

SUMMARY
Information in mRNA has largely been thought to be confined to its nucleotide sequence. However, the advent of mapping techniques to detect modified nucleotides has revealed that mRNA contains additional information in the form of chemical modifications. The most abundant modified nucleotide is N6-methyladenosine (m6A), a methyl modification of adenosine. Although early studies viewed m6A as a dynamic and tissue-specific modification, it is now clear that the mRNAs that contain m6A and the location of m6A in those transcripts are largely universal, and are influenced by gene architecture, i.e., the size and location of exons and introns. m6A can affect nuclear processes such as splicing and epigenetic regulation, but the major effect of m6A on mRNAs is to promote degradation in the cytoplasm. m6A marks a functionally related cohort of mRNAs linked to certain biological processes, including cell differentiation and cell fate determination. m6A is also enriched in other cohorts of mRNAs and can therefore affect their respective cellular processes and pathways. Future work will focus on understanding how the m6A pathway is regulated to achieve control of m6A-containing mRNAs.
eTOC blurb
In this review, Murakami et. al. discuss the recently recognized concept of epitranscriptomic regulation with a focus on N6-methyladenosine (m6A), the most abundant internal mRNA modification. They describe the early views of m6A, how these views have changed, and highlight emerging concepts that fundamentally change our understanding of how m6A functions.
INTRODUCTION
The search for regulatory elements in mRNA has primarily focused on sequence-specific RNA-binding proteins, which influence processes such as splicing, mRNA stability, and translation. However, some of the regulatory elements in mRNA are not evident simply by looking at the sequence. Instead, they are encoded by chemical modifications of nucleotides in mRNA. The most abundant chemically modified nucleotide in mRNA is N6-methyladenosine (m6A), which is also found in ribosomal RNA (rRNA) and small nuclear RNA (snRNA). m6A was identified as a component of mRNA in the 1970s (Desrosiers et al., 1974; Perry and Kelley, 1974), but interest in the functional and physiological roles of m6A, especially in mRNA, was stimulated by a study in Arabidopsis. In this study, the plant homolog of the mammalian m6A-synthesizing enzyme METTL3 was depleted, resulting in marked developmental defects (Zhong et al., 2008). These pioneering studies revealed the first phenotypes of m6A depletion in a multicellular organism, and demonstrated that m6A has key roles in development.
Over the past 10 years, with the advent of transcriptome-wide mapping of m6A and the identification of m6A readers, a variety of functions and mechanisms of m6A have been identified. Highly divergent models have been proposed to explain how mRNAs are selected for methylation and the effect of m6A on mRNAs (Zaccara et al., 2019). Recently, the answers to these questions have become much clearer, resulting in a more coherent model for the role of m6A in gene expression.
The major question is the function of m6A on mRNAs that contain it. It is clear that m6A has a major effects on splicing in a small number of transcripts (Wei et al., 2021). m6A can also influence epigenetic silencing, e.g. in the case of endogenous retrotransposons (Liu et al., 2021; Xu et al., 2021). However, the major function of m6A is to confer mRNA instability (Lasman et al., 2020; Zaccara and Jaffrey, 2020).
An early concept was that m6A is a dynamic modification (Roundtree et al., 2017a). In this model, an mRNA could be methylated and demethylated during its lifecycle, much like phosphorylation and dephosphorylation of a protein. The transcriptome-wide distribution of m6A was also described as tissue specific, thus potentially explaining tissue-specific patterns of gene expression (Zhang et al., 2021). However, as described below, it is likely that the transcriptome-wide distribution of m6A is largely “hard-wired” by the gene architecture (i.e., length and distribution of exons and introns) of each gene. Although a subset of m6A sites might be variable, the majority of m6A sites are found in the same transcripts in the same location in every tissue (Garcia-Campos et al., 2019).
The major implication of the hard-wired distribution of m6A is that m6A marks a specific cohort of mRNAs whose stability can be jointly regulated. Many of the mRNAs that contain multiple annotated m6A sites (e.g. four or more m6A sites) are linked to pathways that control cell fate, differentiation, and morphogenesis (Ries et al., 2019). These results are consistent with earlier studies in yeast and Arabidopsis which showed m6A deficiency leads to developmental defects (Agarwala et al., 2012; Zhong et al., 2008), as well as subsequent studies which showed that m6A controls embryonic stem cell differentiation (Batista et al., 2014; Geula et al., 2015; Wang et al., 2014). Thus, the ability of m6A to influence development is evolutionarily conserved.
m6A can regulate biology by enhancing degradation of specific transcripts that play central roles in the cellular pathways. In many cases, m6A-containing mRNAs are master regulators of specific signaling pathways. m6A-mediated degradation of these mRNAs can alter signaling or transcription, leading to indirect increases or decreases in gene expression. For example, m6A residues are found on mRNAs encoding the tumor suppressor APC and transcription factors Nanog and Klf4. m6A-mediated repression of APC attenuates cancer progression (Wang et al., 2021a), while the decreased expression of Nanog and Klf4 enables differentiation of naïve embryonic stem cells (Geula et al., 2015).
Here we describe how the current views of m6A have emerged and we discuss the outstanding questions about m6A pathway regulation (Figure 1).
Figure 1. Writing and reading m6A.
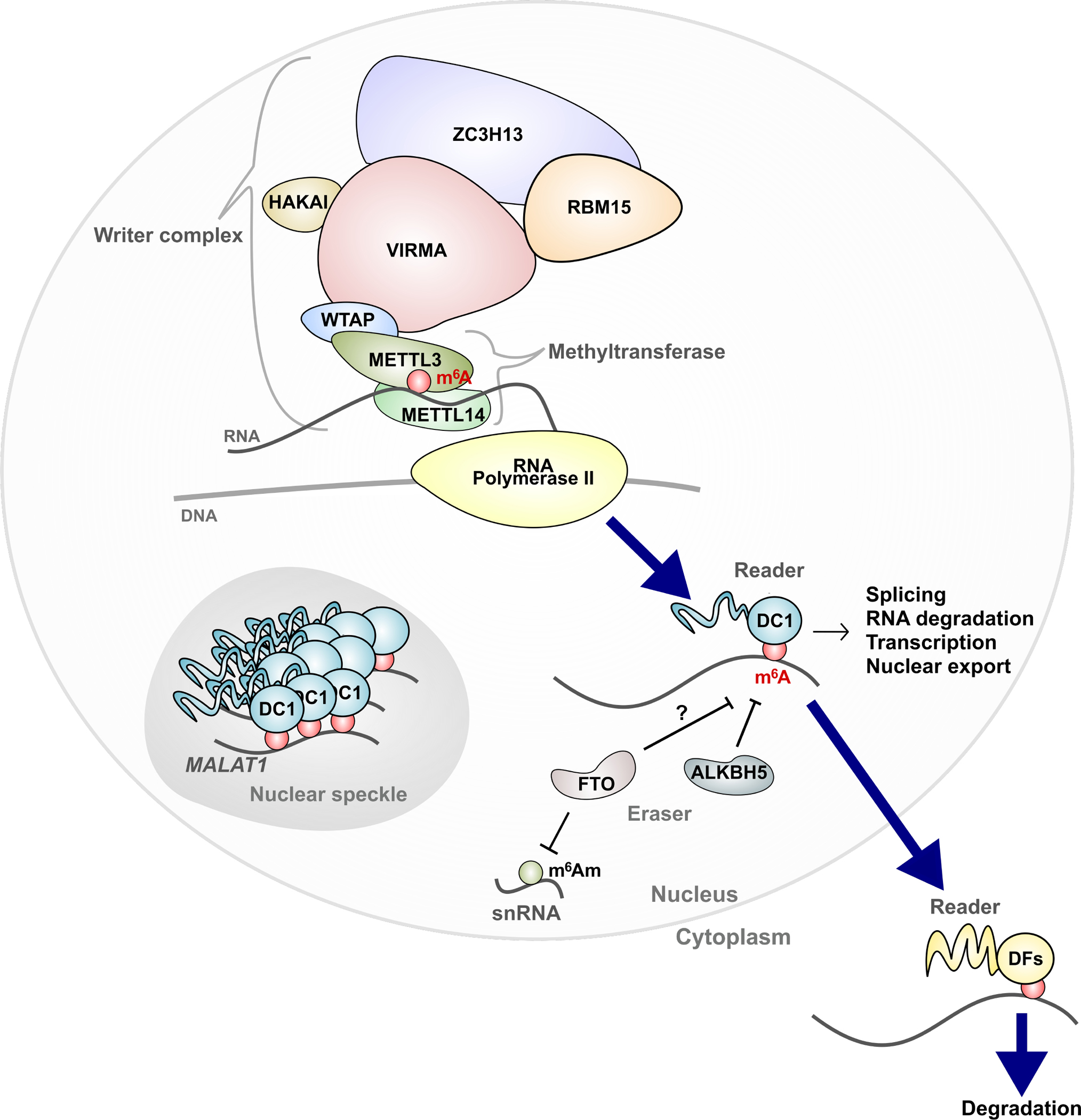
m6A is deposited co-transcriptionally by a writer complex. The catalytic subunit METTL3 forms a heterodimer with METTL14 to form the minimal methyltransferase core complex. METTL3/14 is a component of a larger writer complex, and each protein is required for m6A deposition in the cell. YTHDC1 (DC1), a nuclear m6A-binding protein, is assembled into nuclear condensates via m6A-containing RNA, including the nuclear noncoding RNA MALAT1. These condensates, such as nuclear speckles, are important for various processing steps that most mRNAs undergo in the nucleus. DC1 also binds and regulates specific m6A RNA to regulate splicing in some transcripts, or to influence epigenetic marks, transcription, RNA stability, or nuclear export. In the nucleus, m6A may be erased, particularly in testes where the nuclear m6A eraser ALKBH5 is enriched. FTO is an efficient demethylase for m6Am in snRNA, and may also demethylate m6A. Once exported to the cytoplasm, m6A is bound by DF proteins, which comprise YTHDF1, YTHDF2, and YTHDF3, which all promote mRNA degradation (Bawankar et al., 2021; Schöller et al., 2018; Knuckles et al., 2018).
m6A is a low stoichiometry modification
m6A mapping revealed the fundamental features of m6A, including which transcripts contain m6A and potential mechanisms by which certain sites are selected for methylation. The initial mapping methods (MeRIP-seq and m6A-seq) revealed ~13,000 thousand m6A sites in ~6000 mRNAs (Dominissini et al., 2012; Meyer et al., 2012). These methods used m6A antibodies to immunoprecipitate m6A-containing fragments after shearing mRNA. These fragments were then sequenced by next-generation sequencing and mapped to the transcriptome. Subsequent mapping methods (miCLIP) crosslinked m6A antibodies to RNA to achieve antibody-RNA adducts that created signature mutations adjacent to m6A, providing the first single-nucleotide resolution maps of m6A (Linder et al., 2015). These mapping methods have been used in many studies to show that m6A is dynamic and markedly altered in different cellular contexts and disease states (Liu et al., 2020b). However, as will be described below, more recent studies have argued that m6A dynamics may have been overestimated, or at least were performed with data that contained insufficient replicates to detect such differences (McIntyre et al., 2020).
m6A mapping also revealed that identifying the methylation consensus sequence is not enough to reveal the sites of methylation in the transcriptome. Mapping identified the major m6A consensus sequence, designated DRACH (D = A, G, U; R = A, G; H = A, C, U) (Linder et al., 2015). This consensus sequence is very similar to the sequence context predicted in the 1970s based on nuclease mapping experiments with metabolically labeled RNA (Wei et al., 1976). However, the DRACH sequence motifs are ubiquitous in mRNA, with the statistical frequency of one in every ~57 nucleotides. However, m6A is much more rare (~1 in every thousand nucleotides) (Perry et al., 1975), indicating that most DRACH sites are not methylated. Thus, additional factors are needed beyond the consensus motif to target methylation to a DRACH site.
One mechanism that could account for the methylation of some DRACH sites versus others is local structure or other contextual features, as seen in yeast (Garcia-Campos et al., 2019; Schwartz et al., 2013). In the case of structure, methylation appears to be more likely to occur in less structured RNA (Sun et al., 2019), likely due to the greater accessibility to the m6A writer complex. However, as described below, gene architecture (i.e. the size and location of exons and introns in a gene) appears to be particularly important for determining which DRACH sites will be methylated in mammals.
Designating adenosine residues or DRACH sites as either “methylated” or “not methylated” is an oversimplification. It is now clear that m6A is a substoichiometric modification. In other words, at any given adenosine site in an mRNA, only a fraction of the copies of the mRNA actually contain m6A at that site. The low m6A stoichiometry was first seen in early studies of Rous sarcoma virus using nuclease digestion and chromatography of Rous sarcoma virus RNA (Kane and Beemon, 1985). More recent methods, such as SCARLET, have allowed precise quantification of m6A stoichiometry (Liu et al., 2013). In SCARLET, an mRNA of interest is site-specifically cleaved using a “SCARLET oligo.” This oligo hybridizes with the target mRNA to create a region of RNA-DNA hybridization that allows RNase H to cleave precisely at the 5’ side of the adenosine residue of interest. Next, this adenosine is selectively radiolabeled, and the RNA is then digested to single nucleotides and, analyzed by thin-layer chromatography. The radiolabeled A and m6A derived from the exact position in the mRNA will appear as two separately resolved spots. The signal intensity from each spot can be used to calculate the m6A stoichiometry at this position. These experiments generally showed that most methylated sites exhibit low methylation stoichiometry with the highest stoichiometry of 20% at one m6A residue in the ß-actin mRNA (Liu et al., 2013).
A recent m6A mapping method, called MAZTER-seq, maps m6A and provides estimates of m6A stoichiometry on a transcriptome-wide scale (Garcia-Campos et al., 2019). MAZTER-seq takes advantage of a unique ribonuclease called MazF that cleaves mRNA at ACA sequence motifs only if the first adenosine is not N6-methylated. The MAZTER-seq method thus examines global patterns of ACA site resistance to MazF cleavage. This analysis is complicated because ACA motifs are relatively widespread, and therefore MazF treatment leads to high fragmentation of RNA, which prevents unambiguous mapping of sequence data to the reference genome. However, careful analysis can reveal the percent of cleaved and uncleaved ACA sites at ~16% of all previously known m6A In a MAZTER-seq analysis of mouse mRNA, a set of previously mapped m6A sites showed a predicted median stoichiometry of ~26% (Garcia-Campos et al., 2019).
However, the median stoichiometry of m6A may be even lower. As the sensitivity of m6A mapping increases, more m6A sites can be identified, typically reflecting m6A sites of low stoichiometry. For example, DART-seq maps considerably more m6A sites than other methods (Meyer, 2019). This method involves expressing a fusion protein comprising an m6A-binding YTH domain fused to the APOBEC RNA-modifying enzyme. This fusion protein causes C→U conversion at the C next to m6A in proportion to m6A stoichiometry. Unlike MAZTER-seq, DART-seq allows stoichiometric measurement of m6A in any sequence context. DART-seq analysis revealed that as many as ~100,000 sites contain m6A, but most have very low stoichiometry (Figure 2A).
Figure 2. Deposition and detection of m6A.
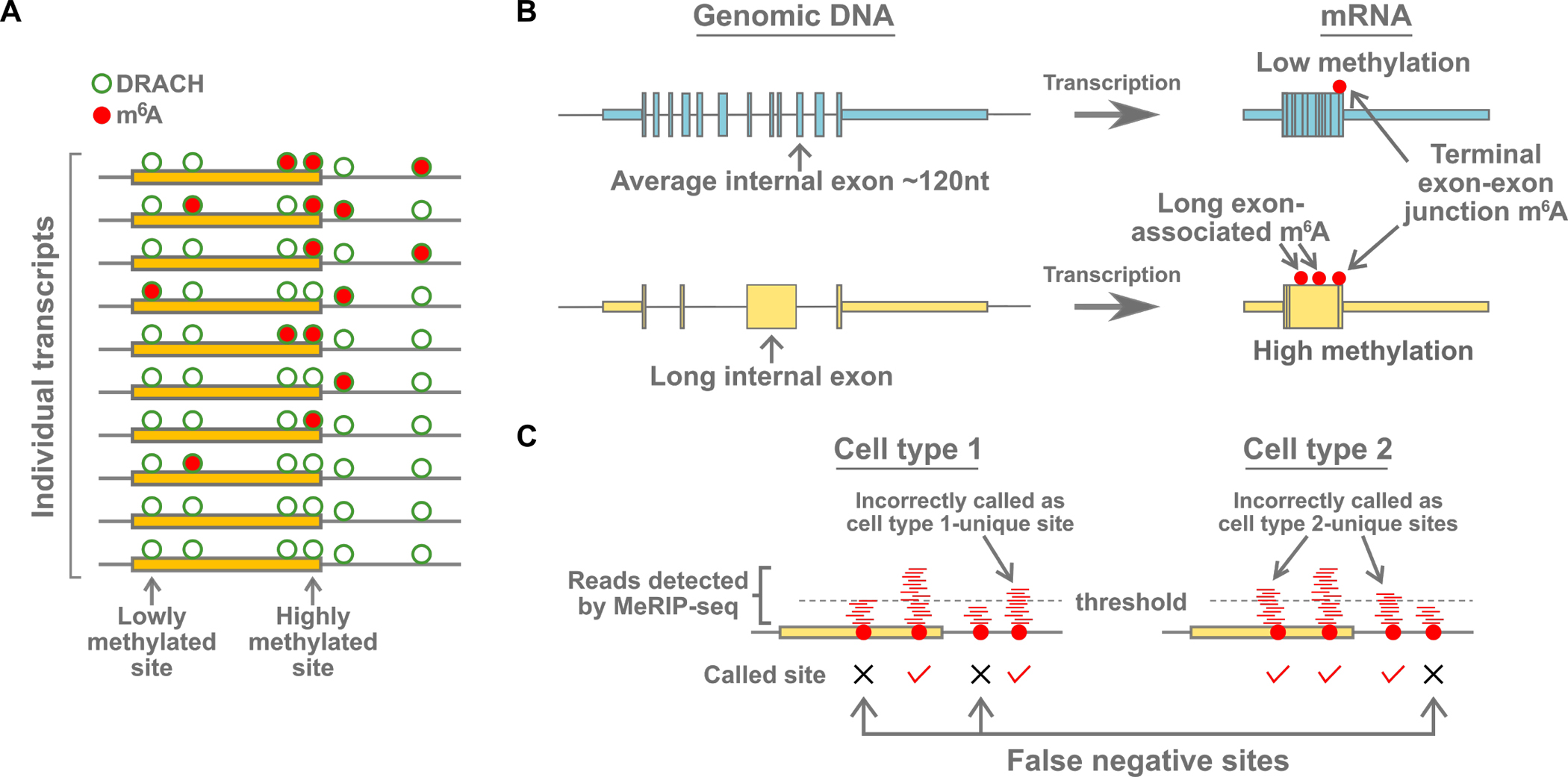
(A) m6A is a low stoichiometry modification. At any given m6A site in a specific mRNA, only a small number (usually less than 20%) of the transcript copies in the cell will have m6A at that site. Most of the DRACH m6A consensus sites are not methylated (green circle). Additionally, some DRACH sites are more methylated than others, and all DRACH sites are likely methylated to some degree, although the stoichiometry is likely very low. The molecular mechanism that causes some DRACH sites to be more methylated than others, even in the same transcript, are not fully understood. Although m6A mapping may reveal multiple m6A sites for a given mRNA, the individual transcripts that comprise the m6A annotation are typically methylated only at a subset of sites. Newer single-transcript analysis methods have revealed the stochastic nature of methylation on individual transcripts.
(B) Gene architecture influences m6A deposition. Shown (left) are examples of two genes, one containing a long internal exon. After transcription (right), m6A is preferentially formed on the regions of transcripts corresponding to long exons. m6A is also enriched near the terminal exon-exon junction, particularly in mRNAs with long 3’UTRs. The correlation of m6A with these genomic features suggests that the writer complex is regulated by other events that are responsive to gene architecture.
(C) Putative differences in m6A are sometimes artifacts of the methods used to call m6A sites from mapping data. Many studies have used mapping methods such as MeRIP-seq to map m6A and to compare transcriptome-wide distributions of m6A between two different cell conditions. These methods map reads (red lines) immunoprecipitated by an m6A-binding antibody. m6A sites are often called based on whether there are a sufficient number of reads above a threshold. However, the number of reads that map to any m6A sites can be very variable, even between replicates (McIntyre et al., 2020). Thus, the same m6A site in one sample might be called in one sample, but not the other, if the number of reads just passes or misses the threshold. For this reason, the differences in the transcriptome-wide distribution of m6A in many experiments may have been highly overestimated.
Thus, it is likely that every DRACH site could be methylated to some degree, and DRACH sites should not be considered methylated or unmethylated, but instead should be considered along a wide spectrum of methylation. At any given site, the stoichiometry is likely to be relatively low, but there may be a small subset of DRACH sites that have methylation as high as ~20%, and possibly even higher.
The low stoichiometry of m6A has important implications for how m6A could affect mRNA. Since current thinking is that the major function of m6A in cytosolic mRNA is to induce mRNA degradation (Zaccara and Jaffrey, 2020), the expression of a gene with a single mapped m6A site would likely not be meaningfully reduced by m6A unless the stoichiometry is very high, i.e, most of the encoded transcripts indeed contain m6A at that site. Instead, m6A is likely to be important primarily for genes that have multiple mapped m6A sites, since multiple mapped sites would make it more likely that most transcripts have at least one m6A site for m6A-mediated mRNA degradation.
Methylation patterns are linked to gene architecture
Mapping studies have also shown that gene architecture (i.e., length and distribution of exons and introns within a gene) is directly linked to global patterns of mRNA methylation. In one of the initial m6A mapping studies, the authors noted that methylation was disproportionately enriched in the regions of transcripts that corresponded to long internal exons (Figure 2B) (Dominissini et al., 2012). Internal exons are on average ~120 nucleotides, but in some cases can be several hundred or even several thousand nucleotides (Movassat et al., 2019). These long internal exons have a disproportionate likelihood of containing m6A and thus a higher density of m6A when corrected for their longer length (Dominissini et al., 2012). Thus, long internal exons may create signals that recruit the methyltransferase complex. Notably, some m6A sites are located in normal sized exons. These m6A sites may exhibit lower stoichiometry than m6A in long internal exons, but this has not been specifically demonstrated.
In addition to long internal exons, m6A is enriched near the stop codon (Meyer et al., 2012) (Dominissini et al., 2012). The stop codon itself is unlikely to trigger m6A formation in the nucleus since stop codons are presumably only recognized by translating ribosomes in the cytosol. Although every spliced mRNA has a terminal exon-exon junction, not all mRNAs have mapped m6A sites near their terminal exon-exon junction. This suggests that additional features lead cause some terminal exon-exon junctions to be more likely to be methylated than others. This feature might be the length of the terminal exon, since the length of the terminal exon is correlated to the levels of m6A near the stop codon (Ke et al., 2015). Thus, the process of m6A formation near the terminal exon-exon junction is likely to be caused by events related to the splicing of the last exon or transcription termination.
Overall, methylation of DRACH sites in mRNA is driven, at least in part, by gene architecture. Currently the major question is the precise mechanism by which gene architecture is linked to methylation. This process may involve RNA polymerase II, whose C-terminal domain undergoes phosphorylation in a manner that is related to its location in the gene body, including its location in exons (Nojima et al., 2015). The C-terminal domain has been shown to bind numerous proteins involved in mRNA processing in a phosphorylation-dependent manner (Harlen and Churchman, 2017). Conceivably, RNA polymerase II could bind the m6A writer complex as well. Alternatively, the m6A writer complex may bind to genomic regions corresponding to long internal exons. Long internal exons are enriched in the H3K36me3 histone modification (Huang et al., 2019). H3K36me3 was shown to bind METTL14 and proposed to account for selective methylation of the encoded transcripts (Huang et al., 2019). However, H3K36me3 binding to METTL14 was not observed in a separate study (Vermeulen et al., 2010). Ultimately resolving how gene architecture is related to recruitment of the m6A writer complex will provide insights into the pathways that regulate m6A levels, and will reveal why only some DRACH sites are preferentially methylated.
Is m6A dynamic?
m6A is widely described as a dynamic modification, but the evidence for its dynamics is highly limited and has been challenged. A potential caveat is that the term dynamic may have different meanings. For example, dynamic may refer to the ability of m6A to appear on a transcript, be removed, reappear, then removed in multiple cycles during the lifetime of the single mRNA molecule. This seems unlikely. Methylation occurs only once, i.e. co-transcriptionally (Ke et al., 2017c; Salditt-Georgieff et al., 1976), and demethylation, if it can occur, would be limited to the nucleus as the putative erasers, such as ALKBH5, are predominantly in the nucleus (Zaccara et al., 2019). Thus, rather than being dynamic in this respect, m6A acquires a specific pattern in the nucleus, and retains that pattern throughout its subsequent cytosolic life. In this respect, m6A is not dynamic.
Another definition for dynamic can refer to an increase in m6A levels in mRNA after a stimulus. Importantly, this type of dynamics would not be due to methylation of existing cytosolic transcripts. Since m6A formation is co-transcriptional and nuclear, a stimulus would only change the methylation of any newly made transcripts and would therefore alter the fate of only the newly methylated mRNAs, rather than the pre-existing mRNAs. A variety of different conditions have been suggested to lead to an increase in methylation, including heat shock and DNA damage (Xiang et al., 2017; Dominissini et al., 2012; Zhang et al., 2020).
However, assessing these dynamics by simply measuring total m6A levels in mRNA is not trivial. m6A can be quantified by mass spectrometry, but this requires extensive purification of mRNA from ribosomal RNA and small nuclear RNA, which contain m6A and comprise >90% of m6A in the cell (Legrand et al., 2017). Evidence is usually not presented to document mRNA purity. Thus, differences in m6A between two samples may reflect different levels of contaminating ribosomal RNA. Dot blotting and ELISA-based methods are also widely used due to their simplicity but these methods also suffer from rRNA contamination. Surprisingly, many studies measure m6A in total RNA, with researchers not realizing that they are measuring predominantly m6A from rRNA. These problems make many of the claims of m6A dynamics unsubstantiated.
Another way to detect m6A dynamics is to use m6A mapping datasets that compare different cellular conditions. m6A mapping involves calling m6A peaks based on thresholds that indicate whether m6A-immunoprecipitated reads are above background levels (Figure 2C). In principle an m6A site that shows a larger peak in one condition compared to another would indicate an m6A site that has changed and is thus may be dynamic. However, traditional m6A mapping method produce m6A peaks with variable heights even between replicates (Dierks et al., 2021; McIntyre et al., 2020). In some cases, and m6A site might not be called because the peak height is just below the threshold for calling these m6A sites in one dataset, but is just above the threshold and called in another dataset. This may give the false appearance of a complete gain or loss of an m6A site. However, based on a statistical analysis of m6A peak heights in previously published studies, Mason and colleagues argued that the vast majority of those reported alterations in m6A were simply due to the noise in peak height measurements rather than true differences in m6A (McIntyre et al., 2020).
To overcome this noise, as well as variability in sample-to sample preparation, Schwartz and colleagues developed m6A-Seq2, which uses barcoded samples that are processed in a single reaction (Dierks et al., 2021). This markedly improves detection of true differences in m6A between samples. Notably, neither m6A-Seq2 nor MAZTER-Seq showed clear differences in m6A distribution on commonly expressed genes between different tissues or in differentiating embryonic stem cells. Approximately 10% of m6A sites showed potential tissue specificity, but these differences may have reflected low expression levels of the mRNAs harboring these m6A sites. Thus, these highly quantitative methods suggest that most m6A sites are conserved, at least in the tested tissues and experimental conditions.
The overall similarity of the m6A maps between cell types and samples supports the idea that m6A is largely “hard-wired,” as described above. An important implication is that m6A mapping may not therefore be needed for most studies. A high-resolution m6A map from one tissue may be used to reasonably predict the likely location of m6A in any other tissue.
Because of the data suggesting that m6A is largely similar between tissues, any proposed differentially regulated m6A sites need to be rigorously documented. In particular, the exact m6A stoichiometry in each condition needs to be measured using methods such as SCARLET. This type of data is generally lacking in the field but is needed to convincingly identify specific dynamic m6A sites and the magnitude of the difference in their methylation status. Identification of these sites will provide a readout that can be used to test molecular pathways that lead to induction of m6A.
m6A reversibility
m6A dynamics could occur through m6A demethylases in the nucleus before being transported to the cytosol. Although this model is appealing, the relevance of demethylases to m6A biology is unclear and inconsistent, except in testes which show marked spermatogenesis defects in Alkbh5 knockout mice (Zheng et al., 2013). Alkbh5 knockout mice otherwise appear normal, suggesting that ALKBH5-mediated m6A demethylation is not needed for normal cellular functions or development outside germ cells. Indeed, quantitative m6A mapping assays have suggested that only ~2% of m6A sites might change stoichiometry in somatic cells upon depletion of ALKBH5 (Koh et al., 2019). Conversely, overexpression of ALKBH5 only showed slight decreases of m6A in a small number of transcripts (Garcia-Campos et al., 2019). However, even for these slightly affected sites, it is not clear why these sites and not other m6A sites are affected by ALKBH5. Additionally, it is not clear if the small change in m6A stoichiometry is meaningful for any transcript. Precise measurements of m6A stoichiometry in control and ALKBH5-depleted conditions will help address the importance of ALKBH5-mediated demethylation on specific mRNAs.
The connection between another demethylase, FTO, and m6A is also unclear. Although FTO was first shown to demethylate m6A (Jia et al., 2011), this activity is exceptionally low. It is now known that FTO exhibits markedly more efficient demethylation of m6Am (Mauer et al., 2017), a modified nucleotide that can be found at the first transcribed nucleotide position in mRNA and snRNA. The major target of FTO is the m6Am residues in snRNA, where FTO maintains snRNAs in a demethylated state (Mauer et al., 2019). In FTO-deficient cells, snRNAs have highly m6Am-methylated snRNAs, which can affect snRNA-dependent process, such as splicing.
Since FTO can demethylate both m6A and m6Am, several groups have tried to detect which of these are regulated by FTO in cells. To address this, quantitative m6A mapping methods have been used. These studies have clearly shown that m6A sites are not affected in FTO-depleted cells (Garcia-Campos et al., 2019; Koh et al., 2019). In contrast, transcriptome-wide analysis show clear increases in the methylation of snRNAs, which reflect increased m6Am, after FTO depletion. (Koh et al., 2019; Mauer et al., 2019). Thus, these transcriptome-wide quantitative m6A measurements fail to show that FTO appreciably demethylates m6A.
However to definitively determine if the effects of FTO depletion are due to increases in m6A or m6Am (or both), genetic approaches could be used. FTO knockout mice exhibit diverse phenotypes including partial neonatal lethality (Hess et al., 2013; Kim et al., 2021) and FTO-deficient cells show altered gene expression and 3’UTR lengths (Bartosovic et al., 2017) (Mauer et al., 2017). To determine if the effects of FTO are mediated by increasing m6A or m6Am, parallel experiments should be performed with FTO depletion in combination with either METTL3 or PCIF1 depletion. If the effects of FTO are mediated by increasing m6A, then FTO depletion should have no effect in METTL3 deficient cells. Similarly, if the effects of FTO depletion are due to increased m6Am, then FTO depletion effects would be lost in PCIF1-depleted cells. These controls are important to determine if the biological effects are mediated by m6A or m6Am.
A multiprotein m6A writer complex
m6A dynamics could be mediated by regulation of the ~1 MDa m6A writer complex, which contains METTL3 as its catalytic component (Bokar et al., 1997). METTL3 and its adapter component WTAP were first shown to assemble into a multiprotein writer complex in yeast (Agarwala et al., 2012) and plants (Zhong et al., 2008), and subsequently confirmed in mammalian cells (Liu et al., 2014; Ping et al., 2014; Schwartz et al., 2014; Wang et al., 2014). Several additional components were identified, including VIRMA, ZC3H13, RBM15, HAKAI, and METTL14 (Guo et al., 2018; Patil et al., 2016; Ruzicka et al., 2017; Schwartz et al., 2014). Except for METTL14, little is known about these proteins. METTL14 was initially proposed to be a separate methyltransferase that catalyzes m6A alongside METTL3 (Liu et al., 2014). However, it is now clear that METTL3 and METTL14 form a single functional heterodimeric enzyme (Wang et al., 2014); (Sledz and Jinek, 2016; Wang et al., 2016a; Wang et al., 2016b). Structural studies showed that METTL3 comprises the catalytic SAM-binding component, while METTL14 appears to position the RNA for methylation and allosterically activates METTL3. Thus, m6A catalysis is mediated by a single heterodimeric methyltransferase which comprises METTL3 and METTL14.
The mechanism by which the other writer complex components control m6A levels is not clear. Depletion of any of these proteins results in delocalization of METTL3 from nuclear speckles and loss of most, if not all, m6A in mRNA (Bawankar et al., 2021; Knuckles et al., 2018; Ping et al., 2014; Yue et al., 2018). Thus, these components may have a role in placing METTL3 near nascent RNA for co-transcriptional methylation.
It will be important to determine if the writer complex components can be regulated by signaling pathways to activate or inactivate the writer complex to achieve different levels of methylation throughout the transcriptome, or possibly even on unique subsets of mRNAs.
Cytoplasmic readers: YTHDF1, 2, and 3 bind all m6A sites
m6A in mRNA exerts its effects by binding to m6A “reader” proteins. The most well-established m6A readers are YTHDC1 (“DC1”), which is found in the nucleus, and YTHDF1, 2, and 3 (DF1, DF2, DF3), which are cytosolic. Besides these four proteins, YTHDC2 is the only other mammalian protein to contain the YTH domain, which is thought to bind m6A. However, YTHDC2 is primarily found in germ cells (Wojtas et al., 2017) and may not bind or regulate m6A sites in vivo (Saito et al., 2022) (Li et al., 2022). In contrast, DC1 is poised to mediate nuclear processing events due to its nuclear localization, while DFs can regulate cytoplasmic mRNA processing.
DF1, DF2, and DF3 are paralogs that have high amino acid identity across their entire length. These proteins are primarily composed of low complexity domain sequence, comprising several P/Q/N-rich patches and other low-complexity regions (Patil et al., 2017). The C-terminal region contains the YTH domain (Figure 3A). The high proportion of low-complexity domain sequence is consistent with these proteins functioning in intracellular condensates. Recent studies have demonstrated that the three DF proteins are enriched in P-bodies, stress granules, and other cytosolic condensates, and that the DF proteins undergo phase separation when they interact with polymethylated RNAs (Ries et al., 2019; Zaccara and Jaffrey, 2020). Thus, DF protein function and regulation is likely related to condensate biology.
Figure 3. YTHDF proteins function together to promote degradation of m6A-marked mRNAs.
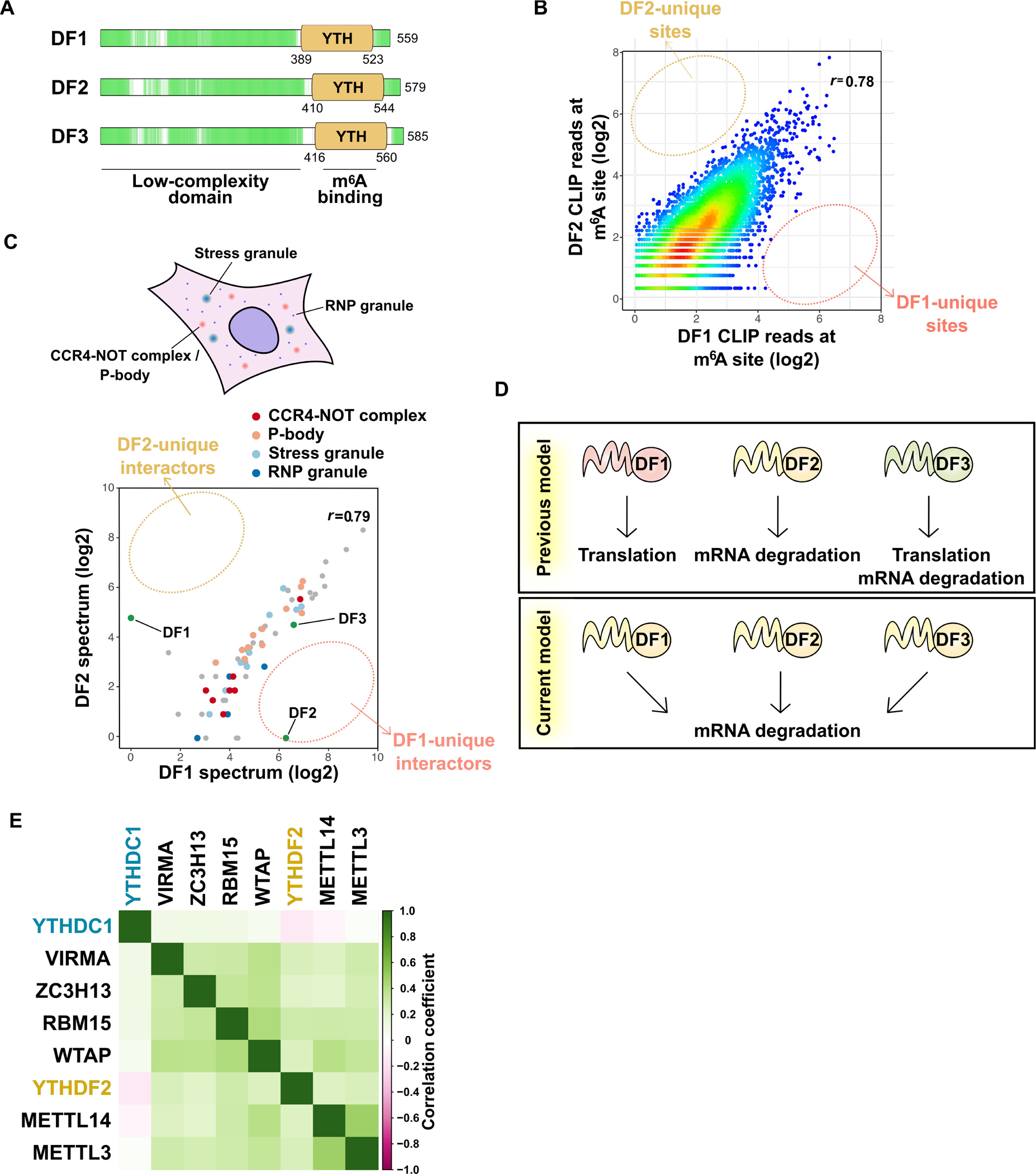
(A) DF proteins are low-complexity domain-containing “m6A reader” proteins. All three paralogs are highly similar and contain a proline-, glutamine-, and asparagine-rich low-complexity domain (green) followed by m6A-binding YTH domain. The low-complexity domain allows these three DF proteins to localize to granule structures such as RNP granules, P-body, and stress granules.
(B) All DF paralogs bind to all m6A sites, with no DF paralog showing preferential binding to any m6A site. Shown is a pairwise comparison of DF1 and DF2 iCLIP coverage at each single nucleotide-resolved m6A site in HEK293T cells. The color indicates the density. r, Pearson correlation coefficient. Similar results are seen when comparing DF3 to either DF1 or DF2 (Zaccara and Jaffrey, 2020).
(C) All three DF paralogs likely serve the same function since they interact with same sets of proteins. Pairwise comparison of the probabilities of interaction for DF1- and DF2-interacting proteins determined by proximal labeling and proteomics (Youn et al., 2018). Proximal proteins were detected by DF1 or DF2 C-terminally tagged with promiscuous biotin ligase (BirA). The average probability of interaction for either DF1 or DF2 or both higher than 0.9 are shown. r, Pearson correlation coefficient.
(D) Revised model of the YTHDF protein function. DF1 and DF3 were previously shown to promote translation, while DF2 and DF3 enhance mRNA degradation. More recent studies show all three DF proteins redundantly promote mRNA degradation.
(E) DF2 is the major protein mediating the effects of m6A on cell survival and proliferation. DepMap analysis reveals genes that show similar patterns of gene dependency as METTL3 across over 1000 different cell types. Members of the writer complex show the highest similarity to METTL3 in the different cell lines. The DF2 also shows a similar type of gene dependency as METTL3. In contrast, the pattern of cell dependency on DC1 shows poor correlation with METTL3. Dependency scores for m6A writers and readers for each cell line were analyzed by pairwise comparison. Pearson correlation coefficients were visualized in the matrix heatmap upon hierarchical clustering.
Recently, the binding properties and function of DF proteins have undergone a major revision, which has reshaped our understanding of both the DF proteins and m6A more generally. The prevailing model was that each of these proteins binds a subset of m6A sites throughout the transcriptome (Shi et al., 2017; Wang et al., 2015). That is, some of these m6A sites are uniquely bound by DF1, some by DF2, some by DF3, and some sites are bound by combinations of two or three of the YTHDF proteins. In this model, there was no clear pattern or mechanistic basis for the distinct binding events. Thus, the actions of any single m6A site are unpredictable until the DF reader(s) that bind to it are identified.
Importantly, the finding that the DF proteins can bind different m6A sites is perplexing since the YTH domains in the three DF proteins are nearly identical. The crystal structures of YTH domains of three DF proteins show identical interactions with m6A. A biophysical study of all three YTH domains shows similar intrinsic molecular dynamics and affinity to m6A (Li et al., 2020a). Thus, the reported differences in binding to m6A was contradictory to the similar sequence and binding properties of the DF proteins. No mechanism was proposed for these putative differences in binding preference.
After extensive reanalysis of the CLIP data from the previous studies, we recently showed that the DF paralogs bind all m6A sites in an essentially equivalent manner (Zaccara and Jaffrey, 2020). Our study concluded that there were no sites that were uniquely bound by one DF protein versus another DF protein. Several problems were identified in the earlier studies in which m6A sites were designated as “DF1 unique” or “DF2 unique” (Wang et al., 2015). Each of these unique sites were shown to have considerable reads for the other DF proteins based on the same PAR-CLIP datasets used to identify sites as unique (Zaccara and Jaffrey, 2020). Direct comparison of CLIP datasets showed that peaks derived from DF1, DF2, DF3, and m6A CLIP datasets show peaks that are essentially indistinguishable on essentially all genes.
The main problem with the earlier analyses was that they relied on calling binding sites based on thresholds. In this approach, many peaks are just above or below the threshold, and thus these borderline peaks are essentially arbitrarily called or eliminated as binding sites (Figure 2C). In the more recent studies, the CLIP datasets were reanalyzed without using thresholds. Instead, the analysis was based on the binding of each DF paralog at each m6A site in the transcriptome. To measure binding, the CLIP reads that overlapped each m6A site was used (Figure 3B). When analyzed this way, it was clear that all m6A sites show essentially equivalent binding to DF1, DF2, and DF3. Another independent study in mouse embryonic stem cells also reached the same conclusion based on strong correlations of the binding properties of all three DF paralogs (Lasman et al., 2020). Thus, different m6A sites are not linked to specific readers—each m6A shows equivalent ability to bind any DF protein.
Although the m6A directly binds each DF paralog (Patil et al., 2018), m6A can indirectly affect the RNA-binding of other proteins. m6A has a tendency to reduce RNA structure due to the low stability of m6A•U basepairs (Liu et al., 2015). The subsequent unfolding of RNA near m6A sites has been proposed to explain the m6A-dependent binding activity of several RNA-binding proteins, such as hnRNPC, hnRNPA2B1, and IGF2BP3 (Sun et al., 2019) (Wu et al., 2018) (Liu et al., 2015). Conceivably, any RNA-binding protein that binds to a single-stranded motif will show enhanced binding if m6A is near the binding site. It is currently unclear if any of these m6A-dependent “structure switches” have an impact on physiology.
Redundant functions for YTHDF1, 2, and 3
Another fundamental change in the m6A field was the recognition that the DF proteins do not have distinct functions, but instead serve the same overall function of promoting mRNA degradation. The prevailing idea was that DF1 and DF3 enhance translation, while DF2, typically the most abundant of these paralogs, promotes mRNA degradation (Shi et al., 2017; Wang et al., 2015). Thus, the model suggested that m6A can either cause mRNA degradation or enhanced translation depending on the bound DF protein. The conclusion that DF1 promotes translation was based on ribosome profiling data in DF1-depleted cells, which showed reduced translation of DF1-binding mRNAs. Additionally, DF1 was shown to bind translation initiation factor eIF3 (Wang et al., 2015). eIF3 binds in the 5’ UTR to position the ribosome near start codons for translation. The overall mechanism was problematic since eIF3 recruitment to mRNA stop codons and 3’UTRs, which is where m6A is generally located, is not consistent with the mechanism by which eIF3 promotes translation (Lee et al., 2015).
The function of the DF proteins, and thus the view of the function of m6A, was fundamentally revised in recent studies (Kontur et al., 2020; Lasman et al., 2020; Zaccara and Jaffrey, 2020). Technical problems were identified in the original ribosome profiling studies of DF1 (Wang et al., 2015). The reanalysis showed that the individual replicates, when analyzed separately, were divergent in their conclusions. (Zaccara and Jaffrey, 2020). Therefore, the ribosome profiling studies were replicated, which showed that there was no effect on m6A mRNA translation upon depletion of any or all DF proteins (Lasman et al., 2020; Zaccara and Jaffrey, 2020). In a recent study, He and colleagues re-examined the effect of DF1 depletion on the translation of m6A-contaiing mRNAs, which were defined using a new method for mapping m6A sites. In this study they showed no statistically significant change in translation of methylated vs. non-methylated mRNAs (Hu et al., 2022). Overall, these studies suggest that DF1 does not promote translation of m6A RNAs.
To determine the function of the DF proteins, protein interactome analysis was used. This approach showed that all three DF paralogs have highly similar binding partners, most notably proteins associated with the CCR4-NOT RNA degradation complex (Figure 3C) (Zaccara and Jaffrey, 2020). Interactions with eIF3 were not detected as significant interactors. The finding that the DF proteins have the same interactors suggested that they may have the same function. When the DF proteins were depleted individually, m6A mRNA stabilization was generally weak. However, when all three paralogs were depleted, there was considerable stabilization of m6A mRNAs (Lasman et al., 2020; Zaccara and Jaffrey, 2020). Notably, this effect was in proportion to the number of annotated m6A sites. These studies supported the new model that DF proteins function together in a redundant manner to promote mRNA degradation.
Studies using mouse genetics came to the same overall conclusion, showing that the DF proteins function together to mediate the essential role of m6A in embryogenesis (Lasman et al., 2020). Lasman and colleagues expressed DF1, DF2, or DF3 separately in DF triple knockout mouse embryonic stem cells. Re-expression of any DF rescued the differentiation defect of the triple knockout cells. This result showed that DF proteins interchangeably mediate the effects of m6A in promoting embryonic stem cell differentiation. Furthermore, they concluded that the tissue-specific phenotypes seen upon knockout of different DF paralogs during mouse development can be explained by the expression pattern of each DF protein in these different tissues (Lasman et al., 2020). In another study, DF protein redundancy was also shown to promote zebrafish embryonic development (Kontur et al., 2020). Overall, these studies support the physiologic relevance of DF protein redundancy in mediating the effects of m6A.
Although the overall function of the DFs is redundant, this does not mean they are identical or completely interchangeable. These are slight differences in the amino acid sequences in the low-complexity domains of these paralogs. These differences could result in differences in phase separation behavior, interaction with specific proteins, or regulation by post-translational modification. If any DF-specific function is observed, it will be important to determine the precise mechanism for these differences given the high similarity of these paralogs.
Does this mean that m6A does not affect mRNA translation rates at all? Indirect mechanisms following m6A-mediated mRNA degradation could lead to translational changes. For example, the expression of ribosomal genes were elevated in single or triple DF knockout embryonic stem cells (Lasman et al., 2020). This could lead to increases in translation. In neurons, DF1 depletion does not affect translation under normal conditions, but affects translation induced by potassium depolarization (Shi et al., 2018). These effects can readily be explained by m6A-mediated degradation of specific mRNAs that encode regulators of these translation pathways. In the case of AML (acute myeloid leukemia), reduced translation was seen upon depletion of m6A (Vu et al., 2017). However, depletion of m6A causes AML cells to undergo differentiation, which itself leads to changes in the expression levels and translation of diverse transcripts (Spevak et al., 2020). Thus, in the absence of a plausible molecular mechanism that links DF proteins to translation, effects of m6A on translation are more likely to be indirect effects of m6A. It should be noted that m6A in the 5’UTR has also been shown to promote translation by directly interacting with eIF3, a major translation initiation factor (Meyer et al., 2015). However, the number of mRNAs with 5’UTR is low (Boulias et al., 2019), making it likely that this mechanism is only relevant for a few mRNAs.
Together, these newer studies have revealed a unified model in which the primary function of m6A is to bind to DF proteins and to promote the degradation of m6A-modified mRNAs (Figure 3D). Although the function of m6A is now clearly linked to controlling mRNA stability, questions remain regarding whether all m6A sites are equally able to induce mRNA degradation, and how DF-mediated degradation could be activated.
Regulation of nuclear mRNA processing with a nuclear m6A reader
A major question is whether the effects of m6A are primarily mediated by the nuclear m6A reader, DC1, or the DF cytosolic readers. Insights into this question can be seen in the using datasets such as the Broad Institute’s Dependency Map (DepMap) (Tsherniak et al., 2017). DepMap is a large scale loss-of-function database in which the degree of dependency of over 10,000 genes was assessed in over 1000 cell lines using CRISPR-mediated knockout. Every gene is assigned a dependency score for each cell line which relates to the degree that the gene knockout reduces cell proliferation or survival. Using DepMap datasets, genes whose effects on growth are strongly correlated with a gene of interest across 1000+ cell lines can be readily identified. This co-dependency analysis can identify genes with similar functions or which operate in similar pathways (Shimada et al., 2021). In the case of METTL3, the genes that show the most similar profile of gene dependency across the thousand cell lines are the other components of the m6A writer complex, i.e., METTL14, VIRMA, ZC3H13, RBM15 and WTAP. However, the next most correlated gene is YTHDF2, the most abundant of the three DF paralogs in the majority of cell lines (Figure 3E). Importantly, DC1 is not identified among the top 100 genes that show co-dependency with METTL3. These data suggest that DF2, likely in concert with the other redundant DF paralogs, mediates the effects of METTL3 depletion, especially with respect to cell growth and proliferation assayed in the DepMap. Importantly, this does not rule out roles for DC1 in mediating other effects of m6A which are not linked to cell proliferation or survival.
Although DF proteins appear to mediate major aspects of m6A biology, DC1 also regulates m6A-dependent functions in the nucleus. Prior to its connection to m6A, DC1 was identified in a yeast two-hybrid screen as an interactor of TRA-2β, a component of the spliceosomal complex (Hartmann et al., 1999; Imai et al., 1998). Nearly all nuclear mRNA processing events have been linked in some way to m6A and DC1, including splicing, nuclear export, polyadenylation site selection, nuclear RNA degradation, and epigenetic regulation (Figure 4) (Mirza and Jaffrey, 2020; Roundtree et al., 2017b; Xiao et al., 2016). Like the DF proteins, DC1 is primarily composed of low-complexity sequence and forms biomolecular condensates upon binding m6A RNA (Cheng et al., 2021). This property likely accounts for the localization of DC1 in nuclear condensates. The DC1 condensates were originally referred to as YT bodies (Nayler et al., 2000), and partially overlap with nuclear speckles (marked by SC35) and super enhancer condensates (marked by BRD4), and some structures that overlap with neither (Cheng et al., 2021).
Figure 4. DC1 regulates diverse nuclear processing events through m6A.
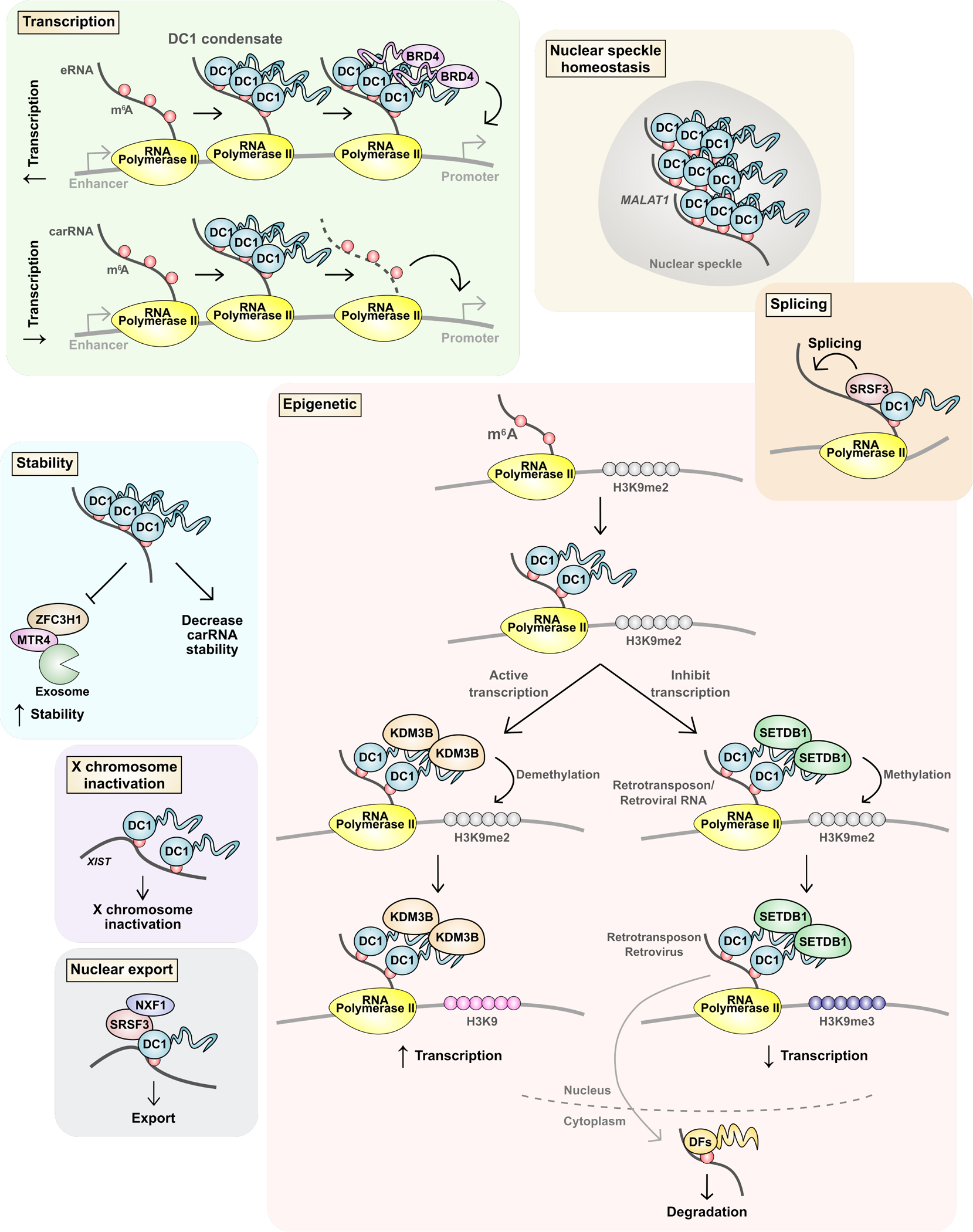
DC1 has been reported to exert a wide range of effects on mRNA. Some studies suggest transcription regulation by DC1. DC1 can enhance transcription by binding m6A-containing enhancer RNAs (eRNAs). DC1 forms a condensate with eRNAs that recruits BRD4. DC1 has also been shown to bind “chromatin-associated regulatory RNAs” (carRNAs), which broadly refers to many types of nuclear RNAs including eRNAs. DC1 was shown to mediate the degradation of carRNAs and subsequently reduce nearby gene expression. DC1 also has been shown to increase and decrease methylation of H3K9. In some studies, DC1 binds m6A mRNA and recruits KDM3B to demethylate H3K9me2 and enhance transcription. Other studies show that DC1 binds retrotransposon RNA and recruits SETDB1 to form H3K9me3 to reduce transcription. DC1 has also been shown to control splicing, RNA stability, nuclear export, and to promote X chromosome inactivation. DC1 also binds to the non-coding RNA MALAT1 which allows DC1 to have an important role in maintaining the protein composition of nuclear speckles.
A major target of DC1 are nuclear noncoding RNAs, such as MALAT1 and XIST, based on CLIP studies (Patil et al., 2016). This preferential enrichment of DC1 on nuclear ncRNA likely reflects the long residence time of ncRNA in the nucleus compared to the short time that mRNAs are found in the nucleus. Nevertheless, DC1 could still have access to mRNA during or shortly after transcription, which can thus provide the opportunity for DC1 to regulate nuclear mRNA processing.
The function of DC1 has been studied by depleting DC1 in cells. Although mRNA processing has been shown to be affected by DC1 depletion, some of these effects could be indirect. Since DC1 is bound to noncoding RNAs that contribute to nuclear structures like nuclear speckles (Patil et al., 2018), depletion of DC1 could impact nuclear architecture and nuclear processing bodies. In this way, depletion of DC1 would appear to affect different aspects of mRNA metabolism.
Consistent with the idea that DC1 depletion can have indirect effects are studies of DC1 interactions with MALAT1 (Patil et al., 2016; Wang et al., 2021b). In this study, DC1 was found to localize to nuclear speckles in part by its ability to bind MALAT1. When the m6A-containing region of MALAT1 was deleted, DC1 was no longer efficiently localized to nuclear speckles. Importantly, DC1 depletion also caused the nuclear speckles to show altered protein composition, indicating that DC1 is needed for the proper assembly of nuclear speckles. These studies highlight the potentially broad impact of depleting DC1 in the nucleus.
One potential way to determine if an effect of DC1 on an mRNA is direct, is to determine if the effect of DC1 is proportional to the number of m6A sites in the mRNA. This is known to be the case with the DF proteins which promote m6A-mediated degradation in proportion to the number of m6A sites in the transcript. In contrast, the proposed direct effect of m6A and DC1 in mediating nuclear export or nuclear degradation (Widagdo et al., 2021) has not been shown to correlate with the number of m6A sites. The idea that m6A is needed for nuclear export is problematic since non-methylated mRNAs are also clearly exported from the nucleus. It should be noted that there is data supporting the idea these effects of DC1 are direct, including data showing that DC1 binds components of the mRNA splicing and nuclear degradation complex (Imai et al., 1998; Roundtree et al., 2017b).
An additional function for DC1 is to control splicing. In Drosophila, m6A promotes the splicing of sex-lethal (Sxl) by binding Drosophila DC1 (Haussmann et al., 2016; Kan et al., 2017; Lence et al., 2016). In mammals, DC1 was originally found to promote exon skipping of Srsf3 splicing reporter gene (Hartmann et al., 1999). More recent study found DC1 depletion influences >2,000 alternative splicing events in a manner that was dependent on its YTH domain (Xiao et al., 2016). These effects may reflect DC1 recruiting SRSF3 to m6A sites to promote exon inclusion. Binding of DC1 to m6A simultaneously leads to displacement of SRSF10, a factor that promotes exon skipping (Xiao et al., 2016).
However, other studies have suggested that m6A has a very limited role in splicing regulation. In Mettl3−/− mouse embryonic stem cells, approximately 3% of all exons exhibit alternative splicing (Ke et al., 2017a), and of these less than 100 contain an m6A site (Geula et al., 2015; Ke et al., 2017b). These data suggest a minimal role for m6A as a direct regulator of splicing.
To distinguish direct from indirect effects of m6A a recent study developed a chemogenetically destabilized METTL3 that allows METTL3 protein to rapidly degraded after addition of a small molecule (Wei et al., 2021). This acute METTL3 depletion blocks co-transcriptional m6A formation but does not give time for indirect effects to occur, such as changes in gene expression or alteration in nuclear architecture. Acute METTL3 depletion caused pronounced effects on the splicing of a small number of transcripts, including several related to the m6A pathway. In particular, DC1 and WTAP both contain an m6A-containing exon that becomes alternatively spliced upon m6A depletion, allowing production of full-length functional protein in a DC1-dependent manner (Ke et al., 2017c; Wei et al., 2021). This study emphasizes how acute depletion of METTL3, using either chemogenetic approaches or newly described METTL3 inhibitors (Yankova et al., 2021), can help identify direct effects of m6A on nuclear mRNA processing events.
Connecting m6A to epigenetic regulation and transcription
DC1 may also have important roles in regulating epigenetic silencing (Figure 4). DC1 is bound to m6A sites on XIST, a noncoding RNA that mediates X chromosome inactivation in female mammalian cells (Engreitz et al., 2013). DC1 promotes XIST-mediated gene silencing (Patil et al., 2016), although notably the effects of m6A and DC1 are not as pronounced as other XIST regulators such as SPEN (Nesterova et al., 2019). Notably, XIST forms liquid-like condensates associated with low-complexity domain proteins (Cerase et al., 2019). DC1 may thus contribute to the formation of these XIST biomolecular condensates.
DC1 has also been shown to contribute to epigenetic silencing in other contexts, such as silencing of highly m6A-methylated retrotransposons (Liu et al., 2021) and endogenous retroviruses (Xu et al., 2021). In both cases, DC1 facilitates the deposition of the repressive H3K9me3 mark by facilitating recruitment of the histone 3 lysine 9 (H3K9) tri-methyltransferase SETDB1 and its cofactor TRIM28 (Xu et al., 2021) to nascent mRNA. It should be noted that another study found that the same retrovirus family is also degraded by DF proteins in cytoplasm (Chelmicki et al., 2021). These results suggest that m6A suppresses retroviruses both through epigenetic suppression and mRNA degradation.
m6A and DC1 have also been shown to have the opposite effect and promote gene activation. YTHDC1 recruits KDM3B to nascent m6A mRNA to promote H3K9me2 demethylation and enhance gene expression of approximately 30% of m6A-containing transcripts (Li et al., 2020b). However, non-m6A genes were also regulated by this pathway, and the relationship between m6A number and gene activation was not addressed. It remains unclear how DC1 can promote H3K9 methylation in one context and suppress H3K9 methylation in another.
m6A can also regulate gene expression through methylation of enhancer RNAs (eRNAs) and other chromatin-associated regulatory RNAs (carRNAs). Upon binding DC1, these RNAs can regulate gene expression from nearby promoters. However, the effects of DC1 binding to these RNAs were opposite in different studies (Lee et al., 2021; Liu et al., 2020a) . In one study, DC1 binding to m6A on carRNAs, which normally upregulate transcription from the nearby genes. As a result, DC1 attenuated transcription from the nearby promoter (Liu et al., 2020a). In another study, DC1 interacted with eRNA to enhance transcription by forming enhancer-associated condensates that recruited the transcriptional coactivator BRD4 (Lee et al., 2021). As with the studies of DC1 and H3K9 methylation, it is not clear why the effects of m6A appear to be different in these different contexts.
A major goal moving forward is to understand the mechanism for the divergent findings of m6A in different assays. Since the observed effects of DC1 depletion could be indirect consequences of alteration of nuclear structure, the use of acute inhibition approaches will be useful to distinguish direct from indirect effects of m6A. These approaches may help to create a more coherent model of how m6A effects transcription.
Future directions and perspectives
Our understanding of m6A has advanced considerably in the past few years. m6A was previously viewed as a dynamic modification that showed tissue-specificity, thus allowing m6A to encode unique patterns of gene expression. Although there is clear evidence for regulation, the overall m6A landscape appears highly similar across tissues. This conservation of m6A sites is likely because gene architecture, which is conserved in all tissues, is a major driver of m6A formation. m6A-marked mRNAs define a cohort of functionally related transcripts that are coordinately regulated, via control of m6A writing or reading, as part of various developmental and other processes. If m6A induction exists, it will be important to determine if these pathways affect all m6A sites, e.g. by globally increasing the efficiency of methylation, or if m6A induction can be targeted to specific sites. In either case, clear documentation of the exact change in stoichiometry of specific adenosine residues will needed. As the field adopts quantitative mapping of m6A, coupled with site-specific validation, we expect that questions of whether m6A is dynamic, whether it can be demethylated, as well as the circumstances, can be answered. Another major question is whether the m6A writer complex is regulated. The writer complex comprises many proteins of unknown function. It seems likely that these proteins could be regulated in ways to influence m6A writing. It will be important to determine if the writer complex is regulated at the level of its assembly, or its recruitment to the transcription machinery, or through other mechanisms.
It remains possible that m6A writing is relatively constant, but the regulation occurs primarily at the level of m6A reading. In this way, the degradation of m6A-marked mRNAs is increased or decreased depending on specific cellular pathways. In this case, the overall m6A level can change depending on the efficiency of m6A-mediated degradation. A major goal is to identify the signaling pathways that can regulate the degradation activity of DF proteins.
Part of understanding the m6A pathway and how m6A reading is regulated is to examine the potential function of protein interactors of DF proteins. DF proteins have a large number of interactors based on proximity labeling experiments, including IGF2BP proteins, PRRC2A, FMRP and others (Wu et al., 2019; Zaccara and Jaffrey, 2020; Zhang et al., 2018; Youn et al., 2018). Thus, DF proteins may exist as a “reader complex” and could be regulated via these interaction partners. Notably, these same interactors have also been shown to be m6A readers in some studies based on their ability to be pulled down with m6A-containing RNA probes (Edupuganti et al., 2017; Huang et al., 2018; Wu et al., 2019). Thus, it is not clear if their binding to m6A mRNA was due to their ability to bind to DF proteins, which are known m6A interactors. Further understanding of these DF-binding proteins will likely reveal insights into the regulation of m6A reading.
Another major question is to understand the molecular basis for the unusual topology of m6A, most notably its enrichment near stop codons and in long internal exons. It will be important to understand the mechanism for the specificity of m6A deposition and if gene structure is guiding this effect. It will be important to determine if the location of m6A in an mRNA influences the functional effects of m6A, or if all m6A re functionally equivalent in mediating m6A-mediated degradation.
Although it is clear that the major effect of m6A is to mediate degradation of a cohort of m6A-marked mRNAs, an important future direction is to resolve the nuclear effects of m6A, especially in light of the numerous contradictory studies regarding m6A and epigenetic regulation (Figure 4). The indirect effect of m6A depletion on nuclear architecture could be profound and thus the use of newer methods for acute m6A depletion using chemogenetic approaches (Wei et al., 2021) or new specific METTL3 inhibitors (Yankova et al., 2021), should help to resolve direct from indirect effects.
In addition to the basic molecular mechanisms of m6A reading and writing, an important future direction is to understand how m6A affects cellular physiology. Since there are so many different m6A-containing mRNAs, deregulation of diverse m6A-containing mRNAs have been used to explain the phenotypes of m6A depletion in cells and tissues. However, it is possible that the effects of m6A can be rationalized based on its ability of regulate the expression of a handful of master regulatory proteins that control major signaling pathways. As mentioned above, m6A is found on transcripts that encode the tumor suppressor APC and the pluripotency transcription factor Nanog and Klf4, which regulate tumor progression and differentiation, respectively. Control of these pathways could contribute to the proliferative and differentiation effects associated with m6A depletion. m6A is also found on transcripts encoding interferon-ß (Rubio et al., 2018; Winkler et al., 2019) as well as Stat1 and Irf1 (Wang et al., 2020), all of which play central roles in the type I interferon pathway. Thus, by controlling these master regulators, m6A suppresses an entire interferon-regulated gene expression network. It will be important to determine the major pathways controlled by m6A and how these act together to mediate the physiological effects of m6A.
ACKNOWLEDGEMENTS
We thank members of the Jaffrey lab for comments and suggestions and Anthony Olarerin-George for preparing the DepMap dependency figure. This work is supported by NIH grants R35NS111631, R01CA186702 and R01MH121072 (S.R.J.) and a postdoctoral fellowship to S.M. from the U.S. Department of Defense (DOD) Breast Cancer Research Program (BC180715).
Footnotes
DECLARATION OF INTERESTS
S.R.J is a scientific founder of, advisor to, and owns equity in 858 Therapeutics and Gotham Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agarwala SD, Blitzblau HG, Hochwagen A, and Fink GR (2012). RNA methylation by the MIS complex regulates a cell fate decision in yeast. PLoS Genet 8, e1002732. 10.1371/journal.pgen.1002732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- PGENETICS-D-12–00168 [pii].
- Bartosovic M, Molares HC, Gregorova P, Hrossova D, Kudla G, and Vanacova S (2017). N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3’-end processing. Nucleic Acids Res 45, 11356–11370. 10.1093/nar/gkx778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. (2014). m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bawankar P, Lence T, Paolantoni C, Haussmann IU, Kazlauskiene M, Jacob D, Heidelberger JB, Richter FM, Nallasivan MP, Morin V, et al. (2021). Hakai is required for stabilization of core components of the m6A mRNA methylation machinery. Nat Commun 12, 3778. 10.1038/s41467-021-23892-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokar JA, Shambaugh ME, Polayes D, Matera AG, and Rottman FM (1997). Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3, 1233–1247. [PMC free article] [PubMed] [Google Scholar]
- Boulias K, Toczydłowska-Socha D, Hawley BR, Liberman N, Takashima K, Zaccara S, Guez T, Vasseur JJ, Debart F, Aravind L, et al. (2019). Identification of the m6Am Methyltransferase PCIF1 Reveals the Location and Functions of m6Am in the Transcriptome. Mol Cell 75, 631–643.e638. 10.1016/j.molcel.2019.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerase A, Armaos A, Neumayer C, Avner P, Guttman M, and Tartaglia GG (2019). Phase separation drives X-chromosome inactivation: a hypothesis. Nature structural & molecular biology 26, 331–334. [DOI] [PubMed] [Google Scholar]
- Chelmicki T, Roger E, Teissandier A, Dura M, Bonneville L, Rucli S, Dossin F, Fouassier C, Lameiras S, and Bourc’his D (2021). m6A RNA methylation regulates the fate of endogenous retroviruses. Nature 591, 312–316. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Xie W, Pickering BF, Chu KL, Savino AM, Yang X, Luo H, Nguyen DT, Mo S, and Barin E (2021). N6-Methyladenosine on mRNA facilitates a phase-separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell 39, 958–972. e958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, and Rottman F (1974). Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A 71, 3971–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierks D, Garcia-Campos MA, Uzonyi A, Safra M, Edelheit S, Rossi A, Sideri T, Varier RA, Brandis A, and Stelzer Y (2021). Multiplexed profiling facilitates robust m6A quantification at site, gene and sample resolution. Nature methods 18, 1060–1067. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- Edupuganti RR, Geiger S, Lindeboom RGH, Shi H, Hsu PJ, Lu Z, Wang SY, Baltissen MPA, Jansen P, Rossa M, et al. (2017). N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat Struct Mol Biol 24, 870–878. 10.1038/nsmb.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, et al. (2013). The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 341, 1237973. 10.1126/science.1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Campos MA, Edelheit S, Toth U, Safra M, Shachar R, Viukov S, Winkler R, Nir R, Lasman L, Brandis A, et al. (2019). Deciphering the “m6A Code” via Antibody-Independent Quantitative Profiling. Cell 178, 731–747 e716. 10.1016/j.cell.2019.06.013. [DOI] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006. 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- Guo J, Tang HW, Li J, Perrimon N, and Yan D (2018). Xio is a component of the Drosophila sex determination pathway and RNA N(6)-methyladenosine methyltransferase complex. Proc Natl Acad Sci U S A 115, 3674–3679. 10.1073/pnas.1720945115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlen KM, and Churchman LS (2017). The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nature reviews Molecular cell biology 18, 263–273. [DOI] [PubMed] [Google Scholar]
- Hartmann AM, Nayler O, Schwaiger FW, Obermeier A, and Stamm S (1999). The interaction and colocalization of Sam68 with the splicing-associated factor YT521-B in nuclear dots is regulated by the Src family kinase p59(fyn). Molecular biology of the cell 10, 3909–3926. 10.1091/mbc.10.11.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, Fray RG, and Soller M (2016). m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature 540, 301–304. 10.1038/nature20577. [DOI] [PubMed] [Google Scholar]
- Hess ME, Hess S, Meyer KD, Verhagen LA, Koch L, Bronneke HS, Dietrich MO, Jordan SD, Saletore Y, Elemento O, et al. (2013). The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci 16, 1042–1048. nn.3449 [pii] 10.1038/nn.3449. [DOI] [PubMed] [Google Scholar]
- Hu L, Liu S, Peng Y, Ge R, Su R, Senevirathne C, Harada BT, Dai Q, Wei J, Zhang L, et al. (2022). m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome. Nat Biotechnol. 10.1038/s41587-022-01243-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al. (2018). Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol 20, 285–295. 10.1038/s41556-018-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, Chen Z, Deng X, Xiao G, and Auer F (2019). Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature 567, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Matsuo N, Ogawa S, Tohyama M, and Takagi T (1998). Cloning of a gene, YT521, for a novel RNA splicing-related protein induced by hypoxia/reoxygenation. Brain Res Mol Brain Res 53, 33–40. [DOI] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, and He C (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7, 885–887. 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan L, Grozhik AV, Vedanayagam J, Patil DP, Pang N, Lim KS, Huang YC, Joseph B, Lin CJ, Despic V, et al. (2017). The m6A pathway facilitates sex determination in Drosophila. Nat Commun 8, 15737. 10.1038/ncomms15737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane SE, and Beemon K (1985). Precise localization of m6A in Rous sarcoma virus RNA reveals clustering of methylation sites: implications for RNA processing. Mol Cell Biol 5, 2298–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, Haripal B, Zucker-Scharff I, Moore MJ, Park CY, et al. (2015). A majority of m6A residues are in the last exons, allowing the potential for 3’ UTR regulation. Genes Dev 29, 2037–2053. 10.1101/gad.269415.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vågbø CB, Geula S, Hanna JH, Black DL, Darnell JE, and Darnell RB (2017a). m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31, 990–1006. 10.1101/gad.301036.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vagbo CB, Geula S, Hanna JH, Black DL, Darnell JE Jr., and Darnell RB (2017b). m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes & development 31, 990–1006. 10.1101/gad.301036.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Pandya-Jones A, Saito Y, Fak JJ, Vagbo CB, Geula S, Hanna JH, Black DL, Darnell JE Jr., and Darnell RB (2017c). m(6)A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev 31, 990–1006. 10.1101/gad.301036.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Lee Y. s., Kim S-M, Jang S, Choi H, Lee J-W, Kim T-D, and Kim VN (2021). RNA demethylation by FTO stabilizes the FOXJ1 mRNA for proper motile ciliogenesis. Developmental Cell 56, 1118–1130. e1116. [DOI] [PubMed] [Google Scholar]
- Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, Masiello I, Hares T, Villaseñor R, Hess D, et al. (2018). Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m. Genes Dev 32, 415–429. 10.1101/gad.309146.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CWQ, Goh YT, and Goh WSS (2019). Atlas of quantitative single-base-resolution N(6)-methyl-adenine methylomes. Nat Commun 10, 5636. 10.1038/s41467-019-13561-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontur C, Jeong M, Cifuentes D, and Giraldez AJ (2020). Ythdf m6A readers function redundantly during zebrafish development. Cell reports 33, 108598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasman L, Krupalnik V, Viukov S, Mor N, Aguilera-Castrejon A, Schneir D, Bayerl J, Mizrahi O, Peles S, and Tawil S (2020). Context-dependent functional compensation between Ythdf m6A reader proteins. Genes & development 34, 1373–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, Kranzusch PJ, and Cate JH (2015). eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature 522, 111–114. 10.1038/nature14267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-H, Wang R, Xiong F, Krakowiak J, Liao Z, Nguyen PT, Moroz-Omori EV, Shao J, Zhu X, and Bolt MJ (2021). Enhancer RNA m6A methylation facilitates transcriptional condensate formation and gene activation. Molecular Cell 81, 3368–3385. e3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand C, Tuorto F, Hartmann M, Liebers R, Jacob D, Helm M, and Lyko F (2017). Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs. Genome Res 27, 1589–1596. 10.1101/gr.210666.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, Ho CH, Kreim N, Andrade-Navarro MA, Poeck B, Helm M, and Roignant JY (2016). m6A modulates neuronal functions and sex determination in Drosophila. Nature 540, 242–247. 10.1038/nature20568. [DOI] [PubMed] [Google Scholar]
- Li L, Krasnykov K, Homolka D, Gos P, Mendel M, Fish RJ, Pandey RR, and Pillai RS (2022). The XRN1-regulated RNA helicase activity of YTHDC2 ensures mouse fertility independently of m. Mol Cell 82, 1678–1690.e1612. 10.1016/j.molcel.2022.02.034. [DOI] [PubMed] [Google Scholar]
- Li Y, Bedi RK, Moroz-Omori EV, and Caflisch A (2020a). Structural and dynamic insights into redundant function of YTHDF proteins. Journal of Chemical Information and Modeling 60, 5932–5935. [DOI] [PubMed] [Google Scholar]
- Li Y, Xia L, Tan K, Ye X, Zuo Z, Li M, Xiao R, Wang Z, Liu X, and Deng M (2020b). N6-Methyladenosine co-transcriptionally directs the demethylation of histone H3K9me2. Nature Genetics 52, 870–877. [DOI] [PubMed] [Google Scholar]
- Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, and Jaffrey SR (2015). Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 12, 767–772. 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, Zhao S, Shen B, Gao Y, and Han D (2020a). N 6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Gao M, He J, Wu K, Lin S, Jin L, Chen Y, Liu H, Shi J, and Wang X (2021). The RNA m6A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature 591, 322–326. [DOI] [PubMed] [Google Scholar]
- Liu J, Li K, Cai J, Zhang M, Zhang X, Xiong X, Meng H, Xu X, Huang Z, Peng J, et al. (2020b). Landscape and Regulation of m6A and m6Am Methylome across Human and Mouse Tissues. Mol Cell 77, 426–440.e426. 10.1016/j.molcel.2019.09.032. [DOI] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N(6)-adenosine methylation. Nat Chem Biol 10, 93–95. nchembio.1432 [pii] 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, and Pan T (2015). N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564. 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Parisien M, Dai Q, Zheng G, He C, and Pan T (2013). Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19, 1848–1856. rna.041178.113 [pii] 10.1261/rna.041178.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, et al. (2017). Reversible methylation of m6Am in the 5’ cap controls mRNA stability. Nature 541, 371–375. 10.1038/nature21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur JJ, Rentmeister A, Gross SS, Pellizzoni L, Debart F, et al. (2019). FTO controls reversible m6Am RNA methylation during snRNA biogenesis. Nature Chemical Biology 15, 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre AB, Gokhale NS, Cerchietti L, Jaffrey SR, Horner SM, and Mason CE (2020). Limits in the detection of m6A changes using MeRIP/m6A-seq. Scientific reports 10, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD (2019). DART-seq: an antibody-free method for global m6A detection. Nat Methods 16, 1275–1280. 10.1038/s41592-019-0570-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, and Jaffrey SR (2015). 5’ UTR m(6)A Promotes Cap-Independent Translation. Cell 163, 999–1010. 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, and Jaffrey SR (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149, 1635–1646. S0092–8674(12)00536–3 [pii] 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza A, and Jaffrey SR (2020). SCARPET: Quantitative m6A stoichiometry method reveals the scope of m6A dyanamics in normal and disease states. In review.
- Movassat M, Forouzmand E, Reese F, and Hertel KJ (2019). Exon size and sequence conservation improves identification of splice-altering nucleotides. RNA 25, 1793–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayler O, Hartmann AM, and Stamm S (2000). The ER repeat protein YT521-B localizes to a novel subnuclear compartment. J Cell Biol 150, 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesterova TB, Wei G, Coker H, Pintacuda G, Bowness JS, Zhang T, Almeida M, Bloechl B, Moindrot B, and Carter EJ (2019). Systematic allelic analysis defines the interplay of key pathways in X chromosome inactivation. Nature communications 10, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Grosso ARF, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, and Proudfoot NJ (2015). Mammalian NET-seq reveals genome-wide nascent transcription coupled to RNA processing. Cell 161, 526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, and Jaffrey SR (2016). m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373. 10.1038/nature19342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil DP, Pickering BF, and Jaffrey SR (2017). Reading m(6)A in the Transcriptome: m(6)A-Binding Proteins. Trends Cell Biol. 10.1016/j.tcb.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil DP, Pickering BF, and Jaffrey SR (2018). Reading m 6 A in the Transcriptome: m 6 A-Binding Proteins. Trends Cell Biol 28, 113–127. 10.1016/j.tcb.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RP, and Kelley DE (1974). Existence of methylated messenger RNA in mouse L cells. Cell 1, 37–42. [Google Scholar]
- Perry RP, Kelley DE, Friderici K, and Rottman F (1975). The methylated constituents of L cell messenger RNA: evidence for an unusual cluster at the 5’ terminus. Cell 4, 387–394. 0092–8674(75)90159–2 [pii]. [DOI] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. cr20143 [pii] 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries RJ, Zaccara S, Klein P, Olarerin-George A, Namkoong S, Pickering BF, Patil DP, Kwak H, Lee JH, and Jaffrey SR (2019). m6A enhances the phase separation potential of mRNA. Nature 571, 424–428. 10.1038/s41586-019-1374-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, and He C (2017a). Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P, et al. (2017b). YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife 6. 10.7554/eLife.31311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio RM, Depledge DP, Bianco C, Thompson L, and Mohr I (2018). RNA m(6) A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes Dev 32, 1472–1484. 10.1101/gad.319475.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruzicka K, Zhang M, Campilho A, Bodi Z, Kashif M, Saleh M, Eeckhout D, El-Showk S, Li H, Zhong S, et al. (2017). Identification of factors required for m(6) A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol 215, 157–172. 10.1111/nph.14586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Hawley BR, Puno MR, Sarathy SN, Lima CD, Jaffrey SR, Darnell RB, Keeney S, and Jain D (2022). YTHDC2 control of gametogenesis requires helicase activity but not m6A binding. Genes & Development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salditt-Georgieff M, Jelinek W, Darnell JE, Furuichi Y, Morgan M, and Shatkin A (1976). Methyl labeling of HeLa cell hnRNA: a comparison with mRNA. Cell 7, 227–237. 0092–8674(76)90022–2 [pii]. [DOI] [PubMed] [Google Scholar]
- Schöller E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A, Feederle R, Bruckmann A, and Meister G (2018). Interactions, localization, and phosphorylation of the m. RNA 24, 499–512. 10.1261/rna.064063.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, Tabach Y, Mikkelsen TS, Satija R, Ruvkun G, et al. (2013). High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155, 1409–1421. 10.1016/j.cell.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. (2014). Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell reports 8, 284–296. 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, and He C (2017). YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res 27, 315–328. 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Zhang X, Weng Y-L, Lu Z, Liu Y, Lu Z, Li J, Hao P, Zhang Y, and Zhang F (2018). m6A facilitates hippocampus-dependent learning and memory through YTHDF1. Nature 563, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Bachman JA, Muhlich JL, and Mitchison TJ (2021). shinyDepMap, a tool to identify targetable cancer genes and their functional connections from Cancer Dependency Map data. Elife 10, e57116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz P, and Jinek M (2016). Structural insights into the molecular mechanism of the m(6)A writer complex. Elife 5. 10.7554/eLife.18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spevak CC, Elias HK, Kannan L, Ali MAE, Martin GH, Selvaraj S, Eng WS, Ernlund A, Rajasekhar VK, Woolthuis CM, et al. (2020). Hematopoietic Stem and Progenitor Cells Exhibit Stage-Specific Translational Programs via mTOR- and CDK1-Dependent Mechanisms. Cell Stem Cell 26, 755–765.e757. 10.1016/j.stem.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Fazal FM, Li P, Broughton JP, Lee B, Tang L, Huang W, Kool ET, Chang HY, and Zhang QC (2019). RNA structure maps across mammalian cellular compartments. Nat Struct Mol Biol 26, 322–330. 10.1038/s41594-019-0200-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, and Krill-Burger JM (2017). Defining a cancer dependency map. Cell 170, 564–576. e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, and Stunnenberg HG (2010). Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142, 967–980. [DOI] [PubMed] [Google Scholar]
- Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al. (2017). The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med 23, 1369–1376. 10.1038/nm.4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, Yuan J, and Rana TM (2020). m6A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. The EMBO journal 39, e104514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Doxtader KA, and Nam Y (2016a). Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell 63, 306–317. 10.1016/j.molcel.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Shao F, Yang X, Wang J, Zhu R, Yang Y, Zhao G, Guo D, Sun Y, Xue Q, et al. (2021a). METTL3 promotes tumour development by decreasing APC expression mediated by APC mRNA N. Nat Commun 12, 3803. 10.1038/s41467-021-23501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, et al. (2016b). Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534, 575–578. 10.1038/nature18298. [DOI] [PubMed] [Google Scholar]
- Wang X, Liu C, Zhang S, Yan H, Zhang L, Jiang A, Liu Y, Feng Y, Li D, and Guo Y (2021b). N6-methyladenosine modification of MALAT1 promotes metastasis via reshaping nuclear speckles. Developmental Cell 56, 702–715. e708. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 161, 1388–1399. 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, and Zhao JC (2014). N(6)-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16, 191–198. ncb2902 [pii] 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CM, Gershowitz A, and Moss B (1976). 5’-Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry 15, 397–401. [DOI] [PubMed] [Google Scholar]
- Wei G, Almeida M, Pintacuda G, Coker H, Bowness JS, Ule J, and Brockdorff N (2021). Acute depletion of METTL3 implicates N6-methyladenosine in alternative intron/exon inclusion in the nascent transcriptome. Genome research 31, 1395–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widagdo J, Anggono V, and Wong JJ-L (2021). The multifaceted effects of YTHDC1-mediated nuclear m6A recognition. Trends in Genetics. [DOI] [PubMed] [Google Scholar]
- Winkler R, Gillis E, Lasman L, Safra M, Geula S, Soyris C, Nachshon A, Tai-Schmiedel J, Friedman N, and Le-Trilling VTK (2019). m6A modification controls the innate immune response to infection by targeting type I interferons. Nature immunology 20, 173–182. [DOI] [PubMed] [Google Scholar]
- Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, and Pillai RS (2017). Regulation of m(6)A Transcripts by the 3’-->5’ RNA Helicase YTHDC2 Is Essential for a Successful Meiotic Program in the Mammalian Germline. Mol Cell 68, 374–387 e312. 10.1016/j.molcel.2017.09.021. [DOI] [PubMed] [Google Scholar]
- Wu B, Su S, Patil DP, Liu H, Gan J, Jaffrey SR, and Ma J (2018). Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat Commun 9, 420. 10.1038/s41467-017-02770-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Li A, Sun B, Sun J-G, Zhang J, Zhang T, Chen Y, Xiao Y, Gao Y, and Zhang Q (2019). A novel m6A reader Prrc2a controls oligodendroglial specification and myelination. Cell research 29, 23–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, et al. (2017). RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576. 10.1038/nature21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al. (2016). Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell 61, 507–519. 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Xu W, Li J, He C, Wen J, Ma H, Rong B, Diao J, Wang L, Wang J, and Wu F (2021). METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature 591, 317–321. [DOI] [PubMed] [Google Scholar]
- Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, Pilka ES, Aspris D, Leggate D, Hendrick AG, et al. (2021). Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601. 10.1038/s41586-021-03536-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JY, Dunham WH, Hong SJ, Knight JDR, Bashkurov M, Chen GI, Bagci H, Rathod B, MacLeod G, Eng SWM, et al. (2018). High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol Cell 69, 517–532 e511. 10.1016/j.molcel.2017.12.020. [DOI] [PubMed] [Google Scholar]
- Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H, et al. (2018). VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov 4, 10. 10.1038/s41421-018-0019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccara S, and Jaffrey SR (2020). A unified model for the function of YTHDF proteins in regulating m6A-modified mRNA. Cell 181, 1582–1595. e1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccara S, Ries RJ, and Jaffrey SR (2019). Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 20, 608–624. 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- Zhang C, Chen L, Peng D, Jiang A, He Y, Zeng Y, Xie C, Zhou H, Luo X, Liu H, et al. (2020). METTL3 and N6-Methyladenosine Promote Homologous Recombination-Mediated Repair of DSBs by Modulating DNA-RNA Hybrid Accumulation. Mol Cell 79, 425–442.e427. 10.1016/j.molcel.2020.06.017. [DOI] [PubMed] [Google Scholar]
- Zhang F, Kang Y, Wang M, Li Y, Xu T, Yang W, Song H, Wu H, Shu Q, and Jin P (2018). Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum Mol Genet 27, 3936–3950. 10.1093/hmg/ddy292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhang S-W, Zhang S-Y, Gao S-J, Chen Y, and Huang Y (2021). m6A-express: uncovering complex and condition-specific m6A regulation of gene expression. Nucleic acids research 49, e116–e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. (2013). ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol Cell 49, 18–29. S1097–2765(12)00892–1 [pii] 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Li H, Bodi Z, Button J, Vespa L, Herzog M, and Fray RG (2008). MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 20, 1278–1288. tpc.108.058883 [pii] 10.1105/tpc.108.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]