

SUMMARY
Faithful DNA replication is critical for the maintenance of genomic integrity. While the DNA replication machinery is highly accurate, the process of DNA replication is constantly challenged by DNA damage and other intrinsic and extrinsic stresses throughout the genome. A variety of cellular stresses interfering with DNA replication, which are collectively termed replication stress, pose a threat to genomic stability in both normal and cancer cells. To cope with replication stress and maintain genomic stability, cells have evolved a complex network of cellular responses to alleviate and tolerate replication problems. This review will focus on the major sources of replication stress, the impacts of replication stress in cells, and the assays to detect replication stress, offering an overview of the hallmarks of DNA replication stress.
eTOC blurb
This review discusses the major sources of DNA replication stress, the direct and indirect effects of replication stress on the genome, and the assays to detect replication stress in human cells, providing an overview of the hallmarks of DNA replication stress.
In eukaryotic cells, DNA replication is a fundamental cellular process in which the entire genome is duplicated once and only once during S phase of the cell cycle. Accurate DNA replication is essential for the faithful transmission of genetic information through cell divisions. DNA replication is initiated from numerous genomic loci termed replication origins (Bell and Dutta, 2002; Fragkos et al., 2015; Gilbert, 2010). The replisome, a multi-protein machinery containing the replicative DNA helicase and polymerases, unwinds template DNA and synthesizes nascent DNA on both leading and lagging strands, generating DNA replication forks (Burgers and Kunkel, 2017; Waga and Stillman, 1998). The progression of replication forks on DNA duplicates both strands of template DNA in a semi-conservative manner. DNA replication is finished by replication termination, which involves the convergence of replication forks, disassembly of the replisome and resolution of the daughter DNA molecules (Dewar and Walter, 2017).
While the DNA replication machinery is highly accurate, the fidelity of this process is often threatened by stresses of both exogenous and endogenous origins, resulting in altered replication fork progression, reduced replication fidelity, and DNA breaks (Techer et al., 2017; Zeman and Cimprich, 2014). This phenomenon, which is broadly termed replication stress, is an important source of genome instability in pre-neoplastic lesions and a hallmark of cancer cells (Bester et al., 2011; Burrell et al., 2013; Hanahan and Weinberg, 2011; Macheret and Halazonetis, 2015). Hence, elucidating the molecular basis of replication stress is essential to reaching a comprehensive understanding of tumorigenesis. In recent years, tremendous efforts have been made to define the causes of replication stress, ranging from oncogene activation to depletion of nucleotide pools, transcription-replication conflicts, and inherently difficult-to-replicate regions (Magdalou et al., 2014; Zeman and Cimprich, 2014). Depending on the type of stress interfering with replication forks, cells activate multiple response pathways to stabilize, repair and restart the forks, in order to ensure timely genome duplication and maintain genome stability.
In general, DNA replication stress induces three major intertwined responses at replication forks: (1) activation of the replication checkpoint, (2) remodeling of stressed or stalled forks, and (3) engagement of DNA repair or tolerance pathways. For instance, replication stress can perturb the coupling between the replicative helicase and polymerases, increasing the exposure of single-stranded DNA (ssDNA) at forks (Byun et al., 2005). The ssDNA at stressed forks is coated by replication protein A (RPA) and serves as a platform to recruit and activate the ATR checkpoint kinase (Zou and Elledge, 2003). The ATR activated by replication stress stabilizes stressed forks, suppresses origin firing, and promotes cell cycle arrests, thereby alleviating the deleterious effects of replication stress (Saldivar et al., 2017). Replication forks also undergo remodeling in response to stress. For example, through a remodeling process called fork reversal, stressed forks are converted into four-way junctions to regulate fork speed and facilitate DNA damage tolerance (DDT) and repair (Quinet et al., 2017b). Furthermore, several DNA repair or tolerance pathways are activated at stressed or stalled forks to enable them to bypass or recover from the stress. These pathways include translesion synthesis (TLS), PrimPol-mediated repriming, template switching (TS), break-induced replication (BIR), homologous recombination (HR), and others (Berti et al., 2020; Cortez, 2019). Together, these concerted responses at replication forks help ensure the timely completion of DNA replication and keep genomic instability at a tolerable level in the presence of replication stress.
Here, we review the various causes of replication stress in eukaryotic models, with an emphasis on evidence from human cells. In addition to the known barriers to fork progression, we discuss recently emerged sources of replication stress, such as dysregulated origin firing, excessive repair intermediates, and aberrant structures arising from DNA replication. We also discuss the cascading impacts of replication stress, from local effects at stressed replication forks, to global effects on DNA synthesis and chromosome segregation, and to trans cell-cycle effects caused by persistent DNA lesions. Finally, we provide an overview of the assays to detect and quantify replication stress in human cells.
SOURCES OF REPLICATION STRESS
Replication stress arises from a variety of sources. These include direct barriers to fork progression such as DNA lesions and secondary DNA structures, R-loops, nucleotide imbalance, etc. (reviewed in (Zeman and Cimprich, 2014)). Indeed, the prevailing model has historically considered that replication stress acts in cis at replication forks. However, recent studies have identified replication-associated stresses that are spatially and/or temporally separate from active replication forks, raising the possibility that replication stress can also act in trans. Another emerging concept is that the intermediates or products of certain DNA repair processes act as roadblocks to DNA replication, suggesting that dysregulated repair can also be a source of replication stress. Here, we discuss these distinct types of replication stress in detail.
Replication fork barriers
Any obstacle that perturbs the progression of replication forks is generally considered to be a form of replication stress. Among the most commonly recognized replication blocks are DNA lesions or adducts (Ashour and Mosammaparast, 2021; Ciccia and Elledge, 2010). These DNA lesions/adducts arise from a variety of endogenous and exogenous sources, including chemical mutagens, UV radiation, reactive oxygen species (ROS), non-canonical nucleotides, or byproducts of cellular metabolism (Tubbs and Nussenzweig, 2017). Many different types of DNA lesions/adducts can interfere with replication forks either directly or indirectly. DNA lesions induced by UV or DNA alkylating agents (e.g. methyl methane sulfonate) present roadblocks to replicative DNA polymerases, forcing forks to bypass these lesions using TLS (Lehmann et al., 2007). DNA adducts and inter-strand crosslinks (ICLs) generated by reactive aldehydes or DNA crosslinkers (e.g. cisplatin, mitomycin C), as well as DNA-protein crosslinks (DPCs) formed by various endogenous, environmental, and chemotherapeutic agents that trap proteins on DNA (e.g. poly [ADP-ribose] polymerase (PARP) and topoisomerase I (TOP1) inhibitors), also act as potent blocks to fork progression (Ide et al., 2011; Vare et al., 2012; Voulgaridou et al., 2011). In addition, encounter of forks with DNA single-stranded breaks (SSBs) generated by ROS and other causes can lead to replication fork run-off and formation of one-ended double-strand breaks (DSBs) (Vrtis et al., 2021) (Figure 1A).
Figure 1. Sources of DNA replication stress.
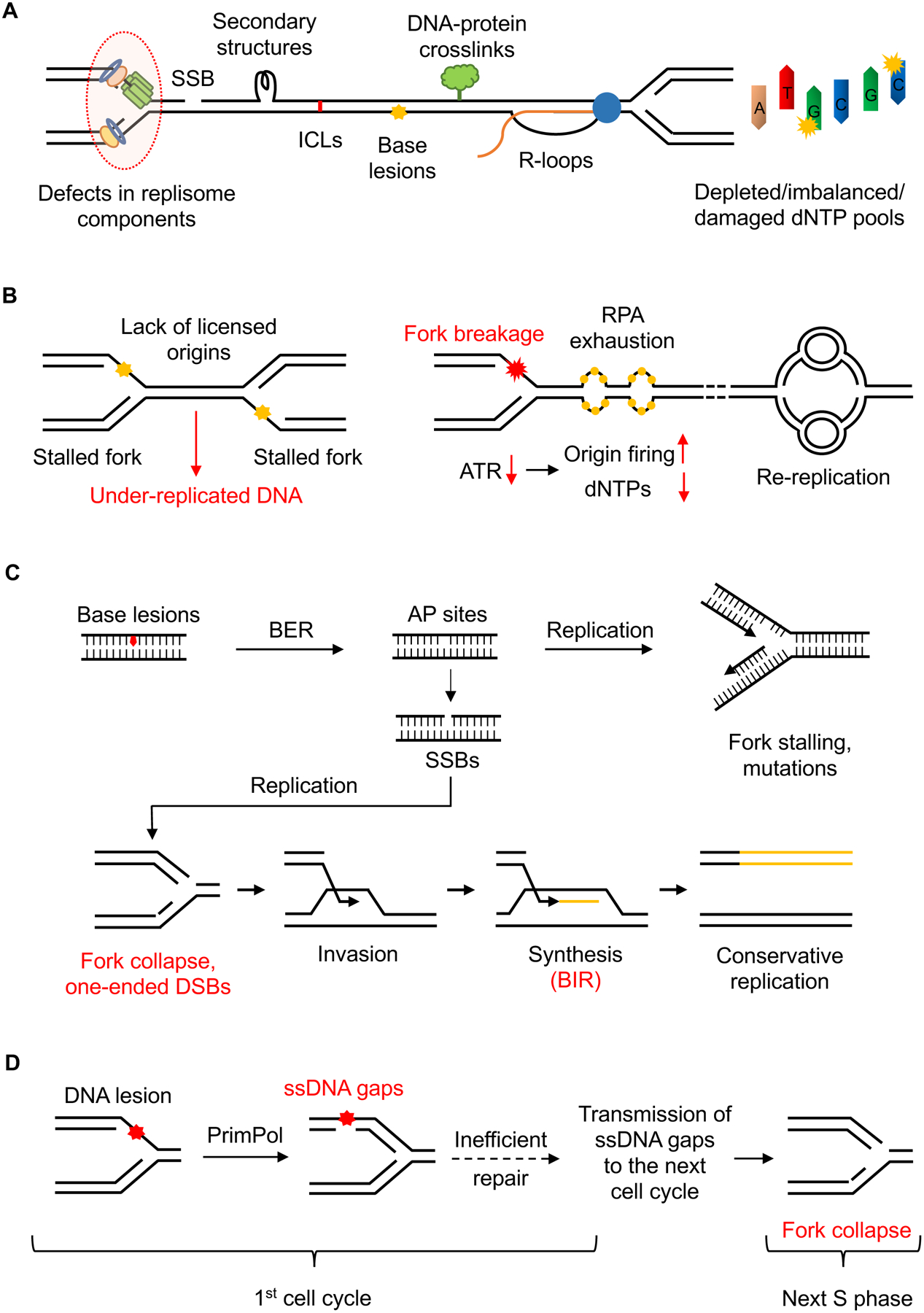
(A) The progression of replication forks can be impeded by several obstacles including SSBs, DNA secondary structures, ICLs, etc. Defects in replisome components or alterations in dNTP pools can also reduce fork speed, compromise fork stability, and promote mutagenesis. (B) Lack of licensed origins leads to reduced rescue of stalled forks, resulting in under-replicated regions. Increased origin firing reduces fork speed by depleting dNTPs and other replisome factors. It also generates excessive amount of ssDNA, which exhausts the nuclear pool of RPA and induces genome-wide breakage of replication forks. Re-replication forks progress slowly and undergo “head-to-tail” collisions with previously initiated replication forks, resulting in DSBs. (C) Excessive repair intermediates like AP sites and SSBs act as roadblocks to replication forks. Collisions of active forks with SSBs cause fork collapse and one-ended DSBs, which can be recovered by BIR, an aberrant replication process that is error-prone and generates replication stress. (D) Replication forks can bypass DNA lesions by PrimPol-mediated repriming, which generates ssDNA gaps behind the forks. If unrepaired, these ssDNA gaps can persist into the next cell cycle, where they encounter active forks and cause fork collapse, generating trans-cell cycle replication stress.
Secondary DNA structures such as hairpins, cruciform structures and G-quadruplexes can form in the genomic DNA during physiological processes that generate ssDNA, such as replication, transcription, and repair. When encountered by replication forks, these structures pose a physical obstacle, causing replication stress (Kumar et al., 2021; Mirkin and Mirkin, 2007; Voineagu et al., 2008) (Figure 1A). Some of the common fragile sites (CFSs), which are regions of the genome that exhibit chromosomal breakage under conditions of mild replication stress, are predicted to form stable DNA secondary structures (Thys et al., 2015). Notably, repetitive DNA sequence elements with high structure-forming potential, such as microsatellites, inverted retroelement repeats, and quasi-palindromic AT-rich repeats, are more reliant on ATR for stability, possibly owing to their inhibitory effects on DNA replication (Shastri et al., 2018). A number of specialized helicases and structure-specific nucleases are involved in dismantling these structures to allow replication fork progression, and the loss of these enzymes is associated with a wide range of genetic disorders characterized by chromosomal instability (Sharma, 2011; van Wietmarschen et al., 2020).
The transcription-replication conflict (TRC) is another source of replication stress. R-loops, which are three-stranded polynucleotide structures consisting of DNA-RNA hybrids and displaced ssDNA, also pose a barrier for fork progression (Aguilera and Garcia-Muse, 2012; Helmrich et al., 2013) (Figure 1A). R-loops levels are affected by the orientation of transcription-replication collisions; head-on collisions increase R-loops, whereas co-directional collisions decrease R-loops (Hamperl et al., 2017). R-loops impede replication forks and are cleaved by specific nucleases, which underlies their deleterious effects on the genome (Gan et al., 2011; Hamperl et al., 2017; Matos et al., 2020; Sollier et al., 2014). Hence, cells have evolved multiple pathways to resolve R-loops and avoid TRC. For instance, TOP1 prevents the collision of replication forks with R-loops at transcription termination sites by removing DNA supercoiling (Promonet et al., 2020). R-loops are also suppressed by RNA processing/splicing factors that prevent DNA-RNA hybridization during transcription (Stirling et al., 2012). Once R-loops are formed, they can be removed by the RNase H family of enzymes and DNA-RNA helicases (Skourti-Stathaki et al., 2011; Wahba et al., 2011), or excised by XPF and XPG nucleases (Sollier et al., 2014). Mutations in the genes involved in these pathways result in increased R-loops and genomic instability, and mutations in some of the R-loop removing factors are associated with neurological disorders and cancers (Richard and Manley, 2017). Aberrant R-loop accumulation is also associated with mutations in certain RNA splicing factors and loss of certain DNA repair proteins, which may contribute to the replication stress and genomic instability in cancer cells (Crossley et al., 2019; Nguyen et al., 2019).
In addition to secondary structures and R-loops in the template DNA, compacted chromatin could also pose a barrier for DNA replication and repair. Heterochromatin regions of the genome such as centromeres, peri-centromeres, and telomeres are inherently hard-to-replicate, and require specialized mechanisms for chromatin remodeling to ensure normal replication fork progression (Ivessa et al., 2003; Miller et al., 2006; Sfeir et al., 2009; Zaratiegui et al., 2011). Loss of a chromatin-remodeling activity in cells harboring mutations in SMARCA4/BRG1, a core component of the SWI/SNF chromatin-remodeling complex, increases heterochromatin-associated replication stress and confers susceptibility to ATR inhibition (Gupta et al., 2020; Kurashima et al., 2020). Loss of ARID1A, a core subunit of the BAF chromatin-remodeling complex, increases replication stress through TRC and R-loops (Tsai et al., 2021). ATRX, another chromatin-remodeling protein of the SWI/SNF family, is required for protecting stalled replication forks in heterochromatin (Huh et al., 2016), and loss of ATRX increases telomere replication stress and instability (Flynn et al., 2015; Li et al., 2019). Paradoxically, loss of chromatin compaction can also induce replication stress. For example, reduced chromatin compaction in histone H1-depleted cells elevates TRC, leading to accumulation of stalled forks (Almeida et al., 2018). Thus, while increased chromatin compaction could interfere with replication fork progression, loss of repressive chromatin could also compromise fork progression, possibly by elevating transcription or origin firing.
Defects in replication forks
Perturbations in cellular nucleotide pools are another source of replication stress (Figure 1A). Intracellular deoxyribonucleoside triphosphate (dNTP) pools must be tightly aligned with replication dynamics to preserve genome integrity. Indeed, alterations in dNTP pools are associated with increased mutagenesis, genomic instability, and tumorigenesis (Bester et al., 2011). High dNTP levels compromise the fidelity of DNA replication by reducing the proofreading efficiency of polymerases (Weinberg et al., 1981). In contrast, low dNTP levels caused by hydroxyurea (HU), an inhibitor of the ribonucleotide reductase, or by excessive origin firing following inhibition of ATR, Chk1, or WEE1, induce replication stress by compromising polymerase activities (Beck et al., 2012; Buisson et al., 2015). Intermediate levels of dNTP starvation, while not posing a global block to DNA replication, can have a more pronounced effect on specific genomic regions such as fragile sites (Pai and Kearsey, 2017). Notably, not only the overall levels of dNTPs, but also the quality of dNTP pools and the balance among individual dNTPs, is critical for genome stability. Incorporation of oxidized dNTPs caused by inefficient sanitation of dNTP pools generates replication-associated DNA damage (Gad et al., 2014), and dNTP imbalance induced by 5-FU or deregulation of enzymes involved in dNTP synthesis induces fork slowing (Saxena et al., 2018). Furthermore, DNA replication under suboptimal dNTP levels may lead to increased misincorporation of ribonucleoside monophosphates (rNMPs). If unrepaired, the rNMPs in DNA can cause replication stress in the subsequent cell cycle as DNA polymerases tend to stall at such sites (Reijns et al., 2012).
Mutations of the replisome components also compromise genome stability by reducing the number, stability, or fidelity of replication forks (Bellelli and Boulton, 2021) (Figure 1A). For instance, in both mouse and human cells, DNA polymerase Polε hypomorphic mutants cause inefficient replication initiation, resulting in replication stress, growth retardation, and tumor predisposition (Bellelli et al., 2018). Similarly, hypomorphic alleles of the genes encoding subunits of the MCM helicase are associated with a reduction in active forks, leading to increased replication stress and embryonic lethality (Alvarez et al., 2015; Kunnev et al., 2010; Shima et al., 2007). Haploinsufficiency of RPA, the ssDNA-binding protein at replication forks, also causes defects in DNA replication and high rates of cancer in mice (Hass et al., 2010). Mutations in the FEN1 nuclease, which is critical for Okazaki fragment maturation, generate replication stress and promote tumorigenesis (Zheng et al., 2011). In addition to the core replisome components, loss of accessory replisome factors TIMELESS, TIPIN and CLASPIN (also known as the fork protection complex) results in replication fork slowdown (Petermann et al., 2008; Somyajit et al., 2017). Similarly, loss of AND-1/CTF4, which bridges the MCM helicase and DNA polymerase α, slows down forks and increases ssDNA (Abe et al., 2018). Furthermore, loss of CAF-1, which is essential for replication-coupled chromatin assembly, compromises replication and activates the replication checkpoint (Hoek and Stillman, 2003).
Dysregulated origin firing
It has been long appreciated that origin firing is regulated by the cellular response to DNA damage and replication stress in eukaryotes (Ge and Blow, 2010; Yekezare et al., 2013). However, recent studies suggest that alterations in the origin firing program are not just a consequence, but also a source of replication stress.
To achieve complete and timely duplication of large genomes, metazoan cells license many folds more origins than are used during each round of replication. These licensed but normally dormant origins can be activated if replication forks are stalled in their vicinity, providing a ‘backup’ mechanism to complete DNA replication (McIntosh and Blow, 2012). Hence, regions in the genome without sufficient licensed origins lack this backup mechanism and are prone to under-replication (Figure 1B). Indeed, in very large and highly transcribed genes poor in replication origins, replication forks cannot cope with TRC adequately, resulting in CFSs (Brison et al., 2019). Moreover, MCM mutants that decrease the number of licensed origins and reduce the rescue of stalled forks (Kawabata et al., 2011) are shown to cause cancer in mouse models (Shima et al., 2007), suggesting that reduced origin licensing and usage could promote tumorigenesis.
Excessive and dysregulated origin firing also results in catastrophic genomic instability and, potentially, tumorigenesis (Thakur et al., 2021). The level of CDK2 kinase activity is a key determinant of origin firing efficiency (Fagundes and Teixeira, 2021). During S phase, CDK2 activity is restricted by ATR, Chk1 and WEE1 kinases, which prevent excessive origin firing. Compromised activities of these kinases increase origin firing and reduce fork speed (Beck et al., 2012; Buisson et al., 2015; Petermann et al., 2010b), possibly due to limitations in dNTPs or replication factors (Zhong et al., 2013) (Figure 1B). Moreover, in checkpoint-defective cells, increased origin firing in the presence of replication stress generates an excessive amount of ssDNA that exhausts the nuclear pool of RPA. This exhaustion of RPA causes insufficient protection of replication intermediates and induces genome-wide breakage of replication forks, a phenomenon termed replication catastrophe (Toledo et al., 2017; Toledo et al., 2013) (Figure 1B). There is also evidence that the forks initiated from newly fired origins in checkpoint-defective cells are prone to collapse, leading to rapid recruitment of non-homologous end joining (NHEJ) factors such as DNA-PKcs, KU70, and KU80 to these forks (Dungrawala et al., 2015). The importance of regulated origin firing for genome stability is further supported by the findings that activation of oncogenes such as Cyclin E alters the spatial and temporal regulation of origin firing (Bester et al., 2011; Dominguez-Sola et al., 2007). This type of dysregulated origin firing elevates TRC, impairing replication fork progression and generating DNA damage (Jones et al., 2013).
The complete and precise genome duplication in proliferating cells requires the entire genome to be replicated once and only once per cell cycle. To avoid re-replication in the same cell cycle, eukaryotic organisms have established several conserved mechanisms to maintain a strict temporal separation of origin licensing and firing (Blow and Dutta, 2005; Hook et al., 2007). Loss of these regulatory mechanisms leads to extensive re-replication of DNA in human cells (Fujita, 2006). Re-replication forks exhibit slow elongation rate, and even re-firing of multiple origins does not result in full replication of the genome (Fu et al., 2021). Moreover, cells undergoing rereplication exhibit DSBs and activate the DNA damage checkpoint. If the checkpoint is abolished, cells enter mitosis with a partially re-replicated genome, resulting in chromosome breaks and fusions and dying cells with sub-G1 ploidy (Melixetian et al., 2004). The DSB formation and chromosome fragmentation during re-replication are consistent with “head-to-tail” collisions of replication forks (Davidson et al., 2006), supporting the idea that re-replication is a source of replication stress (Figure 1B).
DNA repair intermediates
Mammalian cells have evolved multiple mechanisms to maintain fork stability and promote the repair and restart of stalled replication forks, and loss of these protective pathways is associated with elevated replication stress (Liao et al., 2018). For instance, HR is critical for the restart of stalled replication forks, and HR deficient cells exhibit genome-wide slow replication kinetics and genome instability (Spies et al., 2021; Wilhelm et al., 2014). The ATR kinase and its downstream effectors such as Chk1 help stabilize and restart stalled replication forks (Dungrawala et al., 2015; Saxena et al., 2019). Consequently, loss or inhibition of ATR or Chk1 increases fork collapse and DNA damage (Couch et al., 2013; Petermann et al., 2006; Ragland et al., 2013). Loss of proteins involved in the fork protection pathway, such as BRCA1/2, FANCD2 and RAD51 paralogs, results in increased fork degradation and elevated levels of spontaneous replication stress (Hashimoto et al., 2010; Schlacher et al., 2011; Schlacher et al., 2012; Somyajit et al., 2015). Inhibition of PARP1 impairs lagging strand maturation, causes accelerated fork progression, and prevents the timely repair of ssDNA gaps (Genois et al., 2021; Hanzlikova et al., 2018; Maya-Mendoza et al., 2018; Simoneau et al., 2021).
While repair pathways help to alleviate replication stress, excessive and dysregulated DNA repair can become a source of replication stress. An example of this type of replication stress comes from base excision repair (BER). Base modifications and adducts in the genome are primarily repaired by the BER pathway, which creates several intermediates, such as abasic (AP) sites and SSBs. When the levels of DNA lesions are tolerable and BER occurs efficiently, these intermediates are beneficial to the maintenance of genomic stability. However, when the levels of damage are too high or BER is incomplete, these intermediates become roadblocks to replication forks (Fugger et al., 2021; Thompson and Cortez, 2020) (Figure 1C). Moreover, due to their reactive chemistry, AP sites can give rise to other fork-blocking lesions including DSBs, ICLs and DPCs (Dutta et al., 2007; Prasad et al., 2014). Being ‘faceless’ lesions, which provide no coding information, AP sites are also a potent source of mutation (Simonelli et al., 2005). Similarly, in the absence of efficient ribonucleotide repair (RER), ribonucleotides misincorporated into the genome can be cleaved by BER glycosylases to form mutagenic abasic ribonucleotides (rAP-sites), which are further excised by the BER endonuclease APE1, resulting in SSBs (Malfatti et al., 2017).
DDT pathways employed by replication forks to bypass DNA lesions also generate intermediates conferring replication stress. For instance, when replication forks encounter bulky DNA lesions that block DNA polymerase on the leading strand, they use PrimPol, an RNA/DNA primase-polymerase, to reprime for DNA synthesis ahead of the stalled polymerase, thereby preventing fork stalling but leaving ssDNA gaps behind (Garcia-Gomez et al., 2013). Alternatively, stalled forks can undergo reversal to generate a stable ‘chicken foot’ structure. The backtracking and re-annealing of nascent DNA strands prevent fork progression across DNA lesions, thus avoiding fork collapse (Quinet et al., 2017b). Reversed forks can also be processed by structure-specific nucleases such as MUS81, providing intermediates for fork restart (Hanada et al., 2007). Although replication fork reversal is beneficial to cells when DNA damage is tolerable, it becomes a source of replication stress when DNA damage is excessive or fork protection is compromised (Fugger et al., 2013). In this situation, reversed forks are degraded by MRE11 and EXO1 nucleases or cleaved by MUS81 (Lemacon et al., 2017), leading to the loss of nascent DNA or toxic levels of DSBs.
Collapsed replication forks with one-ended DSBs can be recovered by BIR (Kramara et al., 2018). During BIR, one nascent DNA strand is synthesized by the extension of DNA ends in D-loops, whereas the second DNA strand is generated using the first nascent strand as a template (Figure 1C). Thus, the DNA replication complex functioning in BIR is significantly different from the canonical replication forks. BIR is known to be error-prone and induce genomic duplications in cancer cells (Costantino et al., 2014; Deem et al., 2011). At telomeres, BIR is associated with increased replication stress and contributes to telomere fragility (Yang et al., 2020; Zhang et al., 2021). Hence, BIR can be viewed as an aberrant replication process that is both a consequence and a cause of replication stress.
Temporal and spatial separation of stress from replication forks
Recent evidence suggests that the replication stress generated at replication forks can be temporally and spatially separated from DNA replication. As discussed above, while PrimPol-mediated repriming allows replication forks to avoid stalling at DNA lesions, it generates post-replicative ssDNA gaps behind the forks. These gaps are repaired by TS and TLS pathways in S and G2 phases, respectively (Taglialatela et al., 2021; Tirman et al., 2021). However, in the absence of efficient gap filling, these gaps can persist into mitosis and even the next cell cycle. Unrepaired ssDNA gaps can interfere with chromosome segregation in mitosis (Ait Saada et al., 2017). Upon encountering active replication forks in the next S phase, the ssDNA gaps cause fork collapse and one-ended DSBs, resulting in trans cell-cycle replication stress (Simoneau et al., 2021).
IMPACTS OF REPLICATION STRESS
Replication stress interferes with the process of DNA replication, thus compromising the fidelity and timely completion of genome duplication. Replication stress can generate DNA breaks in S phase and subsequent cell cycle stages, impair chromosome segregation during mitosis, and even induce genomic instability in the following cell cycle. Here, we discuss the major consequences of replication stress, ranging from local effects on the forks directly affected by replication barriers, to checkpoint activation and its distal effects on origin firing and genome-wide DNA replication, and finally to the global effects on genomic stability and cell survival.
Local effects at stressed replication forks
Obstructions to replication fork progression cause fork stalling, which has been recognized as a major contributor to genomic instability (Gaillard et al., 2015) (Figure 2A). Some types of DNA damage such as ICLs simultaneously block DNA synthesis on both leading and lagging strands, whereas others affect one of the strands stochastically. Some leading strand damage uncouple the replicative helicase and polymerase, resulting in long stretches of ssDNA (Byun et al., 2005). In contrast, lagging strand damage prevents Okazaki fragment maturation, generating smaller ssDNA gaps (Marians, 2018; Pasero and Vindigni, 2017). When both leading and lagging strands are blocked, ssDNA can be generated through fork remodeling and nucleolytic processing (Quinet et al., 2017b). Stalling of replication forks triggers activation of the ATR-mediated replication checkpoint, remodeling of stalled forks, and activation of DDT pathways (Berti and Vindigni, 2016; Yeeles et al., 2013). These responses help stabilize stalled forks and promote their recovery. Alternatively, replication forks from origins adjacent to stalled forks can help complete duplication of the replicons, alleviating the deleterious effects of fork stalling (Cortez, 2015).
Figure 2. Consequences of DNA replication stress.
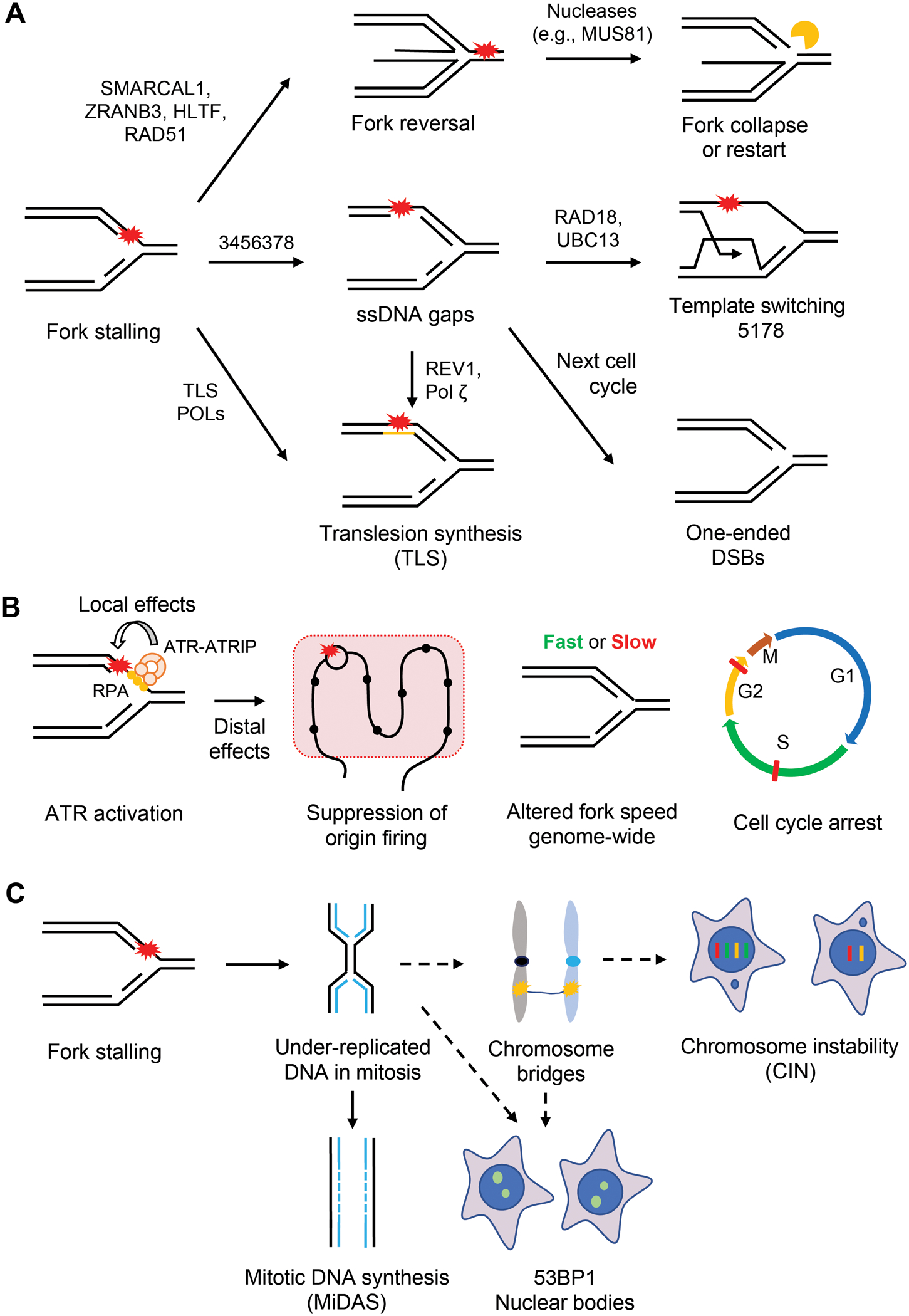
(A) Obstructions to replication progression cause fork stalling. Several replication-coupled repair mechanisms are used to promote the bypass and/or repair of DNA damage. Stalled forks can be reversed by the action of DNA translocases such as ZRANB3, HLTF, and SMARCAL1 and recombinase RAD51. The resulting four-way structure can be cleaved by nucleases like MUS81 to form DSBs. The DSBs at reversed forks can promote fork restart, but can also be toxic when they accumulate at high levels. Alternatively, stalled forks can bypass DNA lesions by PrimPol-mediated repriming. Repriming generates ssDNA gaps behind the fork, which can be repaired by TS or TLS in S and G2 phases. In the event of inefficient repair, these gaps persist into the next S phase and induce one-ended DSBs. Stalled forks can also directly bypass DNA lesions through TLS. (B) Fork stalling generates stretches of ssDNA at stressed forks, which is coated by the RPA complex. ssDNA-RPA in turn recruits the ATR-ATRIP kinase complex to stalled forks. Once activated, ATR phosphorylates substrates at stalled forks to promote fork stabilization and restart (local effects). Furthermore, ATR suppresses origin firing, alters fork speed genome-wide, and induces cell cycle arrests in S and G2/M phases (distal effects). (C) Under-replicated DNA (UR-DNA) can persist into mitosis and undergo MiDAS. If not replicated by MiDAS, UR-DNA forms chromosome or DNA bridges in anaphase. If these bridges are not resolved by repair proteins, they give rise to chromosome instability (CIN). If UR-DNA is segregated into daughter cells, it is sequestered into 53BP1 nuclear bodies (53BP1 NBs).
ssDNA is a physiological intermediate at replication forks and is coated by RPA. While RPA-ssDNA has critical roles in replication, it is a transient structure at progressing forks due to the rapid conversion of ssDNA into dsDNA by DNA polymerases. However, stalling of DNA polymerases increases the exposure of ssDNA, allowing RPA-ssDNA to serve as a platform for recruiting the ATR-ATRIP kinase complex to stalled forks (Cortez et al., 2001; Zou and Elledge, 2003) (Figure 2B). The activation of ATR at stalled forks relies on two parallel pathways involving TOPBP1 and ETAA1, both of which harbor ATR-activating domains (AADs) (Bass et al., 2016; Haahr et al., 2016; Kumagai et al., 2006). The TOPBP1-mediated ATR activation depends on the RFC-like RAD17 complex and the PCNA-like RAD9-RAD1-HUS1 (9-1-1) complex, which recruit TOPBP1 to ssDNA-dsDNA junctions (Delacroix et al., 2007; Lee et al., 2007). In contrast, ETAA1 binds RPA-ssDNA directly (Bass et al., 2016; Haahr et al., 2016). Once activated, ATR phosphorylates numerous substrates, including the effector kinase Chk1 (Liu et al., 2000; Matsuoka et al., 2007). This ATR-Chk1 signaling cascade orchestrates the replication stress response through several distinct mechanisms, such as arresting the cell cycle, suppressing origin firing, stabilizing replication forks, and promoting fork repair and restart (Simoneau and Zou, 2021) (Figure 2B).
Another common effect of replication stress is the formation of DSBs at stalled replication forks. Prolonged stalling of replication forks or inhibition of ATR or Chk1 leads to fork collapse and DSBs (Dungrawala et al., 2015; Petermann et al., 2010a). DNA replication across SSBs and ssDNA gaps generates one-ended DSBs; two-ended DSBs arise when two replication forks converge at SSBs or ssDNA gaps. DSBs are also generated by structure-specific nucleases at stalled or reversed forks (Hanada et al., 2007; Lemacon et al., 2017; Pepe and West, 2014) (Figure 2A). When the levels of DSBs at stalled forks exceed repair activities, they cause genomic instability and even cell death. Consistent with this idea, difficult-to-replicate chromosomal loci such as CFSs, poly dA/dT tracts, and R-loops cause fork stalling and breakage, representing a common source of genomic instability with significant ramifications for human diseases (Glover et al., 2017; Ozeri-Galai et al., 2011).
Repair-associated effects at forks
Some of the local effects of replication stress at replication forks are generated by replication-coupled remodeling or repair processes. Reversal of replication forks slows fork progression but helps forks bypass barriers (Marians, 2018; Zellweger et al., 2015). DNA translocases including SMARCAL1, ZRANB3, and HLTF are recruited to stalled forks to catalyze fork reversal (Bai et al., 2020; Kolinjivadi et al., 2017; Vujanovic et al., 2017) (Figure 2A). The recombinase RAD51 is also required for fork reversal (Quinet et al., 2017b; Zellweger et al., 2015). The RECQ1 helicase can resolve reversed forks to allow resumption of fork progression (Berti et al., 2013). The activity of RECQ1 at reversed forks is inhibited by PARP1, which stabilizes reversed forks (Berti et al., 2013). Reversed forks can be cleaved by the MUS81-EME2 nuclease, which may promote fork restart through the POLD3-mediated BIR pathway (Lemacon et al., 2017; Pepe and West, 2014). A DNA2 and WRN-mediated mechanism also promotes the processing and restart of reversed forks (Thangavel et al., 2015). Notably, reversed forks contain a ‘regressed’ arm formed by both nascent DNA strands, which resembles a DSB and must be protected by BRCA1/2, RAD51, RAD51 paralogs and other factors to prevent degradation of nascent DNA (Schlacher et al., 2011; Schlacher et al., 2012; Somyajit et al., 2015; Thakar and Moldovan, 2021). The cleavage of reversed forks by MUS81 and other SLX4-associated nuclease activities is also a source of DSBs when ATR is compromised (Couch et al., 2013; Ragland et al., 2013) (Figure 2A).
Another mechanism to bypass DNA damage is through repriming by PrimPol. As discussed above, PrimPol generates de novo DNA primers ahead of stalled polymerases, restarting DNA synthesis beyond the lesion (Garcia-Gomez et al., 2013; Mouron et al., 2013). Repriming at replication forks leaves ssDNA gaps behind, which can be repaired by TS or TLS (Tirman et al., 2021). During TS, the stalled nascent strand dissociates from the damaged template and anneals with the complementary nascent strand, allowing extension of the stalled strand (Ashour and Mosammaparast, 2021). During TLS, specialized DNA polymerases are used to bypass DNA lesions or fill the gaps (Sale, 2013) (Figure 2A). Recent evidence also implicates HR proteins in the repair of replication-born ssDNA gaps (Somyajit et al., 2021; Tirman et al., 2021). Some of the repair or replication proteins, such as BRCA1/2 and PCNA, are involved in multiple stress response pathways. The choice of stress response pathways may be influenced by the causes of fork stalling and the length or position of ssDNA at or behind forks, but the details remain to be elucidated.
ATR also contributes to the effects of replication stress at forks. ATR phosphorylates many replisome components and accessory factors in response to replication stress (Errico and Costanzo, 2012; Lossaint et al., 2013; Matsuoka et al., 2007). The phosphorylation of some of these ATR substrates, such as RPA, BLM, WRN, and FANCI, facilitates fork recovery from stress (Ammazzalorso et al., 2010; Chen et al., 2015; Davies et al., 2004; Murphy et al., 2014). The phosphorylation of SMARCAL1 by ATR restricts its fork remodeling activity (Couch et al., 2013). The function of PrimPol is promoted by Chk1-mediated phosphorylation (Mehta et al., 2022). ATR is also required for suppressing DSB formation at stalled forks (Dungrawala et al., 2015).
Distal effects on origin firing and fork speed
One key outcome of replication stress and subsequent ATR activation is the suppression of origin firing (Zeman and Cimprich, 2014). Under low levels of replication stress, ATR preferentially inhibits the activation of new replication factories while allowing dormant origins to fire within the existing factories experiencing replication stress, thus minimizing fork stalling (Ge and Blow, 2010). Upon high replication stress, ATR suppresses origin firing within the same replication initiation zones, preventing the accumulation of stalled forks in damaged regions (Moiseeva et al., 2019) (Figure 2B). ATR suppresses origin firing through several mechanisms. The activation of Chk1 negatively regulates CDK-dependent phosphorylation events at origins, blocking the loading of CDC45 and other pre-initiation complex (pre-IC) factors (Zhao et al., 2002). The inhibition of CDK by Chk1 also indirectly reduces CDC7 kinase activity through the phosphatase PP1 (Moiseeva et al., 2019). Chk1 also phosphorylates Treslin, which limits CDC45 binding to origins (Guo et al., 2015). Furthermore, ATR phosphorylates and stabilizes the MLL protein, which methylates histone H3 Lys4 (H3K4me) at origins and blocks CDC45 loading (Liu et al., 2010). The suppression of origin firing by ATR limits the number of replication forks, thereby preventing excessive ssDNA accumulation, exhaustion of the nuclear RPA pool, and replication catastrophe (Buisson et al., 2015; Toledo et al., 2013). Whether and how ATR exerts its effects on distal and local origins through distinct mechanisms still needs further investigations.
Another general manifestation of replication stress is the slowdown of DNA replication throughout the genome (Techer et al., 2017) (Figure 2B). Both the replication stress itself and the ATR checkpoint response contribute to the reduction in DNA synthesis. In S phase cells, inactivation of the ATR pathway elevates overall DNA synthesis, which is primarily a result of increased origin firing (Petermann et al., 2010b). In addition, ATR promotes dNTP synthesis through RRM2 and the deoxycytidine kinase dCK (Buisson et al., 2015; Le et al., 2017). ATR inhibition causes fork slowing, possibly due to the reduced availability of dNTPs and certain replication proteins when more origins are fired (Buisson et al., 2015). However, there is also evidence that ATR promotes slowing of replication forks upon genotoxic stress. For example, ATR is shown to slow forks genome-wide by promoting fork reversal in response to ICLs, and through RAD51 paralogs after dNTP depletion (Mutreja et al., 2018; Saxena et al., 2018). Thus, the effects of ATR on fork speed could be influenced by the level and nature of replication stress.
Global effects during the cell cycle
In the event of replication stress, cell cycle arrest, which is initiated by the checkpoint activation at stalled forks, is a crucial cellular response (Figure 2B). Once activated by ATR, Chk1 phosphorylates the CDC25A phosphatase and promotes its degradation, preventing the removal of inhibitory phosphorylation from CDK2 (Mailand et al., 2000; Sorensen et al., 2003). In S phase cells, activation of ATR and Chk1 reduces CDK2 activity, restricting origin firing and allowing time to resolve the replication stress (Zhang and Hunter, 2014). In addition, the phosphorylation of CDC25C by Chk1 induces its nuclear export and cytoplasmic sequestration, preventing the activation of CDK1 and the G2/M transition (Peng et al., 1997; Sanchez et al., 1997). Thus, Chk1 is a key checkpoint mediator in both S and G2 phases, which prevents the entry of cells with under-replicated or damaged DNA into mitosis (Xiao et al., 2003).
Despite the functions of cell-cycle checkpoints, under-replicated DNA (UR-DNA) and unresolved replication or repair intermediates can escape checkpoint surveillance and get transmitted to subsequent cell-cycle phases and even to the next cell cycle (Mankouri et al., 2013). Normally, cells deal with such UR-DNA in early prophase through mitotic DNA synthesis (MiDAS), a BIR-like mechanism of conservative DNA synthesis that involves DNA polymerase δ and its accessory subunit POLD3, the nuclease scaffold SLX4, the MUS81-EME1 endonuclease, and the recombinase RAD52 (Bhowmick et al., 2016). Persistent UR-DNA manifests as ultra-fine bridges (UFBs) in late mitosis (Lezaja and Altmeyer, 2021). If segregated into daughter cells, UR-DNA is sequestered into 53BP1 nuclear bodies (53BP1-NBs), which restrain the replication of the embedded genomic loci until they can be repaired by a dedicated RAD52-mediated repair pathway in the next S phase (Harrigan et al., 2011; Lukas et al., 2011; Spies et al., 2019) (Figure 2C).
Genomic alterations resulting from replication stress
The DSBs induced by replication stress can cause mutations and several types of chromosomal alterations, such as deletions, duplications, and translocations. The genomic instability induced by replication stress is at least in part attributed to the error-prone repair pathways activated by stalling or collapse of replication forks, such as TLS, BIR, NHEJ, and microhomology mediated end joining (MMEJ). In addition, replication stress is closely linked to chromosome instability (CIN), another common feature of cancer cells (Burrell et al., 2013). Replication stress leads to UR-DNA persisting into mitosis, resulting in mitotic defects including lagging chromosomes, bulky chromosome bridges, and UFBs (Chan et al., 2009; Wilhelm et al., 2020). These mitotic defects can lead to aneuploidy (numerical CIN) and chromosomal rearrangements (structural CIN) (Burrell et al., 2013). Furthermore, chromosome missegregation gives rise to micronuclei and induces chromothripsis, a process of severe DNA fragmentation and rearrangement in confined genomic regions (Zhang et al., 2015) (Figure 2C).
DETECTION OF REPLICATION STRESS
A number of molecular and cellular assays have been developed to analyze the phenotypes and effects of replication stress. Some of these assays directly analyze the alterations of replication forks, whereas others monitor the indirect effects of replication problems. It is important to note that each of these assays only reflects a specific aspect of replication stress or the stress response. The combination of multiple assays is generally required to obtain a comprehensive understanding of the replication stress and stress responses in specific contexts.
Assays for cellular responses to replication stress
Several assays are used to detect global cellular responses to replication stress, including accumulation of ssDNA, suppression of DNA synthesis, and cell cycle arrests. Several other assays are used to detect replication stress-induced DNA damage and damage responses, such as phosphorylation and focus formation of DNA repair proteins. These assays provide indirect but quantitative measurements of replication stress in cell populations or individual cells.
The thymidine analogue 5-ethynyl-2’-deoxyuridine (EdU) is a convenient tool to monitor DNA synthesis in cultured cells and in vivo (Salic and Mitchison, 2008). Pulse labeling of cells with EdU allows its incorporation into newly synthesized DNA. The EdU intensity in individual cells accurately reflects the levels of DNA synthesis and can be used to identify replicating cells in cell populations. The activation of the ATR-mediated replication checkpoint by replication stress typically results in a reduction of EdU intensity in S phase cells (Fig. 3A). In addition, pulse EdU labeling can be combined with immunofluorescence of specific proteins to study how these proteins behave or function in replicating cells. For example, combined EdU and MCM2 staining was used to reveal how MCM levels affect DNA synthesis (Matson et al., 2019).
Figure 3. Assays for the detection of replication stress.
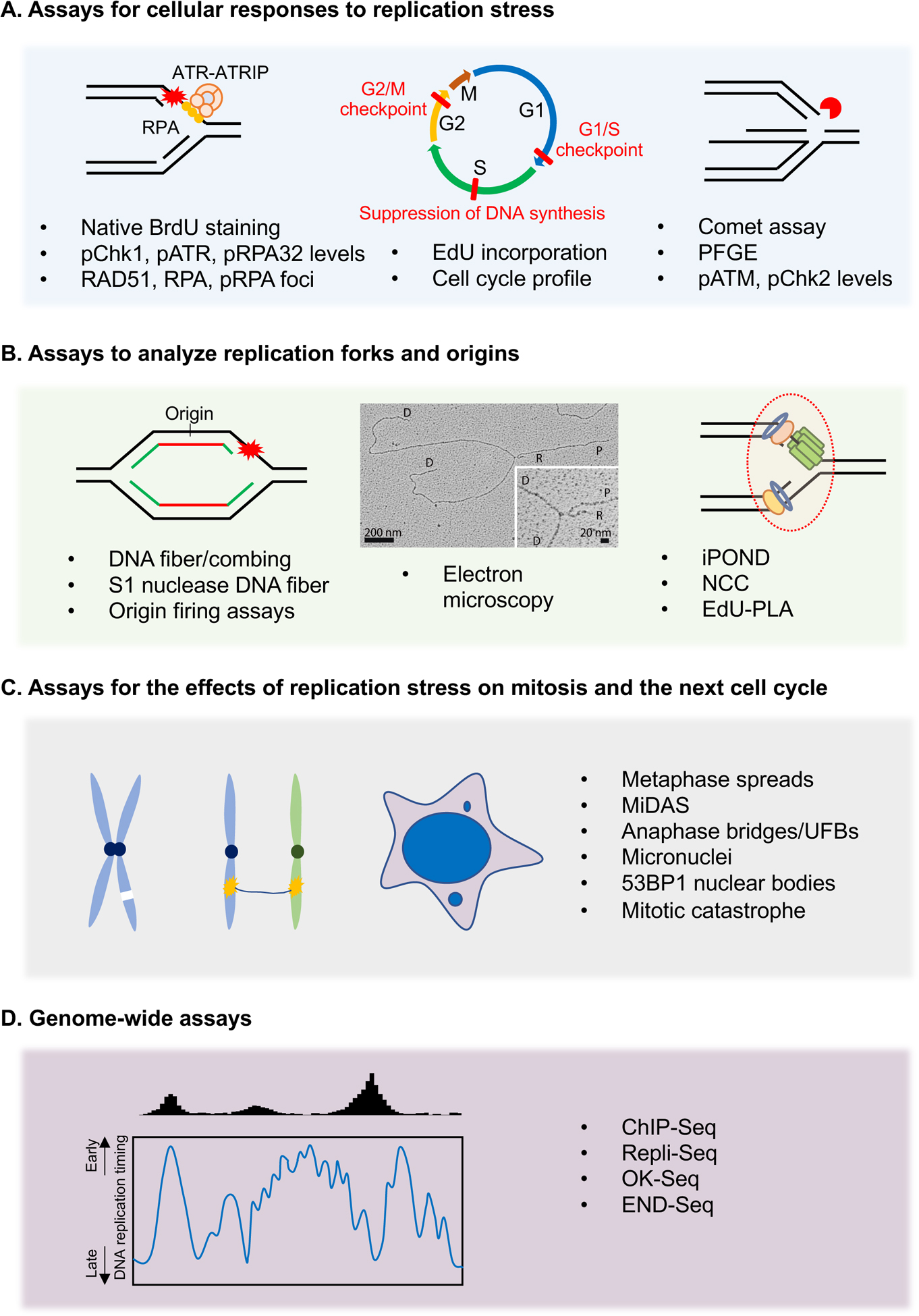
(A) Cellular assays to study the global effects of replication stress by measuring the accumulation of ssDNA, phosphorylation of ATR substrates, suppression of DNA synthesis, cell cycle arrests, and generation of DSBs. (B) DNA fiber-based assays directly study the impact of replication stress on progressing forks and origin firing. Replication forks can be directly visualized by EM to study fork reversal and ssDNA gap generation. The EM image is reproduced from (Genois et al. 2021). Other assays including iPOND and PLA are used to analyze factors associated with nascent DNA. (C) Cellular assays that use mitotic defects as a proxy to measure the replication stress in the preceding S phase. These assays study the effects of replication stress on mitosis and the next cell cycle in the form of anaphase bridges, micronucleation, chromosomal aberrations, and 53BP1 NBs. (D) Genomic assays that use next-generation sequencing (NGS) to obtain a whole-genome view of the effects of replication stress. Theses assays are used to map the regions of replication perturbation across the genome and to understand the contributions of specific chromatin environments and DNA sequence elements to replication stress.
As discussed above, replication stress can induce the accumulation of RPA-ssDNA at or behind stalled forks. Hence, the levels of RPA foci and chromatin-bound RPA can be used as markers for elevated replication stress (Buisson et al., 2015; Toledo et al., 2013; Zou and Elledge, 2003). The exposure of ssDNA in the genome can also be detected by native BrdU staining (Buisson et al., 2015; Couch et al., 2013; Toledo et al., 2013) (Fig. 3A). In this assay, cells are cultured in the presence of BrdU for two cell cycles to label the genomic DNA. If BrdU-labeled DNA is exposed as ssDNA under replication stress, it can be visualized by anti-BrdU immunofluorescence under non-denaturing conditions (Fig. 3A).
The activation of ATR pathway also serves as a reliable marker for replication stress. Several ATR-dependent phosphorylation events, such as ATR p-T1989, Chk1 p-S317/S345, and RPA32 p-S33, can be detected by western blot and immunofluorescence using phospho-specific antibodies (Liu et al., 2000; Liu et al., 2011; Shiotani et al., 2013) (Fig. 3A). The phosphorylation of H2AX is also induced by replication stress, which may reflect the activation of ATR at stalled forks or the activation of ATM and DNA-PKcs at replication-associated DSBs (Ward and Chen, 2001). These phosphorylated proteins are commonly used as a proxy for replication stress in cells proficient for replication and DNA damage checkpoints. In addition, ATR-mediated slowdown of the G2/M transition and accumulation of G2/M cells can be detected by propidium iodide staining of DNA and fluorescence-activated cell sorting (FACS) (Fig. 3A). If the S phase cells exposed to replication stress are given enough time to progress to mitosis, a G2/M arrest can be detected by the reduction of mitotic cells marked by histone H3 p-S10 (Liu et al., 2000). If cells with mild replication stress escape the G2/M checkpoint and progress into the next cell cycle, it can trigger a p53-dependent prolongation of G1 (Lezaja and Altmeyer, 2018).
Several DNA replication and repair proteins involved in the replication stress response, such as RPA, RAD51, and FANCD2, form discrete foci in the nucleus (Andreassen et al., 2004; Petermann et al., 2010a). These foci may reflect the remodeling or repair events at stalled or collapsed replication forks (Fig. 3A). The number and intensity of these foci are used as indirect markers of replication stress. Furthermore, nuclear foci or protein levels of ATM p-S1981, Chk2 p-T68, KAP1 p-S824, RPA2 p-S4/8, and RPA2 p-T21 can be used to indirectly monitor the DSBs formed by prolonged stalling and nucleolytic cleavage of replication forks (Marechal and Zou, 2013; Nakamura et al., 2021; Toledo et al., 2013). The DSBs induced by replication stress can also be directly detected by non-denaturing comet assay or pulsed field gel electrophoresis (PFGE) (Olive and Banath, 2006) (Fig. 3A). It should be noted that the levels of replication-associated DSBs are affected by the levels and duration of replication stress. Prolonged but mild replication stress does not trigger a robust DSB response, but increases DSBs at CFSs (Durkin and Glover, 2007).
Assays to analyze replication forks and origins
A number of DNA fiber-based assays are developed to analyze the behaviors of replication tracts and alterations in nascent DNA in cells. Several EdU-based assays are used to analyze the proteins associated with replication forks in cell populations or individual cells. These assays provide direct measurements of the effects of replication stress on replication forks.
DNA fiber and combing assays directly measure replication fork dynamics at a single-molecule resolution in cell populations. In these assays, newly synthesized DNA in cells is sequentially labeled with two thymidine analogs—e.g., 5-iodo-2′-deoxyuridine (IdU) and 5-chloro-2′deoxyuridine (CldU). Cells are lysed on tilted glass slides and genomic DNA is stretched into fibers by gravity. Immunostaining of these fibers enables microscopic visualization of individual replication tracts (Pasero et al., 2002; Techer et al., 2013) (Fig. 3B). These assays can be used under various conditions to monitor fork progression, fork stalling, fork restart, and nucleolytic degradation of nascent DNA (Quinet et al., 2017a). Replication stress is commonly associated with reduced fork speed, which may reflect a reduction in fork function or stability, an increase of barriers in DNA, or defects in fork restart. Acceleration of replication forks can also result in genomic instability, which may be attributed to the aberrant stress response at forks (Genois et al., 2021; Maya-Mendoza et al., 2018; Saxena et al., 2018). DNA fiber assay can also be used to detect newly fired origins and measure the overall levels of origin firing in cell populations. When cells are sequentially labeled with CldU and IdU and subjected to DNA fiber analysis, continuous IdU-CldU-IdU tracts reflect the origins fired during CldU labelling, and IdU-only tracts indicate origins fired during the IdU pulse (Nieminuszczy et al., 2016). In cells with a proficient ATR checkpoint, replication stress typically leads to a reduction in overall origin firing.
While the DNA fiber assay directly analyzes DNA fibers spread by gravity, the DNA combing assay requires a combing apparatus to stretch DNA fibers and hence offers a more accurate measurement of the length of replication tracts. This assay can be used to directly measure origin density by estimating the distance between adjacent initiation events along a DNA fiber, termed inter-origin distance (IOD) (Techer et al., 2013). The activation of ATR by replication stress reduces origin firing, leading to an increase of IOD. However, the IOD determined by DNA fiber/combing assay likely overestimates the actual distance between origins because only the origins fired during the labeling periods can be detected. DNA combing assay can be coupled with site-specific hybridization probes, sequence enrichment strategies, or labeled DNA lesions to analyze the replication tracts in specific genomic regions, such as telomeres and rDNA repeats, or at sites of ICLs (Huang et al., 2013; Pasero et al., 2002; Sfeir et al., 2009).
The ssDNA gaps induced by replication stress can be detected in DNA fibers by the S1 nuclease, which specifically cleaves ssDNA, leading to shortening of labeled replication tracts (Quinet et al., 2016). The S1 nuclease assay can be used to monitor the formation and repair of ssDNA gaps during and after replication. However, this approach has certain limitations. For instance, S1 nuclease can also cleave ssDNA at secondary DNA structures. Also, ssDNA gaps can only be visualized if they are present on both leading and lagging strands (Mehta et al., 2022). Alternatively, electron microscopy (EM) can be used to directly visualize ssDNA gaps at forks or in the replicated DNA close to forks (Vindigni and Lopes, 2017; Zellweger and Lopes, 2018) (Fig. 3B). This assay has also been successfully used to detect and quantify reversed replication forks (Quinet et al., 2017b; Ray Chaudhuri et al., 2012). However, because EM specifically analyzes the DNA at or close to replication forks, changes in the DNA distal to forks or loss of forks cannot be directly measured by EM.
iPOND (Isolation of proteins on nascent DNA) permits the isolation and analysis of the proteins associated with nascent DNA in cell populations (Fig. 3B). This assay involves pulse labeling of DNA with EdU, followed by the isolation of EdU-labeled DNA and associated proteins (Dungrawala and Cortez, 2015; Sirbu et al., 2011). iPOND can be used to monitor the association of replication proteins with forks during the stress response, as well as the recruitment of repair and signaling proteins to stalled or collapsed forks (Dungrawala et al., 2015). A similar approach, NCC (nascent chromatin capture), relies on biotin-dUTP labelling of nascent DNA (Alabert et al., 2014). In yeast cells, eSPAN (enrichment and sequencing of proteins associated with nascent DNA) offers a strategy to analyze the recruitment of proteins to leading and lagging strands of replication forks (Yu et al., 2014). Another commonly used approach to detect fork-associated proteins is PLA (proximity ligase assay), which combines antibody–oligo conjugates, enzymatic ligation, and rolling-circle amplification to detect the proximity between proteins in situ (Soderberg et al., 2008). Recent studies have combined PLA with EdU pulse labeling (Roy and Schlacher, 2019), allowing detection of the proteins in close proximity to nascent DNA in individual cells. Moreover, PLA between RNA polymerase II (RNAPII) and replisome components has been used to analyze TRC (Kim et al., 2020; Matos et al., 2020). It should be noted that iPOND, NCC and eSPAN analyze the association of many proteins with nascent DNA in cell populations, whereas PLA detects the proximity of individual proteins to nascent DNA or replisomes in single cells.
Assays for the effects of replication stress on mitosis and the next cell cycle
As under-replicated or unresolved DNA structures induced by replication stress can persist into mitosis and jeopardize chromosome segregation (Gelot et al., 2015), some of the mitotic assays can serve as a proxy for replication stress in the preceding S phase.
Chromosomal abnormalities resulting from replication stress, such as chromatid breaks, acentric chromosomes, and radial chromosomes, can be measured in metaphase spreads (Wilhelm et al., 2019) (Fig. 3C). DNA breaks at difficult-to-replicate regions such as CFSs manifest as DAPI-negative breaks, gaps, and constrictions on metaphase chromosomes (Glover et al., 2017). Also, MiDAS-dependent replication of UR-DNA in early mitosis can be visualized as EdU foci on metaphase chromosomes (Bhowmick et al., 2016). Incomplete resolution of replication intermediates can be detected as chromosome bridges. These bridges can be classified into DAPI-positive bulky anaphase bridges and DAPI-negative UFBs, which are characterized by the binding of PICH across the bridge and FANCD2-FANCI at the termini (Chan and Hickson, 2011).
Mitotic catastrophe (MC) is a cell death mechanism in mitosis. MC occurs as a consequence of chromosome nondisjunction or mitotic entry with multiple spindle poles, and is characterized by the formation of giant multinucleated cells (Vitale et al., 2011). Replication stress promotes chromosome nondisjunction and favors mitotic extra centrosomes and multipolar mitosis, thus increasing mitotic catastrophes (Masamsetti et al., 2019). Replication stress also induces micronuclei, which are generated by the entrapment of lagging chromosomes, acentric chromosomes, or chromatid fragments in their own nuclear envelopes upon the completion of mitosis (Xu et al., 2011) (Fig. 3C). Micronuclei serve as a reliable biomarker of replication stress and genomic instability in mammalian cells. Another marker of unresolved replication stress is the presence of large 53BP1 NBs in G1 cells, which contain γH2AX, MDC1 and OPT (Oct1, PTF, transcription) domains (Harrigan et al., 2011; Lukas et al., 2011; Spies et al., 2019).
Genome-wide assays
In addition to the mechanistic insights from molecular and cell biology approaches, it is critical to obtain a genomic view of replication stress. Genomics approaches are particularly useful to understand the contribution of specific DNA sequence elements to replication stress and map the sites of replication fork perturbation and collapse across the genome.
In human cells DNA replication initiates within broad zones of ~150 kb, termed initiation zones (IZs), which contain clusters of origins used at varying efficiencies (Petryk et al., 2016). Induction of replication stress leads to characteristic changes in replication profiles, including the loss of specific IZs within early replication domains, and global disappearance of the replication timing domain structures (Hayakawa et al., 2021). These genome-wide changes in the replication timing domains can be detected by Repli-seq (Hansen et al., 2010) (Fig. 3D). Another widely used approach to study replication initiation, elongation, and termination in yeast and mammalian cells is OK-seq (Kit Leng Lui et al., 2021). OK-seq detects transitions in fork polarity, and it has produced comprehensive maps of fork polarities in several mammalian cell lines (Petryk et al., 2018; Petryk et al., 2016). In addition, a method called ORM (optimal replication mapping) was developed recently to reconstruct genome-wide replication profiles from extra-long DNA fibers (Wang et al., 2021). This approach will also be useful for analyzing the changes in replication under stress.
As an indirect approach, chromatin immunoprecipitation sequencing (ChIP-seq) of replication and repair proteins can be used to identify difficult-to-replicate regions in the genome. For example, ChIP-seq analysis of RPA, BRCA1, and SMC5 revealed that these proteins colocalize at early replicating fragile sites (ERFS) in the genome (Barlow et al., 2013) (Fig. 3D). Alternatively, END-Seq is a highly sensitive technique which allows quantitative mapping of DSBs at nucleotide resolution across the genome (Canela et al., 2016). This assay has been successfully used to map the sites of replication fork collapse throughout the genome (Tubbs et al., 2018).
CONCLUDING REMARKS AND FUTURE PERSPECTIVES
While replication stress is detrimental to the genome, it is an inseparable part of DNA replication. Even in proliferating normal cells, DNA replication forks inevitably encounter transcription machineries, DNA secondary structures, and intrinsic DNA damage. The cellular responses to replication stress, which involve the checkpoint and DNA repair pathways, are critical for maintaining genomic integrity. In cancer cells, replication stress is increased by a wide variety of cellular alterations, ranging from elevated ROS to mutations in DNA repair proteins. While our general understanding of replication stress has improved dramatically during the past decade, how replication stress arises in different types of cancers remains poorly understood. Several concepts emerging from recent studies will help us better understand the replication stress in normal and cancer cells. First, we now appreciate that replication stress comes in multiple flavors. Several sources of replication stress, such as TRC, R-loops, AP sites, ssDNA gaps, and DPCs, may be upregulated in specific cell types and tissues, during specific developmental stages, or in subsets of cancers. Second, cells use multiple mechanisms to deal with replication stress, and the choice among these mechanisms can vary in different contexts. For example, the choice among fork reversal, repriming, TS, and TLS can be influenced by several factors including PARP, DNA translocases and PCNA ubiquitination. Third, replication stress is a quantitative trait with multiple possible outcomes. For example, the levels of ssDNA at replication forks may dictate which repair or cell death pathway to activate. Fourth, it is important to investigate when and where replication stress occurs in the genome. Different types of replication stress may manifest in distinct regions of the genome, and genome-wide assays such as END-seq will be useful for pinpointing these effects. Finally, replication stress can be spatially and temporarily uncoupled from replication forks in some contexts. For example, the ssDNA gaps generated during replication can persist into later phases of the cell cycle and even the next S phase, allowing them to affect genomic stability in different ways. It is conceivable that future studies will benefit from methods that quantitatively analyze different types of replication stress in the genomic space, in cell populations, and across multiple cell cycles. A better understanding of replication stress will provide a basis to improve the treatment of cancer and other diseases associated with replication-born genomic instability.
Acknowledgements
We apologize to those authors whose work could not be cited due to space constraints. S.S. is supported by the James A. Harting Scientific Scholar Award from the Rivkin Center. L.Z. is the James & Patricia Poitras Endow Chair for Cancer Research and support by the NIH (CA263934).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests. L. Z. is a member of the advisory board of Molecular Cell.
References
- Abe T, Kawasumi R, Giannattasio M, Dusi S, Yoshimoto Y, Miyata K, Umemura K, Hirota K, and Branzei D (2018). AND-1 fork protection function prevents fork resection and is essential for proliferation. Nat Commun 9, 3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A, and Garcia-Muse T (2012). R loops: from transcription byproducts to threats to genome stability. Mol Cell 46, 115–124. [DOI] [PubMed] [Google Scholar]
- Ait Saada A, Teixeira-Silva A, Iraqui I, Costes A, Hardy J, Paoletti G, Freon K, and Lambert SAE (2017). Unprotected Replication Forks Are Converted into Mitotic Sister Chromatid Bridges. Mol Cell 66, 398–410 e394. [DOI] [PubMed] [Google Scholar]
- Alabert C, Bukowski-Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, and Groth A (2014). Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol 16, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida R, Fernandez-Justel JM, Santa-Maria C, Cadoret JC, Cano-Aroca L, Lombrana R, Herranz G, Agresti A, and Gomez M (2018). Chromatin conformation regulates the coordination between DNA replication and transcription. Nat Commun 9, 1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez S, Diaz M, Flach J, Rodriguez-Acebes S, Lopez-Contreras AJ, Martinez D, Canamero M, Fernandez-Capetillo O, Isern J, Passegue E, et al. (2015). Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat Commun 6, 8548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammazzalorso F, Pirzio LM, Bignami M, Franchitto A, and Pichierri P (2010). ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J 29, 3156–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen PR, D’Andrea AD, and Taniguchi T (2004). ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev 18, 1958–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashour ME, and Mosammaparast N (2021). Mechanisms of damage tolerance and repair during DNA replication. Nucleic Acids Res 49, 3033–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Kermi C, Stoy H, Schiltz CJ, Bacal J, Zaino AM, Hadden MK, Eichman BF, Lopes M, and Cimprich KA (2020). HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol Cell 78, 1237–1251 e1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow JH, Faryabi RB, Callen E, Wong N, Malhowski A, Chen HT, Gutierrez-Cruz G, Sun HW, McKinnon P, Wright G, et al. (2013). Identification of early replicating fragile sites that contribute to genome instability. Cell 152, 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass TE, Luzwick JW, Kavanaugh G, Carroll C, Dungrawala H, Glick GG, Feldkamp MD, Putney R, Chazin WJ, and Cortez D (2016). ETAA1 acts at stalled replication forks to maintain genome integrity. Nat Cell Biol 18, 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Nahse-Kumpf V, Larsen MS, O’Hanlon KA, Patzke S, Holmberg C, Mejlvang J, Groth A, Nielsen O, Syljuasen RG, et al. (2012). Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol Cell Biol 32, 4226–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, and Dutta A (2002). DNA replication in eukaryotic cells. Annu Rev Biochem 71, 333–374. [DOI] [PubMed] [Google Scholar]
- Bellelli R, Borel V, Logan C, Svendsen J, Cox DE, Nye E, Metcalfe K, O’Connell SM, Stamp G, Flynn HR, et al. (2018). Polepsilon Instability Drives Replication Stress, Abnormal Development, and Tumorigenesis. Mol Cell 70, 707–721 e707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellelli R, and Boulton SJ (2021). Spotlight on the Replisome: Aetiology of DNA Replication-Associated Genetic Diseases. Trends Genet 37, 317–336. [DOI] [PubMed] [Google Scholar]
- Berti M, Cortez D, and Lopes M (2020). The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol 21, 633–651. [DOI] [PubMed] [Google Scholar]
- Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. (2013). Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol 20, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berti M, and Vindigni A (2016). Replication stress: getting back on track. Nat Struct Mol Biol 23, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, and Kerem B (2011). Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145, 435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick R, Minocherhomji S, and Hickson ID (2016). RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol Cell 64, 1117–1126. [DOI] [PubMed] [Google Scholar]
- Blow JJ, and Dutta A (2005). Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol 6, 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brison O, El-Hilali S, Azar D, Koundrioukoff S, Schmidt M, Nahse V, Jaszczyszyn Y, Lachages AM, Dutrillaux B, Thermes C, et al. (2019). Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide. Nat Commun 10, 5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Boisvert JL, Benes CH, and Zou L (2015). Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol Cell 59, 1011–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers PMJ, and Kunkel TA (2017). Eukaryotic DNA Replication Fork. Annu Rev Biochem 86, 417–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, et al. (2013). Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun TS, Pacek M, Yee MC, Walter JC, and Cimprich KA (2005). Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 19, 1040–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canela A, Sridharan S, Sciascia N, Tubbs A, Meltzer P, Sleckman BP, and Nussenzweig A (2016). DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol Cell 63, 898–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KL, and Hickson ID (2011). New insights into the formation and resolution of ultra-fine anaphase bridges. Semin Cell Dev Biol 22, 906–912. [DOI] [PubMed] [Google Scholar]
- Chan KL, Palmai-Pallag T, Ying S, and Hickson ID (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 11, 753–760. [DOI] [PubMed] [Google Scholar]
- Chen YH, Jones MJ, Yin Y, Crist SB, Colnaghi L, Sims RJ 3rd, Rothenberg E, Jallepalli PV, and Huang TT (2015). ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol Cell 58, 323–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, and Elledge SJ (2010). The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D (2015). Preventing replication fork collapse to maintain genome integrity. DNA Repair (Amst) 32, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D (2019). Replication-Coupled DNA Repair. Mol Cell 74, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, and Elledge SJ (2001). ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716. [DOI] [PubMed] [Google Scholar]
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, and Halazonetis TD (2014). Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Betous R, Carroll CM, Jung SY, Qin J, Cimprich KA, et al. (2013). ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27, 1610–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley MP, Bocek M, and Cimprich KA (2019). R-Loops as Cellular Regulators and Genomic Threats. Mol Cell 73, 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson IF, Li A, and Blow JJ (2006). Deregulated replication licensing causes DNA fragmentation consistent with head-to-tail fork collision. Mol Cell 24, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SL, North PS, Dart A, Lakin ND, and Hickson ID (2004). Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol 24, 1279–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, and Malkova A (2011). Break-induced replication is highly inaccurate. PLoS Biol 9, e1000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, and Karnitz LM (2007). The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev 21, 1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar JM, and Walter JC (2017). Mechanisms of DNA replication termination. Nat Rev Mol Cell Biol 18, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, Galloway DA, Gu W, Gautier J, and Dalla-Favera R (2007). Non-transcriptional control of DNA replication by c-Myc. Nature 448, 445–451. [DOI] [PubMed] [Google Scholar]
- Dungrawala H, and Cortez D (2015). Purification of proteins on newly synthesized DNA using iPOND. Methods Mol Biol 1228, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, and Cortez D (2015). The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol Cell 59, 998–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin SG, and Glover TW (2007). Chromosome fragile sites. Annu Rev Genet 41, 169–192. [DOI] [PubMed] [Google Scholar]
- Dutta S, Chowdhury G, and Gates KS (2007). Interstrand cross-links generated by abasic sites in duplex DNA. J Am Chem Soc 129, 1852–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico A, and Costanzo V (2012). Mechanisms of replication fork protection: a safeguard for genome stability. Crit Rev Biochem Mol Biol 47, 222–235. [DOI] [PubMed] [Google Scholar]
- Fagundes R, and Teixeira LK (2021). Cyclin E/CDK2: DNA Replication, Replication Stress and Genomic Instability. Front Cell Dev Biol 9, 774845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RL, Cox KE, Jeitany M, Wakimoto H, Bryll AR, Ganem NJ, Bersani F, Pineda JR, Suva ML, Benes CH, et al. (2015). Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347, 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkos M, Ganier O, Coulombe P, and Mechali M (2015). DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 16, 360–374. [DOI] [PubMed] [Google Scholar]
- Fu H, Redon CE, Thakur BL, Utani K, Sebastian R, Jang SM, Gross JM, Mosavarpour S, Marks AB, Zhuang SZ, et al. (2021). Dynamics of replication origin over-activation. Nat Commun 12, 3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugger K, Bajrami I, Silva Dos Santos M, Young SJ, Kunzelmann S, Kelly G, Hewitt G, Patel H, Goldstone R, Carell T, et al. (2021). Targeting the nucleotide salvage factor DNPH1 sensitizes BRCA-deficient cells to PARP inhibitors. Science 372, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugger K, Chu WK, Haahr P, Kousholt AN, Beck H, Payne MJ, Hanada K, Hickson ID, and Sorensen CS (2013). FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress. Nat Commun 4, 1423. [DOI] [PubMed] [Google Scholar]
- Fujita M (2006). Cdt1 revisited: complex and tight regulation during the cell cycle and consequences of deregulation in mammalian cells. Cell Div 1, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad H, Koolmeister T, Jemth AS, Eshtad S, Jacques SA, Strom CE, Svensson LM, Schultz N, Lundback T, Einarsdottir BO, et al. (2014). MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 508, 215–221. [DOI] [PubMed] [Google Scholar]
- Gaillard H, Garcia-Muse T, and Aguilera A (2015). Replication stress and cancer. Nat Rev Cancer 15, 276–289. [DOI] [PubMed] [Google Scholar]
- Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, and Li X (2011). R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev 25, 2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, Terrados G, Powell C, Salido E, Mendez J, Holt IJ, et al. (2013). PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 52, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge XQ, and Blow JJ (2010). Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol 191, 1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelot C, Magdalou I, and Lopez BS (2015). Replication stress in Mammalian cells and its consequences for mitosis. Genes (Basel) 6, 267–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genois MM, Gagne JP, Yasuhara T, Jackson J, Saxena S, Langelier MF, Ahel I, Bedford MT, Pascal JM, Vindigni A, et al. (2021). CARM1 regulates replication fork speed and stress response by stimulating PARP1. Mol Cell 81, 784–800 e788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert DM (2010). Evaluating genome-scale approaches to eukaryotic DNA replication. Nat Rev Genet 11, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover TW, Wilson TE, and Arlt MF (2017). Fragile sites in cancer: more than meets the eye. Nat Rev Cancer 17, 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Kumagai A, Schlacher K, Shevchenko A, Shevchenko A, and Dunphy WG (2015). Interaction of Chk1 with Treslin negatively regulates the initiation of chromosomal DNA replication. Mol Cell 57, 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Concepcion CP, Fahey CG, Keshishian H, Bhutkar A, Brainson CF, Sanchez-Rivera FJ, Pessina P, Kim JY, Simoneau A, et al. (2020). BRG1 Loss Predisposes Lung Cancers to Replicative Stress and ATR Dependency. Cancer Res 80, 3841–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haahr P, Hoffmann S, Tollenaere MA, Ho T, Toledo LI, Mann M, Bekker-Jensen S, Raschle M, and Mailand N (2016). Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat Cell Biol 18, 1196–1207. [DOI] [PubMed] [Google Scholar]
- Hamperl S, Bocek MJ, Saldivar JC, Swigut T, and Cimprich KA (2017). Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 170, 774–786 e719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, Maas A, Essers J, Hickson ID, and Kanaar R (2007). The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol 14, 1096–1104. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, and Stamatoyannopoulos JA (2010). Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc Natl Acad Sci U S A 107, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, and Caldecott KW (2018). The Importance of Poly(ADP-Ribose) Polymerase as a Sensor of Unligated Okazaki Fragments during DNA Replication. Mol Cell 71, 319–331 e313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, and Jackson SP (2011). Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 193, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, and Costanzo V (2010). Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass CS, Gakhar L, and Wold MS (2010). Functional characterization of a cancer causing mutation in human replication protein A. Mol Cancer Res 8, 1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa T, Suzuki R, Kagotani K, Okumura K, and Takebayashi SI (2021). Camptothecin-Induced Replication Stress Affects DNA Replication Profiling by E/L Repli-Seq. Cytogenet Genome Res 161, 437–444. [DOI] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Nudler E, and Tora L (2013). Transcription-replication encounters, consequences and genomic instability. Nat Struct Mol Biol 20, 412–418. [DOI] [PubMed] [Google Scholar]
- Hoek M, and Stillman B (2003). Chromatin assembly factor 1 is essential and couples chromatin assembly to DNA replication in vivo. Proc Natl Acad Sci U S A 100, 12183–12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook SS, Lin JJ, and Dutta A (2007). Mechanisms to control rereplication and implications for cancer. Curr Opin Cell Biol 19, 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, and Seidman MM (2013). The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell 52, 434–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh MS, Ivanochko D, Hashem LE, Curtin M, Delorme M, Goodall E, Yan K, and Picketts DJ (2016). Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis 7, e2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide H, Shoulkamy MI, Nakano T, Miyamoto-Matsubara M, and Salem AM (2011). Repair and biochemical effects of DNA-protein crosslinks. Mutat Res 711, 113–122. [DOI] [PubMed] [Google Scholar]
- Ivessa AS, Lenzmeier BA, Bessler JB, Goudsouzian LK, Schnakenberg SL, and Zakian VA (2003). The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell 12, 1525–1536. [DOI] [PubMed] [Google Scholar]
- Jones RM, Mortusewicz O, Afzal I, Lorvellec M, Garcia P, Helleday T, and Petermann E (2013). Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 32, 3744–3753. [DOI] [PubMed] [Google Scholar]
- Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, Buske T, Botchan MR, and Shima N (2011). Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol Cell 41, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kang N, Park SH, Wells J, Hwang T, Ryu E, Kim BG, Hwang S, Kim SJ, Kang S, et al. (2020). ATAD5 restricts R-loop formation through PCNA unloading and RNA helicase maintenance at the replication fork. Nucleic Acids Res 48, 7218–7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kit Leng Lui S, Keegan S, Tonzi P, Kahli M, Chen YH, Chalhoub N, Coleman KE, Fenyo D, Smith DJ, and Huang TT (2021). Monitoring genome-wide replication fork directionality by Okazaki fragment sequencing in mammalian cells. Nat Protoc 16, 1193–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 67, 867–881 e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramara J, Osia B, and Malkova A (2018). Break-Induced Replication: The Where, The Why, and The How. Trends Genet 34, 518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, and Dunphy WG (2006). TopBP1 activates the ATR-ATRIP complex. Cell 124, 943–955. [DOI] [PubMed] [Google Scholar]
- Kumar C, Batra S, Griffith JD, and Remus D (2021). The interplay of RNA:DNA hybrid structure and G-quadruplexes determines the outcome of R-loop-replisome collisions. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunnev D, Rusiniak ME, Kudla A, Freeland A, Cady GK, and Pruitt SC (2010). DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene 29, 3630–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurashima K, Kashiwagi H, Shimomura I, Suzuki A, Takeshita F, Mazevet M, Harata M, Yamashita T, Yamamoto Y, Kohno T, et al. (2020). SMARCA4 deficiency-associated heterochromatin induces intrinsic DNA replication stress and susceptibility to ATR inhibition in lung adenocarcinoma. NAR Cancer 2, zcaa005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TM, Poddar S, Capri JR, Abt ER, Kim W, Wei L, Uong NT, Cheng CM, Braas D, Nikanjam M, et al. (2017). ATR inhibition facilitates targeting of leukemia dependence on convergent nucleotide biosynthetic pathways. Nat Commun 8, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kumagai A, and Dunphy WG (2007). The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem 282, 28036–28044. [DOI] [PubMed] [Google Scholar]
- Lehmann AR, Niimi A, Ogi T, Brown S, Sabbioneda S, Wing JF, Kannouche PL, and Green CM (2007). Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair (Amst) 6, 891–899. [DOI] [PubMed] [Google Scholar]
- Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, et al. (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat Commun 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezaja A, and Altmeyer M (2018). Inherited DNA lesions determine G1 duration in the next cell cycle. Cell Cycle 17, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezaja A, and Altmeyer M (2021). Dealing with DNA lesions: When one cell cycle is not enough. Curr Opin Cell Biol 70, 27–36. [DOI] [PubMed] [Google Scholar]
- Li F, Deng Z, Zhang L, Wu C, Jin Y, Hwang I, Vladimirova O, Xu L, Yang L, Lu B, et al. (2019). ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. EMBO J 38, e96659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao H, Ji F, Helleday T, and Ying S (2018). Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Takeda S, Kumar R, Westergard TD, Brown EJ, Pandita TK, Cheng EH, and Hsieh JJ (2010). Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature 467, 343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. (2000). Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14, 1448–1459. [PMC free article] [PubMed] [Google Scholar]
- Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, Glover JN, Yang XH, and Zou L (2011). ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell 43, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lossaint G, Larroque M, Ribeyre C, Bec N, Larroque C, Decaillet C, Gari K, and Constantinou A (2013). FANCD2 binds MCM proteins and controls replisome function upon activation of s phase checkpoint signaling. Mol Cell 51, 678–690. [DOI] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al. (2011). 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13, 243–253. [DOI] [PubMed] [Google Scholar]
- Macheret M, and Halazonetis TD (2015). DNA replication stress as a hallmark of cancer. Annu Rev Pathol 10, 425–448. [DOI] [PubMed] [Google Scholar]
- Magdalou I, Lopez BS, Pasero P, and Lambert SA (2014). The causes of replication stress and their consequences on genome stability and cell fate. Semin Cell Dev Biol 30, 154–164. [DOI] [PubMed] [Google Scholar]
- Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, and Lukas J (2000). Rapid destruction of human Cdc25A in response to DNA damage. Science 288, 1425–1429. [DOI] [PubMed] [Google Scholar]
- Malfatti MC, Balachander S, Antoniali G, Koh KD, Saint-Pierre C, Gasparutto D, Chon H, Crouch RJ, Storici F, and Tell G (2017). Abasic and oxidized ribonucleotides embedded in DNA are processed by human APE1 and not by RNase H2. Nucleic Acids Res 45, 11193–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankouri HW, Huttner D, and Hickson ID (2013). How unfinished business from S-phase affects mitosis and beyond. EMBO J 32, 2661–2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A, and Zou L (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marians KJ (2018). Lesion Bypass and the Reactivation of Stalled Replication Forks. Annu Rev Biochem 87, 217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masamsetti VP, Low RRJ, Mak KS, O’Connor A, Riffkin CD, Lamm N, Crabbe L, Karlseder J, Huang DCS, Hayashi MT, et al. (2019). Replication stress induces mitotic death through parallel pathways regulated by WAPL and telomere deprotection. Nat Commun 10, 4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos DA, Zhang JM, Ouyang J, Nguyen HD, Genois MM, and Zou L (2020). ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol Cell 77, 514–527 e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson JP, House AM, Grant GD, Wu H, Perez J, and Cook JG (2019). Intrinsic checkpoint deficiency during cell cycle re-entry from quiescence. J Cell Biol 218, 2169–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160–1166. [DOI] [PubMed] [Google Scholar]
- Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, and Bartek J (2018). High speed of fork progression induces DNA replication stress and genomic instability. Nature 559, 279–284. [DOI] [PubMed] [Google Scholar]
- McIntosh D, and Blow JJ (2012). Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harb Perspect Biol 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta KPM, Thada V, Zhao R, Krishnamoorthy A, Leser M, Lindsey Rose K, and Cortez D (2022). CHK1 phosphorylates PRIMPOL to promote replication stress tolerance. Sci Adv 8, eabm0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melixetian M, Ballabeni A, Masiero L, Gasparini P, Zamponi R, Bartek J, Lukas J, and Helin K (2004). Loss of Geminin induces rereplication in the presence of functional p53. J Cell Biol 165, 473–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KM, Rog O, and Cooper JP (2006). Semi-conservative DNA replication through telomeres requires Taz1. Nature 440, 824–828. [DOI] [PubMed] [Google Scholar]
- Mirkin EV, and Mirkin SM (2007). Replication fork stalling at natural impediments. Microbiol Mol Biol Rev 71, 13–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiseeva TN, Yin Y, Calderon MJ, Qian C, Schamus-Haynes S, Sugitani N, Osmanbeyoglu HU, Rothenberg E, Watkins SC, and Bakkenist CJ (2019). An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc Natl Acad Sci U S A 116, 13374–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouron S, Rodriguez-Acebes S, Martinez-Jimenez MI, Garcia-Gomez S, Chocron S, Blanco L, and Mendez J (2013). Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20, 1383–1389. [DOI] [PubMed] [Google Scholar]
- Murphy AK, Fitzgerald M, Ro T, Kim JH, Rabinowitsch AI, Chowdhury D, Schildkraut CL, and Borowiec JA (2014). Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol 206, 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutreja K, Krietsch J, Hess J, Ursich S, Berti M, Roessler FK, Zellweger R, Patra M, Gasser G, and Lopes M (2018). ATR-Mediated Global Fork Slowing and Reversal Assist Fork Traverse and Prevent Chromosomal Breakage at DNA Interstrand Cross-Links. Cell Rep 24, 2629–2642 e2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Kustatscher G, Alabert C, Hodl M, Forne I, Volker-Albert M, Satpathy S, Beyer TE, Mailand N, Choudhary C, et al. (2021). Proteome dynamics at broken replication forks reveal a distinct ATM-directed repair response suppressing DNA double-strand break ubiquitination. Mol Cell 81, 1084–1099 e1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Zou L, and Graubert TA (2019). Targeting R-loop-associated ATR response in myelodysplastic syndrome. Oncotarget 10, 2581–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminuszczy J, Schwab RA, and Niedzwiedz W (2016). The DNA fibre technique - tracking helicases at work. Methods 108, 92–98. [DOI] [PubMed] [Google Scholar]
- Olive PL, and Banath JP (2006). The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1, 23–29. [DOI] [PubMed] [Google Scholar]
- Ozeri-Galai E, Lebofsky R, Rahat A, Bester AC, Bensimon A, and Kerem B (2011). Failure of origin activation in response to fork stalling leads to chromosomal instability at fragile sites. Mol Cell 43, 122–131. [DOI] [PubMed] [Google Scholar]
- Pai CC, and Kearsey SE (2017). A Critical Balance: dNTPs and the Maintenance of Genome Stability. Genes (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasero P, Bensimon A, and Schwob E (2002). Single-molecule analysis reveals clustering and epigenetic regulation of replication origins at the yeast rDNA locus. Genes Dev 16, 2479–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasero P, and Vindigni A (2017). Nucleases Acting at Stalled Forks: How to Reboot the Replication Program with a Few Shortcuts. Annu Rev Genet 51, 477–499. [DOI] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, and Piwnica-Worms H (1997). Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 277, 1501–1505. [DOI] [PubMed] [Google Scholar]
- Pepe A, and West SC (2014). MUS81-EME2 promotes replication fork restart. Cell Rep 7, 1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Helleday T, and Caldecott KW (2008). Claspin promotes normal replication fork rates in human cells. Mol Biol Cell 19, 2373–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Maya-Mendoza A, Zachos G, Gillespie DA, Jackson DA, and Caldecott KW (2006). Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol 26, 3319–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, and Helleday T (2010a). Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 37, 492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Woodcock M, and Helleday T (2010b). Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci U S A 107, 16090–16095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryk N, Dalby M, Wenger A, Stromme CB, Strandsby A, Andersson R, and Groth A (2018). MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 361, 1389–1392. [DOI] [PubMed] [Google Scholar]
- Petryk N, Kahli M, d’Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, Silvain M, Thermes C, Chen CL, and Hyrien O (2016). Replication landscape of the human genome. Nat Commun 7, 10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad R, Horton JK, Chastain PD 2nd, Gassman NR, Freudenthal BD, Hou EW, and Wilson SH (2014). Suicidal cross-linking of PARP-1 to AP site intermediates in cells undergoing base excision repair. Nucleic Acids Res 42, 6337–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promonet A, Padioleau I, Liu Y, Sanz L, Biernacka A, Schmitz AL, Skrzypczak M, Sarrazin A, Mettling C, Rowicka M, et al. (2020). Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites. Nat Commun 11, 3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Carvajal-Maldonado D, Lemacon D, and Vindigni A (2017a). DNA Fiber Analysis: Mind the Gap! Methods Enzymol 591, 55–82. [DOI] [PubMed] [Google Scholar]
- Quinet A, Lemacon D, and Vindigni A (2017b). Replication Fork Reversal: Players and Guardians. Mol Cell 68, 830–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Martins DJ, Vessoni AT, Biard D, Sarasin A, Stary A, and Menck CF (2016). Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Res 44, 5717–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragland RL, Patel S, Rivard RS, Smith K, Peters AA, Bielinsky AK, and Brown EJ (2013). RNF4 and PLK1 are required for replication fork collapse in ATR-deficient cells. Genes Dev 27, 2259–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, and Lopes M (2012). Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19, 417–423. [DOI] [PubMed] [Google Scholar]
- Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, et al. (2012). Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149, 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, and Manley JL (2017). R Loops and Links to Human Disease. J Mol Biol 429, 3168–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, and Schlacher K (2019). SIRF: A Single-cell Assay for in situ Protein Interaction with Nascent DNA Replication Forks. Bio Protoc 9, e3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale JE (2013). Translesion DNA synthesis and mutagenesis in eukaryotes. Cold Spring Harb Perspect Biol 5, a012708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salic A, and Mitchison TJ (2008). A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 105, 2415–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, and Elledge SJ (1997). Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277, 1497–1501. [DOI] [PubMed] [Google Scholar]
- Saxena S, Dixit S, Somyajit K, and Nagaraju G (2019). ATR Signaling Uncouples the Role of RAD51 Paralogs in Homologous Recombination and Replication Stress Response. Cell Rep 29, 551–559 e554. [DOI] [PubMed] [Google Scholar]
- Saxena S, Somyajit K, and Nagaraju G (2018). XRCC2 Regulates Replication Fork Progression during dNTP Alterations. Cell Rep 25, 3273–3282 e3276. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, and Jasin M (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, and de Lange T (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S (2011). Non-B DNA Secondary Structures and Their Resolution by RecQ Helicases. J Nucleic Acids 2011, 724215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastri N, Tsai YC, Hile S, Jordan D, Powell B, Chen J, Maloney D, Dose M, Lo Y, Anastassiadis T, et al. (2018). Genome-wide Identification of Structure-Forming Repeats as Principal Sites of Fork Collapse upon ATR Inhibition. Mol Cell 72, 222–238 e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima N, Alcaraz A, Liachko I, Buske TR, Andrews CA, Munroe RJ, Hartford SA, Tye BK, and Schimenti JC (2007). A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat Genet 39, 93–98. [DOI] [PubMed] [Google Scholar]
- Shiotani B, Nguyen HD, Hakansson P, Marechal A, Tse A, Tahara H, and Zou L (2013). Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep 3, 1651–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoneau A, Xiong R, and Zou L (2021). The trans cell cycle effects of PARP inhibitors underlie their selectivity toward BRCA1/2-deficient cells. Genes Dev 35, 1271–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoneau A, and Zou L (2021). An extending ATR-CHK1 circuitry: the replication stress response and beyond. Curr Opin Genet Dev 71, 92–98. [DOI] [PubMed] [Google Scholar]
- Simonelli V, Narciso L, Dogliotti E, and Fortini P (2005). Base excision repair intermediates are mutagenic in mammalian cells. Nucleic Acids Res 33, 4404–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, and Cortez D (2011). Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25, 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Proudfoot NJ, and Gromak N (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 42, 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, and Landegren U (2008). Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45, 227–232. [DOI] [PubMed] [Google Scholar]
- Sollier J, Stork CT, Garcia-Rubio ML, Paulsen RD, Aguilera A, and Cimprich KA (2014). Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell 56, 777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somyajit K, Gupta R, Sedlackova H, Neelsen KJ, Ochs F, Rask MB, Choudhary C, and Lukas J (2017). Redox-sensitive alteration of replisome architecture safeguards genome integrity. Science 358, 797–802. [DOI] [PubMed] [Google Scholar]
- Somyajit K, Saxena S, Babu S, Mishra A, and Nagaraju G (2015). Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res 43, 9835–9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somyajit K, Spies J, Coscia F, Kirik U, Rask MB, Lee JH, Neelsen KJ, Mund A, Jensen LJ, Paull TT, et al. (2021). Homology-directed repair protects the replicating genome from metabolic assaults. Dev Cell 56, 461–477 e467. [DOI] [PubMed] [Google Scholar]
- Sorensen CS, Syljuasen RG, Falck J, Schroeder T, Ronnstrand L, Khanna KK, Zhou BB, Bartek J, and Lukas J (2003). Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 3, 247–258. [DOI] [PubMed] [Google Scholar]
- Spies J, Lukas C, Somyajit K, Rask MB, Lukas J, and Neelsen KJ (2019). 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat Cell Biol 21, 487–497. [DOI] [PubMed] [Google Scholar]
- Spies J, Polasek-Sedlackova H, Lukas J, and Somyajit K (2021). Homologous Recombination as a Fundamental Genome Surveillance Mechanism during DNA Replication. Genes (Basel) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling PC, Chan YA, Minaker SW, Aristizabal MJ, Barrett I, Sipahimalani P, Kobor MS, and Hieter P (2012). R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev 26, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Leuzzi G, Sannino V, Cuella-Martin R, Huang JW, Wu-Baer F, Baer R, Costanzo V, and Ciccia A (2021). REV1-Polzeta maintains the viability of homologous recombination-deficient cancer cells through mutagenic repair of PRIMPOL-dependent ssDNA gaps. Mol Cell 81, 4008–4025 e4007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Techer H, Koundrioukoff S, Azar D, Wilhelm T, Carignon S, Brison O, Debatisse M, and Le Tallec B (2013). Replication dynamics: biases and robustness of DNA fiber analysis. J Mol Biol 425, 4845–4855. [DOI] [PubMed] [Google Scholar]
- Techer H, Koundrioukoff S, Nicolas A, and Debatisse M (2017). The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat Rev Genet 18, 535–550. [DOI] [PubMed] [Google Scholar]
- Thakar T, and Moldovan GL (2021). The emerging determinants of replication fork stability. Nucleic Acids Res 49, 7224–7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur BL, Ray A, Redon CE, and Aladjem MI (2021). Preventing excess replication origin activation to ensure genome stability. Trends Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA, et al. (2015). DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208, 545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PS, and Cortez D (2020). New insights into abasic site repair and tolerance. DNA Repair (Amst) 90, 102866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thys RG, Lehman CE, Pierce LC, and Wang YH (2015). DNA secondary structure at chromosomal fragile sites in human disease. Curr Genomics 16, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirman S, Quinet A, Wood M, Meroni A, Cybulla E, Jackson J, Pegoraro S, Simoneau A, Zou L, and Vindigni A (2021). Temporally distinct post-replicative repair mechanisms fill PRIMPOL-dependent ssDNA gaps in human cells. Mol Cell 81, 4026–4040 e4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo L, Neelsen KJ, and Lukas J (2017). Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol Cell 66, 735–749. [DOI] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, and Lukas J (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103. [DOI] [PubMed] [Google Scholar]
- Tsai S, Fournier LA, Chang EY, Wells JP, Minaker SW, Zhu YD, Wang AY, Wang Y, Huntsman DG, and Stirling PC (2021). ARID1A regulates R-loop associated DNA replication stress. PLoS Genet 17, e1009238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, and Nussenzweig A (2017). Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 168, 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubbs A, Sridharan S, van Wietmarschen N, Maman Y, Callen E, Stanlie A, Wu W, Wu X, Day A, Wong N, et al. (2018). Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 174, 1127–1142 e1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wietmarschen N, Sridharan S, Nathan WJ, Tubbs A, Chan EM, Callen E, Wu W, Belinky F, Tripathi V, Wong N, et al. (2020). Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 586, 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vare D, Groth P, Carlsson R, Johansson F, Erixon K, and Jenssen D (2012). DNA interstrand crosslinks induce a potent replication block followed by formation and repair of double strand breaks in intact mammalian cells. DNA Repair (Amst) 11, 976–985. [DOI] [PubMed] [Google Scholar]
- Vindigni A, and Lopes M (2017). Combining electron microscopy with single molecule DNA fiber approaches to study DNA replication dynamics. Biophys Chem 225, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale I, Galluzzi L, Castedo M, and Kroemer G (2011). Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol 12, 385–392. [DOI] [PubMed] [Google Scholar]
- Voineagu I, Narayanan V, Lobachev KS, and Mirkin SM (2008). Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci U S A 105, 9936–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voulgaridou GP, Anestopoulos I, Franco R, Panayiotidis MI, and Pappa A (2011). DNA damage induced by endogenous aldehydes: current state of knowledge. Mutat Res 711, 13–27. [DOI] [PubMed] [Google Scholar]
- Vrtis KB, Dewar JM, Chistol G, Wu RA, Graham TGW, and Walter JC (2021). Single-strand DNA breaks cause replisome disassembly. Mol Cell 81, 1309–1318 e1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang JW, Holland CL, Zwicky K, et al. (2017). Replication Fork Slowing and Reversal upon DNA Damage Require PCNA Polyubiquitination and ZRANB3 DNA Translocase Activity. Mol Cell 67, 882–890 e885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waga S, and Stillman B (1998). The DNA replication fork in eukaryotic cells. Annu Rev Biochem 67, 721–751. [DOI] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, and Vuica-Ross M (2011). RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell 44, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Klein KN, Proesmans K, Yang H, Marchal C, Zhu X, Borrman T, Hastie A, Weng Z, Bechhoefer J, et al. (2021). Genome-wide mapping of human DNA replication by optical replication mapping supports a stochastic model of eukaryotic replication. Mol Cell 81, 2975–2988 e2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, and Chen J (2001). Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 276, 47759–47762. [DOI] [PubMed] [Google Scholar]
- Weinberg G, Ullman B, and Martin DW Jr. (1981). Mutator phenotypes in mammalian cell mutants with distinct biochemical defects and abnormal deoxyribonucleoside triphosphate pools. Proc Natl Acad Sci U S A 78, 2447–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm T, Magdalou I, Barascu A, Techer H, Debatisse M, and Lopez BS (2014). Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc Natl Acad Sci U S A 111, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm T, Olziersky AM, Harry D, De Sousa F, Vassal H, Eskat A, and Meraldi P (2019). Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat Commun 10, 3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm T, Said M, and Naim V (2020). DNA Replication Stress and Chromosomal Instability: Dangerous Liaisons. Genes (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, and Zhang H (2003). Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem 278, 21767–21773. [DOI] [PubMed] [Google Scholar]
- Xu B, Sun Z, Liu Z, Guo H, Liu Q, Jiang H, Zou Y, Gong Y, Tischfield JA, and Shao C (2011). Replication stress induces micronuclei comprising of aggregated DNA double-strand breaks. PLoS One 6, e18618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Takai KK, Lovejoy CA, and de Lange T (2020). Break-induced replication promotes fragile telomere formation. Genes Dev 34, 1392–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles JT, Poli J, Marians KJ, and Pasero P (2013). Rescuing stalled or damaged replication forks. Cold Spring Harb Perspect Biol 5, a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekezare M, Gomez-Gonzalez B, and Diffley JF (2013). Controlling DNA replication origins in response to DNA damage - inhibit globally, activate locally. J Cell Sci 126, 1297–1306. [DOI] [PubMed] [Google Scholar]
- Yu C, Gan H, Han J, Zhou ZX, Jia S, Chabes A, Farrugia G, Ordog T, and Zhang Z (2014). Strand-specific analysis shows protein binding at replication forks and PCNA unloading from lagging strands when forks stall. Mol Cell 56, 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaratiegui M, Vaughn MW, Irvine DV, Goto D, Watt S, Bahler J, Arcangioli B, and Martienssen RA (2011). CENP-B preserves genome integrity at replication forks paused by retrotransposon LTR. Nature 469, 112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, and Lopes M (2018). Dynamic Architecture of Eukaryotic DNA Replication Forks In Vivo, Visualized by Electron Microscopy. Methods Mol Biol 1672, 261–294. [DOI] [PubMed] [Google Scholar]
- Zeman MK, and Cimprich KA (2014). Causes and consequences of replication stress. Nat Cell Biol 16, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CZ, Spektor A, Cornils H, Francis JM, Jackson EK, Liu S, Meyerson M, and Pellman D (2015). Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Genois MM, Ouyang J, Lan L, and Zou L (2021). Alternative lengthening of telomeres is a self-perpetuating process in ALT-associated PML bodies. Mol Cell 81, 1027–1042 e1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, and Hunter T (2014). Roles of Chk1 in cell biology and cancer therapy. Int J Cancer 134, 1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Watkins JL, and Piwnica-Worms H (2002). Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci U S A 99, 14795–14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Jia J, Finger LD, Guo Z, Zer C, and Shen B (2011). Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res 39, 781–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Nellimoottil T, Peace JM, Knott SR, Villwock SK, Yee JM, Jancuska JM, Rege S, Tecklenburg M, Sclafani RA, et al. (2013). The level of origin firing inversely affects the rate of replication fork progression. J Cell Biol 201, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, and Elledge SJ (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]