

ABSTRACT
Congenital hypothyroidism (CH) is the most common neonatal endocrine disorder and one of the most common preventable causes of intellectual disability in the world. CH may be due to developmental or functional thyroid defects (primary or peripheral CH) or be hypothalamic‐pituitary in origin (central CH). In most cases, primary CH is caused by a developmental malformation of the gland (thyroid dysgenesis, TD) or by a defect in thyroid hormones synthesis (dyshormonogenesis, DH). TD represents about 65% of CH and a genetic cause is currently identified in fewer than 5% of patients. The remaining 35% are cases of DH and are explained with certainty at the molecular level in more than 50% of cases. The etiology of CH is mostly unknown and may include contributions from individual and environmental factors. In recent years, the detailed phenotypic description of patients, high‐throughput sequencing technologies, and the use of animal models have made it possible to discover new genes involved in the development or function of the thyroid gland. This paper reviews all the genetic causes of CH. The modes by which CH is transmitted will also be discussed, including a new oligogenic model. CH is no longer simply a dominant disease for cases of CH due to TD and recessive for cases of CH due to DH, but a far more complex disorder.
Keywords: Congenital hypothyroidism, Development, Genetic, High‐throughput sequencing, Thyroid dysgenesis, Dyshormonogenesis, Oligogenism
Proposed model of congenital hypothyroidism: complex and multi‐factor pathology. Dark blue: scientifically proven factor; light blue: factor as yet unproven or unclear.

INTRODUCTION
Thyroid hormones (THs), produced by the thyroid gland, are essential to the development, growth, and metabolism of practically all human tissues. TH production (T4 and T3) is regulated by the hypothalamic‐pituitary‐thyroid axis. Low serum TH levels lead to increased release of thyroid‐stimulating hormone (TSH) by the pituitary, under the influence of thyrotropin‐releasing hormone (TRH) by the hypothalamus. TSH, a key regulator of thyroid function, stimulates the synthesis and secretion of THs by the thyroid (Figure 1). A TH deficiency, at birth, congenital hypothyroidism (CH) results in severe retardation of growth and neuropsychomotor development in the absence of replacement therapy initiated quickly from the neonatal period.
FIGURE 1.
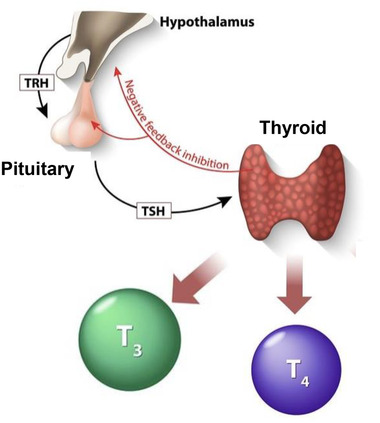
Hormonal regulation of the thyroid gland. TRH, thyrotropin‐releasing hormone; TSH, thyroid‐stimulating hormone; T3, triiodothyronine; T4, thyroxine.
CH affects about 1 out of 2500–3000 newborns worldwide. Since the end of the 1970s, CH can be systematically screened on the 3rd day of life by assaying TSH, allowing treatment with L‐thyroxine (L‐T4). 1 CH may be due to a developmental or functional defect of the thyroid gland (primary/peripheral CH), or to hypothalamic‐pituitary axis malformations (central CH), but also altered action, transport, or metabolism of THs.
In the majority of cases (65%), primary CH is due to a developmental defect of the thyroid gland or thyroid dysgenesis (TD) (CH due to TD, CHTD, online Mendelian inheritance in man [OMIM] #218700). 2 CHTD may be isolated or associated with extra‐thyroidal signs. When the thyroid gland is normally sized or hyperplasic (goiter) (35% of CH cases), hypothyroidism is due to a defect in TH synthesis or dyshormonogenesis (CH due to dyshormonogenesis, CHDH, OMIM #274400–274900) (Figure 2). 3
FIGURE 2.
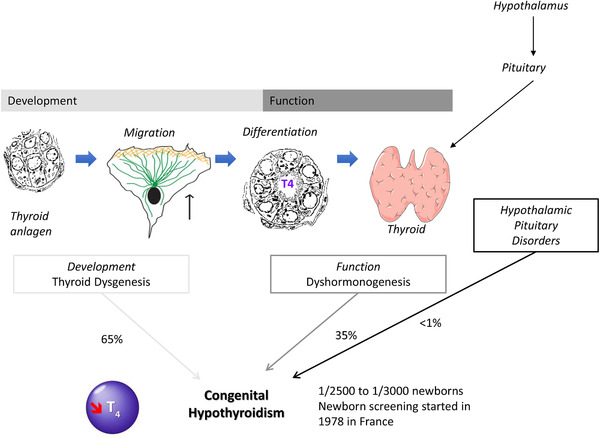
Diagrammatic representation of thyroid development and function, as well as associated congenital hypothyroidism.
There is now a well‐established correlation between the occurrence of CH and the alteration of thyroid development. The primitive thyroid develops from a median anlage that appears around the 3rd gestational week (GW) in Man, as a thickening then evagination of the floor of the endodermis of the buccopharyngeal cavity. This median anlage then migrates and fuses with the ultimobranchial bodies derived from the posterior recess of the 4th pharyngeal pouch (sometimes called the 5th pharyngeal pouch), positioned in front of the trachea to give the primary thyroid from the 7th GW. 4 The Nkx2‐1, Foxe1, Pax8, and HHex transcription factors are essential for the specification of the thyroid. Migration of the progenitor cells is a crucial stage for development and thyroid function. After migration and fusion of the anlages, the cells differentiate mostly into thyroid follicular cells (thyrocytes). The markers of terminal differentiation are the TSH receptor (TSHR), the iodine transporter (NIS, sodium/iodine symporter coded by the SLC5A5 gene), thyroglobulin (TG), thyroperoxidase (TPO), DUOX2 and DUOXA2, involved in hormone synthesis (Figure 3). Iodine, drawn from the bloodstream, enters the thyrocyte through the iodine transporter (NIS). The iodine is then oxidized by TPO and the H2O2 DUOX2/DUOXA2 producer complex to TG, TH matrix protein, T4, and T3. 5 Malformations at any stage of thyroid development (such as the specification, proliferation, migration, growth, organization, differentiation, and survival) can result in a congenital abnormality and/or an alteration to hormone synthesis leading to varying degrees of hypothyroidism.
FIGURE 3.
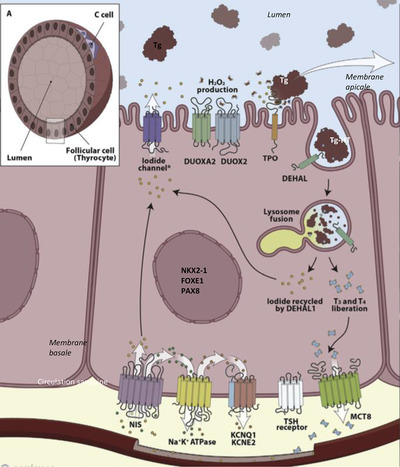
Production of thyroid hormones in the thyrocytes. NIS, sodium/iodide symporter; KCNQ1 and KCNE2, voltage‐gated K+ channels; TSH, thyroid‐stimulating hormone; MCT8, monocarboxylate transporter 8; DUOX2, dual oxidase 2; DUOXA2, maturation factor of dual oxidase 2; TPO, thyroperoxidase; DEHAL, iodotyrosine dehalogenase. (Reprinted with permission 5 Carvalho 2007)
In recent years, the use of new genetic approaches such as high‐throughput sequencing (next‐generation sequencing, NGS; whole‐exome sequencing, WES) and the detailed phenotypic description of patients and/or families affected by CH have given new genetic avenues for CH research. Furthermore, they have made it possible to extend the thyroid phenotype associated with certain mutated genes.
This paper presents all the known genes and recently discovered genes responsible for CH, as well as the modes of transmission of this complex pathology.
RECENTLY DISCOVERED GENES INVOLVED IN PRIMARY CH
Thyroid dysgenesis
There is a broad spectrum of TD ranging from athyreosis (21% of CH cases) due to failure of the thyroid progenitor cells, to thyroid ectopia (41% of CH cases) to the failure of the anlages to migrate during development, in most cases in a sublingual position, or to hypoplasia, hemithyroid/single lobe (3% of CH cases). 2
A genetic cause is identified in fewer than 5% to 10% of TD cases, including mutations in the TSHR 6 and in the genes coding for the transcription factors involved in thyroid development (NKX2‐1/TTF1, PAX8, FOXE1/TTF2, NKX2‐5, and GLIS3). 7 , 8 In general, the mode of transmission of CHTDs is dominant, except for GLIS3 and FOXE1. Based on the thyroid phenotype of transgenic animal models, the involvement of these genes has been validated by the search for mutations in cohorts of patients with CH, whether or not associated with syndromic forms. The phenotypes associated with the different mutated genes are described in Table 1.
TABLE 1.
Genes associated with primary congenital hypothyroidism with thyroid dysgenesis
| Gene (OMIM) | Thyroid phenotype | Transmission mode | Associated pathologies |
|---|---|---|---|
| TSHR (603372) | Complete or partial resistance to TSH: evident athyreosis, a gland in place, and variable hypothyroidism | AD/AR | No |
| NKX2‐1 (600635) | Variable | AD | Respiratory distress, choreoathetosis |
| FOXE1 (602617) | Athyreosis, severe hypoplasia | AR | Cleft palate, choanal atresia, hair standing on end |
| PAX8 (167415) | Variable | AD | Urogenital tract defects |
| NKX2‐5 (600584) | Gland in place, variable hypothyroidism | Unknown | Congenital heart defects |
| GLIS3 (610192) | Gland in place | AR | Neonatal diabetes, polycystic kidneys, cholestasis |
| JAG1 (601920) | Thyroid hypoplasia | AD | Heart defects, hepatic cholestasis |
| NTN1 (601614) | Thyroid ectopia | unknown | Arthrogryposis |
| BOREALIN (609977) | Thyroid ectopia, hemithyroid, thyroid asymmetry | AD/AR | No |
| TUBB1 (612901) | Variable (mainly ectopia) | AD | Macroplatelets and platelet hyperaggregation |
| TRPC4AP (608430) | Thyroid hypoplasia | AD | No |
| GBP1 (600411) | Thyroid dysgenesis (not described) or normal thyroid size | AD/AR | No |
Abbreviations: AD, dominant autosomal; AR, recessive autosomal; OMIM, Online Mendelian Inheritance in Man (https://www.ncbi.nlm.nih.gov/omim/).
In addition, below we describe the genes identified described in the last 5 years as responsible for cases of CHTD.
CDCA8/BOREALIN (OMIM #609977)
Using WES in the case of family TD, we have demonstrated the involvement of BOREALIN/CDCA8 in the migration and adhesion of thyrocytes explaining certain thyroid ectopia cases. 9 Borealin is a major component of the chromosomal passenger complex involved in various stages of mitosis. Biallelic mutations of BOREALIN have been identified in one blood‐related family and two distinct monoallelic mutations in two other families (Figure 4). Patients carrying these mutations have either athyreosis (n = 1), ectopia (n = 2), some family members had normal thyroid function with a hemithyroid, an asymmetric thyroid (n = 3), or nodules (n = 2). Furthermore, one of the patients with asymmetric lobes developed papillary thyroid cancer. At present, it is difficult to associate BOREALIN mutations with nodules or papillary thyroid cancer. This work made it possible to show: 1) expression of BOREALIN in the thyrocytes of the human fetal gland before and after the onset of TH synthesis (8 and 12 GW), unlike very weak expression in the adult thyroid; and 2) the involvement of BOREALIN in the human thyrocyte migration and adhesion process, corresponding to the ectopic phenotypes of certain patients. Since then, another mutation BOREALIN/CDCA8 has been described, c.585‐1G>C, to the heterozygous state in a patient with TD. 10 The 4 mutations described up to now are found in exons 6, 7, and 8 and intron 8, which are in the same region of the gene coding for one part of the protein that is probably not essential for mitosis but is important for the adhesion and migration of thyrocytes.
FIGURE 4.
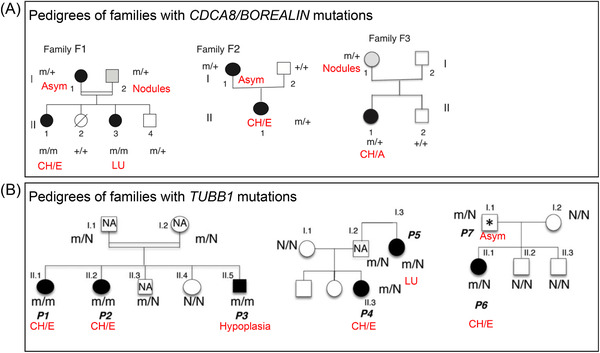
Family trees of families carrying BOREALIN and TUBB1 mutations. 9 , 16 CH/E, congenital hypothyroidism with ectopia; CH/A, congenital hypothyroidism with athyreosis; Asym, asymmetry of the lobes; LU, single lobe; m: disease allele; N or +, normal allele; NA, not available, thyroid ultrasonography.
NTN1/Netrin‐1 (OMIM #601614)
NTN1 codes for Netrin 1, which is involved in regulating various developmental processes, such as angiogenesis, the migration of non‐neuronal cells, and epithelial morphogenesis. 11 Mutations have been identified in patients presenting congenital mirror movement disorders. 12 A patient with a congenital heart defect and TD (ectopia) has been described with a de novo deletion of NTN1. 13 Embryos of the zebrafish with the ntn1a gene disabled have abnormal morphogenesis of the thyroid, probably due to abnormal vascularisation not enabling the thyroid progenitor cells to be correctly guided by the vessels.
JAG1 (OMIM #601920)
JAG1 codes for the protein Jagged1, ligand of the Notch signaling pathway. Heterozygous mutations have been described in Alagille syndrome, a rare genetic disorder that can affect multiple organ systems. The Notch pathway is involved in thyroid development and disabling jag1a/b results in hypothyroidism in the zebrafish. 14 , 15
Thyroid function in 21 patients with JAG1 mutations was analyzed and genetic analysis of JAG1 was carried out in an Italian cohort of 100 CH patients. 15 De Filippis et al. reported the predominance of CH in 6/21 patients with Alagille syndrome, two of which had thyroid hypoplasia. In the Italian cohort of 100 patients with CH, two JAG1 variants to the heterozygous state were found in 4/100 cases (three with TD, two with heart malformations). These data suggest a role for JAG1 in the etiopathology of TDs, principally thyroid hypoplasia.
TUBB1 (OMIM #612901)
Three mutations in the TUBB1 gene (tubulin beta 1 class VI) have recently been identified by our team in patients belonging to three families affected by TD (mainly ectopia) associated with abnormal platelet morphology and aggregation. 16 The first mutation was found using WES in a blood‐related family then two other mutations were detected in two other families from a cohort of 270 patients with CHTD, analyzed using a panel of target genes (Figure 4). TUBB1 mutations were initially found in patients with macrothrombocytopaenia. TUBB1 codes for a protein in the β‐tubulins family. β‐tubulins combine with α‐tubulins in dimers, which auto‐assemble into microtubules, one of the main structures of the cell cytoskeleton. Each of the three TUBB1 mutations results in non‐functional α/β‐tubulin dimers that cannot be incorporated into microtubules. The three mutations, therefore, cause loss of function. Tubb1 is expressed in the thyroid during development and in the adult thyroid in Man and mice. Mice with disabled Tubb1 have microplatelets and hypothyroidism, matching the phenotype found in TUBB1‐mutated patients (Figure 5). The thyroids of Tubb1‐/‐ mice show growth defects during the early stages of development (embryo day E9.5), altered migration at E11.5 and E13.5, and a TH secretion defect at E17.5. Thus, all these mechanisms – growth, migration, TH secretion – require appropriate microtubule function. Furthermore, two of the TUBB1 mutations were associated with increased basal cell activation and platelet hyper‐aggregation in mutated patients.
FIGURE 5.
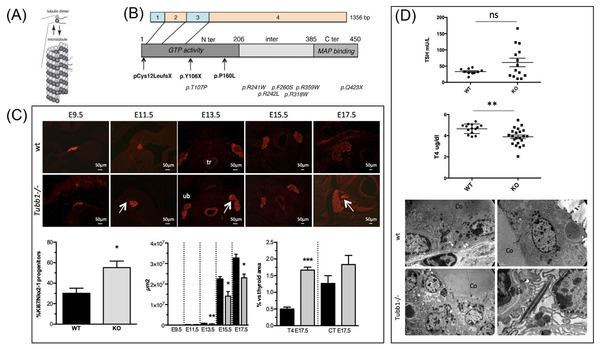
(A) Diagram of a microtubule with tubulin dimers (α/β). (B) Location of TUBB1 mutations in cDNA (exons from 1 to 4) and corresponding protein changes in the GTP (guanosine triphosphate), MAP (protein binding activity domains associated with microtubules). In bold: mutations associated with congenital hypothyroidism; in italics: mutations associated with macrothrombocytopaenia. (C) Phenotype analysis of Tubb1‐/‐ mice. Top: Morphology of the thyroid analyzed by immunohistochemistry with Nkx2‐1 in red at E9.5 and E11.5 (sagittal sections) and at E13.5, E15.5, and E17.5 (transverse sections) of Tubb1‐/‐ and wild‐type (wt) mice. Delays in migration of the thyroid were observed at E11.5 and E13.5 (arrow) in Tubb1‐/‐ mice. tr: trachea; ub: ultimobranchial body. Scale bar: 50 μm. Bottom, from left to right: percentage of growth: proportion of Nkx2‐1‐positive cells marked with Ki67 at E9.5 in proportion to the total number of Nkx2‐1‐positive cells. The total area of the thyroid (μm2). Percentage of the T4 or calcitonin (CT) surface area in proportion to the total surface area of the thyroid at the E17.5 stage. (D) Top: Serum assays of TSH and T4 in 3‐month‐old Tubb1‐/‐ and wild‐type mice. Tubb1‐/‐ mice have hypothyroidism with a high level of TSH and reduced T4. Bottom: Ultrastructural alterations highlighted by electron microscopy in the thyroids of Tubb1‐/‐ and wild‐type mice: disorganization of secretion vesicles (white asterisks) and rods with identical density to the secretion vesicles (white arrow). 16 TSH, thyroid‐stimulating hormone; Co, colloid. The results are given in the form of mean ± SEM. Student t‐test, *P < 0.05, **P < 0.01 and ***P < 0.001.
TRPC4AP (OMIM #608430)
A first de novo TRPC4AP mutation has been identified using WES in a child with TD. 17 Next, 179 patients with CHTD were sequenced using a panel of target genes making it possible to find four variants in TRPC4AP. During development, Choukair et al. showed that Trpc4ap is expressed in the brain, the thyroid bud, and the kidney of the African clawed frog (Xenopus laevis). This team showed that disabling Trpc4ap in the African clawed frog leads to thyroid hypoplasia during development. It was also shown that TRPC4AP interacted with IKBKG which activates the NF‐κB signaling pathway and regulates the genes involved in the growth and survival of thyrocytes. Furthermore, the NF‐kB would control the expression of NKX2‐1, PAX8, TPO, NIS, and TG. 18 The authors conclude that TRPC4AP would be a new candidate gene for TDs, the specific role of which remains to be elucidated.
GBP1 (OMIM #600411)
By analyzing the exome of 98 patients with CH, four GBP1 (Guanylate‐binding protein 1) mutations were identified in three patients with CH, two of whom had TD and the third a gland in place. 19 The mutations were found in the heterozygous state, or composite heterozygote for one of the patients with TD. By using a methylation‐specific PCR, it was shown that a CpG site (cg12054698) of GPB1 was hypermethylated in the genome DNA of two patients, carriers of a heterozygous GBP1 mutation, compared with their euthyroid parents, who had the same variant. Pyrosequencing also revealed hypermethylation of the CpG site in both patients but no difference with their parents. Analysis of thyroid tissues by immunohistochemistry and quantitative PCR showed a reverse correlation between the degree of cg12054698 methylation and GBP1 expression. Thus, genetic and epigenetic factors could participate in the penetrance of CHs in these two patients. Furthermore, Yang et al. showed that zebrafish with disabled Gbp1 had thyroid hypoplasia during development and hypothyroidism. In addition, disabling Gbp1 could disrupt thyrocyte adhesion complexes, including the E‐Cadherin and β‐catenin proteins, probably leading to abnormal thyroid development.
Syndromic CH with TD
Patients with CHTD or a gland in place can have extra‐thyroid abnormalities. The commonest forms of syndromic CH with TD are Bamforth‐Lazarus syndrome (FOXE1) and brain‐lung‐thyroid syndrome (NKX2‐1). Genetic research has identified new genes associated with syndromic CHs, such as TBX1 (Di George syndrome), SALL1 (Townes‐Brocks syndrome), URB1 (Johanson‐Blizzard syndrome), ELN and BAZ1B (Williams‐Beuren syndrome), KMT2D and KDM6A (Kabuki syndrome), KAT6B (Ohdo syndrome) 20 and CDC42 (Takenouchi‐Kosaki syndrome). 21 It has also been shown that apical constriction during the formation of the thyroid anlage would be dependent on Cdc42. 22 In addition, two new genes, CDH1 (coding for E‐cadherin) and CTNND1 (coding for catenin delta 1 or p120ctn) have been found mutated in patients with Blepharo‐cheilo‐dontic syndrome following a dominant mode of transmission. This syndrome combines facial malformations, including ectropion of the lower eyelids, a cleft lip/palate, and ectodermal dysplasia. According to the authors, the CDH1 mutations seem to be more severe. The spectrum of pathologies associated with this syndrome has been enlarged since patients carrying CDH1 or CTNND1 mutations have been reported with CH and thyroid hypoplasia or athyreosis. 23 Other patients also have synpolydactyly, anal atresia, and neural tube defects. E‐cadherin is known for its role in the intercellular junctions of epithelial cells, including thyrocytes. E‐cadherin is linked to p120ctn, stabilizing E‐cadherin at the membrane.
Newborns and very young infants with Down's syndrome often have subclinical hypothyroidism or primary CH. 24 Thyroid malfunction is still poorly understood. However, it was shown that Dyrk1a+/++ mice have abnormal thyroid development and function. 25 Transgenic mice (Dyrk1a+/++) have been studied, as they have neuron, synaptic, learning, and memory disorders like those found in Down's syndrome. Chromosome 21 in Man corresponds to chromosome 18 in the mouse and includes DYRK1a. At E15.5, Dyrk1a+/++ mice have a larger thyroid with reduced T4 and TG levels, deregulation of transcription factors involved in thyroid development, ultimately leading to morphological and functional abnormalities of the thyroid. Indeed, the thyroids of adult Dyrk1a+/++ mice are disorganized with large regions made up of small follicles. These abnormalities are like those found in the thyroids of human fetuses affected by Down's syndrome. Thus, DYRK1A, a gene located in the critical region of Down's syndrome, is therefore implicated in CHTD. Table 1 summarizes the genes associated with CHTD.
Thyroid DH
Defects in one of the proteins essential for TH synthesis result in DH. In these patients, development and differentiation of the thyroid are normal, but one of the critical stages of TH synthesis in the thyrocytes is altered. An ultrasound scan is used to determine if the thyroid gland is normal size or hyperplasic (goiter). Cases of CHDH can be classified using scintigraphy to measure the uptake of radioactive iodine by the thyroid and the perchlorate test:
Defects in iodine absorption by the thyrocytes with little or no radioactive iodine uptake due to NIS/SLC5A5 mutations,
Partial or total iodine organification defects (P/TIOD) dues to mutations in the TPO, DUOX2, DUOXA2, and PENDRIN (SLC26A4) genes. Iodine uptake is normal but the perchlorate test is positive. In a healthy patient, during the perchlorate test, >90% of the radioactive iodine is immediately organified in the thyroid follicles and therefore bound to TG, the perchlorate test is negative. Conversely, in patients with iodine organification defects, 10% to 90% of the radioactive iodine is not organified and immediately comes out in the blood, and the perchlorate test is recorded as positive,
Defects in synthesis, storage, or release of TG, or IYD defects (DEHAL1) result in disruption of iodide recycling by the thyrocytes. 26 In these cases, iodine uptake is normal, and the perchlorate test is negative, but where a DEHAL1 defect exists there is a characteristic very rapid decrease in iodine fixation in the thyroid gland.
Unlike TDs, DHs have a recessive autosomal mode of transmission and are generally without associated malformation. Pendred syndrome, due to SLC26A4 mutations, is the exception, patients have a thyroid goiter and neurosensory hearing loss.
SLC26A7 (OMIM #608479)
Biallelic SLC26A7 mutations have recently been reported in patients with CH, goiter, or a partial iodine organification defect. 10 , 27 SLC26A7 is a member of the same family of transporters as SLC26A4 (Pendrin), an anion exchanger with an affinity for iodide and chloride. However, SL26A7 does not alter iodide efflux in the thyrocytes and people affected have normal hearing. 10 , 27
We have recently shown that the rate of genetic diagnosis of CHDH is 50% in the previously‐cited known genes, in a cohort of patients selected for CHDH (CH with goiter or CH with gland in place and a positive perchlorate test). 28
New thyroid phenotypes in known genes
SLC26A4, 29 DUOX2 30 and TPO 31 mutations have been found unexpectedly in patients with CH without goiter or with thyroid hypoplasia. Recently, DUOX2 mutations have also been found in patients having CH with thyroid ectopia; however, other studies are needed to explain this phenotype and the involvement of DUOX2 in the migration of the thyroid.32 These results show a possible overlap in the genes involved in the etiology and etiopathology between TDs and DHs. The first patients with CH carrying DUOX1 and DUOX2 mutations have been reported, suggesting a possible digenic cause of CH. 33 Table 2 summarises the genes associated with DHs.
TABLE 2.
Genes associated with thyroid dyshormonogenesis
| Gene (OMIM) | Ultrasound scan/scintigraphy of the thyroid | TG assay | Severity of CH, associated pathology | Transmission mode |
|---|---|---|---|---|
|
SLC5A5 (601843) |
Goitre, iodine uptake absent or – | High serum TG | Variable hypothyroidism | AR |
|
SLC26A4/PDS (605646) |
Goitre, iodine uptake +, PIOD (perchlorate +) | High serum TG | Mild to moderate hypothyroidism, Pendred syndrome: deafness | AR |
|
DUOX1/DUOX2 (606758/606759) |
Goiter iodine uptake +, PIOD or TIOD (perchlorate +) |
High serum TG | Variable severity, transient or permanent hypothyroidism | AR/AD |
|
DUOXA2 (612772) |
Goiter iodine uptake +, PIOD or TIOD (perchlorate +), |
High serum TG | Variable severity, transient or permanent hypothyroidism | AR |
|
TPO (606765) |
Goitre, iodine uptake +, TIOD (perchlorate +) | High serum TG | Severe hypothyroidism | AR |
|
TG (188450) |
Congenital or fast growth goiter, iodine uptake +, perchlorate negative | Low serum TG | Variable hypothyroidism | AR |
|
IYD/DEHAL1 (612025) |
Normal size gland in place, goiter, iodine uptake +, perchlorate negative | High serum TG, serum MIT/DIT, and urine | Variable hypothyroidism | AR (incomplete penetrance) |
|
SLC26A7 (608479) |
Goiter, PIOD (perchlorate +) | High serum TG | Variable hypothyroidism | AR |
Abbreviations: AD, dominant autosomal; AR, recessive autosomal; CH, congenital hypothyroidism; DIT, diiodityrosine; Perchlorate, perchlorate test; MIT, monoiodotyrosine; OMIM, Online Mendelian Inheritance in Man; PIOD, partial iodine organification defect; TG, thyroglobulin; TIOD, total iodine organification defect (https://www.ncbi.nlm.nih.gov/omim/). MIT and DIT derive from catabolism/recycling of thyroid hormones.
NEW GENES IN CENTRAL CH
Central CH (CeCH) may be isolated or be found in circumstances of multiple/combined pituitary hormone deficiency (CPHD). The number of probable genetic causes of isolated or syndromic CeCH has increased due to progress in genetic analyses, for primary CH.
CeCH is frequently part of CPHD and can be associated with one or more other pituitary hormone deficiencies. In addition, certain affected patients show morphological abnormalities of the pituitary or hypothalamus, or other neurological abnormalities. 34 , 35
Isolated CeCH is due mainly to biallelic mutations of TSHβ 36 , 37 and TRHR, 38 affecting few families. Below we present the new genes identified in patients affected by CeCH. Table 3 shows the genes involved in CeCH.
TABLE 3.
Genes involved in central hypothyroidism and their associated phenotypes
| Category | Gene (OMIM) | Phenotype | Transmission mode |
|---|---|---|---|
| Isolated CeCH | TSHβ (188540) | Neonatal onset with low TSH level, high glycoprotein alpha subunit and normal PRL, reversible pituitary hyperplasia on L‐T4 | AR |
| TRHR (188545) | Normal TSH and low PRL levels, inadequate TSH/PRL responses in the TRH stimulation test | AR | |
| TBL1X (300196) | Isolated CeCH in men with normal serum TSH levels and normal response to the TRH stimulation test; associated hearing disorders | Linked to the X | |
| IRS4 (300904) | Isolated CeCH in men with normal serum TSH levels, inadequate TSH response to TRH | Linked to the X | |
| CeCH associated with other pituitary abnormalities | IGSF1 (300137) | Normal serum TSH and inadequate response to the TRH stimulation test; low PRL level, variable GH deficit, transient moderate hypocortisolism, metabolic syndrome; post‐puberty macrorchidism | Linked to the X |
| PROP1 (601538) | Variable age of onset, deficiency combined with GH, PRL LH/FSH, and ACTH deficiencies, variable pituitary volume | AR | |
| POU1F1 (173110) | Variable age of onset, associated with a GH and PRL deficiency, frontal bossing, median facial hypoplasia, nasal lordosis | AD/AR | |
| HESX1 (601802) | Hypopituitarism associated with septo‐optic dysplasia | AD/AR | |
| SOX3 (313430) | Anterior pituitary hypoplasia with ectopic post‐hypophysis (EPH), persistent craniopharyngeal canal, and learning difficulties | Linked to the X | |
| OTX2 (600037) | Anterior pituitary hypoplasia with EPH and ocular abnormalities (an‐/micro‐phthalmia/retinal dystrophy) | AD | |
| LHX3 (600577) | Hypopituitarism with inconstant corticotroph deficiency, variable pituitary volume, variable, short and rigid cervical spurs, and variable hearing defect | AR | |
| LHX4 (602146) | Variable hypopituitarism, anterior pituitary hypoplasia with EPH, Arnold‐Chiari syndrome, hypoplasia of the corpus callosum | AD/AR | |
| LEPR (601007) | Hyperphagia, obesity, hypogonadotropic hypogonadism | AR | |
| SOX2 (184429) | Variable hypopituitarism, pituitary hypoplasia, microphthalmia, variable learning difficulties | AD | |
| PCSK1 (162150) | Hyperphagia, early‐onset obesity, hypogonadotropic hypogonadism, corticotroph deficiency | AR | |
| CeCH in the context of a syndrome | PROKR2 (607123) | Variable hypopituitarism associated with septo‐optic dysplasia or a pituitary stem interruption syndrome | AD/AR |
| NFKB2 (164012) | Anterior pituitary deficiency with variable immune deficiency (DAVID) syndrome associated with corticotroph deficiency and variable GH and TSH deficiencies | AD | |
| CHD7 (608892) | CHARGE syndrome (coloboma, heart defects, choanal atresia, growth retardation, genital and ear abnormalities) with EPH and variable LH/FSH, TSH, and GH deficiencies | AD | |
| FGFR1 (136350) | Kallman syndrome (KS) and normosomal congenital hypogonadotropic hypogonadism (nCHH), variable association with other pituitary hormone deficiencies including TSH, septo‐optic dysplasia and EPH | AD | |
| FGF8 (600483) | KS and nCHH, variable associations with other pituitary hormone deficiencies, including TSH, holoprosencephaly, and agenesis of the corpus callosum | AR | |
| FOXA2 (600288) | Hypopituitarism with craniofacial abnormalities and multi‐organ malformation, hyperinsulinism | AD | |
| RNPC3 (618016) | Growth hormone deficiency, congenital cataract, retarded development, retarded puberty | AR |
Abbreviations: ACTH, adrenocorticotropic hormone; AD, dominant autosomal; AR, recessive autosomal; CeCH, central congenital hypothyroidism; GH, growth hormone; L‐T4, L‐thyroxine; OMIM, Online Mendelian Inheritance in Man (https://www.ncbi.nlm.nih.gov/omim/); PRL, prolactin; TRH, thyrotropin‐releasing hormone; TSH, thyroid‐stimulating hormone.
IGSF1 (OMIM #300137)
Mutations of the gene coding for member 1 of the immunoglobulins superfamily (IGSF1) are the cause of a syndrome linked to the X including a moderately severe CeCH. The associated characteristics are abnormal testicular growth leading to macro‐orchidism at adult age, sometimes delayed puberty, low prolactin level, and sometimes a reversible growth hormone deficiency. 39 Some women carriers can have altered thyroid function. Recent data indicate that IGSF1 is now the gene most frequently implicated in cases of CeCH. 40
TBL1X (OMIM #300196)
Mutations in the TBL1X gene are the second most common cause responsible for X‐linked cases of CeCH. TBL1X, transducin‐like protein 1, participates in the corepressor complex of the thyroid hormone's receptor. This protein is expressed in Man from the hypothalamus (paraventricular nucleus) and pituitary. The transgenic mouse model with inactivation of the nuclear receptor corepressor protein leads to a thyrotroph deficiency. Hearing loss is often an associated clinical characteristic in men. 41
IRS4 (OMIM #300904)
Recently, IRS4 mutations have been identified in cases of X‐linked CeCH in five families. The substrate family of the insulin receptor (IRS) acts as an interface between tyrosine kinase receptors, including receptors for insulin, leptin, and insulin‐like growth factor 1 (IGF‐1), and several intracellular signaling pathways. Given that IRS4 is involved in leptin signaling, the proposed mechanism of this CeCH could be disrupted by leptin signalling. 42
PCSK1 (OMIM #162150)
The proprotein convertase 1/3 (PC1/3) deficiency caused by PCSK1 mutations, based on a recessive mode of transmission, leads to hyperphagia, early‐onset obesity, hypogonadotropic hypogonadism, CeCH, and hypocortisolism. 43 Since then, other patients have been described with chronic diarrhea, malabsorption, and diabetes insipidus. To date, 26 patients have been reported with PC1/3 deficiency, 56% of whom have CeCH.
RNPC3 (OMIM #618016)
Recently, biallelic variants of RNPC3 have been identified in two families whose children have a growth hormone deficiency, CeCH, congenital cataracts, learning deficiencies, and delayed puberty. 44 These cases show the relationship between the biallelic RNPC3 variants and severe postnatal growth retardation due to growth hormone deficiency. RNPC3 codes for a protein U11/U12‐65K, a component of the minor spliceosome, alterations of which would result in a pituitary development defect.
TRANSMISSION MODE
The etiopathology of CH is largely unknown and may include the contribution of individual and environmental factors. Although CH is typically reported as sporadic, several studies in Man and experimental models support a genetic origin. Favoring the genetic origin, a greater frequency of developmental thyroid abnormalities has been shown in first‐degree relatives of CH cases 45 and increased incidence of CH in blood‐related families and certain ethnic groups. 46 In addition, CH is associated with a 20‐times greater risk of having other congenital abnormalities 47 and several syndromes have been associated with variable thyroid abnormalities, such as in Alagille, Di George, Williams‐Beuren, and Kabuki syndromes and genito‐patellar syndrome.
In addition, CHTD has always been considered a monogenic disorder with a dominant autosomal mode of transmission, caused by rare variants. It has been shown that the proportion of family CHTD cases is estimated at 2%, suggesting the involvement of genetic factors. 48 Fewer than 5% of CHTD patients carry a mutation in one of the ten causal or predisposing genes (NKX2‐1, FOXE1, PAX8, NKX2‐5, TSHR, JAG1, NTN1, GLIS3, BOREALIN, TUBB1); these patients are sporadic or family cases, with incomplete penetrance of certain genes. 20
Recently, Luca Persani's team showed, by analysis with a panel of NGS target genes containing 11 causal genes in a cohort of 177 patients with CH, an oligogenic origin of sporadic CH cases. 49 This oligogenic model could explain the variable expressiveness and penetrance of genetic abnormalities reported in several family cases of hypothyroidism and correspond well with the previously proposed animal model: the CH could have a multi‐factor cause. 50 , 51 It has been proposed that in addition to rare monogenic forms, CH could arise more frequently in sporadic cases such as multi‐factor disorders with a genetic predisposition. These data suggest a more complex genetic model of CHTDs involving different variants of variable rarity. 52 In this context, the polymorphic nature of the polyalanine of the FOXE1 gene aroused great interest as a susceptibility factor. Several groups, including ours, have studied the relationship between the length of the FOXE1 polyalanine and the TD type, suggesting a role as a susceptibility factor in TDs. 53 , 54 , 55 , 56
Furthermore, the high degree of inconsistency (92%) between monozygous twins and the sex difference with a higher feminine prevalence of CHTD are two arguments against classic Mendelian transmission. 57 The following somatic mechanisms could be involved: 1) early post‐zygotic somatic mutations, 2) specific tissue mutations or epimutations or 3) tissue‐specific autosomal monoallelic expression of specific genes involved in the thyroid. 58 , 59 , 60 , 61 Unfortunately, to date, no firm evidence of these mechanisms has been established.
Moreover, the described TD linked to homozygous mutation of pendrin, peroxidase, and DUOX mutation remained to be fully explained. 29 , 31 , 32 This raised the interesting possibility that genes involved in thyroid function do actually play a role in thyroid development.
To conclude, the cause of CH is still not well defined and has shown its complexity in recent years by the description of new models such as oligogenism, the role of epigenetics, and probably also the involvement of extrinsic factors, such as the environment.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Stoupa A, Kariyawasam D, Polak M, Carré A. Genetics of congenital hypothyroidism: Modern concepts. Pediatr Investig. 2022;6:123–134. 10.1002/ped4.12324
Funding source
Athanasia Stoupa received a research grant from the European Society for Paediatric Endocrinology (ESPE). Aurore Carré, Dulanjalee Kariyawasam, and Michel Polak received funding from Sandoz SAS and Merck Serono France, the French Society of Endocrinology and Paediatric Diabetology (SFEDP), and the French National Research Agency (ANR‐21‐CE14‐0055‐01‐ MITHYPLA).
REFERENCES
- 1. Léger J. Dépistage de l'hypothyroïdie congénitale [Neonatal screening for congenital hypothyroidism]. Med Sci (Paris). 2021;37:474‐481. (in French). [DOI] [PubMed] [Google Scholar]
- 2. Barry Y, Bonaldi C, Goulet V, Coutant R, Léger J, Paty AC, et al. Increased incidence of congenital hypothyroidism in France from 1982 to 2012: a nationwide multicenter analysis. Ann Epidemiol. 2016;26:100‐105.e4. DOI: 10.1016/j.annepidem.2015.11.005 [DOI] [PubMed] [Google Scholar]
- 3. Cavarzere P, Castanet M, Polak M, Raux‐Demay MC, Cabrol S, Carel JC, et al. Clinical description of infants with congenital hypothyroidism and iodide organification defects. Horm Res. 2008;70:240‐248. DOI: 10.1159/000151597 [DOI] [PubMed] [Google Scholar]
- 4. Trueba SS, Augé J, Mattei G, Etchevers H, Martinovic J, Czernichow P, et al. PAX8, TITF1, and FOXE1 gene expression patterns during human development: new insights into human thyroid development and thyroid dysgenesis‐associated malformations. J Clin Endocrinol Metab. 2005;90:455‐462. DOI: 10.1210/jc.2004-1358 [DOI] [PubMed] [Google Scholar]
- 5. Carvalho DP, Dupuy C. Thyroid hormone biosynthesis and release. Mol Cell Endocrinol. 2017;458:6‐15. DOI: 10.1016/j.mce.2017.01.038 [DOI] [PubMed] [Google Scholar]
- 6. Schoenmakers N, Chatterjee VK. Thyroid gland: TSHR mutations and subclinical congenital hypothyroidism. Nat Rev Endocrinol. 2015;11:258‐259. DOI: 10.1038/nrendo.2015.27 [DOI] [PubMed] [Google Scholar]
- 7. Stoupa A, Kariyawasam D, Carré A, Polak M. Update of thyroid developmental genes. Endocrinol Metab Clin North Am. 2016;45:243‐254. DOI: 10.1016/j.ecl.2016.01.007 [DOI] [PubMed] [Google Scholar]
- 8. Fernández LP, López‐Márquez A, Santisteban P. Thyroid transcription factors in development, differentiation and disease. Nat Rev Endocrinol. 2015;11:29‐42. DOI: 10.1038/nrendo.2014.186 [DOI] [PubMed] [Google Scholar]
- 9. Carré A, Stoupa A, Kariyawasam D, Gueriouz M, Ramond C, Monus T, et al. Mutations in BOREALIN cause thyroid dysgenesis. Hum Mol Genet. 2017;26:599‐610. DOI: 10.1093/hmg/ddw419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zou M, Alzahrani AS, Al‐Odaib A, Alqahtani MA, Babiker O, Al‐Rijjal RA, et al. Molecular analysis of congenital hypothyroidism in Saudi Arabia: SLC26A7 mutation is a novel defect in thyroid dyshormonogenesis. J Clin Endocrinol Metab. 2018;103:1889‐1898. DOI: 10.1210/jc.2017-02202 [DOI] [PubMed] [Google Scholar]
- 11. Levy‐Strumpf N, Culotti JG. Netrins and Wnts function redundantly to regulate antero‐posterior and dorso‐ventral guidance in C. elegans . PLoS Genet. 2014;10:e1004381. DOI: 10.1371/journal.pgen.1004381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Méneret A, Franz EA, Trouillard O, Oliver TC, Zagar Y, Robertson SP, et al. Mutations in the netrin‐1 gene cause congenital mirror movements. J Clin Invest. 2017;127:3923‐3936. DOI: 10.1172/JCI95442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Opitz R, Hitz MP, Vandernoot I, Trubiroha A, Abu‐Khudir R, Samuels M, et al. Functional zebrafish studies based on human genotyping point to netrin‐1 as a link between aberrant cardiovascular development and thyroid dysgenesis. Endocrinology. 2015;156:377‐388. DOI: 10.1210/en.2014-1628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marelli F, Persani L. Role of Jagged1‐Notch pathway in thyroid development. J Endocrinol Invest. 2018;41:75‐81. DOI: 10.1007/s40618-017-0715-x [DOI] [PubMed] [Google Scholar]
- 15. de Filippis T, Marelli F, Nebbia G, Porazzi P, Corbetta S, Fugazzola L, et al. JAG1 loss‐of‐function variations as a novel predisposing event in the pathogenesis of congenital thyroid defects. J Clin Endocrinol Metab. 2016;101:861‐870. DOI: 10.1210/jc.2015-3403 [DOI] [PubMed] [Google Scholar]
- 16. Stoupa A, Adam F, Kariyawasam D, Strassel C, Gawade S, Szinnai G, et al. TUBB1 mutations cause thyroid dysgenesis associated with abnormal platelet physiology. EMBO Mol Med. 2018;10:e9569. DOI: 10.15252/emmm.201809569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Choukair D, Eberle B, Vick P, Hermanns P, Weiss B, Paramasivam N, et al. Identification of transient receptor potential channel 4‐associated protein as a novel candidate gene causing congenital primary hypothyroidism. Horm Res Paediatr. 2020;93:16‐29. DOI: 10.1159/000507114 [DOI] [PubMed] [Google Scholar]
- 18. Reale C, Iervolino A, Scudiero I, Ferravante A, D'Andrea LE, Mazzone P, et al. NF‐κB essential modulator (NEMO) is critical for thyroid function. J Biol Chem. 2016;291:5765‐5773. DOI: 10.1074/jbc.M115.711697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang RM, Zhan M, Zhou QY, Ye XP, Wu FY, Dong M, et al. Upregulation of GBP1 in thyroid primordium is required for developmental thyroid morphogenesis. Genet Med. 2021;23:1944‐1951. DOI: 10.1038/s41436-021-01237-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abu‐Khudir R, Larrivée‐Vanier S, Wasserman JD, Deladoëy J. Disorders of thyroid morphogenesis. Best Pract Res Clin Endocrinol Metab. 2017;31:143‐159. DOI: 10.1016/j.beem.2017.04.008 [DOI] [PubMed] [Google Scholar]
- 21. Motokawa M, Watanabe S, Nakatomi A, Kondoh T, Matsumoto T, Morifuji K, et al. A hot‐spot mutation in CDC42 (p.Tyr64Cys) and novel phenotypes in the third patient with Takenouchi‐Kosaki syndrome. J Hum Genet. 2018;63:387‐390. DOI: 10.1038/s10038-017-0396-5 [DOI] [PubMed] [Google Scholar]
- 22. Loebel DA, Plageman TF Jr, Tang TL, Jones VJ, Muccioli M, Tam PP. Thyroid bud morphogenesis requires CDC42‐ and SHROOM3‐dependent apical constriction. Biol Open. 2016;5:130‐139. DOI: 10.1242/bio.014415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ghoumid J, Stichelbout M, Jourdain AS, Frenois F, Lejeune‐Dumoulin S, Alex‐Cordier MP, et al. Blepharocheilodontic syndrome is a CDH1 pathway‐related disorder due to mutations in CDH1 and CTNND1 . Genet Med. 2017;19:1013‐1021. DOI: 10.1038/gim.2017.11 [DOI] [PubMed] [Google Scholar]
- 24. Kariyawasam D, Carré A, Luton D, Polak M. Down syndrome and nonautoimmune hypothyroidisms in neonates and infants. Horm Res Paediatr. 2015;83:126‐131. DOI: 10.1159/000370004 [DOI] [PubMed] [Google Scholar]
- 25. Kariyawasam D, Rachdi L, Carré A, Martin M, Houlier M, Janel N, et al. DYRK1A BAC transgenic mouse: a new model of thyroid dysgenesis in Down syndrome. Endocrinology. 2015;156:1171‐1180. DOI: 10.1210/en.2014-1329 [DOI] [PubMed] [Google Scholar]
- 26. Szinnai G. Clinical genetics of congenital hypothyroidism. Endocr Dev. 2014;26:60‐78. DOI: 10.1159/000363156 [DOI] [PubMed] [Google Scholar]
- 27. Cangul H, Liao XH, Schoenmakers E, Kero J, Barone S, Srichomkwun P, et al. Homozygous loss‐of‐function mutations in SLC26A7 cause goitrous congenital hypothyroidism. JCI Insight. 2018;3:e99631. DOI: 10.1172/jci.insight.99631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stoupa A, Al Hage Chehade G, Chaabane R, Kariyawasam D, Szinnai G, Hanein S, et al. High diagnostic yield of targeted next‐generation sequencing in a cohort of patients with congenital hypothyroidism due to dyshormonogenesis. Front Endocrinol. 2020;11:545339. DOI: 10.3389/fendo.2020.545339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kühnen P, Turan S, Fröhler S, Güran T, Abali S, Biebermann H, et al. Identification of PENDRIN (SLC26A4) mutations in patients with congenital hypothyroidism and “apparent” thyroid dysgenesis. J Clin Endocrinol Metab. 2014;99:E169‐E176. DOI: 10.1210/jc.2013-2619 [DOI] [PubMed] [Google Scholar]
- 30. Srichomkwun P, Takamatsu J, Nickerson DA, Bamshad MJ, Chong JX, Refetoff S. DUOX2 gene mutation manifesting as resistance to thyrotropin phenotype. Thyroid. 2017;27:129‐131. DOI: 10.1089/thy.2016.0469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stoupa A, Chaabane R, Guériouz M, Raynaud‐Ravni C, Nitschke P, Bole‐Feysot C, et al. Thyroid hypoplasia in congenital hypothyroidism associated with thyroid peroxidase mutations. Thyroid. 2018;28:941‐944. DOI: 10.1089/thy.2017.0502 [DOI] [PubMed] [Google Scholar]
- 32. Kizys MML, Louzada RA, Mitne‐Neto M, Jara JR, Furuzawa GK, de Carvalho DP, et al. DUOX2 mutations are associated with congenital hypothyroidism with ectopic thyroid gland. J Clin Endocrinol Metab. 2017;102:4060‐4071. DOI: 10.1210/jc.2017-00832 [DOI] [PubMed] [Google Scholar]
- 33. Aycan Z, Cangul H, Muzza M, Bas VN, Fugazzola L, Chatterjee VK, et al. Digenic DUOX1 and DUOX2 mutations in cases with congenital hypothyroidism. J Clin Endocrinol Metab. 2017;102:3085‐3090. DOI: 10.1210/jc.2017-00529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Persani L, Cangiano B, Bonomi M. The diagnosis and management of central hypothyroidism in 2018. Endocr Connect. 2019;8:R44‐R54. DOI: 10.1530/EC-18-0515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Persani L, Brabant G, Dattani M, Bonomi M, Feldt‐Rasmussen U, Fliers E, et al. 2018 European Thyroid Association (ETA) guidelines on the diagnosis and management of central hypothyroidism. Eur Thyroid J. 2018;7:225‐237. DOI: 10.1159/000491388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Miyai K, Azukizawa M, Kumahara Y. Familial isolated thyrotropin deficiency with cretinism. N Engl J Med. 1971;285:1043‐1048. DOI: 10.1056/NEJM197111042851902 [DOI] [PubMed] [Google Scholar]
- 37. Bonomi M, Proverbio MC, Weber G, Chiumello G, Beck‐Peccoz P, Persani L. Hyperplastic pituitary gland, high serum glycoprotein hormone alpha‐subunit, and variable circulating thyrotropin (TSH) levels as hallmark of central hypothyroidism due to mutations of the TSH beta gene. J Clin Endocrinol Metab. 2001;86:1600‐1604. DOI: 10.1210/jcem.86.4.7411 [DOI] [PubMed] [Google Scholar]
- 38. Bonomi M, Busnelli M, Beck‐Peccoz P, Costanzo D, Antonica F, Dolci C, et al. A family with complete resistance to thyrotropin‐releasing hormone. N Engl J Med. 2009;360:731‐734. DOI: 10.1056/NEJMc0808557 [DOI] [PubMed] [Google Scholar]
- 39. Sun Y, Bak B, Schoenmakers N, van Trotsenburg AS, Oostdijk W, Voshol P, et al. Loss‐of‐function mutations in IGSF1 cause an X‐linked syndrome of central hypothyroidism and testicular enlargement. Nat Genet. 2012;44:1375‐1381. DOI: 10.1038/ng.2453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Joustra SD, Heinen CA, Schoenmakers N, Bonomi M, Ballieux BE, Turgeon MO, et al. IGSF1 deficiency: Lessons from an extensive case series and recommendations for clinical management. J Clin Endocrinol Metab. 2016;101:1627‐1636. DOI: 10.1210/jc.2015-3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heinen CA, Losekoot M, Sun Y, Watson PJ, Fairall L, Joustra SD, et al. Mutations in TBL1X are associated with central hypothyroidism. J Clin Endocrinol Metab. 2016;101:4564‐4573. DOI: 10.1210/jc.2016-2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heinen CA, de Vries EM, Alders M, Bikker H, Zwaveling‐Soonawala N, van den Akker E, et al. Mutations in IRS4 are associated with central hypothyroidism. J Med Genet. 2018;55:693‐700. DOI: 10.1136/jmedgenet-2017-105113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pépin L, Colin E, Tessarech M, Rouleau S, Bouhours‐Nouet N, Bonneau D, et al. A new case of PCSK1 pathogenic variant with congenital proprotein convertase 1/3 deficiency and literature review. J Clin Endocrinol Metab. 2019;104:985‐993. DOI: 10.1210/jc.2018-01854 [DOI] [PubMed] [Google Scholar]
- 44. Verberne EA, Faries S, Mannens M, Postma AV, van Haelst MM. Expanding the phenotype of biallelic RNPC3 variants associated with growth hormone deficiency. Am J Med Genet A. 2020;182:1952‐1956. DOI: 10.1002/ajmg.a.61632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Léger J, Marinovic D, Garel C, Bonaïti‐Pellié C, Polak M, Czernichow P. Thyroid developmental anomalies in first degree relatives of children with congenital hypothyroidism. J Clin Endocrinol Metab. 2002;87:575‐580. DOI: 10.1210/jcem.87.2.8268 [DOI] [PubMed] [Google Scholar]
- 46. Stoppa‐Vaucher S, Van Vliet G, Deladoëy J. Variation by ethnicity in the prevalence of congenital hypothyroidism due to thyroid dysgenesis. Thyroid. 2011;21:13‐18. DOI: 10.1089/thy.2010.0205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Passeri E, Frigerio M, De Filippis T, Valaperta R, Capelli P, Costa E, et al. Increased risk for non‐autoimmune hypothyroidism in young patients with congenital heart defects. J Clin Endocrinol Metab. 2011;96:E1115‐E1119. DOI: 10.1210/jc.2011-0057 [DOI] [PubMed] [Google Scholar]
- 48. Castanet M, Lyonnet S, Bonaïti‐Pellié C, Polak M, Czernichow P, Léger J. Familial forms of thyroid dysgenesis among infants with congenital hypothyroidism. N Engl J Med. 2000;343:441‐442. DOI: 10.1056/NEJM200008103430614 [DOI] [PubMed] [Google Scholar]
- 49. de Filippis T, Gelmini G, Paraboschi E, Vigone MC, Di Frenna M, Marelli F, et al. A frequent oligogenic involvement in congenital hypothyroidism. Hum Mol Genet. 2017;26:2507‐2514. DOI: 10.1093/hmg/ddx145 [DOI] [PubMed] [Google Scholar]
- 50. Amendola E, De Luca P, Macchia PE, Terracciano D, Rosica A, Chiappetta G, et al. A mouse model demonstrates a multigenic origin of congenital hypothyroidism. Endocrinology. 2005;146:5038‐5047. DOI: 10.1210/en.2005-0882 [DOI] [PubMed] [Google Scholar]
- 51. Medici M, Visser TJ, Peeters RP. Genetics of thyroid function. Best Pract Res Clin Endocrinol Metab. 2017;31:129‐142. DOI: 10.1016/j.beem.2017.04.002 [DOI] [PubMed] [Google Scholar]
- 52. Stoupa A, Kariyawasam D, Muzza M, de Filippis T, Fugazzola L, Polak M, et al. New genetics in congenital hypothyroidism. Endocrine. 2021;71:696‐705. DOI: 10.1007/s12020-021-02646-9 [DOI] [PubMed] [Google Scholar]
- 53. Macchia PE, Mattei MG, Lapi P, Fenzi G, Di Lauro R. Cloning, chromosomal localization and identification of polymorphisms in the human thyroid transcription factor 2 gene (TITF2). Biochimie. 1999;81:433‐440. DOI: 10.1016/s0300-9084(99)80092-3 [DOI] [PubMed] [Google Scholar]
- 54. Carré A, Castanet M, Sura‐Trueba S, Szinnai G, Van Vliet G, Trochet D, et al. Polymorphic length of FOXE1 alanine stretch: evidence for genetic susceptibility to thyroid dysgenesis. Hum Genet. 2007;122:467‐476. DOI: 10.1007/s00439-007-0420-5 [DOI] [PubMed] [Google Scholar]
- 55. Pimentel CP, Cortinhas‐Alves EA, de Oliveira E, Santana‐da‐Silva LC. Does the polymorphism in the length of the polyalanine tract of FOXE1 gene influence the risk of thyroid dysgenesis occurrence. J Thyroid Res. 2017;2017:2793205. DOI: 10.1155/2017/2793205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Szczepanek E, Ruchala M, Szaflarski W, Budny B, Kilinska L, Jaroniec M, et al. FOXE1 polyalanine tract length polymorphism in patients with thyroid hemiagenesis and subjects with normal thyroid. Horm Res Paediatr. 2011;75:329‐334. DOI: 10.1159/000322874 [DOI] [PubMed] [Google Scholar]
- 57. Perry R, Heinrichs C, Bourdoux P, Khoury K, Szöts F, Dussault JH, et al. Discordance of monozygotic twins for thyroid dysgenesis: implications for screening and for molecular pathophysiology. J Clin Endocrinol Metab. 2002;87:4072‐4077. DOI: 10.1210/jc.2001-011995 [DOI] [PubMed] [Google Scholar]
- 58. Abu‐Khudir R, Magne F, Chanoine JP, Deal C, Van Vliet G, Deladoëy J. Role for tissue‐dependent methylation differences in the expression of FOXE1 in nontumoral thyroid glands. J Clin Endocrinol Metab. 2014;99:E1120‐E1129. DOI: 10.1210/jc.2013-4414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Abu‐Khudir R, Paquette J, Lefort A, Libert F, Chanoine JP, Vassart G, et al. Transcriptome, methylome and genomic variations analysis of ectopic thyroid glands. PLoS One. 2010;5:e13420. DOI: 10.1371/journal.pone.0013420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Magne F, Ge B, Larrivée‐Vanier S, Van Vliet G, Samuels ME, Pastinen T, et al. Demonstration of autosomal monoallelic expression in thyroid tissue assessed by whole‐exome and bulk RNA sequencing. Thyroid. 2016;26:852‐859. DOI: 10.1089/thy.2016.0009 [DOI] [PubMed] [Google Scholar]
- 61. Magne F, Serpa R, Van Vliet G, Samuels ME, Deladoëy J. Somatic mutations are not observed by exome sequencing of lymphocyte DNA from monozygotic twins discordant for congenital hypothyroidism due to thyroid dysgenesis. Horm Res Paediatr. 2015;83:79‐85. DOI: 10.1159/000365393 [DOI] [PMC free article] [PubMed] [Google Scholar]