

Introduction
The incidence of cancer and cardiovascular disease (CVD) increase with age, and there are a substantial number of individuals who suffer from both conditions.1,2 Advances in cancer therapy over the past few decades have resulted in a large and growing population of long-term cancer survivors. Indeed, approximately half of patients diagnosed with cancer in high-income settings are now expected to survive for ten years or longer.3,4 This new aging population presents novel clinical challenges requiring further attention and investigation. Specifically, patients with cancer and cancer survivors display increased risks for CVD, including coronary artery disease (CAD) and heart failure, due in part to the side-effects of genotoxic therapies.3,5 To date, the acute effects of cancer drugs on cardiac myocytes have been extensively investigated.6,7 However, it is difficult to reconcile how this mechanism can explain the medium- to long-term risk of cancer therapies on the cardiovascular system. Recently, clonal hematopoiesis (CH), a pre-cancerous state in the hematopoietic system, has been recognized as a new causal risk factor for CVD.8–13 This review introduces the concept of CH, summarizes recent epidemiological and experimental studies linking CH to CVD, and discusses recent experimental studies demonstrating that cancer therapy-related CH (t-CH) can induce cardiac dysfunction. These studies suggest a new mechanism linking cancer therapy to cardiac toxicity.
1. What is clonal hematopoiesis?
Hematopoietic stem and progenitor cells (HSPC) typically reside in the bone marrow, produce multiple blood cell types, and replenish themselves through a self-renewal process.14 The progeny immune cells of the HSPC fulfill critical roles in maintaining homeostasis and are also implicated in a myriad of disease states.15 The rapid turnover of blood cells can contribute to the accumulation of mutant HSPC clones with age.16 While most mutations have little or no effect on cellular fitness, some mutations will occur in “driver” genes, such as oncogenes or tumor suppressor genes, that can enable the positive selection of mutant cells.17–20 In this context, mutant cells can outcompete neighboring wild-type cells leading to their clonal expansion.21 Notably, these mutant HSPC give rise to circulating immune cells that harbor the same mutant allele, and it has been shown that these mutations can alter the function of the progeny immune cells.22 Clonal expansions in blood cells have been found to occur in relatively healthy individuals who lack overt signs of hematologic transformation. This condition has historically been referred to as clonal hematopoiesis (CH) and more recently as CH of indeterminate potential (CHIP) to distinguish it from the clonal expansions that occur in malignant blood cancers.23
CH can be detected by bulk DNA sequencing for driver gene mutations in the blood, bone marrow, or tissues infiltrated by blood cells.24 Large-scale, next-generation sequencing (NGS) analyses of blood samples have identified somatic mutations in leukemia-associated genes of asymptomatic individuals who are void of any known hematologic disease.18–20,25 These mutations are most prevalent in the epigenetic regulators DNA methyltransferase-3A (DNMT3A), ten-eleven translocation-2 (TET2), and additional sex combs like 1 (ASXL1). At lower frequencies, CH has also been associated with mutations in janus kinase 2 (JAK2), tumor protein 53(TP53), protein phosphatase, Mg2+/Mn2+ dependent 1D (PPM1D), BCL6 Corepressor (BCOR), guanine nucleotide binding protein, alpha stimulating complex locus (GNAS), Splicing Factor 3b Subunit 1 (SF3B1), and others.23 In addition to single nucleotide variants (SNVs) and small insertions and deletions (Indels) in driver genes, CH can also be assessed cytogenetically by detecting the large mosaic chromosomal alterations in blood.26,27
Variant allele fraction (VAF) refers to the proportion of mutant allele copies relative to total copies sequenced, and it is directly proportional to the percentage of mutant clones within a cell population. Typical NGS has a sequence limitation of 1–2% due to the intrinsic error rate of DNA sequencing. This sequencing shortcoming traditionally limited the threshold for CHIP to a VAF above 2% (meaning 2% of the sequenced alleles or 4% of cells assuming a heterozygous somatic mutation). Using this relatively insensitive VAF criteria, detectable mutations are rarely detected in the young individuals but highly prevalent in the elderly, with between 10 and 20% of people older than 70 years of age harboring a clones of appreciable size.18–20,28,29 More recently, error-corrected DNA sequencing has been developed to detect clones with VAF values as low as 0.03% or below.30,31 Using this technology, smaller clones can be detected in the blood of younger individuals. These finding have led to the notion that relatively small clones are ubiquitous by middle-age, and that these clones expand with age.
2. Clonal hematopoiesis is associated with cardiovascular disease
Many recent studies have linked CH with reduced survival.18–20,27 While CH increases the risk of a hematologic malignancy, as would be expected, blood cancer alone cannot account for the relatively large increase in mortality that is associated with this condition. Surprisingly, Jaiswal et al. initially reported that individuals with CH displayed significantly increased incident coronary heart disease and ischemic stroke risks after adjusting for age and traditional risk factors.8,18 For these analyses, whole exome sequencing of peripheral blood from 4,726 individuals with coronary heart disease and 3,529 control individuals was performed.8 They reported that carriers of CH had an approximately two-fold greater risk of coronary heart disease and a four-fold greater incidence of early-onset myocardial infarction compared to noncarriers. In these analyses, mutations in DNMT3A, TET2, ASXL1, and JAK2 were each individually associated with coronary heart disease and CVD mortality. The risk ratio for CH is similar to or greater than that of conventional risk factors, underscoring the importance of CH in CVD.32
Other studies have confirmed and extended these findings. Bick et al. analyzed whole exome sequences from 35,416 individuals in the UK Biobank without prevalent CVD to identify participants with CH.9 They identified 1,079 (3.0%) individuals with DNMT3A or TET2 CH and found that CH was associated with increased incident CVD event risk (HR 1.27) during a 6.9-year median follow-up. CVD risk was greater for larger clones compared to smaller clones (HR 1.59). More recently, Nachun et al. reported that a combination of CH and epigenetic aging has a strong predictive value in identifying patients with high risk of all-cause mortality and coronary heart disease.10 In this regard, epigenetic age acceleration manifests when the individual’s estimated epigenetic age is older than that of his/her chronological age. This study examined 5,522 human samples from four cohorts within the Trans-omics for Precision Medicine (TOPMed) program and found that ~40% of CHIP carriers exhibited epigenetic age acceleration. Individuals with both CH and aging acceleration displayed greater risks of all-cause mortality (HR 2.90) and coronary heart disease (HR 3.24) compared to individuals without CH and ageing acceleration.10 Notably, CH-positivity was not associated with mortality or CVD if it was independent of epigenetic age acceleration. Saiki et al. performed a combination of targeted DNA sequencing for 23 CH-related genes and array-based copy number alterations (CNAs) detection in blood-derived from 11,234 individuals from the BioBank Japan cohort.11 Driver gene SNVs and Indels or CNAs were detected in ~40% of individuals who were 60 years of age or greater. CH-related SNVs/indels and CNAs exhibited statistically significant co-occurrence within individuals. Notably, this study found that co-occurrence of CNAs and SNVs/indels in DNMT3A, TET2, JAK2, or TP53 genes was associated with much higher cardiovascular mortality.11 Collectively, these findings document a robust and frequent association between CH and CVD, and they are summarized in Table 1.
Table 1.
Summary of studies showing associations between clonal hematopoiesis and cardiovascular disease
| Source | Population | Seq method | Findings |
|---|---|---|---|
| Jaiswal et al 2017 | 4,726 CHD patients 3,529 controls | Whole exome sequence | CHIP was associated with a greater risk of CHD (HR 2.0) and early-onset MI (HR 4.0). |
| Bick et al, 2020 | 35,416 individuals without prevalent CVD | Whole exome sequence | CHIP (DNMT3A, TET2) was associated with increased risk of incident CVD. Genetically reduced IL-6 signaling was associated with an attenuated CVD risk in CHIP. |
| Nachun et al, 2021 | 5,522 individuals | Whole genome sequence DNA methylation array |
CHIP and age acceleration were associated with increased risk of all-cause mortality (HR 2.9) and CHD (HR 3.2). |
| Saiki et al, 2021 | 11,234 individuals | Targeted sequence of 23 CH genes Array-based CNAs detection |
Co-occurrence of SNVs/indels and CNAs (DNMT3A, TET2, JAK2, and TP53) was associated with higher CVD mortality. |
| Yu et al, 2021 | 56,597 individuals without HF and hematological malignancy | Whole exome sequence Whole genome sequence |
Mutations in TET2, ASXL1, and JAK2 were prospectively associated with a 25% increased risk of HF. |
| Bhattacharya et al, 2021 | 7426 incident stroke cases 78752 controls | Whole exome sequence Whole genome sequence |
CHIP was associated with an increased risk of total stroke (HR 1.14). |
| Dorsheimer et al, 2019 | 200 patients undergoing autologous BMT for AMI | Error-corrected targeted exome sequencing | CH (DNMT3A, TET2) was associated with significantly worse long-term clinical outcomes, including mortality and mortality combined with re-hospitalization for HF. |
| Assmus et al, 2021 | 419 patients with chronic HF | Error-corrected targeted exome sequencing | CH (DNMT3A and TET2) was an independent predictor of mortality in HF patients Optimized VAF for TET2 and DNMT3A were 0.73% and 1.13%, respectively. |
| Cremer et al, 2020 | 419 patients with chronic HF | Error-corrected targeted exome sequencing | CH (DNMT3A, TET2, PHF6, SMC1A, PPM1D, EZH2, CEBPA, SRSF2, SETBP) was an independent predictor of mortality in patients with chronic HF. |
| Pascual-Figal et al, 2021 | 62 patients with HF (EF < 45%) | Error collected targeted-exome sequencing for 54 genes | CHIP in either DNMT3A or TET2 exhibited accelerated HF progression irrespective of ischemic/nonischemic etiology. |
CHD: coronary heart disease, MI: myocardial infarction, CVD: cardiovascular disease, CNAs: copy number alterations, BMT: bone marrow transplantation
Because epidemiological studies are correlative by nature, they generally cannot distinguish whether CH and cardiovascular disease are causally linked or are simply an epiphenomenon of the aging process. Thus, potential mechanistic links between CH and CVD have been explored in model systems. Most of these studies have focused on the TET2 epigenetic regulator that is frequently mutated in CH. Two independent groups originally reported that TET2-mediated CH could be causal in the development of atherosclerosis using murine models.8,33 In the study conducted by our lab, a competitive bone marrow transplantation approach was performed to introduce a small number of Tet2-deficient HSPC to a recipient mouse, effectively mimicking the human scenario of individuals carrying a TET2 somatic mutation that gradually expands over time.33 Tet2-deficient HSPC displayed progressive expansion into all immune cell progeny with a bias toward cells of myeloid lineage. This model of Tet2-mediated clonal expansion led to a marked increase in plaque size in hyperlipidemic low-density lipoprotein receptor–deficient (Ldlr−/−) mice.33 Consistent with these observations, it was independently reported that the complete bone marrow transplantation of Tet2-deficient cells to hyperlipidemic mice also led to an increase in vascular plaque size.8 Both studies showed that myeloid-specific ablation of Tet2 was sufficient to promote atherosclerotic development.8,33 Mechanistically, TET2 activates the NLR family pyrin domain containing 3 (NLRP3) inflammasome in immune cells to elevate proinflammatory cytokine release through a HDAC-dependent mechanism.33 The NLRP3 inflammasome is a critical component of the innate immune system that controls the activation and secretion of interleukin-1β(IL-1β) and interleukin-18 (IL-18).34 Notably, it was shown that treatment with a small molecule NLRP3 inflammasome inhibitor reversed the accelerated atherosclerosis that was caused by the expansion of Tet2-deficient hematopoietic cells.33 Subsequent experimental studies also found that JAK2-mediated CH may have a causal role in thrombosis, atherosclerosis, and pulmonary hypertension.35–38 JAK2 is a non-receptor tyrosine kinase and associates with various receptors during signal transduction. JAK2V617F is a constitutively active mutant form that activates STAT transcription factors.
3. Clonal hematopoiesis is associated with heart failure
Recently it was reported that CH, particularly sequence variations in ASXL1, TET2, and JAK2, represents a risk factor for incident heart failure (HF).12 In this study, CH status was determined from whole exome or genome sequence data of blood DNA in participants without prevalent HF or hematological malignancy from 5 large cohorts. Of the 56,597 individuals (41% men, mean age 58 years at baseline), CH could be detected in 3,406 individuals (6%). During the flow up period of up to 20 years 4,694 developed HF (8.3%). After adjusting for demographic and clinical risk factors, CH was prospectively associated with a 25% increased risk of HF in the meta-analysis with consistent associations across all cohorts (HR 1.25).12 In addition, a number of studies have associated CH with poor outcomes in patients with pre-existing HF. Dorsheimer et al. analyzed 200 patients (median age 65 years) who had undergone autologous bone marrow treatment for acute myocardial infarction for mutations in candidate CH genes by deep-targeted amplicon sequencing.39 Using a VAF threshold of 2%, the prevalence of CH was 18.5% with DNMT3A and TET2 being the most prevalent mutant genes. Patients with these forms of CH displayed significantly worse long-term clinical outcomes, including mortality and mortality combined with re-hospitalization for HF.39 Furthermore, when patients were grouped based on VAF, there was a dose-dependent relationship between clone size and clinical outcome, indicating that smaller clone sizes (between 1 and 2% VAF) were associated with worse prognoses.39 Subsequently, Assmus et al. analyzed CH in 419 patients with heart failure (stable HF symptoms, NYHA≥2, ischemic origin, and left ventricular dysfunction). Receiver operating characteristic (ROC) curve analysis of the data was employed to identify prognostic VAF cut-off values.40 Survival ROC analyses revealed optimized VAF cut-off values of 0.73% and 1.15% for TET2 and DNMT3A clones, respectively. Using these respective cutoffs, 5-year mortality was increased in both TET2 and DNMT3A carriers.40 In the same cohort, Cremer et al. reported that in addition to TET2 and DNMT3A, clones with mutations in PPM1D, Serine And Arginine Rich Splicing Factor 2 (SRSF2), CCAAT Enhancer Binding Protein Alpha (CEBPA), PHD Finger Protein 6 (PHF6), Structural Maintenance Of Chromosomes 1A (SMC1A), Enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit (EZH2), and SET Binding Protein (SETBP) were also associated with worse clinical outcomes.41,42 While the aforementioned studies focused on HF of ischemic origin, Fuster et al. recently reported that CH is associated with accelerated disease progression regardless of the HF etiology.43 In this study, blood samples from 62 HF patients with left ventricular ejection fractions below 45% underwent deep sequence analysis for 54 candidate CH genes. After adjusting for risk factors, patients with mutations in either DNMT3A or TET2 exhibited accelerated HF progression as assessed through death (HR 2.79), death or HF hospitalization (HR 3.84), and HF-related death or HF hospitalization (HR 4.41). This association remained significant irrespective of ischemic/nonischemic etiology.43
Experimental studies indicate that CH can causally contribute to HF. Various mouse models have been employed to show that the CH models of TET2, DNMT3A, and JAK2 genes promote adverse cardiac remodeling, and these findings are summarized in detail in recent reviews.22,44 Briefly, in a model of TET2 CH, mice were transplanted with Tet2-mutant HSPC and underwent left anterior descending artery (LAD) ligation or transverse aortic constriction (TAC) surgery. Mice transplanted with Tet2-mutant cells exhibited worse cardiac remodeling compared to mice transplanted with wild-type bone marrow.45 As with the atherosclerosis model, inhibition of the NLRP3 inflammasome alleviated the adverse effects of CH in these heart failure models.33 In another model, Tet2- or Dnmt3a-mutant HSPC were generated through lentivirus transfection of Cas9 protein and sgRNA, and transplanted into recipient mice. Mice harboring Tet2- or Dnmt3a-mutant HSPC displayed worse cardiac remodeling following angiotensin II infusion compared to mice transplanted with wild-type HSPC.46 It has also been shown that the adoptive transfer of Tet2-deficient HSPC to unconditioned mice (i.e. no myeloablation to promote engraftment) leads to the development of age-related, spontaneous cardiomyopathy in the absence of surgical or pharmacological stress.47 Finally, it was found that lentivirus-mediated transduction of HSPC, leading to myeloid-specific expression of Jak2V617F, promoted greater cardiac remodeling following TAC and LAD ligation.48 These CVD features were observed in the absence of changes to hematocrit or blood cell counts that can occur when Jak2V617F is expressed in hematopoietic cells (but is not feature of CH). Collectively, multiple experimental findings illustrate that CH can contribute to the development HF and mechanistic features involving inflammatory cytokines have been defined.
4. Therapy-associated CH is prevalent in cancer patients
While CH spontaneously arises with aging, specific environmental perturbations can augment the fitness of mutant HSPC and contribute to clonal expansions in the hematopoietic system. Patients with cancers display higher prevalence of CH. Coombs et al. analyzed of candidate CH target genes in tumor and blood samples from 8,810 individuals (median age of 58.3 years), and found a high prevalence of CH (25%).49 In addition to increasing age, CH was found to also be associated with tobacco use and prior radiation therapy or chemotherapy. Compared to the age-associated CH described previously, this population displayed an overlapping but distinct mutational landscape that is referred to as therapy-related CH (t-CH).49 This form of CH occurs as a result of the selective pressure that the genotoxic stressors exert on HSPC. Mutations are found in the DNA damage-response pathway (DDR) genes including Ataxia-telangiectasia-mutated (ATM), Checkpoint kinases 1,2 (CHK1, CHK2), TP53, and PPM1D.50,51 In the study of Coombs et al., while CH was found to be associated with increased incidence of hematologic cancers, the diminished survival could not be attributed to hematologic cancer alone, suggesting the presence of unknown contributors.49
To expand upon these findings, Bolton et al. reported an association between cancer treatments and CH mutations in 24,146 cancer patients with variable primary tumor types (n=56) and ages.52 CH mutations were identified in 7,216 individuals, representing 30% of the patient cohort.52 Of all treatment modalities, external beam radiation therapy, cytotoxic chemotherapy, and radionuclide therapy were most strongly associated with CH. Mutations in PPM1D were most strongly associated with previous exposure to platinum or radionuclide therapy.52 There was also a strong enrichment in other DDR genes, including TP53 and CHK2.
Gibson et al. performed targeted sequencing on cryopreserved aliquots of autologous stem-cells from 401 patients with non-Hodgkin lymphoma (NHL).53 These patients underwent autologous stem cell transplantation (ASCT), an extreme hematopoietic stress in which autologous stem cells are harvested before the administration of myeloablative chemotherapy and then reinfused into the patient to repopulate the hematopoietic system. In this cohort, 120 patients (29.9%) had CH at the time of ASCT, and these patients had significantly lower overall survival compared to those without CH (10-year overall survival, 30.4% v 60.9%).53 The increase in death in this population resulted from therapy-related myeloid neoplasm and CVD.53 A further investigation of this patient cohort by Kahn et al. revealed that PPM1D mutations were 60 times more likely to be present in the chemotherapy-exposed lymphoma patients than 28,418 individuals who were unselected for malignancy and adjusted for age.18,19, 54 Recently, Miller et al. reported the effect of t-CH on clinical outcomes in patients with NHL and multiple myeloma (MM).55 In this study, 154 patients with NHL or MM receiving chimeric antigen receptor T cells (CAR T-cells) were assessed by NGS of peripheral blood for mutations in known CH driver genes. It was found that CH was present in 48% of patients. While CH was not associated with a difference in overall survival, it was associated with increased responsiveness to immune-targeting therapies and increased severity of cytokine release syndrome in patients younger than age 60 years. These data suggest that CH can influence inflammatory pathways across numerous therapeutic contexts in cancer patients and cancer survivors. These studies are summarized in Table 2.
Table 2.
Summary of the studies examining clonal hematopoiesis in cancer patients
| Source | Population | Seq method | Findings |
|---|---|---|---|
| Coombs et al 2017 | 8,810 individuals with non-hematologic cancers | Whole exome sequence Paired (Blood and cancer) |
25% of patients had CH. 4.5% of patients had CH-PD. CH was associated with increased age, prior radiation therapy, and tobacco use. CH-PD was associated with diminished patient survival. |
| Gibson et al, 2017 | 12 NHL patients before ASCT 401 NHL patients after ASCT |
Whole exome sequence Targeted-NGS for 86 genes |
29.9% of patients have CHIP. CHIP was associated with inferior survival from tMN and CVD. |
| Kahn et al, 2018 | 401 NHL patients 28,418 individuals unselected for cancer |
Whole exome sequence Targeted-NGS |
PPM1D mutations were 60 times more prevalent in chemotherapy-exposed NHL patients. |
| Bolton et al, 2020 | 24,146 cancer patients (56 types of primary tumor) | Deep targeted amplicon sequence | 30% of patients had CH. Cancer treatment was associated with CH with enrichments in DDR genes (PPM1D, TP53, CHK2). |
| Miller et al, 2021 | 154 NHL or MM patients receiving CAR T-cells | NGS of the driver genes | 48% of patients had CHIP. CHIP was associated with increased CR rate and CRS severity. |
CH-PD: clonal hematopoiesis of putative driver genes, NHL: non-Hodgkin lymphoma, ASCT: autologous stem-cell transplantation, NGS: next-generation sequencing, tMN: therapy-related myeloid neoplasms, CVD: cardiovascular disease, MM: multiple myeloma, CR: complete response, CRS: cytokine release syndrome
Recent experimental studies have investigated the mechanisms that contribute to the clonal expansions induced by genotoxic agents. These studies have shown that the genotoxic stresses of chemotherapy or radiation therapy create selective pressures causing expansion of clones with mutations in DDR genes that confer resistance to these stressors.54,56–58 PPM1D is a member of the PP2C family of serine-threonine phosphatases. PPM1D is induced by TP53 activation which, in turn, dephosphorylates and inactivates TP53 through negative feedback mechanism. In addition, several other proteins in the DDR pathway, including H2A Histone Family Member X (H2AX), ATM, and CHK2, are dephosphorylated by PPM1D. Therefore, PPM1D is a critical negative regulator of many components of the DDR pathway.59 Most PPM1D mutations are truncating mutations in the terminal exon.52,54,56 These exon 6 mutations are gain-of-function mutations that display elevated expression and activity due to loss of a C-terminal degradation domain. The truncating PPM1D mutations confer a chemoresistance phenotype, resulting in the selective expansion of PPM1D-mutant HSPC in the presence of chemotherapeutic agents. The transcription factor TP53, which can be activated by DNA damage,60 is also commonly mutated in t-CH.52 The majority of somatic TP53 mutations are missense mutations in the DNA-binding domain of the TP53 protein, which leads to an inability to conduct DNA damage repair.57,61 It has been reported that DNA damage by radiation triggers TP53-dependent cell competition in HSPC. This competition appears to be mediated by an induction of growth arrest and senescence-related genes in wild-type cells that become outcompeted by cells with mutations in TP53.58 It has also been reported that mutant TP53 interacts with EZH2 to promote HSPC self-renewal and differentiation.57 Collectively, these clinical and experimental studies showed that chemotherapy and/or radiation therapy exert powerful selective pressure on mutant clones in the hematopoietic system, and this can lead to robust clonal expansions.
5. Possible links between therapy-associated clonal hematopoiesis and cardiovascular disease
As noted previously, patients with cancer and cancer survivors display an increased risk of CVD development.3,62 It is widely appreciated that radiation and/or chemotherapy contribute to the development of non-ischemic heart disease. For example, a recent large-scale electronic health record analysis of multiple databases examined CVD outcomes in 126,120 patients with cancer and 630,144 control individuals.63 Increased risks of HF or cardiomyopathy was observed in patients with 10 of the 20 cancers examined, including hematological (HR 1.94), esophageal (HR 1.96), lung (HR 1.82), kidney (HR 1.73), and ovarian (HR 1.59) cancer. Furthermore, it has been reported that mortality due to CVD in cancer survivors is greater than that of the cancer itself after a ten-year follow-up3,62,63 Therefore, given the recent proliferation of new cancer therapies and the longer survival of cancer patients, there is an increasing need to identify the molecular mechanisms that contribute to the CVD observed in these patients.
Given the mechanistic links between age-related CH and CVD, we speculated that the DDR genes commonly mutated in t-CH could contribute to the HF that is prevalent in cancer patients and cancer survivors (Figure 1). Thus, we tested whether causal relationships exist between t-CH and cardiac dysfunction by focusing on hematopoietic cell mutations in PPM1D64 and TP53,65 that represent the most enriched t-CH genes in patients receiving cancer therapy.52,53 In one of these studies, we developed a mouse model using CRISPR-Cas9 technology to generate clonal hematopoiesis-associated mutations in exon 6 of Ppm1d, leading to its overactivation.64 This model of t-CH was then employed to examine whether somatic mutations of PPM1D in hematopoietic cells can increase the heart’s susceptibility to stress. This Ppm1d mouse model of CH revealed worse cardiac remodeling as measured by echocardiography and greater cardiac fibrosis and myocyte hypertrophy following chronic infusion of angiotensin II.64 Mechanistically, Ppm1d-mutant macrophages display impaired DDR pathway activation, greater DNA damage, higher reactive oxygen species production, and an augmented proinflammatory profile with elevations in IL-1β and IL-18.64 It was further shown that the NLRP3 inflammasome inhibitor MCC950 effectively reverses the cardiac remodeling caused by transplantation of Ppm1d-mutant HSPC.64 These data suggest that inflammasome activation and elevated cytokine production are critical for the enhanced pathological cardiac phenotype associated with this form of t-CH.
Figure 1.
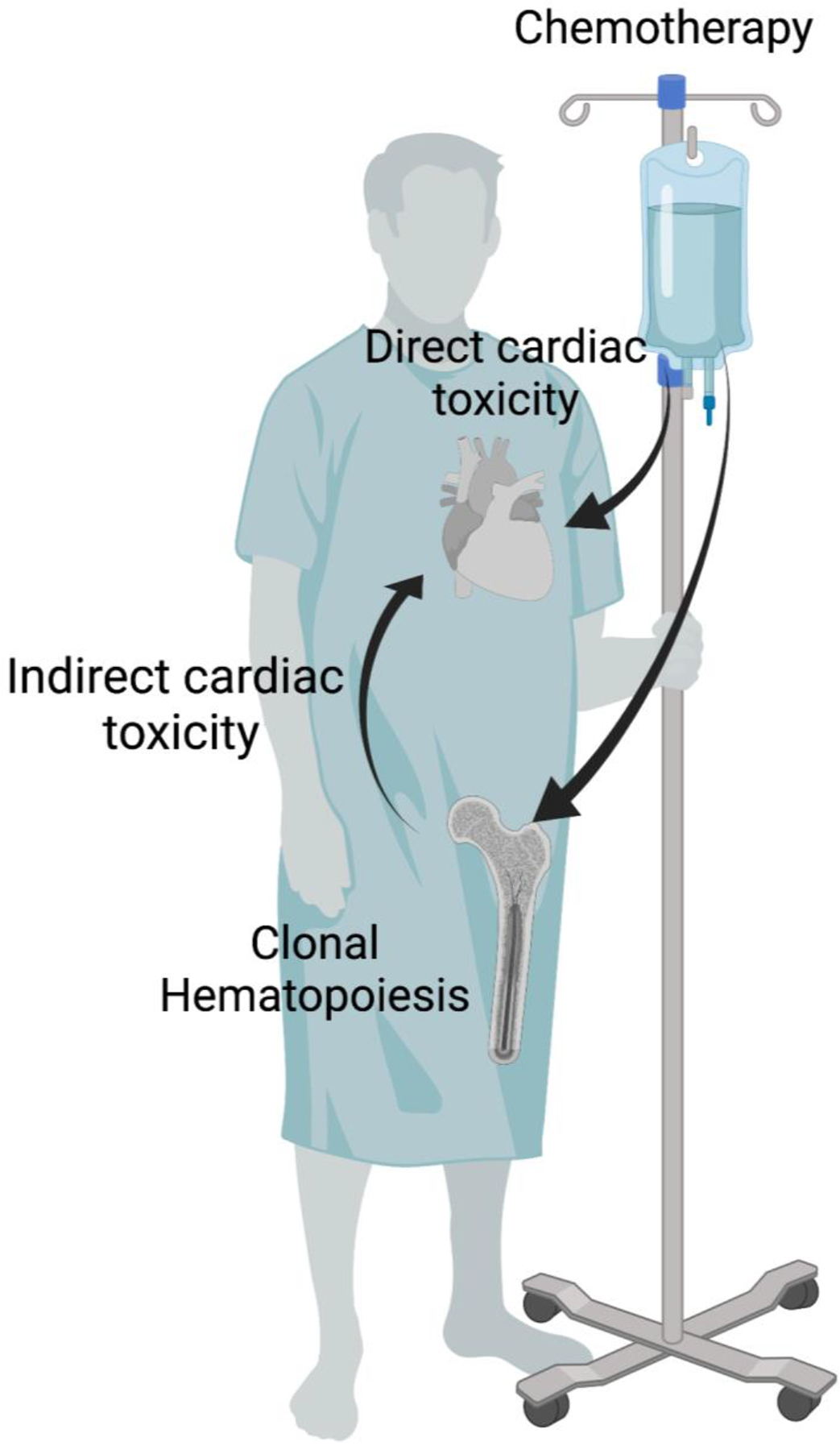
The cardiac toxicity associated with genotoxic therapies may stem from both direct and indirect mechanisms. Cancer patients treated with genotoxic therapies display a higher prevalence of clonal hematopoiesis, which may cause indirect cardiac toxicity.
Created with BioRender.com.
In a second study we examined a model of t-CH involving mutations in TP53.65 This study employed three distinct mouse strains to examine the relationship between t-CH of TP53 clones and cardiac function. The models included mice transplanted with Trp53 heterozygous-knockout bone marrow cells, myeloid-specific Trp53-deficient mice, and mice transplanted with bone marrow cells harboring a common TP53 missense mutation, Trp53R270H. To establish a model of TP53-mediated t-CH, these mice were treated with a course of the chemotherapeutic agent doxorubicin (Dox). As expected, treatment of mice with dox accelerated the expansion of hematopoietic TP53-mutant clones in the adoptive transfer model that avoids myeloablation. It was also found that mice transplanted with HSPC lacking TP53, or with mutant TP53, display worse Dox-induced cardiotoxicity compared to mice transplanted with wild-type HSPC. Mechanistically, treatment with Dox leads to exuberant and persistent neutrophil infiltration into the myocardium. In this context, the Tp53-mutant neutrophil produced greater ROS, leading to further damage of the myocardium and greater inflammatory cytokine production.65
Collectively, these findings provide precedence for the concept that t-CH can contribute to the development of CVD in cancer patients and cancer survivors who have undergone therapy with genotoxic agents (Figure 1). Currently, there are no clinical studies to directly address this hypothesis; however, this possibility could be investigated through a prospective study of clinical outcomes in cancer patients treated with genotoxic therapies. If validated by further studies, these data would suggest that t-CH is predictive of cardiac dysfunction in cancer survivors and that the cardiac pathology in these patients could be responsiveness to anti-inflammatory therapies. As mentioned above, t-CH due to mutations in multiple DDR pathway genes, beyond PPM1D and TP53, are frequently observed in cancer patients and cancer survivors, and these t-CH driver genes could potentially also affect the CVD outcomes. Thus, many future clinical and experimental studies are warranted.
Conclusions and future directions
In closing, CH is an emerging risk factor for both cancer and CVD. Cancer patients and survivors exhibit prevalent t-CH, a form of CH that is associated with DDR gene mutations in HSPC that enable clone expansion in response to genotoxic agent exposure. Given the growing appreciation of its role in CVD, an individual’s t-CH status could become a critical consideration in assessing the cancer patient’s risk and prognosis. Furthermore, recent experimental findings suggest that anti-inflammatory therapies have utility for treating the cardiac toxicities that develop in cancer survivors with t-CH. Thus, additional clinical and experimental studies are warranted to understand the mechanisms that give rise to t-CH and its effects on the cardiovascular system.
Key Points:
Clonal hematopoiesis is a new risk factor for cardiovascular disease.
Both cardiovascular disease and clonal hematopoiesis are frequent in patients with cancer and cancer survivors who have been treated with genotoxic agents.
Experimental studies have shown that cancer therapy-related clonal hematopoiesis contributes to the worse cardiovascular disease outcome.
Synopsis:
Clonal hematopoiesis is a pre-cancerous state that is recognized as a new causal risk factor for cardiovascular disease. Therapy-related clonal hematopoiesis is a condition that is often found in cancer survivors. These clonal expansions are caused by mutations in DNA damage-response pathway genes that allow hematopoietic stem cells to undergo positive selection in response to the genotoxic stress. These mutant cells increasingly give rise to progeny leukocytes that display enhanced pro-inflammatory properties. Recent experimental studies suggest that therapy-related clonal hematopoiesis may contribute to the medium- to long-term risk of genotoxic therapies on the cardiovascular system.
Clinics Care Points:
Although there are no existing guidelines on screening and treating patients with CH, we should recognize that individuals with CH are at higher risk of CVD.
Currently, it is reasonable to suggest that modifiable conventional risk factors be aggressively controlled in patients with CH. Also, additional CVD screening could be considered in individuals with confirmed CH and a suspicion of CVD based upon age, symptoms and prior treatment by anti-cancer therapies.
Abbreviations:
- ASCT
autologous stem-cell transplantation
- ASXL1
additional sex combs like 1
- ATM
Ataxia–telangiectasia-mutated
- BCOR
BCL6 Corepressor
- CAD
coronary artery disease
- CAR T-cells
chimeric antigen receptor T cells
- CEBPA
CCAAT Enhancer Binding Protein Alpha
- CH-PD
clonal hematopoiesis of putative driver genes
- CH
clonal hematopoiesis
- CHIP
clonal hematopoiesis of indeterminate potential
- CHK1
checkpoint kinase 1
- CHK2
checkpoint kinase 2
- CNAs
copy number alterations
- CVD
cardiovascular disease
- DDR
DNA damage response
- DNMT3A
DNA methyltransferase-3A
- Dox
doxorubicin
- EZH2
Enhancer Of Zeste 2 Polycomb Repressive Complex 2 Subunit
- GNAS
GNAS (guanine nucleotide binding protein, alpha stimulating) complex locus
- H2AX
H2A Histone Family Member X
- HF
heart failure
- HR
Hazard ratio
- HSPC
hematopoietic stem and progenitor cells
- IL-18
interleukin-18
- IL-1b
interleukin-1β
- Indels
insertions and deletions
- JAK2
janus kinase 2
- LAD
left anterior descending artery
- LDLR
low-density lipoprotein receptor
- MM
multiple myeloma
- NGS
next-generation sequencing
- NHL
non-Hodgkin lymphoma
- NLRP3
NLR family pyrin domain containing 3
- NYHA
New York Heart Association
- PHF6
PHD finger protein 6
- PPM1D
protein phosphatase, Mg2D/Mn2D dependent 1D
- ROC
receiver operating characteristic
- SETBP
SET Binding Protein
- SF3B1
Splicing Factor 3b Subunit1
- SMC1A
Structural Maintenance Of Chromosomes 1A
- SNVs
single nucleotide variants
- SRSF2
Serine and Arginine Rich Splicing Factor 2
- STAT
signal transducer and activator of transcription
- t-CH
therapy-related clonal hematopoiesis
- TAC
transverse aortic constriction
- TET2
ten-eleven translocation-2
- TOPMed
Trans-omics for Precision Medicine
- TP53
tumor protein 53
- TP53
tumor protein 53
- VAF
variant allele fraction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement: The authors do not have anything to disclose.
References
- 1.Handy CE et al. Synergistic Opportunities in the Interplay Between Cancer Screening and Cardiovascular Disease Risk Assessment: Together We Are Stronger. Circulation 138, 727–734 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Calvillo-Argüelles O et al. Connections Between Clonal Hematopoiesis, Cardiovascular Disease, and Cancer: A Review. JAMA Cardiol. 4, 380–387 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Zamorano JL et al. The cancer patient and cardiology. Eur. J. Heart Fail (2020). doi: 10.1002/ejhf.1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnold M et al. Progress in cancer survival, mortality, and incidence in seven high-income countries 1995–2014 (ICBP SURVMARK-2): a population-based study. Lancet. Oncol 20, 1493–1505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curigliano G et al. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol 31, 171–190 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moslehi JJ Cardiovascular Toxic Effects of Targeted Cancer Therapies. N. Engl. J. Med 375, 1457–1467 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Zhang S et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med 18, 1639–1642 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Jaiswal S et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med 377, 111–121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bick AG et al. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 141, 124–131 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nachun D et al. Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell 20, e13366 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saiki R et al. Combined landscape of single-nucleotide variants and copy number alterations in clonal hematopoiesis. Nat. Med 27, 1239–1249 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Yu B et al. Supplemental Association of Clonal Hematopoiesis With Incident Heart Failure. J. Am. Coll. Cardiol 78, 42–52 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhattacharya R et al. Clonal Hematopoiesis Is Associated With Higher Risk of Stroke. Stroke STROKEAHA121037388 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seita J & Weissman IL Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med 2, 640–653 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaiswal S Clonal hematopoiesis and nonhematologic disorders. Blood 136, 1606–1614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welch JS et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 150, 264–278 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Busque L et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat. Genet 44, 1179–1181 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaiswal S et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med 371, 2488–2498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Genovese G et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie M et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med 20, 1472–1478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shlush LI Age-related clonal hematopoiesis. Blood 131, 496–504 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Yura Y, Sano S & Walsh K Clonal Hematopoiesis: A New Step Linking Inflammation to Heart Failure. JACC. Basic to Transl. Sci 5, 196–207 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steensma DP et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 126, 9–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shumilov E et al. Genetic alterations crossing the borders of distinct hematopoetic lineages and solid tumors: Diagnostic challenges in the era of highthroughput sequencing in hemato-oncology. Crit. Rev. Oncol. Hematol 126, 64–79 (2018). [DOI] [PubMed] [Google Scholar]
- 25.McKerrell T & Vassiliou GS Aging as a driver of leukemogenesis. Sci. Transl. Med 7, 306fs38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forsberg LA et al. Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat. Genet 46, 624–628 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loh P-R et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 559, 350–355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaiswal S & Ebert BL Clonal hematopoiesis in human aging and disease. Science 366, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bick AG et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 586, 763–768 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young AL, Challen GA, Birmann BM & Druley TE Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat. Commun 7, 12484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watson CJ et al. The evolutionary dynamics and fitness landscape of clonal hematopoiesis. Science 367, 1449–1454 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Jaiswal S & Libby P Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat. Rev. Cardiol 17, 137–144 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuster JJ et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swanson KV, Deng M & Ting JP-Y The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol 19, 477–489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolach O et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang W et al. Macrophage Inflammation, Erythrophagocytosis, and Accelerated Atherosclerosis in Jak2 (V617F) Mice. Circ. Res 123, e35–e47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fidler TP et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 592, 296–301 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kimishima Y et al. Clonal hematopoiesis with JAK2V617F promotes pulmonary hypertension with ALK1 upregulation in lung neutrophils. Nat. Commun 12, 6177 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dorsheimer L et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 4, 25–33 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Assmus B et al. Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT3A- and TET2-driver gene mutations. Eur. Heart J 42, 257–265 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Cremer S et al. Multiple Somatic Mutations for Clonal Hematopoiesis Are Associated With Increased Mortality in Patients With Chronic Heart Failure. Circulation. Genomic and precision medicine 13, e003003 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Kiefer KC et al. Full spectrum of clonal haematopoiesis-driver mutations in chronic heart failure and their associations with mortality. ESC Hear. Fail 8, 1873–1884 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pascual-Figal DA et al. Clonal Hematopoiesis and Risk of Progression of Heart Failure With Reduced Left Ventricular Ejection Fraction. J. Am. Coll. Cardiol 77, 1747–1759 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Wang Y et al. Murine models of clonal hematopoiesis to assess mechanisms of cardiovascular disease. Cardiovasc. Res (2021). doi: 10.1093/cvr/cvab215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sano S et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1beta/NLRP3 Inflammasome. J. Am. Coll. Cardiol 71, 875–886 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sano S et al. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res 123, 335–341 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI insight 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sano S et al. JAK2 (V617F) -Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC. Basic to Transl. Sci 4, 684–697 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coombs CC et al. Therapy-Related Clonal Hematopoiesis in Patients with Non-hematologic Cancers Is Common and Associated with Adverse Clinical Outcomes. Cell Stem Cell 21, 374–382.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jackson SP & Bartek J The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hakem R DNA-damage repair; the good, the bad, and the ugly. EMBO J. 27, 589–605 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bolton KL et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet (2020). doi: 10.1038/s41588-020-00710-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gibson CJ et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J. Clin. Oncol 35, 1598–1605 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kahn JD et al. PPM1D-truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 132, 1095–1105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller PG et al. Clonal hematopoiesis in patients receiving chimeric antigen receptor T-cell therapy. Blood Adv. 5, 2982–2986 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hsu JI et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 23, 700–713.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen S et al. Mutant p53 drives clonal hematopoiesis through modulating epigenetic pathway. Nat. Commun 10, 5649 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bondar T & Medzhitov R p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell 6, 309–322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uyanik B, Grigorash BB, Goloudina AR & Demidov ON DNA damageinduced phosphatase Wip1 in regulation of hematopoiesis, immune system and inflammation. Cell death Discov. 3, 17018 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brosh R & Rotter V When mutants gain new powers: news from the mutant p53 field. Nat. Rev. Cancer 9, 701–713 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Boettcher S et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599–604 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stoltzfus KC et al. Fatal heart disease among cancer patients. Nat. Commun 11, 2011 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Strongman H et al. Medium and long-term risks of specific cardiovascular diseases in survivors of 20 adult cancers: a population-based cohort study using multiple linked UK electronic health records databases. Lancet (London, England) 394, 1041–1054 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yura Y et al. The Cancer Therapy-Related Clonal Hematopoiesis Driver Gene Ppm1d Promotes Inflammation and Non-Ischemic Heart Failure in Mice. Circ. Res (2021). doi: 10.1161/CIRCRESAHA.121.319314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sano S et al. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI insight 6, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]