

Abstract
Conventional chemical and even electrochemical Birch-type reductions suffer from a lack of chemoselectivity due to a reliance on alkali metals or harshly reducing conditions. This study reveals that a simpler avenue is available for such reductions by simply altering the waveform of current delivery, namely rapid alternating polarity (rAP). The developed method solves these issues, proceeding in a protic solvent, and can be easily scaled up without any metal additives or stringently anhydrous conditions.
The addition of hydrogen to an aromatic nucleus represents a widely used strategy for rapidly introducing complexity in synthesis.1 As such, the impact of the Birch reduction and related dearomatization strategies in organic synthesis cannot be overemphasized.2–5 This foundational reaction of organic chemistry, taught at the undergraduate level, is also notorious for its harsh reaction conditions: liquid ammonia, elemental alkali metal (Li, Na, K), and a judicially chosen proton source (Figure 1A).2 Owing to the hazardous nature of these conditions, the search for more practical variants is still an active research topic in modern organic synthesis 70+ years following its original disclosure. These efforts can be placed into two categories: (1) conditions that ablate inherent safety hazards (elemental alkali metal and liquid ammonia) and (2) improving the chemoselectivity and scope. Within the former category, various solid-supported reagent systems6–8 have been disclosed as well as mineral oil dispersions with crown-ether additives.9 In 2019 a practical electrochemical Birch variant inspired by Li-ion batteries was developed for ammonia and elemental Li-free arene reduction (Li-ion electroreduction, LER).10 Photochemical variants have also been described, although the scope and reaction times required are suboptimal.11,12 Recently the Koide group demonstrated that ammonia could be replaced by ethylenediamine (e.g., Benkesser modification13–15) to further enhance practicality.16 Although some of these studies enabled practical and scalable Birch reduction, the chemoselectivity was revealed to be analogous to conventional Birch reduction. Regarding chemoselectivity, the procedure employing LiDBB is notable, since it is widely used for reduction of electron-deficient heteroarenes with good tolerance of esters.17 This Communication discloses the finding that simply modifying the waveform of electrolysis (rapid alternating polarity, rAP) can lead to a new level of chemoselectivity for (hetero)arene reduction. This operationally simple protocol proceeds at ambient temperature in protic solvent without the need of sacrificial anode or metal additives, and tolerates functional groups that are notoriously challenging to accommodate by conventional methods.
Figure 1.
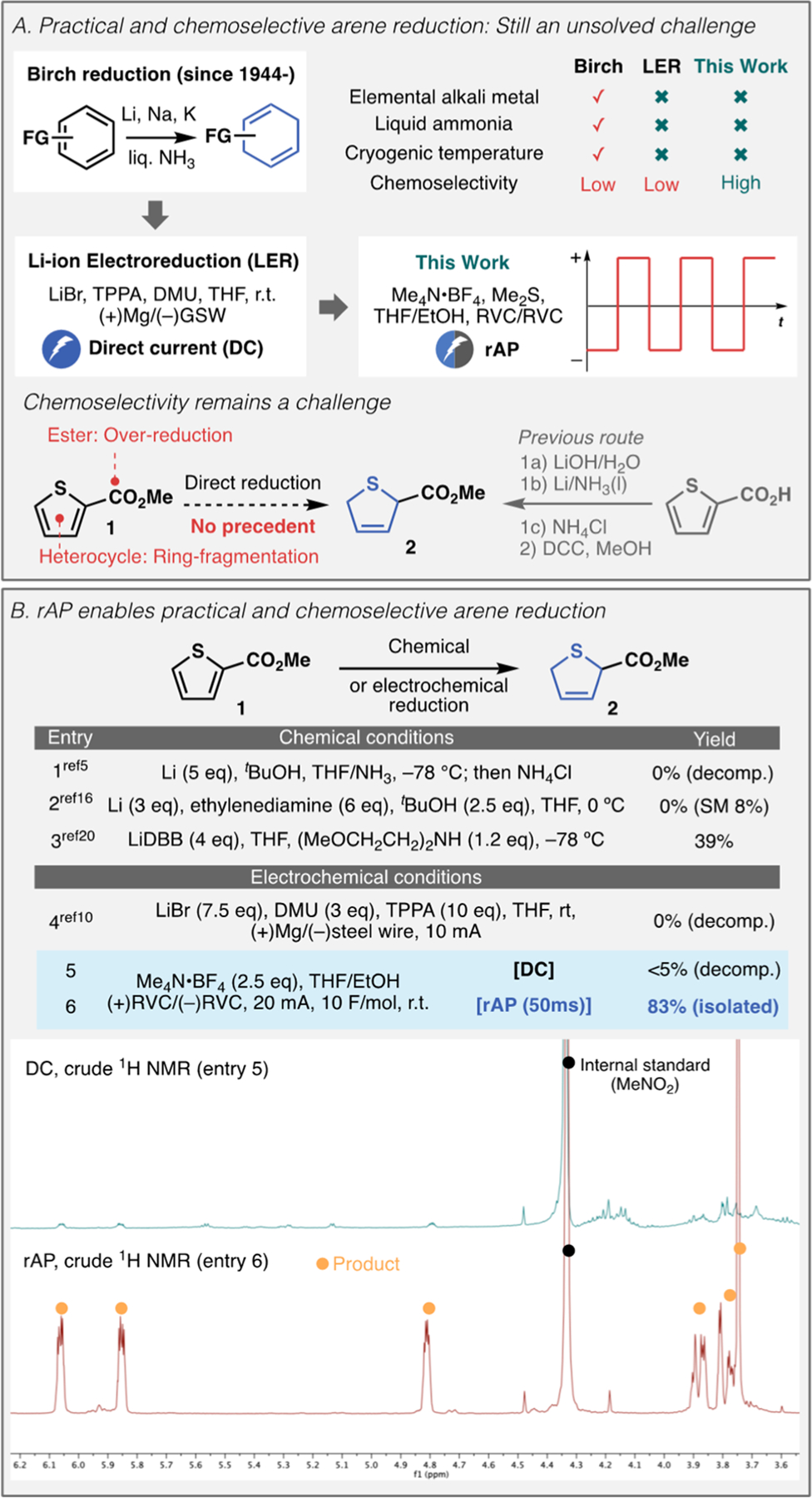
Background of arene reduction and discovery of efficient electroreductive dearomatization by rAP. (A) Practical and chemoselective arene reduction is an unsolved challenge in organic synthesis. (B) Case study with challenging chemoselectivity. rAP gave clean arene reduction without neccesitating special additives, whereas DC electrolysis under the identical conditions resulted in decomposition (Y-axis of the crude NMRs was adjusted to the same scaling).
The canonical Birch reduction is typically applicable to only a limited number of heterocycles. The lack of chemoselectivity of the process is evident in the absence of a literature precedent for reduction of a trivial heterocycle methyl 2-thiophenecarboxylate 1 (Figure 1A). This is because Birch reduction of thiophenecarboxylic acid poses an issue of ring fragmentation.18 The ester functionality could cause a chemoselectivity issue such as overreduction. Accordingly, the reduction product 2 was previously accessed by careful Birch reduction of thiophene-2-carboxylate Li salt, followed by mild esterification.19
Intrigued by the absence of such an example, several representative Birch reduction conditions were applied on this simple substrate 1 (Figure 1B). Not surprisingly, standard Birch conditions5 (entry 1) resulted in complete decomposition of the starting material. The most recent modification16 (entry 2) showed attenuated reactivity, yet extensive decomposition was still observed. Although Donohoe’s LiDBB method20 (entry 3) afforded the product in 39% yield, the highly sensitive nature of DBB (necessitating stringent degassing) and its removal are problematic. Additionally, in this particular case the cost of DBB ($915/mol, TCI) surpasses the cost of the starting material itself ($571/mol, TCI). Regarding the electrochemical conditions, LER10 (entry 4) also led to decomposition. Although simple electrolysis of 1 under DC current in THF/EtOH with RVC electrodes resulted in decomposition (entry 5), it actually showed some peaks in the crude NMR indicative of trace dearomatization products. In striking contrast, under otherwise identical conditions, simply changing the waveform to rapid alternating polarity (rAP, entry 6) afforded 83% of the desired product (crude NMR shown in Figure 1). Sinusoidal waveform was found to be less effective than rAP (see Supporting Information (SI)).
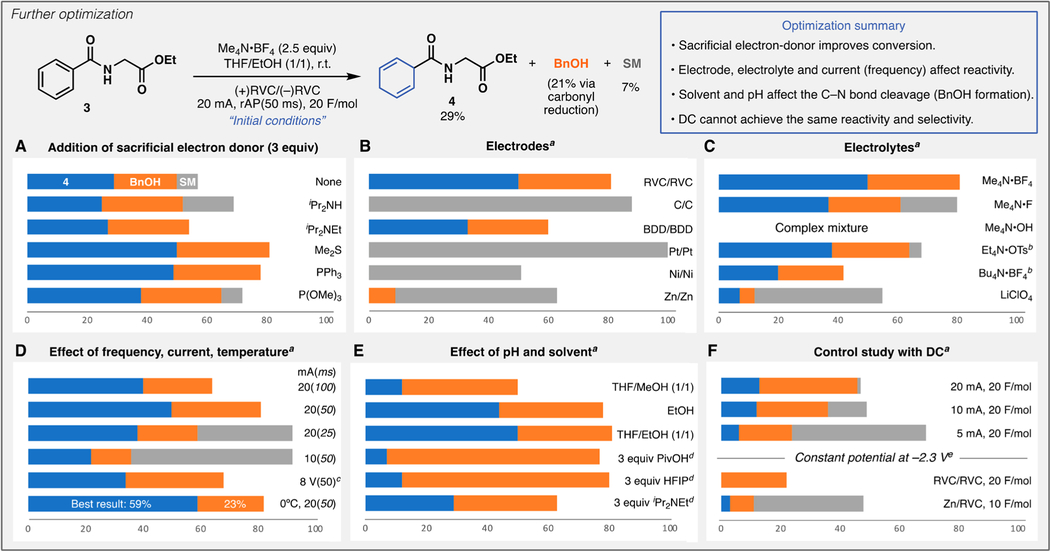
Subsequent optimization was pursued using a less reactive substrate 3 as shown in Table 1. In the initial attempt, a diminished yield (29%) was observed due to the higher aromaticity of this compound compared to 1, along with the formation of benzyl alcohol (BnOH) presumably formed via carbonyl reduction. Standard Na- or Li-based chemical conditions as well as LER on 3 did not afford any 1,4-diene product 4 (see SI). Instead, various over-reduced products and fragmentation-derived products predominated. A systematic study of chemical and electrochemical parameters revealed that dimethylsulfide (DMS) could be employed as a sacrificial electron-donor to improve the yield (Table 1, Panel A). Thus, the rest of the optimization was conducted using 3.0 equiv of DMS. Electrodes and electrolytes were found to be critical to achieve good reactivity (Table 1, Panel B and C). In particular, the combination of RVC electrodes with tetraalkylammonium salts exhibited superior reactivity among various conditions screened. As demonstrated in our previous rAP study,21 the electrochemical driving force delivered is a function of both current and frequency (Table 1, Panel D). Thus, either decreasing pulse width (increasing frequency) or reducing current resulted in decreased conversion. Constant potential rAP22 gave a similar product distribution to constant current rAP. Since a slight warming of the reaction was noted (from 23 to 35 °C; see SI), submerging the reaction vessel in a 0 °C ice bath further increased the desired product yield to 59% (best yield) by suppressing BnOH formation. The nature of solvent and pH of the reaction (Table 1, Panel E) also affected the ratio of the arene reduction vs the carbonyl reduction. Lastly, several control experiments under direct current conditions were performed to provide a comparison with the rAP-based method (Table 1, Panel F). Although constant current as well as constant potential experiments at −2.3 V (see SI for cyclic voltammogram of 3) afforded a small amount of 4 in some cases, the yield and the extent of carbonyl reduction were by no means similar to the case of the rAP-based method. These results are supportive of the documented fundamental reactivity difference between rAP and DC in other studies.23,24
Table 1.
Optimization Study
|
All the reactions were performed on 0.1 mmol scale under the initial conditions with a deviation indicated. Product distribution was analyzed by 1H NMR. aReactions were performed with 3 equiv of Me2S. b1 equiv of electrolyte was used. c5 equiv of Me4N·BF4 was used. dIndicated additive was added to THF/EtOH (1/1). eBu4N·BF4 (0.2 M) was used as an electrolyte.
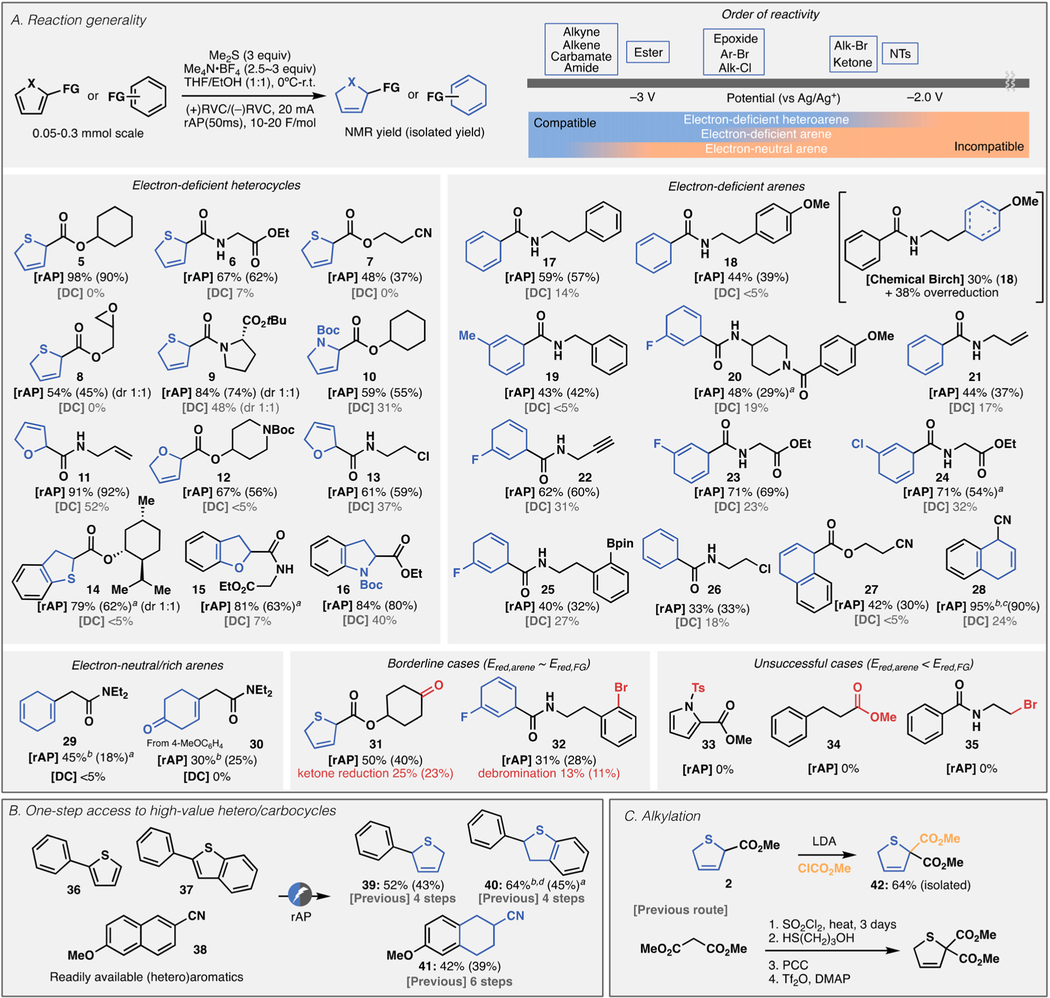
Table 2 demonstrates the reaction generality. Various (hetero)arenes can be reduced chemoselectively under operationally simple conditions without the need for any expensive reagents or additives (Table 2A). Complementary to standard Birch reduction, rAP reduction is most suitable for electron-deficient (hetero)arenes. The success of the reaction is predictable based on the reduction potential of an arene and a functional group that can be easily measured using CV (empirical guidance for FG tolerance is summarized in Table 2A). Thus, synthetically useful yields are obtained when a target arene has a more positive reduction potential than those of other functional groups (Ered,arene > Ered,FG). As such, electron-deficient heteroarenes exhibit the broadest functional group tolerance with ester (5–10, 12, 14–16), nitrile (7), allyl group (11), epoxide (8), and even alkyl chloride (13). Electron-deficient arenes are slightly less reducible than electron-deficient heterocycles, and the functional group compatibility of this class is more limited. Nevertheless, synthetically useful handles such as allyl (21), alkyne (22), ester (23, 24, 27), boronate ester (25), alkyl chloride (26), and nitrile (27, 28) were well-tolerated. A halogen atom directly connected to an arene was also tolerated (20, 22–25). Notably, differentiation of two arenes was possible as exemplified in 17–20 and 25. Achieving such chemoselectivity was found to be challenging under conventional Birch reduction conditions as a mixture of products was obtained even if the two arenes are most electronically differentiated (Table 2A, chemical Birch for obtaining 18). Nonconjugated arenes can be reduced to afford 29 and 30 in this method, though the yields were moderate due to the low reactivity. Borderline cases also exist when Ered,arene is close to Ered,FG, and such cases were exemplified in the reduction of 31 and 32. In accord with this simple rubric, when a functional group is more easily reducible, the method is unable to favor the reduction of (hetero)arenes (33–35). To demonstrate the utility of this method, a collection of readily available heteroarenes 36–38 were enlisted for accessing partially desaturated heterocycles (Table 2B). Subsequent alkylation could also be a useful derivatization as exemplified in 42 (Table 2C). Throughout 39–42, previous syntheses necessitate 4–6 steps involving denovo ring construction;19,25–27 this method subverts conventional multistep ring construction by repurposing existing, readily available heteroarenes.
Table 2.
Chemoselective Reduction of (Hetero)arenes by rAP: (A) Reaction Generality and Empirical Guide for Functional Group Tolerance; (B) Access to High-Value (Hetero)cyclic Systems from Readily Available (Hetero)arenes (See SI for Individual Reaction Conditions); (C) Concise Access to Dihydrothiophene 42 via Alkylation of 2
|
aLarge drop of isolated yield was due to purification loss. b100 ms pulse was used instead of 50 ms. cThe reaction was performed in MeOH with PivOH as an additive. dAmount of Me2S was 10 equiv.
The mechanistic details of this reaction are of great interest due to the notable reactivity difference observed between DC and rAP. Although detailed mechanistic study is beyond the scope of this communication, several pieces of empirical evidence are presented in Figure 2 that help to rationalize the differences in bulk reactivity.28 For instance, the reduction potential of 43 (<–3 V)29 resides far outside the solvent electrochemical window (Figure 2A, solvent CV), yet rAP delivered the product in 45% NMR yield. In contrast, under otherwise identical conditions employing a standard DC waveform resulted in mostly recovered starting material, together with the observation of active gas evolution from the cathode. This gas evolution was also noted at the working electrode after measuring the CV of the solvent. We hypothesized that hydrogen gas was being formed on the cathode (reductive) in the protic medium. To prove the existence of H2 qualitatively, simple hydrogenation experiments were performed; namely, either DC or rAP was applied to the reaction solvent including cyclooctene in the presence of Pd/C catalyst (Figure 2B).30 A considerable quantity of hydrogenated product 45 was observed in the DC electrolysis, while 45 was below the GC/MS detection limit in the rAP experiment. The extent of proton reduction could also be reflected to the basicity of the reaction medium; transesterification product 46 was observed under DC conditions, whereas no such product was observed under rAP conditions when the reduction of 1 was interrupted. Consistent with these findings are literature reports of CO2 or CO reduction using pulsed potential instead of DC that produces less H2.31–36 Collectively, these experiments support the mechanism summarized in Figure 2C. During the cathodic phase, (hetero)arene reduction is taking place through direct SET, leading to the same regioselectivity outcome as conventional Birch reduction. Chemoselectivity follows the reduction potential of the (hetero)arene and those of existing functionalities. Proton reduction, a pathway that normally competes to diminish reactivity toward arene reduction, is largely suppressed by applying rAP. This effect also explains improved chemoselectivity under rAP conditions by removing side reactions promoted under a highly basic environment. During the anodic phase, DMS is presumably oxidized to generate an α-ethoxy derivative. Analogous species 47 was detected in GC analysis of crude reaction mixture when heavier Et2S was used instead of DMS. A small amount of solvent oxidation may also take place during the anodic phase.
Figure 2.
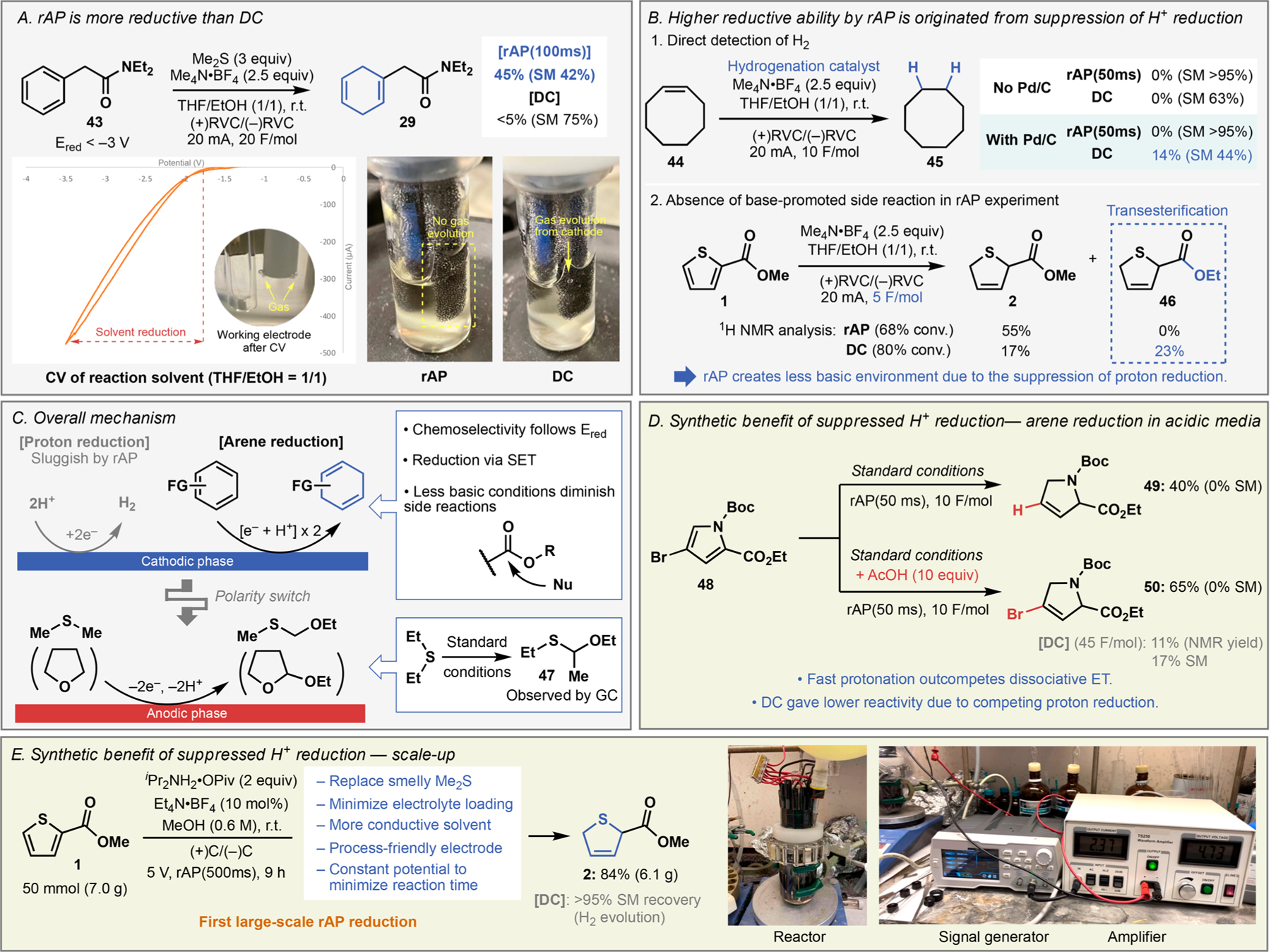
Mechanistic studies and synthetic advantage of outcompeting proton reduction. (A) Gas evolution was notable during DC electrolysis (including CV), whereas little gas evolution was observed with rAP. (B) Direct detection of H2 gas and resulting pH change in the reaction probed by transesterification. (C) Overview of cathodic and anodic reactions. (D) rAP enables efficient arene reduction in the presence of acid with unique chemoselectivity, whereas DC electrolysis suffers proton reduction. (E) First example of large-scale electrolysis with rAP. Corresponding DC reduction resulted in complete recovery of the starting material due to the competing H2 evolution.
The suppression of proton reduction may enable new types of chemoselective (hetero)arene reduction by running reactions deliberately under acidic conditions (Figure 2D), which is challenging under conventional DC electrolysis or Birch reduction. An interesting case was indeed found in the reduction of 48. Standard rAP reduction furnished debrominated dihydropyrolidine 49 in 40% yield via dissociative electron transfer, a common phenomenon during single-electron reduction of halogenated arenes. In striking contrast, running the same reaction in the presence of 10 equiv of AcOH furnished dihydropyrrolidine 50 in 65% yield, maintaining the usef ul C–Br bond. As expected, the corresponding DC electrolysis under identical conditions required a much longer reaction time (6 h, 45 “equiv” of electrons) due to the competing proton reduction, and only delivered 11% of 50 with incomplete conversion. Another dramatic benefit for suppressing proton reduction was found during scale-up efforts (Figure 2E). After introducing several modifications to be more process-friendly (see SI for reoptimization), rAP-based conditions successfully afforded the product 2 in 84% isolated yield on 50 mmol scale, whereas DC electrolysis resulted in full recovery of 1 under identical reaction conditions.37 Again, a large volume of gas evolution was observed during DC electrolysis, rendering the arene reduction unfavorable under such simple conditions.
This study demonstrates another compelling example of how the outcome of an electrochemical transformation can be completely altered simply by changing the waveform of current delivery. This time, chemoselective (hetero)arene reduction under rAP is shown to complement the scope of conventional chemical or electrochemical arene reductions. The enhanced reactivity as well as higher chemoselectivity can be explained by a suppression of competing proton reduction by the easily accessible rAP waveform. On preparative scales, no specialized equipment or engineering is required. On larger scales, a simple signal amplifier and signal generator can be employed (see SI). Although further in-depth analysis of the mechanism is necessary, the implications of this unique mode of reaction control may hold great promise not only for chemoselective reductions of organic compounds in protic media but also for electrochemical CO2 reduction as well as nitrogen fixation in which proton reduction often competes.
Supplementary Material
ACKNOWLEDGMENTS
Financial support for this work was provided by the National Science Foundation Center for Synthetic Organic Electrochemistry (CHE-2002158) and the National Institutes of Health (Grant Number GM-118176). We are grateful to D.-H. Huang and L. Pasternack (Scripps Research) for NMR spectroscopic assistance, and M. Gembicky (UCSD) for X-ray analysis. We also thank A. F. Garrido-Castro, M. D. Palkowitz, and S. B. J. Kan for helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c02102.
Experimental procedures, additional experimental data, NMR characterization data, X-ray and cyclic voltammogram. (PDF)
Accession Codes
CCDC 2122303 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c02102
The authors declare no competing financial interest.
Contributor Information
Kyohei Hayashi, Department of Chemistry, Scripps Research, La Jolla, California 92037, United States;.
Jeremy Griffin, Abbvie Process Research and Development, North Chicago, Illinois 60064, United States.
Kaid C. Harper, Abbvie Process Research and Development, North Chicago, Illinois 60064, United States;.
Yu Kawamata, Department of Chemistry, Scripps Research, La Jolla, California 92037, United States;.
Phil S. Baran, Department of Chemistry, Scripps Research, La Jolla, California 92037, United States;
REFERENCES
- (1).Huck CJ; Sarlah D Shaping Molecular Landscapes: Recent Advances, Opportunities, and Challenges in Dearomatization. Chem 2020, 6, 1589–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Birch AJ The Birch Reduction in Organic Synthesis. Pure Appl. Chem 1996, 68, 553–556. [Google Scholar]
- (3).Donohoe TJ; Garg R; Stevenson CA Prospects for Stereocontrol in the Reduction of Aromatic Compounds. Tetrahedron Asymmetry 1996, 7, 317–344. [Google Scholar]
- (4).Hook JM; Mander LN Recent Developments in the Birch Reduction of Aromatic Compounds: Applications to the Synthesis of Natural Products. Nat. Prod. Rep 1986, 3, 35–85. [Google Scholar]
- (5).Rabideau PW; Marcinow Z The Birch Reduction of Aromatic Compounds. Organic React. 1992, 42, 1–334. [Google Scholar]
- (6).Dye JL; Cram KD; Urbin SA; Redko MY; Jackson JE; Lefenfeld M Alkali Metals Plus Silica Gel: Powerful Reducing Agents and Convenient Hydrogen Sources. J. Am. Chem. Soc 2005, 127, 9338–9339. [DOI] [PubMed] [Google Scholar]
- (7).Nandi P; Redko MY; Petersen K; Dye JL; Lefenfeld M; Vogt PF; Jackson JE Alkali Metals in Silica Gel (M-SG): A New Reagent for Desulfonation of Amines. Org. Lett 2008, 10, 5441–5444. [DOI] [PubMed] [Google Scholar]
- (8).Costanzo MJ; Patel MN; Petersen KA; Vogt PF Ammonia-Free Birch Reductions with Sodium Stabilized in Silica Gel, Na-SG(I). Tetrahedron Lett. 2009, 50, 5463–5466. [Google Scholar]
- (9).Lei P; Ding Y; Zhang X; Adijiang A; Li H; Ling Y; An J A Practical and Chemoselective Ammonia-Free Birch Reduction. Org. Lett 2018, 20, 3439–3442. [DOI] [PubMed] [Google Scholar]
- (10).Peters BK; Rodriguez KX; Reisberg SH; Beil SB; Hickey DP; Kawamata Y; Collins M; Starr J; Chen L; Udyavara S; Klunder K; Gorey TJ; Anderson SL; Neurock M; Minteer SD; Baran PS Scalable and Safe Synthetic Organic Electroreduction Inspired by Li-Ion Battery Chemistry. Science 2019, 363, 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cole JP; Chen D-F; Kudisch M; Pearson RM; Lim C-H; Miyake GM Organocatalyzed Birch Reduction Driven by Visible Light. J. Am. Chem. Soc 2020, 142, 13573–13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chatterjee A; König B Birch-Type Photoreduction of Arenes and Heteroarenes by Sensitized Electron Transfer. Angew. Chem., Int. Ed 2019, 58, 14289–14294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Benkeser RA; Robinson RE; Sauve DM; Thomas OH Reduction of Organic Compounds by Lithium in Low Molecular Weight Amines. I. Selective Reduction of Aromatic Hydrocarbons to Monoölefins. J. Am. Chem. Soc 1955, 77, 3230–3233. [Google Scholar]
- (14).Benkeser RA; Arnold C; Lambert RF; Thomas OH Reduction of Organic Compounds by Lithium in Low Molecular Weight Amines. III. Reduction of Aromatic Compounds Containing Functional Groups. J. Am. Chem. Soc 1955, 77, 6042–6045. [Google Scholar]
- (15).Benkeser RA; Agnihotri RK; Burrous ML; Kaiser EM; Mallan JM; Ryan PW Highly Selective Lithium—Amine Reducing Systems. The Selective Reduction of Aromatic Compounds by Lithium in Mixed Amine Solvents. J. Org. Chem 1964, 29, 1313–1316. [Google Scholar]
- (16).Burrows J; Kamo S; Koide K Scalable Birch Reduction with Lithium and Ethylenediamine in Tetrahydrofuran. Science 2021, 374, 741–746. [DOI] [PubMed] [Google Scholar]
- (17).Donohoe TJ; Thomas RE Partial Reduction of Pyrroles: Application to Natural Product Synthesis. Chem. Rec 2007, 7, 180–190. [DOI] [PubMed] [Google Scholar]
- (18).Blenderman WG; Joullié MM; Preti G The Birch Reduction of Thiophine-2-Carboxylic Acid. Tetrahedron Lett. 1979, 20, 4985–4988. [Google Scholar]
- (19).Altenbach H-J; Brauer DJ; Merhof GF Synthesis of 1-Deoxy-4-Thio-D-Ribose Starting from Thiophene-2-Carboxylic Acid. Tetrahedron 1997, 53, 6019–6026. [Google Scholar]
- (20).Donohoe TJ; House D Ammonia Free Partial Reduction of Aromatic Compounds Using Lithium Di-tert-Butylbiphenyl (LiDBB). J. Org. Chem 2002, 67, 5015–5018. [DOI] [PubMed] [Google Scholar]
- (21).Kawamata Y; Hayashi K; Carlson E; Shaji S; Waldmann D; Simmons BJ; Edwards JT; Zapf CW; Saito M; Baran PS Chemoselective Electrosynthesis Using Rapid Alternating Polarity. J. Am. Chem. Soc 2021, 143, 16580–16588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).The same terminal potential observed under constant current with 5 equiv of Me4N·BF4 was used.
- (23).Rodrigo S; Um C; Mixdorf JC; Gunasekera D; Nguyen HM; Luo L Alternating Current Electrolysis for Organic Electrosynthesis: Trifluoromethylation of (Hetero)Arenes. Org. Lett 2020, 22, 6719–6723. [DOI] [PubMed] [Google Scholar]
- (24).Rodrigo S; Gunasekera D; Mahajan JP; Luo L Alternating Current Electrolysis for Organic Synthesis. Current Opinion in Electrochemistry 2021, 28, 100712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chatterjee A; Roy D; Chatterjee S An Efficient General Method for the Synthesis of Some Intermediates for Heterocyclic Steroids. Synthesis 1981, 1981, 449–451. [Google Scholar]
- (26).Liao Q; You W; Lou Z-B; Wen L-R; Xi C Copper-Catalyzed Tandem S-Alkylation and S-Alkenylation of Sodium Sulfide: Synthesis of 2,3-Dihydrothiophenes and Thiophenes. Tetrahedron Lett. 2013, 54, 1475–1477. [Google Scholar]
- (27).Rogers E; Araki H; Batory LA; McInnis CE; Njardarson JT Highly Selective Copper-Catalyzed Ring Expansion of Vinyl Thiiranes: Application to Synthesis of Biotin and the Heterocyclic Core of Plavix. J. Am. Chem. Soc 2007, 129, 2768–2769. [DOI] [PubMed] [Google Scholar]
- (28).A truly authoritative and complete understanding of this phenomenon would likely entail the use of various electroanalytical methods involving microelectrodes as well as rotating disc electrodes to dissect fast electron transfer kinetics under stationary and non-stationary electrolysis conditions. Relevant techniques are described here. Bamford, C. H. Tipper, C. F. H. Compton, R. G. Electrode Kinetics: Principles and Methodology; Elsevier: 1986. [Google Scholar]
- (29).Mortensen J; Heinze J The Electrochemical Reduction of Benzene—First Direct Determination of the Reduction Potential. Angew. Chem., Int. Ed 1984, 23, 84–85. [Google Scholar]
- (30).To be sure, cyclooctene is unreactive under the arene reduction conditions, and redox perturbation of Pd/C catalyst by electrodes is unlikely since Pd/C does not dissolve in the solvent.
- (31).Kimura KW; Fritz KE; Kim J; Suntivich J; Abruña HD; Hanrath T Controlled Selectivity of CO2 Reduction on Copper by Pulsing the Electrochemical Potential. ChemSusChem 2018, 11 (11), 1781–1786. [DOI] [PubMed] [Google Scholar]
- (32).Kim C; Weng L-C; Bell AT Impact of Pulsed Electrochemical Reduction of CO2 on the Formation of C2+ Products over Cu. ACS Catal. 2020, 10, 12403–12413. [Google Scholar]
- (33).Strain JM; Gulati S; Pishgar S; Spurgeon JM Pulsed Electrochemical Carbon Monoxide Reduction on Oxide-Derived Copper Catalyst. ChemSusChem 2020, 13, 3028–3033. [DOI] [PubMed] [Google Scholar]
- (34).Casebolt R; Levine K; Suntivich J; Hanrath T Pulse Check: Potential Opportunities in Pulsed Electrochemical CO2 Reduction. Joule 2021, 5, 1987–2026. [Google Scholar]
- (35).Casebolt R; Kimura KW; Levine K; DaSilva JAC; Kim J; Dunbar TA; Suntivich J; Hanrath T Effect of Electrolyte Composition and Concentration on Pulsed Potential Electrochemical CO2 Reduction. Chemelectrochem 2021, 8, 681–688. [Google Scholar]
- (36).Tang Z; Nishiwaki E; Fritz KE; Hanrath T; Suntivich J Cu(I) Reducibility Controls Ethylene vs Ethanol Selectivity on (100)-Textured Copper during Pulsed CO2 Reduction. ACS Appl. Mater. Interfaces 2021, 13, 14050–14055. [DOI] [PubMed] [Google Scholar]
- (37).The DC electrolysis comparison was carried out on 2.0 mmol scale under otherwise identical reaction conditions and substrate concentration.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.