

Abstract
Glutamyl-tRNA synthetase 2 (encoded by EARS2) is a mitochondrial aminoacyl-tRNA synthetase required to translate the 13 subunits of the electron transport chain encoded by the mitochondrial DNA. Pathogenic EARS2 variants cause combined oxidative phosphorylation deficiency, subtype 12 (COXPD12), an autosomal recessive disorder involving lactic acidosis, intellectual disability, and other features of mitochondrial compromise. Patients with EARS2 deficiency present with variable phenotypes ranging from neonatal lethality to a mitigated disease with clinical improvement in early childhood. Here, we report a neonate homozygous for a rare pathogenic variant in EARS2 (c.949G>T; p.G317C). Metabolomics in primary fibroblasts from this patient revealed expected abnormalities in TCA cycle metabolites, as well as numerous changes in purine, pyrimidine, and fatty acid metabolism. To examine genotype-phenotype correlations in COXPD12, we compared the metabolic impact of reconstituting these fibroblasts with wild-type EARS2 versus four additional EARS2 variants from COXPD12 patients with varying clinical severity. Metabolomics identified a group of signature metabolites, mostly from the TCA cycle and amino acid metabolism, that discriminate between EARS2 variants causing relatively mild and severe COXPD12. Taken together, these findings indicate that metabolomics in patient-derived fibroblasts may help establish genotype-phenotype correlations in EARS2 deficiency and likely other mitochondrial disorders.
Keywords: EARS2, genotype-phenotype correlation, inborn errors of metabolism, lactic acidosis, metabolomics, mitochondria
1 |. INTRODUCTION
Mitochondrial diseases are among the largest and most common categories of inborn errors of metabolism (IEMs). Severe mitochondrial diseases manifest in early childhood and are associated with high morbidity and mortality.1 Mitochondrial diseases can result from mutations in either the nuclear or the mitochondrial genome because the electron transport chain (ETC) requires contributions from both. These disorders can impact essentially any organ system, or many organ systems simultaneously, with involvement of the nervous system, muscle, heart, and liver being particularly prominent.
Mitochondrial aminoacyl-tRNA synthetases encoded by nuclear genes are essential components of the mitochondrial translation machinery. Pathogenic variants in mitochondrial aminoacyl-tRNA synthetases can compromise ETC function, because charging tRNAs with their cognate amino acids is required to synthesize the 13 subunits of ETC complexes I, III, and IV and ATP synthase (complex V) encoded by the mitochondrial genome. Patients with mitochondrial aminoacyl-tRNA synthetase deficiencies are clinically heterogeneous, with remarkable diversity in their phenotypic severity and age of onset.2 As with other mitochondrial diseases, aminoacyl-tRNA synthetase defects manifest in numerous tissues. Neurological features include early-onset encephalopathy, seizures, leukodystrophy, and sensorineural hearing loss.3 Involvement of the liver, kidney, eye, and heart has also been reported.4 Establishing a specific molecular cause of the disease often requires advanced diagnostics including next-generation sequencing (NGS).
Biallelic variants in all 19 mitochondrial aminoacyl-tRNA synthetase genes have been associated with human disease. This includes EARS2, a gene on chromosome 16p12.2 encoding the mitochondrial glutamyl-tRNA synthetase.3 Human EARS2 is a non-discriminating glutamyl-tRNA synthetase that not only aminoacylates tRNAGlu, but misaminoacylates tRNAGln with Glu.5 The Glu moiety of Glu-tRNAGln is then transaminated by glutamic-tRNAGln amidotransferase with Gln as an amide donor to generate Gln-tRNAGln in human mitochondria. Previous genetic studies have identified pathogenic EARS2 variants as the cause of COXPD12 (MIM:614924), also known as leukoencephalopathy with Thalamus and Brainstem involvement and high lactate (LTBL).6 Biochemical and neuroimaging features of this disease include characteristic lactate elevation in the brain and body fluids, symmetric abnormalities in the cerebral white matter, thalami, and brainstem, and variable involvement of the corpus callosum.
Pathogenic EARS2 variants have been reported in about 30 patients. These patients fall into two clinical groups with either severe or relatively mild phenotypes.7–10 Complex irreversible neurological features with progressive psychomotor disabilities and/or early lethality characterize the severe group. Patients in the other group lack the typical neuroradiological features and display clinical improvement in early childhood. Such marked phenotypic heterogeneity may reflect residual EARS2 activity and therefore better mitochondrial function in patients with the milder disease.
NGS has made it increasingly common to detect rare variants in patients suspected to have monogenic disorders. However, interpreting the pathogenic significance of missense variants still largely relies on computational prediction rather than functional validation.11 Distinguishing neutral or benign variants from disease-causing ones remains a significant challenge, as does predicting disease severity for different pathogenic variants. Broad profiling of metabolite abundance (metabolomics) provides a deep phenotyping approach with the potential to deliver extensive phenotype-relevant information not achievable using conventional diagnostics. Furthermore, because metabolomics can be performed in intact cells or tissues, the resulting data complement in vitro assays of enzyme activity and may be more predictive of a variant’s impact on physiology. We and others have used metabolomics to help interpret genomic variants whose relationship to the phenotype was unclear.12,13
Here we report a patient with severe COXPD12 caused by a pathogenic EARS2 variant. We obtained fibroblasts from this patient, complemented them with wild-type EARS2 or a series of human-derived EARS2 variants, and derived metabolomic signatures of severe and milder forms of COXPD12.
2 |. RESULTS
2.1 |. Homozygous EARS2 variant in a newborn with fatal lactic acidosis, respiratory failure, and agenesis of corpus callosum
Patient P3-041 was a female born at 40 weeks of gestation after an uneventful pregnancy to nonconsanguineous Mexican parents. The APGAR score was 9 at 1 and 5 min. The weight and length were normal, but the head circumference was below the third percentile and the baby was brachycephalic. Shortly after birth, the baby developed persistent pulmonary hypertension and lactic acidosis (Figure 1A,B). Due to the concern for an oxidative phosphorylation defect, biotin, thiamine, and riboflavin were administered, but this did not improve the blood pH or lactate levels. A modified ketogenic diet was initiated on Day 3, but the lactate rose further, peaking at 57.6 mmol/L. Mildly elevated liver transaminases, lactate dehydrogenase, and ammonia were noted in the blood. The patient was too unstable for a brain MRI, but a brain ultrasound revealed colpocephaly and absence of the corpus callosum. Despite aggressive supportive care, the baby developed worsening bradycardia, hypotension and respiratory depression, and died on the 5th day of life. Analysis of the electron transport chain in skeletal muscle obtained shortly after death revealed decreased activities of multiple complexes (Table 1).
FIGURE 1.
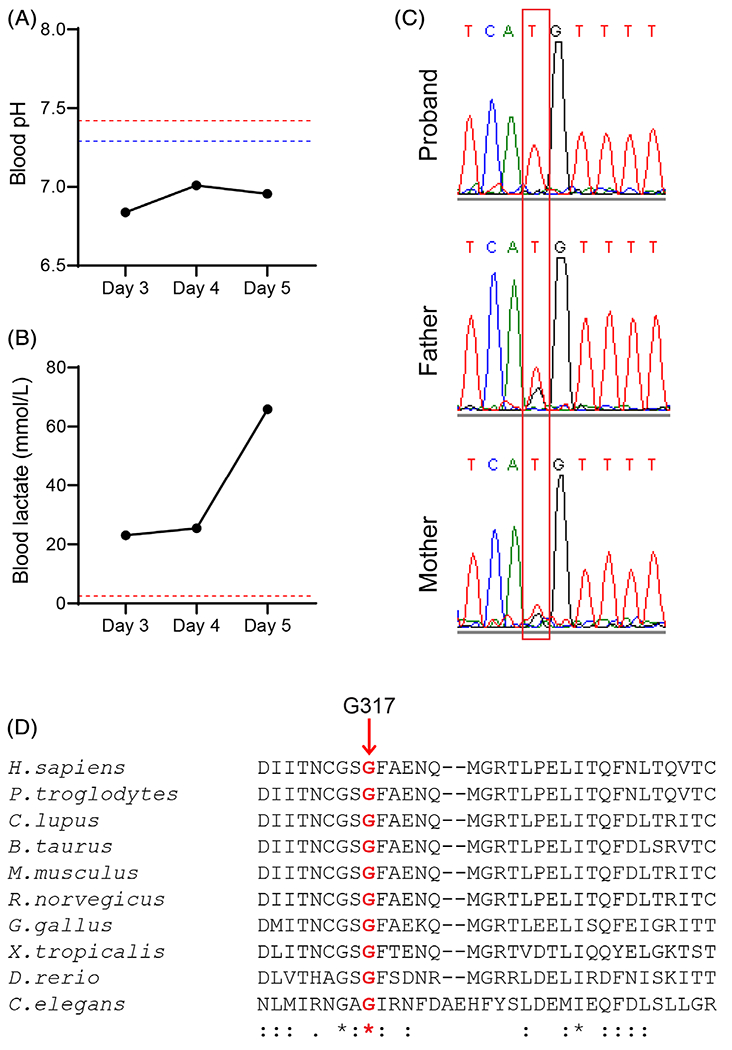
Homozygous EARS2 variants in a newborn with severe lactic acidosis. A, B, Clinical results for blood pH and lactate levels throughout the disease course. The upper and lower limits of the reference ranges are indicated in red or blue, respectively. C, Chromatogram of EARS2 sequences from the proband and her parents. The mutation is highlighted by the red box. D, Conservation of the G317 residue (red bolded) across 10 species
TABLE 1.
Clinical results of electron transport chain activities in skeletal muscle biopsy by Baylor Miraca Genetics Laboratory
| Electron transport chain activities | ETC complexes | Patient (% of mean) (nmol min−1 mg−1 protein) | Control ± SD (nmol min−1 mg−1 protein) |
|---|---|---|---|
| NADH:Ferricyanide dehydrogenase | I | 78 (22) low | 360.4 ± 96.3 |
| NADH:cytochrome c reductase | I + III | ||
| Total | 16.1 (57) | 28.4 ± 6.1 | |
| Rotenone sensitive | 0.94 (11) low | 8.7 ± 3.9 | |
| Succinate dehydrogenase | II | 7.17 (75) | 9.6 ± 3.0 |
| Succinate: cytochrome c reductase | II + III | 3.3 (79) | 4.2 ± 1.2 |
| Cytochrome c oxidase | IV | 10.4 (26) low | 40.3 ± 15.5 |
| Citrate synthase | 320 (109) | 293.1 ± 68.0 |
Molecular analysis ruled out mitochondrial DNA depletion in the skeletal muscle. WES revealed a homozygous c.949G>T (p.G317C) variant in EARS2 in the proband; both parents were heterozygous for the variant, which was confirmed in all three by Sanger sequencing (Figure 1C). G317C is a rare allele (allelic frequency 0.0008 in Latinos and 0.0001 overall in the gnomAD database) and was previously reported in a COXPD12 patient.6 G317 is conserved across multiple species (Figure 1D) and is located within the catalytic domain of mitochondrial glutamyl-tRNA synthetase. In silico structural analyses by polymorphism phenotyping v.2 (PolyPhen-2) and sorting intolerant from tolerant (SIFT) predict that the G317C variant compromises EARS2 function.
2.2 |. Metabolomic profiling of patient-derived fibroblasts with EARS2 variants
The full scope of metabolic abnormalities arising from EARS2 deficiency is unknown. To characterize the metabolic alterations associated with homozygosity for G317C, we used fibroblasts derived from the patient described above. The endogenous EARS2 expression was similar in the patient’s fibroblasts compared to fibroblasts from five control subjects (Figure 2A), indicating that G317C does not affect EARS2 protein stability. A targeted metabolomics assay analyzed 290 metabolites in these fibroblast lines (Table S1, Supporting Information) and the data were processed through a statistical workflow for quality control filtering, missing data imputation and normalizing each metabolite to the total ion count. An unsupervised analysis revealed clustering of the biological replicates derived from each cell line, and differentiated P3-041 fibroblasts from the control cells (Figures 2B and S1), indicating a distinct metabolic profile in the cells from the patient. A set of 117 metabolites was found to discriminate between P3-041 cells and fibroblasts from healthy controls, using cutoffs of t-test p < 0.05 and fold changes ≥1.5 or ≤0.67. Metabolic pathway enrichment analysis14 revealed perturbation of the TCA cycle and related metabolites, with α-ketoglutarate, 2-hydroxyglutarate, cis-aconitate, and malate accumulating in P3-041 fibroblasts (Figure 2C,D).
FIGURE 2.
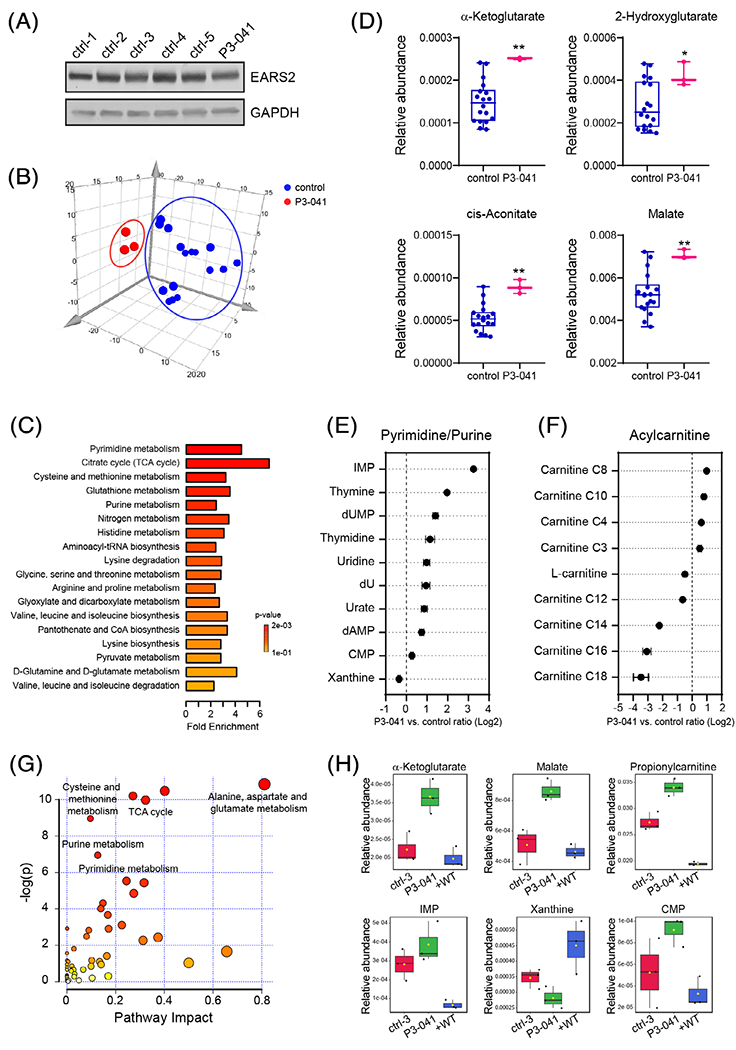
Metabolic abnormalities in patient-derived fibroblasts with the EARS2 G317C variant. A, Immunoblot analysis of primary human fibroblasts from five healthy subjects and patient P3-041. B, Principal component analysis (PCA) of the metabolomes of healthy control and P3-041 fibroblasts. The metabolomic profiling was performed on primary human fibroblasts from six healthy subjects and patient P3-041. C, Metabolite set enrichment analysis (MSEA) of metabolites differentially changed in the P3-041 fibroblasts compared to the control primary fibroblasts. The significantly altered metabolites were filtered by p < 0.05 and fold changes ≥1.5 or ≤0.67. D, Comparison of several metabolites related to the TCA cycle between control and P3-041 primary fibroblasts. **p < 0.01, *p < 0.05, calculated by unpaired 2-tailed t test. E, Metabolic abnormalities related to purine and pyrimidine pathways in P3-041 fibroblasts. Metabolites are ordered by mean log2-transformed P3-041 versus control ratio. F, Acylcarnitine abnormalities in P3-041 fibroblasts compared to controls. Metabolites are ordered by mean log2-transformed P3-041 versus control ratio. G, Pathway enrichment of metabolites that are normalized by ectopic expression of WT EARS2 in P3-041 fibroblasts. The significantly downregulated metabolites by WT EARS2 expression (p < 0.05 and fold changes ≤0.67) were used in MetaboAnalyst pathway analysis. The circle size corresponds to impact values (x-axis) and the color reflects −log(p) values (y-axis). H,Representative metabolites of the significantly affected pathways in (C) but normalized by expression of WT EARS2. The control (ctrl-3) in this experiment is a human neonatal fibroblast line from ATCC
Many of the altered metabolites were from nucleotide-related pathways, including both purines and pyrimidines (Figure 2C,E). Mitochondrial dysfunction has been linked to aberrant synthesis of ribonucleotides and deoxyribonucleotides due to insufficient synthesis of ATP and aspartate, and suppression of mitochondrial enzymes required for pyrimidine and purine synthesis.15,16 In de novo purine synthesis, inosine monophosphate (IMP) is the precursor for both AMP and GMP. Thus, the disproportionate accumulation of IMP relative to AMP and GMP may indicate an imbalance between IMP production through de novo synthesis or salvage pathway, and the conversion of IMP to other nucleotides, including AMP and GMP. Pyrimidine-related metabolites altered in P3-041 fibroblasts included nucleosides and bases. A limitation of metabolomics is that changes in metabolite levels do not necessarily distinguish between activation and suppression of the pathway. Nevertheless, these data indicate widespread perturbation of nucleotide homeostasis impacting both purines and pyrimidines. Several acylcarnitines were also altered in P3-041 fibroblasts (Figure 2F). These included increases of short-and medium-chain acylcarnitines and reduced levels of long-chain acylcarnitines. These may reflect alterations in fatty acid and amino acid degradation in the mitochondria, both of which require a functional ETC to accept reducing equivalents from oxidation of these substrates.
To directly assess the role of the G317C EARS2 variant in these perturbations, we transduced P3-041 fibroblasts with a vector expressing wild-type (WT) EARS2. Ectopic expression of WT EARS2 rescued many of the metabolic abnormalities detected in P3-041 fibroblasts, including acylcarnitines, nucelotides, and TCA cycle metabolites (Figure 2G,H and Table S2). These findings confirm EARS2’s role in regulating these pathways and implicate loss of EARS2 function in the metabolic aberrations observed in the patient’s fibroblasts.
2.3 |. Structural considerations of human pathogenic EARS2 variants
To explore the relationship between EARS2 genotypes and metabolic phenotypes, we first chose several variants observed in COXPD12 patients with varying degrees of clinical severity (Table 2). Four patients, including ours, had homozygous variants and the fifth was a compound heterozygote for missense and start-loss (p.Met1?) variants.8 All five missense variants are located within the conserved aminoacyl-tRNA synthetase domain (Figure 3A,B). Structural modeling of human EARS2 was obtained from the SWISS-MODEL repository17 based on the crystal structure of the glutamyl-tRNA synthetase from Elizabethkingia meningosepticum (PDB: 6brl), and superimposed on the crystal structure of the glutamyl-tRNA synthetase from Escherichia coli (PDB: 1qtq). G317, the residue mutated in our patient with early-lethal COXPD12, is located in a loop region on the surface of the catalytic domain in close proximity to the tRNA substrate (Figure 3B). The G317C substitution may change the local backbone conformation and impair the interaction between EARS2 and tRNA due to cysteine’s increased size and hydrophobicity. Both I62F and K65E occur at highly conserved residues and resulted in lethal COXPD12. I62 is buried in the core of the catalytic domain, and interacts with several other hydrophobic residues (Figure 3B). Mutation of I62 to F, a larger hydrophobic residue, may cause steric hindrance of van der Waals interactions with surrounding hydrophobic residues and is postulated to destabilize the tertiary structure of the catalytic domain. A decrease of EARS2 protein abundance was observed in this patient’s fibroblasts, suggesting that I62F might also affect EARS2 stability.8 K65 and R108 are both surface residues that form salt bridges with conserved residues nearby in the homology model (Figure 3B). K65E introduces a negatively charged residue interfering with hydrogen bond formation and sidechain interactions with D103. Changing R108 to the larger hydrophobic residue Trp abolishes ionic interactions with E92 and E96, and perturbs hydrogen bond formation. G301 is located in a tight turn region between two helices and lies in the interface between the catalytic domain and the anticodon-binding domain. The G301A mutation could destabilize the local backbone conformation, increase the regional hydrophobicity and induce a local misfolding. Altogether, the localization of these amino acid substitutions suggest that all of them might impair catalytic function due to weakened substrate binding or destabilization of the protein, but it is unclear from the structural analysis how these variants relate to phenotypic severity.
TABLE 2.
Clinical presentations of patients with the indicated EARS2 mutations that were selected for functional analysis
| Variant | Age of onset | Phenotype | Disease course | Reference |
|---|---|---|---|---|
| G317C/G317C (severe) | Neonate | Lactic acidosis, agenesis of corpus callosum, respiratory failure | Died at 5 days old | This study |
| I62F/Met1? (severe) | Neonate | Lactic acidosis, hypotonia, agenesis of corpus callosum, hepatomegaly | Died at 5 months old | Oliveira et al.17 |
| K65E/K65E (severe) | Neonate | Lactic acidosis, hypotonia, dysgenesis of corpus callosum, hepatomegaly | Died at 3 months old | Talim et al.23 |
| R108W/R108W (mild) | Infancy | Mild elevation of lactate, seizure, ataxia, leukoencephalopathy | Follow up at 3 years old with improvement | Taskin et al.24 |
| G301A/G301A (mild) | Infancy | Mild elevation of lactate, hypotonia, failure to thrive, leukoencephalopathy, developmental delay | Follow up at 6 years old with improvement | Biancheri et al.2 |
FIGURE 3.
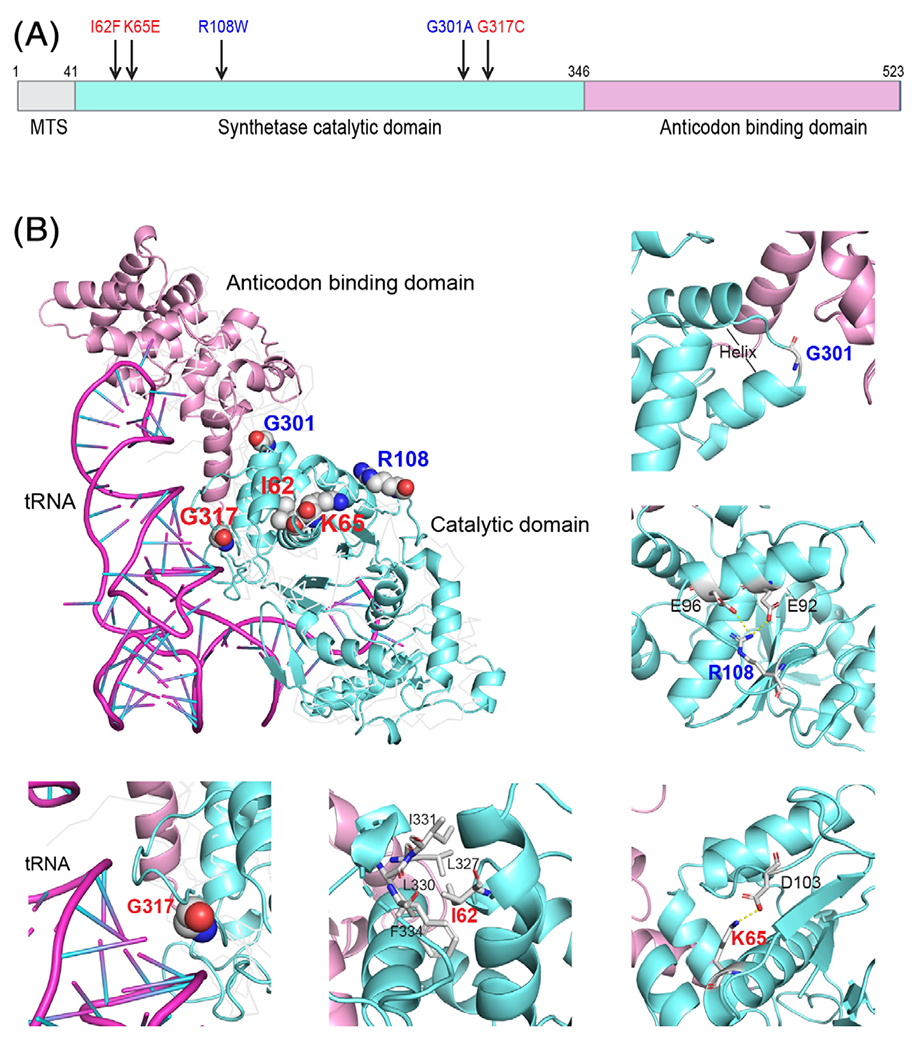
Structural representation of selected EARS2 residues with pathogenic variants. A, Schematic of EARS2 protein with functional domains. The selected disease-associated variants are indicated, with mild variants in blue and severe variants in red. MTS, mitochondrial targeting sequence. B, Three-dimensional structure of EARS2 homology model with highlighted variant positions (upper left panel). The enlargements show the local interaction for each residue as described in the text. The residues associated with mild COXPD12 are in blue and with severe COXPD12 in red
2.4 |. Metabolomic impact of human pathogenic EARS2 variants
We next compared the metabolomic impact of complementing P3-041 cells with each of five MYC-tagged EARS2 variants or WT EARS2. Each variant was expressed in excess compared to endogenous EARS2, but all alleles including WT expressed at comparable levels (Figure 4A). Thus, in these models, most EARS2 activity is predicted to arise from the over-expressed MYC-tagged form. A targeted metabolomics approach detected 352 metabolites in these fibroblast lines (Table S3). Principal component analysis (PCA) identified distinct metabolic profiles for cells expressing WT EARS2 and variants from mild or severe COXPD12 patients (Figure 4B and S2). We then identified 105 metabolites whose abundance varied among cells expressing WT EARS2 and cells expressing variants from patients with mild or severe COXPD12 (Table S4). These metabolites mapped to several pathways, including amino acid catabolism, the TCA cycle, purine and pyrimidine metabolism (Figure 4C,D). TCA cycle-related intermediates, including citrate, α-ketoglutarate and 2-hydroxyglutarate, were higher in cells expressing I62F, K65E, and G317C variants and were normalized by expressing EARS2 variants from patients with mild COXPD12 or WT EARS2 (Figure 4D and Table S4). Propionylcarnitine and isovalerylcarnitine, two intermediates in branched-chain amino acid oxidation, were also elevated in cells expressing variants from severe COXPD12, but were progressively depleted by expression of mild variants and wild-type EARS2 (Figure 4D and Table S4). Cells expressing EARS2 variants from severe COXPD12 had prominent elevations in ophthalmate (Figure 4E and Table S4), a marker of oxidative stress.18 In summary, these data demonstrate metabolomic differences correlating with different degrees of EARS2 dysfunction caused by the expression of EARS2 variants associated with distinct clinical features (Figure 4E).
FIGURE 4.
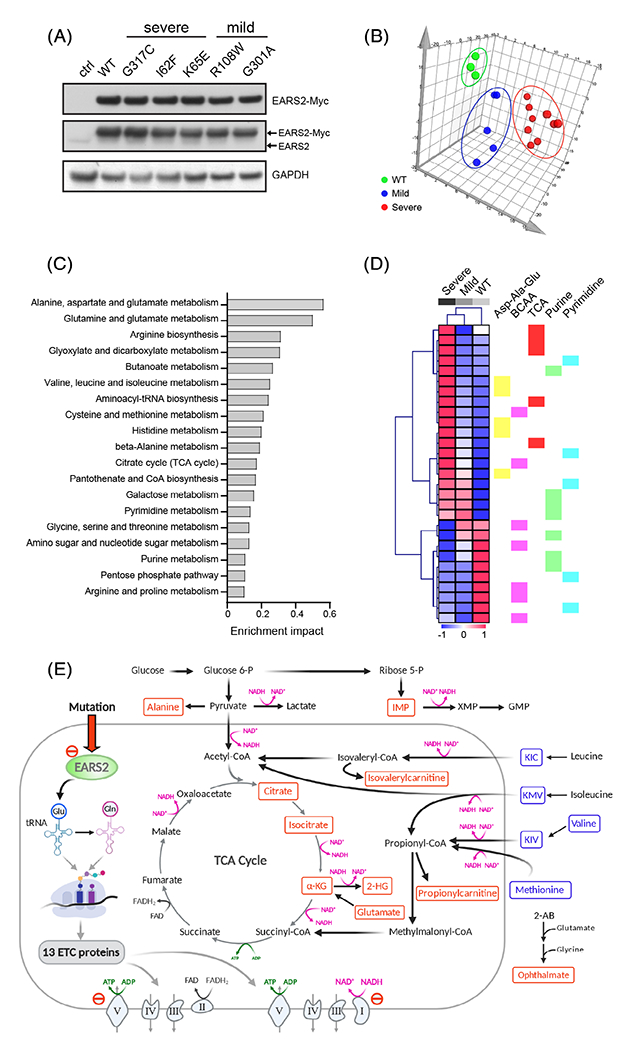
Metabolomic analysis of EARS2 variants associated with differential disease severity. A, Immunoblot analysis of P3-041 fibroblasts with ectopic expression of the indicated EARS2 variants. The upper blot was probed with an anti-MYC antibody, and the middle blot was probed with an anti-EARS2 antibody. B, Principal component analysis of the P3-041 fibroblast lines with ectopic expression of WT (green) EARS2 or EARS2 variants associated with mild (blue) or severe (red) COXPD12. C, Pathway analysis of the metabolites associated with expression of variants from severe COXPD12 in P3-041 fibroblasts. The metabolites significantly associated with disease severity were defined by correlation analysis (coefficient cutoff 4) and one-way ANOVA test (f value ≥1), followed by MetaboAnalyst pathway analysis. D, Heatmap displaying changes in metabolite abundance upon expression of WT EARS2 or EARS2 variants associated with mild or severe COXPD12 in P3-041 fibroblasts. Metabolites whose abundance correlated with disease severity and belonging to the KEGG pathways indicated in (C) are displayed in the heat map. The color scale corresponds to z-score values. E, Metabolic alterations in fibroblasts expressing EARS2 variants. The scheme emphasizes pathways related to the perturbations in the TCA cycle and amino acid catabolism. Red metabolites are elevated and blue metabolites are depleted in cells expressing EARS2 variants from severe COXPD12
3 |. DISCUSSION
Predicting clinical features from variants in mitochondrial aminoacyl tRNA synthetases is challenging. The clinical outcome in COXPD12 ranges from neonatal death to a prolonged clinical course, sometimes including the acquisition of developmental milestones and improvement of MRI abnormalities.7,10,19 Disease severity correlates somewhat with the severity of neuroimaging abnormalities detected early in life,6,8,9,20 but these features are nonspecific and can also appear in mitigated forms of the disease.7,10 Metabolomic studies on clinical specimens provide a broad assessment of the metabolic network and can help evaluate pathogenic variants well beyond conventional diagnostic methods. We speculate that such methods may also help predict the clinical course, as our data on a small number of human EARS2 variants report discrete metabolic phenotypes associated with variants causing mild or severe COXPD12.
Although many metabolic consequences of OXPHOS dysfunction are predictable from our current understanding of the electron transport chain, an advantage of metabolomics is that it uncovers unanticipated anomalies in addition to the well-characterized ones. This is relevant to OXPHOS disorders because we generally do not know the full scope of metabolic abnormalities that arise in these patients, how these abnormalities relate to the phenotype, and whether perturbations of particular pathways predict clinical severity. We determined that fibroblasts expressing EARS2 variants display expected abnormalities in central metabolic pathways linked to OXPHOS, including the TCA cycle, but also widespread perturbation of other pathways such as amino acid and nucleotide metabolism. Cells with EARS2 variants from severe COXPD12 also had elevated ophthalmate, a tripeptide analog of GSH with a methyl group replacing the cysteinyl-SH group. Opthalmate is synthesized using the same enzymes that produce GSH, but with 2-aminobutyrate (2-AB) instead of cysteine combining with glutamate in the initial reaction catalyzed by γ-glutamylcysteine synthetase.21 Its abundance may reflect oxidative stress. Resolution of distinct metabolic pathways varied with expression of different EARS2 variants. Levels of TCA cycle metabolites resolved almost completely upon expression of variants associated with mild COXPD12, whereas abnormalities in pathways related to nucleotide and branched-chain amino acid metabolism required expression of WT EARS2 (Figure 4D). This may indicate differential involvement of these pathways across the spectrum of EARS2 variants. Therefore, metabolomics of pathogenic EARS2 variants in fibroblasts identified signature metabolites to discriminate mild and severe COXPD12 and may represent an opportunity to establish genotype-phenotype correlations. Further analysis of samples from COXPD12 patients will help determine the value of using metabolomic signatures to predict disease severity. Given that several weeks are required to establish and analyze fibroblast samples, a more practical approach will be to define and quantify metabolomic abnormalities in blood samples from patients with COXPD12 and related disorders.
It is noteworthy that many altered metabolites in the EARS2-defecient fibroblasts are involved in oxidoreductase reactions using NAD+ as a cofactor (Figure 4E), including purine biosynthesis in the cytosol and TCA cycle and amino acid oxidation in the mitochondria. NAD+ and its reduced form NADH play a key role in regulating cellular metabolism and energy production.22 The NADH/NAD+ ratio thus regulates many enzymes including ketoacid dehydrogenases in the mitochondria, which convert NAD+ to NADH. Mitochondrial NADH is oxidized by furnishing reducing equivalents to ETC complex I, initiating a series of redox reactions that ultimately generate ATP from ADP by OXPHOS. Therefore, EARS2 defects are predicted to impair NADH oxidation and disturb the balance between NAD+ and NADH in the mitochondria. It is unknown whether alterations in the NADH/NAD+ ratio initiated in the mitochondria by EARS2 deficiency also compromise redox-sensitive cytosolic reactions in purine synthesis and other pathways. This may be clinically relevant, because primary defects in nucleotide metabolism are sufficient to cause central nervous system dysfunction in children. It will be interesting to define the scope of redox perturbation induced by EARS2 dysfunction, determine which of these perturbations can be corrected by providing alternative sources of electron acceptors, and test whether the NADH/NAD + ratio itself predicts clinical severity in OXPHOS disorders.
4 |. MATERIALS AND METHODS
4.1 |. Patient samples
The patient and her parents were enrolled in a clinical research study (NCT02650622) approved by the Institutional Review Board (IRB) at University of Texas Southwestern Medical Center (UTSW). Informed consent was obtained from the patient’s family. Whole blood samples were collected from the family. Punch biopsies of the skin for fibroblast culture were obtained postmortem from the patient, following the standard clinical protocols.
4.2 |. Whole exome sequencing
Genomic DNA of the patient and her parents was extracted from whole blood using the standard QIAmp DNA Blood Midi Kit DNA (Qiagen). DNA concentrations and quality were determined using a Nanodrop (ND-100) spectrophotometer (Thermo Fisher Scientific). Three micrograms of DNA were subjected to library preparation using the SureSelect V4 kit (Agilent) following the manufacturer’s instructions. Sequencing was performed on a HiSeq2500 sequencing system (Illumina) with 150 bases of paired-end reads. Median on-target coverage was 124.6, 102.4, and 110.7-fold for the patient, father and mother, respectively. Sequences were aligned to the human reference genome b37. Variants were called using the Genome Analysis Toolkit23 and annotated by SnpEff24 against the human RefSeq database. Known variants and allele frequencies were annotated with dbSNP 151, and all ethnicities from the Exome Aggregation Consortium (ExAC).25 Nonsynonymous variants were annotated with functional prediction scores from PolyPhen-2, SIFT, MutationTaster, MutationAssessor, and FATHMM using VarSeq (Golden Helix).
4.3 |. Genotyping
To verify the EARS2 variant, a 410 bp region spanning the G317 site was PCR amplified from genomic DNA using the primer pairs (Forward: 5′-CCAAGCACCTGCTCCTCTAC-3′, Reverse: 5′-GGGCAGGGATCATGTCTATC-3′). The genotypes were then determined by Sanger sequencing (Genewiz).
4.4 |. Molecular cloning of EARS2 and variants
The full-length EARS2 open reading frame (wild-type or G317 variant) was PCR amplified using the primer pairs (Forward: 5′-CCGGCGATCGCATGGCGGCGCTCCTGAGGAG-3′, Reverse: 5′-CGGACGCGTGCTGGAAACCACCTTCTGGATCCGTTCCCGT-3′) from cDNA prepared from primary human neonatal fibroblasts or the patient’s fibroblasts, respectively. The PCR products were purified using Qiagen PCR purification kit and then digested by AsiS1 and MluI at 37° C for 4 h followed by alkaline phosphatase treatment for 30 min. DNA fragments were purified from agarose gels using the Qiagen Gel extraction kit, then ligated with pLenti-C-Myc-DDK-IRES-Puro vector (OriGene). The positively selected clones were confirmed by Sanger sequencing. To create additional sequence variants, the Q5 Site-directed mutagenesis kit was used to generate point mutations on the wild-type plasmid following the manufacturer’s protocol. The following primers were designed to introduce each variant: I62F, 5′-GTACAACTACtTCTTTGCTAAGAAGTACCAGGGG-3′ and 5′-AAGGCAGTGCGGAGGCCA-3′; K65E, 5′-CATCTTTGCTgAGAAGTACCAG-3′ and 5′-TAGTTGTACAAGGCAGTG-3′; R108W, 5′-GAGCCCCCGCtGGGGCGGTCC-3′ and 5′-TCATCAGGCGGGATGCCTGC CCAC-3′; G301A, 5′-GCTGCTGATGcCTTCCTGCCC-3′ and 5′-AAAGTGCTCCAGGAAAACG-3′.
4.5 |. Fibroblast culture and lentiviral transduction
Primary patient fibroblasts were cultured from the skin biopsy specimen. Control human fibroblast lines were obtained from ATCC or from the pathology biobank of Children’s Medical Center at Dallas and the cell line information was listed in Figure S1. Fibroblasts were maintained in low-glucose DMEM (Sigma, D6046) supplemented with 5% heat-inactivated fetal bovine serum (FBS, GeminiBio, 100106), and cultured at 37° C with 5% CO2. Lentivirus was produced by transfection of HEK293T cells using the LENTI-Smart reagent (Invivogen, ItsInt-10). Viral supernatants were harvested at 48 and 72 h, filtered through a 0.45 um filter and concentrated using PEG-it Virus Precipitation Solution (System Biosciences, LV810A-1). For transduction, the lentiviral pellets were suspended in culture medium and added to fibroblasts at 70-80% confluency per well in 6-well plates. After 48 h of transduction, the fibroblasts were selected under puromycin for 1 week for stable expression of wild-type or variant EARS2.
4.6 |. QTOF metabolomics
For metabolomics, fibroblasts were cultured in low-glucose (5.5 mM) DMEM with 5% FBS for 3 days without penicillin-streptomycin or additional nutrients, such as pyruvate or uridine. Three biological replicates of each cell line were used in metabolomics analysis. About 500 000 subconfluent cells cultured in a 10-cm plate were washed with saline, and collected and lysed with ice-cold 80% LC/MS-grade methanol. After three freeze-thaw cycles in liquid nitrogen, the lysate was centrifuged to remove debris, and the supernatant was collected and dried in a SpeedVac. The metabolite pellets obtained from fibroblast lines were analyzed by targeted metabolomics using Quadrupole Time-of-Flight (QTOF) Mass Spectrometry. Briefly, data acquisition was performed by reverse-phase chromatography on a 1290 UHPLC liquid chromatography (LC) system interfaced to a high-resolution mass spectrometry (HRMS) 6550 iFunnel Q-TOF mass spectrometer (MS) (Agilent Technologies). The MS was operated in both positive and negative (ESI+ and ESI−) modes. Analytes were separated on an Acquity UPLC HSS T3 column (1.8 pm, 2.1 × 150 mm, Waters). The column was kept at room temperature. Mobile phase A composition was 0.1% formic acid in water and mobile phase B composition was 0.1% formic acid in 100% acetonitrile. The LC gradient was 0 min: 1% B; 5 min: 5% B; 15 min: 99%; 23 min: 99%; 24 min: 1%; 25 min: 1%. The flow rate was 250 μL/min−1. The sample injection volume was 5 μL. ESI source conditions were set as follows: dry gas temperature 225°C and flow 18 L/min−1, fragmentor voltage 175 V, sheath gas temperature 350°C and flow 12 L/min−1, nozzle voltage 500 V, and capillary voltage +3500 V in positive mode and −3500 V in negative. The instrument was set to acquire over the full m/z range of 40-1700 in both modes, with the MS acquisition rate of 1 spectrum per second in profile format. Raw data files were processed using Profinder B.08.00 SP3 software (Agilent Technologies) with an in-house database containing retention time and accurate mass information on 600 standards from Mass Spectrometry Metabolite Library (IROA Technologies) analyzed under the same conditions. The in-house database matching parameters were: mass tolerance 10 ppm; retention time tolerance 0.5 min. Peak integration results were manually curated in Profinder for improved consistency. For some compounds, when standards were analyzed by the same experimental setup, more than one chromatographic peak with similar qualification but different intensity were detected, most likely due to interaction with the column and high pressure; in these cases, each chromatographic peak is denoted by a suffix (−1 or −2, etc.) in the supplemental tables. The peak area for each detected metabolite was normalized against the total ion count of each sample. The normalized areas were used as variables for the multivariate and statistical analyses. The pathway enrichment analysis of differential metabolites among groups was performed using Metaboanalyst 3.0 (http://www.metaboanalyst.ca).
4.7 |. Immunoblotting
Whole cell lysates were extracted from fibroblasts in RIPA buffer followed by three freeze/thaw cycles. The protein supernatants were quantified using the BCA protein assay (Pierce, 23227). Proteins were separated on 4-20% SDS-PAGE gels, transferred to PVDF membranes, and probed with the following primary antibodies: anti-EARS2 (Thermo Fisher, MA5-27312), anti-GAPDH (Cell Signaling, #3683), anti-Myc-tag (Cell Signaling, #2276); secondary antibodies were from Santa Cruz Biotechnology (sc-2005 and sc-2357). Immunoreactive proteins were visualized by enhanced chemiluminescence (Pierce, 32 106).
Supplementary Material
ACKNOWLEDGMENTS
We thank the family who participated in this study. Ralph J. DeBerardinis is funded by the Howard Hughes Medical Institute, Once Upon a Time Foundation and Robert L. Moody, Sr. Faculty Scholar endowment. We thank the Baldridge family for their support of the CRI Metabolomics Facility. We thank Gerardo Guevera for assistance in metabolomics analysis and the University of Texas Southwestern Medical Center Bioinformatics Core Facility, funded by the Cancer Prevention and Research Institute of Texas (CPRIT, RP150596) for the genomics analysis pipeline used in this study.
Funding information
Howard Hughes Medical Institute; Once Upon A Time Foundation
Footnotes
CONFLICT OF INTEREST
Ralph J. DeBerardinis is a member of the Scientific Advisory Board for Agios Pharmaceuticals and Vida Ventures. The other authors have no potential conflict of interest.
ETHICS STATEMENT
The patient and her parents were enrolled in a study focused on developmental and metabolic anomalies and approved by the Institutional Review Board (IRB) at University of Texas Southwestern Medical Center (UTSW). Informed consent was obtained from the patient’s family.
SUPPORTING INFORMATION
Additional supporting information may be found online in the supporting information section at the end of this article.
REFERENCES
- 1.Murayama K, Shimura M, Liu Z, Okazaki Y, Ohtake A. Recent topics: the diagnosis, molecular genesis, and treatment of mitochondrial diseases. J Hum Genet. 2019;64:113–125. [DOI] [PubMed] [Google Scholar]
- 2.Fuchs SA, Schene IF, Kok G, et al. Aminoacyl-tRNA synthetase deficiencies in search of common themes. Genet Med. 2019;21:319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez-Serrano LE, Chihade JW, Sissler M. When a common biological role does not imply common disease outcomes: disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases. J Biol Chem. 2019;294:5309–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sissler M, Gonzalez-Serrano LE, Westhof E. Recent advances in mitochondrial aminoacyl-tRNA synthetases and disease. Trends Mol Med. 2017;23:693–708. [DOI] [PubMed] [Google Scholar]
- 5.Nagao A, Suzuki T, Katoh T, Sakaguchi Y, Suzuki T. Biogenesis of glutaminyl-mt tRNAGln in human mitochondria. Proc Natl Acad Sci USA. 2009;106:16209–16214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steenweg ME, Ghezzi D, Haack T, et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate “LTBL” caused by EARS2 mutations. Brain. 2012;135:1387–1394. [DOI] [PubMed] [Google Scholar]
- 7.Biancheri R, Lamantea E, Severino M, et al. Expanding the clinical and magnetic resonance spectrum of leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL) in a patient harboring a novel EARS2 mutation. JIMD Rep. 2015;23:85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliveira R, Sommerville EW, Thompson K, et al. Lethal neonatal LTBL associated with biallelic EARS2 variants: case report and review of the reported neuroradiological features. JIMD Rep. 2017;33:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Talim B, Pyle A, Griffin H, et al. Multisystem fatal infantile disease caused by a novel homozygous EARS2 mutation. Brain. 2013;136:e228. [DOI] [PubMed] [Google Scholar]
- 10.Taskin BD, Karalok ZS, Gurkas E, et al. Early-onset mild type leukoencephalopathy caused by a homozygous EARS2 mutation. J Child Neurol. 2016;31:938–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farwell Hagman KD, Shinde DN, Mroske C, et al. Candidategene criteria for clinical reporting: diagnostic exome sequencing identifies altered candidate genes among 8% of patients with undiagnosed diseases. Genet Med. 2017;19:224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alaimo JT, Glinton KE, Liu N, et al. Integrated analysis of metabolomic profiling and exome data supplements sequence variant interpretation, classification, and diagnosis. Genet Med. 2020;22:1560–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ni M, Solmonson A, Pan C, et al. Functional assessment of lipoyltransferase-1 deficiency in cells, mice, and humans. Cell Rep. 2019;27:1376–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0—making metabolomics more meaningful. Nucl Acids Res. 2015; 43:W251–W257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desler C, Lykke A, Rasmussen LJ. The effect of mitochondrial dysfunction on cytosolic nucleotide metabolism. J Nucl Acids. 2010;2010:701518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell. 2015;162:552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bienert S, Waterhouse A, de Beer TA, et al. The SWISS-MODEL repository-new features and functionality. Nucl Acids Res. 2017;45:D313–D319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soga T, Baran R, Suematsu M, et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J Biol Chem. 2006;281:16768–16776. [DOI] [PubMed] [Google Scholar]
- 19.Gungor O, Ozkaya AK, Sahin Y, Gungor G, Dilber C, Aydin K. A compound heterozygous EARS2 mutation associated with mild leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL). Brain Dev. 2016;38:857–861. [DOI] [PubMed] [Google Scholar]
- 20.Danhauser K, Haack TB, Alhaddad B, et al. EARS2 mutations cause fatal neonatal lactic acidosis, recurrent hypoglycemia and agenesis of corpus callosum. Metab Brain Dis. 2016;31:717–721. [DOI] [PubMed] [Google Scholar]
- 21.Orlowski M, Wilk S. Synthesis of ophthalmic acid in liver and kidney in vivo. Biochem J. 1978;170:415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fang EF, Lautrup S, Hou Y, et al. NAD(+) in aging: molecular mechanisms and translational implications. Trends Mol Med. 2017;23:899–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of proteincoding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.