

Abstract
The development of Crohn’s disease is characterized by a breakdown of homeostatic immune-bacterial communication, which takes place at the intestinal mucosa when environmental triggers impact genetically predisposed individuals. Converging lines of evidence support the hypothesis that this pathogenetic model develops through sequential, although interrelated, steps that indicate failure of mucosal defense mechanisms at various stages. In this context, immunological phenomena that mediate the initial appearance of inflammatory lesions across the intestinal tissue may substantially differ from those that mediate and perpetuate chronic inflammatory responses. A compromise in the integrity of the epithelial barrier is among the earliest events and leads to accelerated influx of intraluminal antigens and intact microorganisms within the immunologically rich lamina propria. Inadequate clearance of invading microorganisms may also occur due to defects in innate immunity, preventing the timely and complete resolution of acute inflammatory responses. The final step is the development of persistent adaptive responses, which also differ between early and late Crohn’s disease. Current progress in our ability to delineate single cell transcriptomics and proteomics has allowed the discovery of cellular and molecular mechanisms that participate in each sequential step of CD development. This will not only advance our understanding of CD pathogenesis but also facilitate the design of targeted therapeutic approaches.
In recent years, great progress has been made regarding our understanding of Crohn’s disease (CD) pathogenesis. This has led to the currently accepted hypothesis that the fundamental, underlying abnormality in this condition is dysregulated crosstalk between commensal microflora and the gut mucosal-associated immune system. This defect, however, is only revealed when harmful environmental pressures impact individuals with a CD risk-conferring genetic predisposition. Thus, the occurrence of CD-specific intestinal inflammation at the individual level signifies the rare, unfortunate convergence of genetic, environmental, immunological, and microbiome-related triggers. This multifactorial process greatly impedes the discovery of globally-applied mechanisms to provide a uniform explanation for the development of CD and simplifies its therapeutic management.
Further adding to this complexity, CD, like several other chronic inflammatory conditions, most likely evolves through separate, although interrelated, phases. Such diversity is well-recognized at the clinical level, wherein an initial inflammatory stage is typically followed by stricturing and/or penetrating phenotypes.1 Similar diversity appears to also occur at the immunological level. Indeed, CD has traditionally been considered as a paradigm of dysregulated adaptive immunity, a concept which reflected the need to discriminate between the chronic nature of CD-related intestinal inflammation and inflammatory states associated with infectious or other acute insults. This approach, however, has been recently challenged, as it does not take into account important, earlier stages that occur at the intestinal mucosa of patients with CD.2 Such events are the direct result of injury caused by CD-triggering factors and precede the transition to the chronic stage. These early phases of CD are critically dependent upon the breakdown of epithelial defenses and deficiencies in the innate arm of immunity. More importantly, not only do such early phenomena initiate acute inflammation, but they also dictate the fate of subsequent adaptive pathways. Therefore, intestinal damage in CD needs to be viewed as a continuum that is initiated by defective epithelial barrier function, amplified due to compromised bacterial clearance by innate immunity, and perpetuated via dysregulated adaptive immune responses. It is obvious that such diversity is important, not only from a pathogenic perspective, but may also have critical therapeutic implications. In this narrative review, we will focus on immunological events that take place during the early stages of CD. The involvement of the microbiome in the development of intestinal inflammation has been extensively reviewed in several publications and will not be included in this review.
What is early Crohn’s disease?
Although, conceptually, there is little doubt for the existence of an early stage of CD, its definition, and more importantly, its recognition, have proved to be a very challenging task. There are several reasons for this. Clinicians tend to define early CD as its purely inflammatory form of short duration, without stenotic and/or penetrating complications and no previous need for disease-modifying therapies, such as immunomodulators or biologics.3, 4 This definition facilitates uniformity of patients for clinical trials, but at the same time, suffers from important limitations.5 First, it assumes that “early” coincides with the initial clinical presentation of inflammation leading to disease diagnosis. This, however, may oftentimes not be the case as low-grade subclinical inflammation may go unnoticed for long periods before a definitive diagnosis is reached. More importantly, several studies clearly demonstrated that, before symptoms manifest, endoscopic and histologic inflammation may already be evident within the gut mucosa. This was elegantly shown by the pioneering work of Rutgeerts et al., who, regarding the postoperative recurrence of Crohn’s disease, demonstrate that one year after right hemicolectomy, mucosal ulcerations are detected in more than 70% of the patients at the nonterminal ileum, although only 20% report symptoms.6 Similarly, in mechanistic studies using the fecal diversion and re-infusion model, D’Haens et al. shows that histologic inflammation is already evident within 8 days following exposure of the nonterminal ileum to autologous luminal contents7. Second, the definition of early is difficult to apply to patients, who from the start, present with disease complications (i.e., stenosis or internal fistula) without recalling a clinically-evident inflammatory phase; this, in fact, is the case in almost 20% of patients with CD.8 This is particularly true for ileal CD, as oftentimes inflammation in this location produces no or subtle symptoms, which may easily be overlooked or attributed to IBS in young persons, only to present afterwards with obstruction due to intestinal strictures or perforating events, such as abscess formation and/or internal fistulae. Even when diagnosis is made shortly after such complications, disease is way too advanced, pathologically and immunologically, and should not be considered as ‘early”. Furthermore, this distinction has specific and significant clinical implications as clinicians are inclined to think that, following initial diagnosis, CD is presumably at an early stage, particularly if the patient does not report previous intestinal clinical manifestations. Such downgrading of disease progression and severity, however, may lead to the undertreatment of patients. In such cases, the “window of opportunity” for changing the natural course of CD disease may be irreversibly missed. Finally, longitudinal studies from Israel and the Netherlands have shown that serological indicators of mucosal immunoreactivity precede the clinical presentation of CD by several years, indicating that immunological phenomena may occur much earlier that “early” CD.9, 10
The identification of early CD from the basic research standpoint also is not devoid of controversy. Such studies almost exclusively rely on animal models of intestinal inflammation, which allow for manipulations that are not possible in humans. Early stages of inflammation can be relatively easily traced in these models. Moreover, the triggering factors are usually clearly defined, being chemical injury or targeted genetic manipulation, etc. Nevertheless, currently used animal models of inflammation are rarely fully representative of the human condition.11 In addition, what these experimental models show us is that several diverse immunological pathways may all lead to the common phenotypic outcome of mucosal inflammation. Such results are difficult to fit into a single, uniformly-applied model for early CD.
Failure of mucosal defense mechanisms as the early event in the immunopathogenesis of intestinal inflammation in Crohn’s disease
The intestinal mucosa is a unique environment wherein an enormous amount of potentially harmful intraluminal antigens lie in close proximity to the largest immunological machinery of the body. This renders inflammation a constant possibility in the gut, one that is only avoided via the existence of the intestinal mucosal barrier. This is a complex and multilayered anatomical and functional structure, which ensures the physical and biological separation of the intestinal microflora and food antigens from lamina propria immunocytes; it also regulates the occasional and, from the developmental standpoint, necessary encounter between them. Included in this defense network are the mucus layer and natural antimicrobial peptides, the epithelial monolayer with interconnecting tight junctions, as well as innate and adaptive immunity constituents of the lamina propria. Integrity of the epithelial barrier is determined by both genetic factors and environmental challenges, most commonly enteric infections, toxins and medications, which may act either directly, or indirectly by inducing alterations of the bacterial flora or epigenetic modifications.12 Most likely, development of IBD signifies the failure of one or more aspects of the mucosal barrier in individual patients13 that takes place in a stepwise fashion, with some events occurring earlier than others (Figure 1).
Figure 1. Early steps in the pathogenesis of Crohn’ s Disease.
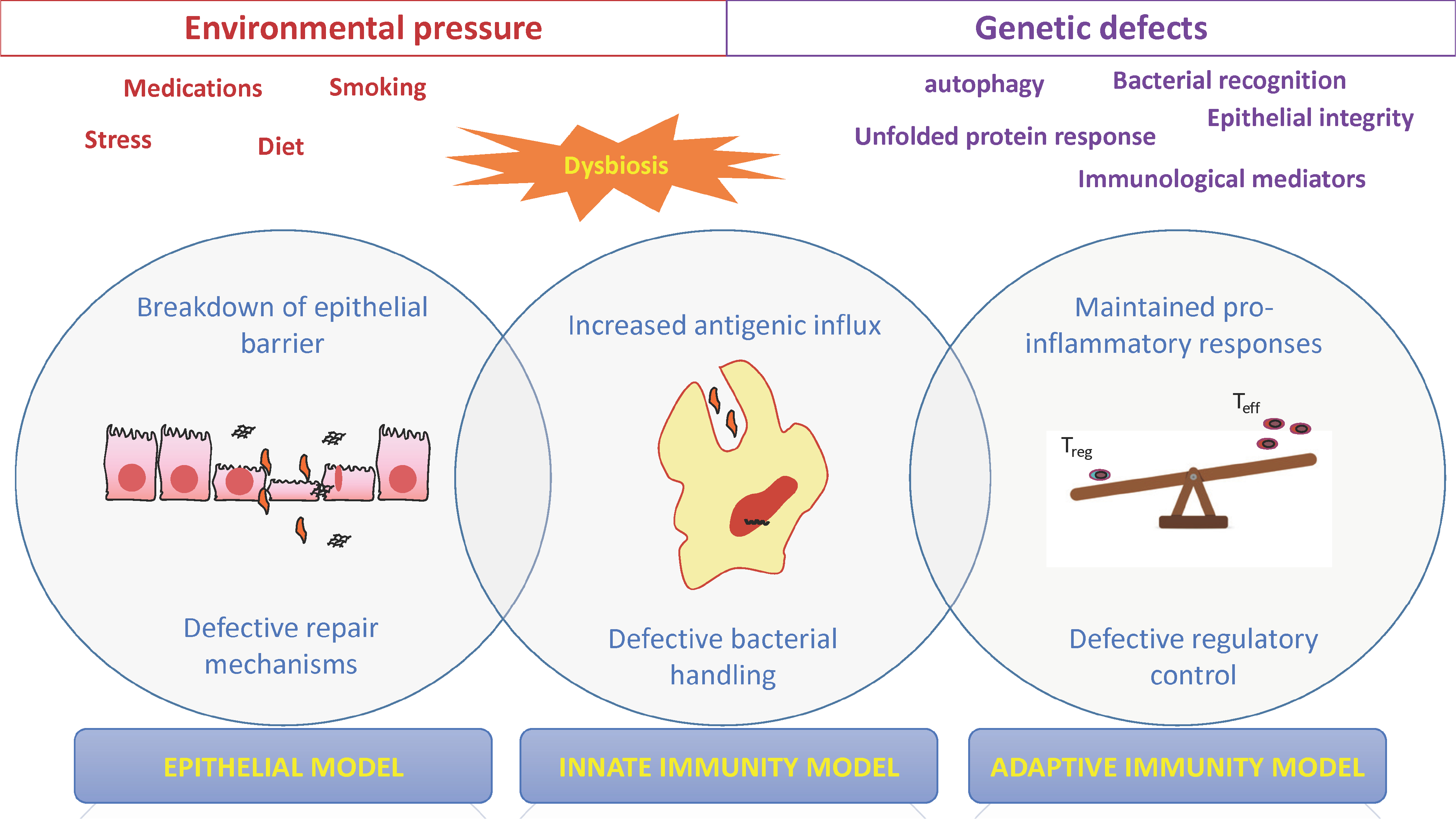
Crohn’s disease develops due to sequential failure of defense mechanisms within the intestinal mucosa that requires the unfavorable combination of genetic defects, caused by one or more CD-risk modifying polymorphisms, and mucosal injury, inflicted by harmful environmental factors. The latter also intervenes with the composition and metabolic properties of the commensal flora, leading to structural or functional dysbiosis, which critically contributes to the loss of mucosal homeostasis. Breaches in the integrity of the epithelial barrier are among the earliest pathogenetic events in Crohn’s disease, which, in combination with defective repair mechanisms, allow for a constant flux of bacterial antigens from the intestinal lumen into the immunologically-rich lamina propria (epithelial model). In healthy states, such bacterial-related products are effectively removed by phagocytes of the innate arm of immunity. In contrast, patients with CD demonstrate defective “clearance” mechanisms induced by genetically-determined deficiencies that include, but are not limited to, bacterial-sensing, autophagy and unfolded protein response. These processes culminate to inadequate removal of antigenic material and relentless stimulation of inflammatory responses (innate-immunity model). A final effect of such constant inflammatory input is the unbalanced expansion of pro-inflammatory, Teff-type adaptive immunity, which is dominated by Th1 cells and cytokines, along with inefficient suppressive activity, either due to Treg defects and/or inefficient apoptosis of Teff cells (early adaptive immunity model).
Breakdown of the epithelial barrier: the Trojan horse for initiating inflammation?
Converging lines of evidence support the notion that one of the earlier events that take place in patients with CD is the breakdown of the epithelial barrier, which is demonstrated by increased small intestinal permeability (Table 1). In patients with active disease, it is not possible to discriminate whether this is the cause or the result of chronic inflammation. On the other hand, such defects may, in fact, precede the appearance of clinical, macroscopic, or even histologic inflammation. Among various genetic determinants for CD susceptibility, a risk locus has been detected in MUC19 and MUC1 genes, which code for components of the intestinal mucous, thus contributing to integrity of the barrier.14 Furthermore, patients with quiescent CD also have increased paracellular permeability15 which importantly, appears to increase the risk for subsequent disease flares.16 Moreover, a plethora of studies report altered small intestinal permeability in first-degree relatives of patients with CD17, 18. This may be genetically-determined, since relatives who carry the CD risk-conferring NOD2 3020insC frameshift mutation display increased intestinal permeability more often and to a higher degree than noncarriers.19 A primary barrier defect was recently proposed by Keita et al., based on their finding of increased baseline and induced permeability in healthy monozygotic, but not dizygotic, twins of patients with CD.20 Such findings indicate that ongoing inflammation is not a prerequisite for dysregulated epithelial permeability; rather, the latter is an early event that facilitates the influx of luminal antigens into the immunologically-rich lamina propria, thus initiating inflammation. In fact, when first degree relatives are subjected to video capsule endoscopy, no increase is observed in the number of ulcerations compared to patients with normal permeability.21 This “permeability-to-inflammation” sequence is strongly supported by mechanistic studies in experimental models. In CD-like ileitis-prone SAMP1/YitFc mice, increased paracellular permeability is detected as early as 3-wks of age, preceding histologic evidence of disease 22. This is followed by dysregulated mucosal immunological responses, which culminate to overt ileitis by 8–10 weeks of age23. Strikingly, a very similar sequence of events was also reported in IL10−/− mice with detectable permeability defects occurring by 2-wks of age, followed by mucosal upregulation of IFNγ and TNFα and eventually, development of inflammation.24 The ability of barrier defects to propagate inflammatory responses is further supported by the development of colitis in mice deficient for either the xenobiotic transporter, mdr1a 25, or the tight junction protein, claudin-7 26, which both affect the integrity and function of the epithelial barrier.
Table 1.
Evidence for failure of the integrity of the intestinal epithelial as an early event in the pathogenesis of Crohn’s disease
| ❑ Genetic polymorphisms in Muc19 increase risk for Crohn’s disease |
| ❑ Increased intestinal permeability in first degree relatives of CD patients |
| ❑ Increased intestinal permeability in CD patients in remission (which predicts subsequent flare) |
| ❑ Mice deficient for mdr1a, claudin-7, or muc2 develop colitis |
| ❑ In longitudinal studies in mice, permeability defect precedes development of inflammation |
Alterations in epithelial permeability may also be a manner in which environmental factors trigger intestinal inflammation.27, 28 In fact, a recent study examined the in depth contribution of 167 IBD-associated SNPs and found no associations with increased permeability in first degree relative of patients, indicating that environmental triggers are more important than genetic background29. Furthermore, of interest, a certain percentage of CD patients’ spouses also report increased permeability.30 Along these lines, certain studies show that both non-steroidal anti-inflammatory drugs (NSAIDs) and aspirin exert a damaging effect on the integrity of tight junctions and intestinal permeability, which are more prominent in first-degree relatives of CD patients than in unrelated healthy controls.31 Similarly, studies in rats show that environmental stress increases epithelial permeability that is dependent on adrenal corticosteroids,32 thus providing a candidate mechanism for stress-induced CD flares.33 Of specific importance to IBD is the finding that cigarette smoking is associated with intestinal barrier dysfunction in the small intestine, but not the large intestine, of mice.34
Taken together, these experimental and clinical observations provide strong proof-of-concept for the etiological significance of early permeability defects in IBD. Nevertheless, only longitudinal studies with careful follow-up will eventually answer the question as to whether increased intestinal permeability in relatives of CD patients occurs at an early, pre-inflammatory step or conversely, represents an indicator of subclinical inflammation, which disrupts the integrity of the barrier. It should be mentioned, however, that the recent RISK study identified that seropositivity for CBir1, a marker of immune response against bacterial flagellin, has the strongest effect on disease progression towards complications.35 Positivity for this marker provides indirect evidence for a compromised intestinal barrier being a critical early step in CD patients, leading to failure to separate commensal bacteria from the immunologically-rich lamina propria and initiating proinflammatory reactivity within the intestinal mucosa.
Over-reactivity in the face of defective innate immunity. An immunological “paradox”?
The noted early increase in permeability leads to unrestricted entry of intraluminal bacteria and their products into the lamina propria and encounter with the immunologically-enriched compartment of this area. Under homeostatic conditions, this would result in rapid and complete elimination of intruding microorganisms by the innate arm of immunity. In contrast, strong evidence indicates that failure of this “clearance” process is an early, critical event in the natural history of CD, pointing to “defective immunity” as the core abnormality that initiates chronic inflammation (Table 2).
Table 2.
Evidence for defective bacterial clearance by innate immunity as an early event in the pathogenesis of Crohn’s disease
| ❑ Genetic polymorphisms in various genes associated with bacterial handling modify the risk for developing Crohn’s disease |
| ❑ Monogenic disorders that affect innate immunity pathways are associated with development of CD-like intestinal inflammation |
| ❑ Defective and delayed acute response to injury in patients with sporadic CD |
| ❑ Protective roles of innate cytokine during acute injury/repair (as opposed to proinflammatory roles in chronic inflammation) |
| ❑ De novo development of IBD-like lesions in patients treated with targeted biological therapies |
First, among more than 150 CD risk-modifying polymorphisms described so far in genetic association studies, the majority consists of genes that encode for proteins that are critically involved in the recognition of, and response to, microbe associated molecular patterns.14, 36 Although none of these polymorphisms suffices to induce a phenotypic effect by itself, they indicate that a subset of individuals, who are preconditioned to develop CD, are deficient in their ability to eliminate intruding microorganisms. Polymorphisms in the genes for the intracellular bacterial sensor, NOD2, and the autophagy gene, ATG16L1, are among the most prominent, and may act in synergy to create a state of immunological compromise that particularly affects Paneth cells.37 This leads to reduced secretion of natural antimicrobial peptides, which further diminishes the anti-bacterial efficacy of the intestinal mucosa. For example, patients with CD have been reported to display suppressed expression of ileal α-defensins and the inability to upregulate colonic β-defensin production, whereas CD-derived monocytes fail to induce Paneth cell defensins.38, 39
Second, it has long been recognized that patients who suffer from monogenic disorders that affect phagocyte function very often develop chronic intestinal inflammation. Most prominent among these are chronic granulomatous disease (CGD) and glycogen storage disease type 1b. Careful analysis of the clinical and histological characteristics of the intestinal lesions from patients with such conditions fail to identify any differences with CD, a finding that points to a common immunopathogenesis, as well.40 As the genetic defect in such individuals is present at birth, it precedes the development of intestinal inflammation, thus providing direct proof that deficient handling of invading microorganisms is a sufficient early step that initiates mucosal inflammatory pathways.
Third, studies in patients with sporadic CD have shown that a compromised response to acute insults may be a common abnormality independent of genetic background. In elegant earlier studies, Marks et al. studied the mucosal reactivity of healthy controls as well as patients with CD and UC to acute injury inflicted by biopsy during colonoscopy. They were able to show that post-injury, mucosal elevations of MPO, a marker of neutrophil infiltration and IL-8, an epithelial-derived monocyte-chemotactic protein, are significantly suppressed in patients with CD.41 Nevertheless, genetic control is always present as peripheral blood mononuclear cells are also deficient in their capacity to secrete IL-1β after stimulation, but only if they are derived from patients with NOD2 mutations. In subsequent experiments, it was shown that inoculation with E. coli induced attenuated local responses in patients with CD. The latter demonstrate defective initial clearance and substantially delayed total clearance of bacteria.42, 43 Taken together, these converging data indicate that it may be possible for a subset of CD patients to suffer from an inherent lack of bacterial clearance in the lamina propria due to defective innate immunity.
Fourth, animal studies provide definitive proof for the paramount importance of intact innate immunity in the containment of harmful mucosal insults and prevention of chronic inflammation. This is clearly shown in the DSS colitis model of acute injury and repair, wherein several studies with knockout mice display the same “paradoxical” effect. Specifically, mice that are rendered deficient for established pro-inflammatory mediators and thus, expected to demonstrate amelioration of inflammation, instead suffer from substantially more severe colitis. Included in this list are innate cytokines and signaling molecules, such as IL-1β, IL-33, TL1A/DR3, IL-18, NF-kB and MyD88, but also TNFα itself.44 In murine models of intestinal inflammation, such factors appear to be protective during acute inflammation, but exert pure pro-inflammatory functions in later stages. This “cytokine paradox” may be explained if we consider that, when barrier integrity is compromised, there is accelerated entry of bacterial-associated factors into the lamina propria. Failure to mount a strong pro-inflammatory response against them would lead to inadequate bacterial clearance and persistence of antigenic material, a form of functional “dysbiosis” that may lead to unrelenting activation of immunological pathways and injury of the intestinal mucosa. In fact, this was mechanistically shown in SAMP1/YitFc mice. Ileitis in this strain is almost completely prevented by administration of the probiotic mixture, VSL#3, an effect that is mediated by the probiotic’s ability to upregulate TNFα, which in this case, acts in a prophylactic manner by fortifying the intestinal epithelial barrier.45, 46 The specificity of this finding is demonstrated by abrogation of the beneficial effect of VSL#3 when anti-TNF neutralizing antibodies are administered. Interestingly, these outcomes are not observed in mice with fully established ileitis.45, 46 These studies emphasize the complexity of mucosal inflammatory pathways, demonstrating a clear distinction between mechanisms that mediate early vs. late ileitis, even if the same molecules participate in both phases. This dichotomous functionality of innate cytokines is most probably explained by the specifics of each clinical and/or experimental setting, including differences in cellular sources, antigenic loads and specificities, mucosal immunological milieu, as well as temporal and spatial associations. Τhe discovery and characterization of innate lymphoid cells (ILCs) further adds to the complexity of homeostatic vs. inflammatory signaling at the gut mucosa. ILCs are innate lymphocytes that are selectively enriched at mucosal sites and, currently include NK, ILC1, ILC2, and ILC3 and LTi cells, which all bare distinct immunotypes and exert divergent functions.47 Their involvement in the pathogenesis of IBD remains unclear, but phenotype and functional diversity between acute and chronic immunological responses has been demonstrated. In particular, under non-inflammatory conditions, ILC3s are prominent producers of homeostatic IL-22 in response to DC-derived IL-23 or epithelial cell-derived IL-7.48 During chronic inflammation, however, ILC3s may contribute to mucosal injury in a variety of ways. In particular, ILC3s may produce IL-22 with pro-inflammatory function,49 secrete pathogenic IL-1750 or produce GM-CSF, which recruits inflammatory monocytes into the intestine and sustains local inflammation.51 Other possibilities also include a decrease of protective ILC3s in chronic inflammatory states and an increased ILC1/decreased ILC3 balance. In fact, re-distribution with an elevated ILC1/ILC3 ratio has been reported both in patients with CD and in experimental colitis, which may also allude to the possibility of transdifferentiation of ILC3s to ILC1s via myeloid-cell produced IL12+/−IL-23.52 In a similar fashion, ILC2s may also exert diverse roles in the intestinal mucosal environment. ILC2s are pivotal participants in anti-helminthic immunity but also contribute to the maintenance of the intestinal barrier.53 Nevertheless, their role in the development of experimental colitis has been controversial as they have been assigned either a pro-inflammatory, IL-25 dependent or a protective, IL-33 mediated role.54, 55
Finally, the de novo development of IBD-like intestinal inflammation in patients receiving anti-cytokine therapies for other immune-mediated disorders provides in vivo proof-of-principle for the failure of immunological surveillance by innate immunity as an early step in IBD pathogenesis. This, in fact, is repeatedly reported for patients that are treated with anti-TNF agents.56, 57 Furthermore, new onset of IBD is also described in patients receiving immunosuppression after organ transplantation58, once again indicating that intestinal inflammation can be associated not only with immune reactivity, but also with immunosuppression. Lastly, the rapidly expanding field of immune-related adverse effects following immune checkpoint inhibition (such as anti-CTLA4 or anti-PDL1/PD1 blockade) in patients with malignancies has opened a new field for the study of pathogenesis of IBD.59, 60 Not only do such cases provide proof-of-principle for the critical importance of early, antigen presentation-related immunological effects in the preservation of homeostasis, but they also offer a unique opportunity to study in vivo early events in IBD development.
In all, the aforementioned studies indicate that during an acute insult, gut-associated lymphoid tissue (GALT) reacts by augmenting local innate immune functions, which are necessary for effectively eliminating the plethora of bacterial-driven and potentially harmful stimuli. It follows that genetically-determined impairment of innate immunity pathways in patients with CD may render them unable to handle disease-inducing environmental factors. Consequently, CD is increasingly considered a state of immunodeficiency of the innate arm of immunity, underlying defective bacterial recognition, autophagy and/or antigen presentation. As such, IBD may belong to a group of disorders associated with bacterial processing, in which the far end of the spectrum is represented by diseases, such as CGD or glycogen storage disease 1b. This “innate immunodeficiency” may therefore represent a critical, early point of mucosal regulatory failure, which is inevitably followed by persistent setting of the mucosal immunostat to “inflammation” and the development of chronic disease.
The evolving pattern of adaptive immunity in Crohn’s disease: early is not the same as late
Following the breakdown of the epithelial barrier and/or inadequate ‘bacterial” clearance, the adaptive immune system faces the task of handling a diverse and abundant antigenic load within the lamina propria. This leads to lymphocyte activation and the generation of effector immunological pathways that eventually maintain intestinal injury via the constant generation of inflammatory mediators. There is now compelling evidence to support the notion that initial reactivity of adaptive immunity to antigen penetration into the lamina propria is quite different from that observed during late phases of CD.
In an earlier study, Kugathasan et al. examined the immunophenotype of pediatric patients with CD, comparing patients with recent diagnosis (0–6 months, first attack, i.e., presumably early disease) to those with long-standing disease (5–8 years, presumably late disease)61. The authors found that early CD resembles an acute infectious colitis with an IL-12-induced, polarized Th1-type response, as determined by high IFNγ production and IL-12Rβ2 chain expression in isolated T-cell clones. Such changes are absent in children with late CD, wherein the development of Th2-specific, IL-4 producing clones is noted. In addition, elevated mucosal levels of IL-12p40 and IL-12Rβ2 mRNA is observed in early, but not late CD. In another study, Nakajima et al. characterized CD lesion–specific clonal TCR bands that were obtained either from apthoid lesions (early disease) or areas with frank ulceration (late disease). Although considerable overlap exists between the two types of lesions, the authors also note the presence of discrete early-lesion-specific TCR patterns. Interestingly, the latter are conserved among separated apthae, but absent in the intervening, non-inflamed mucosa, pointing to the fact that they may correspond to T cell clones that are specifically activated in the early phases of CD.62 The importance of early immunological alterations in CD is also emphasized in a recent publication showing that altered barrier function and blood T cell phenotype (as well as intestinal microbiota changes) are shared by patients with CD and their unaffected siblings63. Along the same line, newly diagnosed, untreated adult patients with IBD display unique patterns of maturation and activation patterns of T-cells along with distinct cytokine responses.64 Moreover, in a recent publication, the relative contribution of Th1 and Th17 responses in peripheral blood immunophenotypes were compared and found significantly different between patients with early vs. late CD.65 Similar diversity is also seen at the mucosal level when lymphocytic cytokine mRNA levels were compared and found to be disparate between patients with longstanding inflammation and early post-operative recurrence.66 The translational implications of such findings were shown in a recent study reporting certain candidate serum biomarkers that discriminated newly diagnosed, untreated CD patients from healthy controls.67
These earlier reports are remarkably recapitulated in studies of immunological characterization of the natural history of experimental chronic intestinal inflammation. Animal models of colitis or ileitis offer a unique opportunity to clearly discriminate between immunological phenomena that precede the development of mucosal damage (pre-clinical stage), those that occur concomitantly to the initial presentation of inflammation (early disease), and those that persist after disease has been present for an extended period of time (late disease). Work utilizing the SAMP1/YitFc strain has pioneered this concept in that this model does not require any chemical, genetic or immunological manipulations, and develops small intestinal inflammation with remarkable similarity to CD, including localization to the terminal ileum, presence of lymphoplasmatocytic infiltrates and formation of granulomas.23 Extensive, time-course-based immunophenotyping reveal the clear distinction of two immunological stages in this model, an early Th1 dominant and a late mixed Th1/Th2. SAMP1/YitFc mice do not display histological disease at week 4, but gradually demonstrate overt inflammation by week 10. Thus, the interval between weeks 4–10 weeks should be considered an early stage, whereby prevailing immunological pathways eventually lead to ileitis. It has been shown, in fact, that this early stage is dominated by increased IFNγ and TNFα, bearing the characteristics of a typical Th1 response.68 Interestingly, this disease phase is clearly dependent on bacterial triggers, as it is greatly prevented by either probiotic or antibiotic therapies.45, 69 Following the establishment of chronic ileitis, however, the mucosal milieu in SAMP1/YitFc mice is completely altered and dominated by huge increases of IL-33, IL-13 and IL-5,68, 70 indicating the appearance of a strong Th2 response, whereas Th1 responses remain preserved. Interestingly, this “early/Th1” to “late Th2” transition appears to be a common pattern in chronic inflammation. A similar pattern is also reported in IL-10 knock-out mice, in which colitis development is also characterized by early Th1 immunity evolving to Th2 later on.71 Furthermore, chronic TNBS colitis starts as a pure, Th1-mediated immunological response that is dependent on elevated IL-12p70 and IFNγ, only to be replaced at later time points with exaggerated Th2 and Th17 immunity dominated by increases in IL-13 and IL-23, respectively.72. Taken together, it appears that mucosal immunoregulation varies with the course of human IBD. It also indicates that early disease development may depend on limited, bacterial-driven mechanisms that may also respond more optimally to therapeutic intervention. This is actually supported by the well-established notion that patients with a short duration of CD demonstrate higher response rates to anti-TNF agents. Such positive indications for early intervention has also been recently suggested for anti-integrin therapy.73 The latter may also be linked to observations in the SAMP1/YitFc mice, demonstrating that, during the induction phase of ileitis, lymphocytes utilize homeostatic trafficking mechanisms that are similar to those in the healthy mucosa. Therefore, it makes sense that drugs that target the α4β7/MadCAM-1 pathway, such as vedolizumab or etrolizumab, may be more efficient in early disease. In contrast, upon commencement of the chronic phase, not only are homeostatic pathways substantially upregulated, but alternative pathways that involve proinflammatory chemokines, adhesion molecules, and integrins are also triggered.74 These may be more resistant to currently utilized anti-integrin interventions. Overall, these studies strongly imply that patients with the initial manifestations of IBD may represent an ideal population in which immunomodulation may have optimal therapeutic efficacy. Recently, monoclonal antibodies that target IL-23, the central Th17 cytokine, have evolved as reliable therapeutic options for CD. The currently approved mAb, ustekinumab, targets p40, a chain that is shared by IL-12 and IL-23, thus interfering with both Th1 and Th17 immunity. On the other hand, novel monoclonal antibodies also target p19, the IL-23-specific chain. Interestingly, a recent report pointed to temporally distinct involvement of IL-12 and IL-23 in the pathogenesis of chronic murine colitis, with the former triggering the initiation of colitis and the latter driving the chronic disease phase.75 At the moment, it is not known whether these experimental findings translate into the human disease setting; nevertheless, if this proves to be the case, it may affect the selection of biologics in clinical practice.
Translational implications: can we intervene early in CD?
There is now indisputable evidence to support a distinction between at least two phases in the natural history of CD. Early disease consists of the initial events that take place when homeostatic mechanisms fail and acute inflammatory responses cannot be resolved. In contrast, late disease is established after adaptive immunity has been irreversibly primed towards dysregulated effector phenotypes. Temporal themes in disease evolution cannot be overemphasized, given the fact that inflammatory mediators, primarily innate cytokines and ILCs, oftentimes play dichotomous roles during early and late disease states. The translational implications of these immunological phenomena are obvious. While late disease has been traditionally treated with anti-inflammatory approaches, patients with early disease may benefit more from enhancement of innate immune responses, which may require “thinking outside the box”.76 Treatment that specifically aims to correct defective epithelial barrier in IBD remains in its infancy, and the few reported efforts that have reached the clinical stage have been applied to patients with UC rather than CD. It should be noted that one of the properties of mesalamine is fortification of the epithelial barrier, most probably via its function as an agonist for peroxisome proliferator-activated receptor-γ (PPAR-γ)77. Interestingly, although mesalamine is not currently recommended for the treatment of CD, its sole potential indication may be the prevention of post-operative recurrence in low-risk patients, thus, interfering with early pathogenetic events in CD. In recent studies, hyperbaric oxygen treatment and phosphatidylcholine (PC), which both act by augmenting intestinal barrier function, have shown positive results in UC, but so far, have not been tested in CD 78–80. Targeting defective clearance in CD is an equally difficult task. Based on the resemblance between CD and inherent deficiencies of phagocytosis, treatment with haemopoietic growth factors has been proposed for sporadic cases as well. Such treatment aims to correct defective phagocytosis, thus, enhancing innate immunity. Nevertheless, a sustained benefit has not been shown in clinical trials.81, 82 Another therapeutic approach targets defective autophagy, via administration of rapamycin, an mTOR inhibitor that is widely used for the prevention of post-transplant rejection, and has also been shown to upregulate autophagy in vitro.83 Proof-of-principle is provided by findings of abnormal mTOR activation in the inflamed colon of IBD patients and the development of a murine model of inflammation-induced colon cancer that depends on mTOR-Stat3 signaling84. In addition, rapamycin has been reported to induce clinical response in a patient with severe refractory CD.85
Although there is large theoretical support for therapeutic approaches that target epithelial barrier and/or innate immunity in CD, there are several obstacles that need to be overcome before such efforts reach the clinical arena (Table 3). First, patients whose disease is in early stages are rarely recognized in clinical practice, making properly designed clinical trials difficult, if not impossible, at the present time. Second, several of the “early” pathogenetic mechanisms are intertwined with pathways of more advanced stages. A typical example is TNF overproduction that is a characteristic of the late adaptive immune response, but also, a strong inducer of epithelial barrier compromise. Finally, most probably, CD represent a spectrum of overlapping, but not identical disorders. Thus identification of groups of patients with absolutely matching profiles remains a very challenging task.
Table 3.
Obstacles in the treatment of early Crohn’s disease
| ❑ Difficulty in obtaining adequate number of patients with truly early disease |
| ❑ Overlap between early and late mechanisms |
| ❑ Variability of the clinical, pathological and immunological spectrum of CD |
| ❑ Lack of reliable biomarkers associated with early or late disease |
The aforementioned problems are further exemplified by the current lack of reliable biomarkers that can discriminate between different immunological profiles that may be expected to respond in a uniform manner to common therapies. This is, however, prone to change in the near future as results from single cell protein and mRNA analyses are beginning to accumulate.86 Single-cell RNA-sequencing (scRNA-seq) allows for full transcriptomic analysis at a single-cell level; hence, the cellular composition of intestinal tissues are depicted in detail, including rare cell types that may not contribute to expression profiling via bulk RNA analysis. This is followed by bioinformatic approaches that incorporate computational analysis to further categorize cells by their genomic signatures and expands analysis to lineage decisions and intercellular networks. Such approaches have already started to provide fruitful results and have uncovered unique cellular components of the inflammatory milieu in IBD.87, 88 In parallel to scRNA-seq, single-cell proteomic analysis has also advanced. Traditional flow cytometry has been gradually replaced by cytometry by time of flight (CyTOF), which allows simultaneous quantification of over 40 cellular parameters (soon to exceed 100). This again, is followed by computational analysis, which enables the characterization of proteomic heterogeneity at the single-cell level. By applying this new methodology, a remarkable variability of cellular composition was recently shown across various locations of the normal human colon, a finding with obvious implications for appropriate tissue sampling in IBD research.89 Finally, single cell RNA and protein characterization can now be combined in individual samples, further enhancing the accuracy of cellular compartmentalization in intestinal tissue during homeostasis and inflammation.90
The current recognition of early clinical, or even pre-clinical, stages of CD will clearly affect both the design and therapeutic targets of future clinical trials. In this sense, there is currently a shift in clinical research towards epidemiological studies aimed to delineate factors that determine the initial development of CD. In a recent trial of this type, archived serum samples from the United States Defense Medical Surveillance System were screened by proteomics for the presence of various soluble proteins.91 Patients who later developed CD were compared with healthy controls, and the predictive value of various proteins was evaluated. The authors were able to identify 51 proteins that could predict the development of CD after 1 or 5 years. Interestingly, no predictive biomarkers were found for UC. In a different approach, Canadian scientists have launched the Genetic Environmental Microbial (GEM) project, focusing on first degree relatives of patients with IBD in an effort to identify predictive factors for the future development of IBD.92 As several individuals of this population are expected to develop IBD later on in life, the comparative analysis of their environmental and genetic signatures will definitely further elucidate the pathogenesis of the early steps in CD. Since these early events appear to differ from later ones, this will also affect our interventional strategies. Although effector immunological pathways will continue to offer cellular and molecular targets, earlier steps may be amenable to quite different approaches, among which microbiome-related therapies may be at the very center. For example, the Exploring MEChanisms Of disease traNsmission In Utero through the Microbiome (MECONIUM) study reported the establishment of dysbiosis in infants born to mothers with IBD, as opposed to babies of healthy mothers.93 Furthermore, the dysbiotic IBD-related flora induced immunological alterations in germ-free mice, indicating a transmittable abnormality. It follows that both the mother and infant microflora offer a therapeutic opportunity to restore a healthy flora and prevent the development of a pro-inflammatory-prone immune system. This appealing hypothesis would require therapeutic approaches that are quite disparate from the currently implemented immunosuppression. Probiotics may induce beneficial modifications of the microbiome, although the particular microorganisms, dosage and duration of therapies are far from known at the moment. Alternatively, diet interventions may induce an anti-inflammatory state, either in a direct manner or indirectly via microbiome changes. Similarly, fecal material transplantation offers the opportunity for broad remodeling of the microbiome and correction of dysbiosis, although several technical and clinical issues remain to be resolved. It should be noted, however, that targeting the microbiome is only one of several possible interventions in high risk individuals for developing CD. In this sense, drugs that are commonly used for the treatment of unrelated conditions, such as statins and metformin, may offer unexpected benefits in high-risk individuals or during early disease stages. Recognizing patients with these early forms of CD via clinical or laboratory biomarkers will be critical for the design of clinical trials that will test these unconventional therapeutic approaches.
Footnotes
The authors report no conflict of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cosnes J, Gower-Rousseau C, Seksik P, et al. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology 2011;140:1785–1794. [DOI] [PubMed] [Google Scholar]
- 2.Bamias G, Nyce MR, De La Rue SA, et al. New concepts in the pathophysiology of inflammatory bowel disease. Ann Intern Med 2005;143:895–904. [DOI] [PubMed] [Google Scholar]
- 3.Peyrin-Biroulet L Why should we define and target early Crohn’s disease? Gastroenterol Hepatol (N Y) 2011;7:324–326. [PMC free article] [PubMed] [Google Scholar]
- 4.Peyrin-Biroulet L, Billioud V, D’Haens G, et al. Development of the Paris definition of early Crohn’s disease for disease-modification trials: results of an international expert opinion process. Am J Gastroenterol 2012;107:1770–1776. [DOI] [PubMed] [Google Scholar]
- 5.Peyrin-Biroulet L, Loftus EV Jr, Colombel JF, et al. Early Crohn disease: a proposed definition for use in disease-modification trials. Gut 2010;59:141–147. [DOI] [PubMed] [Google Scholar]
- 6.Rutgeerts P, Geboes K, Vantrappen G, et al. Predictability of the postoperative course of Crohn’s disease. Gastroenterology 1990;99:956–963. [DOI] [PubMed] [Google Scholar]
- 7.D’Haens GR, Geboes K, Peeters M, et al. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology 1998; 114:262–267. [DOI] [PubMed] [Google Scholar]
- 8.Rieder F, Zimmermann EM, Remzi FH, et al. Crohn’s disease complicated by strictures: a systematic review. Gut 2013; 62:1072–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Israeli E, Grotto I, Gilburd B, et al. Anti-Saccharomyces cerevisiae and antineutrophil cytoplasmic antibodies as predictors of inflammatory bowel disease. Gut 2005;54:1232–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Schaik FD, Oldenburg B, Hart AR, et al. Serological markers predict inflammatory bowel disease years before the diagnosis. Gut 2013;62:683–688. [DOI] [PubMed] [Google Scholar]
- 11.Valatas V, Bamias G, Kolios G. Experimental colitis models: insights into the pathogenesis of inflammatory bowel disease and translational issues. Eur J Pharmacol 2015;759:253–264. [DOI] [PubMed] [Google Scholar]
- 12.Mehta M, Ahmed S, Dryden G. Immunopathophysiology of inflammatory bowel disease: how genetics link barrier dysfunction and innate immunity to inflammation. Innate Immun 2017; 23:497–505. [DOI] [PubMed] [Google Scholar]
- 13.Jager S, Stange EF, Wehkamp J. Inflammatory bowel disease: an impaired barrier disease. Langenbecks Arch Surg 2013; 398:1–12. [DOI] [PubMed] [Google Scholar]
- 14.Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 2011;140:1704–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vivinus-Nebot M, Frin-Mathy G, Bzioueche H, et al. Functional bowel symptoms in quiescent inflammatory bowel diseases: role of epithelial barrier disruption and low-grade inflammation. Gut 2014;63:744–752. [DOI] [PubMed] [Google Scholar]
- 16.Arnott ID, Kingstone K, Ghosh S. Abnormal intestinal permeability predicts relapse in inactive Crohn disease. Scand J Gastroenterol 2000;35:1163–1169. [DOI] [PubMed] [Google Scholar]
- 17.Munkholm P, Langholz E, Hollander D, et al. Intestinal permeability in patients with Crohn’s disease and ulcerative colitis and their first degree relatives. Gut 1994;35:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peeters M, Geypens B, Claus D, et al. Clustering of increased small intestinal permeability in families with Crohn’s disease. Gastroenterology 1997;113:802–807. [DOI] [PubMed] [Google Scholar]
- 19.Buhner S, Buning C, Genschel J, et al. Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation? Gut 2006;55:342–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keita AV, Lindqvist CM, Ost A, et al. Gut barrier dysfunction-a primary defect in twins with Crohn’s disease predominantly caused by genetic predisposition. J Crohns Colitis 2018; 12:1200–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Teshima CW, Goodman KJ, El-Kalla M, et al. Increased intestinal permeability in relatives of patients with Crohn’s disease is not associated with small bowel ulcerations. Clin Gastroenterol Hepatol 2017;15:1413–1418 e1. [DOI] [PubMed] [Google Scholar]
- 22.Olson TS, Reuter BK, Scott KG, et al. The primary defect in experimental ileitis originates from a nonhematopoietic source. J Exp Med 2006;203:541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pizarro TT, Pastorelli L, Bamias G, et al. SAMP1/YitFc mouse strain: a spontaneous model of Crohn’s disease-like ileitis. Inflamm Bowel Dis 2011;17:2566–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Madsen KL, Malfair D, Gray D, et al. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis 1999; 5:262–270. [DOI] [PubMed] [Google Scholar]
- 25.Resta-Lenert S, Smitham J, Barrett KE. Epithelial dysfunction associated with the development of colitis in conventionally housed mdr1a−/− mice. Am J Physiol Gastrointest Liver Physiol 2005;289:G153–G162. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka H, Takechi M, Kiyonari H, et al. Intestinal deletion of Claudin-7 enhances paracellular organic solute flux and initiates colonic inflammation in mice. Gut 2015;64:1529–1538. [DOI] [PubMed] [Google Scholar]
- 27.Soderholm JD, Olaison G, Peterson KH, et al. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn’s disease. Gut 2002;50:307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soderholm JD, Peterson KH, Olaison G, et al. Epithelial permeability to proteins in the noninflamed ileum of Crohn’s disease? Gastroenterology 1999;117:65–72. [DOI] [PubMed] [Google Scholar]
- 29.Kevans D, Turpin W, Madsen K, et al. Determinants of intestinal permeability in healthy first-degree relatives of individuals with Crohn’s disease. Inflamm Bowel Dis 2015;21:879–887. [DOI] [PubMed] [Google Scholar]
- 30.Soderholm JD, Olaison G, Lindberg E, et al. Different intestinal permeability patterns in relatives and spouses of patients with Crohn’s disease: an inherited defect in mucosal defence? Gut 1999;44:96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hilsden RJ, Meddings JB, Sutherland LR. Intestinal permeability changes in response to acetylsalicylic acid in relatives of patients with Crohn’s disease. Gastroenterology 1996; 110:1395–1403. [DOI] [PubMed] [Google Scholar]
- 32.Meddings JB, Swain MG. Environmental stress-induced gastrointestinal permeability is mediated by endogenous glucocorticoids in the rat. Gastroenterology 2000; 119:1019–1028. [DOI] [PubMed] [Google Scholar]
- 33.Bitton A, Dobkin PL, Edwardes MD, et al. Predicting relapse in Crohn’s disease: a biopsychosocial model. Gut 2008; 57:1386–1392. [DOI] [PubMed] [Google Scholar]
- 34.Zuo L, Li Y, Wang H, et al. Cigarette smoking is associated with intestinal barrier dysfunction in the small intestine but not in the large intestine of mice. J Crohns Colitis 2014;8:1710–1722. [DOI] [PubMed] [Google Scholar]
- 35.Kugathasan S, Denson LA, Walters TD, et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. Lancet 2017;389:1710–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012;491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaser A, Blumberg RS. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology 2011;140:1738–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Courth LF, Ostaff MJ, Mailander-Sanchez D, et al. Crohn’s disease-derived monocytes fail to induce Paneth cell defensins. Proc Natl Acad Sci U S A 2015;112:14000–14005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wehkamp J, Harder J, Weichenthal M, et al. Inducible and constitutive beta-defensins are differentially expressed in Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 2003; 9:215–223. [DOI] [PubMed] [Google Scholar]
- 40.Marks DJ, Miyagi K, Rahman FZ, et al. Inflammatory bowel disease in CGD reproduces the clinicopathological features of Crohn’s disease. Am J Gastroenterol 2009;104:117–124. [DOI] [PubMed] [Google Scholar]
- 41.Marks DJ, Harbord MW, MacAllister R, et al. Defective acute inflammation in Crohn’s disease: a clinical investigation. Lancet 2006;367:668–678. [DOI] [PubMed] [Google Scholar]
- 42.Smith AM, Rahman FZ, Hayee B, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med 2009; 206:1883–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Segal AW. Making sense of the cause of Crohn’s - a new look at an old disease. F1000Res 2016;5:2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopetuso LR, Chowdhry S, Pizarro TT. Opposing functions of classic and novel IL-1 family members in gut health and disease. Front Immunol 2013;4:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pagnini C, Saeed R, Bamias G, et al. Probiotics promote gut health through stimulation of epithelial innate immunity. Proc Natl Acad Sci U S A 2010;107:454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corridoni D, Pastorelli L, Mattioli B, et al. Probiotic bacteria regulate intestinal epithelial permeability in experimental ileitis by a TNF-dependent mechanism. PLoS One 2012;7:e42067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spits H, Artis D, Colonna M, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol 2013; 13:145–149. [DOI] [PubMed] [Google Scholar]
- 48.Buela KA, Omenetti S, Pizarro TT. Cross-talk between type 3 innate lymphoid cells and the gut microbiota in inflammatory bowel disease. Curr Opin Gastroenterol 2015;31:449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song C, Lee JS, Gilfillan S, et al. Unique and redundant functions of NKp46þ ILC3s in models of intestinal inflammation. J Exp Med 2015;212:1869–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Powell N, Walker AW, Stolarczyk E, et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 2012; 37:674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pearson C, Thornton EE, McKenzie B, et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife 2016;5:e10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bernink JH, Krabbendam L, Germar K, et al. Interleukin-12 and −23 control plasticity of CD127(þ) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity 2015; 43:146–160. [DOI] [PubMed] [Google Scholar]
- 53.Ochel A, Tiegs G, Neumann K. Type 2 innate lymphoid cells in liver and gut: from current knowledge to future perspectives. Int J Mol Sci 2019;20:1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Camelo A, Barlow JL, Drynan LF, et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J Gastroenterol 2012;47:1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Monticelli LA, Osborne LC, Noti M, et al. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A 2015;112:10762–10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toussirot E, Houvenagel E, Goeb V, et al. Development of inflammatory bowel disease during anti-TNF-alpha therapy for inflammatory rheumatic disease: a nationwide series. Joint Bone Spine 2012;79:457–463. [DOI] [PubMed] [Google Scholar]
- 57.van Dijken TD, Vastert SJ, Gerloni VM, et al. Development of inflammatory bowel disease in patients with juvenile idiopathic arthritis treated with etanercept. J Rheumatol 2011; 38:1441–1446. [DOI] [PubMed] [Google Scholar]
- 58.Ramji A, Owen DA, Erb SR, et al. Post-liver transplant Crohn’s disease: graft tolerance but not self-tolerance? Dig Dis Sci 2002; 47:522–527. [DOI] [PubMed] [Google Scholar]
- 59.Bamias G, Delladetsima I, Perdiki M, et al. Immunological characteristics of colitis associated with anti-CTLA-4 antibody therapy. Cancer Invest 2017;35:443–455. [DOI] [PubMed] [Google Scholar]
- 60.Siakavellas SI, Bamias G. Checkpoint inhibitor colitis: a new model of inflammatory bowel disease? Curr Opin Gastroenterol 2018;34:377–383. [DOI] [PubMed] [Google Scholar]
- 61.Kugathasan S, Saubermann LJ, Smith L, et al. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut 2007;56:1696–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakajima A, Kodama T, Yazaki Y, et al. Specific clonal T cell accumulation in intestinal lesions of Crohn’s disease. J Immunol 1996;157:5683–5688. [PubMed] [Google Scholar]
- 63.Hedin CR, McCarthy NE, Louis P, et al. Altered intestinal microbiota and blood T cell phenotype are shared by patients with Crohn’s disease and their unaffected siblings. Gut 2014; 63:1578–1586. [DOI] [PubMed] [Google Scholar]
- 64.Horjus Talabur Horje CS, Middendorp S, van Koolwijk E, et al. Naive T cells in the gut of newly diagnosed, untreated adult patients with inflammatory bowel disease. Inflamm Bowel Dis 2014;20:1902–1909. [DOI] [PubMed] [Google Scholar]
- 65.Veny M, Esteller M, Ricart E, et al. Late Crohn’s disease patients present an increase in peripheral Th17 cells and cytokine production compared with early patients. Aliment Pharmacol Ther 2010;31:561–572. [DOI] [PubMed] [Google Scholar]
- 66.Desreumaux P, Brandt E, Gambiez L, et al. Distinct cytokine patterns in early and chronic ileal lesions of Crohn’s disease. Gastroenterology 1997;113:118–126. [DOI] [PubMed] [Google Scholar]
- 67.Smids C, Horjus Talabur Horje CS, Nierkens S, et al. Candidate serum markers in early Crohn’s disease: predictors of disease course. J Crohns Colitis 2017;11:1090–1100. [DOI] [PubMed] [Google Scholar]
- 68.Bamias G, Martin C, Mishina M, et al. Proinflammatory effects of TH2 cytokines in a murine model of chronic small intestinal inflammation. Gastroenterology 2005;128:654–666. [DOI] [PubMed] [Google Scholar]
- 69.Bamias G, Marini M, Moskaluk CA, et al. Down-regulation of intestinal lymphocyte activation and Th1 cytokine production by antibiotic therapy in a murine model of Crohn’s disease. J Immunol 2002;169:5308–5314. [DOI] [PubMed] [Google Scholar]
- 70.De Salvo C, Wang XM, Pastorelli L, et al. IL-33 drives eosinophil infiltration and pathogenic type 2 helper T-cell immune responses leading to chronic experimental ileitis. Am J Pathol 2016;186:885–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spencer DM, Veldman GM, Banerjee S, et al. Distinct inflammatory mechanisms mediate early versus late colitis in mice. Gastroenterology 2002;122:94–105. [DOI] [PubMed] [Google Scholar]
- 72.Fichtner-Feigl S, Fuss IJ, Young CA, et al. Induction of IL-13 triggers TGF-beta1-dependent tissue fibrosis in chronic 2,4,6-trinitrobenzene sulfonic acid colitis. J Immunol 2007; 178:5859–5870. [DOI] [PubMed] [Google Scholar]
- 73.Faleck DM, Winters A, Chablaney S, et al. Shorter disease duration is associated with higher rates of response to vedolizumab in patients with Crohn’s disease but not ulcerative colitis. Clin Gastroenterol Hepatol 2019;17:2497–2505.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rivera-Nieves J, Olson T, Bamias G, et al. L-selectin, alpha 4 beta 1, and alpha 4 beta 7 integrins participate in CD4+ T cell recruitment to chronically inflamed small intestine. J Immunol 2005;174:2343–2352. [DOI] [PubMed] [Google Scholar]
- 75.Eftychi C, Schwarzer R, Vlantis K, et al. Temporally distinct functions of the cytokines IL-12 and IL-23 drive chronic colon inflammation in response to intestinal barrier impairment. Immunity 2019;51:367–380 e4. [DOI] [PubMed] [Google Scholar]
- 76.Torres J, Danese S, Colombel JF. New therapeutic avenues in ulcerative colitis: thinking out of the box. Gut 2013;62:1642–1652. [DOI] [PubMed] [Google Scholar]
- 77.Rousseaux C, Lefebvre B, Dubuquoy L, et al. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med 2005;201:1205–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Buchman AL, Fife C, Torres C, et al. Hyperbaric oxygen therapy for severe ulcerative colitis. J Clin Gastroenterol 2001; 33:337–339. [DOI] [PubMed] [Google Scholar]
- 79.Ehehalt R, Wagenblast J, Erben G, et al. Phosphatidylcholine and lysophosphatidylcholine in intestinal mucus of ulcerative colitis patients. A quantitative approach by nanoElectrospraytandem mass spectrometry. Scand J Gastroenterol 2004; 39:737–742. [DOI] [PubMed] [Google Scholar]
- 80.Treede I, Braun A, Sparla R, et al. Anti-inflammatory effects of phosphatidylcholine. J Biol Chem 2007;282:27155–27164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Korzenik JR, Dieckgraefe BK, Valentine JF, et al. Sargramostim for active Crohn’s disease. N Engl J Med 2005; 352:2193–2201. [DOI] [PubMed] [Google Scholar]
- 82.Valentine JF, Fedorak RN, Feagan B, et al. Steroid-sparing properties of sargramostim in patients with corticosteroid-dependent Crohn’s disease: a randomised, double-blind, placebo-controlled, phase 2 study. Gut 2009;58:1354–1362. [DOI] [PubMed] [Google Scholar]
- 83.Garcia-Maurino S, Alcaide A, Dominguez C. Pharmacological control of autophagy: therapeutic perspectives in inflammatory bowel disease and colorectal cancer. Curr Pharm Des 2012; 18:3853–3873. [DOI] [PubMed] [Google Scholar]
- 84.Deng L, Zhou JF, Sellers RS, et al. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am J Pathol 2010; 176:952–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Massey DC, Bredin F, Parkes M. Use of sirolimus (rapamycin) to treat refractory Crohn’s disease. Gut 2008;57:1294–1296. [DOI] [PubMed] [Google Scholar]
- 86.Corridoni D, Chapman T, Antanaviciute A, et al. Inflammatory bowel disease through the lens of single-cell RNA-seq technologies. Inflamm Bowel Dis 2020;26:1658–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kinchen J, Chen HH, Parikh K, et al. Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell 2018;175:372–386 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smillie CS, Biton M, Ordovas-Montanes J, et al. Intra- and intercellular rewiring of the human colon during ulcerative colitis. Cell 2019;178:714–730 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tyler CJ, Guzman M, Lundborg LR, et al. Inherent immune cell variation within colonic segments presents challenges for clinical trial design. J Crohns Colitis 2020;14:1364–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hartmann FJ, Bendall SC. Immune monitoring using mass cytometry and related high-dimensional imaging approaches. Nat Rev Rheumatol 2020;16:87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Torres J, Petralia F, Sato T, et al. Serum biomarkers identify patients who will develop inflammatory bowel diseases up to 5 years before diagnosis. Gastroenterology 2020;159:96–104. [DOI] [PubMed] [Google Scholar]
- 92.Turpin W, Espin-Garcia O, Xu W, et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet 2016;48:1413–1417. [DOI] [PubMed] [Google Scholar]
- 93.Torres J, Hu J, Seki A, et al. Infants born to mothers with IBD present with altered gut microbiome that transfers abnormalities of the adaptive immune system to germ-free mice. Gut 2020; 69:42–51. [DOI] [PubMed] [Google Scholar]