LC/MS and cross-path reactive chromatography (XP-RC) MS. Methods (San Diego, Calif.) 2018, 144, 14–26. [DOI] [PubMed] [Google Scholar]
- (41).Petel BE; Matson EM Oxygen-atom vacancy formation and reactivity in polyoxovanadate clusters. Chem. Commun 2020, 56, 13477–13490. [DOI] [PubMed] [Google Scholar]
- (42).Niu C; Yang Y; Huynh A; Nazy I; Kaltashov IA Platelet factor 4 interactions with short heparin oligomers: implications for folding and assembly. Biophys. J 2020, 119, 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kaltashov IA; Cotter RJ; Feinstone WH; Ketner GW; Woods AS Ferrichrome: Surprising stability of a cyclic peptide Fe-III complex revealed by mass spectrometry. J. Am. Soc. Mass Spectrom 1997, 8, 1070–1077. [Google Scholar]
- (44).Johnson KA; Verhagen MF; Brereton PS; Adams MW; Amster IJ Probing the stoichiometry and oxidation states of metal centers in iron- sulfur proteins using electrospray FTICR mass spectrometry. Anal. Chem 2000, 72, 1410–1418. [DOI] [PubMed] [Google Scholar]
- (45).Kaltashov IA; El Khoury A; Ren C; Savinov SN Ruthenium coordination preferences in imidazole-containing systems revealed by electrospray ionization mass spectrometry and molecular modeling: Possible cues for the surprising stability of the Ru (III)/tris (hydroxymethyl)-aminomethane/imidazole complexes. J. Mass Spectrom 2020, 55, e4435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.