

Abstract

A facile and efficient method has been developed for the synthesis of quinoline-fused fluorescent dihydro/spiro-quinazolinones. A plausible mechanism involving an acid-mediated enaminone intermediate is provided. The reaction proceeded using p-toluene sulfonic acid as a green promoter. The methodology was successful in synthesizing various quinoline-appended spiro-quinazolinones 4a–o. The synthetic utility of compounds 4a–o was demonstrated by synthesizing compounds 6a–d via Suzuki coupling as a key reaction. Significantly, the π–π* electronic transition of compounds 4c and 4k showed a blue shift. The molar extinction coefficient (ε), Stoke’s shift (Δu̅), and quantum yield (Φf)c were calculated for these derivatives (4c and 4k).
Introduction
The chemistry of quinoline scaffolds is well documented. Owing to their biological properties, it leads to great interest among medicinal chemists in the development of drug candidates. Various classical methods such as Skraup, Doebner-von Miller, Friedländer, Pfitzinger, Conrad-Limpach, and Combes synthesis are known for a quinoline ring system.1 Later, due to the importance of the quinoline backbone, a number of new methods have been developed by employing both transition metals and metal-free conditions such as CuCN, LiCl3, RuCl3,2 Yb(OTf)3,3 tungsten vinylidene complex,4 BF3OEt2,5,6 benzotriazoleiminium salts, etc.7 Notably, cabozantinib, and bosutinib are a few FDA-approved marketed anticancer drugs containing a quinoline moiety8 and a quinazolinone skeleton present in many drugs and natural products such as bouchardatine, rutaecarpine, etc. (Figure 1).9 Owing to the diverse range of pharmacological activities, various methods have been developed using copper,10,11 iridium,12 manganese,13 silver,14 vanadium,15 cyanuric chloride,16 cationic Amberlyst-15 resin,17 clays,18p-TSA,19 starch sulfate,20 and TFA21 for the synthesis of quinazolinone derivatives. Indeed, the nitrogen-containing heterocyclic compounds play a significant role in biological activities such as chorismate mutase inhibitors, IRAP inhibitors, etc. (Figure 1).22
Figure 1.

Biologically active molecules having quinoline and quinazolinone cores.
Recently, molecular hybridization has been developed as a tool in the development of hybrid analogues with enhanced potency by combining two or more pharmacophores of bioactive scaffolds. The molecular hybridization of various biologically active pharmacophores resulted in lead compounds with multifaceted biological activity wherein specific as well as multiple targets were involved.23 Thus, we were interested to develop molecular hybridization having quinoline and quinazolinone cores. A few reports are available for the synthesis of quinoline-fused quinazolines. One such example is Luotonin A (Figure 1), a pyrroloquinazolinoquinoline alkaloid extracted from the Chinese medicinal plant Peganum nigellastrum. Nevertheless, although the mechanism is unknown, Luotonin A is cytotoxic toward the murine leukemia P-388 cell line (IC50 1.8 μg/mL).24
To minimize waste and reaction time, “step-economic” and “pot-economic” syntheses have emerged as efficient and sustainable reaction protocols. Since the beginning, one-pot reactions have grown in two directions, namely, multiple orthogonal, irreversible steps are combined, while in the second case, multiple reversible steps are coupled to one irreversible step using an enzymatic catalyst.25 To the best of our knowledge, the synthesis of quinoline-appended quinazolinone in a one-pot manner is reported via thermal electrocyclization of aldimine26 (Scheme 1, equation 1) and the synthesis of 2-hetero-substituted 2,3-dihydroquinazolin-4(3H)-ones is carried out using Mont. K10 clay as a catalyst27 (Scheme 1, equation 2). Thus, we have developed a novel one-pot protocol for the synthesis of quinoline-appended quinazolinones from a reaction of 2-aminoacetophenone, 1,3-cyclohexanedione, and anthranilamide utilizing p-TSA as a reagent, and the products thus formed have been evaluated for photophysical properties (Scheme 1, equation 3). It should be noted that both the quinoline and the quinazolinone rings in the products have been formed simultaneously in the one-pot reaction.
Scheme 1. Synthesis of Quinoline-Fused Quinazolinone.

Results and Discussion
At first, 1.0 equiv of each of 2-aminoacetophenone 1a, 1,3-cyclohexanedione 2a, and anthranilamide 3a were treated with 2.0 equiv of p-TSA in a sealed tube at 100 °C under a neat condition over 12 h, affording 9-methyl-3,4-dihydro-1’H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one 4a in a 17% yield along with an inseparable mixture. Compound 4a was thoroughly characterized by spectroscopic methods. To prevent the formation of undesired byproducts, the reaction was performed in a periodic addition of the reagents. Thus, initially, a mixture of 1.0 equiv of 1a, and 1.0 equiv of 2a was treated with 2.0 equiv of p-TSA in a sealed tube and heated at 100 °C for 3 min. Following this, 1.0 equiv of 3a was added and stirred for 12 h at 100 °C. The reaction yielded 4a in a 17% yield and a new compound 9-methyl-3,4-dihydroacridin-1(2H)-one 5a in a 74% yield as the major product (Table 1, entry 1).
Table 1. Optimization of the Synthesis of Compound 4.

| yieldb |
||||||
|---|---|---|---|---|---|---|
| entry | substrate ratio 1a:2a:3a | reagent (equiv) | solvent | temp. °C initial temp., reaction temp.a | 4a | 5a |
| 1 | 1:1:1 | p-TSA (2.0) | 100, 100 | 17 | 74 | |
| 2 | 1:1:1 | p-TSA (2.0) | 100, 115 | 69 | 23 | |
| 3 | 1:1:1 | p-TSA (2.0) | DMF | 115, 115 | 86 | 7 |
| 4 | 1:1:1 | p-TSA (2.0) | 1,4-dioxane | 100, 115 | 85 | 6 |
| 5 | 1:1:1 | p-TSA (2.0) | toluene | 100, 115 | 83 | 10 |
| 6 | 1:1:1 | p-TSA(2.0) | xylene | 100, 115 | 83 | 10 |
| 7c | 1:1:1 | p-TSA (2.0) | DMSO | 100, 115 | 87 | 6 |
| 8 | 1:1:1 | CuSO4·5H2O (2.0) | DMSO | 100, 115 | 32 | |
| 9 | 1:1:1 | CuI (2.0) | DMSO | 100, 115 | 40 | |
| 10 | 1:1:1 | NiCl2·6H2O(2.0) | DMSO | 100, 115 | 11 | |
| 11 | 1:1:1 | AcOH (2.0) | DMSO | 100, 115 | 16 | |
| 12 | 1:1:1 | FeCl3·6H2O (2.0) | DMSO | 100, 115 | 30 | 63 |
| 13 | 1:1:1 | Ceralite IR120 (100% W/W) | DMSO | 100, 115 | ||
| 14 | 1:1:1 | MK-10 (100%) W/W | DMSO | 100, 115 | ||
| 15 | 1:1:1 | p-TSA (1.0) | DMSO | 100, 115 | d | 73 |
| 16 | 1:1:1 | p-TSA (1.5) | DMSO | 100, 115 | 43 | 33 |
| 17 | 1:1:1 | p-TSA (3.0) | DMSO | 100, 115 | 85 | 10 |
| 18 | 1:1:1.2 | p-TSA (2.0) | DMSO | 100, 115 | 85 | 7 |
| 19 | 1.2:1:1 | p-TSA (2.0) | DMSO | 100, 115 | 84 | 3 |
| 20 | 1:1.2:1 | p-TSA (2.0) | DMSO | 100, 115 | 85 | 6 |
Reaction was initially carried out at 100 °C for 3 min followed by increasing the temperature to 115 °C.
Isolated yield.
Optimized condition.
Trace.
To improve the yield of 4a, parameters such as temperature, reagents, the mole ratio of reactants, and solvents were considered. Thus, a reaction with a 1:1 ratio of compounds 1a and 2a in the presence of 2 equiv of p-TSA at 100 °C for 3 min was carried out, followed by the addition of 1 equiv of 3a, and the temperature was increased to 115 °C. The reaction yielded 69% of 4a and 23% of 5a. (Table 1, entry 2). However, we observed that the solvent-free protocol was not suitable for all of the substrates. Hence, various solvents like DMSO, DMF, 1,4-dioxane, toluene, and xylene were introduced to determine the effect of the solvent in facilitating the reaction. The screening of the solvents revealed that the presence of a solvent in the reaction increased the yield of the reaction, but none of the solvents showed a remarkable superiority in the obtained yield. (Table 1, entries 3–7). To choose the best reagents, reagents such as CuSO4·5H2O, CuI, NiCl2·6H2O·CH3COOH, FeCl3.6H2O, Ceralite IR120, and MK-10 were screened. None of the above reagents improved the yield (Table 1, entries 8–14). Among the various reagents screened, only FeCl3·6H2O gave a 30% yield of 4a. To optimize the amount of p-TSA, various equivalents of p-TSA were used and the highest yield was obtained with 2 equiv of p-TSA (Table 1, entries 15–17). Also, various equivalents of substrates were taken in the presence of 2 equiv of p-TSA at 100 °C for 3 min, followed by the addition of 3a, and the temperature of the reaction system was increased to 115 °C. We observed that no significant improvement in the yield was noticed. However, in all of the cases, a trace of 5a was observed (Table 1, entries 18–20). Furthermore, to remove the intermittent addition of anthranilamide 3 into the reaction, we performed a reaction by adding all of the reactants (1, 2, and 3), p-TSA, and DMSO together and heating at 100 °C for 3 min, followed by heating at 115 °C over 12 h. We observed no change in the yield percentage of the expected products 4a and 5a.
Based on the structure of product 4, a plausible mechanism is proposed in Scheme 2. Thus, 2-aminoacetophenone 1a undergoes Friedländer condensation with 1,3-cyclohexanedione 2a in the presence of p-TSA to form an isolable acridinone intermediate 5. The subsequent reaction of intermediate 5 with anthranilamide 3 forms an imine intermediate, which undergoes intramolecular nucleophilic amide nitrogen attack on the imine, yielding quinoline-appended spiro-quinazolinone 4. The isolated acridinone intermediate 5 supports the proposed reaction pathway.
Scheme 2. Plausible Mechanism for the Formation of Compounds 4 and 5.

The structure of the representative compound 4b was confirmed by spectroscopic data analysis (see SI), and the relative stereochemistry was assigned based on single-crystal X-ray analysis (Figure 5).28
Figure 5.

ORTEP diagram of compound 4b.
Figure 2.

Screening of the starting materials.
Encouraged by the preliminary results, we investigated the scope of the reaction with several 2-aminoacetophenones, 2-aminoamides, and 1,3-cyclohexanedione (Figures 3–5 and Table 2). Under optimized conditions, (Table 1, entry 7) all of the reactions went smoothly to produce the respective quinoline-appended quinazolinones 4 as the major product and acridinone as the minor product 5. It was observed that the unsubstituted 2-aminobenzamide 3a gave a higher yield compared to those with bromine-substituted aminobenzamides 3b and 3c. This might be due to the interaction of the bulky Br group with the quinoline methyl in the imine intermediate. The bulky methyl substitutions on the aliphatic ring of the compound did not affect the yield of the reaction. The reaction gave only acridinone 5 as the sole product when the reaction was performed with 3-aminofuran-2-carboxamide and 2-aminobenzene sulfonamide.
Figure 3.

Isolated intermediates.
Table 2. Scope of the Reaction.

| |
(% Yield)a,b |
||||
|---|---|---|---|---|---|
| Sl. no | starting materials (1:1:1) | 4 | 5 | ||
| 1 | 1a | 2a | 3a | 4a (87) | 5a (6) |
| 2 | 1a | 2b | 3a | 4b (85) | 5b (7) |
| 3 | 1b | 2a | 3a | 4c (82) | 5c (7) |
| 4 | 1a | 2a | 3b | 4d (83) | 5a (6) |
| 5 | 1a | 2a | 3c | 4e (82) | 5a (8) |
| 6 | 1a | 2b | 3b | 4f (64) | 5b (31) |
| 7 | 1a | 2b | 3b | 4g (73) | 5b (21) |
| 8 | 1b | 2b | 3c | 4h (6) | 5d (81) |
| 9 | 1a | 2c | 3a | 4i (88) | 5e (3) |
| 10 | 1a | 2c | 3b | 4j (81) | 5e (8) |
| 11 | 1a | 2c | 3c | 4k (79) | 5e (10) |
| 12 | 1b | 2c | 3a | 4l (82) | 5f (7) |
| 13 | 1b | 2c | 3b | 4m (81) | 5f (10) |
| 14 | 1b | 2a | 3b | 4n (86) | 5c (5) |
| 15 | 1b | 2a | 3c | 4o (19) | 5c (64) |
| 16 | 1c | 2c | 3c | 5g (66) | |
Optimized condition.
Isolated yield.
Figure 4.
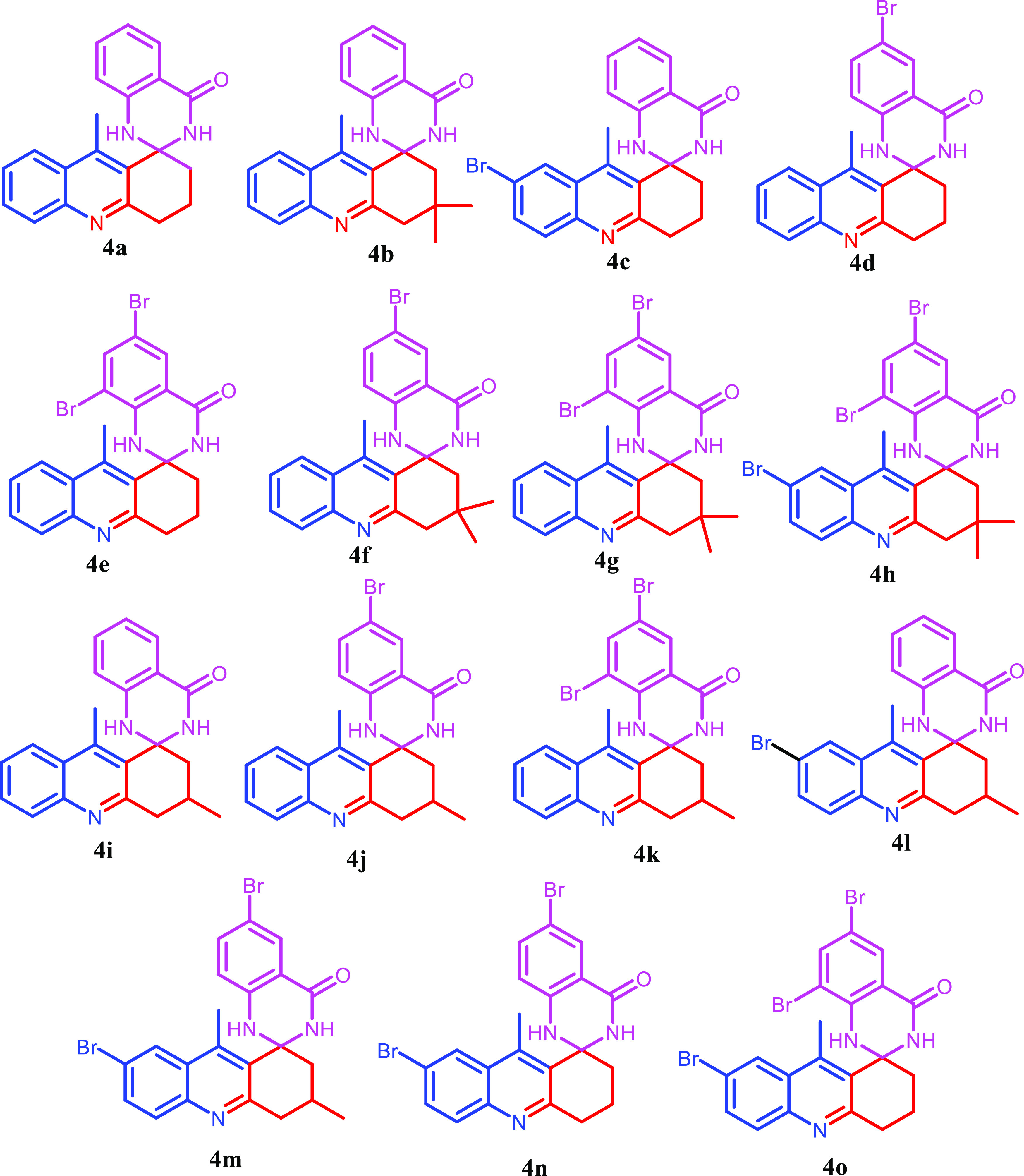
Compounds synthesized.
The bromine substitutions on the derivatives facilitated further synthetic transformations of the molecules. The effectiveness of this methodology was further scrutinized by a gram-scale synthesis of 4a under optimized reaction conditions giving a yield of 81%.
To demonstrate the synthetic utility of compounds synthesized, several biphenyl tethered quinoline-appended spiro-quinazolinones were synthesized via the Suzuki coupling reaction, as shown in Scheme 3. Thus, compounds 4c, 4d, and 4n were treated with various aryl boronic acids in the presence of Pd(OAc)2 as a catalyst and K2CO3 as a base to afford respective arylated products 6a–d in an 89–95% yield.
Scheme 3. Synthetic Transformation of Compounds 4c, 4d, and 4n into Biphenyl Derivatives 6a–d.

Photophysical Studies
The structural uniqueness of quinoline-appended dihydro/spiro-quinazolinones encouraged us to explore their photophysical properties. Thus, compounds 4c and 4k were chosen for the investigation. Initially, to establish solvatochromic property, UV–visible spectra of 4c and 4k were recorded using solvents such as acetonitrile, methanol, tetrahydrofuran, 1,4-dioxane, and toluene. Two absorption bands were observed in all of the solvents, as shown in Figures 6 and 7. A higher energy band in the range 228 to 287 nm begins with π–π* electronic transition [intramolecular charge transfer (ICT)] and other bands with lower energy n−π* electronic transition in the region 345–365 nm were observed. While increasing the solvent polarity from toluene to acetonitrile, a blue shift was observed for both compounds. The wavelength shifted from 287 nm in toluene to 228 nm in acetonitrile to give a shift of 59 nm for compound 4c. For compound 4k, a similar shift of 57 nm was observed in the respective solvent. The hypsochromic (blue) shift observed can be associated due to the decrease in the dipole moment in the excited state as compared to the ground state, stabilizing the ground-state energy in polar solvents.29 Furthermore, the molar extinction coefficient (ε) was calculated using Lambert–Beer’s law (A = εcl). The molar extinction coefficient value of both the derivatives decreased with the decrease in solvent polarity. As the solvent polarity decreased, the value of the molar extinction coefficient of 4c decreased from 5.1295 × 104 to 0.5664 × 104 M–1cm–1 (Table 3). In the case of 4k, the value of the molar extinction coefficient decreases from 10.3089 × 104 to 1.1268 × 104 M–1cm–1 (Table 4). Furthermore, the quantum yield and Stoke’s shift were calculated for 4c and 4k in all of the selected solvents.
Figure 6.

(a) Normalized absorption spectra of compound 4c recorded at a concentration of 2 × 10–5 M at 298 K and (b) normalized emission spectra of compound 4c recorded at a concentration of 2 × 10–5 M at 298 K.
Figure 7.

(a) Normalized absorption spectra of compound 4k recorded at a concentration of 2 × 10–5 M at 298 K and (b) normalized emission spectra of compound 4k recorded at a concentration of 2 × 10–5 M at 298 K.
Table 3. Photophysical Properties of Compound 4c.
| entry | solvent | absorptiona λmax,abs (nm) | emissiona λmax,emi (nm) | molar extinction coefficient × 104 (ε) π–π* | Stoke’s shift Δν̅ (cm–1)b | quantum yield (Φf)c |
|---|---|---|---|---|---|---|
| 1 | CH3CN | 228, 263 | 414 | 5.00985 | 19 705 | 0.3681 |
| 2 | MeOH | 230, 264 | 436 | 5.12955 | 20 542 | 0.3195 |
| 3 | THF | 247, 317 | 409 | 1.67091 | 16 035 | 0.1063 |
| 4 | dioxane | 271, 317 | 408 | 0.97554 | 12 390 | 0.2146 |
| 5 | toluene | 287, 319 | 407 | 0.56647 | 10 273 | 0.2270 |
Recorded at 298 K.
Stoke’s shift = λmax,abs – λmax,emi [cm –1].
Determined with anthracene as a standard Φf = 0.27 at an excitation wavelength of 246 nm.
Table 4. Photophysical Properties of Compound 4k.
| entry | solvent | absorptionaλmax,abs (nm) | emissionaλmax,emi (nm) | molar extinction coefficient × 104 (ε) π–π* | Stoke’s shift Δν̅ (cm–1)b | quantum yield (Φf)c |
|---|---|---|---|---|---|---|
| 1 | CH3CN | 231 | 418 | 7.6462 | 19 366 | 0.1522 |
| 2 | MeOH | 233 | 463 | 10.3089 | 21 320 | 0.1580 |
| 3 | THF | 249, 357 | 408 | 3.41119 | 15 650 | 0.2754 |
| 4 | dioxane | 272, 356 | 407 | 1.54363 | 12 194 | 0.7916 |
| 5 | toluene | 288, 353 | 408 | 1.12686 | 10 212 | 0.8019 |
Recorded at 298 K.
Stoke’s shift = λmax,abs – λmax,emi [cm–1].
Determined with anthracene as a standard Φf = 0.27 at an excitation wavelength of 246 nm.
Quantum yields of compounds were estimated by comparison with the known quantum yields of anthracene in ethanol (Φ = 0.27) at an excitation wavelength of 246 nm using the equation given in the SI. For compound 4c, the quantum yield varied from 0.3681 to 0.1063. The highest quantum yield was obtained in acetonitrile. For compound 4k, the quantum yield varied from 0.8019 to 0.1522 with the highest quantum yield observed in toluene. The Stoke’s shift value of compounds 4c and 4k in different solvents are given in Tables 3 and 4.
Both the compounds 4c and 4k exhibited a large Stoke’s shift in the ranges from 20 542 to 12 390 and 21 320 to 10 212 cm–1, which is associated with highly polarizable π-conjugated systems due to ICT. It has also been noted that a large red shift was observed in the excited state when methanol was used as a solvent, as shown in Figures 6 and 71b, 2b. This might be due to the stronger electron-withdrawing nature of the quinoline ring and the presence of a strong electron-donating amino group in the molecule. Protonation of the compound by the solvent also facilitates the red shift.30
The extended π conjugation induced by the aryl system encouraged us to further investigate the photophysical properties of the Suzuki coupled products 6a–d. Thus, UV–visible and fluorescence spectra of compounds 6a–d were measured in methanol and the spectra are displayed in Figure 8 a,b. The absorption spectra of compounds 6 a–d showed two bands in the region of 240–380 nm. The first absorption band is related to higher energy with a lower wavelength π–π* transition that appeared in the range of 250–280 nm and another medium energy belonging to the n−π* transition of the compounds appeared as a shoulder in the region of 330–380 nm for compounds 6a–d. Also, in fluorescence spectra, the medium energy exhibits emission at 367 nm (6a), 371 nm (6b), 380 nm (6c), and 380 nm (6d).
Figure 8.

(a) Normalized absorption spectra of compounds 6a–d recorded at a concentration of 2 × 10–5 M at 298 K and (b) normalized emission spectra of compounds 6a–d recorded at a concentration of 2 × 10–5 M at 298 K.
Furthermore, Stoke’s shift and the molar extinction coefficient for the π–π* transition were calculated for 6a–d. It was observed that the compounds 6a–d exhibited similar Stoke’s shift values. Monoarylated derivatives 6a–b exhibited higher Stoke’s shift than the biarylated derivatives 6c–d. The complete photophysical data along with fluorescence quantum yield (Φf) for the synthesized biaryls are summarized in Table 5. It was observed that much increase in the quantum yield was not observed when the phenyl ring was tethered to quinoline-appended quinazolinones.
Table 5. Photophysical Properties of Biaryl Derivatives 6a–d.
| entry | product | absorptionaλmax,abs (nm) | emissiona λmax,emi (nm) | molar extinction coefficient × 104 (ε) π–π* | Stoke’s shift Δν̅ (cm–1)b | quantum yield (Φf)c |
|---|---|---|---|---|---|---|
| 1 | 6a | 256, 334 | 367 | 2.0435 | 11 814 | 0.1676 |
| 2 | 6b | 257, 328 | 371 | 5.0952 | 11 956 | 0.1212 |
| 3 | 6c | 276 | 380 | 4.5657 | 9916 | 0.1755 |
| 4 | 6d | 276 | 380 | 6.0996 | 9916 | 0.0359 |
Recorded in MeOH at 298 K.
Stoke’s shift = λmax,abs – λmax,emi [cm –1].
Determined with anthracene as a standard Φf = 0.27 at an excitation wavelength of 246 nm.
It was observed that the quantum yield and Stoke’s shift values obtained for the quinoline-appended quinazolinones were higher compared to other spiro- and cyclic-quinazolinone heterocyclic derivatives.31 This class of quinoline-based compounds with a high quantum yield and Stoke’s shift values is very useful as labels in biochemical and technical applications.32
In conclusion, an efficient one-pot synthesis of quinoline-appended quinazolinone derivatives has been accomplished via Friedländer condensation. A plausible reaction mechanism is provided, and a representative structure of product 4b was confirmed by XRD. The synthetic utility of the products is demonstrated by the Suzuki coupling reaction. Further, photophysical properties of compounds 4c and 4k were evaluated and synthesized biphenyl tethered quinoline-appended quinazolinones were found to be promising blue-emissive fluorescent molecules.
Experimental Section
General Remarks
All of the reactions were carried out in oven-dried glassware. Progress of reactions was monitored by thin-layer chromatography (TLC), while purification of crude compounds was done by column chromatography using silica gel (Mesh size 100–200). The NMR spectra were recorded on a Bruker 400 MHz NMR spectrometer (400 MHz for 1H NMR and 100 MHz for 13C NMR) with CDCl3 or (CD3)2SO as a solvent and TMS as an internal reference. Integrals are in accordance with assignments; coupling constants were reported in Hertz (Hz). All 13C spectra are proton-decoupled. Multiplicity is indicated as follows: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublet), and br s (broad singlet). FTIR spectra were recorded on a Perkin-Elmer RX-I FTIR, and absorbance is reported in cm–1. HRMS analyses were recorded using a Q-Tof Micro mass spectrometer (different mass analyses based on the availability of instruments). Yields refer to quantities obtained after chromatography. Absorption spectra were recorded using a JASCO V-670 spectrophotometer. Steady-state fluorescence spectra were recorded on a Hitachi F-7000 FL Spectro fluorophotometer by excitation at the respective absorption maxima.
Quantum yields of compounds were estimated by comparison with the known quantum yields of anthracene in ethanol (Φ = 0.27) at an excitation wavelength of 246 nm using the following equation:
where Φ is the quantum yield, I is the integrated intensity, OD is the optical density, and n is the refractive index. The subscript R refers to anthracene.
The molar extinction coefficient (ε) was calculated using Lambert–Beer’s law
The Stoke’s Shift was calculated using the following equation:
Experimental Procedures
Compounds 1b, 3b, and 3c were synthesized according to the procedure given in refs (33−35).
General Procedure for the Synthesis of 2-Amino-5-bromoacetophenone (1b)
To a stirred solution of 1-(2-aminophenyl)ethanone (0.5 g, 3.7 mmol) in 5 mL of CH3CN at 0 °C, N-bromosuccinimide (0.66 g, 3.7 mmol) was added dropwise and dissolved in 5 mL of CH3CN. The mixture was allowed to stand at room temperature and continually stirred at room temperature for 3 h. The removal of the solvent under in vacuo and purification through a column of silica gel (petroleum ether/ethyl acetate = 5:1) afforded 1-(2-amino-5-bromophenyl)ethanone.
General Procedure for the Synthesis of 2-Amino-5-bromobenzamide (3b)
In a screw-capped reaction tube, 2-aminobenzamide (0.5 mmol) was dissolved in acetonitrile (2 mL) and N-bromosuccinamide (0.6 mmol) dissolved in CH3CN was added, and the reaction mixture was heated at 60 °C for 10 min. Then, the mixture was diluted with EtOAc and washed with saturated brine. The organic layer separated was dried over anhyd. Na2SO4 and concentrated under reduced pressure. The crude compound was purified through a column of silica gel (petroleum ether), affording 1-(2- amino-5-bromophenyl) ethanone.
General Procedure for the Synthesis of 2-Amino-3,5-dibromobenzamide (3c)
In a stirred solution of 2-aminobenzamide (2.0 mmol) in acetonitrile (10.0 mL), N-bromosuccinimide (0.85 g, 4.8 mmol, 2.0 equiv) was added, and the reaction mixture was stirred at room temperature for 3 h. The reaction mixture was quenched with crushed ice, resulting in a precipitate. The recrystallization of the residue from MeCN afforded 2-amino-3,5-dibromobenzamide (3c).
General Procedure for the Synthesis of Quinoline-Appended Quinazolinone 4a–o
A sealed tube containing 2-aminoacetophenone (1 mmol), 1,3-cyclohexanedione (1.0 mmol), anthranilamide (1.0 mmol), and p-toluenesulfoniconic acid (TSA) (2.0 mmol) was heated initially at 100 °C for 5 min. Next, DMSO (300 μL) was added and then the reaction temperature was increased to 115 °C continuously for 12 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with water, EtOAc, and washed using a 10% NaOH solution. The combined organic layer was dried over anhyd. Na2SO4 and the solvent was evaporated under reduced pressure. The crude mixture was purified by silica gel column chromatography to obtain pure compounds 4a–o.
9-Methyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4a)
Nature: a brown powder; yield: 87%; Rf (50% EtOAc–hexane): 0.46, M. P: 230–231 °C. FTIR(KBr)νmax: 3292, 3176, 3049, 1654, 1610, 1512, 1487, 754 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.31 (s, 1H), 8.16 (d, J = 8.5 Hz, 1H), 7.88 (d, J = 8.4 Hz, 1H), 7.71 (t, J = 7.6 Hz, 1H), 7.65 (d, J = 7.9 Hz, 1H), 7.54 (t, J = 7.6 Hz, 1H), 7.29 (s, 1H), 7.24 (t, J = 7.6 Hz, 1H), 6.65 (dd, J = 7.7, 4.3 Hz, 2H), 3.10–2.95 (m, 2H), 2.91 (s, 3H), 2.35–2.06 (m, 3H), 2.00–1.79 (m, 2H). 13C NMR (101 MHz, DMSO-d6): δ 162.2, 158.1, 146.6, 146.2, 146.0, 133.6, 130.3, 129.6, 128.4, 127.7, 127.3, 125.7, 124.2, 116.4, 114.2, 113.2, 70.2, 34.7, 31.5, 29.4, 16.3. HRMS-ESI: calcd for C21H19N3O [M + H]+m/z: 330.1606; found: 330.1614.
Isolated Intermediate: 5a (6%).
3,3,9-Trimethyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4b)
Nature: a brown powder; yield: 85%; Rf (50% EtOAc–hexane): 0.48, M. P: 240–241 °C. FTIR(KBr)νmax: 3259, 3062, 2922, 1633, 1606, 1510, 1357, 745 cm–1; 1H NMR (400 MHz, DMSO-d6):δ 8.26–8.10 (m, 2H), 7.91 (d, J = 8.1 Hz, 1H), 7.75–7.55 (m, 3H), 7.25 (t, J = 7.1 Hz, 1H), 6.93 (s, 1H), 6.67 (t, J = 7.2 Hz, 2H), 2.97 (s, 3H), 2.89 (s, 2H), 2.35–2.22 (m, 2H), 1.03 (s, 6H). 13C NMR (101 MHz, DMSO-d6): δ 163.4, 157.5, 148.5, 147.4, 145.1, 134.6, 130.5, 129.2, 128.7, 128.5, 127.4, 126.4, 124.3, 119.1, 114.5, 114.1, 77.4, 77.3, 77.1, 71.9, 52.5, 49.3, 30.0, 29.2, 27.6, 17.0. HRMS-ESI: calcd for C23H23N3O [M + H]+m/z: 358.1919; found: 358.1938.
Isolated Intermediate: 5b (7%).
7-Bromo-9-methyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4c)
Nature: a brown powder; yield: 82%; Rf (50% EtOAc–hexane): 0.50, M. P: 265–266°C. FTIR(KBr)νmax: 3284, 3064, 2924, 2852, 1658, 1606, 1481, 759 cm–1; 1H NMR (400 MHz, DMSO-d6) δ: 8.44 (s, 1H), 8.10 (d, J = 8.4 Hz, 1H), 7.82 (d, J = 8.3 Hz, 1H), 7.66 (dd, J = 7.8, 4.8 Hz, 2H), 7.49 (t, J = 7.2 Hz, 2H), 7.33 (dd, J = 8.7, 2.4 Hz, 1H), 6.57 (d, J = 8.7 Hz, 1H), 2.96–2.89 (m, 2H), 2.81 (s, 3H), 2.29–1.99 (m, 2H), 1.83 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 161.0, 158.1, 146.7, 146.3, 145.0, 136.1, 129.8, 129.4, 128.4, 127.7, 125.9, 124.3, 116.6, 114.8, 107.3, 70.3, 62.8, 34.7, 21.1, 17.1, 16.3. HRMS-ESI: calcd for C21H18BrN3O [M + H]+m/z: 408.0711; found: 408.0713.
Isolated Intermediate: 5c (7%).
6′-Bromo-9-methyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4d)
Nature: a brown powder; yield: 83%; Rf (50% EtOAc–hexane): 0.51, M. P: 270–271°C. FTIR(KBr)νmax: 3284, 2922, 2850, 1656, 1604, 1481, 759 cm–1; 1H NMR (400 MHz, DMSO-d6) δ: 8.48 (s, 1H), 8.14 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 8.2 Hz, 1H), 7.73–7.66 (m, 2H), 7.57–7.49 (m, 2H), 7.36 (dd, J = 8.6, 2.4 Hz, 1H), 6.61 (d, J = 8.7 Hz, 1H), 2.98 (dd, J = 14.0, 7.5 Hz, 2H), 2.85 (s, 3H), 2.33–2.04 (m, 2H), 1.87 (t, J = 6.7 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 161.0, 158.1, 146.7, 146.3, 145.0, 136.1, 129.9, 129.8, 129.4, 128.4, 127.7, 125.9, 124.3, 116.6, 114.8, 107.3, 70.3, 62.8, 34.7, 17.1, 16.3. HRMS-ESI: calcd for C21H18BrN3O [M + H]+m/z: 408.0711; found: 408.0741.
Isolated Intermediate: 5a (6%).
6′,8′-Dibromo-9-methyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4e)
Nature: a white powder; yield: 82%; Rf (50% EtOAc–hexane): 0.47, M. P: 265–266 °C. FTIR(KBr)νmax: 3338, 3163, 3037, 2939, 1656, 1600, 1489, 752 cm–1; 1H NMR (400 MHz, DMSO-d6) δ: 8.83 (s, 1H), 8.18 (d, J = 8.2 Hz, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.82 (m, 2H), 7.79–7.70 (m, 1H), 7.61–7.53 (m, 1H), 6.98 (s, 1H), 3.04 (t, J = 6.2 Hz, 2H), 2.85 (s, 3H), 2.51–2.23 (m, 2H), 2.10–1.92 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ: 159.9, 158.4, 146.3, 145.9, 142.5, 138.2, 129.6, 129.2, 128.4, 127.7, 125.6, 124.1, 116.2, 108.3, 107.3, 70.6, 34.6, 16.9, 16.2. HRMS-ESI: calcd for C21H17Br2N3O [M + H]+m/z: 485.9816; found: 485.9814.
Isolated Intermediate: 5a (8%).
6′-Bromo-3,3,9-trimethyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4f)
Nature: white powder; yield: 64%; Rf (50% EtOAc–hexane): 0.46, M. P: 257–258 °C. FTIR(KBr)νmax: 3186, 3066, 2962, 1656, 1608, 1500, 754 cm–1; 1H NMR (400 MHz, DMSO-d6) δ: 8.35 (s, 1H), 8.20 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.78–7.70 (m, 2H), 7.57 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 8.7 Hz, 1H), 7.21 (s, 1H), 6.63 (d, J = 8.7 Hz, 1H), 2.93 (s, 3H), 2.88 (s, 2H), 2.27 (m, 1H), 1.90 (s, 1H), 1.02 (d, J = 1.8 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ: 171.9, 161.1, 157.5, 146.9, 146.6, 144.9, 136.0, 129.7, 129.3, 128.6, 127.9, 127.8, 125.9, 124.3, 116.7, 115.1, 107.3, 70.1, 52.1, 48.7, 29.5, 27.4, 21.0, 16.3. HRMS-ESI: calcd for C23H22BrN3O [M + H]+m/z: 436.1024; found: 436.1037.
Isolated Intermediate: 5b (31%).
6′,8′-Dibromo-3,3,9-trimethyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4g)
Nature: a yellow powder; yield: 73%; Rf (50% EtOAc–hexane): 0.50, M. P: 239–240 °C. FTIR(KBr)νmax: 3423, 3167, 3066, 2920, 1662, 1598, 1487, 750 cm–1; 1H NMR (400 MHz, CDCl3) δ: 8.29 (d, J = 1.7 Hz, 1H), 8.01–7.93 (m, 2H), 7.68–7.61 (m, 2H), 7.44 (t, J = 7.6 Hz, 2H), 7.34 (t, J = 7.4 Hz, 1H), 3.12 (s, 2H), 3.04 (s, 3H), 2.61 (s, 2H), 1.07 (d, J = 4.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ: 161.4, 157.3, 141.4, 139.1, 130.8, 129.1, 128.4, 126.6, 124.3, 116.3, 110.4, 109.4, 72.0, 52.8, 49.0, 30.0, 27.2, 17.0. HRMS-ESI: calcd for C23H21Br2N3O [M + H]+m/z: 514.0129; found: 514.0124.
Isolated Intermediate: 5b (21%).
6′,7,8′-Tribromo-3,3,9-trimethyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4h)
Nature: a brown powder; yield: 6%; Rf (50% EtOAc–hexane): 0.46, M. P: 212–213 °C. FTIR(KBr)νmax: 3192, 3072, 2922, 1662, 1598, 1463, 796 cm–1; 1H NMR (400 MHz, CDCl3) δ: 8.25 (dd, J = 13.1, 1.7 Hz, 1H), 8.10–8.02 (m, 1H), 7.94–7.85 (m, 1H), 7.59 (m, 1H), 7.52–7.44 (m, 1H), 7.40 (dt, J = 9.7, 5.6 Hz, 1H), 7.37–7.25 (m, 1H), 3.16 (t, J = 9.3 Hz, 2H), 3.03 (t, J = 2.6 Hz, 3H), 2.58 (d, J = 5.5 Hz, 2H), 1.05 (d, J = 4.8 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ: 161.6, 157.7, 141.3, 139.3, 138.7, 134.5, 130.7, 129.7, 126.8, 116.2, 110.6, 109.5, 71.8, 52.7, 30.0, 29.8, 27.3, 17.2, 17.1. HRMS-ESI: calcd for C23H20Br3N3O [M + H]+m/z: 591.9235; found: 591.9231.
Isolated intermediate: 5d (81%).
3,9-Dimethyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4i)
Nature: a pale yellow powder; yield: 88%; Rf (50% EtOAc–hexane): 0.48, M. P: 274–275 °C. FTIR(KBr)νmax: 3290, 3174, 2924, 1658, 1612, 1485, 756 cm–1; 1H NMR (400 MHz, DMSO-d6) δ: 8.52 (s, 1H (D2O exchangeable)), 8.18 (d, J = 4 Hz, 1H) 8.17 (s, 1H (D2O exchangeable)), 7.89 (d, J = 12 Hz, 1H), 7.73 (t, J = 7.6Hz, 1H), 7.68–7.63 (m, 1H), 7.56 (t, J = 7.6Hz, 1H), 7.39 (s, 1H (D2O exchangeable)), 7.25 (t, J = 7.6 Hz, 1H), 7.05 (s, 1H (D2O exchangeable)) 6.69–6.61 (m, 2H), 3.13–3.07 (m, 1H), 2.64–2.58 (m, 2H), 2.27–2.23 (m, 1H), 1.65 (t, J = 12.8, 1H), 1.05–1.01 (m, 3H). 13C NMR (101 MHz, DMSO-d6) δ: 162.2, 157.9, 146.5, 146.4, 146.3, 145.7, 133.6, 133.5, 129.6, 129.6, 128.5, 127.7, 127.3, 127.3, 125.7, 124.2, 124.1, 116.51, 116.2, 114.1, 114.0, 113.2, 70.7, 70.6, 48.7, 43.5, 43.3, 40.1, 38.8, 23.8, 22.9, 21.1, 21.1, 16.1. HRMS-ESI: calcd for C22H21N3O [M + H]+m/z: 344.1763; found: 344.1779.
Isolated Intermediate: 5e (3%)
6′-Bromo-3,9-dimethyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4j)
Nature: a white powder; yield: 81%; Rf (50% EtOAc–hexane): 0.50, M. P: 279–280 °C. FTIR(KBr)νmax: 3188, 3062, 2926, 1660, 1610, 1481, 756 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.71 (s, 1H), 8.38 (s, 1H), 8.19 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 8.3 Hz, 1H), 7.76–7.70 (m, 2H), 7,65 (s, 1H), 7.57 (t, J = 7.6Hz, 1H), 7.40 (d, J = 8.8 Hz, 1H), 7.33 (s, 1H), 6.66–6.60 (m, 1H), 3.14–3.09 (m, 1H), 2.89 (d, J = 6 Hz, 3H), 2.67–2.53 (m, 2H), 2.25–2.20 (m, 1H), 1.70–1.63 (m, 1H), 1.04 (t, J = 7.2Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 160.9, 157.7, 146.3, 144.7, 136.0, 129.7, 129.3, 129.2, 128.5, 127.7, 125.8, 124.2, 116.6, 114.9, 107.4, 70.7, 47.9, 43.2, 23.8, 21.0, 16.1. HRMS-ESI: calcd for C22H20BrN3O [M + H]+m/z: 422.0868; found: 422.0900.
Isolated Intermediate: 5e (8%).
6′,8′-Dibromo-3,9-dimethyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4k)
Nature: a brown powder; yield: 79%; Rf (50% EtOAc–hexane): 0.45, M. P: 268–269 °C. FTIR(KBr)νmax: 3192, 3062, 2922, 1664, 1598, 1483, 756 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.93 (s, 1H), 8.65 (s, 1H), 8.17 (d, J = 8.8 Hz, 1H), 7.90 (d, J = 8.4 Hz, 1H), 7.82–7.77 (m, 2H), 7.73 (t, J = 7.2 Hz, 1H), 7.56 (t, J = 7.6Hz, 1H), 7.02 (s, 1H), 6.89 (s, 1H), 3.13–3.05 (m, 1H), 2.83 (d, J = 6.8 Hz, 3H), 2.69–2.58 (m, 2H), 2.28–2.24 (m, 1H), 1.92–1.64 (m, 1H), 1.05–1.01 (m, 3H). 13C NMR (101 MHz, DMSO-d6): δ 159.6, 158.3, 158.2, 146.4, 142.8, 142.1, 138.3, 129.6, 129.5, 129.2, 128.5, 128.4, 125.6, 124.2, 124.1, 116.8, 115.5, 108.5, 108.1, 107.7, 71.2, 70.9, 48.5, 47.9, 43.3, 43.2, 23.6, 22.9, 21.1, 21.0, 16.3, 15.9. HRMS-ESI: calcd for C22H19Br2N3O [M + H]+m/z: 499.9973; found: 499.9970.
Isolated Intermediate: 5e (10%).
7-Bromo-3,9-dimethyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4l)
Nature: a white powder; yield: 82%; Rf (50% EtOAc–hexane): 0.53, M. P: 276–278 °C. FTIR(KBr)νmax: 3194, 3066, 2926, 1660, 1610, 1498, 756 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.71 (s, 1H), 8.38 (s, 1H), 8.19 (d, J = 8.4 Hz, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.72 (d, J = 11.2Hz, 2H), 7.65 (s, 1H) 7.58 (t, J = 8.0Hz, 1H), 7.40 (d, J = 8.0Hz, 1H), 7.33 (s, 1H), 6.66–6.60 (m, 1H), 3.12 (d, J = 15.6 Hz, 1H), 2.89 (s, 3H), 2.68–2.50 (m, 2H), 2.23 (s, 1H), 1.70–1.64 (m, 1H), 1.03 (d, J = 6.2 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 160.9, 157.7, 146.3, 144.7, 136.0, 129.7, 129.3, 129.2, 124.2, 116.6, 114.9, 107.4, 70.7, 23.8, 21.0, 16.1. HRMS-ESI: calcd for C22H20BrN3O [M + H]+m/z: 422.0868; found: 422.0886.
Isolated Intermediate: 5f (7%).
6′,7-Dibromo-3,9-dimethyl-3,4-dihydro-1′H,2H-spiro[acridine- 1,2′-quinazolin]-4′(3′H)-one (4m)
Nature: a yellow powder; yield: 81%; Rf (50% EtOAc–hexane): 0.51, M. P: 280–281 °C. FTIR(KBr)νmax: 3304, 3062, 2924, 1674, 1604, 1479, 831 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.72 (s, 1H), 8.38 (s, 1H), 7.85 (s, 2H), 7.74–7.70 (m, 1H), 7.76 (s, 1H), 7.37 (t, J = 8.0 Hz, 1H), 6.65–6.59 (m, 1H), 5.75 (s, 1H), 3.12–3.06 (m, 1H), 2.86 (d, J = 8.0 Hz, 3H), 2.66–2.56 (m, 2H), 2.26–2.20 (m, 1H), 1.69–1.62 (m, 1H), 1.03 (t, J = 7.0 Hz, 3H). 13C NMR (101 MHz, DMSO-d6): δ 160.6, 158.8, 158.6, 146.1, 145.4, 145.0, 144.6, 136.0, 132.8, 130.7, 130.3, 130.2, 129.3, 129.3, 129.1, 126.4, 126.4, 119.1, 119.0, 116.6, 116.5, 114.8, 107.4, 107.1, 70.7, 70.6, 54.8, 43.1, 23.7, 22.7, 20.9, 20.9, 16.2, 16.1. HRMS-ESI: calcd for C22H19Br2N3O [M + H]+m/z: 499.9973; found: 499.9973.
Isolated Intermediate: 5f (10%).
6′,7-Dibromo-9-methyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (4n)
Nature: a brown powder; yield: 86%; Rf (50% EtOAc–hexane): 0.49, M. P: 260–261 °C. FTIR(KBr)νmax: 3305, 3062, 2933, 1666, 1604, 1492, 1315, 835 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.52 (s, 1H), 8.38 (s, 1H), 8.31 (s, 1H), 7.85 (s, 1H), 7.72 (d, J = 2.4 Hz, 1H), 7.58 (s, 1H), 7.42–7.39 (m, 1H), 6.64 (d, J = 8.4Hz, 1H), 3.07–3.00 (m, 2H), 2.86 (s, 3H), 2.34–2.29 (m, 1H), 2.16–2.14 (m, 1H), 1.94–1.88 (m, 1H) 13C NMR (101 MHz, DMSO-d6): δ 160.8, 158.9, 146.1, 144.9, 144.9, 136.0, 132.8, 130.8, 130.7, 129.3, 129.1, 126.4, 119.0, 116.5, 114.7, 107.3, 79.1, 70.2, 34.6, 16.9, 16.2. HRMS-ESI: calcd for C21H17Br2N3O [M + H]+m/z: 485.9816; found: 488.9835.
Isolated Intermediate: 5c (5%).
6′,7,8′-Tribromo-9-methyl-3,4-dihydro-1′H,2H-spiro [acridine-1,2′-quinazolin]-4′(3′H)-one (4o)
Nature: a brown powder; yield: 19%; Rf (50% EtOAc–hexane): 0.50, M. P: 207–208 °C. FTIR(KBr)νmax: 3194, 3059, 2924, 1666, 1598, 1481,825 cm–1; 1H NMR (400 MHz, DMSO-d6): δ 8.83 (s, 1H), 8.37 (s, 1H), 8.32 (s, 1H), 7.85–7.79 (m, 4H), 7.01 (s, 1H), 3.01 (t, J = 6.0Hz, 2H), 2.81 (s, 3H), 2.32–2.19 (m, 1H), 1.19 (s, 2H). 13C NMR (101 MHz, DMSO-d6): δ 162.2, 158.1, 146.6, 146.2, 146.0, 133.6, 130.3, 129.6, 128.4, 127.7, 127.3, 125.7, 124.2, 116.4, 114.1, 113.2, 70.2, 34.7, 31.5, 17.1, 16.3. HRMS-ESI: calcd for C21H16Br3N3O [M + H]+m/z: 563.8921; found: 563.8931.
Isolated intermediate: 5c (64%).
Gram-Scale Synthesis of 4a
A 100 mL sealed tube containing 1 g (0.900 mL) of 2-aminoacetophenone, 0.830 g of 1,3-cyclohexanedione, 1.007 g of 2-aminobenzamide, and 2.548 g of p-TSA was heated initially at 100 °C for 5 min. Next, DMSO (3 mL) was added and then the reaction temperature was increased to 115 °C continuously for 12 h. The completion of the reaction was monitored by TLC. The reaction mixture was diluted with water, EtOAc, and washed using a 10% NaOH solution. The combined organic layer was dried over anhyd. Na2SO4 and the solvent was evaporated under reduced pressure. The crude mixture was purified by silica gel column chromatography to obtain pure compound 4a (1.973 g) in an 81% yield and compound 5a (0.172 g) in an 11% yield.
General Procedure for the Synthesis of Compounds 6a and 6b
A mixture of compound 4d or 4c (0.191 mmol), aryl boronic acids (0.286 mmol), Pd(OAc)2 (10 mol %), and K2CO3 (0.286 mmol) in 2 mL of DMF–H2O (3:1) was stirred at 100 °C for 3 h in a sealed tube. After the completion of the reaction (TLC), the residue was diluted with EtOAc and washed with HCl (0.25 M, 20 mL), followed by saturated brine. The combined organic layer was dried over anhyd. Na2SO4 and purified through silica gel column chromatography by gradient elution using EtOAc/hexane to afford compounds 6a–6b in good yields.
9-Methyl-7-phenyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (6a)
Nature: a red powder; yield: 95%; Rf (30% EtOAc–hexane): 0.50, M.P: 240–242°C. FTIR(KBr)νmax: 3408, 3192, 2922, 1658, 1608, 1483, 756 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.08 (d, J = 6.8 Hz, 1H), 7.99–7.89 (m, 1H), 7.86 (d, J = 8.8 Hz, 2H), 7.58 (d, J = 6.9 Hz, 2H), 7.41 (t, J = 7.3 Hz, 2H), 7.32 (m, 1H), 7.27 (t, J = 7.6 Hz, 1H), 6.77 (d, J = 3.2 Hz, 1H), 6.61 (d, J = 8.0 Hz, 1H), 6.13 (s, 1H), 3.11 (s, 2H), 3.00–2.87 (m, 3H), 2.36 (m, 2H), 1.88 (s, 2H). 13C NMR (101 MHz, CDCl3): δ 163.4, 157.9, 145.0, 140.5, 134.6, 129.3, 129.1, 128.7, 128.4, 127.9, 127.6, 122.0, 119.0, 114.7, 114.1, 39.7, 29.8, 18.2, 17.2 HRMS-ESI: calcd for C27H23N3O [M + H]+m/z: 406.1919; found 406.1923.
9-Methyl-6′-phenyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (6b)
Nature: a brown powder; yield: 94%; Rf (30% EtOAc–hexane): 0.49, M.P: 259–260 °C. FTIR(KBr)νmax: 3246, 3051, 2935, 1645, 1614, 1481, 754 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.35 (d, J = 8.5 Hz, 2H), 8.07 (d, J = 8.3 Hz, 1H), 7.97 (d, J = 8.7 Hz, 1H), 7.84 (d, J = 7.3 Hz, 2H), 7.65 (m, 1H), 7.51 (t, J = 7.6 Hz, 2H), 7.40 (t, J = 7.3 Hz, 1H), 7.31 (s, 1H), 7.28–7.20 (m, 1H), 6.65 (m, 1H), 3.03 (m, 2H), 3.00 (s, 3H), 2.23 (m, 2H), 2.05–1.81 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 161.4, 157.3, 141.4, 139.1, 130.8, 130.7, 129.1, 128.4, 126.6, 124.3, 116.3, 110.4, 109.4, 72.0, 52.8, 49.0, 27.1, 17.0 HRMS-ESI: calcd for C27H23N3O [M + H]+m/z: 406.1919; found 406.1927.
General Procedure for the Synthesis of Compounds 6c and 6d
A mixture of compound 4n (0.191 mmol), aryl boronic acids (0.573 mmol), Pd(OAc)2 (20 mol %), and K2CO3 (0.573 mmol) in 2 mL of DMF–H2O (3:1) was stirred at 100 °C for 3 h in a sealed tube. After the completion of the reaction (TLC), the residue was diluted with EtOAc and washed with HCl (0.25 M, 20 mL), followed by saturated brine. The combined organic layer was dried over anhydrous Na2SO4 and purified through silica gel column chromatography by gradient elution using EtOAc/hexane to afford compounds 6c–6d in very good yields.
9-Methyl-6′,7-diphenyl-3,4-dihydro-1′H,2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (6c)
Nature: a brown powder; yield: 92%; Rf (30% EtOAc–hexane): 0.40, M.P: 250–252 °C. FTIR(KBr)νmax: 3244, 3057, 2924, 1647, 1481,756 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.06 (s, 1H), 7.91 (s, 1H), 7.84 (t, J = 9.0 Hz, 1H), 7.80–7.72 (m, 1H), 7.54–7.41 (m, 5H), 7.41–7.24 (m, 5H), 7.25–7.14 (m, 1H), 6.71 (t, J = 10.5 Hz, 1H), 6.50 (d, J = 6.2 Hz, 1H), 5.11 (s, 1H), 3.01 (s, 2H), 2.76 (s, 3H), 2.45–2.15 (m, 2H), 1.82 (s, 2H). 13C NMR (101 MHz, CDCl3): δ 163.5, 157.9, 148.5, 146.1, 144.4, 140.4, 140.1, 138.83, 133.1, 131.8, 130.1, 129.2, 129.1, 128.8, 128.2, 127.8, 127.5, 127.3, 127.2, 126.8, 126.4, 121.8, 115.2, 114.2, 71.3, 39.8, 35.0, 27.5, 18.1, 16.9 HRMS-ESI: calcd for C33H27N3O [M + H]+m/z: 482.2232; found 482.223.
9-Methyl-6′,7-bis(3-nitrophenyl)-3,4-dihydro-1′H, 2H-spiro[acridine-1,2′-quinazolin]-4′(3′H)-one (6d)
Nature: a brown powder; yield: 89%; Rf (30% EtOAc–hexane): 39, M.P: 262–263 °C. FTIR(KBr)νmax: 3215, 2922, 2852, 1658, 1514, 1344, 736 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.26 (d, J = 9.1 Hz, 1H), 8.12–7.98 (m, 4H), 7.90 (d, J = 8.7 Hz, 1H), 7.83 (d, J = 7.6 Hz, 1H), 7.75 (d, J = 7.6 Hz, 2H), 7.59–7.49 (m, 2H), 7.46 (t, J = 8.0 Hz, 1H), 7.06 (s, 1H), 6.74 (m, 1H), 5.19 (s, 1H), 3.14 (d, J = 5.8 Hz, 2H), 2.98 (s, 3H), 2.54–2.27 (m, 2H), 1.98–1.88 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 163.4, 158.9, 148.8, 148.7, 145.2, 142.0, 141.6, 136.3, 133.3, 133.1, 132.1, 130.0, 129.8, 129.4, 129.1, 128.3, 127.0, 122.5, 122.5, 122.1, 121.5, 120.9, 115.6, 114.2, 71.4, 40.0, 34.9, 18.1, 17.1 HRMS-ESI: calcd for C33H25N5O5 [M + H]+m/z: 572.1934; found 572.1924.
9-Methyl-3,4-dihydroacridin-1(2H)-one (5a)
Nature: a colorless powder; Rf (15% EtOAc–hexane): 0.50, M. P: 65–66 °C. FTIR(KBr)νmax: 2926, 1676, 1552, 1203, 840, 758, 696 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.14 (d, J = 8.5 Hz, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.70 (t, J = 7.6 Hz, 1H), 7.50 (t, J = 7.7 Hz, 1H), 3.21 (t, J = 6.3 Hz, 2H), 2.98 (s, 3H), 2.74 (t, J = 6.6 Hz, 2H), 2.28–1.97 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 200.8,162.2, 150.1, 148.0, 131.6, 129.2, 127.8, 126.5, 125.6, 125.5, 41.2, 34.9, 21.4, 16.2.
3,3,9-Trimethyl-3,4-dihydroacridin-1(2H)-one (5b)
Nature: a colorless powder; Rf (15% EtOAc–hexane): 0.60, M. P: 105–1076 °C. FTIR(KBr)νmax: 2960, 1691, 1556, 1259, 1014, 763 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J = 8.6 Hz, 1H), 8.05 (d, J = 8.4 Hz, 1H), 7.74 (t, J = 7.6 Hz, 1H), 7.54 (t, J = 7.7 Hz, 1H), 3.18 (s, 2H), 3.03 (s, 3H), 2.60 (s, 2H), 1.07 (s, 6H). 13C NMR (101 MHz, CDCl3): δ 200.0, 160.9, 132.3, 128.9, 128.2, 127.8, 127.0, 126.2, 125.7, 124.3, 54.8, 47.7, 32.2, 28.3, 21.4, 16.3.
7-Bromo-9-methyl-3,4-dihydroacridin-1(2H)-one (5c)
Nature: a colorless powder; Rf (15% EtOAc–hexane): 0.52, M. P: 83–85 °C. FTIR(KBr)νmax: 3251, 1662,1558, 1375, 1172, 1076, 910, 831 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.09 (d, J = 1.4 Hz, 1H), 7.65 (dt, J = 8.9, 5.3 Hz, 2H), 3.11 (t, J = 6.3 Hz, 2H), 2.79 (s, 3H), 2.69 (t, J = 6.6 Hz, 2H), 2.15–2.02 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 200.1, 162.4, 148.5, 146.3, 134.5, 130.7, 128.7, 127.63, 125.6, 120.3, 40.9, 34.6, 21.1, 15.9. HRMS-ESI: calcd for C14H12BrNO [M + H]+m/z: 290.1433; found 290.0160.
7-Bromo-3,3,9-trimethyl-3,4-dihydroacridin-1(2H)-one (5d)
Nature: a colorless powder; Rf (15% EtOAc–hexane): 0.63, M. P: 105–107 °C. FTIR(KBr)νmax: 3072, 2939, 2873, 1670, 1556, 1479, 906, 827 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J = 1.9 Hz, 1H), 7.77 (m, 2H), 3.08 (s, 2H), 2.94 (s, 3H), 2.60 (s, 2H), 1.06 (s, 6H). 13C NMR (101 MHz, CDCl3): δ 200.5, 161.6, 148.7, 147.0, 134.8, 131.0, 129.1, 128.0, 124.8, 120.6, 54.9, 48.6, 32.2, 28.4, 16.0 HRMS-ESI: calcd for C16H16BrNO [M + H]+m/z: 318.0493; found 318.0497.
3,9-Dimethyl-3,4-dihydroacridin-1(2H)-one (5e)
Nature: a colorless powder; Rf (15% EtOAc–hexane): 0.55, M. P: 78–80 °C. FTIR(KBr)νmax: 2947, 1674, 1560, 1211, 759 cm–1; 1H NMR (400 MHz, CDCl3): δ 8.14 (d, J = 8.5 Hz, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.70 (t, J = 7.6 Hz, 1H), 7.49 (t, J = 7.6 Hz, 1H), 3.29 (d, J = 16.0 Hz, 1H), 2.98 (s, 3H), 2.94–2.76 (m, 2H), 2.39 (m, 2H), 1.13 (d, J = 6.1 Hz, 3H). 13C NMR (101 MHz, CDCl3): 200.4, 162.6, 148.9, 146.6, 134.8, 131.0, 129.1, 127.9, 125.9, 120.5, 41.1, 34.8, 21.3, 16.1.
7-Bromo-3,9-dimethyl-3,4-dihydroacridin-1(2H)-one (5f)
Nature: a colorless powder; Rf (15% EtOAc–hexane): 0.56, M. P: 93–94 °C. FTIR(KBr)νmax: 3572, 3452, 3319, 1656, 1558, 1219, 840 cm–1; colorless powder; Rf (15% EtOAc–hexane): 0.56, M. P: 93–94 °C 1H NMR (400 MHz, CDCl3): δ 8.26 (s, 1H), 7.78 (m, 2H), 3.28 (d, J = 16.2 Hz, 1H), 3.00–2.74 (m, 5H), 2.52–2.27 (m, 2H), 1.13 (d, J = 6.2 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 200.3, 162.0, 149.0, 146.5, 135.0, 130.8, 129.0, 127.9, 125.3, 120.7, 49.2, 42.8, 28.5, 21.2, 16.1. HRMS-ESI: calcd for C15H14BrNO [M + H]+m/z: 304.0337; found 304.0375.
10-Methyl-1,6,7,8-tetrahydrofuro[3,4-b]acridin-9(3H)-one (5g)
Nature: a colorless powder; Rf (15% EtOAc–hexane): 0.50, M. P: 112–114 °C. FTIR(KBr)νmax: 1697, 1641, 1589, 1452, 1257, 1174, 1114, 819, 715, 529 cm–1; 1H NMR (400 MHz, CDCl3): δ 7.31 (s, 1H), 7.20 (s, 1H), 6.05 (s, 2H), 3.18–3.08 (m, 2H), 2.83 (s, 3H), 2.68 (t, J = 6.6 Hz, 2H), 2.09 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 200.60, 160.63, 152.31, 148.19, 148.06, 146.99, 124.55, 124.12, 105.40, 102.18, 100.74, 41.14, 34.45, 21.59, 16.53. HRMS-ESI: calcd for C15H13NO3 [M + H]+m/z: 256.0978; found 256.0941.
Acknowledgments
K.G. and P.E. thank VIT, Vellore, for research fellowships. S.K. thanks VIT Management for providing infrastructure facilities for carrying out this work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c00674.
K.S. thanks VIT Management for providing VIT SEED GRANT for carrying out this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Kumar S.; Bawa S.; Gupta H. Biological activities of quinoline derivatives. Mini-Rev. Med. Chem. 2009, 9, 1648–1654. 10.2174/138955709791012247. [DOI] [PubMed] [Google Scholar]
- Cho C. S.; Oh B. H.; Shim S. C. Synthesis of quinolines by ruthenium-catalyzed heteroannulation of anilines with 3-amino-1-propanol. J. Heterocycl. Chem. 1999, 36, 1175–1178. 10.1002/jhet.5570360510. [DOI] [Google Scholar]
- Makioka Y.; Shindo T.; Taniguchi Y.; Takaki K.; Fujiwara Y. Ytterbium (III) triflate catalyzed synthesis of quinoline derivatives from N-arylaldimines and vinyl ethers. Synthesis 1995, 1995, 801–804. 10.1055/s-1995-4002. [DOI] [Google Scholar]
- Sangu K.; Fuchibe K.; Akiyama T. A novel approach to 2-arylated quinolines: electrocyclization of alkynyl imines via vinylidene complexes. Org. Lett. 2004, 6, 353–355. 10.1021/ol036190a. [DOI] [PubMed] [Google Scholar]
- Crousse B.; Bégué J. P.; Bonnet-Delpon D. Synthesis of tetrahydroquinoline derivatives from α-CF3-N-arylaldimine and vinyl ethers. Tetrahedron Lett. 1998, 39, 5765–5768. 10.1016/S0040-4039(98)01202-7. [DOI] [Google Scholar]
- Crousse B.; Bégué J. P.; Bonnet-Delpon D. Synthesis of 2-CF (3)-tetrahydroquinoline and quinoline derivatives from CF (3)-N-aryl-aldimine. J. Org. Chem. 2000, 65, 5009–5013. 10.1021/jo9918807. [DOI] [PubMed] [Google Scholar]
- Katritzky A. R.; Arend M. A convenient and highly regioselective one-pot synthesis of quinolines by addition of a Vilsmeier-type reagent to N-arylimines. J. Org. Chem. 1998, 63, 9989–9991. 10.1021/jo981252+. [DOI] [Google Scholar]
- Abdelsalam E. A.; Zaghary W. A.; Amin K. M.; Abou Taleb N. A.; Mekawey A. A.; Eldehna W. M.; Abdel-Aziz H. A.; Hammad S. F. Synthesis and in vitro anticancer evaluation of some fused indazoles, quinazolines and quinolines as potential EGFR inhibitors. Bioorg. Chem. 2019, 89, 102985 10.1016/j.bioorg.2019.102985. [DOI] [PubMed] [Google Scholar]
- Novanna M.; Kannadasan S.; Shanmugam P. Phosphotungstic acid mediated, microwave assisted solvent-free green synthesis of highly functionalized 2ˈ-spiro and 2, 3-dihydro quinazolinone and 2-methylamino benzamide derivatives from aryl and heteroaryl 2-amino amides. Tetrahedron Lett. 2019, 60, 201–206. 10.1016/j.tetlet.2018.12.011. [DOI] [Google Scholar]
- Liu X.; Fu H.; Jiang Y.; Zhao Y. A Simple and Efficient Approach to Quinazolinones under Mild Copper-Catalyzed Conditions. Angew. Chem. 2009, 121, 354–357. 10.1002/ange.200804675. [DOI] [PubMed] [Google Scholar]
- Xu W.; Jin Y.; Liu H.; Jiang Y.; Fu A. H. Copper-Catalyzed Domino Synthesis of Quinazolinones via Ullmann-Type Coupling and Aerobic Oxidative C– H Amidation. Org. Lett. 2011, 13, 1274–1277. 10.1021/ol1030266. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Fang J. One-pot synthesis of quinazolinones via iridium-catalyzed hydrogen transfers. J. Org. Chem. 2011, 76, 7730–7736. 10.1021/jo201054k. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Wang M.; Zhang C.; Zhang Z.; Lu J.; Wang F. The cascade synthesis of quinazolinones and quinazolines using an α-MnO2 catalyst and tert-butyl hydroperoxide (TBHP) as an oxidant. Chem. Commun. 2015, 51, 9205–9207. 10.1039/C5CC02785C. [DOI] [PubMed] [Google Scholar]
- Zheng J.; Zhang Y.; Wang D.; Cui S. Silver (I)-Mediated Phosphorylation/Cyclization Cascade of N-Cyanamide Alkenes for Divergent Access to Quinazolinones and Dihydroisoquinolinones. Org. Lett. 2016, 18, 1768–1771. 10.1021/acs.orglett.6b00481. [DOI] [PubMed] [Google Scholar]
- Bie Z.; Li G.; Wang L.; Lv Y.; Niu J.; Gao S. A facile vanadium-catalyzed aerobic oxidative synthesis of quinazolinones from 2-aminobenzamides with aldehydes or alcohols. Tetrahedron Lett. 2016, 57, 4935–4938. 10.1016/j.tetlet.2016.09.077. [DOI] [Google Scholar]
- Sharma M.; Pandey S.; Chauhan K.; Sharma D.; Kumar B.; Chauhan P. M. Cyanuric chloride catalyzed mild protocol for synthesis of biologically active dihydro/spiro quinazolinones and quinazolinone-glycoconjugates. J. Org. Chem. 2012, 77, 929–937. 10.1021/jo2020856. [DOI] [PubMed] [Google Scholar]
- Rambabu D.; Kumar S. K.; Sreenivas B. Y.; Sandra S.; Kandale A.; Misra P.; Rao M. B.; Pal M. Ultrasound-based approach to spiro-2, 3-dihydroquinazolin-4 (1H)-ones: their in vitro evaluation against chorismate mutase. Tetrahedron Lett. 2013, 54, 495–501. 10.1016/j.tetlet.2012.11.057. [DOI] [Google Scholar]
- Salehi P.; Dabiri M.; Baghbanzadeh M.; Bahramnejad M. One-Pot, Three-Component Synthesis of 2, 3-Dihydro-4 (1 H)-quinazolinones by Montmorillonite K-10 as an Efficient and Reusable Catalyst. Synth. Commun. 2006, 36, 2287–2292. 10.1080/00397910600639752. [DOI] [Google Scholar]
- Revathy K.; Lalitha A. p-TSA-catalyzed synthesis of spiroquinazolinones. J. Iran. Chem. Soc. 2015, 12, 2045–2049. 10.1007/s13738-015-0680-2. [DOI] [Google Scholar]
- Shaterian H. R.; Rigi F. An efficient synthesis of quinazoline and xanthene derivatives using starch sulfate as a biodegradable solid acid catalyst. Res. Chem. Intermed. 2015, 41, 721–738. 10.1007/s11164-013-1223-z. [DOI] [Google Scholar]
- Yang X.; Cheng G.; Shen J.; Kuai C.; Cui X. Cleavage of the C–C triple bond of ketoalkynes: synthesis of 4 (3 H)-quinazolinones. Org. Chem. Front. 2015, 2, 366–368. 10.1039/C4QO00260A. [DOI] [Google Scholar]
- a Venkateshwarlu R.; Murthy V. N.; Tadiparthi K.; Nikumbh S. P.; Jinkala R.; Siddaiah V.; Madhu babu M. V.; Mohan H. R.; Raghunadh A. Base mediated spirocyclization of quinazoline: one-step synthesis of spiro-isoindolinone dihydroquinazolinones. RSC Adv. 2020, 10, 9486–9491. 10.1039/C9RA09567E. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sharma M.; Pandey S.; Chauhan K.; Sharma D.; Kumar B.; Chauhan P. M. Cyanuric chloride catalyzed mild protocol for synthesis of biologically active dihydro/spiro quinazolinones and quinazolinone-glycoconjugates. J. Org. Chem. 2012, 77, 929–937. 10.1021/jo2020856. [DOI] [PubMed] [Google Scholar]
- a Auti P. S.; George G.; Paul A. T. Recent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids. RSC Adv. 2020, 10, 41353–41392. 10.1039/D0RA06642G. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Han S.; Sang Y.; Wu Y.; Tao Y.; Pannecouque C.; De Clercq E.; Zhuang C.; Chen F. E. Molecular hybridization-inspired optimization of diarylbenzopyrimidines as HIV-1 nonnucleoside reverse transcriptase inhibitors with improved activity against K103N and E138K mutants and pharmacokinetic profiles. ACS Infect. Dis. 2020, 6, 787–801. 10.1021/acsinfecdis.9b00229. [DOI] [PubMed] [Google Scholar]
- Cagir A.; Jones S. H.; Gao R.; Eisenhauer B. M.; Hecht S. M. Luotonin A. A naturally occurring human DNA topoisomerase I poison. J. Am. Chem. Soc. 2003, 125, 13628–13629. 10.1021/ja0368857. [DOI] [PubMed] [Google Scholar]
- Broadwater S. J.; Roth S. L.; Price K. E.; Kobašlija M.; McQuade D. T. One-pot multi-step synthesis: a challenge spawning innovation. Org. Biomol. Chem. 2005, 3, 2899–2906. 10.1039/b506621m. [DOI] [PubMed] [Google Scholar]
- Kwon S. H.; Seo H. A.; Cheon C. H. Total synthesis of luotonin A and rutaecarpine from an aldimine via the designed cyclization. Org. Lett. 2016, 18, 5280–5283. 10.1021/acs.orglett.6b02597. [DOI] [PubMed] [Google Scholar]
- Badolato M.; Aiello F.; Neamati N. 2, 3-Dihydroquinazolin-4 (1 H)-one as a privileged scaffold in drug design. RSC Adv. 2018, 8, 20894–20921. 10.1039/C8RA02827C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCDC-2101995(compound 4b) contains the supplementary crystallographic data for this paper.This data can be obtained free of charge from the Cambridge CrystallographicData Centre via. www.ccdc.cam.ac.uk/datarequest/cif
- Nigam S.; Rutan S. Principles and applications of solvatochromism. Appl. Spectrosc. 2001, 55, 362A–370A. 10.1366/0003702011953702. [DOI] [Google Scholar]
- Allaoui Z. I. M.; Le Gall E.; Fihey A.; Plaza-Pedroche R.; Katan C.; Robin-Le Guen F.; Rodriguez-Lopez J.; Achelle S. Push-pull (iso) quinoline chromophores: synthesis, photophysical properties, and use for white light emission. Chem. - Eur. J. 2020, 26, 8153–8161. 10.1002/chem.202000817. [DOI] [PubMed] [Google Scholar]
- Novanna M.; Kannadasan S.; Shanmugam P. Microwave assisted synthesis and photophysical properties of blue emissive 2-amino-3-carboxamide-1, 1′-biaryls and 4-(arylamino)-[1, 1′-biphenyl]-3-carboxamides via Suzuki and Chan-Evans-Lam coupling. Dyes. Pigm. 2020, 174, 108015–108024. 10.1016/j.dyepig.2019.108015. [DOI] [Google Scholar]
- Pillai S.; Kozlov M.; Marras S. A.; Krasnoperov L. N.; Mustaev A. New cross-linking quinoline and quinolone derivatives for sensitive fluorescent labeling. J. Fluoresc. 2012, 22, 1021–1032. 10.1007/s10895-012-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamidala R.; Subramani M. S.; Samser S.; Biswal P.; Venkatasubbaiah K. Chemoselective Alkylation of Aminoacetophenones with Alcohols by Using a Palladacycle-Phosphine Catalyst. Eur. J. Org. Chem 2018, 2018, 6286–6296. 10.1002/ejoc.201801155. [DOI] [Google Scholar]
- Laha J. K.; Manral N.; Hunjan M. K. Palladium-catalysed regioselective N-arylation of anthranilamides: a tandem route for dibenzodiazepinone synthesis. New. J. Chem 2019, 43, 7339–7343. 10.1039/C9NJ00539K. [DOI] [Google Scholar]
- Rahmannejadi N.; Yavari I.; Khabnadideh S. Synthesis and antitumor activities of novel bis-quinazolin-4 (3H)-ones. J. Heterocycl. Chem. 2020, 57, 978–982. 10.1002/jhet.3749. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.