

SUMMARY
Correct positioning of T cells within infected tissues is critical for T cell activation and pathogen control. Upon tissue entry, effector T cells must efficiently locate antigen-presenting cells (APC) for peripheral activation. We reveal that tissue entry and initial peripheral activation of Th1 effector T cells are tightly linked to perivascular positioning of chemokine-expressing APCs. Dermal inflammation induces tissue-wide de novo generation of discrete perivascular CXCL10+ cell clusters, enriched for CD11c+MHC-II+ monocyte-derived dendritic cells. These chemokine clusters are “hotspots” for both Th1 extravasation and activation in the inflamed skin. CXCR3-dependent Th1 localization to the cluster micro-environment prolongs T-APC interactions and boosts function. Both the frequency and range of these clusters are enhanced via a T helper 1 (Th1)-intrinsic, interferon-gamma (IFNγ)-dependent positive-feedback loop. Thus, the perivascular CXCL10+ clusters act as initial peripheral activation niches, optimizing controlled activation broadly throughout the tissue by coupling Th1 tissue entry with enhanced opportunities for Th1-APC encounter.
Graphical Abstract
In brief
Prizant et. al. identify a perivascular peripheral activation niche for T cells, defined by myeloid cell expression of the chemokine CXCL10. These CXCL10+ perivascular clusters serve as hotspots for T cell entry into the inflamed skin and a niche for early activation.
INTRODUCTION
The spatiotemporal control of effector T cell activation at sites of inflammation and infection is critical for precise delivery of effector molecules and for limiting collateral damage. For pathogen control within infected tissues, CD4+ effector T cells must locate cognate antigen-bearing, antigen-presenting cells (APCs) for peripheral re-activation of effector functions (Mandl et al., 2014; Masopust and Schenkel, 2013). The range and efficiency of tissue search for ligand by CD4+ T cell effectors has an important role in successful anti-pathogen immunity.
Models for how T cells efficiently search the inflamed tissue have been informed by intravital imaging studies that have revealed a surprising degree of apparently random movement of T cells at infection and tumor sites (Filipe-Santos et al., 2009; Harris et al., 2012; Mrass et al., 2006; Overstreet et al., 2013), guided on a micro-anatomical scale by local chemical and physical cues (Gaylo et al., 2016b; Krummel et al., 2016; Sarris and Sixt, 2015) and a paucity of the ligand-bearing targets, APC (Egen et al., 2011; Fowell and Kim, 2021). The scope of T cell search is influenced by the point of tissue entry and the distribution pattern of the relevant APCs. Indeed, the efficiency of the immune responses can be enhanced by reducing random distribution through clustering targets in regions of greater density. In inflamed peripheral tissues, small, self-organizing clusters of T cells with innate effectors have been described: perivascular clustering of APCs and CD8+ T cells in the skin (Natsuaki et al., 2014), clustering of APCs and Th2 cells in the asthmatic lung (Veres et al., 2017), and macrophage clusters that sustain resident tissue memory T cells (Iijima and Iwasaki, 2014). The positional cues that shape the recruitment, composition, and retention of T cells within these niches are not fully understood.
Chemokines and other chemoattractants have important roles in the spatial organization of lymphoid tissues and in the movement of leukocytes within and between lymphoid and peripheral tissues (Griffith et al., 2014; Krummel et al., 2016). How chemokines direct the accumulation of T cells to particular microanatomical regions within tissues remains elusive (Sarris and Sixt, 2015). Chemokine sources within inflamed tissues are speculated to locally tune T cell migration in a form of “guided randomness,” which may result in iterative, small directional changes that result in location-specific T cell accumulation (Weber et al., 2013). This may be achieved by precise spatial display of chemokines through cell- or extracellular matrix (ECM)-associated heparan-sulfate immobilization (Schumann et al., 2010; Sorokin, 2010). Additional positional control comes from the dynamic regulation of chemokine-receptor sensitivity, which can lead to chemokine-mediated arrest (Lämmermann and Kastenmüller, 2019; Sarris et al., 2012). Thus, chemokines may serve as both “stop” and “go” signals within tissues. For example, the acute blockade of chemokines CXCL9 and CXCL10 has been shown to halt T cell interstitial migration (Gaylo-Moynihan et al., 2019; Harris et al., 2012), whereas T cells deficient in the CXCL9/10-receptor CXCR3 have defects in “stopping” at viral infection foci (Hickman et al., 2015). How T cells integrate chemokine signals with other positional cues, such as cognate antigen, is unclear.
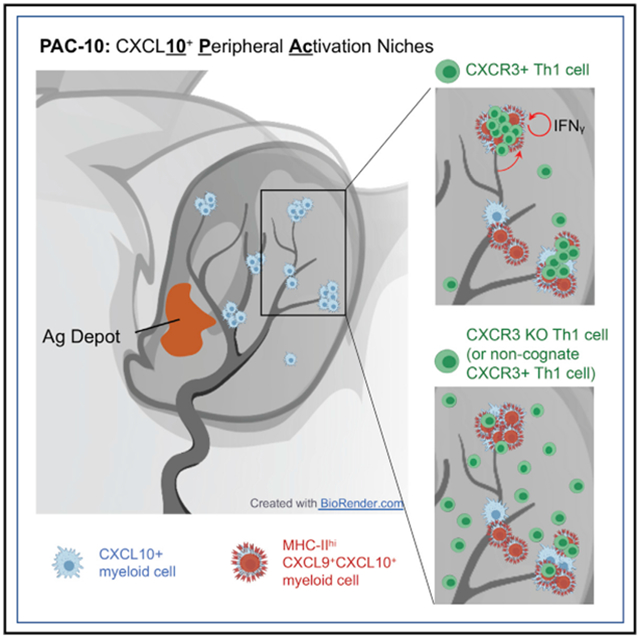
In this report, we used intravital multiphoton microscopy (IV-MPM) and a CXCL9/CXCL10 reporter (Groom et al., 2012) to define the spatial and temporal relationship between CXCR3 chemokine availability and the dynamic range of CD4+ Th1 cell migration in vivo. We reveal that the first step in peripheral activation of Th1 effector T cells is tightly linked to tissue entry through perivascular positioning of chemokine-expressing APCs. After immunization or infection of the skin, CXCL9 and CXCL10 expression was micro-anatomically restricted to distinct perivascular clusters of APCs that “marked” a previously unrecognized spatial preference for Th1 cell accumulation. These clusters were distributed throughout the inflamed ear pinna, often 1,000s of microns from the immunization site, forming distinct CXCL10+ peripheral activation (PAC-10) niches that facilitate tissue-wide immune activation. The CXCL10+ chemokine-rich clusters served to nucleate the recruitment of CXCR3+ Th1 cells from the bloodstream with durable Th1 encounters with cognate Ag-bearing APCs for initial peripheral activation. Th1 dynamics in the PAC-10 niche is regulated by CXCR3-dependent localization to the chemokine-rich cluster and by antigen-dependent retention within the cluster. Moreover, we find a key role for IFNγ-producing CD4+ T cells themselves in shaping the composition, number, and position of PAC-10 niches. These findings suggest incoming effector T cells have a crucial role in dictating the magnitude and tissue range of initial activation at inflamed sites through marshaling chemokine-rich, peripheral activation niches.
RESULTS
De novo generation of perivascular, CXCL10-expressing cell clusters is broadly distributed within the inflamed skin
The CXCR3 chemokines CXCL9 and CXCL10 are strongly upregulated in response to many immune challenges. In response to immunization with protein antigens and complete Freund’s adjuvant (CFA), CXCL10 is one of the dominant chemokines at the site of cutaneous inflammation (Figure 1A). To identify the location and cellular source of CXCR3 chemokines, we used the REX3 reporter mouse (Groom et al., 2012); in which transcriptional expression of CXCL9 and CXCL10 are linked to red fluorescent protein (RFP) and blue fluorescent protein (BFP), respectively. REX3 mice were intradermally (i.d.) immunized with ovalbumin (OVA) protein emulsified in CFA (OVA/CFA) in the ear pinna, and the kinetics of CXCL9 and CXCL10 expression was determined by flow cytometric analysis. BFP-CXCL10 was rapidly and strongly induced within the hematopoietic compartment (Figures 1B, S1A, and S1B), with fewer cells expressing RFP-CXCL9 (Figure 1B). Most RFP-CXCL9+ cells co-expressed BFP-CXCL10 (Figures S1A and S1B). Using non-invasive, intravital multiphoton microscopy (IV-MPM) to visualize the precise location of the BFP-CXCL10+ cells in three dimensions (3D), BFP expression was found in distinct regions adjacent to CD31+ blood vessels of the inflamed tissue (Figures 1C and 1D). RFP-CXCL9 expression was found coincident with BFP-CXCL10 expression in these peri-vascular clusters (Figure S1). Most BFP-CXCL10+ cells were either in contact or close (within 10 μm) to a CD31+ vessel (Figures 1E-1G; Video S1); indeed, the average distance of BFP-CXCL10+ cells from the CD31+ vessels was significantly shorter than the average distance of cells if they were randomly distributed within the same field (Figure 1F). Although BFP-CXCL10+ cells were found close to vessels, we did not find evidence that they surrounded the vessels as a perivascular cuff. Similar proximity analysis of BFP-CXCL10+ cells to LYVE-1+ lymphatic vessels showed no association of BFP-CXCL10+ cells with lymphatics (Figures 1G, S1D, and S1E). Thus, after immunization, clusters of CXCL9- and CXCL10-expressing cells are spatially restricted to perivascular areas.
Figure 1. Th1 accumulation at perivascular BFP-CXCL10+ clusters within the inflamed ear.

(A) Chemokine protein levels from day 3 OVA/CFA-immunized WT ears. Day 5 OVA/CFA immunization of REX3 mice, cells in inflamed ears.
(B) Frequency of BFP-CXCL10 and RFP-CXCL9 expressing hematopoietic cells. Representative from five independent experiments, >20 REX mice per group. Day 5 OVA/CFA immunization of REX3 mice, cells in inflamed ears.
(C) Distribution of BFP-CXCL10+ cells (blue) and CD31+ vessels (red) by IV-MPM in the dermis: gray, SHG; scale bar, 50 μm. Processed image: CD31+ vessels (red) reconstructed and volumetrically rendered in 3D using Imaris; BFP-CXCL10+ cells color coded by distance from vessel. Representative maximal z projection image, four independent experiments, 12 ears, >25 imaging volumes. Day 5 OVA/CFA immunization of REX3 mice, cells in inflamed ears.
(D) 3D surface rendering of BFP-CXCL10+ perivascular cluster (Imaris). Day 5 OVA/CFA immunization of REX3 mice, cells in inflamed ears.
(E and F) Frequency distribution of BFP-CXCL10+ cells from CD31+ vessels (E). BFP-CXCL10+ cell distance from CD31+ vessels (Data) compared to random distribution in same imaging volume (Random) (F). (E and F) Three independent experiments, 10 ears, 20 imaging volumes. Day 5 OVA/CFA immunization of REX3 mice, cells in inflamed ears.
(G) BFP-CXCL10+ cell distance from CD31+ and LYVE-1+ vessels. Two independent experiments, five ears, 10 imaging volumes. Day 5 OVA/CFA immunization of REX3 mice, cells in inflamed ears.
(H) Tiled image of six imaging fields of 512 (x) × 512 (y) × 60 (z) μm; scale bar, 100 μm; raw and processed images as in (C). Right panel, semi-automated 3D cluster reconstruction using DBSCAN-based algorithm: R, radius from cell centroid (μm); N, number of neighbors within the radius. Each dot represents a single detected BFP-CXCL10+ cell within the identified clusters, with each cell in the same cluster designated a cluster-specific color. Day 5 OVA/CFA immunization of REX3 mice, cells in inflamed ears.
(I) Tissue-wide cluster distribution relative to the OVA/CFA emulsion (Ag depot, highly auto-fluorescent), IV-MPM tiled image of region highlighted in upper-left picture using a 10× objective; scale bar, 300 μm. Individual images (1–4) acquired with 25× objective (below); scale bar, 50 μm. Day 5 OVA/CFA immunization of REX3 mice, cells in inflamed ears.
(J and K) Individual cluster distance (J) and frequency of BFP-CXCL10+ cell distance (K) from Ag depot. Cluster identified using DBSCAN-based algorithm (H) and distances calculated between the center of the cluster and emulsion. Three independent experiments, 133 clusters, >20 imaging volumes. Day 5 OVA/CFA immunization of REX3 mice, cells in inflamed ears (J).
For unbiased analysis of chemokine cluster positioning and subsequent analysis of immune cell positioning to those clusters, we employed semi-automated, density-based spatial clustering of application with noise (DBSCAN)(Ester et al., 1996; Figure S2). This computational analysis enabled the accurate 3D identification and reconstruction of high-density BFP-CXCL10+ cell clusters in the inflamed skin (Figures 1H and S2). The BFP-CXCL10+ clusters were not observed in the naive ear dermis before immunization (Figure S2). Similar perivascular clustering of BFP-CXCL10+ cells was observed after cutaneous infection with the intracellular protozoan parasite Leishmania major (Figure S3).
The use of OVA/CFA immunization enabled us to determine the position of the CXCL10+ perivascular clusters relative to the antigen depot (Ag depot; highly auto-fluorescent) by examining a large area of the inflamed ear pinna by MPM (Figure 1I). Unexpectedly, BFP-CXCL10+ cells were found in distinct clusters, scattered throughout the tissue, often 1,000s of micrometers from the emulsion site (Figure 1I). Analysis of BFP+ clusters using our unbiased clustering tool revealed a broad distribution of clusters across the tissue (Figure 1J) with most clusters some 4,000 μm from the Ag depot (Figure 1K). We cannot rule out expression of CXCL10 at the Ag depot site itself, but we are unable to distinguish that from the autofluorescence from the emulsion itself. Thus, immunization in the skin induces the de novo formation of distinct, perivascular clusters of CXCR3-chemokine-producing cells with far-reaching distribution.
Th1 cells enter the skin at sites of BFP-CXCL10+ clusters and accumulate in a CXCR3-mediated and antigen-dependent manner
Once clusters were defined, we were able to quantitate the positioning of Th1 cells relative to the chemokine cluster. In-vitro-generated, adoptively transferred, Th1 cells preferentially accumulated at sites of CXCL10+ cell perivascular clusters (Figures 2A and 2B). We did not find an accumulation of T cells directly at the auto-fluorescent Ag depot site itself. To assess whether chemokines CXCL9 or CXCL10 had an active role in Th1 accumulation, wild-type (WT) and CXCR3-deficient (CXCR3 knockout [KO]), fluorescently labeled Th1 cells were co-transferred. CXCR3 KO Th1 cells failed to accumulate within BFP-CXCL10+ clusters (Figures 2C and 2D; Video S2). Similarly, OT-II Th2 cells, which naturally fail to upregulate the expression of CXCR3 (Griffith et al., 2014; Sallusto and Lanzavecchia, 2000), were also not restricted to the BFP-CXCL10+ clusters in the OVA/CFA dermis (Figure S3). Notably, CXCR3-dependent Th1 accumulation within BFP-CXCL10+ clusters did not occur in the absence of cognate peptide: OVA-specific Th1 cells failed to accumulate within BFP-CXCL10+ clusters formed in the keyhole limpet hemocyanin (KLH)/CFA-immunized dermis (Figures 2E, 2F, and S4; Video S3), suggesting chemokine signals alone are insufficient for maintaining T cells in the perivascular clusters. To formally test the role of antigens in T cell positioning, we acutely blocked T cell receptor (TCR)/peptide:major histocompatibility complex (MHC)-II interactions with an MHC-II-blocking antibody (Ab) and assessed Th1 cell localization. After MHC-II blockade, Th1 cells showed a loss of spatial preference for the BFP-CXCL10+ clusters (Figures 2G and S4). Thus, the de novo induction of perivascular BFP-CXCL10+ cell clusters marked an unrecognized spatial preference for antigen-dependent CXCR3+ Th1 cell accumulation.
Figure 2. Th1 cell spatial preference.

OT-II Th1 cells were transferred i.v. into OVA/CFA-immunized REX3 mice, and their position relative to the BFP-CXCL10+ clusters was determined on day 5 of immunization by IV-MPM of the inflamed ear dermis.
(A) OT-II Th1 cells (green) localized to BFP-CXCL10+ clusters (blue). Maximal z projection by IV-MPM: gray, SHG; scale bar, 100 μm.
(B) Frequency of OT-II Th1 cells within 10 μm of BFP-CXCL10+ cells, with paired data points from the same imaging field. Pooled data from three independent experiments, >15 imaging volumes.
(C and D) Co-transferred WT (green) and CXCR3 KO (magenta) OT-II Th1 cells relative to BFP-CXCL10+ cells (blue). Representative maximal z projection by IV-MPM; scale bar, 50μm. (D) WT and CXCR3-KO OT-II Th1 cell distance from BFP-CXCL10+ cells. Pooled data from three independent experiments, >15 imaging volumes.
(E and F) Immunization with cognate (OVA/CFA) or non-cognate (KLH/CFA) antigen and transfer of OVA-specific OT-II Th1 cells. (E) Representative maximal z projection by IV-MPM; scale bar, 50 μm; and (F) frequency of OT-II Th1s (green) relative to BFP-CXCL10+ clusters (blue).
(G) After Th1 transfer and immunization with OVA/CFA, mice were treated i.d. in the ear with isotype control or anti-MHC-II Ab 6 h before imaging. The position of Th1 cells relative to the BFP-CXCL10+ cluster was analyzed as in (E) and (F). Data from 2–3 independent experiments.
Statistics: Mann-Whitney (D) or paired t test (B, F, and G), *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Much of our knowledge of the dynamics of leukocyte extravasation comes from the intravital study of neutrophils recruited in large numbers at kinetically well-defined times after immune challenge (Nourshargh and Alon, 2014). The relative paucity of CD4+ effector T cells entering the inflamed tissue at any given time has hindered the intravital study of their tissue entry. CXCL10 is strongly induced in the brain endothelium upon infection with Plasmodium berghei and appears to help promote T cell-endothelial adhesion(Sorensen et al., 2018). In contrast, the blood vessels within the OVA/CFA-inflamed dermis did not express detectable levels of BFP-CXCL10 or RFP-CXCL9 by intravital imaging (Figures 1C and 1H). However, the perivascular localization of BFP-CXCL10+ clusters raised the possibility that locally recruited, chemokine-producing hematopoietic cells may direct targeted T cell extravasation. To test that notion, we intravenously (i.v.) injected fluorescently labeled, OVA-specific OT-II TCR transgenic Th1 cells into OVA/CFA-immunized REX3 mice and used IV-MPM to detect the position of incoming Th1 cells relative to the BFP-CXCL10+ clusters. As early as 1–2 h after cell transfer, Th1 cells were observed to enter the dermis through vessels adjacent to BFP-CXCL10+ clusters (Figures 3A and 3B; Videos S4 and S5). By 18 h after cell transfer, OT-II-Th1 cell accumulation in the dermis was notably clustered at perivascular sites of BFP-CXCL10 expression (Figures 3C-3E; Video S6).
Figure 3. Th1 cells preferentially enter the skin at BFP-CXCL10+ clusters in a CXCR3-mediated manner.

Fluorescently labeled OT-II Th1 cells were transferred i.v. into day-5 OVA/CFA-immunized REX3 mice, and IV-MPM of inflamed dermis was performed hours after cell transfer.
(A) 1 h after Th1 cell (green) transfer, CD31+ vessel (magenta), BFP-CXCL10+ cells (blue); scale bar, 50 μm. Right panel, BFP-CXCL10+ cluster identification using the DBSCAN-based algorithm.
(B) 2 h after Th1 cell (green) transfer, CD31+ vessel (magenta), BFP-CXCL10+ cells (blue); scale bar, 50 μm. Insert (upper right): time-lapse images of Th1 cell (arrow) transmigrating a CD31+ vessel. Processed images (lower right): CD31+ vessels reconstructed and volumetrically rendered in 3D using Imaris.
(C–E) 18 h after Th1 cell (green): gray, SHG; scale bar, 200 μm. Representative of two independent experiments, three mice, six imaging volumes (C). (D) Cluster identification in 3D using DBSCAN-based algorithm of BFP-CXCL10+ cells (left) and OT-II Th1 cells (middle) from (C) (R = 50 μm, N = 10 neighbors). Top panels, all cells; bottom panels, clustered cells. Merged clusters (top, right). (E) 3D representation of BFP-CXCL10+:T cell co-clusters from (D).
(F–I) Co-transferred WT (Thy1.2+ GFP+) and CXCR3 KO (Thy1.2+ GFP−) OT-II Th1 cells in OVA/CFA-immunized Thy1-congenic mice, acutely labeled with anti-CD45 Ab 2 min before sacrifice. (F) Acute CD45 labeling of circulating leukocytes (blood) and tissue leukocytes (LN). (G) Representative plots of CD45 labeling in the inflamed ear pinna of endogenous T cells (Thy1.2−) and co-transferred WT(Thy1.2+ GFP+) and CXCR3 KO(Thy1.2+ GFP−) OT-II cells, (H) frequency and (I) number of intravascular (CD45+) versus extravascular Th1 cells. Three independent experiments, three to five mice/experiment.
Stats by paired t test (H and I), ns, p > 0.05, **p ≤ 0.01, ***p ≤ 0.001.
To confirm that the CXCR3-driven Th1 cell accumulation in the perivascular region was indeed a result of local extravasation, as opposed to intravascular accumulation, WT and CXCR3 KO OT-II-Th1 cells were co-transferred to OVA/CFA-immunized mice, and their intravascular versus extravascular location was distinguished by acute i.v. injection of anti-CD45 Ab 2 min before sacrifice, followed by flow cytometry(Anderson et al., 2014). Using this system, on average, 98% of CD45+ cells were labeled in the blood (intravascular), whereas only 0.17% of cells were extravascular (Figure 3F). In the inflamed ear tissue, most of the WT Th1 cells were extravascular (not labeled by the i.v. anti-CD45 Ab) (Figure 3G) consistent with their local exit from the blood. The inflamed ear pinna is heavily vascularized and of the cells harvested from the ear, around 11%–15% are intravascular and are labeled by acute i.v. anti-CD45 Ab (Figure 3G). Fewer CXCR3 KO OT-II-Th1 cells were present extravascularly (Figures 3G-3I), highlighting a role for CXCR3 in tissue entry. In contrast, there was no difference in the number of WT versus CXCR3 KO OT-II-Th1 cells within the intravascular pool (Figures 3G-3I), indicating that CXCR3 deficiency does not impair the representation of CXCR3 KO OT-II-Th1 cells within the circulating pool of transferred Th1 cells. Overall, these data suggest that perivascular clusters of CXCL10 production serve as “hotspots” for CXCR3-mediated Th1 tissue entry.
BFP-CXCL10+ clusters are enriched for myeloid CD11b+CD11c+MHC-II+ cells
To determine the role of these perivascular clusters in shaping Th1 cell function, we next defined the cellular sources of the chemokines using flow cytometry. Greater than 95% of the CD45+ RFP-CXCL9+ and BFP-CXCL10+ cells were CD11b+ myeloid cells and were enriched for a subset with MHC-II expression (Figures 4A and 4B). Using t-distributed stochastic neighbor embedding (tSNE) analysis of a multiparameter flow-cytometry panel aimed at distinguishing major myeloid cell subsets (Guilliams et al., 2016; Nakano et al., 2015; Figure S5), the BFP-CXCL10+ cells were found to be coincident with a subset of the MHC-II+ cells (Figure 4C). Further analysis, defined the predominant BFP-CXCL10+ cells to be within monocyte-derived dendritic cells (DCs) (moDCs: Ly6GlowCD64+F4/80+CD88+CD11c+MHC-II+) and monocyte/macrophage (mono-macs) subsets. There was little CXCL9 or CXCL10 expression within the classical DC (cDC) subsets (Figures 4D-4F), predominantly of the cDC2 subset (Figure S5) or B cells (Figures 4D-4F). Expression levels of BFP extended over a number of logs by flow cytometry, with different innate cell types expressing distinct levels of BFP (Figures 4F and S5), but moDCs and MHC-II+ mono-macs contained cells with the highest levels of BFP-CXCL10 (Figure 4F; histograms in Figure S5), suggesting that the high chemokine expressing cells are also cells capable of presenting antigens and activating Th1 cells. Many of these innate subsets can be found as tissue-resident populations or, during inflammation, can be recruited from the blood. The use of a photo-conversion system, Kaede Tg+ mice (Tomura et al., 2008), enabled us to identify newly recruited cells from the blood into the tissue and determine their CXCL10 and MHC-II status (Figure S6). Approximately 10%–20% of moDC and mono-mac subpopulations were BFP-CXCL10+ in the blood of immunized mice (Figure S6). Those percentages increased to 40% BFP-CXCL10+ for moDC and mono-mac subpopulations within the pool of newly recruited cells to the inflamed tissue (Figure S6). Thus, BFP-CXCL10+ cells with APC potential, or their precursors, can enter via the blood already expressing chemokines and/or can upregulate chemokine-producing potential soon after tissue entry.
Figure 4. BFP-CXCL10+ myeloid cell clusters enriched for CD11c+MHC-H+ cells.

(A) Representative dot plots of BFP-CXCL10 and RFP-CXCL9 expression by CD11b+ cells within the day-5 OVA/CFA-immunized REX3 dermis.
(B) Representative histograms of MHC-II expression by BFP-CXCL10+ (blue) and BFP-CXCL9+ (red) CD45+ cells from (A). Gray: RFP-CXCL9− BFP-CXCL10− CD45+ cells.
(C) tSNE analysis on singlet live CD45+ cells using concatenated multi-color flow cytometry data.
(D) Immune subsets gated by manual flow cytometry analysis displayed on tSNE plots (left). Overlay of BFP-CXCL10+ cells (right).
(E) Frequency of BFP-CXCL10+ cells (left), BFP-CXCL10+RFP-CXCL9+ cells (middle). and BFP-CXCL10−RFP-CXCL9+ cells (right) within each of the immune subsets in (D). Bars represent means ± SEM of three independent experiments, three to four mice/experiment.
(F) Density plots for BFP-CXCL10, RFP-CXCL9, and MHC-II within each subtype in (E).
(G–I) MHC-II+ (orange) staining relative to the BFP-CXCL10+ cluster (G), cluster analysis of BFP-CXCL10+ and MHC-II+ cells (H), MHC-II MFI of cells (I) relative to the BFP-CXCL10+ clusters in (H). Representative maximal z projection images by IV-MPM from two independent experiments, two to three mice/imaging session, >10 imaging volumes; scale bar, 50 μm.
(J) Colocalization of MHC-II+, BFP-CXCL10+ and Th1 cells. Right panel (processed image), cluster analysis of MHC-II and BFP-CXCL10 cells using the DBSCAN-based algorithm. Representative maximal z projection images by IV-MPM from two independent experiments, two to three mice/imaging session, >10 imaging volumes; scale bar, 50 μm.
(K) CD11c+ (yellow) cells relative to BFP-CXCL10+ clusters (blue); day-5 OVA/CFA-immunized REX3 ears.
To spatially correlate the cell subsets with the BFP-CXCL10+ perivascular clusters in situ in the inflamed dermis, Abs against MHC-II or CD11c were i.d. injected directly into the immunized REX3 ears 5 days after immunization, when the BFP-CXCL10+ clusters had formed. Using IV-MPM, we identified dense clusters of MHC-II+ cells (Figure 4G) that co-localized with BFP-CXCL10+ clusters (Figures 4G and 4H), with the MHC-II mean fluorescent intensity (MFI) being greatest on those cells positioned within the BFP-CXCL10+ clusters (Figure 4I). Moreover, BFP-CXCL10+ and MHC-II+/CD11c co-clusters were preferred sites of Th1 cell accumulation (Figures 4J and 4K). Thus, Th1 cells enter and accumulate within the inflamed tissue in CXCL10+ perivascular hubs enriched in both chemokines and APCs.
Distinct roles for chemokines and antigens in the dynamics of chemokine cluster entry and exit
A pattern of random migration of T cells has been observed within a variety of inflamed tissues including the skin, lung, liver, and brain (Egen et al., 2008; Harris et al., 2012; Overstreet et al., 2013). In the absence of evidence for directional migration, the role of chemokines in driving T cell positioning within tissues remains unclear. The CXCL9/10 reporter provided a unique opportunity to study the role of CXCR3 chemokines in Th1 tissue positioning in real time in mice. We used the density-based cluster identification algorithm to define a virtual 3D perimeter of the cluster and then classified T cell tracks according to their interactions with that cluster surface (Figure 5A). While most Th1 cells remained inside or outside of the chemokine perivascular cluster over the 60-min imaging time, co-transferred CXCR3-KO OT-II Th1 cells were much less likely to enter the chemokine-cluster than WT OT-II Th1 cells (Figures 5B and 5C). Therefore, in the absence of CXCR3, Th1 cells are not efficiently recruited into the CXCL10-rich cluster. To determine relative exit rates (independent of entry differences), we followed only those Th1 cells (WT versus CXCR3 KO) present in the cluster at the start of the imaging time. There was no difference in the degree of cluster exit between WT and CXCR3-KO OT-II Th1 cells (Figures 5B and 5C), suggesting CXCR3 chemokines do not have a key role in retention of Th1 cells in the CXCL10-rich cluster. Given the antigen dependency of the cluster association (Figures 2G and S4), we speculated that cognate antigens would be a major driver in retaining Th1 cells within the CXCL10-rich cluster. As predicted, fewer OT-II Th1 cells were present within the non-cognate antigen KLH/CFA clusters than were in the OVA-containing clusters during the imaging time (Figure 5D), but OVA-specific Th1 cells were also far more likely to leave the KLH-containing clusters compared with the OVA-containing clusters (Figures 5D and 5E). These findings indicate that Th1 cells require CXCR3 chemokine signals to efficiently position within CXCL10 clusters but depend on cognate interactions with antigen-bearing cells to be retained.
Figure 5. Distinct roles for CXCR3 and cognate antigen in cluster dynamics.

IV-MPM analysis of Th1 cell movement in the dermis of REX3 day 5 OVA/CFA- or KLH/CFA-immunized mice. Virtual 3D BFP-CXCL10+ clusters defined as in Figure 1. OT-II Th1 entry and exit from BFP-CXCL10+ clusters was tracked over a 60-min imaging period.
(A) Representative semi-automated DBSCAN identification of the CXCL10+ cells (blue) in the chemokine cluster and designation of Th1 cell location in (green) or out (red) of the chemokine cluster at the start of the imaging period.
(B) Movement of co-transferred WT (left) and CXCR3-KO OT-II Th1 (right) cells in the dermis of OVA/CFA-immunized mice. The circle represents the BFP-CXCL10+ cluster. Arrows indicate the movement of cells relative to the BFP-CXCL10+ cluster: green, entered the cluster; red, exited the cluster. Arrow tail position indicates the region in which the track began, whereas arrow head position indicates the region in which the track ended over the 60-min imaging window. Shown are averaged percentages of cells from each genotype that exhibited the same pattern of movement. Numbers in parentheses indicate numbers of cells in each compartment.
(C) Frequency of cell entry and exit from the BFP-CXCL10+ clusters as in (B).
(D and E) Pattern of movement of WT OT-II Th1 cells into and out of BFP-CXCL10+ clusters in the dermis of OVA/CFA (left) or KLH/CFA (right), analyzed as in (B). Pooled data from three independent experiments; three to six mice/group. Bars represent means ± SEM. Statistics by two-way ANOVA, *p < 0.05, **p < 0.01.
Perivascular CXCL10+ APC clusters optimize antigen-specific T cell activation
To determine the functional effect of CD4+ T cell positioning to chemokine-rich regions, we next examined the dynamic behavior of Th1 cells at the BFP-CXCL10 clusters. OT-II Th1 cell movement was more confined within the chemokine-rich cluster than it was outside of the cluster, as measured by the mean-squared displacement (Figure 6A, left panel; Video S7), indicating cues within the cluster that modify Th1 migration. Of note, those CXCR3-deficient Th1 cells that made it into the chemokine-rich cluster were also confined (Figure 6A, middle panel), thus the CXCR3-chemokines do not appear to have a role in the location-specific change in Th1 motility. Rather, the presence of cognate antigen dictated the confinement; with no changes in movement of OVA-specific Th1 cells within or outside of KLH-bearing chemokine-rich clusters (Figure 6A, right panel). Analysis of instantaneous velocity for individual cell tracks, relative to the BFP-CXCL10+ cluster, highlighted remarkable spatiotemporal control of T cell motility (Figures 6B and 6C). T cell speed was instantly reduced as the Th1 cell moved from the parenchyma into the CXCL10-rich cluster (Figure 6B, track 2), and speed increased as soon as the cell moved from the cluster out into the interstitium (Figure 6B, track 3). Further analysis reinforced those observations, Th1 cell motility (average velocity) was reduced within the chemokine-rich cluster (Figures 6D and 6E), independent of CXCR3 chemokines (CXCR3 KO Th1) (Figure 6E) but was dependent on the presence of cognate antigen (OVA versus KLH clusters) (Figure 6E). Similarly, Th1 cells were more likely to arrest within the chemokine-rich cluster when cognate antigens were present (Figure 6F), independent of their ability to respond to the CXCR3 chemokines. These data indicate that, although CXCR3 chemokines optimize movement of the Th1 cells into the chemokine-rich clusters, they are, surprisingly, not major contributors to Th1 dynamics once within the cluster. However, there was an interesting difference in WT versus CXCR3 KO Th1, with CXCR3 KO Th1 cells being less dynamic (slower speed, higher arrest coefficient) than WT Th1 cells (Figures 6E and 6F). The data suggest that, within the cluster, there may still be an active balance between antigen (stop) and chemokine (go) signals that affects movement and that, in the absence of CXCR3-signaling (CXCR3 KO Th1 cells), the dominant antigen stop-signal prevails.
Figure 6. Perivascular BFP-CXCL10+ clusters nucleate antigen-specific T:APC interactions to boost function.

Virtual 3D BFP-CXCL10+ clusters were defined by a DBSCAN-based algorithm, and Th1 cell motility was classified according to their position relative to the cluster perimeter.
(A–D) T cell motility inside and outside of the BFP-CXCL10+ clusters. (A) Mean-squared displacement (MSD) of WT (left) or CXCR3 KO (middle) OT-II Th1 cells in the OVA/CFA-immunized dermis, 30-min IV-MPM. Right panel, WT Th1 cell motility in the OVA/CFA or KLH/CFA immunized dermis. Pooled data from three independent experiments, three to six mice with 6–12 imaging volumes/group. (B) Instantaneous speed of individual Th1 cells in and out of the BFP-CXCL10+ cluster; scale bar, 50 μm. Right panels are enlarged regions from the cluster image at left. (C) Instantaneous speed of individual T cells relative to the BFP-CXCL10+ cluster. (D) Migratory paths (x–y projections) of OT-II Th1 cells relative to BFP-CXCL10+ clusters in the OVA/CFA dermis, 60-min IV-MPM. Representative data from three independent experiments, >12 imaging volumes; >30 tracks per plot.
(E and F) Average velocity (E) and arrest coefficient (F) of WT and CXCR3 KO OT-II Th1 cells described in (A).
(G) Th1:BFP-CXCL10+ cell-contact time, 60-min IV-MPM. Left, representative IV-MPM image of OT-II Th1 (green):BFP-CXCL10+ (blue) cell contacts (white), using the 3D surface overlap rendering tool in Imaris. Right, Th1:BFP-CXCL10+ cell contact duration, inside (In) and outside (Out) of BFP-CXCL10+ clusters.
(H) Th1:BFP-CXCL10+ cell-contact durations for WT and CXCR3 KO Th1 cells, 60-min IV-MPM.
(I) WTOT-II Th1:BFP-CXCL10+ cell-contact durations in the OVA/CFA (cognate antigen)- and KLH/CFA (non-cognate)-immunized dermis. Pooled data from three independent experiments, three to five mice with 6–10 imaging volumes/group.
(J and K) Frequency of CD69+ (J) and IFN-γ+ (K) WT Th1 cells in the OVA/CFA- or KLH/CFA-immunized REX3 dermis and CXCR3 KO Th1 cells in the OVA/CFA immunized dermis.
Violin plots represent the frequency distribution of the data: black dotted line, median; border-colored dotted lines, the first and third quartiles. Bars represent means ± SEM. Statistics by one-way ANOVA (E and F) or two-way ANOVA (G–K), *p < 0.05, **p < 0.01, ****p < 0.0001.
Does location to this niche affect Th1 function? To explore the cognate T:APC interactions in these niches, we used an automated 3D surface-rendering tool in Imaris (Bitplane) (Figure 6G; Gaylo-Moynihan et al., 2019) to calculate the duration of Th1 cell interactions with BFP-CXCL10+ APC within and outside of the chemokine-rich clusters. Th1 cells were much more likely to form prolonged contacts with BFP-CXCL10+ cells within the perivascular cluster (Figure 6G). Consistent with the motility data, it was cognate antigen and not CXCR3-chemokines that affected stable contacts of more than 45 minutes (Figures 6H and 6I). In contrast, functional readouts of the quality of interactions revealed significant contributions from both antigen and CXCR3 chemokines. The upregulation of CD69 as a marker of T cell activation and the frequency of IFNγ producers as a measure of effector function, by ex vivo intracellular cytokine production (McLachlan et al., 2009; Sojka and Fowell, 2011), were predictably compromised in the absence of cognate antigen but were also markedly blunted by the loss of CXCR3 (Figures 6J and 6K). Thus, the CXCR3-dependent ability to position to these chemokine-rich perivascular clusters affects the magnitude of the effector response. We reveal that clustering of CXCL10 chemokine signals and antigen-presenting capacity into specialized niches aids in the targeted recruitment of Th1 cells into tissues and nucleates signals for peripheral T cell activation, which prolongs T:APC contacts and boosts cytokine production. Our data are consistent with the idea that these PAC-10 niches facilitate initial activation of antigen-specific effector T cells without the need for an extensive tissue search.
An IFNγ-dependent feedback loop enables Th1 cells to orchestrate the composition, number, and position of the BFP-CXCL10+ clusters
Given the potent action of IFNγ on the upregulation of CXCR3-chemokines, we hypothesized that early recruitment of Th1 cells themselves may establish a positive-feedback loop that amplifies the pool of chemokine-producing APCs at the perivascular sites. To test that idea, we enhanced or deleted the pool of CD4+ effector cells and determined the effect on PAC-10 niche formation. Boosting the number of effector T cells by transfer of OTII Th1 cells potently enhanced the number and frequency of CXCL10 single-positive (BFP+) and CXCL9/10 double-positive (BFP+RFP+) immune cells in the inflamed skin (Figure 7A), in an antigen-dependent fashion (Figure 7B). Notably, Th1 cells specifically enhanced the numbers of MHC-II+ CXCL10+ moDCs (Figure 7C) and enhanced CXCL9 and CXCL10 chemokine expression by both moDCs and cDCs (Figure 7D). To test the specific requirement for Th1 cells in the CXCL9/10 induction, OTII Th1 or Th2 cells were transferred to OVA/CFA immunized mice and the frequency of BFP+ and BFP+RFP+ cells assessed. The transfer of Th1 cells, but not Th2 cells, led to the induction of BFP+ and BFP+RFP+ expression (Figure 7E), suggesting a Th1-specific factor was responsible for the chemokine induction. Indeed, an Ab-mediated blockade of IFNγ was sufficient to abrogate the Th1 boost in CXCL9 and CXCL10 expression (Figures 7F and 7G) and attenuated BFP+RFP+ expression by innate subsets induced by the endogenous immune response, independent of exogenous Th1 cell transfer (Figure S7). Similarly, the Th1-mediated induction of MHC-II expression on BFP-CXCL10+ cells was lost in the absence of IFNγ (Figure 7G). These data support a model in which the interaction of Th1 effectors and APCs in the PAC-10 niche results in cognate upregulation of CXCR3-chemokine expression by APCs (moDCs and cDCs), which may amplify recruitment and activation of additional Th1 cells. Consistent with this notion, moDCs and T cells shared a similar kinetic pattern of tissue accumulation, with moDC numbers increasing coincident with CD4+ T cell recruitment between day 3 and day 5 (Figures 7I and S7).
Figure 7. IFNγ-dependent T cell amplification of the chemokine-rich niches.

(A) Representative plots of BFP-CXCL10 and RFP-CXCL9 expression of day 5 OVA/CFA-immunization ± OT-II Th1 transfer day 3 after immunization. Three independent experiments; three to four mice/group per experiment.
(B) Frequency (left) and number (right) of CD45+ BFP-CXCL10+ and BFP-CXCL9+ BFP-CXCL10+ cells in mice immunized with OVA/CFA or KLH/CFA ± OT-II Th1 transfer. Three independent experiments; three to four mice/group per experiment.
(C and D) Number (C) and BFP-CXCL10 and RFP-CXCL9 MFI (D) within each immune subset. Three independent experiments; three to four mice/group per experiment.
(E) Frequency of BFP-CXCL10 and RFP-CXCL9 expression of day-5 OVA/CFA immunization ± OT-II Th1 or OT-II Th2 cell transfer of day 3 after immunization. Two independent experiments; three mice/group.
(F–H) IFNγ blockade, 0.5 mg XMG1.2 Ab (or control immunoglobulin G Ab [Ctl IgG]) administered i.p. 1 day before T cell transfer, followed by three daily injections of 1 mg. (F) Representative plots of BFP-CXCL10 and RFP-CXCL9 expression ± IFNγ blockade, day 5 OVA/CFA. (G) Frequency of chemokine expressing cells from (E). (H) Representative histograms of MHC-II expression on BFP-CXCL10+ cells from (E). (F–H) Three independent experiments; three to five mice/group.
(I) Number of CD4+ T cells and moDCs in the inflamed dermis following OVA/CFA immunization over time.
(J–N) CD4 depletion, 0.5 mg GK1.5 Ab (or Ctl IgG) administered i.p. 1 day before OVA/CFA immunization and day 1 and day 3 after immunization. (J) Frequency of RFP-CXCL9+ BFP-CXCL10+ within each of the immune subsets ± CD4 depletion. (K) Frequency of total BFP-CXCL10+ (left) and RFP-CXCL9+ BFP-CXCL10+ (right) cells, day 5 after immunization. Pooled data from two independent experiment; three to four mice/experiment. (L) Representative dermal tiled images of eight imaging fields of 512 (x) × 512 (y) × 60 (z) μm from control (left) and CD4 depleted (right) day-5 OVA/CFA mice, by IV-MPM; scale bar, 200 μm. Bottom, semi-automated 3D cluster reconstruction using DBSCAN-based algorithm. (M) Number of identified clusters per square millimeter of day-5 OVA/CFA-immunized dermis ± CD4 depletion. (N) Frequency distribution of BFP-CXCL10+ cell distance from Ag depot ± CD4 depletion. Three independent experiments, six mice, >15 imaging volumes per condition. Bars represent means ± SEM.
Statistics by two-way ANOVA with Sidak’s multiple comparisons (B–E, G, J, and K) or Mann-Whitney (M and N), *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
To determine the role of CD4+ T cells in the establishment and/or maintenance of the PAC-10 niche, we deleted CD4+ cells using the anti-CD4 Ab GK1.5 (1 day before immunization and on days 1 and 3 after immunization). The loss of CD4+ T cells resulted in a marked loss of the MHC-II+ CXCL9+ CXCL10+ moDC population in particular (Figures 7J and 7K). Moreover, the loss of CD4+ T cells (and co-dependent moDCs) had a profound effect on the PAC-10 niche (Figures 7L-7N). CD4 depletion reduced the overall number of clusters in the inflamed skin (Figures 7L and 7M) and limited their distribution within the tissue (Figure 7N).
Together, these data reveal the induction of CXCL10+ perivascular immune cell clusters that are distributed broadly throughout the inflamed tissue. These PAC-10 niches serve to cluster APC targets at high density in perivascular sites, coupling targeted Th1 recruitment and efficient peripheral activation of Th1 cells. A CD4+ T cell-positive feedback loop shapes the number and distribution of PAC-10 niches, amplifying the surveillance range of incoming Th1 cells.
DISCUSSION
Effector CD4+ T cells entering inflamed tissues use environmental cues to migrate through the tissue interstitium in “search” of cognate ligands (Fowell and Kim, 2021). Recent advances in intravital imaging have led to new insights into the molecular machinery used by T cells for interstitial migration (Lämmermann and Germain, 2014). However, we remain ill-informed regarding points of T cell tissue entry, the range and modality of tissue exploration, and the relative positioning of T cell re-activation events (by ligand-bearing APCs) and the infection foci themselves. The findings presented here suggest clustering of chemokine and antigen presentation signals into sub-anatomical structures, close to vessels , optimizes CD4+ T cell encounter with APCs in the periphery. This first step in peripheral activation promotes early and selective peripheral activation of antigen-specific effector cells, without the need for extensive tissue search. The use of the CXCL9 and CXCL10 reporter mouse (Groom et al., 2012), enabled the identification of an unrecognized spatial preference for Th1-cell accumulation in the inflamed skin at de novo organized perivascular sites of CXCL10 expression that we refer to as a PAC-10 niche. Our studies reveal that, within the inflamed skin, CXCL9 and CXCL10 expression was enriched in MHC-II-expressing cells, predominantly moDCs. Mechanistically, colocalization of chemokine-producing APCs and Th1 cells depended on T cell expression of CXCR3, both for site-specific entry into the inflamed tissues and for Th1 positioning within the PAC-10 niche. Constraints on Th1 motility, prolonged APC contacts, and retention within the PAC-10 niches were dependent on recognition of cognate antigens, independent of CXCR3. Nonetheless, chemokine- and antigen-derived signals were both critical for the optimization of Th1 effector function within the inflamed skin, chemokines helping to position the Th1 cells to these activation niches and antigens helping to keep them there.
Our results provide insight into the relationship between the point of CD4+ T cell tissue entry and the site of initial effector T cell activation in inflamed tissues. Sites of preferred leukocyte entry have been described for neutrophils, in which spatially restricted changes in endothelial expression of adhesins and chemokines or presence of perivascular macrophages mark hotspots of neutrophil extravasation (Abtin et al., 2014; Nourshargh and Alon, 2014). Our findings show that preferred entry sites for CD4+ T cell effectors are shaped by the focal perivascular accumulation of chemokine-producing myeloid cells. The ability to now visualize preferred sites of CD4+ T cell entry into the inflamed skin will enable important studies to define possible location-specific changes in the endothelium that support T cell extravasation. Our data are consistent with the idea that the spatial synchronization of vascular exit cues with antigen presentation in the interstitial tissue reduces the scale of the T cell search for rare APC targets upon initial tissue entry. This mechanism may also confer an early competitive advantage to those antigen-specific effectors that are recruited along with a host of non-specific effector T cells (Chapman et al., 2005). Therefore, we suggest that PAC-10 niches provide a rapid-screening platform for incoming effector CD4+ T cells to promote antigen-driven tissue retention of those effector cells with “useful” specificities. This might be particularly important at early time points after immune challenge, when the number of incoming antigen-specific T cells, initial “tissue pioneers,” will be limiting. Moreover, our data support the notion that such effector T cell tissue pioneers activated within the PAC-10 niche promote the recruitment and/or differentiation of additional chemokine-producing APCs to aid in further Th1 recruitment via an IFNγ-dependent positive-feedback loop.
The de novo induction of leukocytic perivascular clusters in the skin has previously been observed at sites of preferred CD8+ T cell accumulation in a model of contact hypersensitivity (Natsuaki et al., 2014). That prior study elegantly mapped the sequence of events that established the clusters, with dermal macrophages being critical early initiators of DC and T cell clustering. Here, we reveal a Th1-dependent amplification of the number and tissue range of those peripheral activation niches defined by CXCR3-chemokine expression. PAC-10 niches were broadly distributed throughout the inflamed dermis, often thousands of microns from the Ag depot, facilitating a targeted, yet tissue-wide, response to immune challenge. What determines the location of these perivascular clusters relative to the initial immune challenge remains unclear. Location could be random, driven by the stochastic activation of a critical mass of innate and/or incoming T cells or directed by the distribution of the infectious agent. Alternatively, these perivascular sites of APC and T cell accumulation may evolve strategically in anatomically distinct pre-organized niches defined by the steady-state position of resident innate and stromal cells, vasculature, and peripheral nerves (Dahlgren and Molofsky, 2019). Emerging data from a variety of tissues has fueled the notion that homeostatic positioning of immune cell types takes advantage of specific micro-anatomically advantageous locations within tissues to optimize the ability to respond to immune challenge. For example, in the lymph node (LN), naive CD4+ T cells appear to have a peripheral bias in their steady-state position at the paracortical region of the LN, perhaps to be better positioned to interact with incoming cDC2 cells (Baptista et al., 2019). Similarly, group 2 innate lymphoid cells (ILC2s) appear to be located within perivascular adventitial cuffs in the lung, where colocalized DC and stromal cells are poised to respond to local signals that drain from the tissue parenchyma (Dahlgren et al., 2019) and in niches adjacent to adrenergic neurons in the intestine (Moriyama et al., 2018). Therefore, the “scattered” or broad distribution of PAC-10 niches that we have observed after immunization may reflect similar spatially pre-determined sites poised to respond to immune challenge. In a Th1-dominated challenge, these peripheral activation (PAC) niches are defined by CXCL10, but the signature chemokine may differ depending on the type of immune challenge. Future studies to determine how the position of these early activation niches relate to the subsequent requirement for effector function proximal to the infection foci (Filipe-Santos et al., 2009; Hickman et al., 2015) will be important.
Chemokine receptor-deletion approaches clearly highlight a role for chemokines in effector T cell spatial positioning in inflamed tissues; CXCR3-deficient CD8+ T cells fail to end up in the right place and, hence, have reduced ability to clear infections (Hickman et al., 2015; Sung et al., 2012). Mechanistic insight has been limited because of the inability to define 3D chemokine gradients in situ. Estimations based on CCL21-producing lymphatic vessels suggest steeply decaying functional gradients from the cellular source, being undetectable for small cells such as lymphocytes within 25 μm of the cellular source (Weber et al., 2013). Thus, the spatially discrete chemokine-rich platforms illuminated by the REX3 chemokine reporter provided an opportunity to study chemokine and antigen contributions to CD4+ T cell positioning in situ, in the inflamed skin. Functionally, CXCR3-optimized Th1 activation was controlled at two levels: site-specific extravasation at hotspots of perivascular chemokine production, and enhanced interstitial access to these strategically positioned clusters of antigen presentation. In silico modeling of 2D and 3D T cell migration suggests small directional preferences, or “subtle chemotaxis,”enhances the time-to-target location and could account for the dependency on CXCR3 for cluster entry (Ariotti et al., 2015). Additional behavior previously implicated in chemokine sensing (Sarris and Sixt, 2015), such as a reduction in speed, migratory arrest close to the source, and enhanced T:APC interactions, was not observed to be CXCR3 dependent for Th1 cells in the inflamed skin. Instead, cognate antigen drove the confined migratory patterns within the PAC-10 niche, enhanced the duration of T:APC interactions, and promoted Th1 retention within the PAC-10 niche. Therefore, we show that CXCR3 chemokines have a very specific role in fine-tuning the location of Th1 cells: promoting/guiding access to chemokine-rich APC clusters, rather than influencing their dwell time in those regions. Nonetheless, the functional effect of CXCR3 deficiency was profound, having as great an influence on Th1 activation within the tissue as that observed in the absence of cognate antigens. Thus, chemokine-guided positioning is essential for the location of cognate antigen-bearing cells.
The microanatomical activation niche appears to provide additional support for initial peripheral T cell activation. Th1 interactions with CXCL10+ APCs within the cluster were prolonged compared with interactions with CXCL10+ APCs outside the cluster. Chemokines have been implicated in stabilizing T:APC interactions (Friedman et al., 2006; Molon et al., 2005), but the enhanced duration of the interactions with CXCL10+ APCs in the cluster does not appear to be chemokine driven. Nonetheless, chemokine/chemokine receptor interactions could support a qualitatively different activation signal. This may be achieved by locally enriching chemokine or antigen signals, cross talk between neighboring Th1 cells, and/or local accumulation or induction of APCs with superior stimulatory potential.
Many pathogens evade immune cell recognition by modulating the local chemokine milieu, (Antonia et al., 2019; Katzman and Fowell, 2008; Mantovani et al., 2006). Therefore, effector function may be enhanced by a sequential two-step process (Ley, 2014) in which amplification of the frequency or function of incoming antigen-specific T cells (step 1, exemplified here by inducible PAC-10 niches) is separated kinetically and/or spatially from the pathogen-infection foci itself (step 2, movement from activation niches to the pathogen foci). CD8+ T cells were shown to require interactions with interstitial DCs after arriving in the influenza-infected lung for effective antiviral immunity at the infected epithelium (McGill et al., 2008). Activation within these peripheral activation niches may provide tissue-specific signals that reinforce the Th differentiation program (Katzman and Fowell, 2008; Ley, 2014), or “license” cells for better subsequent search of the tissue to locate the infection foci, or enhance function once at the infection foci (Odoardi et al., 2012).
Many immune-pathologies are accompanied by perivascular immune infiltrates. Our work suggests that these immune clusters may reflect a conserved mechanism designed to efficiently boost activation of newly tissue-recruited effector T cells. We identify a spatially distinct peripheral activation niche for Th1 cells, PAC-10, which is regulated by the CXCR3-chemokine system. The perivascular positioning of CXCR3-chemokine-producing APCs provides spatial cues that couple Th1 extravasation with Th1 peripheral activation events, effectively reducing the scale of the initial T cell search for limited ligands. The identification of CXCL10+ peripheral activation sites provides a platform to locate and analyze the functional significance of early Th1 re-activation events in inflamed peripheral tissues. These sites may be important therapeutic targets in chronic inflammation, in which disruption of the activation niche may mitigate local inflammation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and request for resources and reagents should be directed to Dr Deborah J. Fowell: djf273@cornell.edu
Materials availability
All in-house generated mouse strains generated for this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
This study did not generate/analyze datasets. Custom-written Python code available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Rochester. Female and male mice were bred and maintained in pathogen-free conditions in group housing, used between 6-14 weeks of age, and euthanized in accordance with the University of Rochester guidelines. B6(Cg)-Tyrc-2J/J (Albino B6) mice, OVA-specific OT-II TCR transgenic C57BL/6 mice, B6.PL-Thy1a/CyJ (B6 Thy1.1) and Cxcr3tm1Dgen/J (B6 CXCR3 KO) mice were purchased from The Jackson Laboratory. REX3 (Reports the Expression of CXCR3 ligands) B6 mice were provided by A. Luster, Massachusetts General Hospital. WT C57BL/6 mice were purchased from the National Cancer Institute, Bethesda, MD. REX3 mice were crossed to Albino B6 mice. OT-II TCR transgenic mice were crossed to CXCR3 KO mice. Kaede transgenic mice were obtained from RIKEN.
METHOD DETAILS
T cell culture and adoptive transfers
For in vitro generation of effector Th1 cells, naive CD4+ T cells were isolated from lymph nodes and spleens of OT-II TCR transgenic mice using negative selection with a mouse CD4+ T Cell Isolation Kit (Miltenyi Biotec). Splenocytes from C57BL/6 mice were T cell depleted by complement lysis of Thy1.2+ cells (clone J1J) and irradiated with 2500 rads to use as antigen presenting cells (APCs). Naive CD4+ T cells were plated with irradiated APCs, 1μM cognate OVA peptide and with IL-2 (10 U/ml). For skewing to a Th1 phenotype, IL-12 (20 ng/ml; Peprotech), and anti-IL-4 (40 μg/ml; 11B11) were added to the culture. For skewing to a Th2 phenotype, IL-4 (50ng/ml; Peprotech) and anti-IFNγ (50 μg/ml; XMG1.2) were added to the culture. Cells were split in 1:2 ratio three days later and harvested on day five of culture. Cells were labeled with either the fluorescent dye CFSE (carboxyfluorescein succinimidyl ester; Life Technologies) or CellTracker DeepRed dye (Thermo Fisher scientific). Either 7x106 Th1 cells or Th2 cells were adoptively transferred into recipient REX3 or B6 Thy1.1 mice through retro-orbital injection.
In-vivo treatments and reagents
Mice were immunized intradermally (i.d.) in the ear with 10 μg OVA protein or KLH protein (Sigma Aldritch) emulsified in complete Freund’s adjuvant (CFA, Sigma Aldritch). Mice were infected i.d. in the ear pinna with 2x105 infectious (PNA-selected) Leishmania major promastigotes (strain WHOM/IR/-/173) in 10ml PBS as previously described (Fowell et al., 1997). For MHC-II blockade, 70 μg isotype control Ab or anti-MHC-II Ab (M5/114, BioXCell) was administered i.d. in the ear pinna, 6 hours prior to imaging on day 5 post immunization and cell transfer. For in-vivo CD4+ T cell depletion, anti-CD4+ antibody (0.5 mg per mouse, GK1.5) administered i.p. 1 day prior to immunization and on days 1 and 3 post immunization. For IFNγ blockade, anti-IFNγ antibody (XMG1.2) administered i.p. 1 day prior to Th1 cell adoptive transfer (0.5 mg per mouse) followed by 3 daily injections of 1 mg per mouse. For intravascular immune cell staining, anti-CD45 antibody (3μg per mouse, 30-F11, Biolegend) was injected retro-orbitally 2 min before mice were sacrificed and the ears were removed for cell isolation, ex-vivo cytokine staining and flow cytometry analysis as detailed below.
For photoconversion studies, REX3+ Kaede Tg+ mice were immunized with OVA/CFA as described above. Day 4 of immunization, the ear pinna was exposed to violet light (Tomura et al., 2008) for 10 minutes to ensure complete photoconversion from Kaede-green to Kaede-red of all cells in the ear. The rest of the mouse was shielded with aluminum foil and cells in the blood and LN remained Kaede-green. 16 hours after exposure to violet light, the ear tissue, blood and draining-lymph node were harvested for flow cytometry.
Flow cytometry
Mice were euthanized, ears were excised, ventral and dorsal sheets separated and each ear placed in 1ml Liberase DL (1 mg/ml in PBS; Sigma Aldrich) with DNase I (50 μg per ear, Sigma Aldrich), minced and incubated with agitation for 30 min at 37°C. The digested tissues were disrupted by gently pushing the ear through a metal strainer. Insoluble material was removed by filtration and the pellet was washed twice with 2% neonatal calf serum (NCS) in Hank’s balanced-salt solution (HBSS). Single cell suspensions were stained with 1:1000 Ghost dye violet 510 dead cell stain for 405nm excitation (Tonbo biosciences) in PBS for 30 min followed by a wash with 2% NCS HBSS. Next, cells were incubated for 10 min with 1:10 CD16/CD32 Fc block (2.4G2) in FACS buffer (2% NCS/PBS) followed by staining for 30 min at 4°C with fluorochrome-conjugated antibody mix in FACS buffer. Antibodies used include: anti-CD45 BUV395 (1:500, 30-F11, BD Biosciences), anti-CD64 BV605 (1:200, X54-5/7.1, Biolegend), anti-CD11c BV711 (1:450, N418, Biolegend), anti-CD19 BV786 (1:100, 1D3, BD Biosciences), anti-F4/80 PE-Cy5 (1:200, BM8, Invitrogen), anti-CD88 anti-APC (1:800, 20/70, Biolegend), anti-MHC-II APC-ef780 (1:2000, M5/114.15.2, eBioscience), anti-CD26 PE-Cy7 (1:50, H194-112, Biolegend), anti-XCR1 BV650 (1:200, ZET, Biolegend), anti-Ly6G FITC (1:500, 1A8, Biolegend), anti-CD3e Biotin (1:400, 145-2C11, eBioscience) with Streptavidin BV510 (1:1000, Biolegend), anti-CD45 PB (3ug per mouse anti-CD4 BV605 (1:200, RM4-5, Bioleged), anti-CD4 (1:200, RM4-4, Bioleged), anti-IFNγ (1:500, XMG1.2, Invitrogen) anti-Thy-1.2 PE (1:500, 53-2.1, BD biosciences), anti-CD69 PerCP-Cy5.5 (1:500, H1.2F3, BD Biosciences). For ex vivo intracellular cytokine staining, Brefeldin A (1 μg/ml) was added to all digestion and wash buffers to block cytokine secretion (Sojka and Fowell, 2011). Cell were washed, filtered and resuspended in FACS buffer with 25 μg DNase I and 20 μL AccuCheck counting beads (Thermo Fisher scientific) per ear. Samples were collected with LSR Fortessa (BD Biosciences) and analyzed with FlowJo Software (Treestar, Ashland, OR). tSNE analysis: tSNE algorithm in FlowJo (learning configuration, Auto, opt-SNE; iterations, 1000; perplexity, 30) was applied on concatenated non-gated or manually gated multi-color flow cytometry data from inflamed REX3 ears.
Luminex
Ears of mice were excised, digested, and disrupted as above in a total volume of 500μl and kept on ice. Homogenates were centrifuged and the supernatant collected and frozen at −80 C before use. Ear homogenate supernatants were assayed for chemokines and cytokines with the Milliplex MAP Mouse Cytokine/Chemokine Magnetic Bead Panel kit (Millipore). Samples were collected and analyzed using BioPlex 200 reader (BioRad).
Intravital multiphoton imaging
Mice were anesthetized with isoflurane (induction ~4%; maintenance ~1%–2%, in air) with an isoflurane vaporizer-ventilation machine (M3000R; Lei Medical). Once mice were anesthetized, the ventral side of the ear pinna was taped to a coverslip. Mice were placed on a custom-made platform for imaging (Gaylo et al., 2016a). Body temperature was maintained with a heated blanket (Kent Scientific) and a heating block (WPI). The microscope objective was heated (Bioptechs) to 36°C to maintain constant dermal temperature during imaging. Images were acquired with an Olympus FVMPE-RS twin-laser multiphoton system equipped with 4 photomultipliers tubes (PMTs) with blue, green, near-red and far-red filters (Semrock). Fluorescence was collected with an Olympus XLPlanN 25x objective (numerical aperture, 1.05) for deep-tissue multiphoton imaging and was detected with three proprietary photomultipliers. Fluorescence excitation was achieved by a sequential scan of Spectra-Physics DeepSee-MaiTai HP Ti:Sapphire laser tuned to 800 nm and Spectra-Physics InsightX3 laser tuned to 920 nm with DM690-870 and DM690-1050 dichroic mirror. The z-stack images (512 × 512 pixels) were 60μm in thickness acquired with a vertical resolution of 2–5 μm and a lateral pixel size of 994 nm. For time-series analyses, three-dimensional stacks were acquired every ~60 s. For acute detection of the MHC-II+ and CD11c+ cells in the inflamed ear, a 1:1 mixture of anti-CD11c FITC (N418, eBioscience) or anti-MHC-II AF647 antibody (M5/114.15.2, eBioscience) with anti-CD16/32 antibody (Fc Block, 2.4G2) were dialyzed against PBS to remove sodium azide. Immunized mice were injected i.d. in the ear with the antibody mix 2 hours prior to IV-MPM imaging. Blood and lymphatic vessels were stained with anti-CD31 AF647 (10 μg per mouse, retro-orbital 15 min prior to imaging, clone 390, Biolegend) and anti-LYVE-1 AF647 (10 μg per mouse, i.d. 2 hours prior to imaging) respectively.
Analysis of multiphoton data using Imaris and Python
Raw imaging data were processed with Imaris software (Bitplane). A 3x3 median filter was used to diminish noise and T cells were tracked with automated algorithms with manual correction. Cell tracks lasting for less than 8 min were excluded from analyses. No minimum cell-displacement criteria were imposed that would have excluded non-motile cells. Mean squared displacement (MSD) was calculated from raw imaging data using a custom Python script. The MSD was calculated from trajectory data using the following equation: where N is the total length of the data, r is the trajectory vector, and k is the normalized time delay, which is the elapsed time divided by the time per step. Average velocity and instantaneous speed were calculated in Imaris using track speed mean and speed formulas, respectively. Arrest coefficients were calculated as the ratio between the time that a cell was not moving (instantaneous speed < 2 μm/min) and the total time the cell was observed. Cell tracks were linearly interpolated in order to maintain consistent time windows for analysis. All analysis was performed through custom-written Python code available upon request. Movies were created in Imaris, with animation and titles added with Adobe Primer Pro.
T:CXCL10+ cell contacts
cell:cell interactions were detected within the 3D volume reconstituted from all z stacks (over time). T cells and BFP-CXCL10+ cells were reconstructed and volumetrically rendered in 3D using Imaris. We first binary masked the signal from each channel (threshold for masking is based on object size and fluorescence intensity). A separate channel was created based on overlapped pixels (threshold is absolute intensity). These overlapped pixels subjected to surface rendering in 3D to represent and visualize T cell-CXCL10+ cell contacts. To measure duration of the contacts, 3D surfaces were tracked over time.
Relative cell distance
CXCL10+ cell distance from blood vessels or T cell distance from CXCL10+ cells: vessels and cells were volumetrically rendered in 3D using Imaris. Distance transformation Xtension was applied to the CD31+ or CXCL10+ surface, resulted in a new channel with the intensities equal to the distance from the surface object. Minimal distance of each CXCL10+ cell from the closest CD31+ vessel, or T cell from the closest CXCL10+ cell, was calculated using Imaris.
3D CXCL10+ cluster reconstruction and T cell dynamics
Clusters of CXCL10+ cells were defined and reconstituted in 3D using a Python code that utilizes the unsupervised machine learning algorithm DBSCAN (Density-based spatial clustering of applications with noise) (Ester et al., 1996). There are two parameters to the algorithm; maximum distance between points, radius (R) and minimum number of neighbors (N) within R to define as part of the cluster. Density cutoffs of R = 50 μm and N = 10 neighbors were determined by grid-search where combinations of different R and N were plotted to find inflection points after which return on metric of choice is incremental. Cells outside the identified clusters were treated as noise and therefore ignored in cluster analysis. To determine T cell dynamics relative to the identified clusters (entry, exit. re-entry etc.), T cells were registered at the start of the imaging session and thereafter per time point, as positioned inside or outside of the identified clusters. For analysis of T cell motility inside versus outside of clusters, only cells that positioned the entire imaging session inside clusters compared with cells that spent the entire imaging session outside of the clusters, were included. All analysis was performed through custom-written Python code available upon request.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism was used for all statistical tests. Most analyses used the nonparametric Mann-Whitney or Kolmogorov-Smirnov tests to compare 2 treatment groups. ANOVA tests were used across multiple groups, with multiple comparisons. * = p ≤ 0.05, ** = p ≤ 0.01, *** = p ≤ 0.001, **** = p < 0.0001, ns = p > 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD45 BUV395 (clone 30-F11) | BD Biosciences | Cat# 564279; RRID: AB_2651134 |
| anti-mouse CD45 Pacific Blue (clone 30-F11) | Biolegend | Cat# 103126; RRID: AB_493535 |
| anti-mouse CD4 BV605 (clone RM4-5) | Biolegend | Cat# 100548; RRID: AB_11125962 |
| anti-mouse CD4 Pacific Blue (clone RM4-5) | Biolegend | Cat# 100531; RRID: AB_493375 |
| anti-mouse CD4 PE (clone RM4-5) | Biolegend | Cat# 100512; RRID: AB_312715 |
| anti-mouse CD4 Pacific Blue (clone RM4-4) | Biolegend | Cat# 116007; RRID: AB_11147758 |
| anti-mouse CD69 PerCP-Cy5.5 (clone H1.2F3) | BD Biosciences | Cat# 551113; RRID: AB_394051 |
| anti-mouse CD90.2, Thy1.2 PE (clone 53-2.1) | BD Biosciences | Cat# 553006; RRID: AB_394545 |
| anti-mouse CD64 BV605 (clone X54-5/7.1) | Biolegend | Cat# 139323; RRID: AB_2629778 |
| anti-mouse CD19 BV786 (Clone 1D3) | Biosciences | Cat# 563333; RRID: AB_2738141 |
| anti-mouse F4/80 PE-Cy5 (Clone BM8) | Invitrogen | Cat# 25-4801-82; RRID: AB_469653 |
| anti-mouse CD88 APC (Clone 20/70) | Biolegend | Cat# 135808; RRID: AB_10899415 |
| anti-mouse MHC-II APC-ef780 (Clone M5/114.14.2) | eBioscience | Cat# 47-5321-82; RRID: AB_1548783 |
| anti-mouse IFNg APC (clone XMG1.2) | Ebioscience | Cat# 17-7311-82; RRID: AB_469504 |
| anti-mouse CD11c FITC (clone N418) | Ebioscience | Cat# 11-0114-85; RRID: AB_464941 |
| anti-mouse CD11c BV711 (clone N418) | Biolegend | Cat# 117349; RRID: AB_2563905 |
| anti-mouse CD26 PE-Cy7 (clone H194-112) | Biolegend | Cat# 137810; RRID: AB_2564312 |
| anti-mouse XCR1 BV650 (clone ZET) | Biolegend | Cat# 148220; RRID: AB_2566410 |
| anti-mouse Ly6G FITC (clone 1A8) | Biolegend | Cat# 127606; RRID: AB_1236494 |
| anti-mouse CD3e Biotin (clone 145-2C11) | eBioscience | Cat# 13-0031-82; RRID: AB_466319 |
| anti-mouse CD31 AF647 (clone MEC13.3) | Biolegend | Cat# 102416: RRID: AB_493410 |
| anti-mouse MHC-II AF647 (clone M5/114) | Biolegend | Cat# 107618, RRID: AB_493525 |
| anti-mouse LYVE-1 AF647 (clone 223322) | R&D Systems | Cat# FAB2125R |
| Ghost Dye Violet 510 | Tonbo biosciences | Cat# 13-0870 |
| anti-mouse IL-4 (clone 11B11), purified | ATCC | Cat# HB-188; RRID: CVCL_9139 |
| anti-mouse IFNg (clone XMG1.2), purified | N/A | N/A |
| anti-mouse CD4 (clone GK1.5) | BioXcell | Cat# BE0003-1 |
| anti-mouse MHC-II (clone M5/114) | BioXcell | Cat# BE0108 |
| CD16/CD32 Monoclonal antibody (Fc block clone 2.4G2) | N/A | N/A |
| JIJ monoclonal antibody (anti Thy1.2) | ATCC | Cat# TIB-184; RRID: CVCL_9186 |
| Chemicals, peptides, and recombinant proteins | ||
| CellTracker Orange Deep Red Dye | Invitrogen | Cat# C34565 |
| CFSE (5-(and-6)-Carboxyfluroescein Diacetate, Succinimidly Ester) | Invitrogen (Molecular Probes) | Cat# C1157 |
| Complete Freund’s Adjuvant (CFA) | Sigma-Aldrich | Cat# F5881 |
| OVA peptide (Structure: ISQAVHAAHAEINEAGR-OH) | Biopeptide Co., Inc. | Custom peptide |
| OVA protein | Sigma-Aldrich | cat# A5503 |
| KLH protein | Sigma-Aldrich | Cat#H8283 |
| Human rIL-2 | NIH AIDS Reagent Program | Cat# 11697 |
| Recombinant Murine IL-4 | Peprotech | Cat# 214-14 |
| Recombinant Murine IL-12 p70 | Peprotech | Cat# 210-12 |
| Guinea Pig Complement, standard | Cedarlane | Cat# CL5000 |
| Fixation/Permeablization Solution Kit with BD GolgiPlug | BD Biosciences | Cat# 555028 |
| Fluriso (Isoflurane, USP) | VET One | Cat# 502017 |
| Liberase DL Research grade | Sigma Aldrich | Cat# 5466202001 |
| RPMI 1640 | Lonza BioWhittaker | Cat# 12-115F |
| HBSS | Corning Cellgro | Cat# 21-021-CV |
| PBS | Corning Cellgro | Cat# 21-040-CV |
| Fetal Bovine Serum-Premium | Atlanta Biologicals | Cat# S11150 |
| EDTA | Corning Cellgro | Cat# 46-034-CI |
| Streptavidin BV510 | Biolegend | Cat# 405233 |
| Brefeldin A | Invitrogen | Cat# B6542 |
| AccuCheck Counting Beads | Thermo Fisher Scientific | Cat# PCB100 |
| Peanut agglutinin (PNA) | Sigma Aldrich | Cat# L2507 |
| DNase I | Sigma Aldrich | Cat# DN25 |
| Critical commercial assays | ||
| Milliplex MAP Kit, Mouse Cytokine/Chemokine Magnetic Bead Panel | Millipore | Cat# MCYTMAG70PMX32BK |
| Naive CD4+ T cell isolation kit, mouse | Miltenyi | Cat# 130-104-453 |
| Experimental models: Organisms/strains | ||
| Mouse: REX3 mouse | Andrew Luster (MGH) | N/A |
| Mouse: B6.PL674 Thy1a/CyJ (B6 Thy1.1) | The Jackson Laboratory | Stock No: 000406 |
| Mouse: B6(Cg)-Tyrc-2J/J (Albino B6) | The Jackson Laboratory | Stock No: 000058 |
| Mouse: OTII TCR Transgenic B6 | The Jackson Laboratory | Stock No: 004194 |
| Mouse: B6.129P2-Cxcr3tm1Dgen/J (B6 CXCR3 KO) | The Jackson Laboratory | Stock No: 005796 |
| Mouse: Kaede B6 | M. Tomura, Kyoto University, RIKEN | Stock No: RBRC05737 |
| Parasite: Leishmania Major strain WHOM/IR/-/173 | R. Locksley (UCSF) | N/A |
| Software and algorithms | ||
| Imaris | Bitplane | RRID: SCR_007370 |
| Flow Jo | Tree Star | RRID: SCR_008520 |
| Python | Python Software Foundation | RRID: SCR_008394 |
| Prism | Graph Pad | RRID: SCR_002798 |
| Other | ||
| Isoflurane vaporizer-ventilation machine | Lei Medical | Model# M3000R |
| LS Columns | Miltenyi | Cat# 130-042-401 |
| Objective heater | Bioptechs | SKU: 150819-19 |
| Objective controller | Bioptechs | SKU: 150803 |
| Olympus FVMPE-RS | Olympus | N/A |
| Olympus FV-MRVGR/XR, filter set | Olympus | N/A |
| Olympus XLPlanN 25x objective | Olympus | Product# N5165100 |
| BD LSR Fortesa | BD Biosciences | N/A |
| BioPlex 200 reader | BioRad | N/A |
Highlights.
CXCL10 expression is limited to discrete perivascular niches in the inflamed skin
The CXCL10+ niches are hotspots or preferred sites of Th1 tissue entry
The niche is enriched for MHC-II+ moDCs and supports prolonged Th1:APC interactions
IFNγ enhances niche availability, boosting opportunities for Th1:APC encounter
ACKNOWLEDGMENTS
We thank the members of the Fowell laboratory for helpful discussions on the studies. Jim Miller provided insightful comments on the manuscript. We gratefully acknowledge support from the following agency: National Institutes of Health grants P01 AI02851 and R01 AI070826 to D.J.F.; F32 AI138415 to H.P.; R25 GM064133 to S.N.; and R01 CA204028 to A.D.L. We acknowledge the support of the Multiphoton and Flow Cytometry core facilities of the University of Rochester Medical Center.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109523.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abtin A, Jain R, Mitchell AJ, Roediger B, Brzoska AJ, Tikoo S, Cheng Q, Ng LG, Cavanagh LL, von Andrian UH, et al. (2014). Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat. Immunol 15, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, and Masopust D (2014). Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc 9, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonia AL, Gibbs KD, Trahair ED, Pittman KJ, Martin AT, Schott BH, Smith JS, Rajagopal S, Thompson JW, Reinhardt RL, and Ko DC (2019). Pathogen evasion of chemokine response through suppression of CXCL10. Front. Cell. Infect. Microbiol 9, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariotti S, Beltman JB, Borsje R, Hoekstra ME, Halford WP, Haanen JB, de Boer RJ, and Schumacher TN (2015). Subtle CXCR3-dependent chemotaxis of CTLs within infected tissue allows efficient target localization. J. Immunol 195, 5285–5295. [DOI] [PubMed] [Google Scholar]
- Baptista AP, Gola A, Huang Y, Milanez-Almeida P, Torabi-Parizi P, Urban JF Jr., Shapiro VS, Gerner MY, and Germain RN (2019). The chemoattractant receptor Ebi2 drives intranodal naive CD4+ T cell peripheralization to promote effective adaptive immunity. Immunity 50, 1188–1201.e6. [DOI] [PubMed] [Google Scholar]
- Chapman TJ, Castrucci MR, Padrick RC, Bradley LM, and Topham DJ (2005). Antigen-specific and non-specific CD4+ T cell recruitment and proliferation during influenza infection. Virology 340, 296–306. [DOI] [PubMed] [Google Scholar]
- Dahlgren MW, and Molofsky AB (2019). Adventitial cuffs: regional hubs for tissue immunity. Trends Immunol. 40, 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren MW, Jones SW, Cautivo KM, Dubinin A, Ortiz-Carpena JF, Farhat S, Yu KS, Lee K, Wang C, Molofsky AV, et al. (2019). Adventitial stromal cells define group 2 innate lymphoid cell tissue niches. Immunity 50, 707–722.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, and Germain RN (2008). Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity 28, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egen JG, Rothfuchs AG, Feng CG, Horwitz MA, Sher A, and Germain RN (2011). Intravital imaging reveals limited antigen presentation and T cell effector function in mycobacterial granulomas. Immunity 34, 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester M, Kriegel H-P, Sander J, and Xu X (1996). A density-based algorithm for discovering clusters in large spatial databases with noise. Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (KDD-96) (AAAI), Simoudis E, Han J, and Fayyad U, eds. pp. 226–231. [Google Scholar]
- Filipe-Santos O, Pescher P, Breart B, Lippuner C, Aebischer T, Glaichenhaus N, Späth GF, and Bousso P (2009). A dynamic map of antigen recognition by CD4 T cells at the site of Leishmania major infection. Cell Host Microbe 6, 23–33. [DOI] [PubMed] [Google Scholar]
- Fowell DJ, and Kim M (2021). The spatio-temporal control of effector T cell migration. Nat. Rev. Immunol Published online February 24, 2021. 10.1038/s41577-021-00507-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowell DJ, Magram J, Turck CW, Killeen N, and Locksley RM (1997). Impaired Th2 subset development in the absence of CD4. Immunity 6, 559–569. [DOI] [PubMed] [Google Scholar]
- Friedman RS, Jacobelli J, and Krummel MF (2006). Surface-bound chemokines capture and prime T cells for synapse formation. Nat. Immunol 7, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Gaylo A, Overstreet MG, and Fowell DJ (2016a). Imaging CD4 T cell interstitial migration in the inflamed dermis. J. Vis. Exp 109, e53585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaylo A, Schrock DC, Fernandes NR, and Fowell DJ (2016b). T cell interstitial migration: motility cues from the inflamed tissue for micro- and macro-positioning. Front. Immunol 7, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaylo-Moynihan A, Prizant H, Popović M, Fernandes NRJ, Anderson CS, Chiou KK, Bell H, Schrock DC, Schumacher J, Capece T, et al. (2019). Programming of distinct chemokine-dependent and -independent search strategies for Th1 and Th2 cells optimizes function at inflamed sites. Immunity 51, 298–309.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith JW, Sokol CL, and Luster AD (2014). Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol 32, 659–702. [DOI] [PubMed] [Google Scholar]
- Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, von Andrian UH, Moon JJ, Mempel TR, and Luster AD (2012). CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 37, 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilliams M, Dutertre CA, Scott CL, McGovern N, Sichien D, Chakarov S, Van Gassen S, Chen J, Poidinger M, De Prijck S, et al. (2016). Unsupervised high-dimensional analysis aligns dendritic cells across tissues and species. Immunity 45, 669–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TH, Banigan EJ, Christian DA, Konradt C, Tait Wojno ED, Norose K, Wilson EH, John B, Weninger W, Luster AD, et al. (2012). Generalized Lévy walks and the role of chemokines in migration of effector CD8+ T cells. Nature 486, 545–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman HD, Reynoso GV, Ngudiankama BF, Cush SS, Gibbs J, Bennink JR, and Yewdell JW (2015). CXCR3 chemokine receptor enables local CD8+ T cell migration for the destruction of virus-infected cells. Immunity 42, 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima N, and Iwasaki A (2014). T cell memory: a local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 346, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman SD, and Fowell DJ (2008). Pathogen-imposed skewing of mouse chemokine and cytokine expression at the infected tissue site. J. Clin. Invest 118, 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel MF, Bartumeus F, and Gérard A (2016). T cell migration, search strategies and mechanisms. Nat. Rev. Immunol 16, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämmermann T, and Germain RN (2014). The multiple faces of leukocyte interstitial migration. Semin. Immunopathol 36, 227–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lämmermann T, and Kastenmüller W (2019). Concepts of GPCR-controlled navigation in the immune system. Immunol. Rev 289, 205–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K (2014). The second touch hypothesis: T cell activation, homing and polarization. F1000Res. 3, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandl JN, Torabi-Parizi P, and Germain RN (2014). Visualization and dynamic analysis of host-pathogen interactions. Curr. Opin. Immunol 29, 8–15. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Bonecchi R, and Locati M (2006). Tuning inflammation and immunity by chemokine sequestration: decoys and more. Nat. Rev. Immunol 6, 907–918. [DOI] [PubMed] [Google Scholar]
- Masopust D, and Schenkel JM (2013). The integration of T cell migration, differentiation and function. Nat. Rev. Immunol 13, 309–320. [DOI] [PubMed] [Google Scholar]
- McGill J, Van Rooijen N, and Legge KL (2008). Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J. Exp. Med 205, 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan JB, Catron DM, Moon JJ, and Jenkins MK (2009). Dendritic cell antigen presentation drives simultaneous cytokine production by effector and regulatory T cells in inflamed skin. Immunity 30, 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molon B, Gri G, Bettella M, Gómez-Moutón C, Lanzavecchia A, Martínez-A C, Mañes S, and Viola A (2005). T cell costimulation by chemokine receptors. Nat. Immunol 6, 465–471. [DOI] [PubMed] [Google Scholar]
- Moriyama S, Brestoff JR, Flamar AL, Moeller JB, Klose CSN, Rankin LC, Yudanin NA, Monticelli LA, Putzel GG, Rodewald HR, and Artis D (2018). β2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 359, 1056–1061. [DOI] [PubMed] [Google Scholar]
- Mrass P, Takano H, Ng LG, Daxini S, Lasaro MO, Iparraguirre A, Cavanagh LL, von Andrian UH, Ertl HC, Haydon PG, and Weninger W (2006). Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J. Exp. Med 203, 2749–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano H, Moran TP, Nakano K, Gerrish KE, Bortner CD, and Cook DN (2015). Complement receptor C5aR1/CD88 and dipeptidyl peptidase-4/CD26 define distinct hematopoietic lineages of dendritic cells. J. Immunol 194, 3808–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsuaki Y, Egawa G, Nakamizo S, Ono S, Hanakawa S, Okada T, Kusuba N, Otsuka A, Kitoh A, Honda T, et al. (2014). Perivascular leukocyte clusters are essential for efficient activation of effector T cells in the skin. Nat. Immunol 15, 1064–1069. [DOI] [PubMed] [Google Scholar]
- Nourshargh S, and Alon R (2014). Leukocyte migration into inflamed tissues. Immunity 41, 694–707. [DOI] [PubMed] [Google Scholar]
- Odoardi F, Sie C, Streyl K, Ulaganathan VK, Schläger C, Lodygin D, Heckelsmiller K, Nietfeld W, Ellwart J, Klinkert WE, et al. (2012). T cells become licensed in the lung to enter the central nervous system. Nature 488, 675–679. [DOI] [PubMed] [Google Scholar]
- Overstreet MG, Gaylo A, Angermann BR, Hughson A, Hyun YM, Lambert K, Acharya M, Billroth-Maclurg AC, Rosenberg AF, Topham DJ, et al. (2013). Inflammation-induced interstitial migration of effector CD4+ T cells is dependent on integrin αV. Nat. Immunol 14, 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, and Lanzavecchia A (2000). Understanding dendritic cell and T-lymphocyte traffic through the analysis of chemokine receptor expression. Immunol. Rev 177, 134–140. [DOI] [PubMed] [Google Scholar]
- Sarris M, and Sixt M (2015). Navigating in tissue mazes: chemoattractant interpretation in complex environments. Curr. Opin. Cell Biol 36, 93–102. [DOI] [PubMed] [Google Scholar]
- Sarris M, Masson JB, Maurin D, Van der Aa LM, Boudinot P, Lortat-Jacob H, and Herbomel P (2012). Inflammatory chemokines direct and restrict leukocyte migration within live tissues as glycan-bound gradients. Curr. Biol 22, 2375–2382. [DOI] [PubMed] [Google Scholar]
- Schumann K, Lämmermann T, Bruckner M, Legler DF, Polleux J, Spatz JP, Schuler G, Förster R, Lutz MB, Sorokin L, and Sixt M (2010). Immobilized chemokine fields and soluble chemokine gradients cooperatively shape migration patterns of dendritic cells. Immunity 32, 703–713. [DOI] [PubMed] [Google Scholar]
- Sojka DK, and Fowell DJ (2011). Regulatory T cells inhibit acute IFN-γ synthesis without blocking T-helper cell type 1 (Th1) differentiation via a compartmentalized requirement for IL-10. Proc. Natl. Acad. Sci. USA 108, 18336–18341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen EW, Lian J, Ozga AJ, Miyabe Y, Ji SW, Bromley SK, Mempel TR, and Luster AD (2018). CXCL10 stabilizes T cell-brain endothelial cell adhesion leading to the induction of cerebral malaria. JCI Insight 3, e98911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin L (2010). The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol 10, 712–723. [DOI] [PubMed] [Google Scholar]
- Sung JH, Zhang H, Moseman EA, Alvarez D, Iannacone M, Henrickson SE, de la Torre JC, Groom JR, Luster AD, and von Andrian UH (2012). Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell 150, 1249–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A, and Kanagawa O (2008). Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc. Natl. Acad. Sci. USA 105, 10871–10876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veres TZ, Kopcsányi T, van Panhuys N, Gerner MY, Liu Z, Rantakari P, Dunkel J, Miyasaka M, Salmi M, Jalkanen S, and Germain RN (2017). Allergen-induced CD4+ T cell cytokine production within airway mucosal dendritic cell-t cell clusters drives the local recruitment of myeloid effector cells. J. Immunol 198, 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hauschild R, Schwarz J, Moussion C, de Vries I, Legler DF, Luther SA, Bollenbach T, and Sixt M (2013). Interstitial dendritic cell guidance by haptotactic chemokine gradients. Science 339, 328–332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets. Custom-written Python code available from the Lead Contact upon request.