

Abstract
Bispeptide nucleic acids (bis-PNAs; PNA clamps), PNA oligomers, and DNA oligonucleotides were evaluated as affinity purification reagents for subfemtomolar 16S ribosomal DNA (rDNA) and rRNA targets in soil, sediment, and industrial air filter nucleic acid extracts. Under low-salt hybridization conditions (10 mM NaPO4, 5 mM disodium EDTA, and 0.025% sodium dodecyl sulfate [SDS]) a PNA clamp recovered significantly more target DNA than either PNA or DNA oligomers. The efficacy of PNA clamps and oligomers was generally enhanced in the presence of excess nontarget DNA and in a low-salt extraction-hybridization buffer. Under high-salt conditions (200 mM NaPO4, 100 mM disodium EDTA, and 0.5% SDS), however, capture efficiencies with the DNA oligomer were significantly greater than with the PNA clamp and PNA oligomer. Recovery and detection efficiencies for target DNA concentrations of ≥100 pg were generally >20% but depended upon the specific probe, solution background, and salt condition. The DNA probe had a lower absolute detection limit of 100 fg of target (830 zM [1 zM = 10−21 M]) in high-salt buffer. In the absence of exogenous DNA (e.g., soil background), neither the bis-PNA nor the PNA oligomer achieved the same absolute detection limit even under a more favorable low-salt hybridization condition. In the presence of a soil background, however, both PNA probes provided more sensitive absolute purification and detection (830 zM) than the DNA oligomer. In varied environmental samples, the rank order for capture probe performance in high-salt buffer was DNA > PNA > clamp. Recovery of 16S rRNA from environmental samples mirrored quantitative results for DNA target recovery, with the DNA oligomer generating more positive results than either the bis-PNA or PNA oligomer, but PNA probes provided a greater incidence of detection from environmental samples that also contained a higher concentration of nontarget DNA and RNA. Significant interactions between probe type and environmental sample indicate that the most efficacious capture system depends upon the particular sample type (and background nucleic acid concentration), target (DNA or RNA), and detection objective.
The development and application of nucleic acid techniques in applied and environmental microbiology (40, 44) have invigorated the field by liberating researchers from many constraints imposed by laboratory cultivation of microorganisms. The power and utility of molecular biology, however, depend upon our ability to efficiently extract and purify nucleic acids from various sample matrices. In relatively high biomass settings (>108 cells g−1 or ml−1), numerous extraction and purification procedures allow fairly sensitive recovery and detection of rare, spiked targets in complex genetic and chemical backgrounds (17, 23, 34, 41, 42, 50, 51, 53). The carrier effect of nontarget nucleic acids undoubtedly aids in the purification process under these circumstances but is negligible for low-biomass samples such as those recovered from subsurface sediments (2, 4, 7, 12, 19, 20, 25). At 104 cells g−1, for example, only picogram quantities of DNA are available for isolation and subsequent analysis. If we assume 100% extraction and purification efficiency, direct probing methods are too insensitive to detect DNA targets at these concentrations and biomass levels (≤106 cells), which necessitate the use of PCR-based techniques for the detection and quantification of microorganisms in these environments. However, the combined effects of reduced extraction efficiency for native cells (typically ca. 10% for unspiked sediments) (9, 28, 45) and the possible effects of PCR inhibition (47) or bias (10, 43, 46) indicate that microbial or gene detection (let alone quantification) in low-biomass settings is a tremendous challenge that frequently ends in nondetection, even with PCR-based techniques. We are therefore interested in developing new extraction and purification strategies that will increase the utility of molecular techniques for low-biomass environmental samples.
The affinity hybridization and purification principle has been employed for DNA, rRNA, and mRNA isolation from soil extracts (6, 15, 24) and is perhaps the most direct method for recovering nucleic acids from solution in a form that is suitable for PCR analysis. At low target concentrations and for nucleic acid targets containing significant secondary and tertiary structure, however, solution-phase hybridizations are constrained by the thermodynamic, kinetic, and equilibrium binding properties of DNA probes, potentially limiting their efficiency in a hybridization-capture format (i.e., for ribosomal DNA [rDNA] or rRNA isolation). Peptide nucleic acids (PNAs) (Fig. 1A) represent a new class of nucleotide analog containing a neutral, archiral backbone of repeating N-(2-aminoethyl)-glycine units linked by amide bonds, with purine and pyrimidine bases attached by methylene carbonyl linkages (33). The resulting noncharged nature of the PNA backbone is an important feature with many interesting biophysical consequences. For example, a PNA-DNA or PNA-RNA hybrid has much higher thermal stability than the corresponding DNA-DNA or DNA-RNA duplex, and the Tm of PNA duplexes is insensitive to ionic strength. Consequently, PNA hybrids can form under extremely stringent conditions which strongly disfavor DNA or RNA duplex formation (16, 18, 27). PNAs also show greater specificity in binding to complementary DNA, since a PNA-DNA mismatch is more destabilizing than a mismatch in a DNA-DNA duplex (16). Bis-PNAs, or PNA clamps (Fig. 1B), are homopyrimidine sequences that form stable triplexes with single-stranded DNA and “invade” double-stranded DNA without prior heat denaturation (1, 5, 31, 33). Bis-PNA binding to DNA is essentially irreversible due to a “locking” effect of the Hoogstein-binding strand (29), an equilibrium property of great interest and potential value for the capture and purification of dilute nucleic acids from crude environmental lysates.
FIG. 1.

(A) Primary structures of PNA and DNA oligomers. (B) Sequences (bold) and target secondary structure for Geobacter-specific PNA, bis-PNA (clamp), and DNA probes. 16S rRNA secondary structures are based upon those of Guttell (22), utilizing the Escherichia coli numbering system.
The unique properties and hybridization characteristics of PNAs have been explored primarily within the context of antisense applications (26) but also for mutation analysis (38, 48), sequence-specific suicide transcription (27) and transcriptional activation (52), inhibition of restriction enzyme cleavage (32) and telomerase activity (35), and sensor development (55), with some very recent applications to environmental microbiology (54, 57). The physical properties of PNAs, however, suggest that they will be very useful as sequence-specific affinity purification probes (39), with speed, sensitivity, and specificity characteristics superior to those of DNA probes under identical hybridization conditions. However, PNA clamps have not yet been investigated for their affinity purification properties, nor have PNA oligomers been investigated for their binding properties in the presence of soluble soil constituents (humic acids) encountered during environmental molecular analyses. The purpose of this work, then, was to evaluate the efficacy of PNA clamps and PNA oligomers as affinity purification reagents for low-copy targets in crude nucleic acid extracts derived from environmental samples compared to standard DNA probes.
MATERIALS AND METHODS
Control DNA.
A specific nucleic acid target for all experiments was obtained from the iron-reducing organism Geobacter chapelleii. Cultures of G. chapelleii were grown anaerobically in 100-ml serum bottles as described elsewhere (13). Cultures were grown in the dark at ambient temperature for 2 weeks prior to DNA isolation. Geobacter cells were collected by centrifugation, and genomic DNA was isolated by a standard CTAB (hexadecyltrimethylammonium bromide) procedure (3). Genomic DNA was sheared to 2 to 10 kbp in size by ballistic disintegration for 1 min at 5,000 oscillations s−1 in an 8-place bead beater (BioSpec Products, Inc., Bartlesville, Okla.) with 12 μg of DNA, 0.75 g of 0.1-mm glass beads, and 500 μl of water. Residual DNA in the bead void volume was collected with an additional 500-μl water rinse. DNA concentration was determined by fluorometry, and sizes were determined by gel electrophoresis on 1.2% agarose (SeaKem GTG; FMC, Rockland, Maine) gels in 1× Tris-acetate-EDTA (TAE) running buffer, both containing ethidium bromide. Control RNA was isolated from Geobacter cells with a guanidine isothiocyanate-phenol-Sarkosyl extraction and immunomagnetic separation protocol described elsewhere (15) and quantified by UV absorption prior to use.
Nucleic acid extraction from environmental samples.
Three environmental samples were selected for extraction, including a surface soil, subsurface sediment, and street-level class 2 industrial air filter (glass bag, 8.36-m2 surface area, 85% efficiency) from a major metropolitan area, representing several levels of organic C, N, mineral (metal) content, and total biomass. Fifty grams of soil or sediment or 2 g of air filter was homogenized with 25 g of 0.1-mm glass beads and 200 ml of high-salt extraction buffer (0.2 M NaPO4, 0.1 M EDTA, 0.5% sodium dodecyl sulfate [SDS] [pH 8.0]). Solids were removed by centrifugation and extracted once more (without SDS), and like supernatants were combined. Ten-milliliter aliquots of crude lysate were dialyzed against sterile water (3,000 molecular weight cutoff), ethanol precipitated, and reconstituted to their original volume with low-salt buffer (10 mM NaPO4, 5 mM EDTA, 0.025% SDS [pH 8.0]). To estimate the nontarget DNA concentration in crude lysates, 2 ml of each crude DNA extract was desalted on Amicon Centricon-100 cartridges and loaded directly onto a 1% agarose gel, poststained in ethidium bromide. Relative to a Geobacter genomic DNA standard (prepared above), the amount of DNA in the soil, sediment, and air filter extracts was ca. 1.2 μg (soil), <0.05 μg (sediment), or <0.005 μg (air filter) ml−1, with the majority of DNA sheared to ca. 4 to 20 kpb. At least two replicate extractions were performed for each environmental sample.
Solution-phase hybridization and magnetic capture.
DNA oligomer Gbc.1400r (5′ biotin-PEG-PEG-GGACCAATCGACTCCCGT), containing two 18-atom polyethylene glycol (PEG) linkers, was synthesized and purified by high-pressure liquid chromatography (HPLC) by Keystone Labs (Menlo Park, Calif.) and reconstituted in 10 mM Tris–1 mM EDTA (pH 7.8). PNA oligomer Gbc.1400r (5′ biotin-OOOO-GGACCAATCGACTCCCGT) and Gbc.clamp (5′ biotin-OOO-JJTTTTJJ-OOO-CCTTTTCC) were synthesized and HPLC purified by PerSeptive Biosystems (Framingham, Mass.), reconstituted in 0.1% trifluoroacetic acid, and lyophilized in working aliquots of 150 or 15,000 pmol; O's denote the 9-atom hydrophobic spacer 8-amino-3,6-dioxaoctanoic acid, and J's are pseudoisocytosine. For affinity hybridization and capture experiments, lyophilized PNA probes (150 pmol) or 3-μl aliquots of DNA probe (at 50 pmol μl−1) were resuspended in 200 μl of high-salt (0.2 M sodium phosphate, 0.1 M disodium EDTA, 0.25% SDS [pH 8.0]) or low-salt buffer (0.05× high-salt buffer) for 20 min at room temperature. Four microliters of target DNA (1 or 100 ng) or 16S rRNA (1 ng) was then added to the probe with or without exogenous DNA and environmental extracts, and the mixture was heat denatured in boiling water for 5 min. Denatured DNA-probe solution was quick-chilled on ice and incubated at 55°C for 10 min, 4 h, or overnight. The hybridization mixture was then added directly to 0.6 mg of streptavidin-coated paramagnetic particles that had been washed in 0.5× SSC (20× SSC is 3.0 M NaCl plus 0.3 M trisodium citrate · 2H2O [pH 7.0]) according to the manufacturer's instructions (Promega; Madison, Wis.), and biotinylated hybrids were captured for 10 min at room temperature with intermittent mixing. Magnetic beads were washed three times in 0.5 ml of 0.5× SSC at room temperature, and target DNA (or RNA) was eluted in two 50-μl water washes at 90°C for 2 min each. Eluants were lyophilized to dryness and resuspended in 100 μl of water for PCR detection and enumeration. At least two replicate nucleic acid captures were performed for each nucleic acid extract or target.
Absolute detection limit.
Trends from the solution-phase results were used to define a hybridization time for absolute capture and detection limits of the PNA-DNA oligonucleotides and PNA clamp in both high- and low-salt buffer systems. Based on these results, 10-fold serial dilutions of Geobacter genomic DNA (4 to 10 kpb, 1 ng to 100 fg) were prepared and captured as described above for solution-phase hybridizations, using a 10-min hybridization time for PNA and DNA oligonucleotides in high- and low-salt buffers. PCR detection and enumeration proceeded as described below except that for target DNA quantities of ≤100 pg (830 aM) and RNA detection, a simple yes-or-no PCR determination was performed rather than a semiquantitative enumeration of capture efficiency. At least three replicate captures were performed for each estimate of the absolute detection limit.
qPCR.
Geobacter 16S rDNA was detected and enumerated with a dilution-to-extinction quantitative PCR (qPCR) approach (8) to estimate capture efficiency at subfemtomolar concentrations of target DNA. PCR primers S-δ401F-20 [5′ AA(G/C)CCTGACGCAGC(A/G)ACGCC] and S-δ683aR-20 (5′ TCTACGGATTTCACTCCTACAC) (modified from reference 21) were synthesized by Keystone Labs (Camarillo, Calif.). PCR amplification was carried out in 25-μl total volume, utilizing an MJ Research (Watertown, Mass.) Tetrad thermal cycler and 0.2-ml thin-walled reaction tubes. Purified DNA was serially diluted in a fivefold series immediately prior to PCR, such that the first sample in the dilution series represented 2.5% of the purified DNA eluant, or at least 5 pg of target DNA, assuming 100% capture-elution efficiency. Final reaction conditions were 5 μl of purified or diluted DNA, 10 mM Tris (pH 8.3), 50 mM KCl, 2.5 mM MgCl2, 200 μM each of the four deoxynucleoside triphosphates, 0.2 μM each of the primers, 0.375 μg of bacteriophage T4 gene 32 protein (Boehringer Mannheim, Indianapolis, Ind.), and 0.625 U of LD-Taq polymerase (Perkin-Elmer, Foster City, Calif.), which had been pretreated with TaqStart antibody at the recommended concentration (Sigma, St. Louis, Mo.). Assembled reactions were heated to 80°C for 5 min (hot start) and amplified with 5 cycles at 94°C for 40 s, 60°C for 10 s, and 72°C for 75 s, followed by 40 cycles at 94°C for 12 s, 65°C for 10 s, and 72°C for 80 s, with a 2-s extension per cycle. A final 20-min, 72°C extension was performed before chilling the reactions to 4°C. The entire contents of each PCR were analyzed on 1% NuSieve–1% Seakem GTG agarose (FMC Bioproducts) gels in 1× TAE running buffer, both containing ethidium bromide, and gel images were captured with a Bio-Rad (Hercules, Calif.) Fluor-S imager and Molecular Analyst software.
Statistics.
The classical model for dilution-to-extinction qPCR is derived from most-probable number theory and assumes that organisms (or target nucleic acids) are randomly distributed in solution during dilution steps and that a single organism (or nucleic acid copy) gives rise to a positive signal. From a process perspective, the fundamental assumptions of most-probable-number qPCR break down, and competitive PCR techniques become difficult to validate (8). Thus, the qPCR technique employed here makes no assumptions regarding the PCR detection limit and instead recognizes that the probability of detecting a given target will vary from day to day, from experiment to experiment, and with changing environmental or experimental conditions. Under this framework, the probability of a positive PCR result [P(pos)] as a function of copy number in the PCR is given by the function
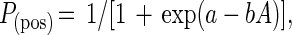 |
where a and b are positive constants that control the shape of the response curve and A is the starting DNA copy number. External calibration runs with samples of known starting copy number are used to estimate the values of a and b.
The experimental design resulted in a 2 × 2 PCR matrix for each sample-variable combination, two replicate dilution series and extinction points from each of two replicate capture experiments. In this manner, our statistical model for qPCR actually accounts for process-level variability that occurs prior to the PCR measurement itself. Four positive control dilution series were also performed for each PCR experiment and used to calibrate the qPCR calculation. The observation and critical value for each dilution series are the last dilution giving rise to a positive result (on an agarose gel), with no attempt to quantify the amplicon (by densitometry) within the last positive dilution. The sum of the four extinction points is a random variable with a discrete unimodal probability distribution that is dependent on the target copy number in the PCR.
We used techniques described by Mood et al. (30) for unimodal probability distributions to construct 95% confidence intervals around the target copy number (or quantity in femtograms) in the PCR. Point estimates for mean recovery of target DNA were calculated by a maximum-likelihood approach. Estimates of target DNA recovery were converted to percent recovery by dividing by 106 fg, the concentration of template DNA for each capture experiment. A log transformation of the data was used to stabilize the variance, and analysis of variance was then used to compare estimates of capture efficiency between treatments, identify interactions between treatment variables, and illuminate trends in the data. Calculations performed in this manner therefore do not require absolute knowledge of target copy number in the genome, but we assume 1 rDNA copy per genome and a genome size of 1 Mb for G. chapelleii (D. Lovley, personal communication).
When interactions were significant, pairwise comparisons of log-transformed treatment means were used to identify statistically significant effects within fixed factor levels. Bonferroni's multiple comparison procedure with a simultaneous confidence level of 90% was used to control the probability of incorrectly declaring at least one pair of means significantly different for a family of comparisons. In addition, the exquisite sensitivity of PCR, a relatively insensitive fivefold dilution series, and day-to-day variations in amplification efficiency led to large levels of uncertainty for estimated DNA capture efficiency. Thus, some calculations of capture efficiency may appear to be intuitively different but are in fact statistically insignificant.
RT-PCR.
Capture and purification of target rRNA from crude environmental extracts was assessed by a simple reverse transcription (RT)-PCR procedure, with PCR product accumulation measured by densitometry using Molecular Analyst (Bio-Rad) software. Primer S-δ683aR-20 (2 pmol) was used to prime the RT reaction from 5 μl of purified rRNA (50 pg, assuming 100% efficiency). Reaction conditions were essentially as described elsewhere (15), except that Moloney murine leukemia virus RT (Life Technologies, Gaithersburg, Md.) was used instead of SuperScriptII RT. Positive controls included 8 fg to 5 pg of purified Geobacter 16S rRNA; the specificity of the RT-PCR was verified by performing reactions with and without added RT and with and without RNase treatment, as described previously (15).
RESULTS AND DISCUSSION
Rationale.
The theoretical and biochemical basis for PNA interaction with nucleic acids has been described in detail elsewhere (18, 29, 49). Our general interest in PNA stems from studies indicating that PNA oligos will bind as well as or better than DNA oligos under identical solution conditions (ionic strength or temperature). PNA clamps further represent a unique two-step binding and locking mechanism with the potential to realize both high target specificity and high target affinity (29). These properties were of particular interest for the affinity purification of very dilute, large nucleic acid targets and eventual automation of nucleic acid purification procedures for such samples. The purpose of this study was therefore to directly compare a PNA clamp, PNA oligomer, and DNA oligomer for the affinity purification of dilute 16S rDNA and rRNA targets in complex environmental samples.
qPCR and statistics.
Evaluating nucleic acid capture and purification efficiency at subfemtomolar concentrations (≤106 copies) required a technique other than scintillation counting of radiolabeled nucleic acid to assess the functional purity and quality of nucleic acids recovered from environmental samples. Thus, we developed and used a qPCR method and statistical algorithms to fulfill these requirements. The premise of the replicative limiting-dilution PCR technique has been described in detail elsewhere (8) but takes into account process-level variation that is not associated only with PCR amplification efficiency. Because a finite volume is drawn from each template dilution prior to PCR and PCR is prone to molecular sampling error at low target concentrations, it is possible to achieve target amplification at a 56 dilution in one series (e.g., the sample) but only a 55 dilution in another (e.g., the positive control). Since the technique is prone to discrete sampling error and is based upon a fivefold dilution curve and calculations of capture efficiency are based upon the external calibration standard curve, it is therefore possible to calculate a capture efficiency in excess of 100%. In these cases, we maintained the 95% confidence intervals in excess of 100% for purposes of statistical comparisons but report the capture efficiency as 100% in the appropriate tables.
Target recovery at 8.3 fM concentration.
Our central hypothesis for this study was that a PNA clamp would purify and recover significantly more target DNA from solution than either PNA or DNA oligomers under identical solution hybridization conditions. For statistical analyses, five probe-salt concentration combinations (probe condition) were considered: DNA in high salt, PNA in high salt, PNA clamp in high salt, PNA in low salt, and PNA clamp in low salt. Three solution hybridization times (10 min, 4 h, and overnight) and two chemical backgrounds (clean and soil) were also considered.
A three-factor analysis of variance between probe condition, hybridization time, and chemical background showed no significance (P = 0.275). However, significant interactions between hybridization time and probe condition (P = 0.012) and between background and probe condition (P = 0.001) were observed. In an attempt to isolate hybridization time and solution background effects individually, the data were collapsed over hybridization time (Table 1) and background (Table 2), and a separate analysis-of-variance model was used to investigate treatment effects in each table. The relative standard errors for the values in Tables 1 and 2 are 42.6 and 34.3%, respectively.
TABLE 1.
Capture and elution efficiencies averaged over different hybridization timea
| Salt condition | Probe | Capture efficiency (%)
|
|
|---|---|---|---|
| Clean | Soil | ||
| High | Gbc.clamp | 3a | 30a |
| PNA.1400r | 8ab | 42ab | |
| DNA.1400r | 24b | 31a | |
| Low | Gbc.clamp | 100c | 100b |
| PNA.1400r | 100c | 48ab | |
| DNA.1400r | ND | ND | |
Pairwise comparisons of probe conditions were made separately within each column. Letters indicate values within a data column that are statistically similar but are not transferable across columns. Capture efficiencies calculated >100% are reported as 100%, as described in the Discussion. The soil background contained approximately 240 ng of nontarget DNA and 5.8 μg equivalent of soluble soil constituents in 200 μl of lysate. ND, not determined.
TABLE 2.
Capture and elution efficiencies averaged over different chemical backgroundsa
| Salt condition | Probe | Capture efficiency (%)
|
||
|---|---|---|---|---|
| 10 min | 4 h | Overnight | ||
| High | Gbc.clamp | 11a | 3a | 28a |
| PNA.1400r | 15a | 13ab | 29a | |
| DNA.1400r | 52ab | 25bc | 16a | |
| Low | Gbc.clamp | 100b | 100c | 100a |
| PNA.1400r | 32ab | 100c | 73a | |
| DNA.1400r | ND | ND | ND | |
See Table 1, footnote a.
The most striking result from Tables 1 and 2 is that oligomer Gbc.clamp in low-salt buffer captured significantly more target DNA than oligomer DNA.1400r in high salt (Table 1), even after accounting for the highly variable PCR enumeration strategy (above). Relative to oligomer PNA.1400r, however, the clamp showed no statistically significant advantage with respect to the environmental background (Table 1) or hybridization time (Table 2), and the capture efficiencies with the PNA and DNA oligomers were statistically similar. Conclusions of nonsignificance in these cases may be related solely to the variability in the qPCR strategy, which encompasses variation in both the solution-phase capture and PCR. However, trends in the data (Tables 1 and 2) further suggest that (i) target recovery under low-salt hybridization conditions is enhanced with Gbc.clamp over PNA.1400r, (ii) PNA.1400r recovers more target than Gbc.clamp under high-salt conditions, (iii) PNA clamps and PNA oligomers in high salt recover more target DNA in the presence of excess nontarget DNA than they do in the clean background, and (iv) both PNA probes tended to recover more target DNA under low-salt conditions than they did under high-salt conditions. These results and trends are consistent with our earlier findings (14) and provide further evidence that PNA capture efficiency is salt dependent, regardless of the independence of PNA Tm on ionic strength (16). The reason for enhanced PNA performance in the equivalent of a high-biomass background relative to a low-biomass background is unknown.
Under high-salt hybridization conditions, capture efficiencies with the DNA.1400r oligo were significantly greater than with the Gbc.clamp and PNA.1400r capture probes, consistent with our previous findings (14). This result and conclusion run counter to widely held perceptions of PNA efficiency and our principal hypothesis but should not be construed as conflicting data. That is, PNA clamps and oligomers have never been tested against DNA probes under solution hybridization conditions that mimic a low-biomass environmental nucleic acid extract and practical purification method; namely, very dilute target concentrations (femtomolar), relatively large nucleic acid targets (to 10 kb), and functional assay (e.g., affinity purification procedure instead of Tm determinations). The choice of PNA clamp, PNA oligo, or DNA oligo capture probes therefore depends upon the objectives of the entire analytical process (sample extraction through detection). Nonetheless, the assumption that a PNA clamp or PNA oligomer will outperform a DNA oligomer under all conditions is clearly unwarranted.
Absolute detection limits.
Absolute recovery and detection limits were assessed with a 10-min solution-hybridization of 16S rDNA targets in sheared genomic DNA. For target DNA concentrations of ≤100 pg, recovery and detection efficiencies were generally >20% but depended upon the specific probe, solution background, and salt condition. For example, DNA.1400r capture efficiencies in high-salt buffer alone (clean background) were 25 to 134% at 100 ng to 100 pg of target, whereas the Gbc.clamp and PNA.1400r capture efficiencies under the same conditions ranged from 6 to 56% and 18 to 32%, respectively.
Differences in PNA probe performance relative to DNA.1400r also extended into subfemtomolar concentrations, as summarized in Table 3. In high salt and the absence of a soil background, the DNA.1400r oligomer had a lower absolute detection limit of 100 fg of target, representing approximately 100 copies of 16S rDNA at 830 zM concentration (1 zM = 10−21 M). At a 1.6-fg detection limit for the PCR assay, the efficiency of target capture was therefore ≥1.6%. In the absence of a soil background (i.e., exogenous DNA), neither the Gbc.clamp nor the PNA.1400r oligomer achieved the same absolute detection limit, even under the low-salt condition clearly favorable to PNA performance at 1 ng of target DNA (Tables 1 and 2). In the presence of a soil background (i.e., with exogenous DNA), both PNA probes provided more sensitive absolute detection than the DNA.1400r oligomer. This result is in agreement with our previous study of PNA probes targeting universal 16S rRNA regions 519r and 786r (14).
TABLE 3.
Capture and detection limits of different probesa
| Background | Salt condition | Probe | Detection at input level:
|
Detection limit (fg) | ||
|---|---|---|---|---|---|---|
| 10 pg | 1 pg | 100 fg | ||||
| Clean | High | Gbc.clamp | + | − | − | 1.6 |
| PNA.1400r | + | + | − | |||
| DNA.1400r | + | + | + | |||
| Low | Gbc.clamp | − | − | − | 8 | |
| PNA.1400r | − | − | − | |||
| DNA.1400r | − | − | − | |||
| Soil | High | Gbc.clamp | − | − | − | 3.6 |
| PNA.1400r | + | + | + | |||
| DNA.1400r | − | − | − | |||
| Low | Gbc.clamp | + | + | + | 6.8 | |
| PNA.1400r | + | + | + | |||
| DNA.1400r | + | + | − | |||
Absolute capture and detection limit of G. chapelleii genomic DNA with bis-PNA, PNA, and 16S rDNA probes in 200-μl volumes for 10 min at 55°C. The PCR detection limit for a group of results is shown. One-nanogram capture efficiencies for all salt and background conditions are reported separately in Tables 1 and 2.
PNA.1400r exhibited a 100-fg recovery-detection limit under both high- and low-salt conditions, with at least 6.8% capture efficiency. The Gbc.clamp, on the other hand, only achieved the 100-copy detection limit in the low-salt buffer. Under the high-salt condition, the Gbc.clamp was titrated by nonspecific interactions with endogenous soil DNA, as evidenced by multiple, nonspecific PCR products that were not present under the clean background conditions (not shown). As with the Gbc.clamp in high salt and a soil background, DNA.1400r oligomer was titrated off the Geobacter target by related organisms (not shown). Under the low-salt condition, however, all PNA and DNA oligomers showed greater binding specificity, reflected in a lower absolute detection limit and reduced incidence of nontarget bands arising from the environmental extracts. Differences in absolute detection limits cannot be attributed to differences in PCR detection limits, since PCR detection limits maximally varied 5-fold, whereas differences in capture efficiency and detection limits were measured over a 10-fold range.
Interestingly, oligomer DNA.1400r achieved a 1-pg capture-detection limit (8.3 aM); in the soil background under the low-salt condition (approximately 50 mM in Na+, including EDTA and SDS salts), whereas the detection limit under a high-salt condition was >10 pg. This result was somewhat surprising, as “standard” DNA hybridization protocols typically utilize Na+ concentrations in excess of 200 mM (3), with the assumption that DNA-DNA duplexes will not form under low-ionic-strength conditions. This assumption appears to be unfounded for the solution-phase hybridization conditions employed here, implying that even DNA probes can be used for affinity purification of target nucleic acids under low-ionic-strength conditions that will promote “breathing” or relaxation of large (many kilobase pairs) targets. Preliminary results in our laboratory suggest that the efficacy of DNA-DNA binding at extremely low ionic strength is, however, dependent upon whether the experiment is performed in a solution- or solid-phase hybridization format.
DNA and rRNA recovery from various environmental samples.
The results in Tables 1 and 2 suggest that PNA clamp and PNA oligomer performance in the high-salt buffer is significantly improved by exogenous (nontarget) DNA or (possibly) coextracted contaminants. The affinity purification approach was therefore applied to a street-level industrial air filter and subsurface sediment, each containing different levels of background DNA, organic C, and qualitatively different types of contaminants (e.g., diesel soot and humic acids). Recovery efficiencies at an 8.3 fM target DNA concentration (200-μl volume) in the standard (high-salt) buffer condition are consistent with the background DNA hypothesis, since both PNA probes clearly recovered more Geobacter target from environmental samples in the presence of relatively high background DNA concentrations (Table 4).
TABLE 4.
Effect of environmental background on capture efficiencya
| Background | DNA content (ng/ml) | Organic carbon content (μg/ml) | DNA recovery (%) with probe:
|
||
|---|---|---|---|---|---|
| Gbc.clamp | PNA.1400r | DNA.1400r | |||
| Air filter | 5 | 103.36 | 1 | 2 | 86 |
| Sediment | 50 | 1.54 | 12 | 56 | 94 |
| Soil | 1,200 | 29.34 | 26 | 29 | 68 |
Capture experiments were performed with 1 ng of spiked G. chapelleii sheared genomic DNA in 200 μl for 10 min at 55°C in high-salt buffer. See Results for discussion of statistical significance. DNA content for each environmental sample was estimated by gel electrophoresis against Geobacter sheared genomic DNA standards. Organic carbon was determined per milliliter of water leachate.
An analysis of variance showed a significant interaction between probe type and environmental background (P < 0.0001), indicating that the effect of either factor on capture efficiency depends on the level of the other factor. The relative standard error for the measurements in Table 4 is 78.7%. Pairwise comparisons of capture efficiencies by probe type (Gbc.clamp, PNA.1400r, and DNA.1400r) were performed within each environmental sample type (clean, air filter, sediment, and soil). Statistically significant differences in probe efficacy were only observed in the air filter extract. In this sample, target DNA capture efficiency was significantly higher using the DNA.1400r than either the PNA.1400r or Gbc.clamp probe.
Pairwise comparisons of the environmental sample types within each probe type were performed in the same manner. Within probe type, the efficacy of the DNA.1400r oligomer was statistically insignificant for all sample types; the PNA.1400r oligomer showed significant differences in the air filter-sediment and air filter-soil comparisons; and the Gbc.clamp showed significant differences between the air filter-soil pair only. The rank order for capture probe performance in high-salt buffer over all samples was thus DNA > PNA > clamp.
Recovery of 1 ng of 16S rRNA (1.2 × 109 copies; 2 nM) from environmental samples was similar to the quantitative results for DNA target recovery (Table 5). In high-salt buffer, the DNA.1400r oligomer produced more positive results than either the Gbc.clamp or PNA.1400r probes, regardless of the RT-PCR detection limit. In the presence of environmental backgrounds (i.e., exogenous nucleic acid), the Gbc.clamp was as efficacious as the DNA.1400r oligomer. Using RT-PCR detection limits, we can estimate RNA recovery efficiency for each probe type and environmental background. That is, only half of the total PCR product is detected on a gel; we incurred a 25× dilution of cDNA into the PCR volume; a 5× dilution of 16S rRNA target into the RT-PCR; and only 1/20 of the total 16S rRNA was used as the template in the initial RT-PCR. Thus, the PCR detection limit multiplied by the 5,000× conversion factor yields total femtograms of recovered 16S rRNA. RT-PCR nondetection in clean, air filter, sediment, and soil extracts therefore suggests ≤67, 20, 9, and 100% capture efficiency, respectively. Whereas the results in Table 5 indicate the incidence of rRNA detection, qualitative comparison of RT-PCR product accumulation (e.g., densitometry) suggested that the Gbc.clamp oligomer actually recovered more target rRNA than either the DNA.1400r or PNA.1400r oligomer for all environmental sample extracts (not shown). A more sensitive and precise quantitative RT-PCR assay or technique will be required to verify increased capture efficiency of RNA from environmental samples by PNA clamps under the salt and buffer conditions reported here. Nevertheless, these results show that a PNA clamp can indeed recover RNA targets from environmental samples. Given the exceptional performance of PNA probes (in general) in low-salt conditions (Tables 1 and 2), it is conceivable that a PNA clamp would also be the best affinity purification reagent for recovery of dilute rRNAs in a high-biomass background, although we did not explicitly address this hypothesis in this study.
TABLE 5.
Detection of G. chapelleii 16S rRNAa
| Background | No. of positive samples/4 tested with probe:
|
Detection limit (fg) | ||
|---|---|---|---|---|
| Gbc.clamp | PNA.1400r | DNA.1400r | ||
| Clean | 0 | 1 | 3 | 134 |
| Air filter | 4 | 2 | 4 | 40 |
| Sediment | 4 | 2 | 4 | 18 |
| Soil | 4 | 4 | 4 | 200 |
Two-hundred-microliter aliquots of environmental extracts were spiked with 1 ng of RNA and hybridized for 10 min at 55°C. The numbers of positive samples from four extractions given the RNA detection limit of the RT-PCR assay are reported.
Summary.
More efficient nucleic acid purification procedures are needed not only for routine laboratory handling of low-biomass environmental samples, but also for the development of fully automated, real-time biodetection devices for applications in bioremediation, biological warfare and counter terrorism, microbial ecology, and public health. This study represents the first attempt to apply PNA clamps and PNA oligomers for the affinity purification of DNA and RNA from environmental extracts. Clearly, significantly better solution hybridization conditions for PNA clamps and oligomers relative to DNA oligomers are found in low-ionic-strength buffers. However, the most efficacious nucleic acid extraction buffers for low-biomass samples are currently high-salt formulations (11, 36, 37). First-generation automated systems for environmental analysis, then, will require affinity purification of target nucleic acids from crude environmental extracts in a high-salt buffer, and for this reason the majority of the work reported here is focused on PNA versus DNA probe performance in 0.4 M Na+ buffer. Some of our data (e.g., Table 4), however, indicate that DNA probes can also function as affinity purification reagents in a low-ionic-strength buffer; whether or not PNA capture oligomers will outperform DNA oligomers under these conditions must be tested explicitly.
Significant interactions between probe type and environmental sample indicate that the best capture probe depends upon the particular sample type (and background DNA concentration). This study indicates that a PNA clamp is more efficacious in recovering target DNA under a low-biomass condition than DNA or PNA oligomers under low-salt conditions, an important result for the analysis of subsurface sediments or routine analysis of aerosol samples for biowarfare agents. Our prior studies also suggest that PNA oligomers have distinct advantages over DNA probes when first tethered to a solid support (14). Thus, target binding to immobilized PNA under low-salt conditions may have certain advantages not only within the context of solid-phase nucleic acid purification strategies, but also as capture-detection probes in a microarray format (56). Under high-salt extraction and purification conditions, however, there was no evidence that PNA probes (clamps or oligomers) recover any more 16S rDNA or rRNA target than a comparable DNA oligomer in any of the environmental extracts tested here. The overall efficacy of PNA probes, then, must ultimately be gauged within the context of the entire bioanalytical process, from sample collection through detection. We will therefore continue to investigate and develop PNA-based bioanalytical techniques for low-biomass environmental samples that exploit the unique biophysical properties of PNA.
ACKNOWLEDGMENTS
We thank Ivar Jensen for expert technical advice and assistance with PNA probe design and synthesis.
This work was supported by the U.S. Department of Energy (DOE) Laboratory Technology Research (LTR) Program with in-kind contributions from PerSeptive Biosystems. Pacific Northwest National Lab is operated by Battelle Memorial Institute for the U.S. DOE under contract DE-AC06-76RLO 1830.
REFERENCES
- 1.Almarsson Ö, Bruice T C. Peptide nucleic acid (PNA) conformation and polymorphism in PNA-DNA and PNA-RNA hybrids. Proc Natl Acad Sci USA. 1993;90:9542–9546. doi: 10.1073/pnas.90.20.9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amy P S, Haldeman D L, Ringelberg D, Hall D H, Russell C. Comparison of identification systems for classification of bacteria isolated from water and endolithic habitats within the deep subsurface. Appl Environ Microbiol. 1992;58:3367–3373. doi: 10.1128/aem.58.10.3367-3373.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons, Inc.; 1995. [Google Scholar]
- 4.Balkwill D L. Numbers, diversity, and morphological characteristics of aerobic, chemoheterotrophic bacteria in deep subsurface sediments from a site in South Carolina. Geomicrobiol J. 1989;7:33–52. [Google Scholar]
- 5.Betts L, Josey J A, Veal J M, Jordan S R. A nucleic acid triple helix formed by a peptide nucleic acid-DNA complex. Science. 1995;270:1838–1841. doi: 10.1126/science.270.5243.1838. [DOI] [PubMed] [Google Scholar]
- 6.Bogan B W, Schoenike B, Lamar R T, Cullen D. Manganese peroxidase mRNA and enzyme activity levels during bioremediation of polycyclic aromatic hydrocarbon-contaminated soil with Phanerochaete chrysosporium. Appl Environ Microbiol. 1996;62:2381–2386. doi: 10.1128/aem.62.7.2381-2386.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brockman F J, Kieft T L, Fredrickson J K, Bjornstad B N, Li S W, Spangenburg W, Long P E. Microbiology of vadose zone paleosols in south-central Washington state. Microb Ecol. 1992;23:279–301. doi: 10.1007/BF00164101. [DOI] [PubMed] [Google Scholar]
- 8.Chandler D P. Redefining relativity: quantitative PCR at low template concentrations for industrial and environmental microbiology. J Ind Microbiol. 1998;21:128–140. [Google Scholar]
- 9.Chandler D P, Brockman F J. Estimating biodegradative gene numbers at a JP-5 contaminated site using PCR. Appl Biochem Biotechnol. 1996;57/58:971–982. doi: 10.1007/BF02941777. [DOI] [PubMed] [Google Scholar]
- 10.Chandler D P, Brockman F J, Fredrickson J K. Effect of PCR template concentration on the composition and distribution of total community 16S rDNA clone libraries. Mol Ecol. 1997;6:475–482. doi: 10.1046/j.1365-294x.1997.00205.x. [DOI] [PubMed] [Google Scholar]
- 11.Chandler D P, Brockman F J, Fredrickson J K. Phylogenetic diversity of Archaea and Bacteria in a deep subsurface paleosol. Microb Ecol. 1998;36:37–50. doi: 10.1007/s002489900091. [DOI] [PubMed] [Google Scholar]
- 12.Chandler D P, Brockman F J, Fredrickson J K. Use of 16S rDNA clone libraries to study changes in a microbial community resulting from ex situ perturbation of a subsurface sediment. FEMS Microbiol Rev. 1997;20:217–230. [Google Scholar]
- 13.Chandler D P, Schuck B L, Brockman F J, Bruckner-Lea C J. Automated nucleic acid isolation and purification from soil extracts using renewable affinity microcolumns in a sequential injection system. Talanta. 1999;49:969–983. doi: 10.1016/s0039-9140(99)00074-0. [DOI] [PubMed] [Google Scholar]
- 14.Chandler, D. P., J. R. Stults, K. A. Anderson, S. Cebula, B. L. Schuck, and F. J. Brockman. Affinity capture and recovery of DNA at femtomolar concentrations with peptide nucleic acid probes. Anal. Biochem., in press. [DOI] [PubMed]
- 15.Chandler D P, Wagnon C A, Bolton H., Jr Reverse transcriptase (RT) inhibition of the PCR at low template concentrations and its implications for quantitative RT-PCR. Appl Environ Microbiol. 1998;64:669–677. doi: 10.1128/aem.64.2.669-677.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egholm M, Buchardt O, Christensen L, Behrens C, Freier S, Driver D A, Berg R H, Kim S K, Norden B, Nielsen P E. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen bonding rules. Nature. 1993;365:566–568. doi: 10.1038/365566a0. [DOI] [PubMed] [Google Scholar]
- 17.Ekendahl S, Arlinger J, Ståhl F, Pedersen K. Characterization of attached bacterial populations in deep granitic groundwater from the Stripa research mine by 16S rRNA gene sequencing and scanning electron microscopy. Microbiology. 1994;140:1575–1583. doi: 10.1099/13500872-140-7-1575. [DOI] [PubMed] [Google Scholar]
- 18.Eriksson M, Nielsen P E. PNA-nucleic acid complexes. Structure, stability and dynamics. Q Rev Biophys. 1996;29:369–394. doi: 10.1017/s0033583500005886. [DOI] [PubMed] [Google Scholar]
- 19.Fredrickson J K, Balkwill D L, Zachara J M, Li S W, Brockman F J, Simmons M A. Physiological diversity and distributions of heterotrophic bacteria in deep cretaceous sediments of the Atlantic coastal plain. Appl Environ Microbiol. 1991;57:402–411. doi: 10.1128/aem.57.2.402-411.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fredrickson J K, Brockman F J, Bjornstad B N, Long P E, Li S W, McKinley J P, Wright J V, Conca J L, Kieft T L, Balkwill D L. Microbiological characteristics of pristine and contaminated deep vadose sediments from an arid region. Geomicrobiol J. 1993;11:95–107. [Google Scholar]
- 21.Fry N K, Fredrickson J K, Fishbain S, Wagner M, Stahl D A. Population structure of microbial communities associated with two deep, anaerobic, alkaline aquifiers. Appl Environ Microbiol. 1997;63:1498–1504. doi: 10.1128/aem.63.4.1498-1504.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guttell R R, Larsen N, Woese C R. Lessons from an evolving rRNA: 16S and 23S rRNA structures from a comparative perspective. Microbiol Rev. 1994;58:10–26. doi: 10.1128/mr.58.1.10-26.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hovanec T A, DeLong E F. Comparative analysis of nitrifying bacteria associated with freshwater and marine aquaria. Appl Environ Microbiol. 1996;62:2888–2896. doi: 10.1128/aem.62.8.2888-2896.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobsen C S. Microscale detection of specific bacterial DNA in soil with a magnetic capture-hybridization and PCR amplification assay. Appl Environ Microbiol. 1995;61:3347–3352. doi: 10.1128/aem.61.9.3347-3352.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kieft T L, Fredrickson J K, McKinley J P, Bjornstad B N, Rawson S A, Phelps T J, Brockman F J, Pfiffner S M. Microbiological comparisons within and across contiguous lacustrine, paleosol, and fluvial subsurface sediments. Appl Environ Microbiol. 1995;61:749–757. doi: 10.1128/aem.61.2.749-757.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knudsen H, Nielsen P E. Antisense properties of duplex- and triplex-forming PNAs. Nucleic Acids Res. 1996;24:494–500. doi: 10.1093/nar/24.3.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Larsen H J, Nielsen P E. Transcription-mediated binding of peptide nucleic acid (PNA) to double-stranded DNA: sequence-specific suicide transcription. Nucleic Acids Res. 1996;24:458–463. doi: 10.1093/nar/24.3.458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee S Y, Bollinger J, Bezdicek D, Ogram A. Estimation of numbers of an uncultured soil bacterial strain by a competitive quantitative PCR method. Appl Environ Microbiol. 1996;62:3787–3793. doi: 10.1128/aem.62.10.3787-3793.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lomakin A, Frank-Kamenetskii M D. A theoretical analysis of specificity of nucleic acid interactions with oligonucleotides and peptide nucleic acids (PNAs) J Mol Biol. 1998;276:57–70. doi: 10.1006/jmbi.1997.1497. [DOI] [PubMed] [Google Scholar]
- 30.Mood A M, Graybill F A, Boes D C. Introduction to the theory of statistics. 3rd ed. New York, N.Y: McGraw-Hill; 1974. [Google Scholar]
- 31.Nielsen P E, Christensen L. Strand displacement binding of duplex-forming homopurine PNA to a homopyrimidine duplex DNA target. J Am Chem Soc. 1996;118:2287–2288. [Google Scholar]
- 32.Nielsen P E, Egholm M, Berg R H, Buchardt O. Sequence specific inhibition of DNA restriction enzyme cleavage by PNA. Nucleic Acids Res. 1993;21:197–200. doi: 10.1093/nar/21.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nielsen P E, Egholm M, Berg R H, Buchardt O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991;254:1497–1500. doi: 10.1126/science.1962210. [DOI] [PubMed] [Google Scholar]
- 34.Noble R T, Fuhrman J A. Virus decay and its causes in coastal waters. Appl Environ Microbiol. 1997;63:77–83. doi: 10.1128/aem.63.1.77-83.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Norton J C, Piatyszek M A, Wright W E, Shay J W, Corey D R. Inhibition of human telomerase activity by peptide nucleic acids. Nat Biotechnol. 1996;14:615–619. doi: 10.1038/nbt0596-615. [DOI] [PubMed] [Google Scholar]
- 36.Ogram A, Sayler G S, Barkay T. The extraction and purification of microbial DNA from sediments. J Microbiol Methods. 1987;7:57–66. [Google Scholar]
- 37.Ogram A, Sun W, Brockman F J, Fredrickson J K. Isolation and characterization of RNA from low-biomass deep-subsurface sediments. Appl Environ Microbiol. 1995;61:763–768. doi: 10.1128/aem.61.2.763-768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ørum H, Nielsen P E, Egholm M, Berg R H, Buchardt O, Stanley C. Single base pair mutation analysis by PNA directed PCR clamping. Nucleic Acids Res. 1993;21:5332–5336. doi: 10.1093/nar/21.23.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ørum H, Nielson P E, Jørgensen M, Larsson C, Stanley C, Koch T. Sequence-specific purification of nucleic acids by PNA-controlled hybrid selection. BioTechniques. 1995;19:472–480. [PubMed] [Google Scholar]
- 40.Pace N R, Stahl D A, Lane D J, Olsen G J. The analysis of natural microbial populations by ribosomal RNA sequences. Adv Microb Ecol. 1986;9:1–55. [Google Scholar]
- 41.Picard C, Ponsonnet C, Paget E, Nesme X, Simonet P. Detection and enumeration of bacteria in soil by direct DNA extraction and polymerase chain reaction. Appl Environ Microbiol. 1992;58:2717–2722. doi: 10.1128/aem.58.9.2717-2722.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Powell J L, Loutit M W. Development of a DNA probe using differential hybridization to detect the fish pathogen Vibrio anguillarum. Microb Ecol. 1994;28:365–373. doi: 10.1007/BF00662029. [DOI] [PubMed] [Google Scholar]
- 43.Reysenbach A-L, Giver L J, Wickham G S, Pace N R. Differential amplification of rRNA genes by polymerase chain reaction. Appl Environ Microbiol. 1992;58:3417–3418. doi: 10.1128/aem.58.10.3417-3418.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sayler G S, Layton A C. Environmental application of nucleic acid hybridization. Annu Rev Microbiol. 1990;44:625–648. doi: 10.1146/annurev.mi.44.100190.003205. [DOI] [PubMed] [Google Scholar]
- 45.Steffan R J, Goksoyr J, Bej A K, Atlas R M. Recovery of DNA from soils and sediments. Appl Environ Microbiol. 1988;54:2908–2915. doi: 10.1128/aem.54.12.2908-2915.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki M T, Giovannoni S J. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol. 1996;62:625–630. doi: 10.1128/aem.62.2.625-630.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tebbe C C, Vahjen W. Interference of humic acids and DNA extracted directly from soil in detection and transformation of recombinant DNA from bacteria and yeast. Appl Environ Microbiol. 1993;59:2657–2665. doi: 10.1128/aem.59.8.2657-2665.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thiede C, Bayerdörffer E, Blasczyk R, Wittig B, Neubauer A. Simple and sensitive detection of mutations in the ras proto-oncogene using PNA-mediated PCR clamping. Nucleic Acids Res. 1996;24:983–984. doi: 10.1093/nar/24.5.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tomac S, Sarkar M, Ratilainen T, Wittung P, Nielsen P E, Nordén B, Gräslund A. Ionic effects on the stability and conformation of peptide nucleic acid complexes. J Am Chem Soc. 1996;118:5544–5552. [Google Scholar]
- 50.Toranzos G A, Alvarez A J. Solid-phase polymerase chain reaction: applications for direct detection of enteric pathogens in waters. Can J Microbiol. 1992;38:365–369. doi: 10.1139/m92-062. [DOI] [PubMed] [Google Scholar]
- 51.Tsai Y-L, Olson B H. Rapid method for direct extraction of DNA from soil and sediments. Appl Environ Microbiol. 1991;57:1070–1074. doi: 10.1128/aem.57.4.1070-1074.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vickers T A, Griffith M C, Ramasamy K, Risen L M, Freier S M. Inhibition of NF-κB specific transcriptional activation by PNA strand invasion. Nucleic Acids Res. 1995;23:3003–3008. doi: 10.1093/nar/23.15.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Volossiouk T, Robb E J, Nazar R N. Direct DNA extraction for PCR-mediated assays of soil organisms. Appl Environ Microbiol. 1995;61:3972–3976. doi: 10.1128/aem.61.11.3972-3976.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.von Wintzingerode F, Landt O, Ehrlich A, Göbel U B. Peptide nucleic acid-mediated PCR clamping as a useful supplement in the determination of microbial diversity. Appl Environ Microbiol. 2000;66:549–557. doi: 10.1128/aem.66.2.549-557.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J, Rivas G, Cai X, Chicharro M, Parrado C, Dontha N, Begleiter A, Mowat M, Palecek E, Nielsen P E. Detection of point mutation in the p53 gene using a peptide nucleic acid biosensor. Anal Chem Acta. 1997;344:111–118. [Google Scholar]
- 56.Weiler J, Gausepohl H, Hauser N, Jensen O N, Hoheisel J D. Hybridisation based DNA screening on peptide nucleic acid (PNA) oligomer arrays. Nucleic Acids Res. 1997;25:2792–2799. doi: 10.1093/nar/25.14.2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Worden A Z, Chisholm S W, Binder B J. In situ hybridization of Prochlorococcus and Synechococcus (marine cyanobacteria) spp. with rRNA-targeted peptide nucleic acid probes. Appl Environ Microbiol. 2000;66:284–289. doi: 10.1128/aem.66.1.284-289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]