

Abstract

Small-molecule (SM)-based organic solar cells (OSCs) have dominated the photovoltaic industry on account of their efficient optical and electronic properties. This quantum mechanical study addresses a DFT study of pentacyclic aromatic bislactam (PCL)-based small molecules for extremely proficient OSCs. Five novel small molecules (PCLM1–PCLM5) retaining the A−π–A−π–D−π–A−π–A arrangement were fabricated from the reference PCLR. At the MPW1PW91/6-31G** level of theory, detailed profiling of these novel molecules was performed by accurately following DFT, along with the time-dependent density functional theory (TD-DFT) hypothetical simulations to analyze the UV–visible absorption (λmax), light-harvesting efficiency (LHE), dipole moment (μ), fill factor (FF), open-circuit voltage (VOC), power conversion efficiency (PCE), frontier molecular orbitals (FMOs), binding energy (Eb), density of states (DOS), electrostatic potential (ESP), and transition density matrix (TDM) plots. Alteration of peripheral acceptors in all of the molecular structures drastically modified their charge-transfer properties, such as a strong light-harvesting capability in the range of 0.9993–0.9998, reduced exciton Eb (from 0.34 to 0.39 eV), a reduced bandgap (Eg) in the range of 1.66–1.99 eV, an elevated λmax (775–959 nm) along with a higher μ in the solvent phase (1.934–7.865 D) when studied in comparison with PCLR, possessing an LHE of 0.9986, an Eb of 0.40, an Eg 2.27 eV, λmax at 662 nm, and a μ of 0.628 D. The FMO analysis revealed the uniform dispersal of charge density entirely along the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals in newly constructed moieties. Electron as well as hole mobility rates, VOC, FF, and PCE of all novel molecules (PCLM1–PCLM5) were higher as compared with those of PCLR, ultimately making them exceptional candidates for solar devices. Focusing on the outcomes, terminal acceptor modification was found to be a suitable method for the development of highly tuned OSCs in the future.
1. Introduction
The exhaustion of nonrenewable natural energy resources such as coal, oil, natural gas, and nuclear energy has drastic effects on the natural environment. Besides, these resources also present a risk to human beings as they cause environmental pollution.1 To overcome the ongoing energy crisis, substitute renewable energy supplies such as biomass, hydropower plants, solar energy, etc., are being utilized. Solar energy supply is the most significant and limitless form of energy that can satisfy the energy demands of mankind. The use of photovoltaic cells, which directly convert solar energy into electrical power, is one of the best ways to harness solar energy currently. Earlier, inorganic silicon-based solar cells (SCs) were used owing to their elevated power conversion efficiency (PCE).2 Despite this, inorganic SCs have major drawbacks, including their high cost, rigidity, susceptibility to heat changes, and inability to adjust the energy level.3 These negative features of silicon-based inorganic SCs have given rise to a persistent desire to find suitable alternatives.
Organic solar cells (OSCs) have gained considerable attention recently for converting solar power into electricity owing to their properties of light weight, flexibility, ease of fabrication, and cost-efficient production processes.4 When it comes to portable devices, OSCs have provided new possibilities.5 The development of the bulk heterojunction OSCs caused to an improvement in the efficiency of SCs made from organic materials.6 Because of their elevated photoabsorption coefficient, enhanced stability, and robustness, OSCs may have an enhanced PCE when associated with silicon-based SCs.7 There are several organic molecules that may be altered to construct novel and enhanced photovoltaic materials.8
Over the past few decades, the operating efficiency of fullerene OSCs has increased by 11.7%. Simultaneously, non-fullerene-based organic photovoltaic cells (NF-OPVs) have made significant development due to their tunable energy levels, specific absorption spectra, and electron mobilities.9 These attributes are absent in fullerene derivatives due to their limited chemical modification. The low voltage loss and tunable optoelectronic features of NF-OPVs, together with their synthetic flexibility and broad spectral range, make them easy to adapt architecturally to satisfy the project’s objectives while also allowing PCEs of over 17%.10
Several molecular modeling approaches such as alkyl chain substitution, interfacial alterations of donor–acceptor units, and alteration of end-capped terminal moieties have been exploited to further increase the energy efficiency of NF systems.11 Recently, novel organic materials such as pentacyclic aromatic bislactam,12 benzodifurandione-based poly(phenylene vinylene) (BDPPV),13 and diketopyrrolopyrrole (DPP) acceptors14 have been explored as promising building blocks for OPVs. DPP exhibits a range of desirable optical characteristics and adequate solubility in organic solvents. The DPP unit is a rigid and planar aromatic structure that improves the intermolecular packing in molecules and offers higher charge mobilities.15
In the current project, five novel molecules (PCLM1–PCLM5) were designed from the reference molecule named PCLR. PCLR possesses two monomers, an electron-deficient pentacyclic aromatic bislactam (PCL) central core followed by thiophene rings attached on both sides of PCL, which are further preceded by DPP and a thiophene ring.16 The technique of structural modification was used to design five new small donor groups from the reference PCLR. The five newly designed molecules were modeled by adding end-capped acceptors such as X1 (2-thiophen-2-ylmethylene-malononitrile) in PCLM1, X2 (2-cyano-acrylic acid methyl ester) in PCLM2, X3 (2-(5,6-difluoro-2-methylene-3-oxo-indan-1,1-dicarbonitrile)) in PCLM3, X4 (3-methyl-5-methylene-2-thioxo-thiazolidin-4-one) in PCLM4, and X5 (2-methylene-3-oxo-indan-1,1,5,6-tetracarbonitrile) in PCLM5 after both terminal thiophene rings (π-spacer) in the reference molecule (PCLR). The configuration of the reference molecule was DPP–PCL–DPP, and the five novelly designed molecules had an acceptor–DPP–PCL–DPP–acceptor configuration. ChemDraw structures of the reference (PCLR) and newly developed chromophores (PCLM1–PCLM5) are shown in Figure 1.
Figure 1.

ChemDraw sketches of PCLR and PCLM1–PCLM5.
2. Computational Methodology
The optimization, at the ground state, of the reference molecule PCLR was carried out with four different functionals, i.e., MPW1PW91,17 B3LYP,18 wB97XD,19 and CAM-B3LYP,20 to compare the molar absorptivity (λmax) values with that of the experimental value of PCLR. The λmax obtained with the MPW1PW91 functional for the reference PCLR showed good synchronization with the experimentally reported value. Further computations of newly constructed molecules were executed with MPW1PW91/6-31G**. The designed molecules PCLM1, PCLM2, PCLM3, PCLM4, and PCLM5 were optimized by the above-stated functional and basis set. Time-dependent approach of density functional theory (TD-DFT) calculations in a gaseous medium was accomplished after structural modification to model the absorption graph of the reference PCLR. The influence of the solvent chloroform on IEFPCM21 computations was also investigated. UV–visible spectra of the reference (PCLR) and five newly designed molecules (PCLM1–PCLM5) were assessed by the Swizard program.22 Origin 6.0 software23 was employed for the construction of the absorption graphs. To analyze the contribution of all fragments to the absorption of solar radiations, the density of states (DOS) of PCLR and the newly developed molecules were computed by PyMOlyze 1.1 software.24 TDM analyses were performed using Multiwfn 3.7 software25 to comprehend the electronic excitations with the help of the same functional selected for optimization. To calculate electron, as well as hole mobilities, of the reference PCLR along with the five novelly constructed molecules (PCLM1–PCLM5), eqs 1 and 2 based on the Marcus theory were used. The mathematical expressions of these equations are as follows:
| 1 |
| 2 |
eq 1 expresses the electron mobility (λe) and eq 2 expresses the hole mobility, where E0 is the neutral energy of the optimized molecule and E0– and E0 are the anionic and cationic energies, respectively, at ground states computed successively from optimized neutral molecules. The cation’s neutral energy is represented by E+0 and the anion’s neutral energy is represented by E–. Both were estimated using cationic and anionic optimized geometries sequentially.
3. Results and Discussion
3.1. Validation of the Method and Geometric Optimization
In the current research work, the reference molecule PCLR was optimized with four commonly used functionals, B3LYP, MPW1PW91, wB97XD, and CAM-B3LYP, with a basis set of 6-31G (d,p). The observed absorption maxima (λmax) were at 719, 663, 512, and 529 nm, respectively, in a chloroform (CHCl3) solvent. Figure 2 demonstrates the absorption spectra of PCLR with different functionals in the gaseous (a) as well as solvent (CHCl3) (b) phases.
Figure 2.

UV–vis absorption spectra of PCLR with four discrete functionals in the (a) gaseous and (b) CHCl3 solvent phases.
A comparison of DFT-computed λmax of PCLR was carried out with the experimentally reported λmax of PCLR (675 nm). The λmax displayed at the MPW1PW91 functional level with the above-stated basis set of 6-31G (d,p) (663 nm) was in agreement with experimental value (675 nm), as depicted in Figure 3 and Table 1. Therefore, all DFT calculations of the newly designed molecules (PCLM1–PCLM5) were executed at the MPW1PW91/6-31G** level.
Figure 3.

Bar graph of PCLR with four different functionals in the chloroform solvent with the 6-31G basis set for method validation.
Table 1. Computed λmax of PCLR with Four Different Functionals.
| methods | λmaxcal (nm) | λmaxexp (nm) |
|---|---|---|
| B3LYP | 719 | 675 |
| MPW1PW91 | 663 | |
| WB97XD | 512 | |
| CAM-B3LYP | 529 |
The selected functional with 6-31G** was used to optimize all of the designed compounds after a scrupulous analysis. Optimized frameworks showed that all fragments of molecules arranged themselves in one plane, which facilitates efficient charge transfer. The optimized geometries of all five newly fabricated molecules (PCLM1–PCLM5) and the reference (PCLR) are displayed in Figure 4.
Figure 4.

Ground state optimized geometries of PCLR along with PCLM1–PCLM5.
3.2. Frontier Molecular Orbitals (FMOs)
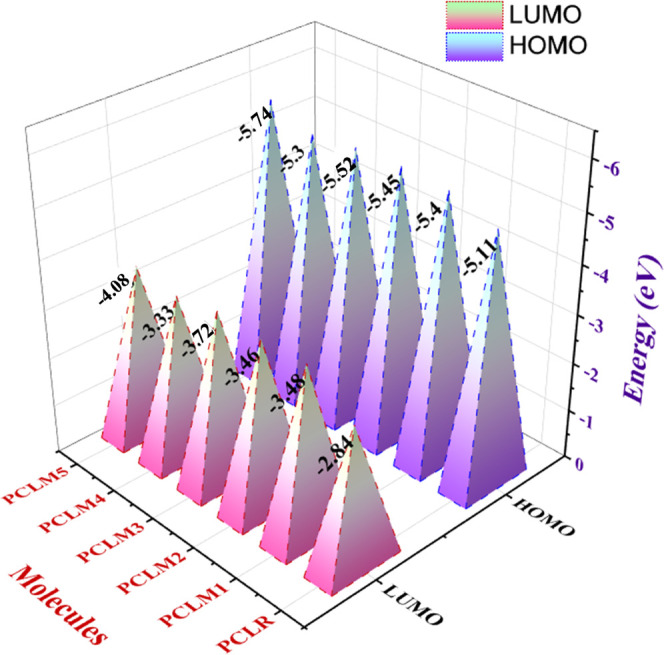
Frontier molecular orbitals (FMOs) are important to describe the distribution of the electron cloud in the highest occupied (HOMO) and the lowest unoccupied (LUMO) molecular orbitals. Both of these are valuable parameters to determine the efficiency of OPVs.26,27 The intermolecular charge-transfer properties are also explored using FMOs. The thermal stability of a molecule is estimated by the bandgap (Eg) between both the FMO energy levels.28 Charge transfer of a molecule becomes more rapid when the Eg is smaller. Hard molecules have a larger Eg, which means that they have more stability and less reactivity, whereas soft molecules have a narrow Eg as they exhibit less stability, more reactivity, and less efficient intermolecular charge-transfer attributes.29 The newly designed chromophores (PCLM1–PCLM5) enhanced the charge transfer from the HOMO to the LUMO due to the presence of strong electron-withdrawing moieties and increased π-conjugation in the system. Computed HOMO/LUMO energies and Eg of the reference PCLR along with those of the newly designed molecules (PCLM1–PCLM5) with the selected functional are tabulated in Table 2.
Table 2. Evaluated HOMO/LUMO Energy as well as Bandgap (Eg) values of PCLR and PCLM1–PCLM5.
| molecules | HOMO (eV) | LUMO (eV) | Eg (eV) |
|---|---|---|---|
| PCLR | –5.11 | –2.84 | 2.27 |
| PCLM1 | –5.40 | –3.48 | 1.92 |
| PCLM2 | –5.45 | –3.46 | 1.99 |
| PCLM3 | –5.52 | –3.72 | 1.80 |
| PCLM4 | –5.30 | –3.33 | 1.97 |
| PCLM5 | –5.74 | –4.08 | 1.66 |
The HOMO and LUMO values of PCLR were seen to be −5.11 and −2.84 eV, respectively, while its bandgap was 2.27 eV. All of the studied chromophores arranged in the increasing order of their HOMO energy levels were as follows: PCLM5 > PCLM2 > PCLM3 > PCLM1 > PCLM4 > PCLR. The trend of their LUMO values was as follows: PCLR > PCLM3 > PCLM1 > PCLM2 = PCLM5 > PCLM4. The HOMO and LUMO energy states of the reference PCLR and newly designed molecules (PCLM1–PCLM5) are demonstrated in Figures 5 and 6. In the present investigation, the Eg values of the reference PCLR, as well as the newly constructed molecules (PCLM1–PCLM5), were as follows: PCLR (2.27 eV) > PCLM2 (1.99 eV) > PCLM4 (1.97 eV) > PCLM1 (1.92 eV) > PCLM3 (1.80 eV) > PCLM5 (1.66 eV). The Eg values of all newly designed molecules (PCLM1–PCLM5) were lower than that of PCLR. Thus, on account of their reduced Eg values, all newly developed structures were characterized as soft, polarizable, and highly reactive molecules.
Figure 5.
Representation of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) at the elected method of DFT/MPW1PW91/6-31G (d,p), of PCLR and PCLM1-–PCLM5.
Figure 6.

FMO representation of PCLR and PCLM1–PCLM5.
The FMO plots of PCLM1–PCLM5 showed a similar pattern of charge density in HOMOs. The charge density in HOMOs was primarily concentrated on the central donor core and π-spacers and minorly on acceptor moieties, with negligible effect on the end-capped acceptors. Similarly, the charge dispersal in LUMOs was majorly concentrated on terminal acceptor moieties and minorly on π-spacers. In case of the HOMO and LUMO of PCLR, the electron density was localized equally on all fragments of the molecule. FMO diagrams of all molecules are demonstrated in Figure 6.
3.3. UV–Visible Absorption Spectral Properties
Examination of UV–visible absorption features helps one to assess the working and light-harvesting efficiency of PV devices. The spectra of UV–visible absorption in all of the scrutinized molecules with MPW1PW91/6-31G in the gaseous phase were analyzed. It was found that in comparison with PCLR, all newly modeled chromophores presented higher values of λmax, which might be because of the influence of delocalized π-electrons on extending the conjugation with most electron-withdrawing acceptor moieties (X1–X5). In the meanwhile, to understand the role of the solvent on the absorption of these molecules, the λmax of all molecules (PCLM1–PCLM5) was evaluated in chloroform (CHCl3). In the presence of the CHCl3 solvent, all newly designed molecules (PCLM1–PCLM5) along with the reference exhibited a red-shift in their absorption bands due to the stabilization of delocalized π-electrons, as shown in Figure 7. The electron-withdrawing ability of acceptor moieties is intrinsically linked to the λmax and indirectly correlated to the bandgap (Eg). The reference (PCLR) indicated a lower λmax as compared with all molecules due to the presence of the least resonance, whereas PCLM5 acquired X5 as an efficient electron-pulling acceptor possessing cyano groups, which resulted in its maximum absorbance. In brief, all of our designed chromophores have peripheral acceptor moieties with a strong electron-withdrawing ability.
Figure 7.

UV–vis spectra of PCLR andPCLM1–PCLM5 at the MPW1PW91/6-31G level (d,p) in the (a) gaseous and (b) solvent (CHCl3) phases.
Tables 3 and 4 display numerous photophysical properties, such as absorption maximum (λmax), oscillator strength (fos), excitation energy (Ex), light-harvesting energy (LHE), and the assignment of all molecules.
Table 3. Computed λmaxcal, Energy of First Excitation (Eopt), Oscillator Strength (fOC), Light-Harvesting Efficiency (LHE), along with the Major Molecular Transitions of PCLR, as well as PCLM1–PCLM5 in the Gaseous Phase.
| molecules | λmaxcal (nm) | λmaxexp (nm) | Eopt (eV) | fOC | LHE | major molecular transitions (HOMO = H, LUMO = L) |
|---|---|---|---|---|---|---|
| PCLR | 642 | 675 | 1.93 | 2.6127 | 0.9975 | 68% |
| PCLM1 | 769 | 1.61 | 3.6541 | 0.9998 | 66% | |
| PCLM2 | 738 | 1.68 | 3.1634 | 0.9993 | 67% | |
| PCLM3 | 824 | 1.50 | 3.4381 | 0.9996 | 65% | |
| PCLM4 | 746 | 1.66 | 3.4726 | 0.9997 | 65% | |
| PCLM5 | 890 | 1.49 | 3.3504 | 0.9995 | 64% |
Table 4. Computed λmaxcal, Energy of First Excitation (Eopt), Oscillator Strength (fOC), Light-Harvesting Efficiency (LHE), as well as the Major Molecular Transitions, of PCLR along with PCLM1–PCLM5 in CHCl3.
| molecules | λmaxcal (nm) | λmaxexp(nm) | Eopt (eV) | fOC | LHE | major molecular transitions (HOMO = H, LUMO = L) |
|---|---|---|---|---|---|---|
| PCLR | 662 | 675 | 1.87 | 2.8808 | 0.9986 | 68% |
| PCLM1 | 796 | 1.56 | 3.9300 | 0.9999 | 65% | |
| PCLM2 | 775 | 1.60 | 3.3894 | 0.9995 | 66% | |
| PCLM3 | 874 | 1.42 | 3.6540 | 0.9998 | 66% | |
| PCLM4 | 777 | 1.59 | 3.7123 | 0.9998 | 65% | |
| PCLM5 | 959 | 1.32 | 3.5487 | 0.9997 | 64% |
Excitation energy (Ex) is another crucial parameter to examine while evaluating photoelectric properties of PV devices. Low Ex values result in higher electron–hole mobilities, more efficient charge migration, and elevated power conversion efficiency (PCE). Tables 3 and 4 illustrate the values of Ex in gaseous and solvent phases. The Ex of the fabricated molecules decreases in the order of PCLR > PCLM2 > PCLM4 > PCLM1 > PCLM3 > PCLM5 both in the gaseous and the solvent medium. All of our newly developed chromophores (PCLM1–PCLM5) indicated lower Ex as compared with PCLR; therefore, they can undergo more excitations from the ground state toward the first excited state and have a higher charge-transport potential, validating their superior photoelectronic features.
The light-harvesting efficiency (LHE) has a critical role in enhancing the efficiency of OSCs. It is important to know the LHE of molecules at a specific wavelength to determine their contribution to photon absorption. The short-circuit current is maximized when the LHE is regulated to a high value, ultimately increasing the efficiency of OSCs.30 The LHE of the reference PCLR and newly designed chromophores (PCLM1–PCLM5) in gaseous as well as CHCl3 solvent phases were estimated through eq 3, and the values are given in Tables 3 and 4.
| 3 |
Here, f illustrates the oscillator strength. From the above equation, it can be observed that a higher value of oscillator strength leads to an elevated value of light-harvesting efficiency. The values of LHE computed for the reference PCLR and newly designed molecules (PCLM1–PCLM5) ranged from 0.9995 to 0.9999, as shown in Table 4. Overall, PCLM1 had the maximum photocurrent response due to the highest LHE value of 0.9999. Graphical representations of the LHE as well as foc are illustrated in Figure 8. The LHE results indicate that the molecules newly constructed by modifying end-capped acceptors are more efficient than the reference PCLR.
Figure 8.

Oscillator strength (foc) and light-harvesting efficiency (LHE) of PCLR and PCLM1–PCLM5.
3.4. Dipole Moment
Dipole moment (μ), a significant aspect, influences the efficiency of OPVs. The molecular solubility in solvents is estimated by the measured electronic density of an organic molecule in a given solvent. The value of μ increases with an increase in the polarity of a molecule, which improves the solubility of any molecule in the respective solvent.31 A molecule’s solubility is intimately associated with a well-ordered geometry of active layer while constructing a solar device. A smooth, thin-film morphology in OSCs leads to efficient charge transfer and enhances its efficiency.27Table 6 reveals the computed μg (gas-phase dipole moment), μs (solvent-phase dipole moment), and Δμ (difference between μg and μs) values for all of the investigated molecules.
Table 6. Role of the Core, Spacer, and Acceptor in Escalating the HOMO/LUMO Energy Levels of PCLR and PCLM1–PCLM5.
| molecules | HOMO = H LUMO = L | core (eV) | spacer (eV) | acceptor (eV) |
|---|---|---|---|---|
| PCLR | H | 23.6 | 36.3 | 40.2 |
| L | 30.1 | 26.2 | 43.7 | |
| PCLM1 | H | 8.1 | 27.9 | 64 |
| L | 31.7 | 25.0 | 43.3 | |
| PCLM2 | H | 11.3 | 34.7 | 54.0 |
| L | 34.5 | 23.9 | 41.5 | |
| PCLM3 | H | 23.6 | 36.3 | 40.2 |
| L | 30.1 | 26.2 | 43.7 | |
| PCLM4 | H | 10.2 | 31.6 | 58.2 |
| L | 29.0 | 24.3 | 46.7 | |
| PCLM5 | H | 5.9 | 9.1 | 85.0 |
| L | 36.9 | 15.6 | 47.5 |
Currently, μ lies in the range of 0.628–8.612 D in the solvent phase. Data presented in Table 5 and Figure 9 illustrate that the μ values of the newly fabricated molecules (PCLM1–PCLM5) are higher than that of the reference PCLR (0.628) in CHCl3. Because of stronger π-interactions in CHCl3 solvents, an increase in the μ value was observed for all molecules, which clarifies their higher solubility in the CHCl3 solvent. PCLM5 displayed the highest μ value due to the enhanced polarity and excellent solubility of the X5 acceptor moiety in the CHCl3 solvent. The dipole moment follows an increasing trend of PCLR < PCLM1 < PCLM4 < PCLM2 < PCLM3 < PCLM5 both in the gaseous phase and in the chloroform solvent. Furthermore, the difference between μg and μs, i.e., Δμ, suggests that PCLM5 has a greater shift of μ in the CHCl3 solvent. Hence the newly fabricated molecules (PCLM1–PCLM5) can be used as efficient molecules and solution-processable materials in designing OSCs for their strong optical results.
Table 5. Theoretically Computed Dipole Moments of PCLR and PCLM1–PCLM5 in the Gaseous (μg) and Excited (μs) States, along with Their Difference (Δμ).
| molecules | μg (D) | μs (D) | Δμ (D) |
|---|---|---|---|
| PCLR | 0.619 | 0.628 | 0.009 |
| PCLM1 | 1.683 | 1.934 | 0.251 |
| PCLM2 | 4.530 | 5.368 | 0.838 |
| PCLM3 | 5.754 | 6.395 | 0.641 |
| PCLM4 | 2.563 | 3.340 | 0.777 |
| PCLM5 | 6.874 | 7.865 | 0.991 |
Figure 9.

Graphical description of dipole moments at the ground (μg) and excited (μs) states and their difference (Δμ) for PCLR and PCLM1–PCLM5.
3.5. Density of States (DOS)
In addition, the Mulliken’s charge distribution through the DOS is assessed for PCLR and the newly constructed moieties PCLM1–PCLM5 at the MPW1PW91/6-31G** level.32 The DOS graphs of the newly fabricated moieties PCLM1–PCLM5 were compared with that of the reference PCLR to analyze the endowment of fragments to the maximum absorption and charge density. In the DOS graphs, the negative values on the x-axis showed the HOMO energy level and positive values showed LUMO energy levels, whereas the relative intensity of charge density was placed along the y-axis. For their DOS analysis, the newly fabricated chromophores (PCLM1–PCLM5) were allocated into three portions, namely, donors (blue), π-spacers (green), and acceptors (red). Table 6 displays the quantitative role of different fragments in forming the HOMOs and LUMOs. The density of states (DOS) graphs showed the equal and significant role of all three fragments in shifting the charge density from the HOMO to the LUMO.
The distribution pattern of all molecules is quite analogous. HOMOs are formed particularly by the contribution of spacer and acceptor fragments and slightly by the central core. The spacer made almost equal contributions to the HOMO and LUMO. Likewise, LUMOs are formed by the equal contribution of all fragments (core, spacer, and acceptor), as displayed in Figure 10.
Figure 10.

DOS graphs of the reference (PCLR) and designed chromophores (PCLM1–PCLM5).
3.6. Binding Energy (Eb) and Transition Density Matrix (TDM) Analysis
The binding energy (Eb) of excitons is another essential property for the interpretation of optical characteristics. The least amount of energy needed for the separation of excitons into free electrons and holes is defined by Eb.33 The exciton Eb is calculated from eq 4.
| 4 |
where, Eb constitutes the binding energy of the exciton, EL-H represents the bandgap between the evaluated FMOs, and Ex denotes the excitation energy for these FMOs.
The exciton dissociation is more advantageous if the Eb is lower, resulting in a higher current charge density.34Figure 11 demonstrates that all of our newly designed molecules showed inferior Eb values compared with that of PCLR, which indicates the presence of weak Coulombic interactions and easy separation of the bound hole–electron pairs.
Figure 11.

Graphical illustration of computed binding energy (Eb) values of PCLR and PCLM1–PCLM5.
The Eb values of the reference PCLR and the newly fabricated molecules (PCLM1–PCLM5) in CHCl3 were 0.4, 0.36, 0.39, 0.38, 0.37, and 0.34 eV, respectively, as indicated in Table 7. Among all molecules, PCLM1 showed the smallest Eb value, which implied its efficient charge-generation properties.
Table 7. Theoretically Computed Values of Binding Energy (Eb) from the Energy Gap (EL-H) between the FMOs and the Excitation Energy (Eopt) of PCLR and PCLM1–PCLM5.
| molecules | EL-H (eV) | Eopt (eV) | Eb (eV) |
|---|---|---|---|
| PCLR | 2.27 | 1.87 | 0.40 |
| PCLM1 | 1.92 | 1.56 | 0.36 |
| PCLM2 | 1.99 | 1.60 | 0.39 |
| PCLM3 | 1.80 | 1.42 | 0.38 |
| PCLM4 | 1.97 | 1.60 | 0.37 |
| PCLM5 | 1.66 | 1.32 | 0.34 |
Transition density matrix (TDM) analysis is another effective method for investigating electronic excitation processes in real time (including the creation and dispersion of excitons, recombination, and separation of charges).35 As a result of excitation measurements, the TDM may be used to investigate and understand the quantum geometry. The interaction between acceptor and donor moieties in the excited state is shown by TDM plots.36 The TDM graphs of the reference PCLR and the newly constructed molecules (PCLM1–PCLM5) are demonstrated in Figure 12. For the TDM analysis, all the molecules were sectioned into three fragments, namely, the donor (D), π-spacer (B), and acceptor (A) moieties. In the TDM plots, the number of atoms partaking are represented by lower abscissa and ordinates on the left side, whereas the right ordinates indicate the flow of electron density.
Figure 12.

TDM plots of the reference (PCLR) and the designed chromophores (PCLM1–PCLM5).
Since hydrogen atoms have minimal influence on the transitions, they have been ignored in the TDM analysis.37 It can be understood from the TDM graph analysis of PCLR, PCLM1, PCLM3, and PCLM4 that the electron density is shifted from D to A via B of the DPP acceptor uniformly. The bright portion moving diagonally in the TDM graph shows the electron density. The examination of the TDM graphs of PCLM2 and PCLM5 showed that the molecule has a uniform diagonal electron density as well as off-diagonal charge transfer from D to A moieties. The brighter portion is more concentrated on A moieties, indicating the excellent charge transfer from D to A via π-conjugation. Hence, it is summarized that the newly fabricated molecules exhibit efficient charge-transmission and charge-dissociation properties.
3.7. Reorganization Energy (RE)
Reorganization energy (RE) is an excellent analytical scheme for designing efficient OSCs and assessing the operating efficiency of OSCs. Electron and hole mobilities in a molecule during excitation are described by this energy.38 Electrons and holes must be separated as they have different charges. The charge transferability shows an inverse relation with RE. When a molecule’s charge mobility is low, it migrates toward the respective electrode readily. The electron and hole mobilities are statistically determined by eqs 1 and 2. The results of RE in terms of electron and hole mobilities of the reference PCLR and the five newly fabricated molecules (PCLM1–PCLM5) are reviewed in Table 8 and a pictorial representation is given in Figure 13.
Table 8. Calculated Hole (λh) and Electron Mobilities (λe) of PCLR and PCLM1–PCLM5.
| molecules | λe (eV) | λh (eV) |
|---|---|---|
| PCLR | 0.00552 | 0.00623 |
| PCLM1 | 0.00428 | 0.00497 |
| PCLM2 | 0.00292 | 0.00550 |
| PCLM3 | 0.00516 | 0.00523 |
| PCLM4 | 0.00561 | 0.00561 |
| PCLM5 | 0.00367 | 0.00497 |
Figure 13.

Plot of hole (λh) and electron (λe) mobilities for PCLR and PCLM1–PCLM5.
After a careful evaluation, it is noted that the newly engineered molecules PCLM1 (0.00428 eV), PCLM2 (0.00292 eV), PCLM3 (0.00516 eV), and PCLM5 (0.00367 eV) have the least values of electron RE (λe) as compared with the reference PCLR (0.00552 eV), hence offering a better electron mobility rate. Also, all of our newly designed molecules have low values of hole RE (λh) compared with the reference PCLR, making them suitable candidates for hole-transport materials. The decreasing order of λh is PCLR > PCLM4 > PCLM2 > PCLM3 > PCLM2 > PCLM1.
3.8. Electrostatic Potential (ESP) Analysis
ESP is also crucial in predicting the relationship between the molecular structure, molecular interaction, and photophysical properties of molecules.39 The ESP simulations performed at the selected functional determines the regions of nucleophiles and electrophiles in the researched molecule. The green color in ESP diagrams denotes the electrically neutral region, the red color shows the electrophilic regions having an abundance of electrons, and finally the blue color is concentrated on the nucleophilic regions where there is a positive electrostatic potential and lack of electrons.38,40
ESP analysis of all doped complexes (PCLM1–PCLM5) along with PCLR was performed theoretically at the MPW1PW91/6-31G** functional level. Figure 14 shows colored ESP maps of the PCLR and the freshly designed molecules (PCLM1–PCLM5). ESP surfaces of all of these molecules illustrate that the red color (negative potential) is concentrated on the acceptor regions of molecules, making them more electrophilic. The region where the donor exists in each molecular framework possesses a blue color (positive potential) and behaves as a nucleophile.
Figure 14.

ESP depiction of PCLR and PCLM1–PCLM5.
3.9. Open-Circuit Voltage (VOC)
Another credible approach to evaluate the OSC’s operating efficiency is the open-circuit voltage (VOC). VOC is basically that maximum amount of current, which is produced when the externally applied voltage is zero.41 Variables such as the donor’s HOMO and acceptor’s LUMO are scaled up. VOC is estimated with the selected MPW1PW91 functional. The mathematical expression used for computing VOC is expressed by eq 5.
| 5 |
Here, VOC is open-circuit voltage (VOC), e is the charge of the molecules in this study, and 0.3 is the empirical factor.
The designed chromophores (PCLM1–PCLM5) act as promising donors, and so their HOMOs were cross-examined with the LUMO of PC61BM. PC61BM is a widely used acceptor. In the present quantum chemical investigation, PC61BM is geometrically optimized at the MPW1PW91 functional level. To obtain a higher VOC value, it should be taken into account that the donor’s HOMO has to be at a higher level and the LUMO of acceptor at a low level of energy. Figure 15 depicts the VOC values of the reference PCLR and the newly investigated chromophores (PCLM1–PCLM5) along with the acceptor PC61BM.
Figure 15.

Pictorial view of the open-circuit voltage (VOC) of PCLR and all of the newly modeled chromophores (PCLM1–PCLM5).
The VOC value of the reference PCLR computed when blended with PC61BM was 1.11 eV. VOC values of the newly fabricated chromophores (PCLM1–PCLM5) were 1.40, 1.45, 1.52, 1.30, and 1.74 eV, respectively. All of our newly constructed chromophores showed a pronounced shift in VOC values when compared with PCLR. It can be predicted that all designed chromophores can contribute to improving the device performance owing to their better charge-conduction properties.
3.10. Fill Factor (FF) and Power Conversion Efficiency (PCE)
The fill factor (FF) is one the most prominent determining element and integral parameter to calculate the PCE of OSCs. VOC of the donor–acceptor interface has a significant influence on FF.42 FF is mathematically computed using eq 6 expressed below:
 |
6 |
In this equation, 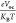 illustrates the normalized
open-circuit
voltage, e represent the fixed charge on each molecule
(taken to be 1), KB represents the Boltzmann
constant, and T is the temperature in Kelvin. Calculated
values for normalized VOC, along with
the FF, are given in Table 9. All newly designed chromophores (PCLM1–PCLM5) have revealed a higher value of FF than PCLR. The greater
FF supports the efficacy of the newly designed structures for future
high-potential solar devices.
illustrates the normalized
open-circuit
voltage, e represent the fixed charge on each molecule
(taken to be 1), KB represents the Boltzmann
constant, and T is the temperature in Kelvin. Calculated
values for normalized VOC, along with
the FF, are given in Table 9. All newly designed chromophores (PCLM1–PCLM5) have revealed a higher value of FF than PCLR. The greater
FF supports the efficacy of the newly designed structures for future
high-potential solar devices.
Table 9. Computed Open-Circuit Voltage (VOC), Normalized VOC, Fill Factor (FF), and Power Conversion Efficiency (PCE) of all Studied Molecules (PCLR and PCLM1–PCLM5).
| molecules | VOC (eV) | normalized VOC (eV) | FF | Jsc (mA cm–2) (this assumed short circuit current was taken from reference paper) | PCE (%) |
|---|---|---|---|---|---|
| PCLR | 1.11 | 43.224 | 0.8918 | 9.7 | 9.603 |
| PCLM1 | 1.40 | 54.517 | 0.9097 | 9.7 | 12.35 |
| PCLM2 | 1.45 | 56.464 | 0.9122 | 9.7 | 12.83 |
| PCLM3 | 1.52 | 58.796 | 0.9149 | 9.7 | 13.49 |
| PCLM4 | 1.30 | 50.623 | 0.9043 | 9.7 | 11.40 |
| PCLM5 | 1.74 | 67.306 | 0.9235 | 9.7 | 15.59 |
The PCE value for a device helps one to predict its working accuracy and mechanism.43 PCE relies on a few essential parameters, which are given in eq 7.
| 7 |
In the above equation, Pin represents an empirical constant, which depicts the power of the incident radiation that strikes the surface of the cell. The remaining quantities, i.e., the short-circuit current (Jsc), VOC, and FF, have an association with the PCE value. Computed PCE values of the studied chromophores are displayed in Table 9. An incremental effect is observed in the PCE for all novelly designed molecules (PCLM1–PCLM5) in comparison with that of the reference molecule (PCLR, 9.603%).
4. Conclusions
To summarize the above results, five pentacyclic aromatic bislactam (PCL)-based small donor groups (PCLM1–PCLM5) were fabricated from the reference PCLR, and their optoelectronic properties were computed at the MPW1PW91/6-31G** level. The newly modeled chromophores (PCLM1–PCLM5) showed satisfactory and competent results from DFT and TD-DFT computations. All of our novel molecules exhibited a lower bandgap and a red-shifted λmax compared with the reference PCLR due to their strong electron-pulling moieties. The newly designed chromophores PCLM1 (796 nm), PCLM2 (775 nm), PCLM3 (874 nm), PCLM4 (777 nm), and PCLM5 (959 nm) displayed higher λmax values than PCLR (662 nm) in a CHCl3 solvent. The bandgaps of the devised molecules ranged from 1.66 to 1.99 eV, whereas that of PCLR was 2.27 eV. Likewise, the binding energies of all molecules were reduced in comparison with that of PCLR. An increase in the dipole moment was seen for all molecules in the CHCl3 solvent, as opposed to the gaseous phase, resulting in better solubility and charge recombination. Moreover, the minimal λe and λh values of all of these newly designed molecules represent their higher electron and hole mobility rates, sequentially. The evaluation of the FMOs disclosed the uniform dispersal of charge density in the HOMOs and LUMOs of the newly fabricated molecules. According to a TDM analysis, the bound electron–hole pairs of the newly designed donor molecules (PCLM1–PCLM5) can be easily dissociated into electrons and holes because of their lower interaction coefficient. Furthermore, when scaled with a PC61BM acceptor, all of the newly proposed molecules exhibited greater VOC values than PCLR, which implies that these donor molecules play an important role in increasing the operational efficiency. Therefore, in this assay, highly productive PCE values in the range of 11.4–15.59% were estimated for the novelly designed molecules based on the normalized Voc and FF, which predict an increase in their photovoltaic efficiency compared with that of the reference PCLR (9.603%). It has been summarized that structural modification is an effective strategy to construct new organic materials for designing OSCs in the near future.
Acknowledgments
The authors acknowledge the financial and technical support from the Punjab Bio-energy Institute (PBI), University of Agriculture, Faisalabad (UAF), Pakistan. The authors gratefully acknowledge the financial support provided from the Taif University Researchers Supporting Project number TURSP-2020/ 106, Taif University, Taif, Saudi Arabia. We are also thankful to Dr. Khurshid Ayub, Department of Chemistry, COMSATS University Islamabad, Abbottabad Campus, for additional resources.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00001.
Cartesian coordinates of internally optimized geometries of all molecules (reference PCLR, PCLM1, PCLM2, PCLM3, PCLM4, and PCLM5) along the X, Y, and Z axes at the MPW1PW91/6-31G (d, p) level of the density functional theory (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Rahman M. M.; Velayutham E. Renewable and non-renewable energy consumption-economic growth nexus: new evidence from South Asia. Renewable Energy 2020, 147, 399–408. 10.1016/j.renene.2019.09.007. [DOI] [Google Scholar]
- a Sun J.; Zhang Z.; Yin X.; Zhou J.; Yang L.; Geng R.; Zhang F.; Zhu R.; Yu J.; Tang W. High performance non-fullerene polymer solar cells based on PTB7-Th as the electron donor with 10.42% efficiency. J. Mater. Chem. A 2018, 6, 2549–2554. 10.1039/C7TA10391C. [DOI] [Google Scholar]; b Gong J.; Sumathy K.; Qiao Q.; Zhou Z. Review on dye-sensitized solar cells (DSSCs): Advanced techniques and research trends. Renewable Sustainable Energy Rev. 2017, 68, 234–246. 10.1016/j.rser.2016.09.097. [DOI] [Google Scholar]
- Yao J.; Zheng X.; Xu J.; Wu K. In Cross-Technology Communication through Symbol-Level Energy Modulation for Commercial Wireless Networks 2020, IEEE International Conference on Pervasive Computing and Communications (PerCom); IEEE, 2020; pp 1–10.
- a Mujahid M.; Chen C.; Hu W.; Wang Z.-K.; Duan Y. Progress of high-throughput and low-cost flexible perovskite solar cells. Sol. RRL 2020, 4, 1900556 10.1002/solr.201900556. [DOI] [Google Scholar]; b Iqbal J.; Enevold J.; Larsen C.; Wang J.; Revoju S.; Barzegar H. R.; Wågberg T.; Eliasson B.; Edman L. An arylene-vinylene based donor-acceptor-donor small molecule for the donor compound in high-voltage organic solar cells. Sol. Energy Mater. Sol. Cells 2016, 155, 348–355. 10.1016/j.solmat.2016.06.018. [DOI] [Google Scholar]
- Riede M.; Spoltore D.; Leo K. Organic solar cells—the path to commercial success. Adv. Energy Mater. 2021, 11, 2002653 10.1002/aenm.202002653. [DOI] [Google Scholar]
- a Zhang G.; Zhao J.; Chow P. C.; Jiang K.; Zhang J.; Zhu Z.; Zhang J.; Huang F.; Yan H. Nonfullerene acceptor molecules for bulk heterojunction organic solar cells. Chem. Rev. 2018, 118, 3447–3507. 10.1021/acs.chemrev.7b00535. [DOI] [PubMed] [Google Scholar]; b Rafique S.; Abdullah S. M.; Sulaiman K.; Iwamoto M. Fundamentals of bulk heterojunction organic solar cells: An overview of stability/degradation issues and strategies for improvement. Renewable Sustainable Energy Rev. 2018, 84, 43–53. 10.1016/j.rser.2017.12.008. [DOI] [Google Scholar]
- Schlachter A.; Marineau-Plante G.; Harvey P. D.; Agrawal A.; Sharma G. D. Efficient ternary bulk heterojunction organic solar cells using a low-cost nonfullerene acceptor. J. Mater. Chem. C 2022, 10, 4372–4382. 10.1039/D1TC05653K. [DOI] [Google Scholar]
- a Liu G.; Jia J.; Zhang K.; Jia Xe.; Yin Q.; Zhong W.; Li L.; Huang F.; Cao Y. 15% efficiency tandem organic solar cell based on a novel highly efficient wide-bandgap nonfullerene acceptor with low energy loss. Adv. Energy Mater. 2019, 9, 1803657 10.1002/aenm.201803657. [DOI] [Google Scholar]; b Ostroverkhova O.Handbook of organic materials for optical and (opto) electronic devices: properties and applications, Elsevier, 2013. [Google Scholar]
- a Nielsen C. B.; Holliday S.; Chen H.-Y.; Cryer S. J.; McCulloch I. Non-fullerene electron acceptors for use in organic solar cells. Acc. Chem. Res. 2015, 48, 2803–2812. 10.1021/acs.accounts.5b00199. [DOI] [PMC free article] [PubMed] [Google Scholar]; b McAfee S. M.; Topple J. M.; Hill I. G.; Welch G. C. Key components to the recent performance increases of solution processed non-fullerene small molecule acceptors. J. Mater. Chem. A 2015, 3, 16393–16408. 10.1039/C5TA04310G. [DOI] [Google Scholar]; c Lin Y.; Zhan X. Oligomer molecules for efficient organic photovoltaics. Acc. Chem. Res. 2016, 49, 175–183. 10.1021/acs.accounts.5b00363. [DOI] [PubMed] [Google Scholar]
- a Cui Y.; Yao H.; Zhang J.; Xian K.; Zhang T.; Hong L.; Wang Y.; Xu Y.; Ma K.; An C.; et al. Single-junction organic photovoltaic cells with approaching 18% efficiency. Adv. Mater. 2020, 32, 1908205 10.1002/adma.201908205. [DOI] [PubMed] [Google Scholar]; b Liu T.; Ma R.; Luo Z.; Guo Y.; Zhang G.; Xiao Y.; Yang T.; Chen Y.; Li G.; Yi Y.; et al. Concurrent improvement in J sc and V oc in high-efficiency ternary organic solar cells enabled by a red-absorbing small-molecule acceptor with a high LUMO level. Energy Environ. Sci. 2020, 13, 2115–2123. 10.1039/D0EE00662A. [DOI] [Google Scholar]; c Ans M.; Paramasivam M.; Ayub K.; Ludwig R.; Zahid M.; Xiao X.; Iqbal J. Designing alkoxy-induced based high performance near infrared sensitive small molecule acceptors for organic solar cells. J. Mol. Liq. 2020, 305, 112829 10.1016/j.molliq.2020.112829. [DOI] [Google Scholar]; d Ma R.; Liu T.; Luo Z.; Gao K.; Chen K.; Zhang G.; Gao W.; Xiao Y.; Lau T.-K.; Fan Q.; et al. Adding a third component with reduced miscibility and higher LUMO level enables efficient ternary organic solar cells. ACS Energy Lett. 2020, 5, 2711–2720. 10.1021/acsenergylett.0c01364. [DOI] [Google Scholar]; e Wang X.; Chen H.; Ran A.-R.; Luo L.; Chan P. P.; Tham C. C.; Chang R. T.; Mannil S. S.; Cheung C. Y.; Heng P.-A. Towards multi-center glaucoma OCT image screening with semi-supervised joint structure and function multi-task learning. Med. Image Anal. 2020, 63, 101695 10.1016/j.media.2020.101695. [DOI] [PubMed] [Google Scholar]
- Zhao W.; Li S.; Yao H.; Zhang S.; Zhang Y.; Yang B.; Hou J. Molecular optimization enables over 13% efficiency in organic solar cells. J. Am. Chem. Soc. 2017, 139, 7148–7151. 10.1021/jacs.7b02677. [DOI] [PubMed] [Google Scholar]
- a Cao Y.; Guo Z.-H.; Chen Z.-Y.; Yuan J.-S.; Dou J.-H.; Zheng Y.-Q.; Wang J.-Y.; Pei J. Pentacyclic aromatic bislactam-based conjugated polymers: constructed by Beckmann rearrangement and application in organic field-effect transistor. Polym. Chem. 2014, 5, 5369–5374. 10.1039/C4PY00272E. [DOI] [Google Scholar]; b Truong N. T. T.; Nguyen L. T.; Mai H. L. T.; Doan B. K.; Tran D. H.; Truong K. T.; Nguyen V. Q.; Nguyen L.-T. T.; Hoang M. H.; Van Pham T.; et al. Phenothiazine derivatives, diketopyrrolopyrrole-based conjugated polymers: synthesis, optical and organic field effect transistor properties. J. Polym. Res. 2020, 27, 223 10.1007/s10965-020-02199-x. [DOI] [Google Scholar]
- a Chen Z.; Huang J.; Zhang W.; Zhou Y.; Wei X.; Wei J.; Zheng Y.; Wang L.; Yu G. Tunable charge-transport polarity in thienothiophene–bisoxoindolinylidene-benzodifurandione copolymers for high-performance field-effect transistors. J. Mater. Chem. C 2022, 10, 2671–2680. 10.1039/D1TC02833B. [DOI] [Google Scholar]; b Tam T. L. D.; Handoko A. D.; Lin T. T.; Xu J. Electron n-doping of a highly electron-deficient chlorinated benzodifurandione-based oligophenylene vinylene polymer using benzyl viologen radical cations. Mater. Chem. Front. 2021, 5, 6182–6191. 10.1039/D1QM00695A. [DOI] [Google Scholar]
- Zhou J.; Zuo Y.; Wan X.; Long G.; Zhang Q.; Ni W.; Liu Y.; Li Z.; He G.; Li C.; et al. Solution-processed and high-performance organic solar cells using small molecules with a benzodithiophene unit. J. Am. Chem. Soc. 2013, 135, 8484–8487. 10.1021/ja403318y. [DOI] [PubMed] [Google Scholar]
- a Hong W.; Sun B.; Aziz H.; Park W.-T.; Noh Y.-Y.; Li Y. A conjugated polyazine containing diketopyrrolopyrrole for ambipolar organic thin film transistors. Chem. Commun. 2012, 48, 8413–8415. 10.1039/c2cc33998f. [DOI] [PubMed] [Google Scholar]; b Sonar P.; Zhuo J.-M.; Zhao L.-H.; Lim K.-M.; Chen J.; Rondinone A. J.; Singh S. P.; Chua L.-L.; Ho P. K.; Dodabalapur A. Furan substituted diketopyrrolopyrrole and thienylenevinylene based low band gap copolymer for high mobility organic thin film transistors. J. Mater. Chem. 2012, 22, 17284–17292. 10.1039/c2jm32376a. [DOI] [Google Scholar]
- Feng G.; Xu Y.; Zhang J.; Wang Z.; Zhou Y.; Li Y.; Wei Z.; Li C.; Li W. All-small-molecule organic solar cells based on an electron donor incorporating binary electron-deficient units. J. Mater. Chem. A 2016, 4, 6056–6063. 10.1039/C5TA10430K. [DOI] [Google Scholar]
- Adamo C.; Barone V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The m PW and m PW1PW models. J. Chem. Phys. 1998, 108, 664–675. 10.1063/1.475428. [DOI] [Google Scholar]
- Civalleri B.; Zicovich-Wilson C. M.; Valenzano L.; Ugliengo P. B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm 2008, 10, 405–410. 10.1039/B715018K. [DOI] [Google Scholar]
- Chai J.-D.; Head-Gordon M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Yanai T.; Tew D. P.; Handy N. C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. 10.1016/j.cplett.2004.06.011. [DOI] [Google Scholar]
- Bjorgaard J. A.; Velizhanin K. A.; Tretiak S. Solvent effects in time-dependent self-consistent field methods. II. Variational formulations and analytical gradients. J. Chem. Phys. 2015, 143, 054305 10.1063/1.4927167. [DOI] [PubMed] [Google Scholar]
- Gorelsky S.SWizard program; University of Ottawa: Ottawa, Canada, 2010. [Google Scholar]
- Deschenes L. A.; David A.. Vanden BoutUniversity of Texas, A. Origin 6.0: Scientific Data Analysis and Graphing Software Origin Lab Corporation (formerly Microcal Software, Inc.), ACS Publications, Web site: www.originlab.com, Commercial price: 595.Academicprice: 446, 2000.
- Tenderholt A. L.; Szilagyi R. K.; Holm R. H.; Hodgson K. O.; Hedman B.; Solomon E. I. Electronic Control of the “Bailar Twist” in Formally d0-d2 Molybdenum Tris (dithiolene) Complexes: A Sulfur K-edge X-ray Absorption Spectroscopy and Density Functional Theory Study. Inorg. Chem. 2008, 47, 6382–6392. 10.1021/ic800494h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T.; Chen F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- Shehzad R. A.; Muhammad S.; Iqbal J.; Al-Sehemi A. G.; Yaseen M.; Aloui Z.; Khalid M. Exploring the optoelectronic and third-order nonlinear optical susceptibility of cross-shaped molecules: insights from molecule to material level. J. Mol. Model. 2021, 27, 12 10.1007/s00894-020-04619-7. [DOI] [PubMed] [Google Scholar]
- Rasool A.; Zahid S.; Shehzad R. A.; Akhter M. S.; Iqbal J. Designing of benzodithiophene (BDT) based non-fullerene small molecules with favorable optoelectronic properties for proficient organic solar cells. Comput. Theor. Chem. 2021, 1203, 113359 10.1016/j.comptc.2021.113359. [DOI] [Google Scholar]
- a Jilani F.; Iqbal J.; Shahid I.; Yaseen M.; Mahr M. S.; Khalid M.; Ayub K. Rational design of naphthalimide based small molecules non-fullerene acceptors for organic solar cells. Comput. Theor. Chem. 2020, 1187, 112916 10.1016/j.comptc.2020.112916. [DOI] [Google Scholar]; b Asif T.; Khera R. A.; Naveed A.; Salim M.; Iqbal J. Tuning the optoelectronic properties of naphthodithiophene (NDT) for designing of ADA type photovoltaic materials. Optik 2021, 247, 167892 10.1016/j.ijleo.2021.167892. [DOI] [Google Scholar]
- Zahid S.; Rasool A.; Shehzad R. A.; Bhatti I. A.; Iqbal J. Tuning the optoelectronic properties of triphenylamine (TPA) based small molecules by modifying central core for photovoltaic applications. J. Mol. Model. 2021, 27, 237 10.1007/s00894-021-04867-1. [DOI] [PubMed] [Google Scholar]
- Khan M. U.; Khalid M.; Ibrahim M.; Braga A. A. C.; Safdar M.; Al-Saadi A. A.; Janjua M. R. S. A. First theoretical framework of triphenylamine–dicyanovinylene-based nonlinear optical dyes: structural modification of π-linkers. J. Phys. Chem. C 2018, 122, 4009–4018. 10.1021/acs.jpcc.7b12293. [DOI] [Google Scholar]
- Zahid S.; Rasool A.; Ayub A. R.; Ayub K.; Iqbal J.; Al-Buriahi M. S.; Alwadai N.; Somaily H. H. Silver cluster doped graphyne (GY) with outstanding non-linear optical properties. RSC Adv. 2022, 12, 5466–5482. 10.1039/D1RA08117A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fonseca Guerra C.; Handgraaf J. W.; Baerends E. J.; Bickelhaupt F. M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. 10.1002/jcc.10351. [DOI] [PubMed] [Google Scholar]; b Naeem M.; Jabeen S.; Khera R. A.; Mubashar U.; Iqbal J. Tuning of optoelectronic properties of triphenylamines-based donor materials for organic solar cells. J. Theor. Comput. Chem. 2019, 18, 1950036 10.1142/S0219633619500366. [DOI] [Google Scholar]
- Clarke T. M.; Durrant J. R. Charge photogeneration in organic solar cells. Chem. Rev. 2010, 110, 6736–6767. 10.1021/cr900271s. [DOI] [PubMed] [Google Scholar]
- Köse M. E. Evaluation of acceptor strength in thiophene coupled donor–acceptor chromophores for optimal design of organic photovoltaic materials. J. Phys. Chem. A 2012, 116, 12503–12509. 10.1021/jp309950f. [DOI] [PubMed] [Google Scholar]
- Naveed A.; Khera R. A.; Azeem U.; Zubair I.; Farhat A.; Ayub A. R.; Iqbal J. Tuning the optoelectronic properties of benzodithiophene based donor materials and their photovoltaic applications. Mater. Sci. Semicond. Process. 2022, 137, 106150 10.1016/j.mssp.2021.106150. [DOI] [Google Scholar]
- Farhat A.; Khera R. A.; Iqbal S.; Iqbal J. Tuning the optoelectronic properties of Subphthalocyanine (SubPc) derivatives for photovoltaic applications. Opt. Mater. 2020, 107, 110154 10.1016/j.optmat.2020.110154. [DOI] [Google Scholar]
- Sabir S.; Khera R. A.; Jabeen S.; Shafiq Z.; Musawwir A.; Iqbal J. Tuning the optoelectronic properties of Benzo Thiophene (BT-CIC) based non-fullerene acceptor organic solar cell. J. Theor. Comput. Chem. 2020, 19, 2050003 10.1142/S0219633620500030. [DOI] [Google Scholar]
- Zahid S.; Rasool A.; Ans M.; Yaseen M.; Iqbal J. Quantum Chemical Approach of Donor– π–Acceptor Based Arylborane–Arylamine Macrocycles with Outstanding Photovoltaic Properties Toward High-Performance Organic Solar Cells. Energy Fuels 2021, 35, 15018–15032. 10.1021/acs.energyfuels.1c02260. [DOI] [Google Scholar]
- Drissi M.; Benhalima N.; Megrouss Y.; Rachida R.; Chouaih A.; Hamzaoui F. Theoretical and experimental electrostatic potential around the m-nitrophenol molecule. Molecules 2015, 20, 4042–4054. 10.3390/molecules20034042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahid S.; Rasool A.; Ans M.; Akhter M. S.; Iqbal J.; Al-Buriahi M.; Alomairy S.; Alrowaili Z. Environmentally compatible and highly improved hole transport materials (HTMs) based on benzotrithiophene (BTT) skeleton for perovskite as well as narrow bandgap donors for organic solar cells. Sol. Energy 2022, 231, 793–808. 10.1016/j.solener.2021.12.010. [DOI] [Google Scholar]
- Rasool A.; Zahid S.; Ans M.; Muhammad S.; Ayub K.; Iqbal J. Bithieno Thiophene-Based Small Molecules for Application as Donor Materials for Organic Solar Cells and Hole Transport Materials for Perovskite Solar Cells. ACS Omega 2022, 7, 844–862. 10.1021/acsomega.1c05504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yaqoob U.; Ayub A. R.; Rafiq S.; Khalid M.; El-Badry Y. A.; El-Bahy Z. M.; Iqbal J. Structural, optical and photovoltaic properties of unfused Non-Fullerene acceptors for efficient solution processable organic solar cell (Estimated PCE greater than 12.4%): A DFT approach. J. Mol. Liq. 2021, 341, 117428 10.1016/j.molliq.2021.117428. [DOI] [Google Scholar]; b Jao M.-H.; Liao H.-C.; Su W.-F. Achieving a high fill factor for organic solar cells. J. Mater. Chem. A 2016, 4, 5784–5801. 10.1039/C6TA00126B. [DOI] [Google Scholar]
- Chen J. D.; Cui C.; Li Y. Q.; Zhou L.; Ou Q. D.; Li C.; Li Y.; Tang J. X. Single-junction polymer solar cells exceeding 10% power conversion efficiency. Adv. Mater. 2015, 27, 1035–1041. 10.1002/adma.201404535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.