

Abstract
Aberrant pro-inflammatory activation of Kupffer cells (KCs) is strongly involved in the pathogenesis of septic liver injury. Recent evidence indicates the crucial roles of excessive stimulator of interferon genes (STING) signaling activation during sepsis. However, the role of STING signaling in septic liver injury remains unclear. In this study, we demonstrated that STING signaling was markedly activated in KCs isolated from wild type mice after lipopolysaccharide (LPS) treatment. STING deficiency effectively protected liver function, attenuated systemic inflammatory response and decreased mortality in LPS-treated mice, which were aggravated by STING agonist (DMXAA). Importantly, STING signaling activation in KCs contributed to LPS-induced liver injury through promoting hepatocyte death. Mechanistically, STING signaling could be activated by release of mitochondrial DNA (mtDNA) through dynamin-related protein 1 (DRP1)-dependent mitochondrial fission in LPS-treated KCs. Additionally, LPS stimulation enhanced DRP1-dependent mitochondrial ROS production, which promoted the leak of mtDNA into the cytosol and subsequent STING signaling activation in KCs. The in vivo experiments showed that pharmacological inhibition of DRP1 with Mdivi-1 partially prevented the activation of STING signaling in KCs isolated from LPS-challenged mice, as well as alleviated liver injury and inhibited systemic inflammatory response. In summary, our study comprehensively confirmed that STING signaling senses the DRP1-dependent release of mtDNA in KCs and its activation might play a key role in LPS-induced liver injury, which offers new sights and therapeutic targets for management of septic liver injury.
Keywords: STING, DRP1, mtDNA, Kupffer cell, LPS, Liver injury
Graphical abstract
Highlights
-
•
STING signaling is activated in Kupffer cells in LPS-induced septic mice.
-
•
STING activation in Kupffer cells promotes hepatocyte death during sepsis.
-
•
DRP1-dependent mtDNA release activates STING in LPS-treated Kupffer cells.
-
•
DRP1-dependent mtROS enhances the leak of mtDNA and the subsequent STING activation.
-
•
Mdivi-1 attenuates LPS-induced STING activation in Kupffer cells and protects liver function.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate transaminase
- ATP
adenosine triphosphoric acid
- DMSO
dimethyl sulfoxide
- DMXAA
5,6-dimethylxanthenone-4-acetic acid
- DRP1
dynamin-related protein 1
- dsDNA
double-stranded DNA
- IRF3
interferon regulatory factor 3
- LPS
lipopolysaccharide
- Mdivi-1
mitochondrial division inhibitor 1
- MitoQ
mitoquinone
- MLKL
mixed lineage kinase domain like protein
- mtDNA
mitochondrial DNA
- nDNA
nuclear DNA
- NF-κB
nuclear factor-kappa B
- PI
propidium iodide
- ROS
reactive oxygen species
- STING
stimulator of interferon genes
- VDAC1
voltage-dependent anion channel 1
1. Introduction
Triggered by infection, sepsis is a life-threatening clinical syndrome with multiple organ dysfunction [1]. Despite advances in early identification and urgent management, sepsis remains the major cause of death in intensive care units [2,3]. During the development of sepsis, the liver plays a critical role in regulating metabolic and immune defense [4,5]. Evidences have shown that liver dysfunction has a great impact on the prognosis of sepsis [6,7]. Kupffer cells (KCs), the liver-resident macrophages, act as gatekeepers for bacteria phagocytosis and endotoxin clearance [8]. At the same time, KCs might initiate local inflammatory cascades by producing a large number of pro-inflammatory mediators, reactive oxygen species (ROS) and lipid peroxides, and recruiting other inflammatory cells to the liver as well, leading to the irreversible injury of hepatocytes and liver damage [9,10]. Therefore, protection of liver function by inhibiting KCs hyperactivation would be a promising therapeutic approach for sepsis.
Stimulator of interferon genes (STING) signaling pathway, a double-stranded DNA (dsDNA) sensing system, initiates powerful type I interferons and pro-inflammatory response [11]. Upon the recognition of cytosolic dsDNA from pathogens or host (including nuclear DNA, nDNA, or mitochondrial DNA, mtDNA) by cytosolic DNA sensor, STING changes to an active state and then promotes phosphorylation of interferon regulatory factor 3 (IRF3) and nuclear factor-kappa B (NF-κB). Phosphorylated IRF3 and NF-κB translocate to nucleus and induce transcription of pro-inflammatory genes [12,13]. Recently, growing evidence has confirmed that aberrant STING signaling activation plays a vital role in the pathogenesis of multiple organ dysfunction during sepsis. Genetic inhibition of STING alleviates cardiomyocyte injury [14], intestinal barrier disruption [15] and coagulation abnormalities [16] in septic mice. However, the role of STING signaling in septic liver injury has not been reported. Our preliminary data indicated that STING was expressed in KCs, but not expressed in hepatocytes and STING signaling was activated in the liver tissues of LPS-challenged mice. During sepsis, persistent immune response causes damage of parenchymal cells, resulting in multiple organ failure [17]. As an innate immune pro-inflammatory pathway, STING signaling activation in KCs might lead to hepatocyte injury and liver dysfunction. Thus, we first wondered whether STING signaling activation in KCs contributes to liver injury in sepsis.
As the sensing system of dsDNA, STING pathway could be activated by increased cytosolic dsDNA, including mtDNA. Mitochondria, as the only organelle that has independent DNA other than the nucleus in the cell, are highly dynamic undergoing continuously fission and fusion to maintain their homeostasis [18,19]. Further, there are evidences that mitochondrial fission is required for macrophages pro-inflammatory differentiation upon LPS stimulation, while inhibition of mitochondrial fission through knock-down the major pro-fission regulator, dynamin-related protein 1 (DRP1) could effectively reduce the activation of NF-κB and the subsequent release of pro-inflammatory factors [20,21]. It is still unclear whether the disturbance of mitochondrial dynamics may lead to the release of mtDNA and exert an effect in the activation of STING signaling. By focusing on the role of DRP1 in triggering mitochondrial fission and subsequent mtDNA release, this study explored whether the STING pathway could sense the release of mtDNA and be activated in LPS-stimulated KCs. The channels of mtDNA release was also investigated in LPS-treated KCs.
Overproduced mitochondrial ROS (mtROS) in activated macrophages could act as an important pro-inflammatory signal to promote mtDNA release and inflammasome activation [22]. Yu Tet al. Revealed that inhibition of mitochondrial fission prevented hyperglycemia-induced mtROS production [23]. However, it is not clear whether mtROS generation is regulated by mitochondrial fission in KCs during sepsis. It also needs to verify whether mtROS, in turn, induces mitochondrial fission-dependent mtDNA release and then lead to STING activation and pro-inflammatory response.
In this study, we aimed to investigate the effect of STING signaling activation in KCs on septic liver injury and explore the mechanism of DRP1-mediated mitochondrial fission in promoting KC STING signaling activation during sepsis.
2. Materials and methods
2.1. Animal studies
Wild-type (WT) C57/BL mice (8–10 weeks old) were purchased from the Animal Lab Center of Southern Medical University. STING-deficient mice (Tmem 173 gt/J, Sting 1 gt, STINGgt) of C57/BL background (Stock No:017,537) were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). All animals were maintained in a specific pathogen-free conditions (temperature 23–25 °C; humidity 50 ± 5%) with 12 h light/12 h dark cycle and fed standard food and water. All animal experiments were approved by the National Institutional Animal Care and Ethical Committee of Southern Medical University.
2.2. Drug administration and experimental design in vivo
Sepsis-associated liver injury mouse models were constructed by an intraperitoneal injection of 10 mg/kg lipopolysaccharide (LPS, E. coli O111:B4, Sigma, USA). Those control groups were received the same volume of PBS intraperitoneally.
To examine the role of STING on LPS-induced liver injury, mice were randomly divided into six groups, the three PBS control groups of WT, STINGgt and DMXAA (abbreviated as DMX, an agonist of STING, Selleck) WT, respectively; and the three LPS-treated groups of WT, STINGgt and DMX WT, respectively. Mice in DMX WT and DMX WT + LPS groups were treated with 10 mg/kg DMXAA intraperitoneally 2 h prior to PBS or LPS injection.
To observe the effects of DRP1-dependent mitochondrial fission on LPS-induced liver injury, mitochondrial division inhibitor 1 (Mdivi-1, Selleck), a selective inhibitor of DRP1, was intraperitoneally administrated at does of 20 mg/kg or equal volume of vehicle DMSO at 2 h prior to PBS or LPS injection.
Each group of mice was divided into two parts. The mice in first part were sacrificed for isolation of KCs and hepatocytes and the mice in second part were subjected for the harvest of blood samples and liver tissues at 12 h after LPS stimulation.
2.3. Cell isolation and treatment
The isolation of primary KCs and hepatocytes was based on a two-step perfusion procedure followed by density gradient centrifugation as previously described [24,25]. A 26G catheter was inserted into the mouse portal vein, and the liver was perfused with 30 mL warmed Hank’s balanced salt solution (HBSS, without calcium or magnesium, GIBCO) containing 1% penicillin streptomycin to flush out the blood, followed by 40 mL DMEM/F12 (GBICO) containing 0.05% collagenase IV (GBICO) and 1% penicillin streptomycin. Then the liver tissue was cut off and transferred into a 6 cm culture dish with 10 mL DMEM/F12. The liver capsule was teared apart by forceps and the liver was gently shaken to release the cells. The cell suspension was filtered through a 70 μm cell strainer and centrifuged at 50 g for 3 min at 4 °C. Hepatocytes were in the pellet. The supernatant was centrifuged at 500 g for 3 min at 4 °C to harvest non-parenchymal cells (including KCs). Non-parenchymal cells were mixed with a density gradient of 20%/50% Percoll (GE Healthcare) and centrifuged at 800 g for 15 min at 4 °C. Enriched KCs fraction was in the gradient interphase layer. KCs were then separated from other non-parenchymal cells by selective adhesion for 1 h. F4/80 antibody (Biolegend) staining for KCs purity measurement by flow cytometry showed that KCs were over 90% pure (Fig. S3).
KCs and hepatocytes isolated from LPS-treated and control mice were used for immediate evaluation. The primary KCs and hepatocytes isolated from untreated mice were used for in vitro studies. Hepatocyts were cultured in DMEM low glucose (GBICO) containing 5% FBS and 1% penicillin streptomycin and KCs were cultured in DMEM/F12 containing 10% FBS and 1% penicillin streptomycin at 37 °C in a humidified 5% CO2 incubator.
To examine the effects of STING activation in KCs on hepatocytes, KCs were isolated from untreated WT and STINGgt mice. WT KCs treated with DMX (25 μg/mL) for 12 h were labeled as DMX WT KCs. Then WT, STINGgt and DMX WT KCs were treated with PBS or LPS (100 ng/mL) for 12 h. The supernatant of KCs was collected as conditional medium (KC-CM) for 12 h incubation of hepatocytes isolated from WT mice (Fig. 3D).
Fig. 3.

STING signaling activation in LPS-treated KCs enhances hepatocyte death. (A–C) Western blot analysis of MLKL, p-MLKL and cleaved-caspase3 expression in hepatocytes in each group. Primary hepatocytes were isolated from WT, STINGgt, or DMX-treated WT mice with or without LPS challenge (n = 6). (D) The protocol of in vitro experiments for detection of hepatocyte death regulated by STING activation in KCs. KCs were isolated from untreated WT and STINGgt mice. WT KCs treated with DMX (25 μg/mL) for 12 h were labeled as DMX WT KCs. Then WT, STINGgt and DMX WT KCs were treated with PBS or LPS (100 ng/mL) for 12 h. The supernatant of KCs was collected as conditional medium (KC-CM) for 12 h incubation of hepatocytes isolated from WT mice. (E–F) The hepatocyte death rate measured by Annexin V and PI staining (n = 4). (G–H) ALT and AST concentrations in the medium of hepatocytes (n = 4). Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ns, no significance.
To assess the effects of mtROS on mtDNA-triggered STING activation, mitoquinone (MitoQ, 500 nM; MCE, HY-100116 A) or DMSO was applied for 1 h prior to LPS (100 ng/mL) administration in KCs.
2.4. Cell transfection
GV119 adenoviral vectors expressing DRP1 short hairpin RNA (shRNA) and its negative control virus (CON098 hU6-MCS-CMV-EGFP), as well as GV314 adenoviral vectors expressing DRP1 and its negative control virus (CON267 CMV-MCS-3FLAG-SV40-EGFP) were constructed by Genechem Co. Ltd (Shanghai, China). Adenovirus were added to KCs with serum-free DMEM/F12 and incubated for 3 h. Subsequently, the cells were rinsed and cultured in fresh DMEM/F12 with 10% FBS for another 48 h before LPS stimulation. The efficiency of DRP1 knockdown and over-expression in KCs was assayed by western blot analysis (Fig. S4).
2.5. Western blot analysis
KCs and hepatocytes were lysed with RIPA lysis buffer (GBCBIO) containing 1 mM PMSF (Ubio), 1 × Phosphatase Inhibitor Cocktail I (GLPBIO) and 1 × EDTA-free Protease Inhibitor Cocktail (TargetMol). The lysates were centrifuged for 15 min at 12,000 rpm at 4 °C and then the supernatant was collected and quantified by the BCA Protein Assay Kit (GBCBIO). Protein samples (30 μg per lane) were separated by 10%–12% SDS-PAGE and transferred to polyvinyl difluoride membranes. The membranes were blocked with TBST containing 5% bovine serum albumin for 1 h and then were incubated with primary antibodies overnight at 4 °C followed by horseradish peroxidase-conjugated (HRP) secondary antibody. Blots were developed using the ECL Chemiluminescence reagents. All samples were normalized against β-actin and analyzed with ImageJ software (NIH). Primary antibodies used for western blotting were β-actin (1:10,000, Ray antibody Biotech, Z0529), STING (1:1000, Abclonal, A3575), IRF3 (1:1000, Abclonal, A2172), p-IRF3-S396 (1:1000, Abclonal, AP0623), p65 (1:1000, Immunoway, YM3111), p-p65-S536 (1:1000, Immunoway, YP0191), DRP1 (1:1000, Cell signaling, #5391), p-DRP1-S616 (1:1000, Cell signaling, #3455), Caspase-3 (1:1000, Cell signaling, #9662), MLKL (1:1000, Abclonal, A13451), p-MLKL-S358 (1:1000, Cell signaling, #91689 S), respectively. Goat anti-mouse IgG-HRP (1:10,000, Ray antibody Biotech, 3001) and Goat anti-Rabbit IgG-HRP (1:10,000, Ray antibody Biotech, 3002) were used as secondary antibodies.
2.6. Quantitative real-time PCR
Total RNA from KCs was isolated with Trizol reagent. 500 ng of total RNA was reverse-transcribed into cDNA by using the RT-PCR Mix for qPCR kit (R1031, GDSBio) in which the genomic DNA was removed by DNase digestion. Real-time qPCR was conducted on the 7500 Real-Time PCR System (Applied Biosystems) with a Power Green qPCR Mix (P2102a, GDSBio) according to the manufacturer’s procedures. The results of relative expression of mRNA of IFN-β, TNF-α and IL-1β were normalized to the Ct value of GAPDH in each sample. The primers for qPCR analysis of sequences were presented in Supplementary Table 1.
2.7. Histopathology and immunohistochemistry
Liver tissues were fixed in 4% paraformaldehyde and embedded in paraffin. 4 μm sections were cut and stained with hemaroxylin and eosin (H&E). The sections were imaged under a light microscope (Carl Zeiss, Germany) and scored in a blinded manner. The scoring standard of liver injury was assessed based on previous articles [26]: spotty necrosis (grade, 0–4), capsular inflammation (grade, 0–3), portal inflammation (grade, 0–3), ballooning degeneration (grade, 0–3), statosis (grade, 0–3). The sum of the scores for each character constitutes the total liver injury pathology score, ranging from 0 (none) to 16 (severe).
For immunohistochemistry staining of STING, the liver sections were dewaxed and antigen retrieved, then the endogenous peroxidase and the non-specific binding sites were blocked by 0.3% H2O2 and 5% BSA, respectively. The sections were incubated with primary antibody of STING (1:100) at 4 °C overnight and with goat anti-rabbit IgG at room temperature for 1 h. The sections were incubated with DAB (Dako, K5007) followed by counterstaining with hematoxylin and dehydration with ethanol. Finally, these sections were observed under a light microscope (Carl Zeiss, Germany).
2.8. Flow cytometry
For the detection of hepatocytes’ death in different groups, annexin V/propidium iodide (PI) staining kit (BestBio, China) was used. Briefly, hepatocytes from different groups were isolated, washed with PBS, and re-suspended with binding buffer. The cells were stained with annexin V-FITC and PI in the dark at 4 °C for 15 min. The samples were detected immediately on the flow cytometer (BD Biosciences) and the results were analyzed by the FlowJo software (v10.6.2).
2.9. Immunofluorescence staining
KCs were seeded on coverslips and stimulated as described. Medium containing 100 nM Mitotracker Red CMXRos (Invitrogen, M7512) was added and cells were incubated for 20 min under cultured conditions. Then cells were fixed 4% paraformaldehyde for 15 min, permeabilized with 0.2% Triton X-100 for 10 min and blocked with goat serum for 60 min. Subsequently, the cells were incubated with respective primary antibodies overnight at 4 °C and stained with Alexa Flour 488 conjugated second antibodies for 60 min at room temperature. DAPI was used to stain the cell nucleus. The cells were washed with PBS buffer between each step. The stained cells were observed under confocal laser-scanning microscopy (Zeiss LSM 780, Carl Zeiss, Germany) with a 63 × oil immersion objective. The primary antibodies used for immunofluorescence were anti-p-DRP1 (1:300, Cell signaling, #3455) and anti-DNA (1:100, Progen, AC-30-10), respectively. Mitochondria with p-DRP1 co-localization assessment were quantified by ImageJ software. Mitochondrial network morphology parameters (mitochondrial length, branch junctions and the numbers of mitochondria) were analyzed by Fiji software through binary and skeleton according to previous literature [27].
2.10. Biochemical analysis
The levels of alanine aminotransferase (ALT) and aspartate transaminase (AST) in mouse serum, as well as in hepatocyte culture medium, were detected by commercial kits (Jiancheng Bioengineering, Nanjing, China) according to the manufacturer’s protocol. The levels of IFN-β, TNF-α and IL-1β in mouse serum and in KC conditional medium were measured with enzyme-linked immunosorbent assay kits (Elabscience Biotechnology, Wuhan, China).
2.11. Measurement of mtDNA in the cytosolic extracts
After adenovirus transfection and LPS stimulation, KCs were harvested and divided into two equal aliquots. One aliquot was used to extract whole-cell DNA by FastPure Cell/Tissue DNA isolation kit (Nanjing Vazyme Biotech, China). To isolate cytosolic DNA, the cells in other aliquot were gently re-suspended in digitonin lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl and 25 μg/mL digitonin, Sigma- Aldrich). Cells were incubated on a rotary mixer for 10 min at 4 °C and centrifuged at 980 g for 3 min to remove intact cells. The supernatants were collected into fresh tubes and centrifuged at 17,000 g for 10 min to remove cytosolic fragments. The cytosolic DNA was extracted by FastPure Cell/Tissue DNA isolation kit. Quantitative PCR using the SYBR Green qPCR Mix (TOYOBO Life Science) was performed on the whole-cell DNA and cytosolic DNA. The copy number of mtDNA (Dloop1-3) obtained from cytosolic extracts was normalized by the copy number of nuclear DNA (Tert) obtained from the whole-cell extracts. The primers’ sequences for qPCR analysis were listed in Supplementary Table 1.
2.12. Measurement of mitochondrial ROS
Mitochondrial ROS generation in KCs after adenovirus transfection and LPS stimulation was assessed with MitoSOX™ Red (Invitrogen, M36008), a fluorogenic dye for highly selective detection of mitochondrial superoxide. KCs in each group were incubated with MitoSOX™ Red (5 μM, 10 min, 37 °C) and then washed three times with PBS buffer. The cells were captured by Zeiss LSM 780 and the mean fluorescence intensities of MitoSOX™ Red were quantified by ImageJ software.
2.13. DNase I treatment
After transfection with GV314 adenoviral vectors for DRP1-overexpression as described previously, the KCs were treated with 3 μg DNaseI (Invitrogen) with PULSin transfected regent (Polyplus transfection) for 4 h at 37 °C. After removing DNaseI/PULSin mixture, the KCs were cultured in DMEM/F12 containing 10% FBS for 2 h, and then treated with LPS for further studies.
2.14. Cell viability assay
Cell viability was evaluated by Cell Counting Kit-8 (CCK-8, Dojindo, Japan) assay. KCs were seeded in 96-well plate at a density of 5000 cells/well. LPS was added at final concentrations of 50, 100, 200, 500, 1000 ng/mL for 12 h or 100 ng/mL for 2, 4, 8, 12, 24 h. 10 μL CCK-8 was added to each well and the cells were incubated for 1 h at 37 °C. The optical absorption values were measured at 450 nm by a SepctraMax M5 Microplate Reader (Molecular Devices, USA).
2.15. VDAC1 cross-linking
KCs were washed with PBS (pH 8.0) after LPS treatment, harvested by scraping, and incubated with 200 μM EGS in PBS (pH 8.0) containing 1 mM PMSF at 30 °C for 10 min, then quenched with 20 mM Tris (pH 7.5) for 15 min at room temperature. The reaction mixture was centrifuged for 10 min at 10,000 rpm at 4 °C and the pellet was resuspended with 1% NP-40 lysis buffer following by sonication. The homogenate was centrifuged for 10 min at 12,000 rpm at 4 °C, and then the supernatant was collected and quantified by the BCA Protein Assay Kit. The samples (50 μg) were subjected to 10% SDS-PAGE and immunoblotting with anti-VDAC1 antibody (1:2000, Ray antibody Biotech, 2010).
2.16. Statistical analysis
The differences between groups were evaluated by two-tailed unpaired Student’s t-test and one way analysis of variance with the least significant difference (LSD) test or Dunnett T3 test for post hoc comparisons. All data were analyzed by GraphPad Prism 8.0 software and presented as the mean ± SEM. P-values less than 0.05 were considered statistically significant.
3. Results
3.1. STING signaling pathway is activated in KCs in LPS-induced septic mice
We first examined STING expression and the phosphorylation of IRF3 and p65, the downstream signals of STING pathway, in the livers of mice treated by an intraperitoneal injection of 10 mg/kg LPS. Immunohistochemistry of liver sections showed that STING protein was highly expressed in LPS group and STING positive areas were concentrated in sinusoids (Fig. 1A–B). Western blot results showed that, compared with those of control mice, the protein levels of STING and the phosphorylation states of IRF3 and p65 were significantly elevated in liver tissues of mice challenged with LPS for 12 h, suggesting that STING signaling pathway is activated in livers of septic mice (Fig. S1). Whether STING is expressed in hepatocytes has not reached a consistent conclusion. It has been reported that STING mainly exists in immune cells [28]. Hence, we isolated primary hepatocytes and KCs from WT mice, then detected the STING expression. The results showed that STING was expressed in KCs, but not expressed in hepatocytes, indicating that STING activation in the livers of septic mice is more likely related to KCs (Fig. 1C). Moreover, we found that STING expression and the phosphorylation of IRF3 and p65 in KCs isolated from LPS-treated mice were significantly increased, compared with KCs isolated from control mice (Fig. 1D–G). Consistent with the activation of STING signaling, downstream inflammatory cytokines including IFN-β, TNF-α and IL-1β mRNA levels were also obviously upregulated in KCs from LPS-treated mice (Fig. 1H–J). Taken together, the above results show that STING signaling pathway is activated in KCs of LPS-induced septic mice, resulting in the phosphorylated activation of IRF3 and p65, and the subsequent expression of cytokines.
Fig. 1.

STING signaling pathway is activated in KCs in LPS-induced septic mice. (A–B) Representative images of immunohistochemistry (IHC) staining for STING protein in Control and LPS (10 mg/kg for 12 h) group. The AOD value of IHC staining was measured by imagePro Plus software (n = 6, 10 + fields per group, bar = 100 μm). (C) Western blot analysis of STING protein expression in KCs and hepatocytes isolated from WT mice (n = 6). (D–G) Western blot analysis of STING, IRF3, p-IRF3, p65 and p-p65 protein expression in KCs isolated from WT mice after Control or LPS treatment (n = 6). (H–J) The mRNA levels of inflammatory cytokines including IFN-β, TNF-α and IL-1β in KCs after Control or LPS treatment (n = 6). Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
3.2. STING signaling activation plays a critical role in LPS-induced systemic inflammatory response and liver injury
STINGgt mice and STING pharmacologic agonist DMXAA (DMX) were applied to investigate the role of STING signaling activation in sepsis-induced liver injury. STINGgt mouse has a point mutation (T596A) in exon 6 of STING gene that results in STING protein expression deficiency (Fig. S2A). DMX administration increased the levels of serum pro-inflammatory cytokines (IFN-β and TNF-α) downstream of STING signaling in WT mice (Figs. S2B–D). Furthermore, we isolated KCs from WT mice or STINGgt mice and treated with DMX, respectively. IFN-β was significantly increased in the culture medium of WT KCs, but not in that of STINGgt KCs, suggesting that type I IFN response is STING dependent (Fig. S2E).
Next, LPS was applied in WT mice, STINGgt mice and DMX-treated WT (DMX WT) mice, and the histology of liver was assessed, and serum ALT, AST as well as cytokines concentrations were detected. H&E staining showed that there were obvious ballooning degeneration of hepatocytes and an increased number of leukocyte infiltration in liver tissues of WT mice challenged with LPS. Compared with WT mice, STINGgt mice challenged with LPS showed less swelling hepatocytes and lighter inflammation in liver tissues (Fig. 2A–B). The serum levels of ALT and AST, as well as IFN-β, TNF-α and IL-1β were all significantly reduced in STINGgt mice. Compared with WT mice treated with LPS alone, the addition of STING agonist DMX worsened liver injury with more severe morphology changes and higher serum concentrations of ALT, AST (Fig. 2C–D) and IFN-β, TNF-α, IL-1β (Fig. 2E–G) as well. Furthermore, we challenged mice with a higher does of LPS (25 mg/kg) to observe whether STING deficiency protect mice from death. Under the same experimental settings, WT mice showed 60% mortality within 96 h after receiving LPS. In comparison, 90% STINGgt mice survived more than 96 h after LPS administration. As we expected, in DMX-treated WT group, the mortality was markedly increased, only 10% of mice challenged with LPS survived within 96 h (Fig. 2H). Altogether, these data indicated that STING signaling played a critical role in sepsis-induced systemic inflammatory response and liver injury.
Fig. 2.

STING signaling activation plays a critical role in LPS-induced systemic inflammatory response and liver injury. (A–B) Representative hematoxylin and eosin (H&E) staining and histology scores of liver sections in WT, STINGgt, or DMX-treated WT mice with or without LPS challenge (n = 6, bar = 20 μm). (C–D) ALT and AST concentrations in mouse serum in each group (n = 6). (E–G) Levels of inflammatory cytokines including IFN-β, TNF-α and IL-1β in mouse serum in each group (n = 6). (H) The Kaplan-Meier survival analysis after LPS (25 mg/kg) treatment in WT, STINGgt, or DMX-treated WT mice (n = 10). Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ns, no significance.
3.3. STING signaling activation in LPS-treated KCs enhances hepatocyte death
Previous reports showed that pro-inflammatory activation in KCs during sepsis was associated with liver injury in sepsis. We speculated that STING signaling activation in KCs mediated LPS-induced liver injury. We first analyzed the role of STING signaling in hepatocytes death in LPS-challenged mice. The apoptotic and necroptotic events in isolated hepatocytes from WT mice were upregulated after LPS treatment, as indicated by the increased cleaved-caspase3 and the phosphorylation of mixed lineage kinase domain-like (MLKL), which was consistent with our previous findings about increased hepatocytes death in a rat model of cecal ligation and puncture (CLP) [29]. Comparatively, apoptotic and necroptotic levels in hepatocytes were significantly reduced in LPS-treated STINGgt mice. The addition of STING agonist DMX further worsened hepatocyte death from WT mice treated with LPS (Fig. 3A–C). Evidences suggested that STING activation contributes to hepatocyte apoptosis and necroptosis in LPS-challenged mice.
STING deficiency mouse in this study is a whole body phenotype, not cell or tissue specific. To further confirm the effects of STING activation in KCs on hepatocytes, we conducted the in vitro study to observe the critical impact of STING activation in KCs on hepatocytes. We treated the isolated hepatocytes from WT mice with the conditional medium (CM) of KCs (KC-CM) from different group to verify the effect of STING activation in KCs on hepatocyte (Fig. 3D). Flow cytometry analysis displayed that, compared with those incubated with LPS-treated WT KC-CM, hepatocytes incubated with LPS-treated STINGgt KC-CM showed lower death rate, while hepatocytes incubated with DMX + LPS-treated WT KC-CM showed significantly higher death rate (Fig. 3E–F). ALT and AST as liver injury biomarkers are passively released from dead hepatocytes. We collected the medium of hepatocytes after incubating with KC-CM to detect ALT and AST concentrations. Compared with those incubated with LPS-treated WT KC-CM, hepatocytes incubated with LPS-treated STINGgt KC-CM released significant less ALT and AST, while hepatocytes incubated with DMX + LPS-treated WT KC-CM released more ALT and AST (Fig. 3G–H). Thus, we further confirmed, in vitro cell experiments, that STING signaling activation in LPS-treated KCs enhanced hepatocyte death.
3.4. DRP1-mediated mitochondrial fission triggers STING signaling activation in LPS-treated KCs
Mitochondrial fission drives macrophage metabolic reprogramming and pro-inflammatory responses. We further investigated whether mitochondrial fission had a direct effect on STING signaling activation in KCs induced by LPS. We found that mitochondrial fission was significantly increased in KCs isolated from LPS-challenged mice, as indicated by the up-regulated phosphorylation of DRP1 at serine 616 site (Fig. 4A–B). In vitro, DRP1 expression was knocked down in KCs with shRNA to further confirm the potential role of DRP1 in STING signaling activation. The confocal images showed that treating KCs with LPS induced the switch of mitochondria from normal reticular to short rod shape, which was improved by knocking down DRP1 with shRNA (Fig. 4C). Compared with the NC LPS group, DRP1 shRNA markedly increased mitochondrial length from 1.667 ± 0.078 μm to 3.936 ± 0.195 μm, mitochondrial branch junction counts from 13.25 ± 0.830 to 25.10 ± 1.807, long mitochondria (>1 μm) percentage from 29.22% to 48.51% upon LPS treatment (Fig. 4D–F). Meanwhile, increased co-location of mitochondria and S616p-DRP1 in KCs induced by LPS was obviously reversed by knocking down DRP1 (Fig. 4G). Importantly, we found that, while treating KCs with LPS induced the up-regulation of STING signal pathway, the knocking down of DRP1 with shRNA partially attenuated the expression of STING protein and the phosphorylation of IRF3 and p65 (Fig. 4H–K). These results suggested that DRP1 could activate STING signaling pathway and mediate excessive mitochondrial fission as well in LPS-treated KCs.
Fig. 4.

DRP1-mediated mitochondrial fission triggers STING signaling activation in LPS-treated KCs. (A–B) Western blot analysis of DRP1 protein expression in KCs isolated from WT mice after Control or LPS treatment (n = 6). (C) Representative of immunofluorescence staining of MitoTracker and p-DRP1 in KCs (bar = 5 μm). (D–F) Mitochondrial network parameters including mitochondrial length, mitochondrial branch junctions and the percentage of mitochondrial with different lengths in KCs transfected with negative control vector (NC) or shDRP1 in the absence or presence of LPS (100 ng/mL for 12 h, n = 40 cells). (G) Co-location analysis of mitochondrial and p-DRP1 in each group (n = 12 cells). (H–K) Western blot analysis of STING, IRF3, p-IRF3, p65 and p-p65 protein expression in KCs transfected with NC or shDRP1 in the absence or presence of LPS (n = 4). Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
3.5. Mitochondrial fission-induced mtDNA release mediates STING signaling activation in LPS-treated KCs
To further investigate the mechanism of mitochondrial fission-mediated STING signaling activation in LPS-treated KCs, we first examined the presence of double-stranded DNA (dsDNA) in the cytosol by observing the dsDNA colocalizing neither with DAPI in nuclei nor with mito-tracker in mitochondrial (Fig. 5A). The results showed that treating KCs with LPS significantly increased the positive number of dsDNA in cytosol, which could be reduced by knocking down DRP1 with shRNA (Fig. 5B). To identify the source of the increase dsDNA in cytosol, the copy numbers of mtDNA D-loops levels in cytosol was measured. Consistently, application of DRP1 shRNA decreased cytosolic D-loops of mitochondrial genome copy numbers in LPS-treated KCs (Fig. 5C). Furthermore, while the overexpression of DRP1 enhanced LPS-induced mtDNA release, the application of DNase I significantly reduced the level of cytosolic mtDNA (Fig. 5D–F), and inhibited the expression of inflammatory factor (IFN-β) downstream of STING (Fig. 5G). These results suggests that DRP1-dependent mitochondrial fission induces the release of mtDNA and then mediates STING signaling activation in LPS-treated KCs.
Fig. 5.

Mitochondrial fission-induced mtDNA release mediates STING signaling activation in LPS-treated KCs. (A) Representative of immunofluorescence staining of Mitotracker and dsDNA in KCs (bar = 5 μm). (B) Relative numbers of cytosolic dsDNA in KCs transfected with NC or shDRP1 in the absence or presence of LPS (n = 15 cells). (C) Cytosolic mtDNA fragments of D-loops levels in KCs transfected with NC or shDRP1 in the absence or presence of LPS (n = 3). (D–F) Cytosolic mtDNA D-loops levels in KCs transfected with empty vector (EV) or GV-314 vector for over-expressing DRP1 followed by incubation of DNase I or heat-inactivated DNase I (HI DNaseI) and then treated with LPS (n = 3). (G) IFN-β mRNA levels in KCs treated as in D (n = 3). Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
3.6. The formation of VDAC1 oligomer pores might mediate the release of mtDNA to cytosol in LPS-treated KCs
During apoptosis, BAX/BAK oligomers formed pores on the mitochondrial outer membrane which induced the release of mtDNA into the cytosol [30]. However, apoptosis is a highly regulated non-inflammatory cell death process, in which mtDNA-dependent activation of STING signaling is actually inhibited [31,32]. To verify whether LPS-mediated increased cytosolic mtDNA was involved in KCs apoptosis, cell viability was detected by CCK-8 assay. The data demonstrated that the cell viability was not significantly affected in KCs treated with 100 ng/mL LPS for up to 24 h in the time-dependent test (Fig. 6A), and was decreased by up to 500 ng/mL LPS in 12 h in a does-dependent detection (Fig. 6B), indicating that there was no apoptosis in our experimental setting of KCs treated with 100 ng/mL LPS for 12 h. It has been reported that VDAC1 oligomer pores mediated mtDNA release under moderate stress [33]. We further analyzed VDAC1 oligomerization in KCs revealed by EGS cross-linking and western blot. The results showed that, compared with the control group, VDAC1 dimers and trimers were markedly increased upon 100 ng/mL LPS stimulation for 12 h (Fig. 6C–E). These results illustrated that cytosolic mtDNA in LPS-treated KCs was not driven by apoptosis but might be released through VDAC1 oligomer pores.
Fig. 6.

The formation of VADC1 oligomer pores might mediate the release of mtDNA to cytosol in LPS-treated KCs. (A–B) Cell viability detection by CCK-8 assay (n = 6). (C–E) Western blot analysis of VDAC1 oligomerization evaluation in KCs isolated from WT mice after Control or LPS treatment (n = 3). Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
3.7. The oxidative stress induced by DRP1-dependent mitochondrial fission enhances the release of mtDNA and subsequent STING activation in LPS-treated KCs
A reciprocal relationship between aberrant mitochondrial fission and mitochondrial ROS (mtROS) generation had been reported under certain conditions [34]. Moreover, mtROS are involved in activating macrophages pro-inflammatory responses [35]. We wondered if DRP1 mediated mitochondrial fission influenced mitochondria oxidative stress. By labeling mtROS with MitoSOX, we found that LPS-induced upregulation of mtROS in KCs was downregulated in DRP1 shRNA-transfected KCs (Fig. 7A–B). It has been reported that increased mtROS triggered mtDNA releasing into the cytosol, which then activated STING signaling. In present study, the application of mitochondria-targeted antioxidant MitoQ prevented the increase of mtROS, resulting in the amelioration of cytosolic mtDNA release (Fig. 7C) and the attenuation of STING downstream cytokine (IFN-β) expression (Fig. 7D). The data demonstrate that DRP1-dependent mitochondrial fission effectively induced mtROS production and this oxidative stress, in turn, enhances the release of mtDNA and subsequent STING activation in LPS-treated KCs.
Fig. 7.

The oxidative stress induced by DRP1-dependent mitochondrial fission enhances the release of mtDNA and subsequent STING signaling activation in LPS-treated KCs. (A–B) Representative of immunofluorescence staining of MitoSOX in KCs transfected with NC or shDRP1 in the absence or presence of LPS (n = 12 cells, bar = 5 μm). (C) Cytosolic mtDNA D-loops levels in KCs pre-treated with DMSO or MitoQ and then stimulated with LPS (n = 3). (D) IFN-β mRNA levels in KCs treated as in C (n = 3). Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
3.8. Inhibition of mitochondrial fission with Mdivi-1 attenuates LPS-induced STING activation in KCs and protects liver function
As a selective DRP1 inhibitor, Mdivi-1 inhibited excessive mitochondrial fission by repressing DRP1 GTPase activity [36]. We applied Mdivi-1 in vivo to confirm its effects on STING activation in KCs. The results showed that Mdivi-1 treatment reduced the phosphorylation states of DRP1 at serine 616 site in KCs isolated from LPS-challenged mice, suggesting that Mdivi-1 inhibited DRP1-mediated mitochondrial fission (Fig. 8A and E). As expected, we found that STING protein expression, IRF3 and p65 phosphorylation in KCs (Fig. 8A–D), as well as the serum concentrations of IFN-β, TNF-α and IL-1β (Fig. 8F–H), were also significantly reduced after Mdivi-1 treatment. Present study further implies that Mdivi-1 has a potential protective role in LPS-induced liver injury. Flow cytometry results showed that the increased percentage of annexin V/PI positive hepatocytes isolated from liver of LPS-challenged mice was markedly reversed in the liver of LPS-challenged mice treated with Mdivi-1 (Fig. 8I–J). Compared with livers in Vehicle + LPS mice, the ballooning degeneration of hepatocytes and leukocyte infiltration were significantly attenuated with a lower histology score in livers from Mdivi-1+LPS group (Fig. 8K-L). The serum levels of ALT and AST in Mdivi-1+LPS mice were reduced as well (Fig. 8M–N). Altogether, these results clearly confirmed the effect of mitochondrial fission and subsequent STING activation in LPS-induced liver injury and the inhibition of mitochondrial fission might be an effective way to prevent this liver injury.
Fig. 8.


Inhibition of mitochondrial fission with Mdivi-1 attenuates LPS-induced STING signaling activation in KCs and protects liver function. Primary hepatocytes and KCs were isolated from WT mice treated with Mdivi-1 20 mg/kg or Vehicle (equal volume of DMSO) in the absence or presence of LPS (10 mg/kg for 12 h). (A–E) Western blot analysis of STING, IRF3, p-IRF3, p65, p-p65, DRP1, p-DRP1 protein expression in KCs (n = 6). (F–H) Levels of inflammatory cytokines including IFN-β, TNF-α and IL-1β in mouse serum in each group (n = 4). (I–J) Hepatocyte death detection by flow cytometry in each group (n = 4). (K–L) H&E staining and histology scores of liver sections in each group (n = 6, bar = 100 μm). (M − N) ALT and AST concentrations in mouse serum in each group (n = 6). Data are expressed as mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001.
4. Discussion
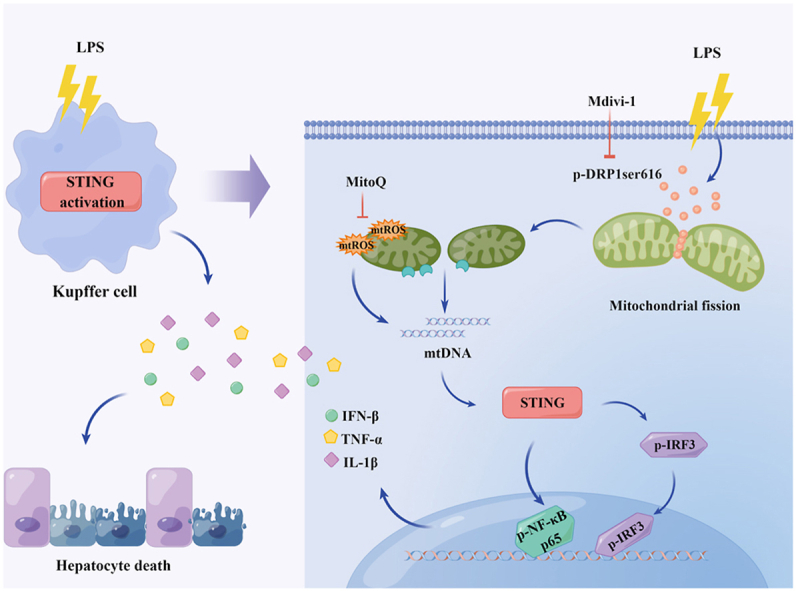
Liver injury often occurs in the early stage of sepsis and it is an independent risk factor for hospitalization and death of sepsis [6]. Hepatocytes, the parenchymal cells of the liver, are mainly responsible for synthesis, metabolism, and detoxification. Irreversible loss of function in hepatocytes leads to liver injury [37]. KCs, the specialized macrophages resident in liver sinusoids, are indispensable for clearing pathogens and mediating inflammatory responses. Excessive pro-inflammatory activation of KCs is the key factor of septic liver injury [9]. In this study, we found that LPS administration promoted STING signaling activation in KCs and STING deficiency markedly alleviated LPS-induced liver injury by inhibiting hepatocyte death. In mechanism, we found that the activation of STING signaling in LPS-treated KCs could be mediated by cytosol mtDNA which was released by DRP1-induced mitochondrial fission and mtROS production. Meanwhile, pharmacologically inhibiting DRP1 with Mdivi-1 significantly blunted STING signaling activation in KCs from LPS-challenged mice and alleviated septic liver injury (Fig. 9). These findings supplies a new sight for the development of septic liver injury and a potential therapeutic target for management of this liver injury.
Fig. 9.

Schematic process of the activation of STING signaling mediated by DRP1-dependent release of mtDNA in KCs in LPS-induced liver injury.
Accumulating evidence suggests that abnormal STING signaling activation in macrophages plays an important role in the developments of sepsis-induced coagulation disorders [16], acute pancreatitis [38] and non-alcoholic fatty liver disease [39]. Herein, our study, for the first time, focused on the role of STING signaling in hepatic macrophages, KCs, in the development of septic-induced liver injury. Previous studies have shown that STING is mainly expressed in immune cells [28]. Consistent with these results, we found that STING was expressed in KCs, but not in hepatocytes. The results confirmed that STING signaling was activated in liver tissue, as well as in KCs isolated from LPS-treated mice. The effect of STING signaling activation on septic-induced liver injury was further revealed that STING deficiency alleviated LPS-induced liver injury with the improvement of liver morphology and function, the inhibition of systemic inflammatory response and the reduction of mortality of septic mice, whereas the enhanced activation of STING with agonist DMX significantly aggravated those LPS-induced liver injury responses. Hepatocyte death is considered to be the fundamental cause of liver dysfunction. Our previous study has found that there were marked apoptotic and necroptotic hepatocytes in septic rats [29]. In present study, we provided evidence that down-regulation of STING pathway with STINGgt effectively inhibited hepatocyte apoptosis and necroptosis in LPS-challenged mice. More directly, the death rate was significantly elevated while the hepatocytes was incubated with KC-CM from DMX + LPS-treated WT KCs, as the death rate was attenuated in hepatocytes incubated with KC-CM from LPS-treated STINGgt KCs, These results unveiled the critical effect of STING activation in KCs in the development of septic-induced liver injury.
The changes of mitochondrial dynamics between fusion and fission play an important role in macrophage function [40,41] and previous studies demonstrated that DRP1-dependent mitochondrial fission could induce the pro-inflammatory activation of macrophages [20]. In this study, mitochondrial fission and the pro-inflammatory response induced by STING activation were all inhibited by transfection of DRP1 shRNA in LPS-treated KCs, indicating that DRP1-mediated mitochondrial fission was required for STING signaling activation. The inhibition of DRP1 GTPase activity with small-molecule Mdivi-1 inhibited the activation of STING signaling in KCs isolated from LPS-challenged mice. And Mdivi-1 treatment partially attenuated liver pathological damage and inhibited systemic inflammatory response at the same time. These results further support the effects of DRP1-dependent mitochondrial fission on STING activation and subsequent LPS-induced liver injury, and also indicate that Mdivi-1 may be a potential agent for treating septic liver injury.
It is reported that the mtDNA escaped from fragmented mitochondria to cytosol is one of the triggers for the activation of STING signaling that senses changes in dsDNA, resulting to the enhanced expression of type I interferon during the infection of dengue virus [42]. In present study, we found that the STING signaling activation in LPS-treated KCs was also related to the release of mtDNA to cytosol, which was dependent on DRP1-mediated mitochondrial fission. Down-regulated DRP1 expression in KCs significantly decreased LPS-induced mtDNA release. Previous study has demonstrated that DRP1-dependent mitochondrial fission could subsequently induce voltage-dependent anion channel 1 (VDAC1) oligomerization in the mitochondrial outer membrane [43]. And the release of mtDNA into cytosol via VDAC1 oligomer pores activated STING signaling in mouse embryo fibroblasts (MEFs) under moderate stress [33]. But there was also a report demonstrated that the release of mtDNA could also be facilitated by the macropores formed by Bcl-2 family proteins-BAX/BAK in the mitochondrial outer membrane during the process of intrinsic apoptosis in MEFs [30]. In present study, the viability of KCs was not significantly altered, but the formations of VDAC1 protein dimer and trimer were markedly elevated in KCs with 100 ng/mL LPS stimulation for 12 h. This result indicated that mtDNA might be released through the channel formed by the oligomerization of VDAC1 during DRP1-dependent mitochondrial fission. The result from degradating mtDNA with DNase I transfection in LPS-treated KCs further revealed that the release of mtDNA from fragmented mitochondria is necessary for STING activation.
It has been reported that mitochondrial fragmentation has a regulatory effect on the generation of mtROS and inhibition of DRP1 with Mdivi-1 also reduced the production of mtROS [36]. In present study, increased mtROS in LPS-activated KCs was remarkably down-regulated by knock-down of DRP1, indicating that DRP1 mediated mitochondrial fission could indeed regulate the level of mtROS. On the other hand, there was a study showed that the release of mtDNA into the cytosol was dependent on mtROS [22], while exogenous ROS (H2O2) induced mtDNA releasing into the cytosol in MEFs [33]. In present study, the inhibition of mtROS with mitochondria-specific antioxidant MitoQ could not only attenuated the release of mtDNA, but also down-regulated the activation of STING, confirming that the increased mtROS, in turn, plays a role in mtDNA release and subsequent STING activation.
In conclusion, our findings revealed that STING signaling activation in KCs played an important role in septic liver injury by promoting hepatocyte death and system inflammatory response. DRP1-mediated mitochondrial fission promoted mtROS generation and mtDNA release into cytosol, which activated STING signaling activation in KCs. These findings provides novel evidences for the involvement of STING activation in the development of sepsis-induced liver injury, as well as new therapeutic strategies for management of septic liver injury.
Funding
This study was supported by the National Natural Science Foundation of China (No. 82172139, 81870210) and Guangdong Basic and Applied Basic Research Foundation (No.2019A1515012022).
Author contributions
Qin Zhang, Jiayi Wei, Zhuanhua Liu, Xiaoxia Huang and Wujiang Lai performed experiments; Qin Zhang, Maomao Sun and Jie Wu analyzed results; Yanjia Chen and Xiaohua Guo provided scientific advice; Qin Zhang and Qiaobing Huang designed the study and wrote the manuscript; Qiaobing Huang supervised the research.
Declaration of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank Biying Zhou for providing technical support in the experiments. We drew the Graphical Abstract by Figdraw (www.figdraw.com).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102367.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Singer M., Deutschman C.S., Seymour C.W., et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3) JAMA. 2016;315(8):801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taeb A.M., Hooper M.H., Marik P.E. Sepsis: current definition, pathophysiology, diagnosis, and management. Nutr. Clin. Pract. 2017;32(3):296–308. doi: 10.1177/0884533617695243. [DOI] [PubMed] [Google Scholar]
- 3.Fleischmann-Struzek C., Mellhammar L., Rose N., et al. Incidence and mortality of hospital- and ICU-treated sepsis: results from an updated and expanded systematic review and meta-analysis. Intensive Care Med. 2020;46(8):1552–1562. doi: 10.1007/s00134-020-06151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Protzer U., Maini M.K., Knolle P.A. Living in the liver: hepatic infections. Nat. Rev. Immunol. 2012;12(3):201–213. doi: 10.1038/nri3169. [DOI] [PubMed] [Google Scholar]
- 5.Sun J., Zhang J., Wang X., et al. Gut-liver crosstalk in sepsis-induced liver injury. Crit. Care. 2020;24(1):614. doi: 10.1186/s13054-020-03327-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woznica E.A., Inglot M., Woznica R.K., et al. Liver dysfunction in sepsis. Adv. Clin. Exp. Med. 2018;27(4):547–551. doi: 10.17219/acem/68363. [DOI] [PubMed] [Google Scholar]
- 7.Jothimani D., Venugopal R., Abedin M.F., et al. COVID-19 and the liver. J. Hepatol. 2020;73(5):1231–1240. doi: 10.1016/j.jhep.2020.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krenkel O., Tacke F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017;17(5):306–321. doi: 10.1038/nri.2017.11. [DOI] [PubMed] [Google Scholar]
- 9.Yan J., Li S., Li S. The role of the liver in sepsis. Int. Rev. Immunol. 2014;33(6):498–510. doi: 10.3109/08830185.2014.889129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shan Z., Ju C. Hepatic macrophages in liver injury. Front. Immunol. 2020;11:322. doi: 10.3389/fimmu.2020.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Decout A., Katz J.D., Venkatraman S., et al. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021;21(9):548–569. doi: 10.1038/s41577-021-00524-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hopfner K.P., Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat. Rev. Mol. Cell Biol. 2020;21(9):501–521. doi: 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 13.Chen Q., Sun L., Chen Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016;17(10):1142–1149. doi: 10.1038/ni.3558. [DOI] [PubMed] [Google Scholar]
- 14.Li N., Zhou H., Wu H., et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019;24 doi: 10.1016/j.redox.2019.101215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Q., Ren H., Li G., et al. STING-mediated intestinal barrier dysfunction contributes to lethal sepsis. EBioMedicine. 2019;41:497–508. doi: 10.1016/j.ebiom.2019.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H., Zeng L., Xie M., et al. TMEM173 drives lethal coagulation in sepsis. Cell Host Microbe. 2020;27(4):556–570. doi: 10.1016/j.chom.2020.02.004. e556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strnad P., Tacke F., Koch A., et al. Liver - guardian, modifier and target of sepsis. Nat. Rev. Gastroenterol. Hepatol. 2017;14(1):55–66. doi: 10.1038/nrgastro.2016.168. [DOI] [PubMed] [Google Scholar]
- 18.Xie L.L., Shi F., Tan Z., et al. Mitochondrial network structure homeostasis and cell death. Cancer Sci. 2018;109(12):3686–3694. doi: 10.1111/cas.13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma K., Chen G., Li W., et al. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 2020;8:467. doi: 10.3389/fcell.2020.00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu W., Wang X., Zhao J., et al. Stat2-Drp1 mediated mitochondrial mass increase is necessary for pro-inflammatory differentiation of macrophages. Redox Biol. 2020;37 doi: 10.1016/j.redox.2020.101761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rambold A.S., Pearce E.L. Mitochondrial dynamics at the interface of immune cell metabolism and function. Trends Immunol. 2018;39(1):6–18. doi: 10.1016/j.it.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 22.Nakahira K., Haspel J.A., Rathinam V.A., et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu T., Robotham J.L., Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. U. S. A. 2006;103(8):2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu M., Xu H.H., Lin Y., et al. LECT2, a ligand for Tie 1, plays a crucial role in liver fibrogenesis. Cell. 2019;178(6):1478–1492. doi: 10.1016/j.cell.2019.07.021. e1420. [DOI] [PubMed] [Google Scholar]
- 25.Charni-Natan M., Goldstein I. Protocol for primary mouse hepatocyte isolation. STAR Protoc. 2020;1(2) doi: 10.1016/j.xpro.2020.100086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang H.W., Choi S., Song H., et al. Effect of hyperbaric oxygen therapy on acute liver injury and survival in a rat cecal slurry peritonitis model. Life. 2020;10(11) doi: 10.3390/life10110283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valente A.J., Maddalena L.A., Robb E.L., et al. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017;119(3):315–326. doi: 10.1016/j.acthis.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Luo X., Li H., Ma L., et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology. 2018;155(6):1971–1984. doi: 10.1053/j.gastro.2018.09.010. e1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Q., Wei S., Lu J., et al. Necrostatin-1 accelerates time to death in a rat model of cecal ligation and puncture and massively increases hepatocyte caspase-3 cleavage. Am. J. Physiol. Gastrointest. Liver Physiol. 2019;316(4):G551–G561. doi: 10.1152/ajpgi.00175.2018. [DOI] [PubMed] [Google Scholar]
- 30.McArthur K., Whitehead L.W., Heddleston J.M., et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. 2018;359(6378) doi: 10.1126/science.aao6047. [DOI] [PubMed] [Google Scholar]
- 31.Rongvaux A., Jackson R., Harman C.C., et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014;159(7):1563–1577. doi: 10.1016/j.cell.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.White M.J., McArthur K., Metcalf D., et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 2014;159(7):1549–1562. doi: 10.1016/j.cell.2014.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J., Gupta R., Blanco L.P., et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366(6472):1531–1536. doi: 10.1126/science.aav4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Picard M., Shirihai O.S., Gentil B.J., et al. Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013;304(6):R393–R406. doi: 10.1152/ajpregu.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brune B., Dehne N., Grossmann N., et al. Redox control of inflammation in macrophages. Antioxidants Redox Signal. 2013;19(6):595–637. doi: 10.1089/ars.2012.4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan C., Wang L., Zhang J., et al. Mdivi-1 attenuates oxidative stress and exerts vascular protection in ischemic/hypoxic injury by a mechanism independent of Drp1 GTPase activity. Redox Biol. 2020;37 doi: 10.1016/j.redox.2020.101706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong V., Nanchal R., Karvellas C.J. Pathophysiology of acute liver failure. Nutr. Clin. Pract. 2020;35(1):24–29. doi: 10.1002/ncp.10459. [DOI] [PubMed] [Google Scholar]
- 38.Zhao Q., Wei Y., Pandol S.J., et al. STING signaling promotes inflammation in experimental acute pancreatitis. Gastroenterology. 2018;154(6):1822–1835. doi: 10.1053/j.gastro.2018.01.065. e1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu Y., Liu Y., An W., et al. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Invest. 2019;129(2):546–555. doi: 10.1172/JCI121842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mehta M.M., Weinberg S.E., Chandel N.S. Mitochondrial control of immunity: beyond ATP. Nat. Rev. Immunol. 2017;17(10):608–620. doi: 10.1038/nri.2017.66. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y., Li N., Zhang X., et al. Mitochondrial metabolism regulates macrophage biology. J. Biol. Chem. 2021;297(1) doi: 10.1016/j.jbc.2021.100904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aarreberg L.D., Esser-Nobis K., Driscoll C., et al. Interleukin-1 beta induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol Cell. 2019;74(4):801–815. doi: 10.1016/j.molcel.2019.02.038. e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou H., Zhang Y., Hu S., et al. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J. Pineal Res. 2017;63(1) doi: 10.1111/jpi.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.