

INTRODUCTION
Soft tissue tumors comprise a wide range of entities, each with distinct diagnostic and biologic features with relevance for clinical management, which can make their diagnostic workup challenging. Recent advances in the molecular diagnostics workup of many benign and malignant mesenchymal neoplasms and the discovery of recurrent genetic aberrations have expanded the diagnostic spectrum and led to the discovery of new tumor types. The 5th edition of WHO Classification of Soft Tissue and Bone Tumors was published in early 20201, seven years after the 4th edition2, and features a number of revisions to existing classification and risk stratification schemes. The update reflects a consensus among an international expert panel including pathologists, geneticists, a medical oncologist, surgeon, and radiologist.3 We here highlight the most relevant changes to the soft tissue chapter in the 2020 World Health Organization Classification by diagnostic category and provide an update on modifications to diagnostic criteria and classification schemes. We further discuss challenging aspects in the diagnosis and/or prognostication of select well-established entities.
UPDATES TO THE 2020 WHO CLASSIFICATION:
ADIPOCYTIC TUMORS
Atypical Spindle Cell/Pleomorphic Lipomatous Tumor
Atypical spindle cell/pleomorphic lipomatous tumor (ASCLT/APLT) is a benign adipocytic neoplasm and mostly affects middle-aged adults with a slight male predominance with predilection for limbs and limb girdle.4,5 ASCLT/APLT is characterized by ill-defined margins (Fig. 1) and nodular to multinodular growth, and can show a range of histologic appearances with varying proportions of atypical spindle cells, adipocytes, lipoblasts, and pleomorphic/multinucleated cells within collagenous and/or myxoid matrix (Fig. 2A, B). The tumors cells express CD34 (Fig. 2C), S100, and desmin to varying extent, but are generally negative for MDM2 (Fig. 2C, inset) and CDK4 given absence of MDM2 or CDK4 amplification.4–6 ASCLT/APLT shows loss of RB1 expression in 50–70% of cases (Fig. 2D) resulting from deletions at 13q14 inactivating RB1 and adjacent genes in a significant subset of cases (Table 1).4,5,7 ASCLT/APLT has an excellent prognosis when completely excised and distant metastases have not been described.
Figure 1.
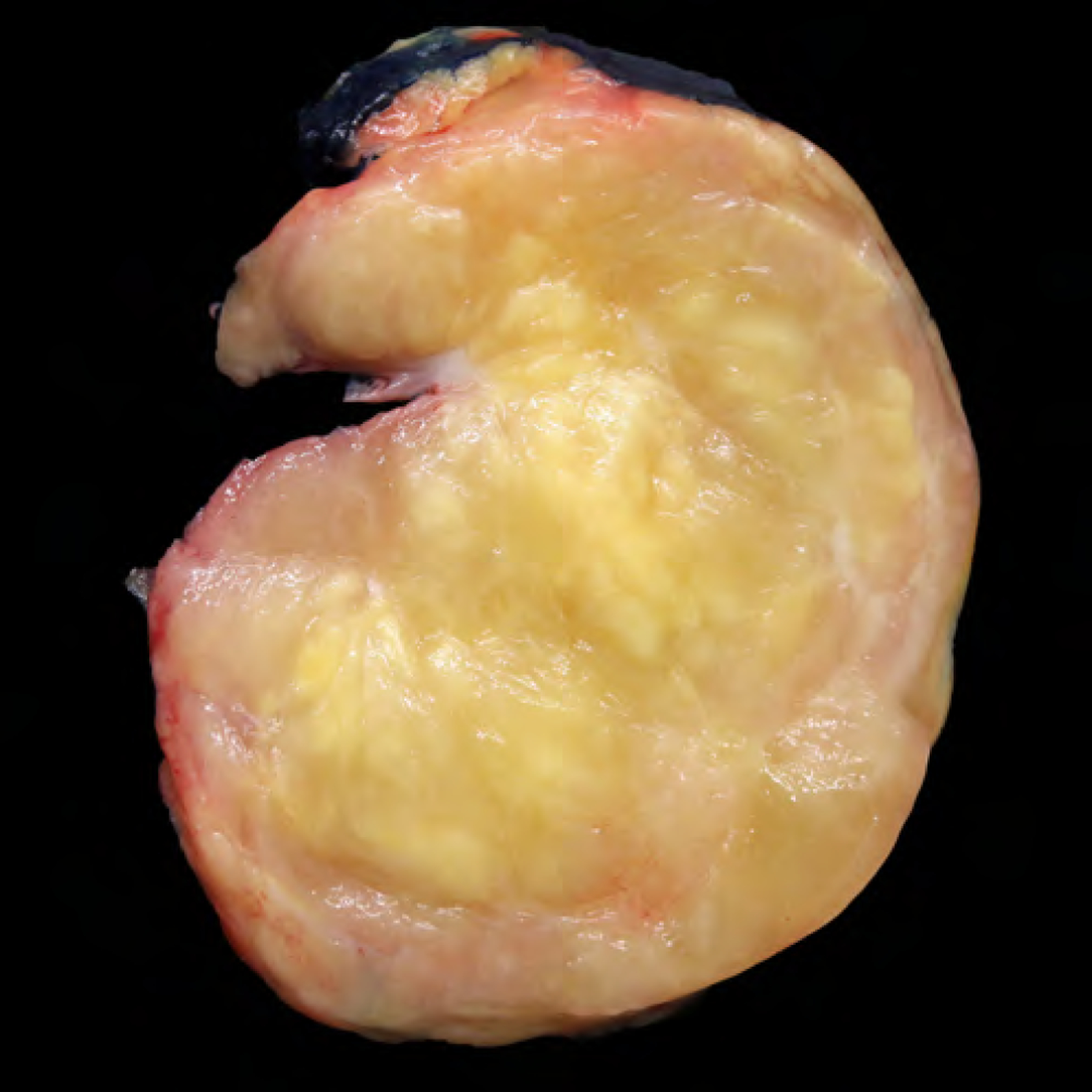
Grossly, atypical pleomorphic lipomatous tumor is unencapsulated with ill-defined tumor margins and nodular growth displaying an admixture of fatty and myxoid to collagenous features.
Figure 2.
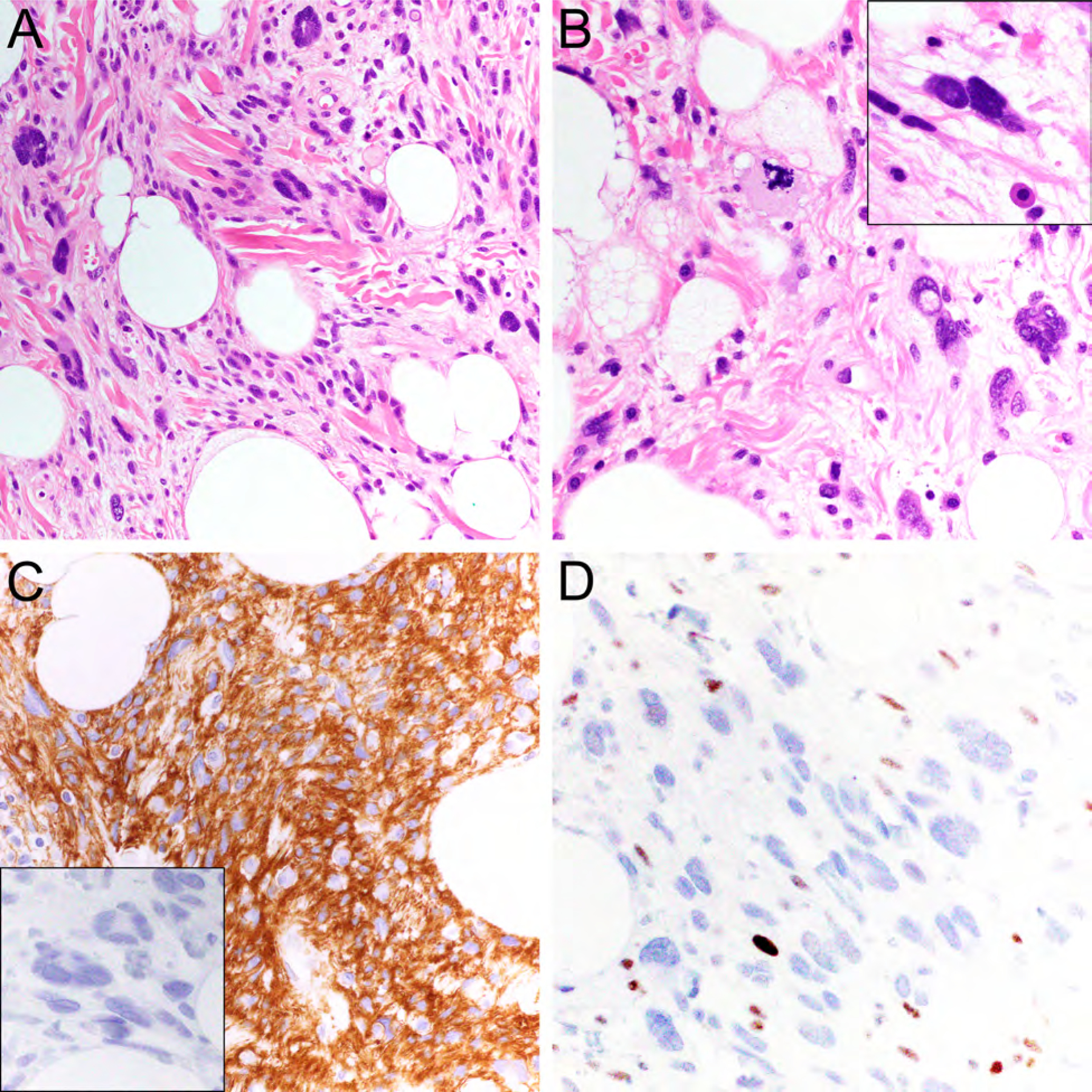
Atypical pleomorphic lipomatous tumor comprised of spindle cells, adipocytic cells, pleomorphic giant cells, and scattered lipoblasts embedded in a myxoid and collagenous extracellular matrix (A, hematoxylin-eosin (HE), 200x; B, HE, x400) with occasional hyperchromatic nuclei (B, inset). The tumor cells express CD34 (C, x400), lack MDM2 expression (C, inset, x400), and show RB1 loss (D, x400).
Table 1.
Summary of diagnostic and prognostic features of select adipocytic neoplasms.
| Tumor Type | Useful Diagnostic IHC Markers | Characteristic Genomic Aberrations | Behavior |
|---|---|---|---|
| Spindle cell/pleomorphic lipoma | RB1 loss; variable expression of CD34, desmin, S100 | 13q14 deletion (RB1) | Benign |
| Atypical spindle cell/pleomorphic lipomatous tumor (ASCLT/APLT) | RB1 loss (50–70%); variable expression of CD34, desmin, S100 | 13q14 deletion (RB1) | Benign; local recurrence in 10–15% if incompletely excised |
| Atypical lipomatous tumor (ALT)/Well-differentiated liposarcoma (WDLPS) | Expression of MDM2 and/or CDK4 | 12q13–15 high-level amplification (MDM2, CDK4) | Locally aggressive; risk of dedifferentiation |
| Dedifferentiated liposarcoma | Expression of MDM2 and/or CDK4 | 12q13–15 high-level amplification (MDM2, CDK4) | Malignant |
| Myxoid liposarcoma | DDIT3 expression in high-grade cases | FUS-DDIT3 gene fusion | Malignant |
| Pleomorphic liposarcoma | None | None | Malignant |
| Myxoid pleomorphic liposarcoma | RB1 loss (subset) | Complex chromosomal alterations | Highly malignant |
Key Features: Atypical Spindle Cell/Pleomorphic Lipomatous Tumor.
Limbs and limb girdle
Middle-aged adults, M≥F
Mild to moderate atypia of spindle cells, adipocytes, lipoblasts, pleomorphic/multinucleated cells; myxoid to collagenous matrix
Loss of RB1; variable expression of CD34, S100, and desmin
Benign; local recurrence in 10–15% if incompletely excised
Myxoid Pleomorphic Liposarcoma
Myxoid pleomorphic liposarcoma (MPLPS) is exceptionally rare and extremely aggressive, mostly arising in the mediastinum of children and young adults with female predominance as a large, deep-seated mass with ill-defined margins.8–11 MPLPS combines histologic features of conventional myxoid liposarcoma (Fig. 3A) and pleomorphic liposarcoma (Fig. 3B). MPLPS lacks both the FUS-DDIT3 gene fusion characteristic of myxoid liposarcoma and MDM2 and/or CDK4 amplification found in dedifferentiated liposarcoma (Table 1) and instead, shows complex chromosomal alterations with occasional losses at 13q14 involving RB1.12,13 MPLPS is extremely aggressive with frequent local recurrences and distant metastases associated with poor survival.8,9
Figure 3.
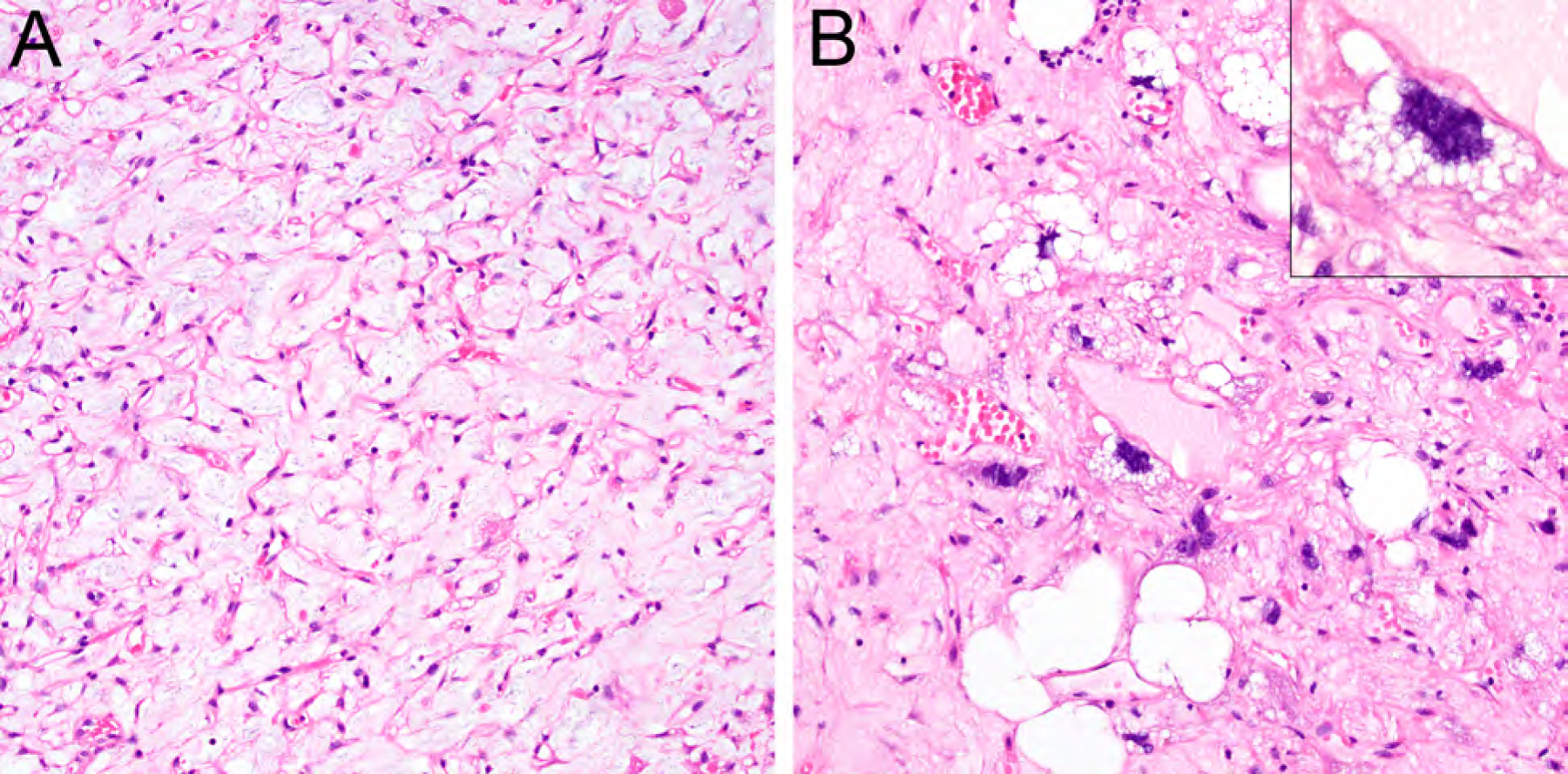
Myxoid pleomorphic liposarcoma combining morphologic features of conventional myxoid liposarcoma (A, HE, x200) and pleomorphic liposarcoma (B, HE, x200; inset, HE, x400).
Key Features: Myxoid Pleomorphic Liposarcoma.
Mediastinum
Children and young adults, F>M
Admixture of areas resembling conventional myxoid liposarcoma and high-grade pleomorphic liposarcoma-like
RB1 loss in subset of cases
Complex chromosomal alterations
Extremely aggressive
FIBROBLASTIC AND MYOFIBROBLASTIC TUMORS
EWSR1-SMAD3-Positive Fibroblastic Tumor
EWSR1-SMAD3-positive fibroblastic tumor is a recently discovered benign neoplasm which has been included in the 2020 WHO classification as emerging entity under a provisional name.1 These tumors show predilection for acral and superficial location, occur over a wide age range with female predominance, and are composed of a centrally located hypocellular hyalinized area (Fig. 4A) and more cellular areas at the periphery containing overlapping fibroblastic spindle cells without atypical features (Fig. 4B).14–17 Diffuse nuclear expression of ERG is characteristic (Fig. 4C), whereas other markers such as CD34 and smooth-muscle actin (SMA), are negative. EWSR1-SMAD3-positive fibroblastic tumor is characterized by a fusion of EWSR1 exon 7 and SMAD3 exon 5 (Fig. 4D).14,15 Their behavior is benign, but local recurrence has been observed if incompletely excised.14,17
Figure 4.
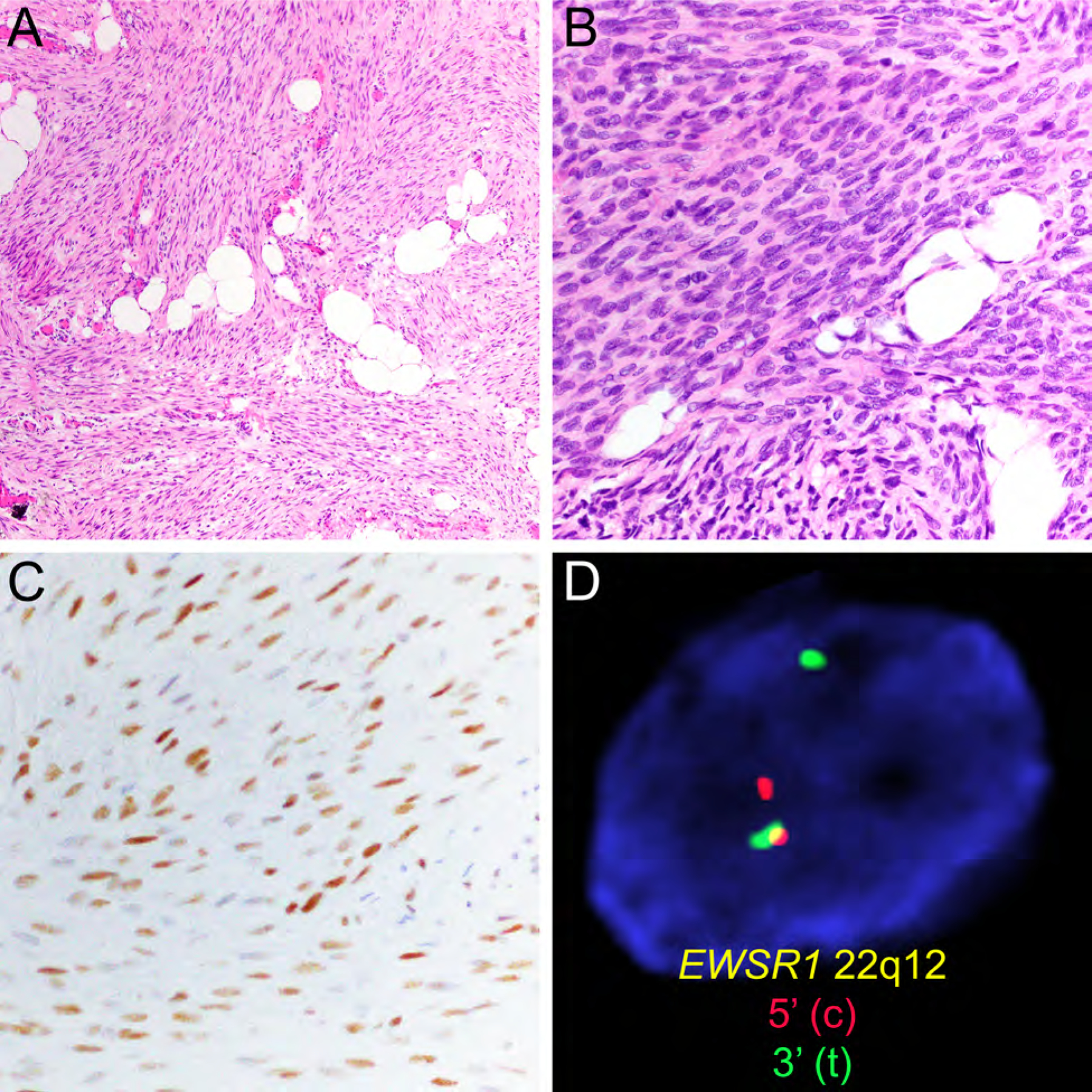
EWSR1-SMAD3-positive fibroblastic tumor with hypocellular hyalinized area (A, HE, x200) and more cellular areas containing fibroblastic spindle cells (B, HE, x400) with expression of ERG (C, x200) and presence of EWSR1 gene rearrangement detected by FISH (D).
Key Features: EWSR1-SMAD3-Positive Fibroblastic Tumor.
Acral location, superficial
Wide age range, F>M
Hypocellular center, hypercellular periphery
Expression of ERG
EWSR1-SMAD3 gene fusion
Benign behavior
Angiofibroma of Soft Tissue
Angiofibroma of soft tissue is a benign tumor composed of uniform bland spindle cells embedded in a network of prominent branching thin-walled blood vessels and fibromyxoid stroma with predilection for the lower extremities of middle-aged adults with slight female predominance.18–20 Expression of CD34 and EMA is variable.18,20 Angiofibroma of soft tissue is characterized by recurrent t(5;8)(p15;q13) resulting in AHRR-NCOA2 gene fusion found in 60–80% of cases.19–21 These tumors behave in a benign fashion with rare local recurrences and no risk of distant metastases.
Key Features: Angiofibroma of Soft Tissue.
Lower extremities
Middle-aged adults, F≥M
Bland spindle cells, network of branching blood vessels, fibromyxoid stroma
Variable expression of CD34 and EMA
AHRR-NCOA2 gene fusion
Benign behavior
Superficial CD34-Positive Fibroblastic Tumor
This distinctive low-grade neoplasm most frequently occurs in skin and subcutis of the lower extremities, especially the thigh, of middle-aged adults.22,23 Superficial CD34-positive fibroblastic tumor is well circumscribed and composed of large spindle cells with abundant eosinophilic cytoplasm (Fig. 5A), marked nuclear pleomorphism (Fig. 5B), but a low mitotic rate.20,23 They typically show strong, diffuse expression of CD34 (Fig. 5C) and focal cytokeratin in about two thirds of cases.20,23 Morphologic overlap exists with tumors described at PRDM10-rearranged soft tissue tumors.24 Superficial CD34-positive fibroblastic tumor has an excellent prognosis with no local recurrences reported and only one case with distant metastasis.22
Figure 5.
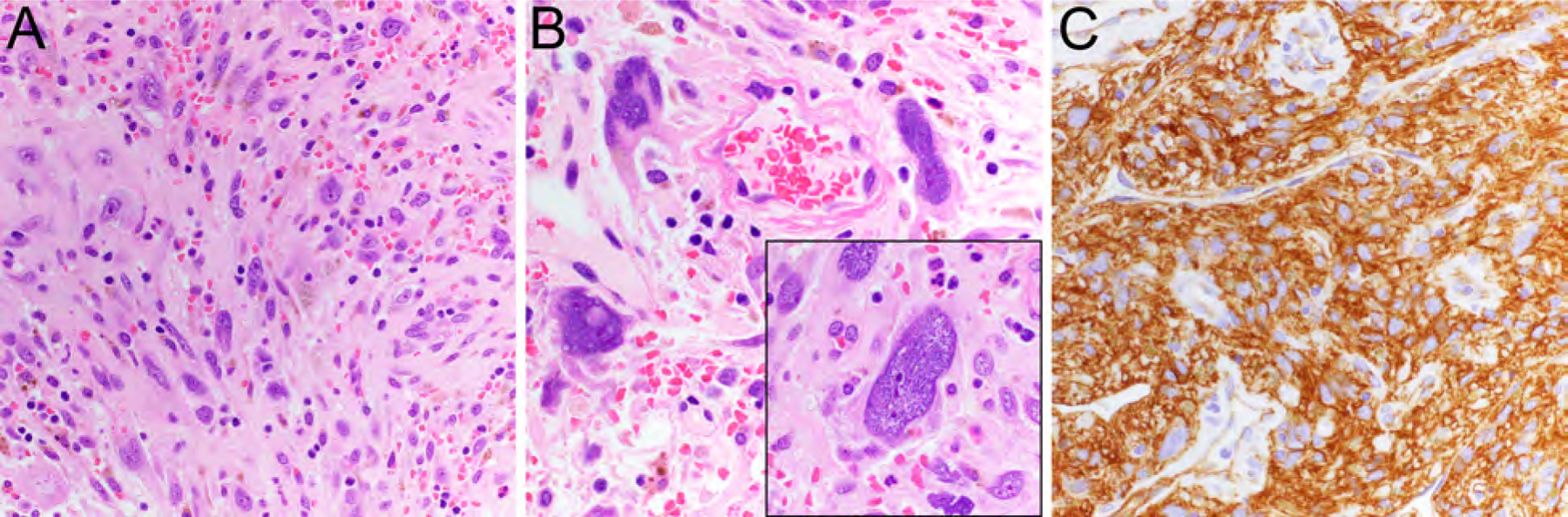
Superficial CD34-positive fibroblastic tumor with sheet-like growth (A, HE, x200) of large eosinophilic tumor cells with marked nuclear pleomorphism (B, HE, x400; inset) and strong, diffuse expression of CD34 (C, x400).
Key Features: Superficial CD34-Positive Fibroblastic Tumor.
Lower extremities, superficial
Middle-aged adults, M≥F
Eosinophilic tumor cells with granular-to-glassy cytoplasm; marked pleomorphism, few mitoses
Expression of CD34, focal cytokeratin
Low-grade behavior, no local recurrences and very low risk of distant metastasis
SMOOTH MUSCLE TUMORS
Inflammatory Leiomyosarcoma
Inflammatory leiomyosarcoma is very rare and most frequently arises in the lower extremity of adults with male predominance.1,25–27 Inflammatory leiomyosarcoma is characterized by eosinophilic spindle cells with blunt-ended elongated nuclei surrounded by a prominent inflammatory infiltrate, consisting mostly of small lymphocytes and occasionally admixed plasma cells or histiocytes with xanthomatous appearance (Fig. 6A, B). The tumor cells express SMA, desmin (Fig. 6C) and/or caldesmon. Inflammatory leiomyosarcoma shows a distinct near-haploid karyotype, with or without subsequent chromosome doubling.26–28 The prognosis appears to be very good, with metastases being documented in only a subset of cases, although long-term follow up data are limited.27,29
Figure 6.
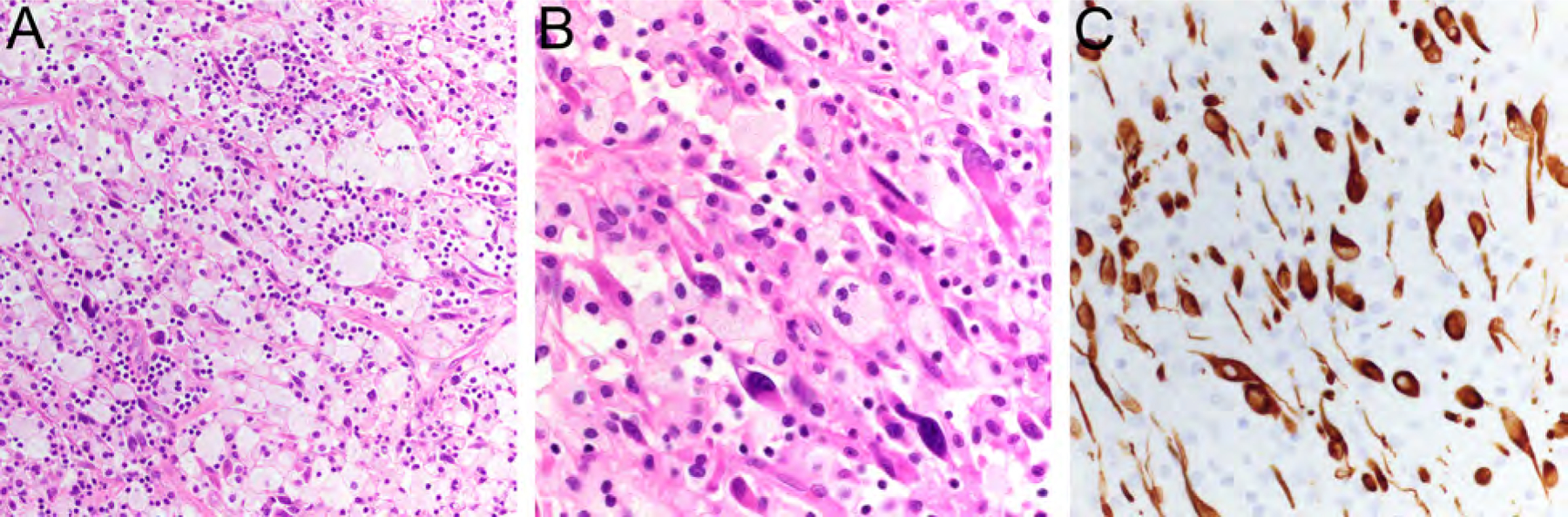
Inflammatory leiomyosarcoma containing spindle cells with blunt-ended elongated nuclei surrounded by a prominent inflammatory infiltrate, consisting of histiocytes with xanthomatous appearance (A, HE, x200; B, HE, x400). The tumor cells are highlighted by strong expression of desmin (C, x400).
Key Features: Inflammatory Leiomyosarcoma.
Lower extremity, trunk, retroperitoneum; deep-seated
Mostly adults, M>F
Eosinophilic spindle cells with blunt ended elongated nuclei; fascicular or storiform growth; mostly low grade
Prominent inflammatory infiltrate
Expression of SMA, desmin and/or caldesmon
Near-haploid karyotype
Good prognosis (data limited)
VASCULAR TUMORS
Anastomosing Hemangioma
Anastomosing hemangioma is most commonly found in the kidney and retroperitoneal adipose tissue, ovary, liver, and other anatomic locations affecting mostly adults and rarely children, without sex predilection.30–33 Anastomosing hemangioma is characterized by a hemorrhagic mahogany spongy appearance, loosely lobulated architecture, and can be associated with a medium-caliber vessel.32 Sinusoidal capillary-sized vessels (Fig. 7A) with scattered hobnail endothelial cells and a framework of nonendothelial supporting cells are characteristic.32 While mild cytologic atypia can be found, mitoses are usually rare or absent and multilayering is not seen, helping in the distinction from angiosarcoma.32,34 Vascular thrombi are frequent (Fig. 7B), extramedullary hematopoiesis and striking hyaline globules are found in a subset of cases.32 The endothelial tumor cells express CD34, CD31, and ERG.32 Recurrent GNAQ34,35 or GNA1434,36 activating mutations are characteristic of anastomosing hemangioma.
Figure 7.
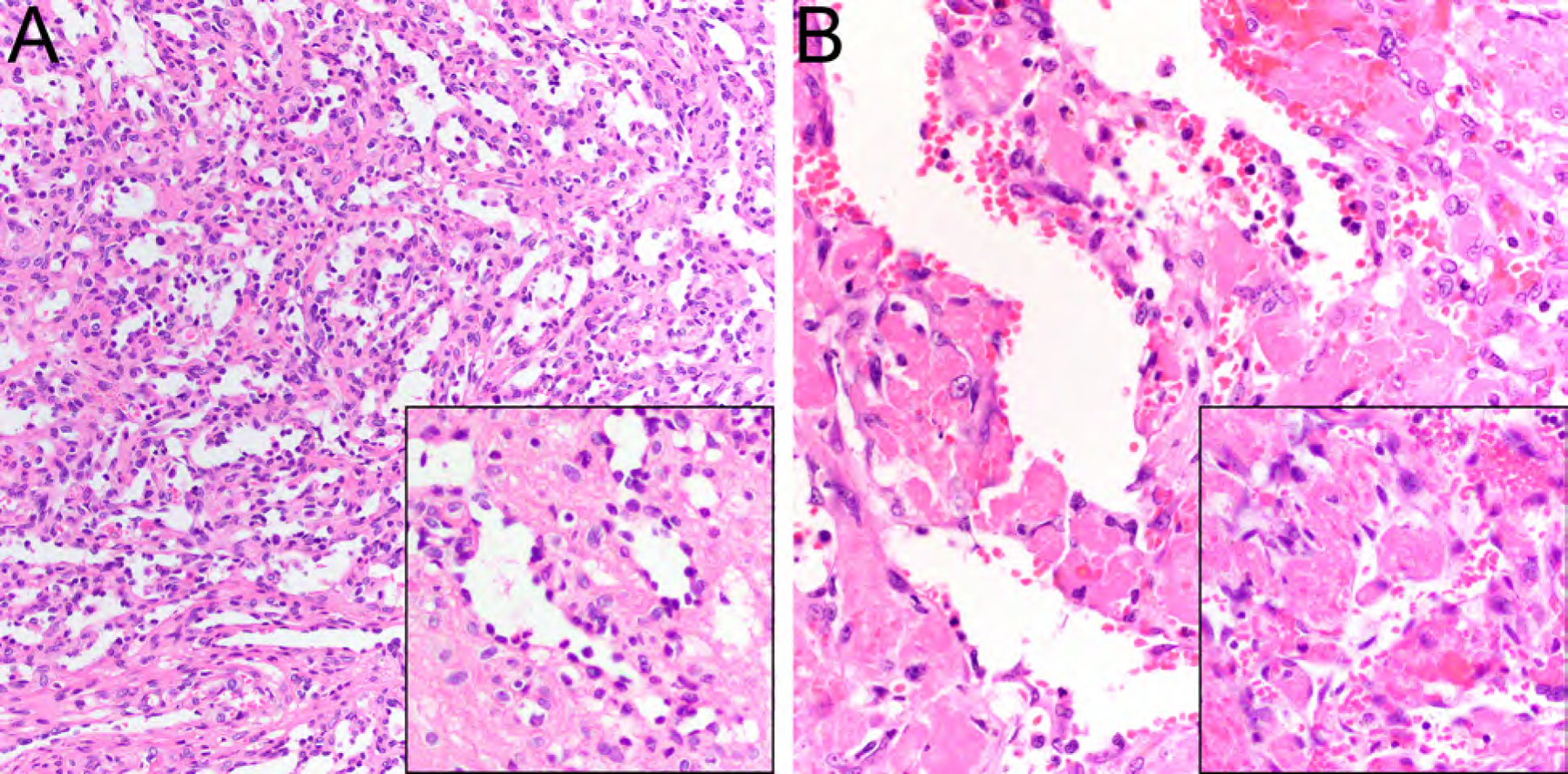
Anastomosing hemangioma consisting of anastomosing sinusoidal capillary-sized vessels (A, HE, x200) with scattered hobnail endothelial cells (A, inset, HE, x400) and vascular thrombi (B, HE, x200; inset, HE, x400).
Key Features: Anastomosing Hemangioma
Mostly kidney, retroperitoneum
Adults, F=M
Lobulated growth, anastomosing sinusoidal capillary-sized vessels with hobnailing endothelium; extramedullary hematopoiesis and hyaline globules in subset of cases
No/rare mitoses, no endothelial multilayering; mild cytologic atypia in some cases
Expression of vascular markers (CD34, CD31, ERG)
Recurrent GNAQ or GNA14 activating mutations
Benign
TUMORS OF UNCERTAIN DIFFERENTIATION
NTRK-Rearranged Spindle Cell Neoplasm (emerging)
NTRK-rearranged spindle cell neoplasm is a molecularly defined category of tumors (outside of infantile fibrosarcoma) which includes the recently described lipofibromatosis-like neural tumor and tumors that closely mimic peripheral nerve sheath tumor.37,38 Most cases harbor NTRK1 gene fusions with various partners (eg., LMNA, PTR, TPM3), and rarely NTRK2 or NRTK3 gene rearrangements representing potential treatment targets for inhibitors of the TRK family of kinases.39 NTRK-rearranged spindle cell neoplasm includes a wide range of morphologic appearances, usually consisting of a population of haphazardly arranged monomorphic spindle cell, distinctive stromal and perivascular keloidal collagen, and infiltration into adipose tissue (Fig. 8A–D).1 The tumor cells frequently express S100, CD34, and pan-TRK (Fig. 8C) but not SOX10.40–42 The prognosis appears to depend on histologic grade. Benign lipofibromatosis-like neural tumor shows infiltrative growth and has a propensity for local recurrence if incompletely excised, but does not metastasize.1,40 Distant metastases may be observed in tumors with high-grade morphologic features.1
Figure 8.
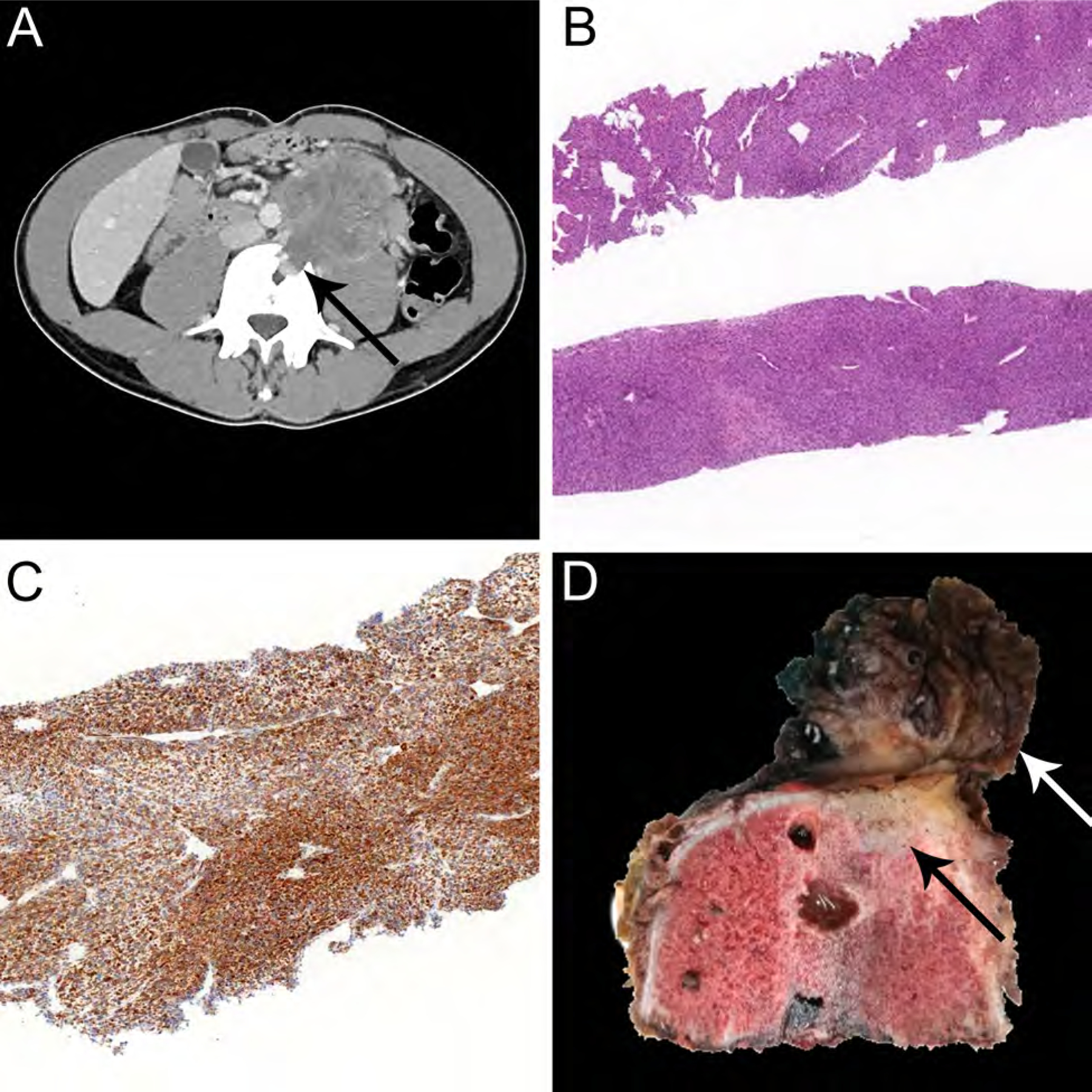
NTRK-rearranged spindle cell neoplasm arising in the retroperitoneum on CT-imaging (A) showing infiltration into adjacent vertebral bone (A, arrow). This tumor consisted of a population of haphazardly arranged monomorphic spindle cells (A, HE, x40) with positive staining for pan-TRK in tumor cells (C, x40) on a pre-treatment biopsy. The patient received neoadjuvant NTRK-inhibitor therapy for 4 months resulting in partial response, before the tumor was surgically resected. The post-treatment specimen (D) reveals residual tumor (white arrow) invading vertebral bone (black arrow).
Key Features: NTRK-Rearranged Spindle Cell Neoplasm
Molecularly defined category of tumors
Mostly children and young adults
Extremities and trunk, superficial or deep location
Wide morphologic range; monomorphic spindle cell, stromal/perivascular keloidal collagen, infiltrating fat
Expression of S100, CD34, and pan-TRK; SOX10-negative
NTRK1, NTRK2 or NTRK3 gene fusion
Wide prognostic range depending on tumor grade
NEW CATEGORY: UNDIFFERENTIATED ROUND CELL SARCOMAS OF BONE AND SOFT TISSUE
A new category has been introduced for “undifferentiated small round cell sarcomas of bone and soft tissue” which includes Ewing sarcoma, sarcoma with EWSR1-non-ETS fusions, CICrearranged sarcoma, and sarcoma with BCOR genetic alterations (Table 2).
Table 2.
Clinico-pathologic features of novel round cell sarcoma subtypes.
| Differential Diagnosis | ||
|---|---|---|
EWSR1-NFATC2 and FUS-NFATC2 Sarcoma
|
C/C-Rearranged Sarcoma
|
BCOR-Rearranged Sarcoma
|
EWSR1-PATZ1 Sarcoma
| ||
Sarcoma with EWSR1-non-ETS Fusions
This group of round and spindle cell sarcomas includes those with fusions of EWSR1 or FUS and fusion partners not belonging to the ETS gene family, specifically EWSR1-NFATC2 and EWSR1PATZ-1. EWSR1-NFATC2 and FUS-NFATC2 sarcomas have a predilection for long bones, and rare cases of EWSR1-NFATC2 sarcoma have also been reported in somatic soft tissue sites (Table 2).43–45 NFATC2-rearranged sarcomas occur in children and adults with male predominance.44,45 EWSR1-PATZ1 sarcomas arise in deep somatic soft tissue, are most common in the chest wall and abdomen, and occur over a wide age range without sex predilection1,46,47 In addition to the EWSR1-PATZ1 gene fusion, frequent inactivation of CDKN2A has been observed.47 NFATC2-rearranged sarcomas are composed of round cell and/or spindle cells with little cytoplasm in a fibrohyaline to myxohyaline background with expression of CD99 in about half of cases, PAX7, NKX2.2, and/or focal dot-like AE1/AE3 in a subset of cases.1 EWSR1-PATZ1 sarcomas consist of small round and/or spindled cells often with fibrous stroma (Fig. 9A, B), can express myogenic and neurogenic markers and CD34 to varying extent.1 NFATC2rearranged sarcomas and EWSR1-PATZ1 sarcomas show variable clinical behavior and may recur locally and/or develop distant metastases43,46–48, but long-term follow up data are limited. Both entities respond poorly to systemic chemotherapies.1
Figure 9.
Examples of EWSR1-PATZ1 sarcoma (A and B, HE, x400) consisting of small round to spindled cells with varying extent of fibrous stroma.
CIC-Rearranged Sarcoma
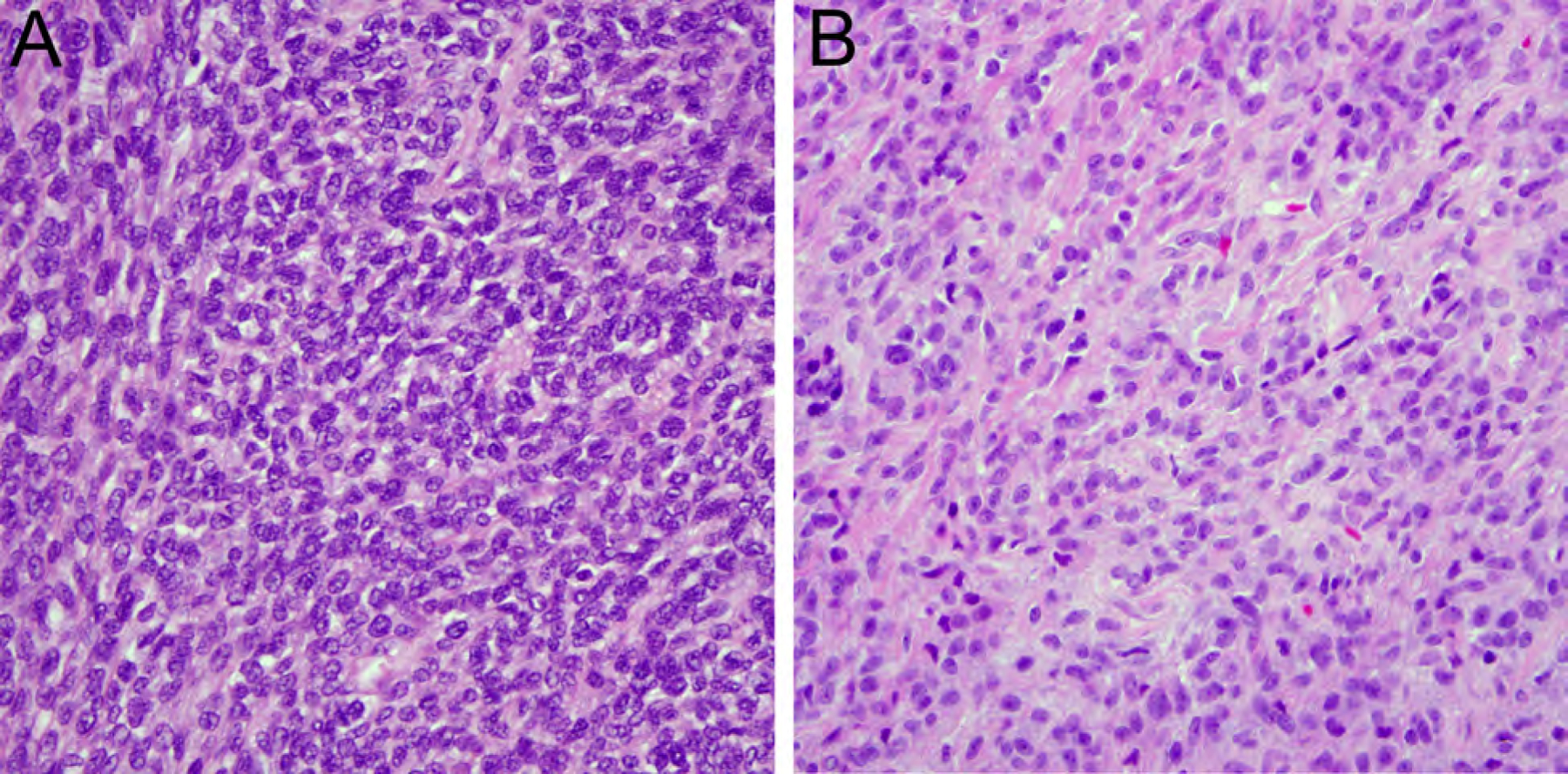
CIC-rearranged sarcoma mostly affects young male adults with predilection for the somatic soft tissue of trunk and extremities and behaves more aggressively than Ewing sarcoma (Table 2).49 CIC-rearranged sarcomas contain moderately pleomorphic round to ovoid cells with frequent mitoses, apoptoses, and necrosis (Fig. 10A), and show expression of WT1 (Fig. 10B) and ETV4 in most cases, whereas CD99 staining is usually limited.50,51 These tumors harbor characteristic
Figure 10.
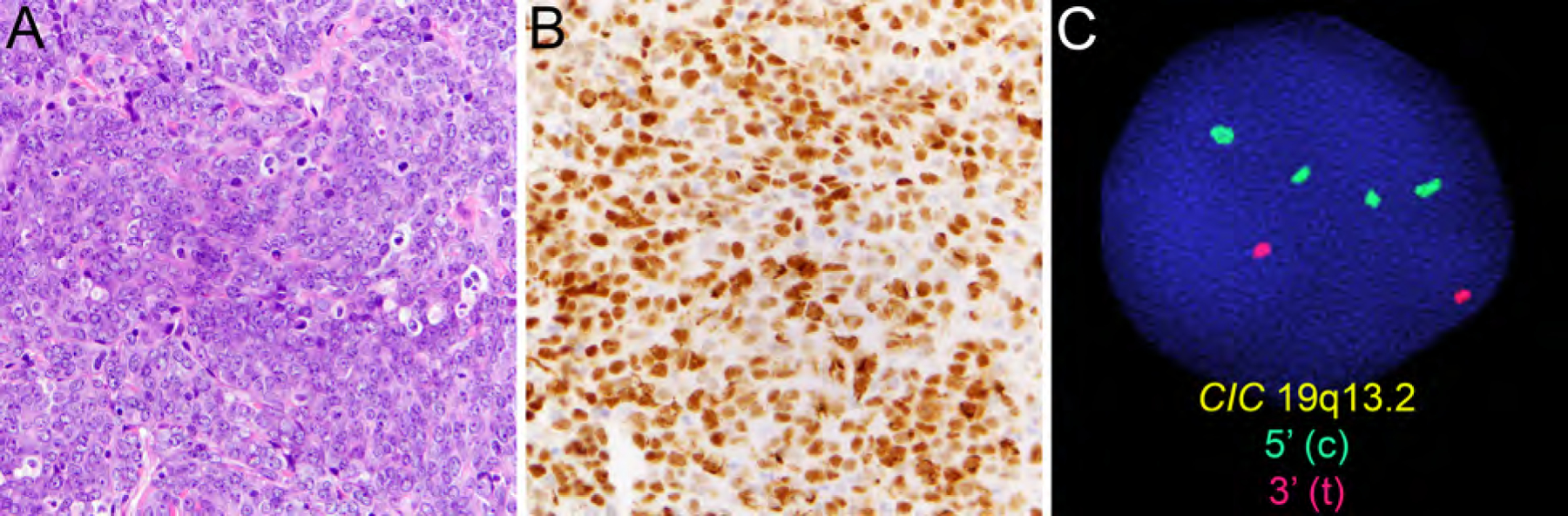
CIC-rearranged sarcoma containing moderately pleomorphic round to ovoid cells with frequent mitoses, apoptoses, and necrosis (A, HE, x400) with expression of WT1 (B, x400) and detection of CIC rearrangement by FISH (C).
CIC-DUX4 fusion resulting from t(4;19)(q35;q13) or t(10;19)(q26;q13) and rarely alternate CIC-FOXO4 fusion (Fig. 10C).52,53CIC-rearranged sarcomas are highly aggressive with frequent distant metastases and a poor prognosis.
Sarcoma with BCOR Genetic Alterations
This group of primitive round cell sarcomas includes BCOR-rearranged sarcoma and tumors with BCOR internal tandem duplication as described in infantile undifferentiated round cell sarcoma and primitive myxoid mesenchymal tumors of infancy. BCOR-rearranged sarcoma shows predilection for bone and soft tissue of male children54,55 and is characterized by BCOR-CCNB3 rearrangement resulting from inv(X)(p11) (Table 2) or rare, alternate rearrangement of BCOR with MAML3 or ZC3H7B.56 BCOR-rearranged sarcomas contain a uniform population of primitive small round to ovoid cells in solid sheets or nested, surrounded by a capillary network (Fig. 11A).55 Expression of BCOR and/or CCNB3 (Fig. 11B, C) can be helpful in the differential diagnostic workup but is not entirely specific.54,55,57 BCOR-rearranged sarcomas are less aggressive than CIC-rearranged sarcoma and have 5-year-overall survival rates of ~75%, comparable to Ewing sarcoma.55,58
Figure 11.
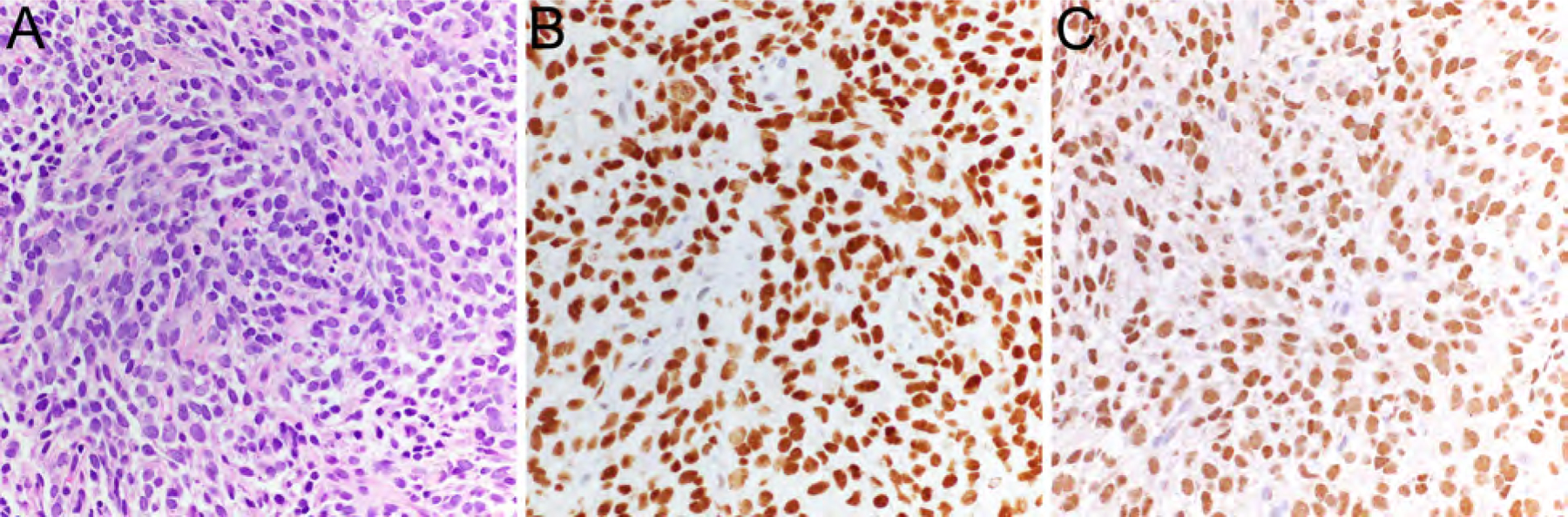
BCOR-rearranged sarcoma consisting of a uniform population of primitive small round to ovoid cells (A, HE, x400) with expression of BCOR (B, x400) and CCNB3 (C, x400).
REVISIONS TO NOMENCLATURE, GRADING, AND RISK STRATIFICATION
Select updates to concepts in nomenclature, grading, and risk stratification for malignant melanotic nerve sheath tumor, dedifferentiated liposarcoma, and solitary fibrous tumor (SFT) are summarized in Table 3. The 2020 WHO Classification discourages the terminology benign/malignant SFT and one of the (several) validated risk models is preferred. Please refer to chapter 9 for details on risk stratification for solitary fibrous tumor.
Table 3.
Select updates to concepts in nomenclature, grading, and risk stratification (modified from: Kallen ME, Hornick JL. The 2020 WHO Classification: What’s New in Soft Tissue Tumor Pathology? Am J Surg Pathol. 2021;45(1):e1-e23.).3
| Key Revision to Nomenclature, Grading, and Risk Stratification |
|---|
Malignant Melanotic Nerve Sheath Tumor (MMNST):
|
Dedifferentiated Liposarcoma (DDLPS):
|
Solitary fibrous tumor (SFT):
|
CHALLENGES IN THE CLASSIFICATION OF WELL-ESTABLISHED ENTITIES:
ATYPICAL LIPOMATOUS TUMOR/WELL-DIFFERENTIATED LIPOSARCOMA
Atypical lipomatous tumor (ALT)/well-differentiated liposarcoma (WDPLPS) is a locally aggressive adipocytic neoplasm consisting either partly of entirely of an adipocytic component with at least focal nuclear atypia in both adipocytes and stromal cells.1 ALT and WDLPS describe the same entity. However, by convention the term ALT is used when referring to tumors arising at anatomic sites amenable to complete surgical resection, such as the extremities or superficial locations. In contrast, WDLPS is used for lesions occurring at deep-seated, central body sites such as the retroperitoneum, for which radical multi-visceral surgery is considered appropriate and where risk of disease progression either due to local spread or dedifferentiation causing systemic spread tends to be higher.1,59 The overall risk of local recurrence for ALT/WDLPS ranges at 30–50%, but distant metastases virtually never occur (unless dedifferentiation develops). Anatomic location of ALT/WDLPS is the major prognostic factor: the 10-year to 20-year overall mortality rates vary by anatomic site and have been estimated as <2% for ALT arising in the extremities and >20% for WDLPS arising in the retroperitoneum.1
PROGNOSTIC IMPACT OF DIFFERENTIATION IN DEDIFFERENTIATED LIPOSARCOMA
Dedifferentiated liposarcoma (DDLPS) describes ALT/WDLPS that progressed to a non-lipogenic sarcoma.60 The non-lipogenic component is generally high-grade and may exhibit a broad spectrum of histologic grades. Morphologically low-grade non-lipogenic components consisting of areas with relatively bland-appearing spindle cells with intermediate cellularity have been recognized and are described as “low-grade dedifferentiation” to emphasize the morphologic difference from conventional “high-grade” components.1,61 Of note, “low grade” here represents a morphologic description (and not, for instance, to FNCLCC grading). The range of histologic appearances of high-grade non-lipogenic components in DDLPS is wide and occasionally, lipoblastic differentiation can be found in otherwise high-grade non-lipogenic areas which is termed “homologous” lipoblastic (pleomorphic liposarcoma-like) differentiation in analogy to the heterologous differentiation frequently observed in DDLPS.62 In addition, it has been demonstrated that myogenic, in particular rhabdomyoblastic, differentiation in DDLPS is associated with worse outcome, as is higher FNCLCC grade .63
RISK STRATIFICATION IN MYXOID LIPOSARCOMA
Myxoid liposarcoma is a malignant adipocytic neoplasm composed of uniform, round to ovoid cells with admixed small lipoblasts, prominent branching capillary vessels surrounded by a myxoid stroma, and characteristic FUS-DDIT3 (or rarely, EWSR1-DDIT3) gene fusion.1 As defined by the WHO Classification of Soft Tissue and Bone Tumors, myxoid liposarcoma is considered high-grade if >5% of the tumor consists of areas of hypercellularity often displaying round cell morphology, with reduced myxoid matrix, less prominent capillary vasculature, increased nuclear grade, increased mitotic activity, frequently with a chorded or trabecular architecture and round cell morphology. While high-grade liposarcoma can histologically mimic other round cell neoplasms and was therefore in the past termed “round-cell liposarcoma”, “high-grade” myxoid liposarcoma is now the widely accepted terminology and significantly hypercellular tumors behave as aggressively as those with round cell morphology. However, a morphologic spectrum exists without easily defined or reproducible cut-offs. The presence of high-grade features is associated with significantly poorer prognosis1 and presence and extent of hypercellularity should therefore be reported. As recently demonstrated, nuclear expression of DDIT3 can be observed in 96% of high-grade liposarcomas and may be helpful in the distinction from other round cell sarcoma.64
DIAGNOSTIC DISTINCTION OF MYXOFIBROSARCOMA AND UNDIFFERENTIATED PLEOMORPHIC SARCOMA
While myxofibrosarcoma and undifferentiated pleomorphic sarcoma may share certain overlapping features, their diagnostic distinction is generally made based on clinic-pathologic features (e.g., anatomic site and depth) and histomorphologic criteria summarized in Table 4
Table 4.
Clinico-pathologic features helpful in the distinction of myxofibrosarcoma and undifferentiated pleomorphic sarcoma.
| Differential Diagnosis | |
|---|---|
Myxofibrosarcoma
|
Undifferentiated pleomorphic sarcoma
|
Myxofibrosarcoma is a malignant fibroblastic neoplasm characterized by multinodular growth, myxoid stroma, pleomorphism and distinctive curvilinear blood vessels occurring mostly in the lower extremity of adults with slight male predominance.1,65 More than half of cases arise in dermal/subcutaneous soft tissue, whereas the remainder arise in fascia or deep skeletal muscle.1,65 The histomorphologic spectrum is wide, but all cases share the features listed above and the diagnosis is generally based on histologic criteria since specific immunohistochemical markers or genetic markers are absent.1 Myxofibrosarcoma demonstrates highly complex karyotypes with intratumor heterogeneity and triploid or triploid chromosome numbers.1 Given their infiltrative growth, local recurrence occurs in 30–50% of cases, and distant metastases develop in 20–35% of cases.1,65
In contrast, undifferentiated pleomorphic sarcoma, belonging to the group of “undifferentiated sarcomas”, represents a diagnosis of exclusion given the absence of an identifiable line of differentiation as determined by histologic examination and available ancillary techniques.1,66 Undifferentiated pleomorphic sarcoma usually affects adults, being most common between 50–70 years of age, and is usually deep-seated.66 These sarcomas closely resemble other types of pleomorphic sarcomas and often show a patternless appearance with frequent bizarre multinucleated giant cells. Their karyotypes are complex without distinctive recurrent genetic aberrations. While long-term follow-up data are relatively limited, the 5-year metastasis-free survival for patients with undifferentiated sarcomas arising in limbs or trunk among the group of pleomorphic sarcomas in adults has been reported as 83%.67
SUMMARY
The 2020 WHO Classification of Soft Tissue and Bone Tumors includes several novel and emerging entities and features a number of revisions to existing classification and risk stratification schemes to incorporate recent advances in the diagnostic workup of these tumors. This update integrates a morphology-based approach combined with evaluation for characteristics cytogenetic/molecular genetic alterations and associated immunohistochemical markers to improve diagnostic precision, reproducibility, and prognostication for state-of-the-art clinical management.
KEY POINTS.
The 2020 WHO Classification of Soft Tissue and Bone Tumors has incorporated a number of changes to reflect recent advances made in the histopathologic and molecular diagnostic workup of soft tissue tumors.
New entities have been added to the categories of adipocytic tumors, fibroblastic and myofibroblastic tumors, smooth muscle tumors, vascular tumors, and tumors of uncertain differentiation.
A new category has been introduced for “undifferentiated round cell sarcomas of bone and soft tissue” which includes Ewing sarcoma, round cell sarcoma with EWSR1-non-ETS gene fusion, CIC-rearranged sarcoma, and sarcoma with BCOR genetic alterations.
EWSR1-SMAD3-positive fibroblastic tumor and NTRK-rearranged spindle cell sarcoma have been included as emerging entities.
Revisions to nomenclature, grading, and risk stratification have been made for malignant melanotic nerve sheath tumor, dedifferentiated liposarcoma, and solitary fibrous tumor.
SYNOPSIS.
The 2020 WHO Classification of Soft Tissue and Bone Tumors features revisions based on recent advances in the histopathologic and molecular diagnostic workup of soft tissue tumors. We herein highlight select new entities in the categories of adipocytic tumors, fibroblastic and myofibroblastic tumors, smooth muscle tumors, vascular tumors, and tumors of uncertain differentiation, a novel category for undifferentiated round cell sarcomas of bone and soft tissue, and revisions to nomenclature, grading, and risk stratification. This paper provides an overview on revised diagnostic criteria, state-of-the-art genetic and immunohistochemical markers, and prognostication with impact on clinical management. In addition, we discuss challenging aspects in the diagnosis and/or prognostication of select well-established entities that will be discussed in more detail in other chapters of this book.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (NIH)/National Cancer Institute (NCI) K08 CA241085 grant (I.-M. Schaefer) and SARC (Sarcoma Alliance for Research through Collaboration) (I.-M. Schaefer). The authors thank Dr. Christopher D. M. Fletcher, Dr. Esther Baranov, and the Division for Cytogenetics, Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, for contributing some of the cases illustrated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST DISCLOSURE
The authors have nothing to disclose.
References
- 1.WHO Classification of Tumours Editorial Board. WHO Classification of Tumours: Soft Tissue and Bone Tumours. Vol 3. 5th ed. IARC Press, Lyon: World Health Organization; 2020. [Google Scholar]
- 2.Fletcher C, Bridge JA, Hogendoorn PCW, Mertens F. WHO Classification of Tumours of Soft Tissue and Bone. IARC Press, Lyon; 2013. [Google Scholar]
- 3.Kallen ME, Hornick JL. The 2020 WHO Classification: What’s New in Soft Tissue Tumor Pathology? Am J Surg Pathol. 2021;45(1):e1–e23. [DOI] [PubMed] [Google Scholar]
- 4.Marino-Enriquez A, Nascimento AF, Ligon AH, Liang C, Fletcher CD. Atypical Spindle Cell Lipomatous Tumor: Clinicopathologic Characterization of 232 Cases Demonstrating a Morphologic Spectrum. Am J Surg Pathol. 2017;41(2):234–244. [DOI] [PubMed] [Google Scholar]
- 5.Creytens D, van Gorp J, Savola S, Ferdinande L, Mentzel T, Libbrecht L. Atypical spindle cell lipoma: a clinicopathologic, immunohistochemical, and molecular study emphasizing its relationship to classical spindle cell lipoma. Virchows Arch. 2014;465(1):97–108. [DOI] [PubMed] [Google Scholar]
- 6.Mentzel T, Palmedo G, Kuhnen C. Well-differentiated spindle cell liposarcoma (‘atypical spindle cell lipomatous tumor’) does not belong to the spectrum of atypical lipomatous tumor but has a close relationship to spindle cell lipoma: clinicopathologic, immunohistochemical, and molecular analysis of six cases. Mod Pathol. 2010;23(5):729–736. [DOI] [PubMed] [Google Scholar]
- 7.Bahadir B, Behzatoglu K, Hacihasanoglu E, Koca SB, Sigirci BB, Tokat F. Atypical spindle cell/pleomorphic lipomatous tumor: A clinicopathologic, immunohistochemical, and molecular study of 20 cases. Pathol Int. 2018;68(10):550–556. [DOI] [PubMed] [Google Scholar]
- 8.Alaggio R, Coffin CM, Weiss SW, et al. Liposarcomas in young patients: a study of 82 cases occurring in patients younger than 22 years of age. Am J Surg Pathol. 2009;33(5):645–658. [DOI] [PubMed] [Google Scholar]
- 9.Coffin CM, Alaggio R. Adipose and myxoid tumors of childhood and adolescence. Pediatr Dev Pathol. 2012;15(1 Suppl):239–254. [DOI] [PubMed] [Google Scholar]
- 10.Boland JM, Colby TV, Folpe AL. Liposarcomas of the mediastinum and thorax: a clinicopathologic and molecular cytogenetic study of 24 cases, emphasizing unusual and diverse histologic features. Am J Surg Pathol. 2012;36(9):1395–1403. [DOI] [PubMed] [Google Scholar]
- 11.Creytens D, van Gorp J, Ferdinande L, Van Roy N, Libbrecht L. Array-based comparative genomic hybridization analysis of a pleomorphic myxoid liposarcoma. J Clin Pathol. 2014;67(9):834–835. [DOI] [PubMed] [Google Scholar]
- 12.Hofvander J, Jo VY, Ghanei I, Gisselsson D, Martensson E, Mertens F. Comprehensive genetic analysis of a paediatric pleomorphic myxoid liposarcoma reveals near-haploidization and loss of the RB1 gene. Histopathology. 2016;69(1):141–147. [DOI] [PubMed] [Google Scholar]
- 13.Creytens D, Folpe AL, Koelsche C, et al. Myxoid pleomorphic liposarcoma-a clinicopathologic, immunohistochemical, molecular genetic and epigenetic study of 12 cases, suggesting a possible relationship with conventional pleomorphic liposarcoma. Mod Pathol. 2021. In Press. [DOI] [PubMed] [Google Scholar]
- 14.Kao YC, Flucke U, Eijkelenboom A, et al. Novel EWSR1-SMAD3 Gene Fusions in a Group of Acral Fibroblastic Spindle Cell Neoplasms. Am J Surg Pathol. 2018;42(4):522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michal M, Berry RS, Rubin BP, et al. EWSR1-SMAD3-rearranged Fibroblastic Tumor: An Emerging Entity in an Increasingly More Complex Group of Fibroblastic/Myofibroblastic Neoplasms. Am J Surg Pathol. 2018;42(10):1325–1333. [DOI] [PubMed] [Google Scholar]
- 16.Habeeb O, Korty KE, Azzato EM, et al. EWSR1-SMAD3 rearranged fibroblastic tumor: Case series and review. J Cutan Pathol. 2021;48(2):255–262. [DOI] [PubMed] [Google Scholar]
- 17.Foot O, Hallin M, Jones RL, Sumathi VP, Thway K. EWSR1-SMAD3-Positive Fibroblastic Tumor. Int J Surg Pathol. 2021;29(2):179–181. [DOI] [PubMed] [Google Scholar]
- 18.Marino-Enriquez A, Fletcher CD. Angiofibroma of soft tissue: clinicopathologic characterization of a distinctive benign fibrovascular neoplasm in a series of 37 cases. Am J Surg Pathol. 2012;36(4):500–508. [DOI] [PubMed] [Google Scholar]
- 19.Yamada Y, Yamamoto H, Kohashi K, et al. Histological spectrum of angiofibroma of soft tissue: histological and genetic analysis of 13 cases. Histopathology. 2016;69(3):459–469. [DOI] [PubMed] [Google Scholar]
- 20.Bekers EM, Groenen P, Verdijk MAJ, et al. Soft tissue angiofibroma: Clinicopathologic, immunohistochemical and molecular analysis of 14 cases. Genes Chromosomes Cancer. 2017;56(10):750–757. [DOI] [PubMed] [Google Scholar]
- 21.Jin Y, Moller E, Nord KH, et al. Fusion of the AHRR and NCOA2 genes through a recurrent translocation t(5;8)(p15;q13) in soft tissue angiofibroma results in upregulation of aryl hydrocarbon receptor target genes. Genes Chromosomes Cancer. 2012;51(5):510–520. [DOI] [PubMed] [Google Scholar]
- 22.Carter JM, Weiss SW, Linos K, DiCaudo DJ, Folpe AL. Superficial CD34-positive fibroblastic tumor: report of 18 cases of a distinctive low-grade mesenchymal neoplasm of intermediate (borderline) malignancy. Mod Pathol. 2014;27(2):294–302. [DOI] [PubMed] [Google Scholar]
- 23.Lao IW, Yu L, Wang J. Superficial CD34-positive fibroblastic tumour: a clinicopathological and immunohistochemical study of an additional series. Histopathology. 2017;70(3):394–401. [DOI] [PubMed] [Google Scholar]
- 24.Puls F, Pillay N, Fagman H, et al. PRDM10-rearranged Soft Tissue Tumor: A Clinicopathologic Study of 9 Cases. Am J Surg Pathol. 2019;43(4):504–513. [DOI] [PubMed] [Google Scholar]
- 25.Merchant W, Calonje E, Fletcher CD. Inflammatory leiomyosarcoma: a morphological subgroup within the heterogeneous family of so-called inflammatory malignant fibrous histiocytoma. Histopathology. 1995;27(6):525–532. [DOI] [PubMed] [Google Scholar]
- 26.Chang A, Schuetze SM, Conrad EU 3rd, Swisshelm KL, Norwood TH, Rubin BP. So-called “inflammatory leiomyosarcoma”: a series of 3 cases providing additional insights into a rare entity. Int J Surg Pathol. 2005;13(2):185–195. [DOI] [PubMed] [Google Scholar]
- 27.Arbajian E, Koster J, Vult von Steyern F, Mertens F. Inflammatory leiomyosarcoma is a distinct tumor characterized by near-haploidization, few somatic mutations, and a primitive myogenic gene expression signature. Mod Pathol. 2018;31(1):93–100. [DOI] [PubMed] [Google Scholar]
- 28.Nord KH, Paulsson K, Veerla S, et al. Retained heterodisomy is associated with high gene expression in hyperhaploid inflammatory leiomyosarcoma. Neoplasia. 2012;14(9):807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michal M, Rubin BP, Kazakov DV, et al. Inflammatory leiomyosarcoma shows frequent co-expression of smooth and skeletal muscle markers supporting a primitive myogenic phenotype: a report of 9 cases with a proposal for reclassification as low-grade inflammatory myogenic tumor. Virchows Arch. 2020;477(2):219–230. [DOI] [PubMed] [Google Scholar]
- 30.O’Neill AC, Craig JW, Silverman SG, Alencar RO. Anastomosing hemangiomas: locations of occurrence, imaging features, and diagnosis with percutaneous biopsy. Abdom Radiol (NY). 2016;41(7):1325–1332. [DOI] [PubMed] [Google Scholar]
- 31.Brown JG, Folpe AL, Rao P, et al. Primary vascular tumors and tumor-like lesions of the kidney: a clinicopathologic analysis of 25 cases. Am J Surg Pathol. 2010;34(7):942–949. [DOI] [PubMed] [Google Scholar]
- 32.Montgomery E, Epstein JI. Anastomosing hemangioma of the genitourinary tract: a lesion mimicking angiosarcoma. Am J Surg Pathol. 2009;33(9):1364–1369. [DOI] [PubMed] [Google Scholar]
- 33.Caballes AB, Abelardo AD, Farolan MJ, Veloso JAD. Pediatric Anastomosing Hemangioma: Case Report and Review of Renal Vascular Tumors in Children. Pediatr Dev Pathol. 2019;22(3):269–275. [DOI] [PubMed] [Google Scholar]
- 34.Joseph NM, Brunt EM, Marginean C, et al. Frequent GNAQ and GNA14 Mutations in Hepatic Small Vessel Neoplasm. Am J Surg Pathol. 2018;42(9):1201–1207. [DOI] [PubMed] [Google Scholar]
- 35.Bean GR, Joseph NM, Gill RM, Folpe AL, Horvai AE, Umetsu SE. Recurrent GNAQ mutations in anastomosing hemangiomas. Mod Pathol. 2017;30(5):722–727. [DOI] [PubMed] [Google Scholar]
- 36.Bean GR, Joseph NM, Folpe AL, Horvai AE, Umetsu SE. Recurrent GNA14 mutations in anastomosing haemangiomas. Histopathology. 2018;73(2):354–357. [DOI] [PubMed] [Google Scholar]
- 37.Suurmeijer AJH, Dickson BC, Swanson D, et al. A novel group of spindle cell tumors defined by S100 and CD34 co-expression shows recurrent fusions involving RAF1, BRAF, and NTRK1/2 genes. Genes Chromosomes Cancer. 2018;57(12):611–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kao YC, Suurmeijer AJH, Argani P, et al. Soft tissue tumors characterized by a wide spectrum of kinase fusions share a lipofibromatosis-like neural tumor pattern. Genes Chromosomes Cancer. 2020;59(10):575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drilon A TRK inhibitors in TRK fusion-positive cancers. Ann Oncol. 2019;30(Supplement_8):viii23–viii30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agaram NP, Zhang L, Sung YS, et al. Recurrent NTRK1 Gene Fusions Define a Novel Subset of Locally Aggressive Lipofibromatosis-like Neural Tumors. Am J Surg Pathol. 2016;40(10):1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis JL, Lockwood CM, Stohr B, et al. Expanding the Spectrum of Pediatric NTRK-rearranged Mesenchymal Tumors. Am J Surg Pathol. 2019;43(4):435–445. [DOI] [PubMed] [Google Scholar]
- 42.Hung YP, Fletcher CDM, Hornick JL. Evaluation of pan-TRK immunohistochemistry in infantile fibrosarcoma, lipofibromatosis-like neural tumour and histological mimics. Histopathology. 2018;73(4):634–644. [DOI] [PubMed] [Google Scholar]
- 43.Diaz-Perez JA, Nielsen GP, Antonescu C, Taylor MS, Lozano-Calderon SA, Rosenberg AE. EWSR1/FUS-NFATc2 rearranged round cell sarcoma: clinicopathological series of 4 cases and literature review. Hum Pathol. 2019;90:45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bode-Lesniewska B, Fritz C, Exner GU, Wagner U, Fuchs B. EWSR1-NFATC2 and FUS-NFATC2 Gene Fusion-Associated Mesenchymal Tumors: Clinicopathologic Correlation and Literature Review. Sarcoma. 2019;2019:9386390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perret R, Escuriol J, Velasco V, et al. NFATc2-rearranged sarcomas: clinicopathologic, molecular, and cytogenetic study of 7 cases with evidence of AGGRECAN as a novel diagnostic marker. Mod Pathol. 2020;33(10):1930–1944. [DOI] [PubMed] [Google Scholar]
- 46.Chougule A, Taylor MS, Nardi V, et al. Spindle and Round Cell Sarcoma With EWSR1-PATZ1 Gene Fusion: A Sarcoma With Polyphenotypic Differentiation. Am J Surg Pathol. 2019;43(2):220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bridge JA, Sumegi J, Druta M, et al. Clinical, pathological, and genomic features of EWSR1-PATZ1 fusion sarcoma. Mod Pathol. 2019;32(11):1593–1604. [DOI] [PubMed] [Google Scholar]
- 48.Wang GY, Thomas DG, Davis JL, et al. EWSR1-NFATC2 Translocation-associated Sarcoma Clinicopathologic Findings in a Rare Aggressive Primary Bone or Soft Tissue Tumor. Am J Surg Pathol. 2019;43(8):1112–1122. [DOI] [PubMed] [Google Scholar]
- 49.Antonescu CR, Owosho AA, Zhang L, et al. Sarcomas With CIC-rearrangements Are a Distinct Pathologic Entity With Aggressive Outcome: A Clinicopathologic and Molecular Study of 115 Cases. Am J Surg Pathol. 2017;41(7):941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Specht K, Sung YS, Zhang L, Richter GH, Fletcher CD, Antonescu CR. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosomes Cancer. 2014;53(7):622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hung YP, Fletcher CD, Hornick JL. Evaluation of ETV4 and WT1 expression in CIC-rearranged sarcomas and histologic mimics. Mod Pathol. 2016;29(11):1324–1334. [DOI] [PubMed] [Google Scholar]
- 52.Sugita S, Arai Y, Tonooka A, et al. A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: a genetically distinct variant of Ewing-like sarcoma. Am J Surg Pathol. 2014;38(11):1571–1576. [DOI] [PubMed] [Google Scholar]
- 53.Solomon DA, Brohl AS, Khan J, Miettinen M. Clinicopathologic features of a second patient with Ewing-like sarcoma harboring CIC-FOXO4 gene fusion. Am J Surg Pathol. 2014;38(12):1724–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. 2012;44(4):461–466. [DOI] [PubMed] [Google Scholar]
- 55.Kao YC, Owosho AA, Sung YS, et al. BCOR-CCNB3 Fusion Positive Sarcomas: A Clinicopathologic and Molecular Analysis of 36 Cases With Comparison to Morphologic Spectrum and Clinical Behavior of Other Round Cell Sarcomas. Am J Surg Pathol. 2018;42(5):604–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Specht K, Zhang L, Sung YS, et al. Novel BCOR-MAML3 and ZC3H7B-BCOR Gene Fusions in Undifferentiated Small Blue Round Cell Sarcomas. Am J Surg Pathol. 2016;40(4):433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kao YC, Sung YS, Zhang L, et al. BCOR Overexpression Is a Highly Sensitive Marker in Round Cell Sarcomas With BCOR Genetic Abnormalities. Am J Surg Pathol. 2016;40(12):1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cohen-Gogo S, Cellier C, Coindre JM, et al. Ewing-like sarcomas with BCOR-CCNB3 fusion transcript: a clinical, radiological and pathological retrospective study from the Societe Francaise des Cancers de L’Enfant. Pediatr Blood Cancer. 2014;61(12):2191–2198. [DOI] [PubMed] [Google Scholar]
- 59.Gronchi A, Lo Vullo S, Fiore M, et al. Aggressive surgical policies in a retrospectively reviewed single-institution case series of retroperitoneal soft tissue sarcoma patients. J Clin Oncol. 2009;27(1):24–30. [DOI] [PubMed] [Google Scholar]
- 60.Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3(6):507–523. [DOI] [PubMed] [Google Scholar]
- 61.Elgar F, Goldblum JR. Well-differentiated liposarcoma of the retroperitoneum: a clinicopathologic analysis of 20 cases, with particular attention to the extent of low-grade dedifferentiation. Mod Pathol. 1997;10(2):113–120. [PubMed] [Google Scholar]
- 62.Marino-Enriquez A, Fletcher CD, Dal Cin P, Hornick JL. Dedifferentiated liposarcoma with “homologous” lipoblastic (pleomorphic liposarcoma-like) differentiation: clinicopathologic and molecular analysis of a series suggesting revised diagnostic criteria. Am J Surg Pathol. 2010;34(8):1122–1131. [DOI] [PubMed] [Google Scholar]
- 63.Gronchi A, Collini P, Miceli R, et al. Myogenic differentiation and histologic grading are major prognostic determinants in retroperitoneal liposarcoma. Am J Surg Pathol. 2015;39(3):383–393. [DOI] [PubMed] [Google Scholar]
- 64.Baranov E, Black MA, Fletcher CDM, Charville GW, Hornick JL. Nuclear expression of DDIT3 distinguishes high-grade myxoid liposarcoma from other round cell sarcomas. Mod Pathol. 2021;34(7):1367–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mentzel T, Calonje E, Wadden C, et al. Myxofibrosarcoma. Clinicopathologic analysis of 75 cases with emphasis on the low-grade variant. Am J Surg Pathol. 1996;20(4):391–405. [DOI] [PubMed] [Google Scholar]
- 66.Widemann BC, Italiano A. Biology and Management of Undifferentiated Pleomorphic Sarcoma, Myxofibrosarcoma, and Malignant Peripheral Nerve Sheath Tumors: State of the Art and Perspectives. J Clin Oncol. 2018;36(2):160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fletcher CD, Gustafson P, Rydholm A, Willen H, Akerman M. Clinicopathologic reevaluation of 100 malignant fibrous histiocytomas: prognostic relevance of subclassification. J Clin Oncol. 2001;19(12):3045–3050. [DOI] [PubMed] [Google Scholar]