

SUMMARY
Genome-wide association studies (GWASs) have identified hundreds of loci associated with psychiatric diseases, yet there is a lack of understanding of disease pathophysiology. Common risk variants can shed light on the underlying molecular mechanisms; however, identifying causal variants remains challenging. We map cis-regulatory elements in human neurons derived from pluripotent stem cells. This system allows us to determine enhancers that activate the transcription of neuronal activity-regulated gene programs, which are thought to be critical for synaptic plasticity and are not possible to identify from postmortem tissues. Using the activity-by-contact model, we create variant-to-gene maps to interpret the function of GWAS variants. Our work nominates a subset of variants to elucidate the molecular mechanisms involving GWAS-significant loci. It also highlights that in vitro human cellular models are a powerful platform for identifying and mechanistic studies of human trait-associated genetic variants in cell states that are inaccessible from other types of human samples.
Graphical abstract
In brief
Sanchez-Priego et al. characterize distinct classes of non-coding regulatory elements in human PSC-derived excitatory and inhibitory neurons. The study reveals enhancers and genetic variants regulating human disease genes.
INTRODUCTION
Neurons have evolved remarkable strategies to respond to sensory information from the external environment on millisecond timescales and store information for long periods. The long-lasting cellular adaptations often required for learning and memory involve converting specific activity patterns to gene transcription programs (Alberini, 2009; Yap and Greenberg, 2018). Non-coding regulatory elements in complex genomes are key players in the dynamic and precise regulation of the transcriptome during responses to external stimuli (Borrelli et al., 2008; Fagiolini et al., 2009; Felling and Song, 2015; Zovkic et al., 2013). Growing evidence indicates that alterations in activity-dependent regulatory elements and transcription can have dire consequences for brain function or strongly affect the risk of psychiatric disorders (Ebert and Greenberg, 2013; Nestler et al., 2016). Large-scale genetic studies have revealed a significant enrichment of common disease risk variants in cis-regulatory elements (CREs), and the accessibility of many of these CREs is determined by activity-dependent transcription factors (TFs) (Maurano et al., 2012). However, identifying disease risk variants located in activity-dependent CREs presents considerable challenges. Epigenomic profiling of postmortem brain tissue lacks a cellular or temporal resolution (de la Torre-Ubieta et al., 2018; Song et al., 2019; Won et al., 2016), precluding the definitive assignment of CREs to specific cell types and unlikely to identify those exhibiting transient activation in response to stimuli (Heinz et al., 2015). Rodent studies revealed that activity-dependent gene expression is specific to neuronal subtypes (Hrvatin et al., 2018; Hu et al., 2017; Lacar et al., 2016; Spiegel et al., 2014; Wu et al., 2017), further underscoring the importance of identifying activity-responsive elements in distinct disease-relevant cell types.
Excitation and inhibition (E-I) are the two fundamental signaling modes within the central nervous system. Perturbation of balance between E-I has been implicated in the etiology and symptomology of multiple psychiatric conditions (Gao and Penzes, 2015; Rubenstein and Merzenich, 2003). Genetic studies have shown that risk variants associated with autism spectrum disorder (ASD) and schizophrenia (SCZ) are enriched for genes expressed in the glutamatergic excitatory (Glu) neurons and GABAergic inhibitory (GABA) neurons (Finucane et al., 2018; Satterstrom et al., 2020; Skene et al., 2018). Therefore, characterizing gene transcription and regulation in human Glu and GABA neurons could provide important insights into biological mechanisms that underlie the risk of psychiatric diseases.
To this end, we obtained human Glu and GABA neurons from pluripotent stem cells (PSCs) and assessed the transcriptome, chromatin landscape, and TF binding before and after membrane depolarization. We identified shared and subtype-specific activity-dependent genes associated with psychiatric diseases and putative CREs critical for neuron-type-specific transcriptional control. Our study nominated TFs that collaborate with broadly expressed activator protein-1 (AP-1) TFs to establish an accessible chromatin state in response to stimuli in specific neuronal types. By applying stratified linkage disequilibrium (LD) score regression (Finucane et al., 2015), we detected significant enrichments for SCZ heritability in activity-inducible enhancers, suggesting that common variants may confer SCZ risk by regulating activity-regulated genes. Our work uncovered human trait-associated genetic variants in cell states that are inaccessible from other types of human samples and provided a resource for the exploration of human brain function and disease.
RESULTS
Activity-induced gene expression in human neurons
To investigate neuronal activity-induced changes in gene expression, we focused on the Glu and GABA neurons generated from human PSCs using the TF-mediated differentiation method (Figure 1A) (Yang et al., 2017; Zhang et al., 2013). Neurons were cultured with mouse glia for 5 weeks, by which time they produced robust action potentials, showed voltage-gated currents, and exhibited spontaneous synaptic activity (Figure 1A). Quantitative RT-PCR (qRT-PCR) analysis confirmed that excitatory and inhibitory neuron cultures were highly enriched for the Glu and GABA neuron markers, respectively, but not the neural progenitor marker NESTIN (NES) (Figure 1A). Similarly, electrophysiological recordings showed that Glu neurons exhibited spontaneous excitatory synaptic currents (sEPSCs) blocked by the AMPA receptor antagonist CNQX; in contrast, the GABA neurons displayed spontaneous inhibitory postsynaptic currents (sIPSCs) blocked by GABAA receptor antagonist picrotoxin (Figure 1A).
Figure 1. Activation of gene expression.
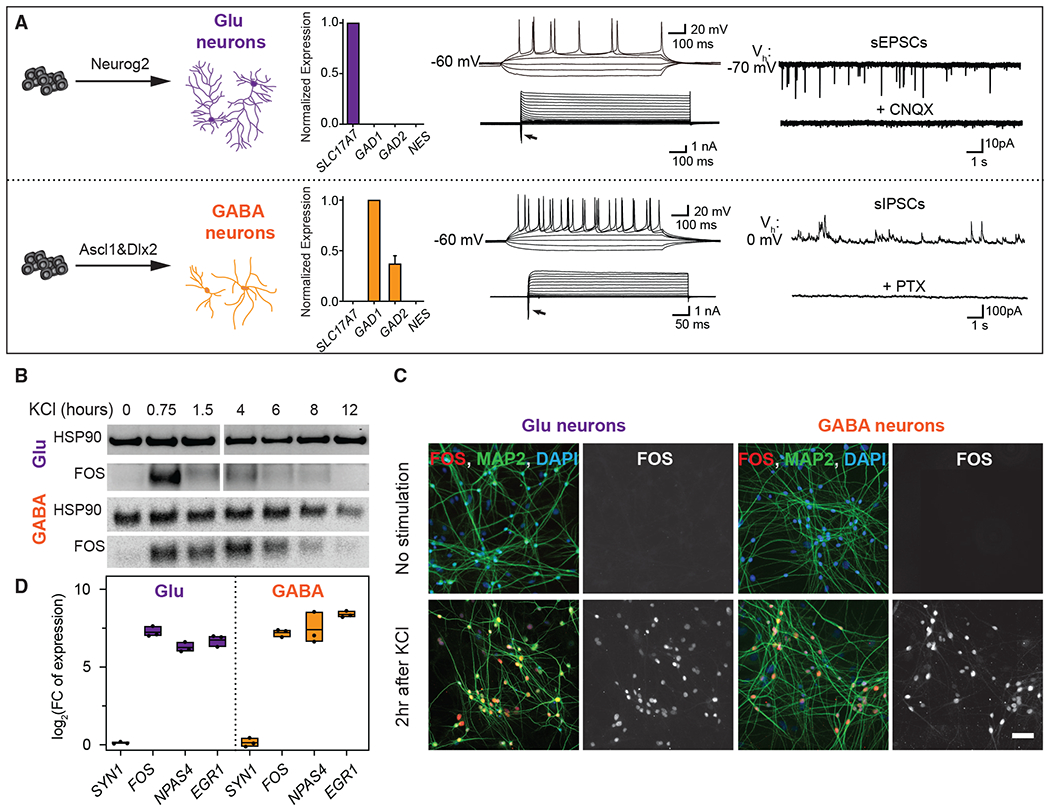
(A) Generation and functional characterization of human neurons. Neurog2 or Ascl1/Dlx2 are delivered to PSCs to generate Glu and GABA neurons. Quantification of selected mRNA levels in neurons after 5 weeks with glia coculture confirmed the neuronal subtype identity. Data are represented as means ± SEMs, n = 3 replicates. Neurons fired repetitive action potentials when depolarized. Whole-cell current response with step current injections was recorded in both neuronal subtypes. sEPSCs were recorded at a holding potential of −70 mV in Glu neurons and blocked by CNQX (20 μM). sIPSCs were detected at a holding potential of 0 mV in GABA neurons and blocked by picrotoxin (50 μM).
(B–D) Expression of activity-responsive genes.
(B) Immunoblot analyses showed FOS activation kinetics. HSP90 was used as the loading control. The marker lane between 1.5 and 4 h was removed.
(C) Neurons immunolabeled for FOS (red) and the pan-neuronal marker MAP2 (green). Images are representative of >20 independent experiments. Scale bar, 50 μm.
(D) Fold induction of mRNA of selected genes at 45 min after KCl measured by qRT-PCR. Data are represented as log2 fold change (n = 3, mean and minimum to the maximum range).
See also Figure S1.
To study activity-induced responses, we exposed neurons to an elevated KCl level. This stimulation paradigm leads to an influx of calcium through L-type voltage-sensitive calcium channels (L-VSCCs), mimicking the response of neurons to sustained synaptic activity and action potential firing (Berridge, 1998; Dolmetsch et al., 2001). Activity-dependent Ca2+ entry via L-VSCCs preferentially activates signaling pathways, including the RAS-MAPK/ERK kinase cascade (Dolmetsch et al., 2001; Tyssowski et al., 2018), which then initiates the transcription of canonical early response genes (ERGs) that in turn regulate a late response gene (LRG) program. We assessed MAPK/ERK pathway activation by immunoblotting for the terminal kinase phospho-ERK (pERK) and found that its level reached the peak magnitude within minutes post-depolarization (Figure S1). FOS was dynamically and uniformly expressed in human neurons upon membrane depolarization resembling primary mouse neurons (Dolmetsch et al., 2001; Sheng et al., 1990) (Figures 1B and 1C). Next, we used species-specific qRT-PCR to measure the expression of prototypical activity-responsive ERGs such as FOS, NPAS4, and EGR1 (Greenberg et al., 1986; Lin et al., 2008). Robust transcriptional responsiveness was readily detected 45 min after depolarization (Figure 1D) and reached peak magnitude 90 min after the start of the activity (Figure S1). The LRGs were maximally expressed 4 h after depolarization (Figure S1). These results demonstrate that human PSC-derived neurons respond robustly to membrane depolarization and express prototypical activity-dependent genes.
Identification of activity-responsive transcriptomic changes
We hypothesized that comprehensively characterizing the membrane depolarization-induced gene expression and regulation programs in Glu and GABA neurons (Figure 2A) could provide insight into the mechanisms of activity-dependent transcription in different neuronal types. We generated Glu and GABA neurons using human embryonic stem cell line H1 (WA01, male) and induced PSC (iPSC) line NSB3188 (F2, female) for RNA sequencing (RNA-seq) profiling. Neurons were harvested without stimulation for 45 min and 4 h after membrane depolarization. We and others (Ichise et al., 2021; Powell et al., 2021; Schrode et al., 2019) have demonstrated that TF(s) induced Glu or GABA neurons with >90% purity from genetically distinct PSC lines. As expected, a high degree of transcriptomic convergence was observed for the F2-derived neurons with the H1-derived neurons that have been comprehensively characterized in our previous reports, confirming their neuronal identities (Figures 2B and S2). Neuronal fate was further confirmed by the demonstrated expression of pan-neuronal, subtype-specific, and synaptic genes and the absence of glial lineage marker genes (Figure S2). Clustering samples by gene expression similarity shows two main groups representing the Glu and GABA neurons (Figure S2), indicating that the small number of other cell types did not affect our downstream studies on subtype-specific gene expression and regulation. Not surprisingly, neurons collected after 45 min clustered with unstimulated cells, as only a small number of genes are induced by depolarization at this time. Principal component analysis (PCA) of the expression data also shows that the samples separate along the first principal component (PC) by cell types and along the second PC by activity state (Figure 2B). Calculating gene expression changes at each stimulation time point compared to the unstimulated condition, we identified a biphasic transcriptional change with a total of 1,430 unique inducible genes (Glu = 1,016; GABA = 1,049) and 1,562 genes (Glu = 980; GABA = 1,079) with reduced expression level after stimulation (Figure S2; Table S1).
Figure 2. Activity-regulated gene transcription and chromatin accessibility.
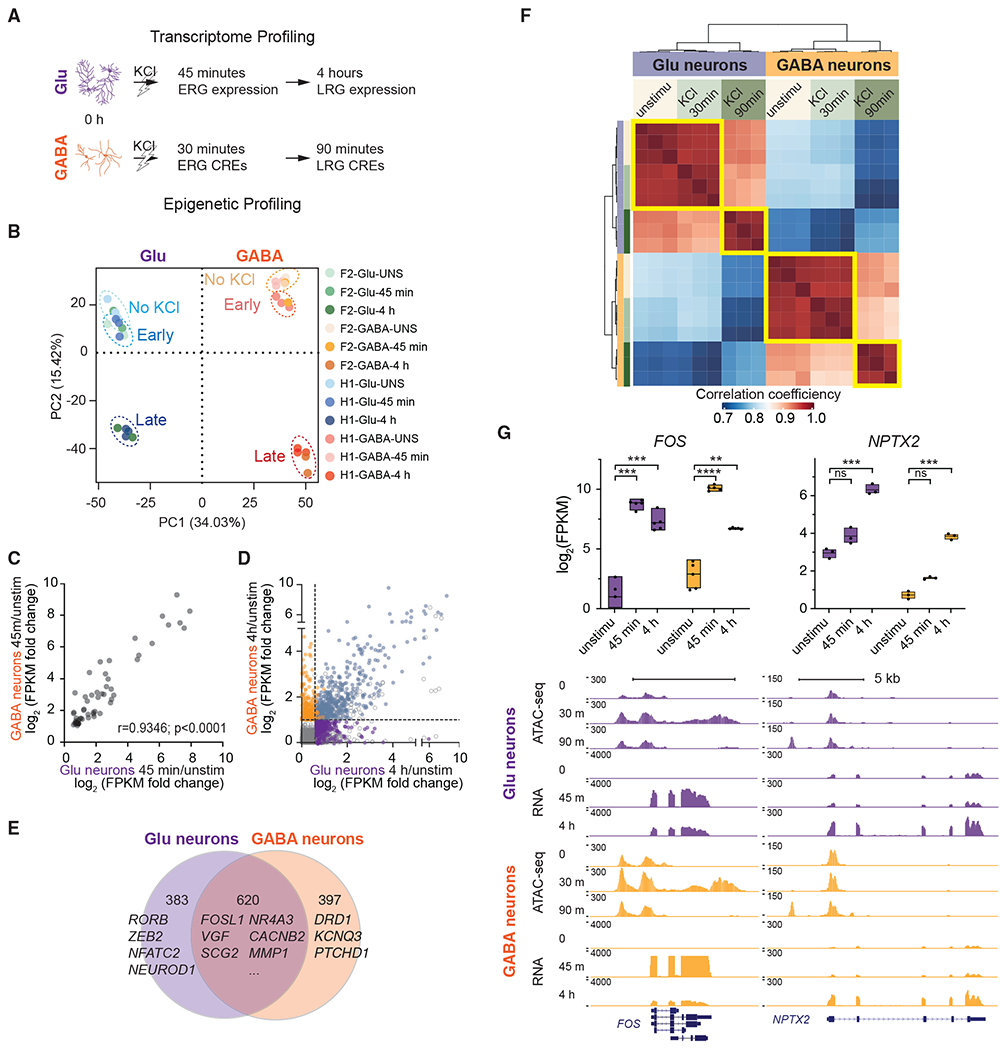
(A) Workflow.
(B) PCA of gene expression data (top 2,000 high-variance genes) from Glu and GABA neurons.
(C) Correlation of expression fold change (log2) values of shared ERGs. Dots represent the mean values of 5 biological replicates. Pearson’s r= 0.9346, p < 0.0001.
(D) Correlation of expression fold change (log2) values of late response genes (LRGs).
(E) Venn diagram displaying the number of genes induced after KCl specifically in Glu (purple), GABA neurons (orange), or commonly in both types (overlapping).
(F) Correlation plot shows the relationship of open chromatin regions among cell types and activity states. Sample-to-sample distance matrix with hierarchical clustering was calculated using the ATAC-seq peak signals.
(G) Expression values measured by RNA-seq of known cell-type-specific or activity-induced genes before or after stimulation. (Upper panel, n = 5; box, minima and maxima, box center = mean; 2-tailed Student’s t test, *p < 0.05 compared with unstimulated (unstimu), ns, not significant.) University of California, Santa Cruz (UCSC) Genome browser tracks for the FOS and NPTX2 loci, indicating chromatin accessibility (ATAC-seq) and gene expression (RNA-seq) in response to KCl.
See also Figures S2 and S3 and Table S1.
Most genes induced after 45 min of depolarization in the Glu neurons (58 of 70) were also induced in the GABA neurons. The expression fold change values of ERGs between the two neuronal types were highly comparable (Pearson’s r = 0.9346, p < 0.0001), demonstrating that the early transcriptional response to activity is very similar (Figure 2C). The ERGs are significantly enriched for TFs, including the immediate-early genes FOS, EGR1, JUN, NPAS4, and NR4A1 (Figure S2; Table S1), known to respond to neuronal activity and mediate essential neuronal functions (Flavell and Greenberg, 2008; Spiegel et al., 2014). Although the essentially identical early-response TFs were detected, a significant fraction of the LRGs (780 of 1,400) are cell-type specific (Figures 2D and 2E), similar to what has been observed in the mouse visual cortex (Hrvatin et al., 2018). We identified 383 LRGs in Glu neurons and 397 in GABA neurons, both cell-type specific and activity induced (Figure 2E). Unsupervised clustering of the differentially expressed genes (DEGs) across cell types and time points revealed four clusters of upregulated genes (Figure S3). Cluster 1 included the ERGs (e.g., FOS, JUNB). Cluster 5 was similarly increased in both neuron types and contained solute transporter and ion channel coding genes, among other known LRGs (e.g., SLC6A17, SLC38A2, KCNA1, KCNJ2). Cluster 6 (e.g., EZH2, ACTN2) was preferentially induced in GABA neurons, and cluster 7 (e.g., MAFB, NPTX2) was more highly expressed in Glu neurons after stimulation. Gene Ontology (GO) enrichment analysis showed that clusters of induced genes were enriched for transcriptional regulation, different signaling pathways, and synaptic transmission as previously described (Pruunsild et al., 2017; Tyssowski et al., 2018), whereas few terms were enriched for genes downregulated by membrane depolarization (Figure S3).
Next, we systematically examined whether activity-regulated genes in human neurons were enriched for disease-associated genes. We collected genome-wide association study (GWAS) data for multiple psychiatric disorders and traits (Table S5) and used MAGMA (multi-marker analysis of GenoMic annotation) to derive risk genes. ASD-associated genes were from the Simons Foundation Autism Research Initiative (SFARI) gene database (Abrahams et al., 2013; Banerjee-Basu and Packer, 2010). Notably, genes shared between Glu and GABA neurons were strongly enriched in risk genes for all of the traits tested (Figure S3). Glu and GABA DEGs were significantly enriched (false discovery rate [FDR] <5%) in risk genes for ASD (odds ratio [OR]: Glu = 2.08; GABA = 2.02) and SCZ (OR: Glu = 1.7; GABA = 1.97), consistent with the recent findings from large-scale sequencing studies (Hauberg et al., 2020; Satterstrom et al., 2020). In addition, GABA neuron DEGs showed enrichment in attention-deficit/hyperactivity disorder (ADHD) (OR = 2.02) and neuroticism (OR = 1.93). These observations underscore the cell-type-dependent expression and regulation of psychiatric disease-associated genes and the importance of identifying and studying the disease risk genes in specific cellular contexts.
Characterization of depolarization-responsive chromatin landscape
To understand the regulatory changes underlying the activity-induced gene expression, we performed assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) (Buenrostro et al., 2013) to identify stimulation-induced early and late responsive chromatin accessibility changes using experimental conditions as described (Malik et al., 2014; Vierbuchen et al., 2017), including the unstimulated state and 30 min and 90 min after stimulation. We discovered a median of 59,656 high-confidence open chromatin regions per condition, totaling 347,237 unique peaks merged across all of the samples. A total of 27,148 peaks are shared across all of the conditions. We performed a correlation analysis of the ATAC-seq peak intensities to define the similarities in chromatin accessibility among samples. Mirroring the RNA-seq results, samples clustered into two main groups representing the Glu and GABA neurons and further clustered by time post-depolarization within the neuronal type (Figure 2F). Of the chromatin-accessible regions in unstimulated Glu and GABA neurons, ~31% (Glu = 17,101) and 33% (GABA = 19,175) coincided with those characterized in the mid-gestation human telencephalon (Markenscoff-Papadimitriou et al., 2020). We noticed that depolarization inducible accessible regions had a much lower representation in the human brain (Glu = 15%, GABA = 13%). This likely indicated that the lack of temporal resolution and the cellular heterogeneity of post-mortem brain tissue could impede the identification of activity-inducible regulatory regions. We also determined the histone modification landscape of open chromatin regions by performing CUT&RUN (Kaya-Okur et al., 2019) for acetylated lysine 27 of histone H3 (H3K27ac), a histone modification associated with active enhancers and promoters (Creyghton et al., 2010; Gorkin et al., 2020; Rada-Iglesias et al., 2011). Using combined data from all of the samples, we identified 180,639 high-confidence H3K27ac peaks, among which 109,416 (60.57%) overlapped brain H3K27ac chromatin immunoprecipitation sequencing (ChIP-seq) peaks obtained from the NIH Epigenomics Roadmap Consortium, the PsychENCODE Consortium, and recent publications (Li et al., 2018; Markenscoff-Papadimitriou et al., 2020; Roadmap Epigenomics et al., 2015). Integration with our ATAC-seq datasets for each sample revealed that 41.17% of H3K27ac peaks overlap ATAC-seq peaks. Reciprocally, 84.87% of ATAC-seq peaks colocalized with H3K27ac peaks.
To annotate CREs functioning as active enhancers, we restricted our analysis to open chromatin regions that do not overlap promoters and further used H3K27ac enrichment to differentiate active from inactive elements (Creyghton et al., 2010; Rada-Iglesias et al., 2011). We identified 127,519 putative enhancers, comprising 86.6% of all high-confidence non-promoter CREs. Approximately 17.5% of the active enhancers respond to neuronal activity. We further investigated the functional relationship between chromatin landscape changes at enhancers and transcription. Activity-inducible enhancers were associated with nearby genes using GREAT (McLean et al., 2010) and were highly enriched for activity-inducible genes (OR = 4.8, p = 4.0 × 10−106). For instance, elevated accessibility at the enhancer regions of FOS was observed. The ATAC-seq signals were most abundant 30 min after KCl and became comparable to the baseline level by 90 min, consistent with its known function as an immediate-early factor (Figure 2G). In contrast, we detected an ATAC-seq peak located ~4 kb upstream of the transcription starting site (TSS) of an LRG NPTX2 induced by activity 90 min after depolarization (Figure 2G). These data confirmed that chromatin dynamics following membrane depolarization accord with gene expression changes.
In vivo activity of putative enhancer elements of human neurons
To benchmark the regulatory sequences identified in human PSC neurons, we leveraged the VISTA Enhancer Browser database (Visel et al., 2007) and tested whether CREs from our unstimulated conditions were enriched for sequences with enhancer activity. VISTA has evaluated sequences overlapping with 504 Glu CREs and 499 GABA CREs. Among them, 320 of 504 and 280 of 499 CRE-overlapping sequences exhibited enhancer activity in mice, considerably more than expected by chance alone (Fisher’s exact test p < 0.05; Figure 3A). Most VISTA-validated human CREs (Glu = 70.3%; GABA = 64.6%) drove reporter gene expression in neural tissues, particularly the brain and neural tube. Next, we investigated the putative enhancers in the unstimulated samples (total = 50,558) and found that they are more enriched for VISTA-validated enhancers than CREs (Figures 3A and 3B).
Figure 3. Prediction and in vivo validation of neuron subtype enhancers by VISTA.
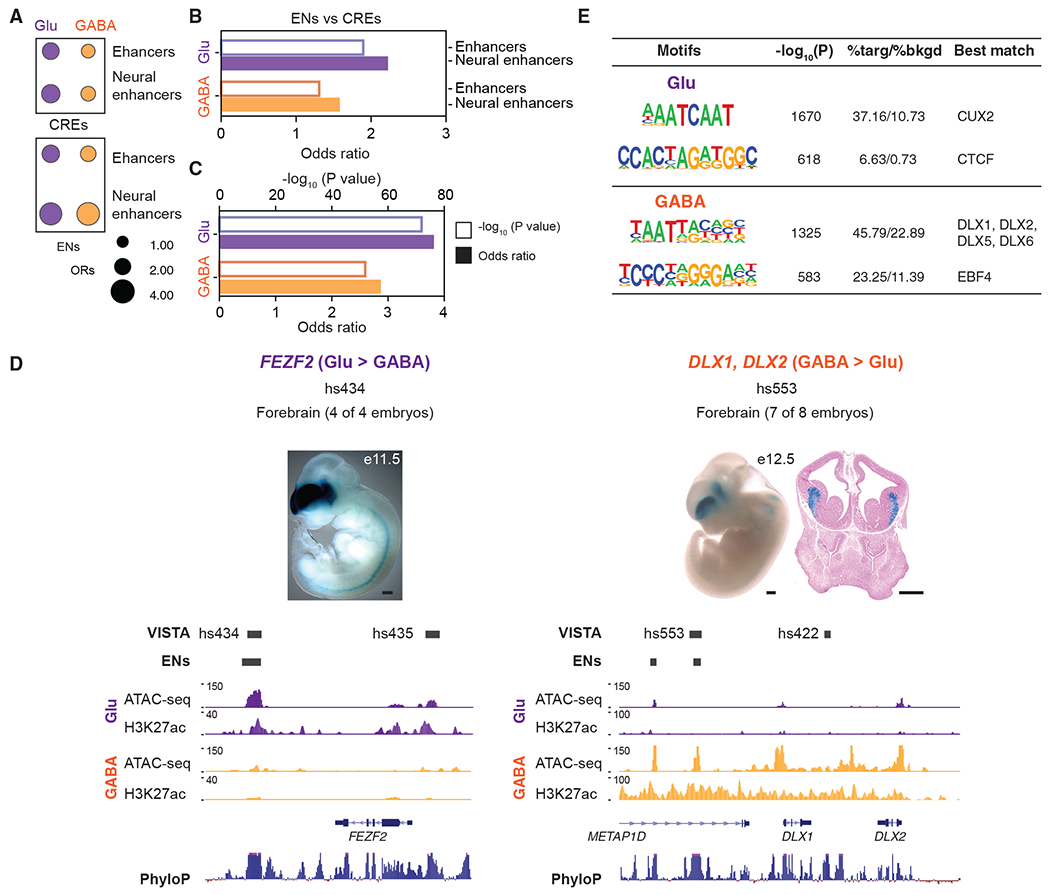
Focusing on subtype-specific differences, we investigated CREs and putative enhancers in the unstimulated samples and found the following.
(A) Neuronal subtype-specific CREs and enhancers are enriched for enhancer sequences validated in mice by VISTA.
(B) The ORs for the enrichment of validated enhancers, computed between subtype CREs and putative enhancers, and reported separately for all validated enhancers by VISTA and enhancers driving reporter expression in the neural tissues.
(C) Glu and GABA enhancers are enriched for DEGs.
(D) ATAC-seq reads and H3K27ac CUT&RUN signals in Glu and GABA neurons at VISTA brain enhancer hs434 and hs553. E11.5 enhancer transgenic mouse hs433 and E12.5 enhancer transgenic mouse hs553 are shown. LacZ expression is stained in blue (from https://enhancer.lbl.gov website). See Visel et al. (2007). Scale bar, 500 μm.
(E) Motif enrichment in neuron-specific CREs.
See also Table S2.
Focusing on neuronal subtype-specific differences, we associated differential enhancers with differences in gene expression. A total of 2,120 DEGs were called between the unstimulated Glu and GABA neurons using the RNA-seq data (log2FC ≥ 1, adjusted p < 0.05). Interrogation of DEGs revealed that enhancers with differential H3K27ac and ATAC-seq signals were highly enriched for DEGs (Figure 3C). A total of 2,048 differential enhancers associated with DEGs between Giu and GABA neurons were identified (Table S2). For example, FEZF2, the gene encoding a transcription repressor required to specify corticospinal motor neurons and other subcerebral projection neurons (Chen et al., 2008), is preferentially expressed in Glu neurons compared to GABA neurons, while DLX1 and DLX2 have the opposite expression pattern. DLXs are expressed in and regulate the development of GABAergic interneurons (Anderson et al., 1997). FEZF2, DLX1, and DLX2 have accompanying differences in chromatin accessibility and H3K27ac signal at enhancer regions (Figure 3D). Some of these enhancers showed strong forebrain enhancer activity supported by in situ hybridization experiments (Visel et al., 2007) (Figure 3D).
To discover TFs likely driving subtype-dependent differences in gene expression, we identified Glu and GABA neuron-specific CREs unique in one neuronal type and looked for enrichment of TF binding motifs in these regions (Figure 3E). Glu-specific peaks were enriched for motifs of CUXs, which have known functions in the developing cortex (Cubelos et al., 2010). GABA-specific regions were enriched for the binding motifs of DLXs and EBFs, TFs with known function in the GABAergic neuron cell fate determination (Garel et al., 1999). Considering that TFs of the same family share DNA-binding domains and recognize similar motifs (Weirauch et al., 2014), we used differential expression levels of individual TFs to refine the observed motif enrichments (Figure 3E). Collectively, the test cases show that enhancer elements identified in human PSC-derived neurons can function in vivo. The neuron-type-specific enhancers accurately distinguish Glu and GABA neurons and underlie cell-type-specific gene expression.
AP-1 TFs collaborate with neuronal-subtype-specific TFs to select LRG enhancers
Given the dramatic differences in LRG gene expression in neurons (Figures 2D and 2E), we sought to understand the molecular mechanisms underlying the cell-type-dependent gene regulatory differences. We focused on activity-induced late responsive enhancers in Glu and GABA neurons and identified 5,052 LRG enhancers in Glu neurons and 12,167 in GABA neurons. Neuronal LRGs typically encode effector proteins that regulate cellular processes such as dendritic growth, synapse remodeling, and a proper E-I balance (Pruunsild et al., 2017; Tyssowski et al., 2018). Consistent with this notion, GREAT analysis of late responsive enhancers yielded GO terms such as “dendrite morphogenesis,” “synapse organization,” and ““learning” (Figure 4A; Table S3). When we integrated the LRG enhancers with LRGs in different neuronal types, we found that LRG enhancers were indeed enriched for genes induced 4 h after the activity (Figures 4B; Table S4).
Figure 4. AP-1 TFs collaborate with neuronal TFs to select LRG enhancers.
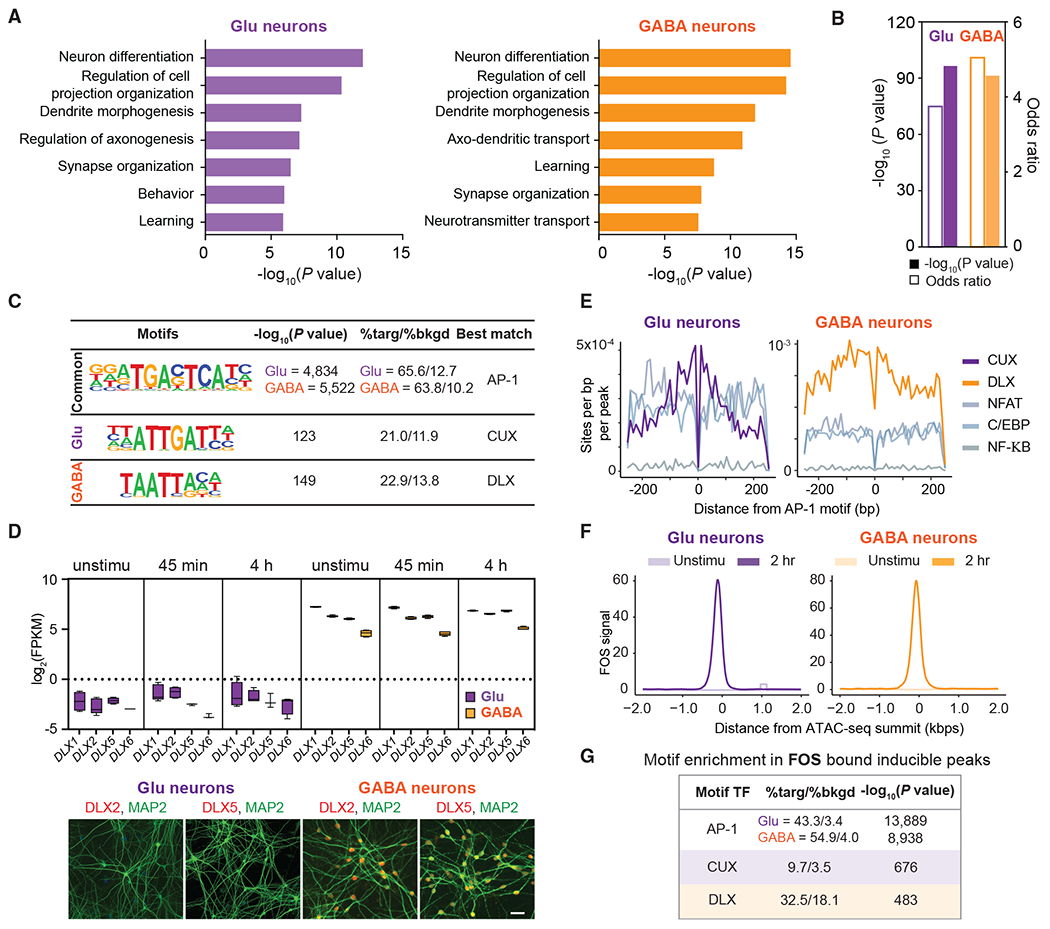
(A) Top-ranked Gene Ontology terms enriched in cell-type-specific enhancers with GREAT analysis; complete list in Table S7.
(B) LRG enhancers in Glu and GABA neurons are enriched for LRGs.
(C) Motif enrichment within late-inducible ATAC-seq peaks.
(D) Expression of DLX family genes. DLX2 and DLX5 proteins are detected in GABA neurons but not Glu neurons. Box center, median; box, 25th to 75th percentiles; whiskers, minima and maxima. Scale bar, 50 μm.
(E) Motif histogram plots of Glu (left) and GABA (right) neuron late-inducible CREs centered over AP-1 binding motif.
(F) Read density aggregate plots of FOS-bound peaks centered over ATAC-seq summits before and after stimulation.
(G) Position weight matrices of top motifs enriched in FOS-bound inducible peaks in Glu (purple) and GABA (orange) neurons. The ratios indicate the proportion of peaks containing respective motifs compared to a guanine-cytosine-matched background set of genomic regions.
Next, we performed TF binding motif enrichment analysis centered on the neuronal subtype-specific activity-induced ATAC-seq peak summits to dissect the molecular mechanisms underlying cell-type-specific LRG enhancer selection. This analysis revealed that AP-1 binding motifs are the most significantly enriched sequences at LRG enhancers in Glu and GABA neurons (Figure 4C). AP-1 TFs, including the FOS/JUN families, are transcriptional effectors in response to various stimuli and are activated by the RAS/MAPK pathway in nearly all cell types, raising the question of how they could contribute to cell-type-specific enhancer selection. One rising model is that AP-1 TFs bind to enhancers collaboratively with lineage-specific TFs (Bevington et al., 2016; Ostuni et al., 2013; Su et al., 2017; Swinstead et al., 2016; Vahedi et al., 2012; Vierbuchen et al., 2017). For example, environmental stimuli-induced AP1-bound enhancers in macrophages and T cells were enriched for lineage-dependent TF motifs proximal to AP-1 motifs. Inducible enhancers in macrophages are enriched for CCAAT-enhancer-binding protein (C/EBP) and nuclear factor κB (NF-κB), whereas induced enhancers in memory T cells are enriched for nuclear factor of activated T cell (NFAT) motifs. Therefore, we asked whether LRG enhancers in neurons were enriched for additional motifs corresponding to neuron-subtype-dependent TFs. Intriguingly, this revealed that the motifs recognized by CUX and DLX family TFs were enriched in LRG enhancers of Glu and GABA neurons, respectively (Figure 4C). CUX protein was detected in the Glu neurons (Song et al., 2019). Multiple DLX members were specifically expressed in the GABA neurons but were not regulated by neuronal activity (Figure 4D). Combinatory and cooperative recruitment of TFs occurs typically in close proximity (e.g., within ~75 bp) (Moyle-Heyrman et al., 2011). Consistent with this, binding motifs for CUXs and DLXs were most highly enriched within 75 bp of the central AP-1 motif (Figure 4E). In contrast, motifs likely bound together with AP-1 TFs in macrophages and T cells were not enriched in proximity to the AP-1 motif in inducible enhancers in neurons (Figure 4E).
To further test whether AP-1 TFs bind together with CUX or DLX to select subtype-specific LRG enhancers, we carried out CUT&RUN to identify the FOS binding regions 2 h after membrane depolarization. We performed motif analysis within 50 bp of the FOS peak summit. CUX and DLX motifs were again retrieved as the second most enriched motif after the consensus sequence for the AP-1 TFs (Figure 4F). The AP-1 motifs were detected in a more significant but comparable fraction (54.9%) of FOS-bound inducible peaks in GABA neurons than the DLX motif (32.5%). However, the abundance of CUX motif-containing peaks was low (9.7%) in FOS-bound inducible peaks in Glu neurons (Figure 4G). This could suggest that additional TFs with more degenerate binding motifs were involved in selecting LRG enhancers specific to Glu neurons. Such motifs may not be readily detectable by the motif search algorithms we used. These findings suggest that the cooperative binding of AP-1 TFs and neuronal-subtype-specific TFs such as CUXs and DLXs could select cell-type-specific enhancers that respond to neuronal activity.
Enrichment for disease risk variants at cis-regulatory elements
A substantial proportion of the heritability for common human diseases and traits partitions to noncoding regulatory elements, particularly regions specific in tissues or cell types related to the trait or disease in question (Finucane et al., 2015; Maurano et al., 2012). To determine whether the proportion of phenotypic variance explained by common single-nucleotide polymorphisms (SNPs) in large psychiatric GWAS (i.e., SNP heritability) was enriched in the CREs identified in Glu and GABA neurons, we performed stratified LD score regression analysis (Finucane et al., 2015). LDSC uses GWAS summary statistics to determine whether genetic heritability for a trait or disease is enriched for SNPs within genome annotations while accounting for LD. We collected GWAS data concerning neurological diseases, psychiatric disorders, personality traits, and non-neural traits and diseases (Table S5). First, we analyzed the disease heritability enrichment in neuronal CREs (shared ATAC-seq regions). Neuronal enrichments at FDR <5% were identified for 8 of the 10 brain-related traits, including ADHD, ASD, bipolar disorder, major depressive disorder (MDD), SCZ, intelligence, education attainment, and neuroticism (Figure 5A). We did not detect enrichment in heritability in non-neural traits and disorders except for body mass index (BMI). Neuronal enrichment for BMI has been previously identified by other genetic analyses (Finucane et al., 2015, 2018), likely reflecting the neural basis for regulating energy homeostasis (Farooqi, 2014). Moreover, Alzheimer’s disease (AD) SNP heritability, known to be most highly enriched in microglia regulatory elements (Nott et al., 2019), was not enriched. When analyzing the differentially enriched ATAC-seq regions in Glu versus GABA neurons (Figure 5A), we detected significant enrichment in GABA-specific ATAC-seq regions for all of the traits except ASD and MDD. Glu-specific ATAC-seq regions were enriched for SCZ. Most of the significant enrichments were found in the H3K27ac regions (Figure S4).
Figure 5. Disease heritability enrichment in CREs.
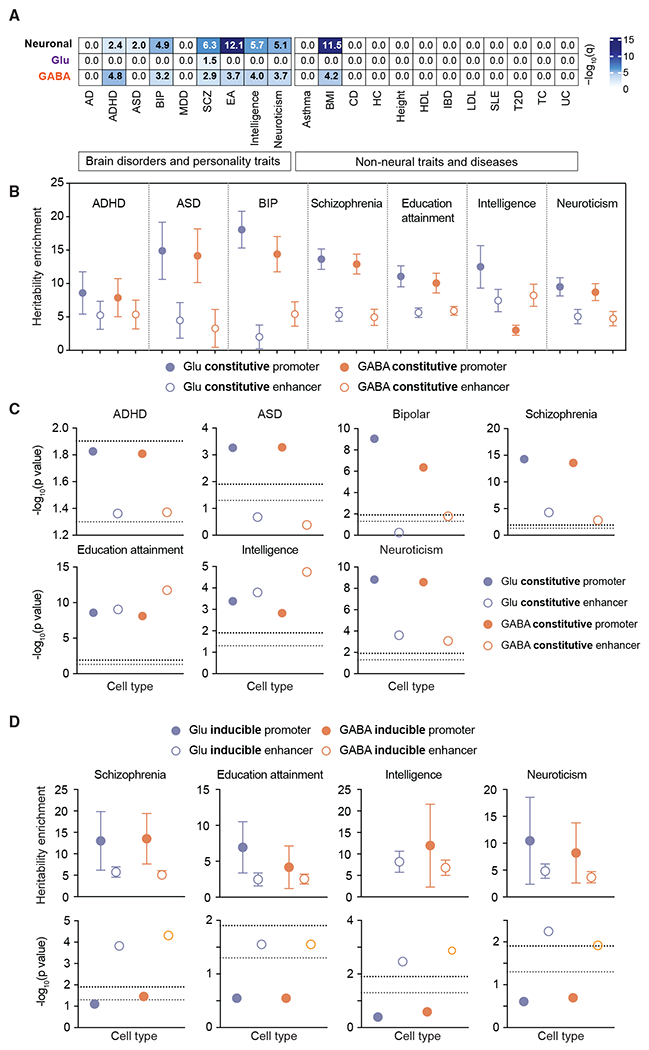
(A) Heatmap of LDSC analysis for genetic variants associated with brain disorders and behavior traits displayed as −log10(q) value for significance of enrichment for shared or cell-type-specific ATAC-seq peak regions in Glu and GABA neurons. Bold text indicates q < 0.05. The top 21,989 peaks from Glu, GABA, and shared neuron datasets were used for the analysis.
(B and C) Heritability enrichment of constitutive promoter regions and enhancer regions across neuropsychiatric disorders and behavioral traits. The black dashed lines in (C) show the cutoff for multi-test corrected p < 0.05. The gray dashed lines in (C) show the cutoff for p < 0.05.
(D) Heritability enrichment of the inducible promoter and enhancer. The black dashed lines show the cutoff for multi-test corrected p < 0.05. The gray dashed lines show the cutoff for p < 0.05. Each heritability enrichment value is provided in the scatterplot (±SE).
Second, we applied stratified LD score regression to constitutive and activity-responsive promoters and enhancers to test for heritability enrichment across brain-related traits. We found a significant heritability enrichment for variants within constitutive promoters and enhancers of Glu and GABA neurons for most psychiatric disorders and behavioral traits (Figures 5B and 5C). Intriguingly, significant enrichment for SCZ and intelligence was found in activity-responsive enhancers of Glu and GABA neurons. Neuroticism also showed enrichments in Glu and GABA neuron-specific inducible enhancers; however, the enrichment was not significant at FDR <5% for the GABA neurons (adjusted p = 7.2 × 10−2) (Figure 5D). Finally, education attainment exhibited enrichment in inducible enhancers but was not significant at FDR <5%. Notably, the promoters and enhancers annotated using ATAC-seq signals, presumably sites of TF binding, showed increased heritability enrichment relative to those defined by H3K27ac signals (Figure S4). This supports the notion that genetic perturbation of TF binding sites is an important mechanism by which SNPs influence the risk for human disease.
Deciphering molecular mechanisms at GWAS loci
Having demonstrated that enhancers identified in human PSC-neurons are enriched for disease heritability, we sought to identify the target genes of disease risk enhancers and gain insights into the regulatory properties of GWAS loci. Leveraging 25,197 unique SNPs associated with at least one of the psychiatric disorders ADHD, ASD, bipolar disorder, and SCZ (at the significance threshold of 5 × 10−8 and the SNPs in high LD with r2 > 0.8), we used the activity-by-contact (ABC) model (Fulco et al., 2019; Nasser et al., 2021) to connect non-coding GWAS signals to target genes. A total of 790 credible sets (enhancergene predictions) were obtained, nominating 272 variants that overlapped enhancers linked to 258 unique genes (Table S6). The distance from the non-coding variant in the ABC enhancer to the TSS of the target gene ranged from 884 to 646,691 bp (median, 20 kb), and 50 of 291 predictions (17.2%) involved a gene that was not the closest (Figure 6A). Of the 272 variants, 74 (27%) were found only in depolarization-inducible enhancers.
Figure 6. Connecting GWAS variants to target genes.
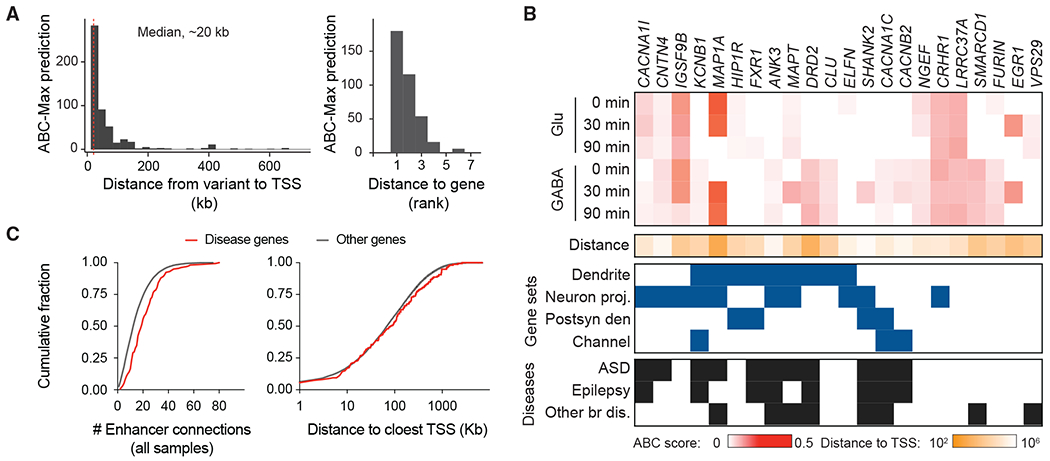
(A) Histogram showing the distances from the disease variants to the TSS of the ABC-Max gene (left) and the distance rank of the gene in the locus (right). Data include predictions for ASD, ADHD, bipolar disorder, and SCZ.
(B) ABC analysis connected variants to target genes. The heatmap shows the ABC scores in 6 samples (maximum value within each condition). Red scale: ABC score; tangerine scale: log10-transformed genomic distance from variant to gene TSS. The ABC genes were enriched for genes that function at the dendrites (adjusted p: 7 × 10−3) and neuron projection (adjusted p: 3.4 × 10−2) based on GO analysis. Blue boxes indicate the ABC disease gene encodes a product located in a specific cellular compartment, including “dendrite,” “neuron projection” (Neuron Proj.), “postsynaptic density” (Postsyn den), and “channel components.” Black boxes indicate that rare variants in the gene have been reported in individuals with ASD, epilepsy, or other brain diseases (Other br dis).
(C) The cumulative distribution plot shows the number of ABC enhancer-gene connections in all of the samples (left) and the distance to the closest TSS for genes linked to diseases and others.
This analysis provides a resource for identifying genes, pathways, and regulatory properties of GWAS loci. Combining all activity states, for SCZ, ABC analysis nominated 176 unique genes (Glu = 84, GABA = 92). The predicted genes were enriched for genes whose products are localized to dendrites (9 genes; OR = 5.8) and neuron projection (11 genes; OR = 3.4) (Figure 6B). Mutations of several of these genes have been identified in subjects with ASD and other brain disorders (Figure 6B). Regarding the regulatory properties of the variants, we found cases for which a variant was predicted to act only in specific neuronal types or cell activity states (Figure 6B). For example, CACNA1C encodes a calcium voltage-gated channel (CaV) subunit that can interact with L-type Ca2+-channel subunits to increase the channel current and regulate their modulation. While CACNA1C is expressed in both subtypes, we identified one SCZ risk variant (rs12424245, p = 1.459 × 10−20) that overlapped an ABC enhancer and was linked to CACNA1C only in GABA neurons. In a similar line, CACNB2 encodes for the β subunit of Cav, and rare CACNB2 variants identified in ASD probands were shown to alter the time-dependent inactivation of Ca2+ channels (Breitenkamp et al., 2014). CACNB2 is a depolarization responsive gene expressed in Glu and GABA neurons; however, ABC analysis linked a variant (rs71497246, p = 5.96 × 10−9) to CACNB2 only in the stimulated GABA neurons. This may reflect that neuronal subtype-specific TFs select cell-type-specific and activity-inducible enhancers to achieve precise transcriptional control and underscore the importance of annotating enhancers in different neuronal types for functional fine-mapping of disease risk variants.
A closer look at the gene structure and surrounding region in the genome revealed that the disease genes identified by the ABC model seemed to have more complex regulatory landscapes. They had more ABC enhancer connections across all samples (median of 19 across all cell types and activity states versus 13 for other genes) and in the samples in which ABC analysis identified variant and gene connections (median of 3.25 versus 2). The disease genes also had more surrounding noncoding sequences (median of 83.3 kb versus 69 kb distance to the closest neighboring TSS) (Figures 6C and S5). We were intrigued by this observation, which indicated that genes with complex enhancer landscapes could be more vulnerable to non-coding variants associated with psychiatric disorders. Consistent with our findings, the recent study on non-neurological conditions (e.g., inflammatory bowel disease [IBD]) revealed that genes with complex enhancer landscapes are more likely to influence multiple traits (Nasser et al., 2021).
DISCUSSION
Neuronal activity-dependent transcriptional regulation plays a fundamental role in regulating neuronal property, synaptic plasticity, cognitive function, and various brain disorders. However, it has been technically challenging to understand the responses in diverse cell types of the human brain and how the various changes contribute to neural plasticity and disease. Here, we generated two major neuronal types using human PSCs and systematically characterized the transcriptional changes and the cis-regulatory elements in response to stimulation. To derive neurons, we used the TF-mediated direct conversion system we pioneered. This paradigm allows us to generate large quantities of neurons with homogenous neurotransmitter specifications from different PSC lines with unprecedented reproducibility. We cannot conclude from our characterization that the cultures of each neuronal subtype consist exclusively of either Glu or GABA neurons. However, based on our assays (Figure S2) and previous work, more than 80%–90% of the cells in our cultures are the appropriate identity. More important, given that our downstream analyses use bulk RNA-seq and chromatin profiling data, it is unlikely that any of our observations can be explained solely by the presence of a small fraction of “off-target” cell types in our cultures.
The RAS/MAPK kinase cascade mediates the cellular responses to external stimuli by activating a similar set of ERGs, including AP1 TFs in most cell types. To initiate cell-type-specific transcription, the AP-1 TFs must strategically select the appropriate enhancers among the ~1 million possible enhancers in the genome (Heintzman et al., 2009). Previous work suggests that AP-1 TFs achieve this by collaboratively binding to enhancers with cell-type-specific TFs (Vierbuchen et al., 2017). Our data identified potential TFs contributing to the cell-type-specific enhancer selection in Glu and GABA neurons. These include proteins that determine neuronal fate commitment. It will be interesting to explore further how they cooperate with AP-1 TFs in distinct neuronal types to shape the dynamic enhancer landscape during cell fate specification.
GWAS has identified thousands of neurological or psychiatric disease-associated variants, but the cellular mechanisms through which these variants drive diseases and traits remain largely elusive. The heterogeneity of the brain and the divergent transcriptional changes occurring across different cell types in response to external stimuli (Hrvatin et al., 2018) make it highly challenging to relate the approximately 95% of common risk variants in non-coding regions to the genes they regulate and to identify the cell type in which these genes are active. Generating and characterizing the transcriptome and regulome of specific neuronal types allows us to distinguish nuanced gene expression and regulatory programs that drive cell-type-specific differences at a static state or in response to membrane depolarization and gain a deeper understanding of psychiatric disease etiology. We found that genes shared between Glu and GABA neurons were strongly enriched in risk genes for all traits tested, including ADHD, ASD, bipolar disorder, SCZ, MDD, and neuroticism. Glu and GABA neuron DEGs were significantly enriched in risk genes for ASD and SCZ. Moreover, activity-dependent genes of Glu and GABA neurons were also enriched for SCZ risk genes. Our observation corroborates previous indications that excitatory and inhibitory neurons are relevant cellular contexts for ASD- and SCZ-associated variations in gene expression (Gandal et al., 2018; Satterstrom et al., 2020; Skene et al., 2018; Velmeshev et al., 2019).
The inferred disease enrichments using cell-type-specific and activity-responsive non-coding regulatory elements revealed that CREs shared between Glu and GABA neurons were enriched in multiple psychiatric disorders and brain traits but not in AD. In addition, we identified that constitutive promoters and enhancers in Glu and GABA neurons carry a significant enrichment for most psychiatric disorders and neural traits we analyzed. However, constitutive promoters, but not the enhancers, were enriched for ASD heritability. This was noted previously using whole-genome sequencing data from quartet families (An et al., 2018). Lastly, we detected significant enrichment for SCZ and intelligence heritability in activity-responsive enhancers in GABA neurons. Notably, common SCZ risk variants have been found to be enriched in CREs of human Glu neurons but not GABA neurons isolated from the human postmortem brains (Hauberg et al., 2020). The discrepancies among different studies could be attributed to the subtype of neurons (Skene et al., 2018) and different data collections (bulk versus single-cell sequencing, postmortem brain tissues versus freshly collected cells). It is worth noting that the stratified LD score regression analysis focused on genome-wide disease heritability rather than specific loci. Similar to studies using mouse (Beagan et al., 2020) and human PSC-derived neurons (Boulting et al., 2021), we found that single-nucleotide variants associated with psychiatric diseases colocalized with activity-regulated promoters and enhancers, which facilitates the fine mapping of disease risk variants in functional regulatory elements and prioritizing variants for further validation.
Limitations of the study
The integrative analysis in this study has several limitations. For example, we did not detect heritability enrichment for MDD in GABA neurons, an association driven by somatostatin (SST)- and parvalbumin (PV)-expressing interneurons (Jagadeesh et al., 2021). However, inhibitory neurons in this study represent a mixed population of GABAergic subtypes, with only a small percentage expressing SST or PV. Notably, the maturation of PV neurons is protracted and exquisitely regulated by experience and cell-extrinsic factors (Donato et al., 2015). Most currently available differentiation methods render immature or fetal cells (Steg et al., 2021; Studer et al., 2015). This sets the stage for optimized methods, in which distinct cell types can be generated for mechanistic investigation. Furthermore, we applied the ABC model to link risk variants to disease genes using cell-type- and activity-specific ATAC-seq data and H3K27ac CUT&RUN data. While the ABC model can make enhancer-gene connection predictions for a given cell type without cell-type-specific Hi-C data, providing such data can increase the accuracy of predictions.
Nevertheless, our study characterized cell-type-specific and activity-regulated gene expression patterns and chromatin landscapes in human PSC-derived neurons widely used in the research community. We anticipate that the future use of these cells will concentrate on complex experimental paradigms impossible to perform using primary human brain cells, including high-throughput approaches to validate the function of cis-regulatory elements or to determine the effect of disease risk alleles on gene expression or cellular phenotypes. Our data can provide guidance for choosing the relevant cell types or experimental conditions to further elucidate the molecular mechanisms involving GWAS-significant loci across the genome.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nan Yang (nan.yang1@mssm.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA-seq, ATAC-seq, and CUT&RUN data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FOS antibody [2H2] | Abcam | Cat # AB208942; RRID: AB_2747772 |
| Anti-MAP2 | Abcam | Cat #ab5392; RRID: AB_2138153 |
| Anti-H3K27ac | Abcam | Cat # AB4729; RRID: AB_2118291 |
| Anti-IgG | Abcam | Cat # AB46540; RRID: AB_2614925 |
| Anti-IgG | Novus | Cat # NBP1-2763 |
| Anti-HSP90 | Cell Signaling | Cat # 4874S; RRID: AB_2121214 |
| Anti-FOS | Abcam | Cat # AB208942; RRID: AB_2747772 |
| Anti-pERK1/2 | Cell Signaling | Cat # 9101S; RRID: AB_331646 |
| Anti-Dlx5 | Wang et al., 2010 | Made and kindly shared by John LR Rubenstein’s group. |
| Anti-Dlx2 | Santa Cruz Biotechnology, Inc | Cat # sc-393879 |
| Chemicals, peptides, and recombinant proteins | ||
| Stemflex supplement | Gibco | Cat # A33492 |
| Stemflex basal media | Gibco | Cat # A33493-01 |
| Geltrex | Gibco | Cat # A14133-02 |
| Poly-L-ornithine hydrobromide | Sigma | Ct # P3655 |
| Accutase | Thermo Scientific | Cat # NC9464543 |
| Thiazovivin | Santa Cruz | Cat # SC-361380 |
| Puromycin dihydrochloride from Streptomyces alboniger | Sigma | Cat # P8833 |
| Hygromycin B (50 mg/mL) | Life Technologies | Cat # 10687010 |
| Cytosine β-D-arabinofuranoside hydrochloride | Sigma | Cat # C1768 |
| TRIzol | Invitrogen | Cat # 15596026 |
| Tetrodotoxin citrate (TTX) | Labome | Cat # 1069/1 |
| DL-2-amino-5-phosphopentnoic acid (DL2-AP5) | Abcam | Cat # ab120271 |
| DMEM media | Gibco | Cat # 11965-092 |
| MEM NEAA (100x) | Gibco | Cat # 11140-050 |
| Sodium Pyruvate (100x) | Gibco | Cat # 11360-070 |
| β-Mercaptoethanol | Sigma | Cat # M3148 |
| Cytiva HyClone™ Cosmic Calf™ Serum | Fisher | Cat # SH3008704 |
| Fetal Bovine Serum (FBS) | Sigma | Cat # F6178 |
| Doxycycline | Sigma | Cat # P3655 |
| MEM media | Gibco | Cat # 11090-081 |
| Glutamax (100×) | Gibco | Cat # 35050-061 |
| B27 supplement | Gibco | Cat # 17504-044 |
| Neurobasal media | Gibco | Cat # 21103-049 |
| Naurobasal A media | Gibco | Cat # 10888-022 |
| Trypsin 0.25% | Gibco | Cat # 25200-114 |
| DMEM/F12 media | Gibco | Cat # 11320-033 |
| N2 supplement | Gibco | Cat # 17502-048 |
| Dimethyl sulfoxide | Sigma | Cat # D2438 |
| Paraformaldehyde solution | Affymetrix | Cat # 19943 |
| D-PBS tablets | Millipore | Cat # 524650-1EA |
| Protease Free Heat Shock Bovine Serum Albumin Powder | Equitech-Bio | Cat # BAH65-0500 |
| Urea | Sigma | Cat # U5128 |
| 1M HEPES/Sodium Hydroxide | Rigaku | Cat # 1008184 |
| RNase A | Thermo Fisher Scientific | Cat #EN0531 |
| Glycogen | Sigma | Cat #10930193001 |
| Spermidine | Sigma | Cat # S0266 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Sigma/Roche | Cat # 11873580001 |
| Concanavalin A (ConA)-coated magnetic beads | Bangs Laboratories | Cat # BP531 |
| Protein AG-MNase | EpiCypher | Cat # 15-1116 |
| AMPure XP beads | Beckman Coulter | Cat # A63881 |
| SYBR green 2x master mix | Applied Biosystems | Cat # 4367659 |
| Critical commercial assays | ||
| Tagment DNA Enzyme and Buffer kit | Illumina | Cat # 20034211 |
| MinElute Reaction Cleanup Kit | Qiagen | Cat # 28206 |
| NEB Next High-Fidelity 2x PCR Master Mix | New England Biolabs | Cat # M0541 |
| Pierce™ BCA Protein Assay Kit | Thermo Scientific | Cat # 23225 |
| TURBO™ DNase | Ambion | Cat # AM1907 |
| SuperScript® IV First-Strand Synthesis System | Thermo Scientific | Cat # 18091050 |
| Bolt™ 4 to 12%, Bis-Tris, 1.0 mm, Mini Protein Gel, 15-well | Thermo Scientific | Cat # MW04125BOX |
| iBlot™ 2 Transfer Stacks, PVDF | Invitrogen | Cat # IB24001 |
| Western Lightning Plus, Chemiluminescent Substrate | Perkin Elmer | Cat # NEL 104001EA |
| NEBNext Ultra II DNA library Prep kit for Ilumina | New England Biolabs | Cat # E7645 |
| NEBNext Multiplex oligos for Ilumina | New England Biolabs | Cat # E7600S and Cat # E7780S |
| Deposited Data | ||
| Raw RNA-sequencing data and Salmon counts | This paper | GEO: GSE196855 |
| Raw ATAC-sequencing data and processed peaks | This paper | GEO: GSE196854 |
| Raw H3K27ac CUT&RUN data and processed peaks | This paper | GEO: GSE196207 |
| Experimental models: cell lines | ||
| H1 (WA01) | WiCell Research Resources | hPSC Reg ID Ae001-A |
| 3188-2A-4N | Hoffman et al., 2017 | GEO: GSM2843584 |
| HEK293T | ATCC | Cat # 11268 |
| Experimental models: organisms/strains | ||
| CD1 mouse strain | Charles River | Cat# CRL:022, RRID: IMSR_CRL:022 |
| Recombinant DNA | ||
| FUW-M2rtTA | Hockemeyer et al., 2008 | RRID: Addgene_20342 |
| TetO-Ascl1-puro | Yang et al., 2017 | RRID: Addgene_97329 |
| TetO-Ngn2-puro | Zhang et al., 2013 | RRID: Addgene_52,047 |
| TeoO-Dlx2-Hygro | Yang et al. 2017 | RRID: Addgene_97330 |
| Software and algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | RRID: SCR_016368; http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Skewer v0.2.2 | Jiang et al., 2014 | https://github.com/relipmoc/skewer |
| Salmon 0.13.1 | Patro et al., 2017 | https://combine-lab.github.io/salmon/ |
| R | Love et al., 2014 | https://www.r-project.org/ |
| RStudio | RStudio Team, 2020 | https://github.com/rstudio/ |
| Apeglm | Zhu et al., 2019 | https://github.com/azhu513/apeglm |
| ENCODE ATAC-seq pipeline v1.4.2 | Consortium, 2012 | https://www.encodeproject.org/atac-seq/ |
| featureCounts | http://subread.sourceforge.net/ | |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| MACS | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| DiffBind | Stark and Brown, 2011 | http://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| ChipSeeker | Yu et al., 2015 | https://guangchuangyu.github.io/software/ChIPseeker/ |
| LDSC | Finucane et al., 2015 | https://github.com/bulik/ldsc |
| Prism | GraphPad | RRID: SCR_005375 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal model
All animal experiments were approved by the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee and were conducted in compliance with the relevant ethical regulations. Mice were maintained in social cages on a 12 h light/dark cycle with free access to food and water; animals were monitored daily for food and water intake. Wild-type CD1 mice were used to isolate primary cell cultures on postnatal day 3 (P03). Animals of both sexes were used in the analyses.
Human cell lines
All human PSCs were maintained on Geltrex-coated plates in feeder-free Stemflex medium and a 5% CO2 environment at 37°C
METHOD DETAILS
Cell culture
All human PSCs were maintained on Geltrex-coated plates in feeder-free Stemflex medium and a 5% CO2 environment at 37°C. Cells were passaged using Accutase in Stemflex supplemented with 2 μM Thiazovivin. Thiazovivin was removed from the media on the following day. Research performed on samples of human origin was conducted according to protocols approved by the institutional review boards of Icahn School of Medicine at Mount Sinai. H1 (WA01) ES cells were obtained from WiCell Research Resources; 3188-2A-4N female iPS cell line (GEO accession number: GSM2843584) was kindly provided by Dr. Kristen Brennand.
Mouse glial cultures were generated from cortical hemispheres at postnatal day 3 (P03). The cortices were incubated in 3 mL of 20 Units/mL Papain, 0.5 μM EDTA, and 1 μM CaCl2 in HBSS for 15 min. After incubation, the tissues were manually dissociated by forceful trituration. The resulting cells were grown at 37°C with 5% CO2 in DMEM media containing 1×NEAA, 1× sodium pyruvate, 10% Cytiva HyClone™ Cosmic Calf™ Serum (CCS), and 0.008% β-mercaptoethanol.
Virus production
Infectious lentiviral particles were produced in HEK293T cells using the third-generation lentiviral packaging plasmids pMD2.G (Addgene plasmid # 12259), pRSV-rev (Addgene plasmid #: 12253), and pMDLg/pRRE (Addgene plasmid #: 12251) (Dull et al., 1998). Lentiviruses were produced as described (Hu et al., 2021; Pang et al., 2011) in HEK293T cells using Polyethy-lenimine (PEI) (Longo et al., 2013). Cellular debris was removed from lentiviral supernatant by centrifugation at 1,000g for 5 min. Lentiviral particles were ultra-centrifuged, resuspended overnight with gentle shaking in DMEM, aliquoted, and stored at −80°C. Only virus preparations with >90% infection efficiency as determined by GFP expression or antibiotic resistance were used for experiments.
Generation of neurons from human PSCs
Glutamatergic neurons were generated by overexpression of the transcription factor Ngn2 as previously described (Zhang et al., 2013). Briefly, human PSCs were dissociated using Accutase and plated at a density of 88,000 cells/cm2. The next day, cells were transduced with FUW-TetO-Ngn2-P2A-puromycin and FUW-rtTA lentiviruses by adding directly to the media. 24 h later, the medium was replaced by N2 media (1×N2 supplement, 1×NEAA in DMEM-F12 media) containing Doxycycline (2 μg/mL) to induce transgene expression. Transduced cells were enriched by applying puromycin (1 μg/mL) for 2–3 days. 5–6 days post doxycycline, neurons were dissociated and plated together with mouse glial cells (104,000 cells/cm2) on Geltrex-coated plates. Two weeks after transgene induction, doxycycline was removed and the neuronal culture was maintained in Neurobasal A media supplemented with 1×B27, 1×Glutamax, and 1% fetal bovine serum. Mature neurons were used for various experiments on day 35.
We followed the same protocol as described above for glutamatergic neurons with minor differences to generate GABAergic neurons (Yang et al., 2017). Briefly, we overexpressed the transcription factors Ascl1 and Dlx2 by infecting cells with FUW-TetO-Ascl1-T2A-puromycin and FUW-TetO-Dlx2-IRES-hygromycin lentiviruses. The transduced cells were selected with N2 media containing hygromycin (200 μg/mL) and puromycin (1 μg/mL) for three days, and the media was replaced with N2 media containing AraC (4 μM) for three days. GABAergic neurons were replated with mouse glial cells on day 8. All the subsequent steps were performed as described to generate glutamatergic neurons.
For the membrane depolarization experiment, neurons were treated with 1 μM of Tetrodotoxin citrate (TTX) and 100 μM of DL-2-amino-5-phosphopentnoic acid (DL-AP5) for 12 h prior to the addition of depolarization buffer (170 mM KCl, 2 mM CaCl2, 1 mM MgCl and 10 mM HEPES) to achieve 55 mM KCl (Malik et al., 2014).
Electrophysiology
Functional analyses of human neurons were conducted using whole-cell patch-clamp as described elsewhere (Pang et al., 2011; Vierbuchen et al., 2010). Briefly, a K-Gluconate internal solution was used, which consisted of (in mM): 126 K-Gluconate, 4 KCl, 10 HEPES, 4 ATP-Mg, 0.3 GTP-Na2, 10 Phosphocreatine. The pH was adjusted to 7.2, and osmolarity was adjusted to 270–290 mOsm. The bath solution consisted of (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 10 Glucose. The pH was adjusted to 7.4. Spontaneous excitatory- and inhibitory postsynaptic currents (sEPSCs and sIPSCs, respectively) were recorded at a holding potential of −70mV and 0 mV, respectively, under voltage-clamp mode. All recordings in cultured human neurons were performed at room temperature.
RNA extraction and quantitative PCR
To determine gene expression level, cells (2 6-well plate wells/time point; 5×105 cells total) were stimulated with depolarization buffer as described above. 3 biological replicates were obtained for each of 3 time points: unstimulated (0 h), 45 min, and 4 h post-membrane depolarization. Cells were washed with PBS and lysed in 2 mL of Trizol for RNA extraction, followed by TURBO DNA-free kit treatment to remove DNA contamination. 500 ng total RNA was reverse transcribed into cDNA using SuperScript® IV First-Strand Synthesis System with Oligo dT primers, according to the manufacturer’s protocol. Quantitative PCR analysis was performed on the Applied Biosystems Quant Studio 7 Flex Real-Time PCR System using an SYBR green-based method. The results were analyzed using Quant Studio Real-Time PCR Software.
Immunocytochemistry and imaging
Cultured cells were fixed with 4% paraformaldehyde (PFA) in D-PBS at room temperature for 5–10 min. Cells were rinsed 3 times with D-PBS and subsequently permeabilized for 5 min at room temperature with 0.2% Triton X-100 in D-PBS. After incubation in blocking buffer (4% bovine serum albumin (BSA) and 1% CCS in D-PBS) for 3 h, primary antibodies were added for incubation overnight at 4°C. After 3 rinses, cells were incubated with secondary antibodies in the blocking buffer for 3 h at room temperature. Images were acquired using the EVOS M5000 imaging system (Life Technologies).
Immunoblotting
Cultured neuron lysates were collected in protein lysis buffer (4M Urea 75 mM Tris-HCl, 3.78% SDS, and 20% Glycerol in H2O) and kept on ice. Protein concentration was determined using the Pierce BSA Protein Assay kit according to the manufacturer’s instructions. For each sample, 10 μg of protein was mixed with NuPAGE LDS Sample buffer and boiled for 5 min at 95°C. Samples were run in a 4–12% Bis-Tris gel and transferred to a PVDF membrane using the iBlot 2 Dry blotting system. PVDF membranes containing the transferred proteins were blocked by incubating with TBST solution of 5% BSA in 1 × Tris-buffered saline, 0.1% Tween® 20 Detergent for 1 h at room temperature. Primary antibodies were diluted in a 1% BSA solution and incubated with the membrane overnight at 4°C. Afterward, the membrane was incubated with secondary antibodies diluted in 1%BSA for 1hr at room temperature. Membranes were washed 3 times with TBST after incubation with primary and secondary antibodies. Incubation of the membrane for 5 min in ECL solution (PerkinElmer NEL 104001EA) was used to visualize the HRP signal.
Library preparation and sequencing
RNA-seq
RNA was extracted with TRIzol following the provider’s instructions and treated with a TURBO DNA-free kit. RNA-seq libraries were prepared and sequenced by GENEWIZ on an Illumina HiSeq 2000 platform to produce 30–35 M paired-end reads of 150 bp uniform length per sample.
ATAC-seq
ATAC libraries were generated as previously described (Buenrostro et al., 2013; Corces et al., 2017) using 75,000 cells per sample. The oligos used for library preparation are provided in Table S7. The libraries were enriched for fragments ranging from 200 to 800 bp using AMPure XP beads and assessed using the Agilent 2200 TapeStation System before high-throughput sequencing using the Illumina HiSeq Platform with 150 bp paired-end reads.
CUT&RUN
CUT&RUN was performed as previously described (Meers et al., 2019) using 50,000 nuclei for the following conditions: all replicates of H3K27ac 0, 30, and 90 min, all replicates of FOS 0 and 2 h. Nuclei bound to Concanavalin-coated magnetic beads were incubated with antibodies. After incubation, the beads were placed on a magnet to remove supernatant, washed, and incubated with Protein AG-MNase. To fragment the DNA, samples were first chilled to 0°C, and the digestion was then activated by adding 2 mM of CaCl2. Reactions were stopped by 2×Stop buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA, 0.05% Digitonin, 100 ug/mL RNaseA, 50 ug/mL Glycogen). DNA fragments released from the nuclei were purified with phenol-chloroform and precipitated ethanol. DNA was quantified using Qubit 4 Fluorometer dsDNA HS assay. 10 ng DNA per sample was used for library preparation using NEBNext Ultra II DNA Library Prep Kit. DNA was enriched for 150–350 bp fragments for transcription factor binding events and 150–800 bp for histone markers using Ampure XP beads. Libraries were quantified with Qubit dsDNA HS assay, and the size distribution was determined by Agilent 4200 TapeStation before being sequenced on an Illumina HiSeq system with 150 bp paired-end reads.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq data processing and gene expression analysis
Raw reads were trimmed for base call quality (PHRED score ≥21) using skewer 0.2.2(Jiang et al., 2014), and transcript quantification was performed using hg38 reference transcriptome and salmon 0.13.1. Count normalization and differential gene expression analysis from three biological replicates were performed using the DESeq2 package in R (Love et al., 2014) applying adaptive t prior shrinkage estimator ‘apeglm’ (Zhu et al., 2019). To identify activity-induced genes, RNA-seq was performed at three time points - unstimulated (0 h), 45 min, and 4 h post-membrane depolarization. Differentially expressed genes (DEGs) were defined as those that change expression at least 1.5-fold in Glu neurons and 2-fold in GABA neurons between any two conditions (adjusted p-value < 0.05).
ATAC-seq data processing and analysis
Primary data processing, including adapter trimming, genomic alignment to hg38 using Bowtie2 (Langmead and Salzberg, 2012), mitochondrial reads and duplicate alignment removal, peak calling and reproducibility analysis using MACS2 and IDR, respectively, were performed using ENCODE ATAC-seq pipeline v1.4.2 (Consortium, 2012) (https://github.com/ENCODE-DCC/atac-seq-pipeline). Reproducible peaks selected using IDR (idr threshold 0.05) were aggregated to the final superset of regulatory elements, accessible at least in one of the conditions. The featureCounts package was then used to obtain ATAC-seq read counts for each of the regions (Liao et al., 2014). To identify activity-induced chromatin accessibility changes, ATAC-seq was performed at three time points - unstimulated (0 h), 30 min, and 90 min post-membrane depolarization. Normalization and differential peak calling was performed using DESeq2 in R (Love et al., 2014). Differentially accessible peaks were defined as those that change accessibility at least 2-fold between any two conditions (adjusted p-value < 0.05).
CUT&RUN data analysis
CUT&RUN data was analyzed as described with minor modifications (Henikoff et al., 2020). In brief, raw sequencing reads were filtered for quality using trimmomatic (Bolger et al., 2014) for H3K27ac and cutadapt (Marcel, 2011) for FOS and Illumina sequencing adapters were removed. Paired-end reads were mapped to the human genome (hg38) using bowtie2 (Langmead and Salzberg, 2012). Aligned reads were sorted and indexed using samtools (Li et al., 2009), and duplicates were removed with Picard (http://broadinstitute.github.io/picard/). Peaks were called using MACS (Zhang et al., 2008). For H3K27ac, we used the set of peaks found in 3 or more samples to determine differential peaks across cell types or time points after stimulation using the DESeq2 option within the DiffBind package in R (Love et al., 2014). For FOS, consistency was determined using an IDR cutoff of 0.1. For cases with more than 2 replicates, we selected the longest peak list from all pairs that passed IDR. Accessible chromatin and H3K27ac-bound peaks that fell within the promoter were identified using ChIPseeker (Yu et al., 2015) in RStudio v1.3.1093 (RStudio Team, 2020). Promoters were defined as upstream 1 kb from the transcription start site (TSS; −1000,0). Any peaks that fell outside the promoter region of genes were considered putative enhancers. The reproducibility of our H3K27ac peaks was determined by identifying the degree of overlap with previously published human brain H3K27ac datasets. We used the bedtools intersect function to intersect our non-regulated and inducible H3K27ac cis-regulatory elements (CREs) with the union of the following: 1) regions marked by H3K27ac in 15, 17, and 18 gestational weeks (GW) human fetal prefrontal cortex (Markenscoff-Papadimitriou et al., 2020); 2) regions marked by H3K27ac in 7, 8.5, and 12 GW human fetal cortex (Reilly et al., 2015); and 3) regions marked by H3K27ac in 17, 19, and 21 GW human fetal cortex (Li et al., 2018). To identify putative enhancer elements, cell-type-specific unique chromatin accessible CREs were intersected with human VISTA enhancers (Visel et al., 2007) using bedtools intersect. We examined the overlap between total VISTA enhancers, validated VISTA enhancers, and VISTA-identified enhancers found in nerve and brain tissue.
Gene Ontology analysis
To identify cellular functions of differentially expressed genes, we used Enrichr (Chen et al., 2013; Kuleshov et al., 2016; Xie et al., 2021). To associate actively responsive predictive enhancer elements with genes and their biological processes, we used GREAT(McLean et al., 2010). Genomic regions were associated with a gene regulatory region if it resided 5 kb upstream and 1 kb downstream (proximal) and up to 100 kb distally.
MAGMA analysis of disease-associated traits
We used MAGMA (v1.08) to generate sets of trait- or disease-associated genes enriched for GWAS variants (de Leeuw et al., 2015). Genes with MAGMA p-values < 10−3 were considered associated.
Motif enrichment and binding analysis
To discover the enrichment of predicted DNA binding motifs in the accessible chromatin peaks and FOS-binding peaks, we used the HOMER v2.29.2 findMotifsGenome.pl function (Heinz et al., 2010). We searched for motifs using the -mask option and a peak size of 500 bp or 100 bp for ATAC or FOS-binding peaks, respectively. The top significant motifs with a fold change > 1.5 were considered for analysis. Motif histograms were generated by first centering inducible ATAC peaks on the AP-1 motif using HOMER annotatePeaks.pl (-center, -size 200). Re-centered datasets were then used to generate specific motif density histograms using a 1,000 bp window and bin size of 10 bp (-size 1000, hist −10). Aggregate plots were generated with HOMER makeTagDirectory where tag directories of biological replicates were merged to create a single tag directory of pooled reads. HOMER annotatePeaks.pl was used to obtain FOS peak read densities in 10 bp bins across 4 kb windows centered at ATAC summits.
Partitioning heritability
Heritability was partitioned using LD score regression as previously described (Finucane et al., 2015). All peaks were extended by ±500 bp and LD scores for each annotation of interest were calculated based on 1000 Genomes phase 3 datasets, using SNPs selected from HapMap3 (International Hapmap3 Consortium, 2012 ) after excluding the major histocompatibility complex (MHC) region (chr6: 26–34Mb) as recommended in the original methodology. We used a baseline model (baseline_v1.2) consisting of 53 functional categories, including Refseq gene models obtained by UCSC [coding, UTRs, promoters, introns], ENCODE functional genomic transcription factor annotations, Roadmap epigenomic annotations, and FANTOM5 enhancers. We added to the baseline model the following pairs of annotations: 1) H3K27ac and open chromatin regions overlapping transcription start sites (TSSs; promoter regions), and H3K27ac and open chromatin regions not overlapping TSSs (enhancer regions) for unstimulated Glu and GABA neurons, and 2) H3K27ac and open chromatin for constitutive (unstimulated) and induced (90 min) promoter and enhancer regions for each neuronal type (Glu and GABA). Each annotation pair was added to the baseline model independently as 6 separate models. Additionally, for each model, we included in the baseline the union of the following H3K27ac fetal brain datasets as a control: 1) regions marked by H3K27ac in 15, 17, and 18 GW human fetal prefrontal cortex (Markenscoff-Papadimitriou et al., 2020); 2) regions marked by H3K27ac in 7, 8.5, and 12 GW human fetal cortex (Reilly et al., 2015); and 3) regions marked by H3K27ac in 17, 19, and 21 GW human fetal cortex (Li et al., 2018). For each model, regions in the control categories that overlapped our annotations included in the model were removed.
We then analyzed cell-type-specific annotations (h2-cts flag). We identified enrichment of disease SNPs in GABA and Glu neuron-specific open chromatin and H3K27ac peaks as well as open chromatin and H3K27ac peaks shared by both cell types (neuronal peaks). The numbers of enhancers used as input for the cell type-specific analysis are as follows: 1) Top 21,989 GABA-specific, Glu-specific and shared ATAC peaks; 2) Top 17,362 GABA-specific, Glu-specific and shared H3K27ac peaks. We used the same fetal brain H3K27ac datasets from the partitioned heritability analysis as a control for each annotation of interest. In analyzing each trait in each dataset, we performed Bonferroni correction for multiple testing and used adjusted p values < 0.05 to indicate significant enrichment for the disease SNPs. p-values were plotted as -log10 (p-adj).
Activity-by-contact (ABC) enhancer-gene interaction model
We used the ABC model (https://github.com/broadinstitute/ABC-Enhancer-Gene-Prediction) to predict enhancer-gene connections for unstimulated, 30 min, and 90mins of stimulation in Glu and GABA neurons. These enhancer predictions were generated using ATAC-seq, H3K27ac CUT&RUN, and gene expression data as previously described (Fulco et al., 2019). For each condition, the ABC model reports an ABC score for each enhancer-gene pair if the enhancer is within 5 Mb of the gene TSS. As suggested by Fulco et al., We performed the following: 1) chromatin accessibility reads in each peak were counted, and the top 150,000 peaks with the most reads were retained. Each peak was resized to 500 bp centered on the peak summit. 500 bp regions centered on all gene TSSs were added to this list, and peaks overlapping blacklisted regions were removed. Any resulting overlapping peaks were merged. 2) Enhancer activity was calculated through quantile normalization of chromatin accessibility and H3K27ac CUT&RUN signals in each candidate enhancer region. 3) The powerlaw.score option was used to estimate contact in lieu of Hi-C data. 4) We removed enhancer-gene connections greater than 2 Mb and removed 150 bp regions from each side of the peak as previously described (Nasser et al., 2021). Only ABC scores ≥0.015 were considered for analysis. Enhancer-gene pairs with the highest ABC score for each unique enhancer were used for all downstream analyses.
To quantify the number of GWAS-identified neuropsychiatric disease SNPs that overlap with ABC enhancers, we lifted SNP chromosomal coordinates from hg19 to hg38 and filtered for SNPs with p value < 5 × 10−8. LDproxy was used to identify SNPs in linkage disequilibrium. The resulting variant list was intersected with our datasets using tools intersect. ABC enhancer-gene pairs that overlapped disease variants were considered ABC-max genes. To determine the number of genes located within the region of variant to predicted ABC-max gene TSS, we intersected with GENCODE annotation (v38). We then used bedtools closest to determine whether the predicted ABC-max gene was the closest gene to the variant. To determine the distance of ABC-max and all ABC-genes to the closest neighboring protein-coding gene TSS, we removed all ABC-genes from the GENCODE annotation list and used bedtools closest.
Statistical analyses
Unless otherwise indicated, all data presented are the average of at least two biological replicates from each of at least two independent experiments. Statistical analysis was matched to the data structure as noted above in the methods details section for RNA-seq, ATAC-seq, and CUT&RUN experiments. Error bars were calculated using the standard deviation of all the replicates. For determining enrichment of accessible chromatin and H3K27ac peaks in VISTA enhancers and eQTLs, we performed Fisher’s Exact test and considered a p-value < 0.05 as significant. Statistical analyses were performed in RStudio (v1.3.1093; RStudio Team, 2020) or GraphPad Prism 9. See figure legends for details on specific statistical tests run for each experiment. Statistical significance is represented by a star (*) and indicates a computed p-value < 0.05. Graphs and plots were generated using Graphpad Prism or RStudio. Figures were generated using Adobe Illustrator and Biolegend.
Supplementary Material
Highlights.
Transcriptomic and epigenetic profiling of human excitatory and inhibitory neurons
Cell-type-specific enrichment of disease heritability in activity-induced enhancers
AP-1 TFs collaborate with neuron subtype TFs to select enhancers
ACKNOWLEDGMENTS
We are grateful to Kayla Townsley for assisting with the GWAS analysis, Xiaoting Zhou for technical supports, Dr. Vierbuchen at Memorial Sloan Kettering Cancer Center for scientific advice and discussions, and Dr. Pang and Le Wang at Rutgers University for assisting with electrophysiology experiments. This work was supported by the Icahn School of Medicine at Mount Sinai (ISMMS) start-up fund to N.Y. L.L.B. was supported by the Training Program in Stem Cell Biology fellowship from the New York State Department of Health (NYSTEM-C32561GG). S.E.W. was supported by the Training Program from the National Institute of Mental Health (T32-MH087004). R.H. was supported by ISMMS and Huazhong Agricultural University.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110877.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We worked to ensure diversity in experimental samples through the selection of the cell lines.
REFERENCES
- Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, Menashe I, Wadkins T, Banerjee-Basu S, and Packer A (2013). SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4, 36. 10.1186/2040-2392-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alberini CM (2009). Transcription factors in long-term memory and synaptic plasticity. Physiol. Rev 89, 121–145. 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JY, Lin K, Zhu L, Werling DM, Dong S, Brand H, Wang HZ, Zhao X, Schwartz GB, Collins RL, et al. (2018). Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362, eaat6576. 10.1126/science.aat6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, and Rubenstein JLR (1997). Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278, 474–476. 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Banerjee-Basu S, and Packer A (2010). SFARI Gene: an evolving database forthe autism research community. Dis. Model. Mech 3, 133–135. 10.1242/dmm.005439. [DOI] [PubMed] [Google Scholar]
- Beagan JA, Pastuzyn ED, Fernandez LR, Guo MH, Feng K, Titus KR, Chandrashekar H, Shepherd JD, and Phillips-Cremins JE (2020). Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression. Nat. Neurosci 23, 707–717. 10.1038/s41593-020-0634-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ (1998). Neuronal calcium signaling. Neuron 21, 13–26. 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bevington SL, Cauchy P, Piper J, Bertrand E, Lalli N, Jarvis RC, Gilding LN, Ott S, Bonifer C, and Cockerill PN (2016). Inducible chromatin priming is associated with the establishment of immunological memory in T cells. EMBO J. 35, 515–535. 10.15252/embj.201592534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrelli E, Nestler EJ, Allis CD, and Sassone-Corsi P (2008). Decoding the epigenetic language of neuronal plasticity. Neuron 60, 961–974. 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulting GL, Durresi E, Ataman B, Sherman MA, Mei K, Harmin DA, Carter AC, Hochbaum DR, Granger AJ, Engreitz JM, et al. (2021). Activity-dependent regulome of human GABAergic neurons reveals new patterns of gene regulation and neurological disease heritability. Nat. Neurosci 24, 437–448. 10.1038/s41593-020-00786-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitenkamp AFS, Matthes J, Nass RD, Sinzig J, Lehmkuhl G, Nurnberg P, and Herzig S (2014). Rare mutations of CACNB2 found in autism spectrum disease-affected families alter calcium channel function. PLoS One 9, e95579. 10.1371/journal.pone.0095579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Wang SS, Hattox AM, Rayburn H, Nelson SB, and McConnell SK (2008). The Fezf2-Ctip2 genetic pathway regulates the fate choice of subcortical projection neurons in the developing cerebral cortex. Proc. Natl. Acad. Sci. U S A 105, 11382–11387. 10.1073/pnas.0804918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U S A 107, 21931–21936. 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubelos B, Sebastian-Serrano A, Beccari L, Calcagnotto ME, Cisneros E, Kim S, Dopazo A, Alvarez-Dolado M, Redondo JM, Bovolenta P, et al. (2010). Cux1 and Cux2 regulate dendritic branching, spine morphology, and synapses of the upper layer neurons of the cortex. Neuron 66, 523–535. 10.1016/j.neuron.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, and Geschwind DH (2018). The dynamic landscape of open chromatin during human cortical neurogenesis. Cell 172, 289–304.e18. 10.1016/j.cell.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw CA, Mooij JM, Heskes T, and Posthuma D (2015). MAGMA: generalized gene-set analysis of GWAS data. Plos Comput. Biol 11, e1004219. 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, and Greenberg ME (2001). Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294, 333–339. 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Donato F, Chowdhury A, Lahr M, and Caroni P (2015). Early- and late-born parvalbumin basket cell subpopulations exhibiting distinct regulation and roles in learning. Neuron 85, 770–786. 10.1016/j.neuron.2015.01.011. [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, and Naldini L (1998).A third-generation lentivirus vector with a conditional packaging system. J. Virol 72, 8463–8471. 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, and Greenberg ME (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327–337. 10.1038/nature11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Jensen CL, and Champagne FA (2009). Epigenetic influences on brain development and plasticity. Curr. Opin. Neurobiol 19, 207–212. 10.1016/j.conb.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi IS (2014). Defining the neural basis of appetite and obesity: from genes to behaviour. Clin. Med. (Lond) 14, 286–289. 10.7861/clinmedicine.14-3-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felling RJ, and Song H (2015). Epigenetic mechanisms of neuroplasticity and the implications for stroke recovery. Exp. Neurol 268, 37–45. 10.1016/j.expneurol.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, Bulik-Sullivan B, Gusev A, Trynka G, Reshef Y, Loh PR, Anttila V, Xu H, Zang C, Farh K, et al. (2015). Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235. 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, Gazal S, Loh PR, Lareau C, Shoresh N, et al. (2018). Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet 50, 621–629. 10.1038/s41588-018-0081-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, and Greenberg ME (2008). Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu. Rev. Neurosci 31, 563–590. 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, et al. (2019). Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet 51, 1664–1669. 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362, eaat8127. 10.1126/science.aat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, and Penzes P (2015). Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med 15, 146–167. 10.2174/1566524015666150303003028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garel S, Marin F, Grosschedl R, and Charnay P (1999). Ebf1 controls early cell differentiation in the embryonic striatum. Development 126, 5285–5294. 10.1242/dev.126.23.5285. [DOI] [PubMed] [Google Scholar]
- Gorkin DU, Barozzi I, Zhao Y, Zhang Y, Huang H, Lee AY, Li B, Chiou J, Wildberg A, Ding B, et al. (2020). An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 583, 744–751. 10.1038/s41586-020-2093-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg ME, Ziff EB, and Greene LA (1986). Stimulation of neuronal acetylcholine receptors induces rapid gene transcription. Science 234, 80–83. 10.1126/science.3749894. [DOI] [PubMed] [Google Scholar]
- Hauberg ME, Creus-Muncunill J, Bendl J, Kozlenkov A, Zeng B, Corwin C, Chowdhury S, Kranz H, Hurd YL, Wegner M, et al. (2020). Common schizophrenia risk variants are enriched in open chromatin regions of human glutamatergic neurons. Nat. Commun 11, 5581. 10.1038/s41467-020-19319-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. (2009). Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459, 108–112. 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, and Glass CK (2015). The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol 16, 144–154. 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG, Kaya-Okur HS, and Ahmad K (2020). Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation. Elife 9, e63274. 10.7554/elife.63274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Cook EG, Gao Q, Mitalipova M, and Jaenisch R (2008). A drug-inducible system for direct reprogramming of human somatic cells to pluripotency. Cell Stem Cell 3, 346–353. 10.1016/j.stem.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman GE, Hartley BJ, Flaherty E, Ladran I, Gochman P, Ruderfer DM, Stahl EA, Rapoport J, Sklar P, and Brennand KJ (2017). Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with post-mortem adult brains. Nat. Commun 8, 2225. 10.1038/s41467-017-02330-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrvatin S, Hochbaum DR, Nagy MA, Cicconet M, Robertson K, Cheadle L, Zilionis R, Ratner A, Borges-Monroy R, Klein AM, et al. (2018). Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat. Neurosci 21, 120–129. 10.1038/s41593-017-0029-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Fabyanic E, Kwon DY, Tang S, Zhou Z, and Wu H (2017). Dissecting cell-type composition and activity-dependent transcriptional state in mammalian brains by massively parallel single-nucleus RNA-seq. Mol. Cell 68, 1006–1015.e7. 10.1016/j.molcel.2017.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Zhu X, and Yang N (2021). Direct differentiation of functional neurons from human pluripotent stem cells (hPSCs). Methods Mol. Biol 2352, 117–126. 10.1007/978-1-0716-1601-7_8. [DOI] [PubMed] [Google Scholar]
- Ichise E, Chiyonobu T, Ishikawa M, Tanaka Y, Shibata M, Tozawa T, Taura Y, Yamashita S, Yoshida M, Morimoto M, et al. (2021). Impaired neuronal activity and differential gene expression in STXBP1 encephalopathy patient iPSC-derived GABAergic neurons. Hum. Mol. Genet 30, 1337–1348. 10.1093/hmg/ddab113. [DOI] [PubMed] [Google Scholar]
- Jagadeesh KA, Dey KK, Montoro DT, Mohan R, Gazal S, Engreitz JM, Xavier RJ, Price AL, and Regev A (2021). Identifying disease-critical cell types and cellular processes across the human body by integration of single-cell profiles and human genetics. Preprint at bioRxiv, 03.19.436212. 10.1101/2021.03.19.436212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Lei R, Ding SW, and Zhu S (2014). Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15, 182. 10.1186/1471-2105-15-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaya-Okur HS, Wu SJ, Codomo CA, Pledger ES, Bryson TD, Henikoff JG, Ahmad K, and Henikoff S (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun 10, 1930. 10.1038/s41467-019-09982-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacar B, Linker SB, Jaeger BN, Krishnaswami SR, Barron JJ, Kelder MJE, Parylak SL, Paquola ACM, Venepally P, Novotny M, et al. (2016). Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun 7, 11022. 10.1038/ncomms11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, Sunkin SM, Li Z, Shin Y, Zhu Y, et al. (2018). Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 362, eaat7615. 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, and Greenberg ME (2008). Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 455, 1198–1204. 10.1038/nature07319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo PA, Kavran JM, Kim MS, and Leahy DJ (2013). Transient mammalian cell transfection with polyethylenimine (PEI). Methods Enzymol. 529, 227–240. 10.1016/b978-0-12-418687-3.00018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK, Harmin DA, and Greenberg ME (2014). Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat. Neurosci 17, 1330–1339. 10.1038/nn.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcel M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMB. Net. J 17, 10–12. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Markenscoff-Papadimitriou E, Whalen S, Przytycki P, Thomas R, Binyameen F, Nowakowski TJ, Kriegstein AR, Sanders SJ, State MW, Pollard KS, and Rubenstein JL (2020). A chromatin accessibility atlas of the developing human telencephalon. Cell 182, 754–769.e18. 10.1016/j.cell.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. (2012). Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195. 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meers MP, Bryson TD, Henikoff JG, and Henikoff S (2019). Improved CUT&RUN chromatin profiling tools. Elife 8, e46314. 10.7554/elife.46314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle-Heyrman G, Tims HS, and Widom J (2011). Structural constraints in collaborative competition of transcription factors against the nucleosome. J. Mol. Biol 412, 634–646. 10.1016/j.jmb.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasser J, Bergman DT, Fulco CP, Guckelberger P, Doughty BR, Patwardhan TA, Jones TR, Nguyen TH, Ulirsch JC, Lekschas F, et al. (2021). Genome-wide enhancer maps link risk variants to disease genes. Nature 533, 238–243. 10.1038/s41586-021-03446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Pena CJ, Kundakovic M, Mitchell A, and Akbarian S (2016). Epigenetic basis of mental illness. Neuroscientist 22, 447–463. 10.1177/1073858415608147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, et al. (2019). Brain cell type-specific enhancer-promoter interactome maps and disease - risk association. Science 366, 1134–1139. 10.1126/science.aay0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, and Natoli G (2013). Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171. 10.1016/j.cell.2012.12.018. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, Citri A, Sebastiano V, Marro S, Sudhof TC, and Wernig M (2011). Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223. 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SK, O’Shea C, Townsley K, Prytkova I, Dobrindt K, Elahi R, Iskhakova M, Lambert T, Valada A, Liao W, et al. (2021). Induction of dopaminergic neurons for neuronal subtype-specific modeling of psychiatric disease risk. Mol. Psychiatry, 1–13. 10.1038/s41380-021-01273-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruunsild P, Bengtson CP, and Bading H (2017). Networks of cultured iPSC-derived neurons reveal the human synaptic activity-regulated adaptive gene program. Cell Rep 18, 122–135. 10.1016/j.celrep.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SK, Yin J, Ayoub AE, Emera D, Leng J, Cotney J, Sarro R, Rakic P, and Noonan JP (2015). Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science 347, 1155–1159. 10.1126/science.1260943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio Team. (2020). RStudio: Integrated Development for R. RStudio. http://www.rstudio.com/. [Google Scholar]
- Rubenstein JLR, and Merzenich MM (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267. 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, Peng M, Collins R, Grove J, Klei L, et al. (2020). Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180, 568–584. 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrode N, Ho SM, Yamamuro K, Dobbyn A, Huckins L, Matos MR, Cheng E, Deans PJM, Flaherty E, Barretto N, et al. (2019). Synergistic effects of common schizophrenia risk variants. Nat. Genet 51, 1475–1485. 10.1038/s41588-019-0497-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, McFadden G, and Greenberg ME (1990). Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron 4, 571–582. 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, Giusti-Rodriguez P, Hodge RD, Miller JA, Munoz-Manchado AB, et al. (2018). Genetic identification of brain cell types underlying schizophrenia. Nat. Genet 50, 825–833. 10.1038/s41588-018-0129-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Yang X, Ren X, Maliskova L, Li B, Jones IR, Wang C, Jacob F, Wu K, Traglia M, et al. (2019). Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet 51, 1252–1262. 10.1038/s41588-019-0472-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel I, Mardinly AR, Gabel HW, Bazinet JE, Couch CH, Tzeng CP, Harmin DA, and Greenberg ME (2014). Npas4 regulates excitatory-inhibitory balance within neural circuits through cell-type-specific gene programs. Cell 157, 1216–1229. 10.1016/j.cell.2014.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R, and Brown G (2011). DiffBind: Differential Binding Analysis of ChIP-Seq Peak Data. [Google Scholar]
- Steg LC, Shireby GL, Imm J, Davies JP, Franklin A, Flynn R, Namboori SC, Bhinge A, Jeffries AR, Burrage J, et al. (2021). Novel epigenetic clock for fetal brain development predicts prenatal age for cellular stem cell models and derived neurons. Mol. Brain 14, 98. 10.1186/s13041-021-00810-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer L, Vera E, and Cornacchia D (2015). Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell 16, 591–600. 10.1016/j.stem.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, Kim D, Ming GL, and Song H (2017). Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat. Neurosci 20, 476–483. 10.1038/nn.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinstead EE, Miranda TB, Paakinaho V, Baek S, Goldstein I, Hawkins M, Karpova TS, Ball D, Mazza D, Lavis LD, et al. (2016). Steroid receptors reprogram FoxA1 occupancy through dynamic chromatin transitions. Cell 165, 593–605. 10.1016/j.cell.2016.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyssowski KM, DeStefino NR, Cho JH, Dunn CJ, Poston RG, Carty CE, Jones RD, Chang SM, Romeo P, Wurzelmann MK, et al. (2018). Different neuronal activity patterns induce different gene expression programs. Neuron 98, 530–546.e11. 10.1016/j.neuron.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, and O’Shea J (2012). STATs shape the active enhancer landscape of T cell populations. Cell 151, 981–993. 10.1016/j.cell.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, Bhaduri A, Goyal N, Rowitch DH, and Kriegstein AR (2019). Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689. 10.1126/science.aav8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol. Cell 68, 1067–1082.e12. 10.1016/j.molcel.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, and Wernig M (2010). Direct conversion of fibroblasts to functional neurons by defined factors. Nature 468, 1035–1041. 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, and Pennacchio LA (2007). VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92. 10.1093/nar/gkl822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Dye CA, Sohal V, Long JE, Estrada RC, Roztocil T, Lufkin T, Deisseroth K, Baraban SC, and Rubenstein JLR (2010). Dlx5 and Dlx6 regulate the development of parvalbumin-expressing cortical interneurons. J. Neurosci 30, 5334–5345. 10.1523/jneurosci.5963-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, Gandal MJ, Sutton GJ, Hormozdiari F, Lu D, et al. (2016). Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527. 10.1038/nature19847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YE, Pan L, Zuo Y, Li X, and Hong W (2017). Detecting activated cell populations using single-cell RNA-seq. Neuron 96, 313–329.e6. 10.1016/j.neuron.2017.09.026. [DOI] [PubMed] [Google Scholar]
- Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene set knowledge discovery with Enrichr. Curr. Protoc 1, e90. 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Chanda S, Marro S, Südhof T, Ng YH, Wernig M, Janas JA, Haag D, Ang CE, Tang Y, et al. (2017). Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 14, 621–628. 10.1038/protex.2017.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap EL, and Greenberg ME (2018). Activity-regulated transcription: bridging the gap between neural activity and behavior. Neuron 100, 330–348. 10.1016/j.neuron.2018.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Ibrahim JG, and Love MI (2019). Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092. 10.1093/bioinformatics/bty895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zovkic IB, Guzman-Karlsson MC, and Sweatt JD (2013). Epigenetic regulation of memory formation and maintenance. Learn. Mem 20, 61–74. 10.1101/lm.026575.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq, ATAC-seq, and CUT&RUN data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-FOS antibody [2H2] | Abcam | Cat # AB208942; RRID: AB_2747772 |
| Anti-MAP2 | Abcam | Cat #ab5392; RRID: AB_2138153 |
| Anti-H3K27ac | Abcam | Cat # AB4729; RRID: AB_2118291 |
| Anti-IgG | Abcam | Cat # AB46540; RRID: AB_2614925 |
| Anti-IgG | Novus | Cat # NBP1-2763 |
| Anti-HSP90 | Cell Signaling | Cat # 4874S; RRID: AB_2121214 |
| Anti-FOS | Abcam | Cat # AB208942; RRID: AB_2747772 |
| Anti-pERK1/2 | Cell Signaling | Cat # 9101S; RRID: AB_331646 |
| Anti-Dlx5 | Wang et al., 2010 | Made and kindly shared by John LR Rubenstein’s group. |
| Anti-Dlx2 | Santa Cruz Biotechnology, Inc | Cat # sc-393879 |
| Chemicals, peptides, and recombinant proteins | ||
| Stemflex supplement | Gibco | Cat # A33492 |
| Stemflex basal media | Gibco | Cat # A33493-01 |
| Geltrex | Gibco | Cat # A14133-02 |
| Poly-L-ornithine hydrobromide | Sigma | Ct # P3655 |
| Accutase | Thermo Scientific | Cat # NC9464543 |
| Thiazovivin | Santa Cruz | Cat # SC-361380 |
| Puromycin dihydrochloride from Streptomyces alboniger | Sigma | Cat # P8833 |
| Hygromycin B (50 mg/mL) | Life Technologies | Cat # 10687010 |
| Cytosine β-D-arabinofuranoside hydrochloride | Sigma | Cat # C1768 |
| TRIzol | Invitrogen | Cat # 15596026 |
| Tetrodotoxin citrate (TTX) | Labome | Cat # 1069/1 |
| DL-2-amino-5-phosphopentnoic acid (DL2-AP5) | Abcam | Cat # ab120271 |
| DMEM media | Gibco | Cat # 11965-092 |
| MEM NEAA (100x) | Gibco | Cat # 11140-050 |
| Sodium Pyruvate (100x) | Gibco | Cat # 11360-070 |
| β-Mercaptoethanol | Sigma | Cat # M3148 |
| Cytiva HyClone™ Cosmic Calf™ Serum | Fisher | Cat # SH3008704 |
| Fetal Bovine Serum (FBS) | Sigma | Cat # F6178 |
| Doxycycline | Sigma | Cat # P3655 |
| MEM media | Gibco | Cat # 11090-081 |
| Glutamax (100×) | Gibco | Cat # 35050-061 |
| B27 supplement | Gibco | Cat # 17504-044 |
| Neurobasal media | Gibco | Cat # 21103-049 |
| Naurobasal A media | Gibco | Cat # 10888-022 |
| Trypsin 0.25% | Gibco | Cat # 25200-114 |
| DMEM/F12 media | Gibco | Cat # 11320-033 |
| N2 supplement | Gibco | Cat # 17502-048 |
| Dimethyl sulfoxide | Sigma | Cat # D2438 |
| Paraformaldehyde solution | Affymetrix | Cat # 19943 |
| D-PBS tablets | Millipore | Cat # 524650-1EA |
| Protease Free Heat Shock Bovine Serum Albumin Powder | Equitech-Bio | Cat # BAH65-0500 |
| Urea | Sigma | Cat # U5128 |
| 1M HEPES/Sodium Hydroxide | Rigaku | Cat # 1008184 |
| RNase A | Thermo Fisher Scientific | Cat #EN0531 |
| Glycogen | Sigma | Cat #10930193001 |
| Spermidine | Sigma | Cat # S0266 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Sigma/Roche | Cat # 11873580001 |
| Concanavalin A (ConA)-coated magnetic beads | Bangs Laboratories | Cat # BP531 |
| Protein AG-MNase | EpiCypher | Cat # 15-1116 |
| AMPure XP beads | Beckman Coulter | Cat # A63881 |
| SYBR green 2x master mix | Applied Biosystems | Cat # 4367659 |
| Critical commercial assays | ||
| Tagment DNA Enzyme and Buffer kit | Illumina | Cat # 20034211 |
| MinElute Reaction Cleanup Kit | Qiagen | Cat # 28206 |
| NEB Next High-Fidelity 2x PCR Master Mix | New England Biolabs | Cat # M0541 |
| Pierce™ BCA Protein Assay Kit | Thermo Scientific | Cat # 23225 |
| TURBO™ DNase | Ambion | Cat # AM1907 |
| SuperScript® IV First-Strand Synthesis System | Thermo Scientific | Cat # 18091050 |
| Bolt™ 4 to 12%, Bis-Tris, 1.0 mm, Mini Protein Gel, 15-well | Thermo Scientific | Cat # MW04125BOX |
| iBlot™ 2 Transfer Stacks, PVDF | Invitrogen | Cat # IB24001 |
| Western Lightning Plus, Chemiluminescent Substrate | Perkin Elmer | Cat # NEL 104001EA |
| NEBNext Ultra II DNA library Prep kit for Ilumina | New England Biolabs | Cat # E7645 |
| NEBNext Multiplex oligos for Ilumina | New England Biolabs | Cat # E7600S and Cat # E7780S |
| Deposited Data | ||
| Raw RNA-sequencing data and Salmon counts | This paper | GEO: GSE196855 |
| Raw ATAC-sequencing data and processed peaks | This paper | GEO: GSE196854 |
| Raw H3K27ac CUT&RUN data and processed peaks | This paper | GEO: GSE196207 |
| Experimental models: cell lines | ||
| H1 (WA01) | WiCell Research Resources | hPSC Reg ID Ae001-A |
| 3188-2A-4N | Hoffman et al., 2017 | GEO: GSM2843584 |
| HEK293T | ATCC | Cat # 11268 |
| Experimental models: organisms/strains | ||
| CD1 mouse strain | Charles River | Cat# CRL:022, RRID: IMSR_CRL:022 |
| Recombinant DNA | ||
| FUW-M2rtTA | Hockemeyer et al., 2008 | RRID: Addgene_20342 |
| TetO-Ascl1-puro | Yang et al., 2017 | RRID: Addgene_97329 |
| TetO-Ngn2-puro | Zhang et al., 2013 | RRID: Addgene_52,047 |
| TeoO-Dlx2-Hygro | Yang et al. 2017 | RRID: Addgene_97330 |
| Software and algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | RRID: SCR_016368; http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Skewer v0.2.2 | Jiang et al., 2014 | https://github.com/relipmoc/skewer |
| Salmon 0.13.1 | Patro et al., 2017 | https://combine-lab.github.io/salmon/ |
| R | Love et al., 2014 | https://www.r-project.org/ |
| RStudio | RStudio Team, 2020 | https://github.com/rstudio/ |
| Apeglm | Zhu et al., 2019 | https://github.com/azhu513/apeglm |
| ENCODE ATAC-seq pipeline v1.4.2 | Consortium, 2012 | https://www.encodeproject.org/atac-seq/ |
| featureCounts | http://subread.sourceforge.net/ | |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| MACS | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| DiffBind | Stark and Brown, 2011 | http://bioconductor.org/packages/release/bioc/html/DiffBind.html |
| ChipSeeker | Yu et al., 2015 | https://guangchuangyu.github.io/software/ChIPseeker/ |
| LDSC | Finucane et al., 2015 | https://github.com/bulik/ldsc |
| Prism | GraphPad | RRID: SCR_005375 |