

Abstract
Ribonucleotide reductase (RR) is an essential multi-subunit enzyme found in all living organisms; it catalyzes the rate-limiting step in dNTP synthesis, namely, the conversion of ribonucleoside diphosphates to deoxyribonucleoside diphosphates. As expression levels of human RR (hRR) are high during cell replication, hRR has long been considered an attractive drug target for a range of proliferative diseases, including cancer. While there are many excellent reviews regarding the structure, function, and clinical importance of hRR, recent years have seen an increase in novel approaches to inhibiting hRR that merit an updated discussion of the existing inhibitors and strategies to target this enzyme. In this review, we discuss the mechanisms and clinical applications of classic nucleoside analog inhibitors of hRRM1 (large catalytic subunit), including gemcitabine and clofarabine, as well as inhibitors of the hRRM2 (free radical housing small subunit), including triapine and hydroxyurea. Additionally, we discuss novel approaches to targeting RR and the discovery of new classes of hRR inhibitors.
Keywords: ribonucleotide reductase, ribonucleotide reductase inhibitors, cancer chemotherapy, small molecule, drug discovery, hydrazone, acyl hydrazone, gemcitabine, fludarabine, clofarabine, cladribine, triapine, COH 29, pancreatic cancer, breast cancer, small cell lung cancer
1. Introduction
Ribonucleotide reductase (RR) is an essential enzyme found in all living organisms; it catalyzes the conversion of ribonucleoside diphosphates (RNDPS) to deoxyribonucleoside diphosphates (DNDPS) [1,2]. As this is the rate-limiting step in dNTP synthesis, human ribonucleotide reductase (hRR) is crucial for maintaining a balanced nucleotide pool within the cell and is integral to DNA synthesis [1,2,3]. During the S-phase of the cell cycle, hRR expression is elevated, making hRR an attractive drug target for proliferative diseases, including cancer [4,5,6]. The majority of cancer drugs targeting hRR are nucleoside analogs that mimic the chemical structure of natural substrates [7,8,9,10,11]. The most successful of these antimetabolites is gemcitabine, which is one of the very few FDA-approved clinical treatments for pancreatic cancers [12,13]. However, antimetabolites such as gemcitabine can inhibit numerous off-target enzymes involved in DNA synthesis and repair, leading to DNA strand breaks and apoptosis [10,14,15,16]. Additionally, gemcitabine confers high rates of therapeutic resistance, where only ~10% of pancreatic cancer patients will respond to single-agent gemcitabine treatment [17]. To overcome these limitations, there were several attempts to identify non-nucleoside inhibitors of hRR in recent years, many of which were designed to inhibit hRR either by targeting the hRRM2 (β subunit) directly or by manipulating its tightly controlled oligomeric regulation. Although there are several excellent reviews on hRR as a cancer drug target [18,19], there have been significant advances in the discovery of inhibitors of hRR since the most recent reviews focused on this subject [4,20], and thus, an update is required. In this review, we discuss the mechanism, activity, and clinical applications of the classic antimetabolite inhibitors clofarabine and gemcitabine, as well as the broad class of inhibitors targeting the small subunit, including triapine and hydroxyurea. Additionally, we discuss novel strategies to target RR, including the discovery of modulators of the hRRM1 (α) oligomeric state, the acylhydrazone class of hRRM1 inhibitors (including the C-site inhibitor NSAH), a high throughput PCR assay method to identify inhibitors of Pseudomonas aeruginosa (P.aeruginosa) RR, and iron chelators as transmetalative inhibitors of hRRM2.
2. Ribonucleotide Reductase
Ribonucleotide reductase was first characterized in 1961 by the Reichard group at the Karolinska Institute in Stockholm, Sweden. The enzyme is composed of two subunits, namely, RR1 and RR2 [1,2]. The large RR1 subunit (also known as the α subunit) contains the catalytic site (C-site) and all allosteric sites, while the small RR2 subunit (also known as the β subunit) contains the diferric-tyrosyl (Y√) radical cofactor that initiates nucleotide reduction via a putative long-range proton-coupled electron transfer (PCET) pathway approximately 35 Å away [1]. Recently, a new form of RR2 was discovered named p53R2 that operates outside of the S-phase of the cell cycle to maintain sufficient levels of dNTP pools for DNA repair. P53R2 is [21,22] expressed in quiescent and post-mitotic cells, where it combines with RR1 to synthesize the dNTPs required for nuclear DNA repair and mitochondrial DNA replication [21]. RRs are divided into three classes by the metal cofactors that generate their free radical electron. In this review, we were interested primarily in hRR, which is a class I enzyme and uses a tyrosyl free radical for catalysis. The class I enzymes are further divided into five subcategories Ia, Ib, Ic, Id, and Ie based on the different radical cofactors used in catalysis [23,24,25,26,27]. Below, we discuss the mechanism of class Ia enzymes, which include human, Escherichia coli (E. coli), and Saccharomyces cerevisiae (S. cerevisiae) RRs.
2.1. The Structure and Allosteric Regulation of Ribonucleotide Reductase
The active form of eukaryotic RR consists of dimeric or multimeric forms of the RR1 (α) and RR2 (β) subunits, such as α2β2 and α6βN (where n = 2, 4, or 6) [28,29,30,31]. Catalysis occurs in the RR1 (α) subunit, which contains the catalytic site (C-site) and two additional allosteric sites, namely, the specificity site (S-site) and the activity site (A-site) (Figure 1).
Figure 1.

Structure of ribonucleotide reductase. (a) Model of the E. coli α2β2 dimer complex (PDB ID: 6W4X) [32]. The α dimer is shown in red and orange. The nucleotide bound at the C—site is represented as green spheres. The nucleotide effect abound at the S—site is represented as blue spheres. The β subunit is shown in magenta and purple; the yellow spheres represent the radical generating di-iron cluster. The iron cluster and C-site are separated by a distance of over 35 Å, shown as a double arrow. (b) Structure of the hRRM1 dimer. The nucleotide bound at the catalytic site (C-site) is rendered as green spheres. The specificity site (S-site) controls substrate selection at the C-site and is represented as blue spheres. The activity site (A-site) controls the overall activity of the enzyme by binding the natural allosteric activator ATP or inhibitor dATP.
The A-site controls the overall activity of the enzyme by binding either ATP or dATP, while the S-site controls which of the four ribonucleoside diphosphate substrates bind to the C-site [2]. A model for the oligomeric equilibrium of hRRM1 and its control via allosteric effector binding to the S-site and A-site is presented in Figure 2a. Crystal structures of both ATP, i.e., the natural activator, and dATP, i.e., the natural inhibitor, in complex with the A-site of hRRM1 were reported by the Dealwis group in 2011 (Figure 2b–e) [30]. ATP was observed to bind deep within a pocket at the tip of the N-terminal domain, which is an area also referred to as the ATP-cone [30]. The natural inhibitor dATP was found to bind the A-site with higher affinity compared with the activator ATP, primarily due to differences in interactions with Ile 18. X-ray crystallography and cryo-EM studies demonstrated that the α6 hexamer consists of hRRM1 trimer of dimers [30,33]. Although there are dATP-bound α6 and α6β2 structures, so far, there are no structures for the ATP hexamer [30,33]. Nevertheless, mutagenesis analysis demonstrated that the active ATP-bound α6 structure is likely to be different from the dATP-inhibited hexamer, possibly explaining the differing activities. The dATP-bound hexamer structures reveal that inhibition occurs due to the positioning of β, which is too far away to transfer the free radical to the α active site.
Figure 2.

(a) Model for dATP-dependent oligomerization of eukaryotic RRs. The binding of effectors to the S-site causes dimerization and the binding of dATP to the A-site causes the formation of hexamers via a hypothesized short-lived tetramer intermediate or the immediate association of three dimers to form a hexamer (question mark). Effectors bound at the S-site are maroon spheres, and the A-site is rendered as a blue ribbon. (b) ATP-hRRM1 complex. The main chain is rendered in gray (helices—red, strands—cyan), while ATP is shown in elemental green. (c) dATP-hRRM1 complex. The main chain is rendered in gray (helices—red, strands—blue), while dATP is shown in elemental green. (d) Superposition of A and B. The ATP-hRRM1 complex is rendered in yellow ribbon with ATP in elemental green. The dATP-hRRM1 complex is rendered as a cyan ribbon with dATP in elemental magenta. (e) The ATP-binding cones of hRRM1 in complex with TTP and ATP (yellow) or TTP and dATP (cyan). ATP is shown in elemental green and dATP in elemental magenta (dTTP is not shown).
For more than four decades, investigators in the RR field have been fascinated by how this enzyme is able to recognize the four nucleotide diphosphates and convert them to their respective deoxy forms that provide the building blocks of life [2]. Most of the model studies with respect to molecular recognition were conducted using the perennial E. coli model of an α2β2 holo-complex. Insights into the molecular basis of the substrate recognition of RR came to light with the availability of crystal structures with their cognate effector–substrate complexes from E. coli, Thermotoga maritima (T. maritima), S. cerevisiae, and human RR, which are described in detail below [30,34,35,36]. Based on biochemical and structural data, a simple model emerged for maintaining balanced nucleotide pools that require elegant effector-based substrate recognition by the RR enzyme. Substrate recognition involves crosstalk between the S- and C-sites that are located in adjacent monomers composing the dimeric α2 molecule (see Figure 1b). The S-site is located >15 Å from the C-site and influences substrate binding by inducing subtle changes to the flexible loop 2 region (residues 285–295, hRRM1 numbering) of the C-site, which promote the binding of a specific substrate [30,31,35,37]. Substrate recognition is controlled by four allosteric effectors, namely, dGTP, dTTP, ATP, and dATP, binding at the S-site. In 1969, Brown and Reichard showed that this regulation is tightly controlled by the concentration of deoxynucleotides in the cell [1,2]. As the concentration of dNTPs decreases, ATP that has an almost constant concentration of 3–5 mM will bind at the A-site, activating RR. ATP will also bind to the S-site, which promotes the binding of CDP and UDP, which are then converted to dCDP and dUDP by RR. Phosphorylation of dCDP gives dCTP. dUDP is reduced to dUMP prior to conversion to dTMP by thymidylate synthase, which leads to an increase in dTTP concentrations [38]. As the concentration of dCTP and dUTP increases, dTTP outcompetes ATP at the S-site, promoting the catalysis of GDP to dGDP. As the concentration of dGTP increases, it will bind the S-site to promote the synthesis of dADP, which then increases the concentration of dATP in the cell. At this point, dATP will bind to the A-site, inhibiting the enzyme. As dATP has a higher binding affinity for the A-site than ATP, RR is only activated when the cellular concentration of dATP is low.
Allosteric effectors that bind to the S-site control substrate binding at the C-site by altering the conformation of a polypeptide chain known as loop 2, located within the C-site [30,35,36,39]. The molecular basis of recognition between effector–substrate pairs was established primarily through X-ray crystallography and site-directed mutagenesis, beginning with the first crystal structure of the substrate/effector pair GDP and dTTP in complex with RR1 by Eriksson et al.in E.Coli in 1997 [34]. Several labs demonstrated that subtle changes in the conformation of loop 2 will preferentially accommodate the binding of one substrate (or two, in the case of ATP) at the C-site [30,35,36,37]. This preference is largely based on steric constraints. The loop 2 residue Arg 293 is observed to form strong π–π stacking and/or π–cation interactions with the nucleoside base of the purines. In E. coli and T. maritima, Arg 293 forms a salt bridge with the phosphate groups of substrates bound in the C-site [35,36]. When purine substrates are bound, the loop 2 residue Gln 288 is observed to swing away from the C-site to accommodate the binding of the larger nucleoside bases. However, the CDP-RR1 complex shows that when smaller pyrimidine substrates bind, Gln 288 will shift further into the C-site. R293A mutants in yeast showed a loss of activity for both ADP and CDP substrates, further indicating that this residue is critical for substrate recognition and RR activity [39].
2.2. The Catalytic Mechanism of Class Ia RRs
The RR2 (β) subunit contains a stable diferric-tyrosyl (Y√) radical cofactor, which is required for catalysis [40]. Prior to catalysis, radical hopping occurs through stable aromatic residues from the diferric-tyrosyl in the RR2 (β) subunit 30–35 Å ito a catalytic cysteine in the C-site by a proton-coupled electron transport (PCET) mechanism [23,40,41,42,43,44,45]. This initiates the conversion of the ribonucleotide to its deoxy form through the mechanism illustrated in Scheme 1, where all residue numbering refers to the human system [42,43,46]. The radical is transferred to the C3′ position of the ribose ring of the substrate, generating a 3′-nucleotide radical. Glutamate 431 (E. coli 441) facilitates radical movement to the C2′ position by functioning as a general base catalyst for deprotonation of the 3′-OH. C443 (E. coli 462) facilitates the removal of the C2′ hydroxy group as a water molecule in a rapid, irreversible step. The subsequently formed 3′-keto-2′ radical is reduced by the PCET mechanism to generate the 3′-ketodeoxynucleotide and the disulfide radical anion (C443–C218 human; C462–C225 E. coli). The disulfide radical anion reduces the 3′-ketone with PCET, where the H atom is donated by E431 (E. coli 441). Finally, the H-atom removed from the C3′ ribose ring is returned to form the dNDP product and the radical is then transferred back to C429 (E. coli 439) and returns to the iron center in RR2 via the PCET pathway [40]. Before additional catalysis can occur, the disulfide bond between C443 and C218 must be reduced. This reduction is dependent upon the thioredoxin and glutaredoxin system, which, in turn, is reduced by thioredoxin reductase and glutathione [47,48]. Both systems are ultimately reduced by NADPH. This cycle of PCET and C-site cysteine reduction is required for each catalytic turnover. The recently reported cryo-EM structure of a trapped radical E. coli holo-complex provided the first glimpses of a structural map of this catalytic mechanism [32].
Scheme 1.

Catalytic mechanism of hRR. Radical movement is shown with red arrows. The movement of electron pairs is shown with blue arrows. Residues are numbered according to human RR.
3. Ribonucleotide Reductase as a Target for Cancer Therapeutics
3.1. The Role of RR in Cancer Biology
The most widely recognized hallmark of cancers is uncontrolled proliferation, which requires a sufficient supply of dNTPs. As hRR plays a crucial role in maintaining dNTP pools, the role of hRR in cancer proliferation has been of scientific interest for decades. In the 1970s, studies of rat hepatomas determined that RR activity is highly correlated with tumor growth rate, where enzymatic activity was approximately 200-fold higher in fast-growing tumors than in slow-growing tumors [49]. Either the hRRM1 or hRRM2 subunits are known to be overexpressed in a wide variety of cancers, including gastric, ovarian, colorectal, brain, breast, liver, and non-small-cell lung (NSCLC) cancers [50,51,52,53,54,55,56,57]. Overexpression of p53R2 is also observed in melanoma, oral carcinoma, esophageal, and NSCLC. In breast and epithelial ovarian cancers, the overexpression of p53R2 is correlated with the tumor grade, and this subunit is also highly expressed in malignant melanoma relative to benign skin lesions [58,59,60]. An analysis of the ONCOMINE dataset identified that the hRRM1 subunit is among the top 10% of most overexpressed genes in 30 of the 170 studies examined, including the brain, central nervous system, lung, and sarcoma datasets [4].
The effects of hRRM1 or hRRM2 overexpression are complex and varied. Dysregulation of hRR results in imbalanced nucleotide pools, which can inhibit DNA synthesis and replication and interfere with DNA damage repair [3]. While elevated dNTP levels can promote mutagenesis by DNA insertion and the escape of DNA polymerase proofreading via the “next-nucleotide effect” or increased frameshift mutations, enhanced hRR activity can also support DNA repair mechanisms. In cancers, these DNA repair mechanisms can be employed to protect against DNA damaging agents, such as the platinum class of chemotherapies. Overexpression of hRRM2 has been associated with increased invasion in human carcinoma cells and increased VEGF production, suggesting that hRRM2 can promote tumor invasion and angiogenesis [60]. Like hRRM1, elevated expression of hRRM2 can confer resistance to genotoxic chemotherapies, such as 5-fluorouracil, platinum drugs, or radiation therapy [61,62,63,64,65,66,67,68,69,70]. Overexpression of either subunit is also predictive of a poor response to hRR inhibitors, such as gemcitabine or triapine [71,72,73,74,75,76].
As the expression of p53R2 can be induced by the tumor suppressor p53 and is known to promote p21 accumulation and G1 arrest, it was originally expected that p53R2 would also play a tumor suppressor role [22]. However, overexpression of p53R2 is found in many cancers and knockdown studies have identified that p53R2 suppression prevents proliferation [77]. Furthermore, hRRM2 and p53R2 were shown to promote NSCLC in vivo, although this has not been replicated for other cancers [78]. The mechanism by which the hRRM2 and p53R2 subunits promote proliferation in cancers needs further examination, although there are several possibilities. Elevated hRRM2 could promote mutagenesis by altering dNTP pools such that base misinsertions, insertion–deletion events, and uracil misincorporation occur at a higher frequency. Several studies have also indicated hRR-driven dNTP alterations impact senescence and apoptosis pathways. hRRM2 and p53R2 overexpression were also linked with increased G→T transversions, an indicator of oxidative DNA damage [78]. As the Y (radical) housed in the hRRM2 subunit is transient and highly reactive, it is possible that overexpression of hRRM2 could result in the generation of reactive oxygen species (ROS) [79,80]. However, it has not yet been demonstrated that hRRM2 is capable of generating such ROS.
While there remains much to be learned about the complex mechanism of hRR’s promotion of cancer proliferation, its role in maintaining dNTP homeostasis has made it an attractive therapeutic target for decades.
3.2. Cancer Chemotherapeutics Targeting hRRs
Inhibiting hRR can lead to mutations, causing the incorporation of incorrect nucleotides, as well as chromosomal instability through replication fork collapses, double-strand breaks, and shortening of telomeres that cause chromosomal fusions [81,82]. Numerous small molecule inhibitors have been developed to inhibit hRR through a variety of mechanisms. The most important anticancer inhibitors can be divided into two classes, those that target the hRRM1 subunit, such as gemcitabine and clofarabine, and those that target the hRRM2 subunit, such as hydroxyurea [20]. Drugs targeting the hRRM1 subunit are primarily antimetabolites, which mimic natural substrates of hRR [7,8,9,10,11,15,18,83,84,85,86]. These drugs are administered in their nucleoside forms in order to be membrane permeable; however, in the cytoplasm, they are phosphorylated by cellular kinases to their mono, di, and triphosphate forms [87]. hRRM2 inhibitors compromise the integrity of the iron-tyrosyl radical center by either scavenging the radical electron, such as with hydroxyurea, or chelating iron, such as with triapine [88,89,90,91,92]. Additionally, recent studies have identified small molecule inhibitors that disrupt the binding of the hRRM1 and hRRM2 subunits. In this review, we focus on the four most clinically important nucleoside analogs, namely, cladribine, fludarabine, clofarabine, and gemcitabine, and provide a broader discussion of attempts to discover and develop non-nucleoside inhibitors of hRR.
4. The Antimetabolite Class of hRRM1 Inhibitors
The majority of clinically used drugs targeting hRR are nucleoside analogs that bind to the C-site or both C- and A-sites of the hRRM1 subunit, and the early development is described in references [86,93]. Among these are fludarabine, cladribine, gemcitabine, and clofarabine [7,8,9,10,11,15,18,83,84,85,86,93]. These four drugs impair DNA synthesis and repair pathways in part through the inhibition of RR, although most are also known to inhibit a variety of off-targets, including DNA polymerase, thymidylate synthase, and both cytidine deaminase and CTP synthase in the case of gemcitabine [10,14,15]. Although hRR is inhibited by these drugs, the major toxicities derive from DNA chain termination. Table 1 contains the structures, mechanisms, and indications of FDA-approved nucleoside analog inhibitors of hRR, as well as the radical scavenger hydroxyurea. The structures and mechanisms of all remaining hRR inhibitors are presented in Table 2 and Table 3.
Table 1.
FDA-approved hRR inhibitors and their anticancer applications.
| Structure | Name | Cellular Mechanism |
RR Subunit Targeted |
Mechanism of RR Inhibition |
Indications: Single Agent | Indications: Combination |
|---|---|---|---|---|---|---|
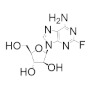
|
Fludarabine | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Induces formation of inactive a6 hexamers | Chronic lymphocytic leukemia |
With cyclophosphamide, mitoxantrone, dexamethasone, and rituximab: non-Hodgkin’s lymphoma With cyclophosphamide, mitoxantrone, dexamethasone, and granulocyte colony-stimulating factor: acute myeloid leukemia |
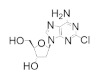
|
Cladribine | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Induces formation of inactive a6 hexamers | Hairy cell leukemia B-cell chronic lymphocytic leukemic | |
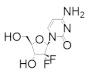
|
Gemcitabine | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Alkylates the C-site irreversibly | Pancreatic cancer Off label: cholangiocarcinoma and other biliary tract cancers | With nab-paclitaxel: pancreatic cancer With cisplatin: advanced or metastatic bladder cancer; advanced or metastatic non-small cell lung cancer With carboplatin: ovarian cancer With paclitaxel: metastatic breast cancer |
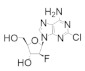
|
Clofarabine | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Induces formation of inactive a6 hexamers | Relapsed or refractory acute lymphoblastic leukemia | |
|
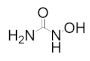
Hydroxyurea | Radical Scavenger | hRRM2 | Quenches the radical center | Chronic myelogenous leukemia |
Table 2.
Structure and mechanism of hRR inhibitors.
| Structure | Name | Cellular Mechanism | RR Subunit Targeted | Mechanism of RR Inhibition |
|---|---|---|---|---|
|
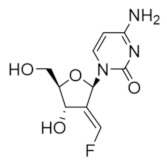
Tezacitabine | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Irreversibly modifies the C-site |
|
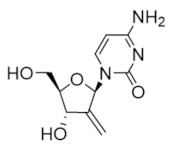
2′-deoxy-2′-methylenecytidine (DMDC) | Interferes with DNA synthesis and repair mechanisms | hRRM1 | Inhibits the C-site |
|
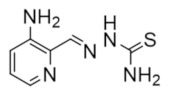
Triapine | Metal chelation | hRRM2 | Chelates FeIII from the radical center |
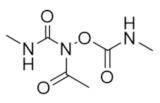
|
Caracemide | Radical Scavenger | hRRM1 | Quenches the radical center |
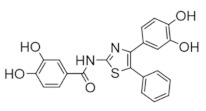
|
COH29 | Interferes with DNA synthesis and repair mechanisms | hRRM2 | Binds hRRM2 near the C-terminal tail to prevent association with hRRM1 |
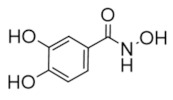
|
Didox | Radical Scavenger | hRRM2 | Chelates FeIII from the radical center |
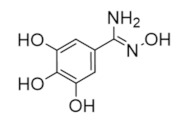
|
Trimidox | Radical Scavenger | hRRM2 | Chelates FeIII from the radical center |
|
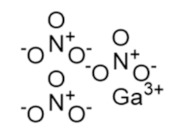
Gallium nitrate | FeIII mimic | hRRM2 | Interferes with FeIII radical center |
|
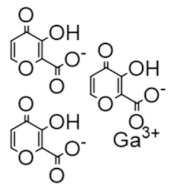
Gallium maltolate | FeIII mimic | hRRM2 | Interferes with FeIII radical center |
|
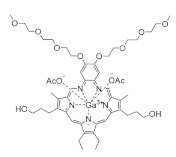
Motexafin gadolinium | FeIII mimic | hRRM2 | Interferes with FeIII radical center |
| GTI-2040 | antisense ologonucleotide | hRRM2 | Inhibits hRRM2 | |
| GTI-2501 | antisense ologonucleotide | hRRM2 | Inhibits hRRM2 | |
| CALAA-01 | siRNA-containing nanoparticle | hRRM2 | Inhibits hRRM2 |
Table 3.
Structure and mechanism of hRR subunit inhibitors.
| Structure | Name | RR Subunit Targeted | Mechanism of RR Inhibition |
|---|---|---|---|
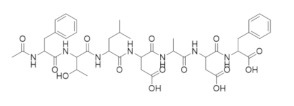
|
P6 | RR1 | Prevents binding of the RR2 subunit |
|
P7 | RR1 | Prevents binding of the RR2 subunit |
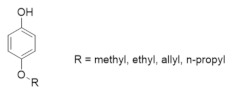
|
p-Alkoxyphenols | RR2 | Quenches the tyrosyl radical |
|
ADP-S-HBES-S-dGTP | RR1 | Targets the S-site |
|
A167 | RR1 | Compeites with ATP to bind the A-site |
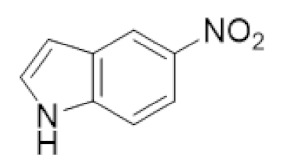
|
5-NITP | RR1 | S-site inhibitor that prevents RR1 hexamerization |
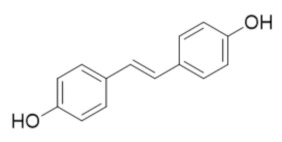
|
DHS | RR2 | Induces degradation of RR2 proteosome degredation pathway |
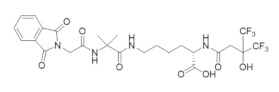
|
OxolsolndoLys | RR1 | Induces formation of inactive a6 hexamers |
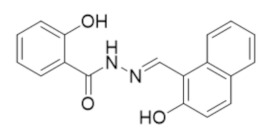
|
NSAH | RR1 | Reversible C-site inhibitor |
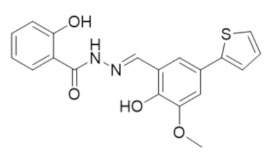
|
TP6 | RR1 | Reversible C-site inhibitor |
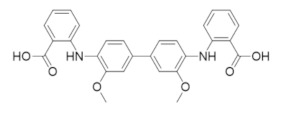
|
NSC73735 | RR1 and RR2 | Prevents alpha subunit hexamerization and quenches the tyrosyl radical of RR2 |
|
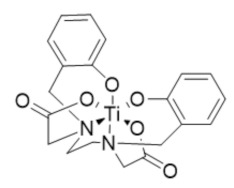
Ti(HBED) | RR2 | Depletes intracellular labile iron pools by transmetalation |
|
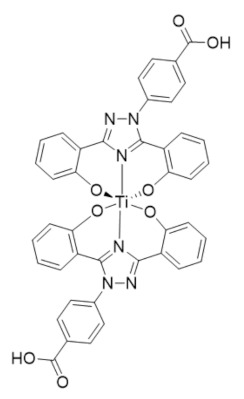
Ti(Deferasirox)2 | RR2 | Depletes intracellular labile iron pools by transmetalation |
4.1. The Antimetabolite Class of RR Inhibitors: Cladribine (CLA) and Fludarabine (FLU)
Fludarabine is approved for the treatment of various leukemias and lymphoma, including chronic lymphocytic leukemia, acute myeloid leukemia, acute lymphocytic leukemia, and non-Hodgkin’s lymphoma [94,95,96,97]. Fludarabine is a structural mimic of ADP with the addition of a fluorine atom on the 2 position of the adenine base and stereochemical inversion of the C2′ hydroxyl of the ribose ring. Typically sold as a monophosphate (Fludara), fludarabine can become further phosphorylated intracellularly to the diphosphate and triphosphate forms, the latter of which is known to inhibit hRR [95], DNA polymerase [98], DNA ligase [99], and DNA primase [100]. Fludarabine is commonly administered in combination with other chemotherapies, such as cyclophosphamide, mitoxantrone, dexamethasone, and rituximab, for the treatment of non-Hodgkin’s lymphomas or as part of the FLAG or FLAMSA treatment regimens for AML, where it is administered with cytarabine and granulocyte colony-stimulating factor. Cladribine is approved for first- and second-line treatment of hairy cell leukemia and for B-cell chronic lymphocytic leukemia [101,102]. Being a structural analog of adenosine, cladribine contains a chlorine atom at the 2 position, which renders the compound partially resistant to breakdown by adenosine deaminase, causing it to accumulate intracellularly and interfere in DNA synthesis and repair. Like other nucleoside analogs, it is phosphorylated intracellularly by deoxycytidine kinase to produce both the diphosphate and triphosphate forms. Incorporation of the triphosphate forms of fludarabine and cladribine into DNA strands by DNA polymerase leads to increased DNA strand breaks and the activation of transcription factor p53, eventually culminating in apoptosis [103,104]. The cellular efficacy of cladribine is dependent upon the ratio of the active triphosphate and the 5′-nucleotidase (5′-NT) family of enzymes, which dephosphorylate cladribine into the inactive prodrug form. As this ratio is highest in T and B lymphocytes, they are particularly susceptible to cladribine-mediated apoptosis, while other non-hematological malignancies are not significantly affected.
The mechanism of inhibition of hRR by fludarabine and cladribine was determined in part via a comparison of their effects on α6 hexamer formation in human cell lines [105]. ClA and FlU nucleotides were determined to promote the formation of α6 hexamers, although the hexamer formed in the presence of FlUDP was only observed in gel filtration studies if FlUDP was supplemented in the running buffer; this unexpected result was not observed for other nucleotides, including FlUTP, and suggests that the hexameric states formed by the diphosphate and triphosphate forms are structurally distinct and maintained post-ligand dissociation. This theory is supported by the additional finding that trypsin protease cleavage of the hexamers induced by different nucleotide inhibitors occurred at different rates. While both FlUDP and ClADP are known to interact with the C-site of hRRM1, this interaction appears to be less relevant than the hexamer-inducing interactions, as inhibition of hRR is dependent upon the ability of the enzyme to form α6 hexamers. The D57N-α mutant was uninhibited by either ClA or FlU because the D57N mutant is unable to form hexamers, which are required for inhibition [105].
4.2. The Antimetabolite Class of hRR Inhibitors: Clofarabine
Clofarabine was approved in 2004 for the treatment of relapsed or refractory acute lymphoblastic leukemia in children [106,107,108]. This drug is a mimic of adenine diphosphate (ADP), with the critical change of the C2′ ribose hydroxyl to a fluorine atom and there is a chlorine substitution at the 2 position of the adenine ring. As the side effects of clofarabine are severe (hematologic toxicity, febrile neutropenia, hepatobiliary toxicity, renal toxicity) and can lead to systemic inflammatory response syndrome, which can cause multiple organ failure, the drug is approved only for patients for whom two other treatment methods have failed [109]. Clofarabine can exist in the monophosphate, diphosphate, and triphosphate forms, with the triphosphate form heavily favored in cells (ratio of ClFDP:ClFTP up to 1:7) [108]. While the diphosphate form binds to the catalytic site to inhibit hRR reversibly in a time-dependent manner, the triphosphate form will inhibit both the A-site of hRR and DNA polymerases α and ε [83]. In contrast to the diphosphate form, ClFTP inhibition of hRR is time independent. Both forms were shown to promote the formation of α hexamers [83,110]. Interestingly, in contrast to the dATP-α6 complex, dissociation of either ClFDP or ClFTP leaves the α subunit trapped in an inactive α6 hexamer conformation. Site-directed mutagenesis studies identified A-site residue D57 to be essential for the formation of this inactive hexamer conformation. The clofarabine triphosphate-induced inactive hexamer structure from mammalian cells was determined using cryo-EM [110]. The conclusion from this study was that by using cell culture, the authors were able to induce hexamer structures of the α subunit in sufficient quantities to be able to capture the cryo-EM image; however, the β subunit appeared to be disordered and could not be assigned in the structure. Clofarabine triphosphate induces apoptosis by incorporating into DNA strands, causing breaks within the strand. The triphosphate form was also shown to directly influence mitochondrial integrity by disrupting the mitochondrial membrane, which leads to apoptosis [111,112].
4.3. The Antimetabolite Class of hRR Inhibitors: Gemcitabine
Gemcitabine is a unique nucleoside drug. Unlike other nucleoside drugs, gemcitabine is efficacious against solid tumors, as well as hematological malignancies. Gemcitabine is approved as a front-line treatment for pancreatic cancer after patients are first treated with a combination of surgical resection and folfiinox [113]. In fact, it is the most commonly used drug against pancreatic cancer. In metastatic non-small-cell lung cancer and metastatic bladder cancer, gemcitabine is combined with cisplatin [114,115]. Gemcitabine is also used as a second-line treatment for ovarian cancer (in combination with carboplatin) [116] and metastatic breast cancer (in combination with paclitaxel) [117]. It is a structural mimic of CDP with the substitution of two fluorine atoms at the C2 position of the ribose ring. Crystal structures of gemcitabine demonstrate that it binds at the catalytic site of the α subunit in an altered conformation to the natural substrate CDP (Figure 3) [118]. The mechanism of inhibition by gemcitabine has been investigated both in the presence and absence of reducing agents (thioredoxin, DTT) [119,120].
Figure 3.

Crystal structure of ScRR1 in complex with gemcitabine. (a) Stereo view of CDP (orange). Interacting atoms: oxygen, red; nitrogen, blue; phosphate, magenta; sulfur, green; substrate carbons, cyan; protein non-Cα carbons, yellow; Cα carbons, as secondary structure, orange. (b) Stereo view of gemcitabine diphosphate. Interacting atoms are colored as in (a), except that sulfur is orange; Cα carbons, as secondary structure, are yellow; and fluorines are gray. (c) Ligand plot of CDP ribose interactions. Colors are as in (a), except that carbons are yellow. (d) Ligand plot of gemcitabine diphosphate interactions. The van der Waals contact with L427 is omitted for clarity. (e) Stereo view of the loop 2 superposition of AMPPNP–CDP (orange) and AMPPNP–gemcitabine diphosphate (yellow). Substrate/inhibitor is seen on the left, and the effector is on the right. The color scheme is the same as in (c), but fluorine is black. Figure reprinted with permission from Xu et al. [118].
In either environment, inhibition is time dependent and irreversible with full hRR inactivation and binds with a stoichiometry of 0.5 equivalent gemcitabine per equivalent hRRM1. When bound at the catalytic site of hRR, the radical is transferred to the C3′ position of the ribose ring but is unable to transfer to the C2′ position as the geminal fluorine atoms cannot react as the hydroxyl group of the natural substrate. At this stage, the ribose ring breaks apart, and in a reducing environment, may alkylate the enzyme, inhibiting it irreversibly [119]. This alkylation was confirmed in Lactobacillus leichmannii (L. leichmannii), where mass spectrometry identified the alkylation of C419 [119,120]. However, this alkylation has not yet been observed in the human enzyme, and gemcitabine’s irreversibility may come from the quenching of the transient thiol radical, which was observed in the absence of reductants [119,120]. This radical scavenging most likely occurs when the enzyme is in its oxidized state. In addition to inhibiting hRR, gemcitabine is phosphorylated in the cell and can replace dCTP in DNA replication, leading to apoptosis [10,11,121]. As the diphosphate form inhibits hRR and depletes the concentration of natural dNTPs in the cell, the levels of dCTP reduce. As dCTP is a natural feedback inhibitor of deoxycytidine kinase, dNTP depletion leads to the enhanced production of gemcitabine triphosphate such that it can compete for incorporation into DNA strands. In this way, gemcitabine’s cytotoxicity is self-potentiating [10,87,122]. Additional studies are needed to fully characterize the effects of this self-potentiating mechanism of cytotoxicity. Other side targets of gemcitabine include thymidylate synthase, cytidine deaminase, deoxycytidine kinase, and DNA polymerase [10,11,15,16,85].
Clinical use of gemcitabine is further limited by the propensity for tumors to develop a resistance to it [123,124,125,126,127]. While this resistance could be attributed to many pathways, gemcitabine-induced upregulation of anti-apoptotic proteins, such as BEX2, Bcl2A1, and CXCR4, could be a major factor [124,128,129]. Increased expression levels of several multidrug-resistant (MDR) genes, such as ABCC1, ABCC3, ABCC5, and ABCB1, in pancreatic cancer cells also promote drug efflux, lowering the concentration of gemcitabine within the tumor over time [130,131,132,133,134,135].
While gemcitabine remains a mainstay of pancreatic cancer chemotherapy, only approximately 10% of patients will respond to gemcitabine treatment. Those that do respond can expect to survive an additional 6–8 weeks after starting treatment, with a 5-year survival rate of only 7% [130]. In the past two decades, other than adjuvant chemotherapy treatment, such as folfirinox, and the combination of nab-paclitaxel with gemcitabine, only one additional drug has been approved to treat pancreatic cancer, namely, the epidermal growth factor receptor (EGFR) inhibitor erlotinib [136,137,138,139,140,141,142,143]. Erlotinib is approved as a combination therapy with gemcitabine for certain local pancreatic tumors and has been shown to expand life expectancy (6.24 months) compared with single-agent gemcitabine (5.91 months), although its use is limited by the high frequency of KRAS2 mutations in pancreatic cancers [142,144]. The lack of progress in treating pancreatic cancers with the limited success rate of adjuvant chemotherapy underscores the dire need for the discovery and development of new chemotherapeutic agents for this disease.
4.4. The Antimetabolite Class of hRR Inhibitors: Tezacitabine
Like gemcitabine, tezacitabine is a structural analog of deoxycytidine, although it is much less susceptible to metabolism by cytidine deaminase [145,146,147]. The diphosphate form irreversibly inhibits the hRRM1 subunit and incorporation of the triphosphate form into DNA strands, leading to strand breaks and apoptosis [145,148,149,150,151,152,153]. Clinical evaluation of tezacitabine has thus far included phase I and II studies of solid refractory tumors as a monotherapy and in combination with cisplatin for the treatment of gastrointestinal cancer and in combination with 5-fluorouracil for the treatment of advanced solid tumors [147,151,154,155,156]. Trials of both the monotherapy and combination regimens have not progressed beyond phase II due to a lack of efficacy.
4.5. Nucleoside Analog DMDC
Like gemcitabine and tezacitabine, 2′-deoxy-2′-methylidenecytidine (DMDC) is a structural mimic of deoxycytidine that is activated by intracellular phosphorylation to the diphosphate and triphosphate forms [157]. The diphosphate inhibits the hRRM1 subunit, while the triphosphate inhibits DNA polymerases, leading to DNA strand breaks and apoptosis [158]. DMDC is resistant to metabolism by cytidine deaminase and was proven effective in xenograft models of cancers, where cytidine deaminase activity is high and gemcitabine response is typically low [159,160,161]. Phase I trials of DMDC indicated a high rate of severe hematological toxicities that terminated its development as a chemotherapeutic [160,162,163].
4.6. hRRM1 Covalent Modifier Caracemide
The hydroxylamine derivative caracemide was demonstrated to irreversibly inhibit active site cysteine residue in E. coli RR1 [164]. Interestingly, although no direct effects on the RR2 subunit are observed, the holocomplex was found to be over 30 times more sensitive to inhibition by caracemide than isolated RR1. Although caracemide demonstrated promising antiproliferative effects in lymphocytic leukemia and Ehrlich ascites carcinoma cells, its efficacy is severely limited by its instability in human plasma and the resulting neurotoxicity of its metabolites [165,166,167]. Caracemide was evaluated in a wide array of phase I trials as an anticancer agent but rarely progressed to phase II due to severe dose-limiting toxicities, including pain, renal failure, respiratory failure, and neurological dysfunction [168,169,170,171,172,173].
5. Non-Nucleoside Inhibitors of hRRM2
To overcome the limitations of the antimetabolite inhibitors of hRRM1, there were attempts to develop non-nucleoside inhibitors that target the hRRM2 subunit or the hRRM1–hRRM2 interface. In this section, we discuss a broad selection of the most common classes of clinically evaluated hRRM2 inhibitors: radical scavengers, metal chelators, and compounds and biologics that disrupt binding between the hRRM1 and hRRM2 subunits.
5.1. The Radical Scavengers: Hydroxyurea
The most important of the radical scavenger class is hydroxyurea, which was one of the first anticancer therapeutics identified in 1953. Hydroxyurea quenches the tyrosyl radical of hRRM2, preventing catalysis of the hRR holocomplex [88,89]. Hydroxyurea has been used clinically for decades to treat a variety of diseases, including chronic myeloid leukemia (CML), polycythemia vera, and cervical cancer, but its use is limited by its poor efficacy, inconvenient dosing schedule, and a high rate of natural resistance in patients [174,175,176,177,178,179,180,181]. Currently, hydroxyurea is more commonly administered as a combination therapy, where it was evaluated in clinical trials for malignant gliomas (with imatinib) [182], glioblastomas (with cisplatin and ara-C or with imatinib) [183,184], and squamous cell carcinoma of the head and neck (with radiation and 5-fluorouracil) [185]. As a monotherapy, hydroxyurea is FDA-approved for the treatment of myeloproliferative disorders and sickle cell disease [186,187].
5.2. Metal Chelators: Triapine, Didox, Trimidox, Desferrioxamine, and Gallium Complexes
Triapine was evaluated as an anticancer therapeutic in phase I and phase II clinical trials and inhibits hRR by chelating FeIII from the radical center in hRRM2 [188]. A member of the heterocyclic carboxyaldehyde thiosemicarbazones (HCTs), triapine coordinates iron in a 2:1 ligand-to-iron ratio through a N*-N*-S* tridentate system, forming an octahedral complex [90,91,92]. In hRR inhibition, the triapine–FeIII complex is quickly reduced to a more favorable triapine–FeII complex, which was shown to activate molecular oxygen to a ROS, quenching the essential tyrosyl radical of hRRM2, [189]. Triapine was also shown to degrade DNA in the presence of hydrogen peroxide and iron, suggesting that this drug is not only an inhibitor of DNA repair/DNA synthesis but also a DNA-damaging agent [190]. Triapine was evaluated in several phase I and II studies for the treatment of leukemia, advanced solid tumors, advanced hematologic malignancies, head and neck cancer, and renal cell carcinoma [191,192,193,194]. In most cases, the response rate to monotherapy triapine was limited (0–7%) and high rates of grade 3 and 4 neutropenias were reported, leading to the termination of the studies [195]. Triapine was also evaluated in combination with various chemotherapies, including fludarabine for refractory acute leukemia and myeloproliferative disorder [191], doxorubicin for advanced solid tumors [196], and gemcitabine for non-small-cell lung cancer and advanced solid tumors [188,197,198,199]. While the combination with gemcitabine was discontinued due to high toxicity, results from a phase II study of combinations with fludarabine for refractory acute leukemia and myeloproliferative disorder were considered promising [200].
The hydroxyl-benzohydroxamic acid derivatives didox and trimidox are radical chelators that complex with Fe to inhibit the hRRM2 subunit [201,202,203,204,205,206,207]. These compounds are known to impair proliferation in a variety of cancer cell lines as monotherapies. Both compounds also showed synergistic effects with approved chemotherapies, both in vitro and in vivo [208,209]. When combined with either carmustine or temozolimide, didox worked synergistically to impair proliferation in brain cancer cell lines [208,209,210]. Didox further reduces tumor growth in Epstein–Barr-virus-positive nasopharyngeal carcinoma xenografts when combined with cidofovir [211]. Phase 1 and 2 studies of didox for various indications were completed, although no study has progressed beyond phase 2 [212,213,214,215]. While didox showed tolerable dosing and toxicity, its efficacy was generally not sufficient to warrant phase 3 evaluation. Didox has not been evaluated in clinical trials as a combination therapy. Trimidox has not been clinically evaluated but has demonstrated synergistic effects with tiazofurin, adriamycin, streptozotocin, ara-C, cisplatin, and cyclophosphamine in leukemia models [204,205,206,216,217].
Desferrioxamine (DFO), also known as deferoxamine (DFOA), is a well-characterized iron and aluminum chelator that is commonly used to treat iron overdose, hemochromatosis, and aluminum toxicity in dialysis patients. DFO is also capable of inhibiting hRR activity by chelating FeIII, forming a 1:1 ligand–metal complex [218]. While DFO does not directly affect the hRRM2 subunit, sequestration of intracellular iron prevents the activation and regeneration of the tyrosyl radical center, thus inhibiting hRR activity [219,220,221]. This inhibition is reversible with the addition of iron and appears to affect p53R2 more potently than hRRM2. DFO showed antiproliferative effects in leukemia and neuroblastoma cells in vitro and in vivo [222,223,224,225]. Clinical trials in neuroblastoma showed some efficacy, but the use of DFO as an antiproliferative agent is limited by its poor cell permeability and very short half-life [222,223,226,227,228]. An additional limitation is the ability of DFO to induce overexpression of hypoxia-inducible factor 1α (HIF1α); a recent report by Lang et al. indicated that co-administration of DFO and the HIF1α inhibitor lificiguat synergistically impaired proliferation in pancreatic cancer cell lines [229]. Encapsulation of DFO in transferrin receptor 1 targeting lysosomes improved cellular uptake and prolonged circulation time in vivo, suggesting that the clinical limitations of DFO could be overcome with novel delivery strategies.
The gallium variants gallium nitrate and gallium maltolate have been clinically evaluated as therapeutics for a wide variety of cancers since the 1980s; in 1991, gallium nitrate was approved for cancer-related hypercalcemia [230]. Gallium mimics ferric iron and is taken up by rapidly proliferating cells, which need iron for DNA synthesis. However, gallium inhibits RR and hence inhibits DNA synthesis, triggering apoptosis [231,232,233]. A recent study by Chitambar et al. found that gallium maltolate treatment of glioblastoma and glioblastoma stem cells led to a loss of mitochondrial reserve capacity which was followed by decreased oxygen consumption and decreased activity of hRRM2, which requires iron for the catalytic ferric-tyrosyl radical cluster [234]. In this way, gallium formulations that interfere with iron metabolism may also indirectly inhibit hRRM2.
Motexafin gadolinium is a synthetic metallotexaphyrin that inhibits both hRR and thioredoxin reductase [235]. Its hRR inhibition is via the replacement of FeIII iron in hRRM2. Motexafin gadolinium accumulates in tumor cells preferentially due to their increased metabolism and generates ROS, resulting in apoptosis. It was evaluated in numerous phase 1 and 2 clinical trials as both a monotherapy and combination therapy for non-small-cell lung cancer [236,237], non-Hodgkin’s lymphoma [238], glioblastoma and other brain cancers [239,240,241,242,243,244], chronic lymphatic leukemia [245], and renal cancer [246]. A phase 3 study of motexafin gadolinium in non-small-cell lung cancer with brain metastasis led to increased disease progression from 10 months to 15 months and suggested improvements to memory and executive function in patients [243]. The drug remains active in phase 3 trials for various other cancers.
5.3. A Disruptor of hRRM1–hRRM2 Binding: COH29, Antisense Oligonucleotides, and siRNAs
The lack of a fully ordered α2β2 complex structure for hRR has limited the options to develop small molecule interference disruptors as a known structure is a prerequisite for knowledge-based drug design. Therefore, most drug design attempts were made with either the hRRM1 structure or the hRRM2 structure. In silico modeling by the Yen group targeting the p53R2 structure identified the non-nucleoside molecule COH29 [247]. Moreover, COH29 inhibits PARP P, suggesting the involvement of polypharmacology [248]. This compound is expected to bind a small pocket on the C-terminal tail of hRRM2 away from the diferric-tyrosyl radical cluster. COH29 reduced proliferation in most of the NIH 60 panel of cancer cell lines and induced S-phase arrest. Twice-daily oral dosing of COH29 (50 and 100 mg/kg) was further found to impair tumor growth and prolong survival in mice with MOLT-4 acute myeloid leukemia or TOV11D ovarian cancer xenografts. In BRCA1-deficient human breast cancer cells, COH29 was found to impair growth by interfering with DNA repair pathways [248]. The drug is currently undergoing phase 2 clinical evaluation.
Additional biologics were developed to inhibit the hRRM2 subunit. GTI-2040, which is an antisense oligonucleotide targeting the hRRM2 mRNA, showed strong inhibition of many cancer types in vitro and in vivo and was studied clinically as both a monotherapy and a combination therapy for renal, prostate, AML, NSCL, and metastatic breast cancers [249,250]. However, GTI-2040 has never progressed beyond phase 2 trials due to limited efficacy. An additional antisense oligonucleotide, namely, GTI-2501, targets the hRRM1 subunit and is currently under evaluation in phase 2 trials [251]. GTI-2501 was shown to decrease mRNA and protein levels of hRRM1 in a variety of tumors and impair tumor progression in vivo [77,252]. A clinical safety study of CALAA-01 (NCT00689065) was terminated for unknown reasons.
Non-nucleoside inhibitors of hRR showed promise in clinical trials as chemotherapeutics for a wide range of cancers. As such, interest remains high in identifying novel non-nucleoside inhibitors of hRR as leads for drug development. In the remainder of the review, we discuss emerging efforts to identify non-nucleoside inhibitors targeting hRR.
6. Continuing Efforts to Identify Novel Inhibitors of hRR
Efforts to develop novel inhibitors of hRR remain very active and have employed a variety of strategies to target either the hRRM1 or hRRM2 subunit. In this section, we review some of the novel classes and methods of hRR inhibition that have not yet achieved clinical evaluation.
6.1. Peptides Targeting the hRRM1 Subunit
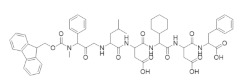
There have been multiple efforts to target the RR1 subunit with peptides that can compete with the RR2 subunit for binding [253,254,255,256,257,258,259]. The first such study was reported in 1986 and demonstrated that the nonapeptide YAGAVVNDL derived from the herpes simplex virus R2 (HSV-RR2) could inhibit HSV-RR [253,254]. Parallel studies established that the majority of the binding affinity could be attributed to the pentapeptide VVNDL, which led to a series of structure–function studies that identified pentapeptides that targeted HSV-RR1 with nanomolar binding affinities.
An N-acetyl heptapeptide derived from the C-terminus of the mouse and human mRR2 and hRRM2 subunits named P7 (N-Ac-FTLDADF) was found to inhibit mammalian RR with an IC50 of ~20 µM [258]. This peptide and its derivatives are stronger inhibitors of the α2β2 dimer complex than the α6βn complex. Crystal structures of P7 and its derivative P6 in complex with Saccharomyces cerevisiae RR1 identified two contiguous binding sites that are highly conserved amongst eukaryotic RR1s (Figure 4) [260]. Subsite A was found to consist of highly hydrophobic residues that housed the N-terminal residues of the peptides, while subsite B carried a highly positively charged surface that favorably interacted with the negatively charged carboxylate terminals. These structural studies support the experimental trend of maximal potency occurring at peptide lengths of at least seven residues. However, a recent crystal structure of a non-peptidyl small molecule inhibitor TAS1553 bound to the hydrophobic site of P7 (PDB ID: 6L3R) was reported, demonstrating that small molecules can bind at the site and can be developed as cancer therapies.
Figure 4.

Structure of the ScR1-P7 complex. (a) The electrostatic potential surface showing the R2 binding site on R1. P7 is shown in yellow. (b) The electrostatic potential surface of the P7 binding site, with subsites (a,b) clearly indicated. (c) Superposition of P7 (orange) with small molecule TAS1553 (green). TAS1553 occupies subsite B of the peptide binding region. Figure adapted with permission from Xu et al. [118].
6.2. Alkoxyphenols Targeting the hRR2 Subunit
Alkoxyphenols were demonstrated to quench the tyrosyl radical of the hRR2 subunit of mouse and herpes simplex virus 2 via a radial redox reaction [261,262]. p-Alkoxyphenols were shown to inhibit RR in Novikoff hepatoma, leukemia, and melanoma cells to impair proliferation [263]. Thus far, these compounds have not advanced to clinical evaluation.
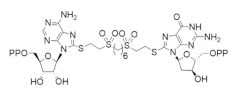
6.3. Bivalent Inhibitors of hRR
Several attempts to develop bivalent inhibitors targeting hRR were undertaken. In 2000, Wu et al. reported the development of a bivalent inhibitor of mouse RR that was designed to target the C-site and S-site of the mRR1 subunit [264]. The compound features ADP and dGDP linked by a 1,6-hexane-(bisethylenesulfone) tether. Despite a promising binding affinity of 12 µM, this compound was found to bind only the S-site. In 2014, Petrelli et al. reported the design of bivalent inhibitors linking 3′-C-methyladenosine and valproic acid, which are known inhibitors of RR and histone deacetylase (HDAC), respectively [265]. While the bivalent compounds remained active inhibitors of RR, they were unable to target HDAC. One analog, namely, A167, demonstrated potent antiproliferative effects in a broad variety of cancer cell models and reduce dNTP pools in HL-60 cells. A167 was found to compete with ATP at the A-site to inhibit the hRRM1 subunit.
6.4. S-Site Inhibition by the Non-Natural Nucleotide 5-NITP
In 2012, Mohammed et al. evaluated the ability of a set of ten 5-substituted indolyl-2′-deoxynucleoside triphosphates (5-NIdR) to inhibit hRRM1 [266]. The 5-NIdR scaffold was selected for evaluation due to its ability to mimic the size and shape of the allosteric inhibitor dATP. One such compound, namely, 5-NITP, was found to inhibit hRR with an IC50 of 170 µM and a Kd of 44 µM. Interestingly no inhibition was observed when CDP was used as the substrate, suggesting that the compound could bind to the S-site as opposed to the C-site. This theory was supported by gel filtration studies, which indicated that the predominant oligomeric form of the hRRM1-5-NITP complex was the dimer complex as opposed to the hexamer complex. A crystal structure of 5-NITP in complex with the Saccharomyces cerevisiae RR1 subunit confirmed that 5-NITP binds to the S-site preferentially and induced cytotoxicity in human leukemia cells (PDB ID: 3RSR). 5-NITP was further demonstrated to induce cell-cycle arrest at the S-phase, consistent with the inhibition of DNA synthesis in human leukemia cells.
6.5. Resveratrol Analog 4,4′-Trans-Dihydroxystiblene (DHS)
The resveratrol analog 4,4′-trans-dihydroxystilbene (DHS) was known to impair the proliferation of a variety of cancer cells, including human promyelocytic leukemia, breast cancer, neuroblastoma, and lung cancer cells although its mechanism of action was unclear [267,268,269,270,271,272]. In 2018, Chen et al. demonstrated that DHS induced cell-cycle arrest and decreased dNTPs pool in a manner consistent with hRR inhibition [273]. Thermal shift assays identified the direct binding of DHS to the hRRM2 subunit and site-directed mutagenesis confirmed that Val 146, Ser 150, Gln 151, and Ile 166 are critical for DHS binding and inhibition. Chen et al. further identified that DHS induced hRRM2 degradation by the proteosome degradation pathway [273]. Xenograft models further established that DHS combination therapies were able to overcome gemcitabine resistance in pancreatic cancer models and cisplatin resistance in ovarian cancer models and delayed tumor progression [273].
6.6. Modulation of the hRRM1 Oligomeric Equilibrium by OxoIsoIndoLys
Initial attempts by Dealwis and colleagues to identify non-nucleoside inhibitors of hRR that could inactivate the enzyme by manipulating its tightly regulated oligomeric equilibrium were reported [274]. The prevailing model of allostery is that hRR is active as an α2β2 complex, but under physiological conditions, exists primarily as an active α6βn hexamer (n = 2, 4, or 6) with a small population of dimers present [28,29]. hRR inhibitors, such as clofarabine and gemcitabine, were shown to induce inactive hRRM1 α6β2 hexamers, supporting the role of oligomeric modulation in the small molecule inhibition of hRR [83,119]. The central hypothesis of the study was that by targeting the hexamer interface with high throughput virtual screening, one could identify small molecules that bind to and stabilize the inactive hexamer conformation preferentially, thus shifting the natural oligomeric equilibrium toward the inactive state. Screening of the University of Cincinnati compound library (formerly the Proctor and Gamble library) identified 91 compounds that were expected to bind and inhibit hRRM1. Docking was directed toward the hexamer interface of the inactive dATP-bound hRRM1 hexamer model constructed from the S. cerevisiae dATP-induced hexamer (PDB: 3PAW) [30]. The docking site was defined as the N-terminal 16 residues from adjacent dimers. The top hits from the docking studies were subject to biophysical fluorescence quenching studies to determine dissociation constants against hRRM1. Out of the 91 candidates, 51 compounds were chosen to have greater than 20% fluorescence quenching and hence deemed as reasonable binders of hRRM1. These compounds were screened at two doses (10 and 1 µM) in multiple cancer cell lines to identify compounds with antiproliferative properties. The 10 most potent inhibitors were verified to inhibit hRRM1 and inhibit the enzymatic activity of recombinant hRR with micromolar potency [274].
A crystal structure of hRRM1 in a complex with a phthalimide-based inhibitor, namely, OxoIsoIndoLys, verified that this compound binds to the hexamer interface at the N-terminus and gel filtration studies demonstrated that this compound promoted hRRM1 hexamers. The structure of the hRRM1–OxoIsoIndoLys complex was determined down to a 3.7 Å resolution [274]. The compound was observed to form non-polar interactions with the N-terminus of one dimer unit, although a polar contact was observed with Ala 49. In the presence of 50 µM dATP, recombinant hRRM1 exists in equilibrium between α2 dimers and α6 hexamers, where the dimer: hexamer ratio is 1:3. Upon addition of 1 mM OxoIsoIndoLys, the dimer to hexamer ratio increased seven-fold, verifying that OxoIsoIndoLys can modulate the hRRM1 oligomeric equilibrium.
6.7. NSAH and Other Acylhydrazones Targeting the C-Site of hRRM1
In 2017, the Dealwis lab identified the first non-nucleoside small molecule capable of binding the C-site of hRRM1, which was a naphthyl salicylic acid hydrazone named NSAH [275]. In an effort to identify unique chemical scaffolds likely to bind the site, an in silico screen targeting the C-site was performed using Glide in the Schrödinger software suite. Cancer cell growth inhibition assays identified two hydrazone derivatives, namely, NSAH and a related naphthyl acyl hydrazone (NAH), with potent cellular activity (IC50 ~250 nM NSAH, 600 nM NAH) and cell-free inhibition against recombinant hRR (IC50 = 32 µM NSAH, 40 µM NAH). The direct binding of NSAH to recombinant hRRM1 was determined using a fluorescent quenching assay, establishing the KD as 37 ± 4 µM. The enzymatic IC50 was established at 32 ± 10 µM. A series of kinetics experiments demonstrated that NSAH inhibited hRR reversibly and competitively. IC50 values of NSAH ranged from 220–500 nM in a broad panel of cancer cells, while no cytotoxicity was observed in normal human blood progenitor cells at concentrations up to 1 µM, indicating NSAH has a much broader therapeutic index than the C-site inhibitor gemcitabine; there are no viable cells at a gemcitabine concentration of 35 nM. A crystal structure of the dimeric form of hRRM1 bound to NSAH at a 2.66 Å resolution confirmed that NSAH binds to the C-site, occupying a similar position to the binding pocket normally observed for diphosphate substrate binding (PDB ID: 5TUS, Figure 5) [275]. NSAH can adopt either a cis or trans conformation, and the cis isomer was observed in the crystal structure.
Figure 5.

Crystal structure of NSAH at the C-site of hRRM1. NSAH is shown in gray, substrate GDP is shown in orange. Figure adapted with permission from Huff et al. [275].
A series of structure–activity relationship studies were undertaken to optimize the potency of NSAH [276]. These efforts initially focused on modification of the substituent groups of the benzene and naphthalene rings while leaving the core scaffold intact. Notably, the synthetic scheme employed in these studies preferentially afforded E-isomers. This initial study identified that ortho-polar substitution to the benzene moiety of the hydrazone scaffold was required for hRR inhibition. An additional series of 13 hydrazones were then designed with modifications focused on disassociating the fused ring system into a biphenyl moiety with various substitutions [277]. All of the modified hydrazones were found to bind hRRM1 with lower KD values than NSAH (KD = 22.0 ± 5.67 µM), where 12 of the 13 NSAH analogs reported KD values below 10 µM. Screening of the compounds in Panc1 cell lines identified three analogs with potent antiproliferative effects, TP 4, 5, and 6 (IC50s > 1.5-0.393 µM). In fact, TP 6 had a twofold lower IC50 compared with NSAH. Interestingly, all three of the most potent analogs contained a biaryl replacement of the naphthalene ring moiety from the parent NSAH structure while maintaining the phenol and hydrazone linker. Further, the biaryl replacement in all three analogs includes a common guaiacol methoxyphenol ring that maintains the polar hydrogen bond donor position of the parent naphthol moiety. The removal of this methoxy group in TP2 and -3 resulted in at least a twofold decrease in cellular potency and induced a different binding orientation when docked at the C-site. These data indicate that including an electronegative element in the biaryl ring system could further increase the potency.
6.8. A Novel High Throughput PCR Assay Identifies Chemically Distinct RR Inhibitors
In an effort to identify RR inhibitors with antiviral activity, the Tholander and Sjöberg groups developed a PCR-based method for screening RR activity in microplate format, allowing for a higher throughput assessment of potential inhibitors than traditional methods [278]. A screen of >1300 compounds against P.aeruginosa RR identified 27 compounds with reported IC50 values between 200 nM and 30 µM. Four of these compounds, namely, NSC36758, NSC45383, NSC361666, and NSC228155, demonstrated antibacterial properties against P.aeruginosa, and both NSC361666 and NSC228155 were shown to deplete dNTP pools, which is consistent with RR inhibition. Both NSC45383 and NSC228155 were found to quench the P.aeruginosa RR2 radical, although these effects could be due to either iron chelation or generation of reactive oxygen species [279]. A comparison of the high throughput assay hits with reported cytotoxicity data from the NCI-60 panel of cancer cell lines revealed a selection of unique hRR inhibitors with anticancer properties, including NSC73735 [280]. Further evaluation of NSC73735 indicated that it prevented α hexamerization, in contrast to other established hRRM1 inhibitors, such as clofarabine. The selective cytotoxicity observed toward leukemia cell lines makes NSC73735 an attractive compound for further development as an anticancer agent. The characteristic phenotype of hRR inhibition, namely, cell cycle arrest and depletion of dNTP pools, was observed in HL-60 cells after treatment with NSC73735. Interestingly, these effects were reversible, in contrast to the less selective hRR inhibitor hydroxyurea. However, this compound is also known to inhibit dihydroorotate dehydrogenase and interfere with the synthesis of pyrimidine nucleotides [281,282]; it is possible that the depletion of dNTP pools can be promoted by both mechanisms.
6.9. Transmetalative Iron Chelators That Inhibit hRRM2
In 2021, Gaur et al. reported an iron chelator transmetalative approach to inhibit the hRRM2 subunit by exchanging a titanium(IV) ion for Fe(III) and depleting the intracellular iron pool [283]. A series of chemical transferrin mimetics (cTfms) were evaluated as the metal chelator due to their thermodynamic preference for binding Fe(III) over Ti(IV), thereby driving the intracellular transmetalation. The hexadentate chelator N,N’-di(o-hydroxybenzyl)-ethylenediamine-N,N’-diacetic acid (HBED) and the FDA-approved tridentate chelator Deferasirox both exhibited potent cytotoxicity to Jurkat cells by decreasing intracellular iron. Both the Ti(HBED) and Ti(Deferasirox)2 complexes were found to alter the nucleotide substrate pool and induce cell-cycle arrest consistent with RR inhibition.
7. Conclusions and Perspectives
Given the number of clinically approved cancer chemotherapeutics that target either the hRRM1 or hRRM2 subunit of ribonucleotide reductase, hRR remains a compelling target for cancer chemotherapy. Currently, hydroxyurea, which targets hRRM2, and the nucleoside analogs that target hRRM1 are the only clinically approved drugs for the treatment of cancer. While most nucleoside analogs were approved for the treatment of leukemia, gemcitabine has demonstrated efficacy against solid tumors and is the most widely used hRR targeting chemotherapy. However, nucleoside analogs suffer from dose-limiting toxicity (possibly due to their chain-terminating activity against the DNA polymerases) and narrow therapeutic windows. Moreover, chemoresistance develops after a few months of treatment, rendering the drug ineffective. Small subunit targeting drugs, such as hydroxyurea, that interfere with the di-iron cluster through scavenging and chelating activity have their own drawbacks due to a lack of target specificity and lack of efficacy. Despite the limitations of these chemotherapies, new on-target therapies, such as kinases and phosphatase inhibitors, have limited use in cancers such as pancreatic cancer, triple-negative breast cancer, and glioblastomas. Therefore, it will be useful to develop new functional “gemcitabine-like” molecules that will maintain their efficacy but lack their current deficiencies. In order to overcome the aforementioned limitations of current hRR drugs, novel strategies were developed that include non-nucleoside small molecule reversible competitive inhibitors that may function more effectively than gemcitabine and other nucleosides and to discover subunit and hexamer interface blocking inhibitors that will not bind at orthostatic sites and hence possess on-target specificity. Future clinical trials will demonstrate whether these new strategies will yield promising therapeutics for the treatment of cancer. After decades of research, RR continues to be a rich and complex target for antiproliferative diseases.
Acknowledgments
The authors would like to acknowledge John Pink of the Case Comprehensive Cancer Center (Cleveland, Ohio, USA) and Hsueh-Yun Lee of Taipei Medical University (Taipei, Taiwan) for their thoughtful discussion.
Author Contributions
Writing—review and editing, S.E.H., J.M.W. and C.G.D. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This publication was made possible by grants from the Clinical and Translational Science Collaborative of Cleveland (UL1TR002548) and from the National Center for Advancing Translational Sciences (NCATS) component of the National Institutes of Health and NIH Roadmap for Medical Research awarded to Dealwis (1R01GM100887-01) and by grants from the National Institutes of Health awarded to Winter (R01 CA212600 and R37CA227865-01A1).
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Brown N.C., Canellakis Z.N., Lundin B., Reichard P., Thelander L. Ribonucleoside diphosphate reductase. Purification of the two subunits, proteins B1 and B2. Eur. J. Biochem. 1969;9:561–573. doi: 10.1111/j.1432-1033.1969.tb00646.x. [DOI] [PubMed] [Google Scholar]
- 2.Brown N.C., Reichard P. Role of effector binding in allosteric control of ribonucleoside diphosphate reductase. J. Mol. Biol. 1969;46:39–55. doi: 10.1016/0022-2836(69)90056-4. [DOI] [PubMed] [Google Scholar]
- 3.Weinberg G., Ullman B., Martin D.W., Jr. Mutator phenotypes in mammalian cell mutants with distinct biochemical defects and abnormal deoxyribonucleoside triphosphate pools. Proc. Natl. Acad. Sci. USA. 1981;78:2447–2451. doi: 10.1073/pnas.78.4.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aye Y., Li M., Long M.J., Weiss R.S. Ribonucleotide reductase and cancer: Biological mechanisms and targeted therapies. Oncogene. 2015;34:2011–2021. doi: 10.1038/onc.2014.155. [DOI] [PubMed] [Google Scholar]
- 5.Greene B.L., Kang G., Cui C., Bennati M., Nocera D.G., Drennan C.L., Stubbe J. Ribonucleotide Reductases: Structure, Chemistry, and Metabolism Suggest New Therapeutic Targets. Annu. Rev. Biochem. 2020;89:45–75. doi: 10.1146/annurev-biochem-013118-111843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wijerathna S.R., Ahmad M.F., Xu H., Fairman J.W., Zhang A., Kaushal P.S., Wan Q., Kiser J., Dealwis C.G. Targeting the Large Subunit of Human Ribonucleotide Reductase for Cancer Chemotherapy. Pharmaceuticals. 2011;4:1328–1354. doi: 10.3390/ph4101328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerqueira N.M., Fernandes P.A., Ramos M.J. Ribonucleotide reductase: A critical enzyme for cancer chemotherapy and antiviral agents. Recent Pat. Anti-Cancer Drug Discov. 2007;2:11–29. doi: 10.2174/157489207779561408. [DOI] [PubMed] [Google Scholar]
- 8.Gandhi V., Mineishi S., Huang P., Chapman A.J., Yang Y., Chen F., Nowak B., Chubb S., Hertel L.W., Plunkett W. Cytotoxicity, metabolism, and mechanisms of action of 2′,2′-difluorodeoxyguanosine in Chinese hamster ovary cells. Cancer Res. 1995;55:1517–1524. [PubMed] [Google Scholar]
- 9.Parker W.B., Shaddix S.C., Chang C.H., White E.L., Rose L.M., Brockman R.W., Shortnacy A.T., Montgomery J.A., Secrist J.A., 3rd, Bennett L.L., Jr. Effects of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl)adenine on K562 cellular metabolism and the inhibition of human ribonucleotide reductase and DNA polymerases by its 5′-triphosphate. Cancer Res. 1991;51:2386–2394. [PubMed] [Google Scholar]
- 10.Heinemann V., Xu Y.Z., Chubb S., Sen A., Hertel L.W., Grindey G.B., Plunkett W. Cellular elimination of 2′,2′-difluorodeoxycytidine 5′-triphosphate: A mechanism of self-potentiation. Cancer Res. 1992;52:533–539. [PubMed] [Google Scholar]
- 11.Baker C.H., Banzon J., Bollinger J.M., Stubbe J., Samano V., Robins M.J., Lippert B., Jarvi E., Resvick R. 2′-Deoxy-2′-methylenecytidine and 2′-deoxy-2′,2′-difluorocytidine 5′-diphosphates: Potent mechanism-based inhibitors of ribonucleotide reductase. J. Med. Chem. 1991;34:1879–1884. doi: 10.1021/jm00110a019. [DOI] [PubMed] [Google Scholar]
- 12.Motoi F., Kosuge T., Ueno H., Yamaue H., Satoi S., Sho M., Honda G., Matsumoto I., Wada K., Furuse J., et al. Randomized phase II/III trial of neoadjuvant chemotherapy with gemcitabine and S-1 versus upfront surgery for resectable pancreatic cancer (Prep-02/JSAP05) Jpn. J. Clin. Oncol. 2019;49:190–194. doi: 10.1093/jjco/hyy190. [DOI] [PubMed] [Google Scholar]
- 13.Sarvepalli D., Rashid M.U., Rahman A.U., Ullah W., Hussain I., Hasan B., Jehanzeb S., Khan A.K., Jain A.G., Khetpal N., et al. Gemcitabine: A Review of Chemoresistance in Pancreatic Cancer. Crit. Rev. Oncog. 2019;24:199–212. doi: 10.1615/CritRevOncog.2019031641. [DOI] [PubMed] [Google Scholar]
- 14.Honeywell R.J., Ruiz van Haperen V.W., Veerman G., Smid K., Peters G.J. Inhibition of thymidylate synthase by 2′,2′-difluoro-2′-deoxycytidine (Gemcitabine) and its metabolite 2′,2′-difluoro-2′-deoxyuridine. Int. J. Biochem. Cell Biol. 2015;60:73–81. doi: 10.1016/j.biocel.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 15.Heinemann V., Plunkett W. Modulation of deoxynucleotide metabolism by the deoxycytidylate deaminase inhibitor 3,4,5,6-tetrahydrodeoxyuridine. Biochem. Pharmacol. 1989;38:4115–4121. doi: 10.1016/0006-2952(89)90693-X. [DOI] [PubMed] [Google Scholar]
- 16.Pourquier P., Gioffre C., Kohlhagen G., Urasaki Y., Goldwasser F., Hertel L.W., Yu S., Pon R.T., Gmeiner W.H., Pommier Y. Gemcitabine (2′,2′-difluoro-2′-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clin. Cancer Res.. 2002;8:2499–2504. [PubMed] [Google Scholar]
- 17.Burris H.A., 3rd, Moore M.J., Andersen J., Green M.R., Rothenberg M.L., Modiano M.R., Cripps M.C., Portenoy R.K., Storniolo A.M., Tarassoff P., et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 18.Tang S.C., Chen Y.C. Novel therapeutic targets for pancreatic cancer. World J. Gastroenterol. 2014;20:10825–10844. doi: 10.3748/wjg.v20.i31.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mannargudi M.B., Deb S. Clinical pharmacology and clinical trials of ribonucleotide reductase inhibitors: Is it a viable cancer therapy? J. Cancer Res. Clin. Oncol. 2017;143:1499–1529. doi: 10.1007/s00432-017-2457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao J., Zhou B., Chu B., Yen Y. Ribonucleotide reductase inhibitors and future drug design. Curr. Cancer Drug Targets. 2006;6:409–431. doi: 10.2174/156800906777723949. [DOI] [PubMed] [Google Scholar]
- 21.Pontarin G., Ferraro P., Rampazzo C., Kollberg G., Holme E., Reichard P., Bianchi V. Deoxyribonucleotide metabolism in cycling and resting human fibroblasts with a missense mutation in p53R2, a subunit of ribonucleotide reductase. J. Biol. Chem. 2011;286:11132–11140. doi: 10.1074/jbc.M110.202283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka H., Arakawa H., Yamaguchi T., Shiraishi K., Fukuda S., Matsui K., Takei Y., Nakamura Y. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature. 2000;404:42–49. doi: 10.1038/35003506. [DOI] [PubMed] [Google Scholar]
- 23.Stubbe J. Di-iron-tyrosyl radical ribonucleotide reductases. Curr. Opin. Chem. Biol. 2003;7:183–188. doi: 10.1016/S1367-5931(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 24.Srinivas V., Lebrette H., Lundin D., Kutin Y., Sahlin M., Lerche M., Eirich J., Branca R.M.M., Cox N., Sjöberg B.M., et al. Metal-free ribonucleotide reduction powered by a DOPA radical in Mycoplasma pathogens. Nature. 2018;563:416–420. doi: 10.1038/s41586-018-0653-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blaesi E.J., Palowitch G.M., Hu K., Kim A.J., Rose H.R., Alapati R., Lougee M.G., Kim H.J., Taguchi A.T., Tan K.O., et al. Metal-free class Ie ribonucleotide reductase from pathogens initiates catalysis with a tyrosine-derived dihydroxyphenylalanine radical. Proc. Natl. Acad. Sci. USA. 2018;115:10022–10027. doi: 10.1073/pnas.1811993115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grāve K., Griese J.J., Berggren G., Bennett M.D., Högbom M. The Bacillus anthracis class Ib ribonucleotide reductase subunit NrdF intrinsically selects manganese over iron. J. Biol. Inorg. Chem. 2020;25:571–582. doi: 10.1007/s00775-020-01782-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hofer A., Crona M., Logan D.T., Sjöberg B.M. DNA building blocks: Keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 2012;47:50–63. doi: 10.3109/10409238.2011.630372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kashlan O.B., Cooperman B.S. Comprehensive model for allosteric regulation of mammalian ribonucleotide reductase: Refinements and consequences. Biochemistry. 2003;42:1696–1706. doi: 10.1021/bi020634d. [DOI] [PubMed] [Google Scholar]
- 29.Rofougaran R., Vodnala M., Hofer A. Enzymatically active mammalian ribonucleotide reductase exists primarily as an alpha6beta2 octamer. J. Biol. Chem. 2006;281:27705–27711. doi: 10.1074/jbc.M605573200. [DOI] [PubMed] [Google Scholar]
- 30.Fairman J.W., Wijerathna S.R., Ahmad M.F., Xu H., Nakano R., Jha S., Prendergast J., Welin R.M., Flodin S., Roos A., et al. Structural basis for allosteric regulation of human ribonucleotide reductase by nucleotide-induced oligomerization. Nat. Struct. Mol. Biol. 2011;18:316–322. doi: 10.1038/nsmb.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ando N., Li H., Brignole E.J., Thompson S., McLaughlin M.I., Page J.E., Asturias F.J., Stubbe J., Drennan C.L. Allosteric Inhibition of Human Ribonucleotide Reductase by dATP Entails the Stabilization of a Hexamer. Biochemistry. 2016;55:373–381. doi: 10.1021/acs.biochem.5b01207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang G., Taguchi A.T., Stubbe J., Drennan C.L. Structure of a trapped radical transfer pathway within a ribonucleotide reductase holocomplex. Science. 2020;368:424–427. doi: 10.1126/science.aba6794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brignole E.J., Tsai K.L., Chittuluru J., Li H., Aye Y., Penczek P.A., Stubbe J., Drennan C.L., Asturias F. 3.3-A resolution cryo-EM structure of human ribonucleotide reductase with substrate and allosteric regulators bound. eLife. 2018;7:e31502. doi: 10.7554/eLife.31502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eriksson M., Uhlin U., Ramaswamy S., Ekberg M., Regnstrom K., Sjoberg B.M., Eklund H. Binding of allosteric effectors to ribonucleotide reductase protein R1: Reduction of active-site cysteines promotes substrate binding. Structure. 1997;5:1077–1092. doi: 10.1016/S0969-2126(97)00259-1. [DOI] [PubMed] [Google Scholar]
- 35.Larsson K.M., Jordan A., Eliasson R., Reichard P., Logan D.T., Nordlund P. Structural mechanism of allosteric substrate specificity regulation in a ribonucleotide reductase. Nat. Struct. Mol. Biol. 2004;11:1142–1149. doi: 10.1038/nsmb838. [DOI] [PubMed] [Google Scholar]
- 36.Zimanyi C.M., Chen P.Y., Kang G., Funk M.A., Drennan C.L. Mol.ecular basis for allosteric specificity regulation in class Ia ribonucleotide reductase from Escherichia coli. eLife. 2016;5:e07141. doi: 10.7554/eLife.07141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu H., Faber C., Uchiki T., Fairman J.W., Racca J., Dealwis C. Structures of eukaryotic ribonucleotide reductase I provide insights into dNTP regulation. Proc. Natl. Acad. Sci. USA. 2006;103:4022–4027. doi: 10.1073/pnas.0600443103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rode W., Jarmuńa A. [Thymidylate synthase-catalyzed reaction mechanism] Postepy. Biochem. 2015;61:274–283. [PubMed] [Google Scholar]
- 39.Ahmad M.F., Kaushal P.S., Wan Q., Wijerathna S.R., An X., Huang M., Dealwis C.G. Role of arginine 293 and glutamine 288 in communication between catalytic and allosteric sites in yeast ribonucleotide reductase. J. Mol. Biol. 2012;419:315–329. doi: 10.1016/j.jmb.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Minnihan E.C., Nocera D.G., Stubbe J. Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc. Chem. Res. 2013;46:2524–2535. doi: 10.1021/ar4000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stubbe J., Riggs-Gelasco P. Harnessing free radicals: Formation and function of the tyrosyl radical in ribonucleotide reductase. Trends Biochem. Sci. 1998;23:438–443. doi: 10.1016/S0968-0004(98)01296-1. [DOI] [PubMed] [Google Scholar]
- 42.Mao S.S., Holler T.P., Yu G.X., Bollinger J.M., Jr., Booker S., Johnston M.I., Stubbe J. A model for the role of multiple cysteine residues involved in ribonucleotide reduction: Amazing and still confusing. Biochemistry. 1992;31:9733–9743. doi: 10.1021/bi00155a029. [DOI] [PubMed] [Google Scholar]
- 43.Licht S., Gerfen G.J., Stubbe J. Thiyl radicals in ribonucleotide reductases. Science. 1996;271:477–481. doi: 10.1126/science.271.5248.477. [DOI] [PubMed] [Google Scholar]
- 44.Reece S.Y., Hodgkiss J.M., Stubbe J., Nocera D.G. Proton-coupled electron transfer: The mechanistic underpinning for radical transport and catalysis in biology. Philos. Trans. R. Soc. B Biol. Sci. 2006;361:1351–1364. doi: 10.1098/rstb.2006.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stubbe J., Nocera D.G., Yee C.S., Chang M.C. Radical initiation in the class I ribonucleotide reductase: Long-range proton-coupled electron transfer? Chem. Rev. 2003;103:2167–2201. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 46.Stubbe J. Ribonucleotide reductases: Amazing and confusing. J. Biol. Chem. 1990;265:5329–5332. doi: 10.1016/S0021-9258(19)39357-3. [DOI] [PubMed] [Google Scholar]
- 47.Lu J., Holmgren A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 48.Arnér E.S., Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000;267:6102–6109. doi: 10.1046/j.1432-1327.2000.01701.x. [DOI] [PubMed] [Google Scholar]
- 49.Elford H.L., Freese M., Passamani E., Morris H.P. Ribonucleotide reductase and cell proliferation. I. Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas. J. Biol. Chem. 1970;245:5228–5233. doi: 10.1016/S0021-9258(18)62745-0. [DOI] [PubMed] [Google Scholar]
- 50.Morikawa T., Maeda D., Kume H., Homma Y., Fukayama M. Ribonucleotide reductase M2 subunit is a novel diagnostic marker and a potential therapeutic target in bladder cancer. Histopathology. 2010;57:885–892. doi: 10.1111/j.1365-2559.2010.03725.x. [DOI] [PubMed] [Google Scholar]
- 51.Wang L.M., Lu F.F., Zhang S.Y., Yao R.Y., Xing X.M., Wei Z.M. Overexpression of catalytic subunit M2 in patients with ovarian cancer. Chin. Med. J. 2012;125:2151–2156. [PubMed] [Google Scholar]
- 52.Morikawa T., Hino R., Uozaki H., Maeda D., Ushiku T., Shinozaki A., Sakatani T., Fukayama M. Expression of ribonucleotide reductase M2 subunit in gastric cancer and effects of RRM2 inhibition in vitro. Hum. Pathol. 2010;41:1742–1748. doi: 10.1016/j.humpath.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 53.Lu A.G., Feng H., Wang P.X., Han D.P., Chen X.H., Zheng M.H. Emerging roles of the ribonucleotide reductase M2 in colorectal cancer and ultraviolet-induced DNA damage repair. World J. Gastroenterol. 2012;18:4704–4713. doi: 10.3748/wjg.v18.i34.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu X., Zhang H., Lai L., Wang X., Loera S., Xue L., He H., Zhang K., Hu S., Huang Y., et al. Ribonucleotide reductase small subunit M2 serves as a prognostic biomarker and predicts poor survival of colorectal cancers. Clin. Sci. 2013;124:567–578. doi: 10.1042/CS20120240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma X.J., Salunga R., Tuggle J.T., Gaudet J., Enright E., McQuary P., Payette T., Pistone M., Stecker K., Zhang B.M., et al. Gene expression profiles of human breast cancer progression. Proc. Natl. Acad. Sci. USA. 2003;100:5974–5979. doi: 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aird K.M., Zhang G., Li H., Tu Z., Bitler B.G., Garipov A., Wu H., Wei Z., Wagner S.N., Herlyn M., et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013;3:1252–1265. doi: 10.1016/j.celrep.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aird K.M., Li H., Xin F., Konstantinopoulos P.A., Zhang R. Identification of ribonucleotide reductase M2 as a potential target for pro-senescence therapy in epithelial ovarian cancer. Cell Cycle. 2014;13:199–207. doi: 10.4161/cc.26953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uramoto H., Sugio K., Oyama T., Hanagiri T., Yasumoto K. P53R2, p53 inducible ribonucleotide reductase gene, correlated with tumor progression of non-small cell lung cancer. AntiCancer Res. 2006;26:983–988. [PubMed] [Google Scholar]
- 59.Duxbury M.S., Ito H., Zinner M.J., Ashley S.W., Whang E.E. RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine. Oncogene. 2004;23:1539–1548. doi: 10.1038/sj.onc.1207272. [DOI] [PubMed] [Google Scholar]
- 60.Zhang K., Hu S., Wu J., Chen L., Lu J., Wang X., Liu X., Zhou B., Yen Y. Overexpression of RRM2 decreases thrombspondin-1 and increases VEGF production in human cancer cells in vitro and in vivo: Implication of RRM2 in angiogenesis. Mol. Cancer. 2009;8:11. doi: 10.1186/1476-4598-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gong W., Zhang X., Wu J., Chen L., Li L., Sun J., Lv Y., Wei X., Du Y., Jin H., et al. RRM1 expression and clinical outcome of gemcitabine-containing chemotherapy for advanced non-small-cell lung cancer: A meta-analysis. Lung Cancer. 2012;75:374–380. doi: 10.1016/j.lungcan.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 62.Nakahira S., Nakamori S., Tsujie M., Takahashi Y., Okami J., Yoshioka S., Yamasaki M., Marubashi S., Takemasa I., Miyamoto A., et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int. J. Cancer. 2007;120:1355–1363. doi: 10.1002/ijc.22390. [DOI] [PubMed] [Google Scholar]
- 63.Ohtaka K., Kohya N., Sato K., Kitajima Y., Ide T., Mitsuno M., Miyazaki K. Ribonucleotide reductase subunit M1 is a possible chemoresistance marker to gemcitabine in biliary tract carcinoma. Oncol. Rep. 2008;20:279–286. [PubMed] [Google Scholar]
- 64.Reynolds C., Obasaju C., Schell M.J., Li X., Zheng Z., Boulware D., Caton J.R., Demarco L.C., O’Rourke M.A., Shaw Wright G., et al. Randomized phase III trial of gemcitabine-based chemotherapy with in situ RRM1 and ERCC1 protein levels for response prediction in non-small-cell lung cancer. J. Clin. Oncol. 2009;27:5808–5815. doi: 10.1200/JCO.2009.21.9766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Akita H., Zheng Z., Takeda Y., Kim C., Kittaka N., Kobayashi S., Marubashi S., Takemasa I., Nagano H., Dono K., et al. Significance of RRM1 and ERCC1 expression in resectable pancreatic adenocarcinoma. Oncogene. 2009;28:2903–2909. doi: 10.1038/onc.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rodriguez J., Boni V., Hernandez A., Bitarte N., Zarate R., Ponz-Sarvise M., Chopitea A., Bandres E., Garcia-Foncillas J. Association of RRM1 -37A>C polymorphism with clinical outcome in colorectal cancer patients treated with gemcitabine-based chemotherapy. Eur. J. Cancer. 2011;47:839–847. doi: 10.1016/j.ejca.2010.11.032. [DOI] [PubMed] [Google Scholar]
- 67.Jordheim L.P., Seve P., Tredan O., Dumontet C. The ribonucleotide reductase large subunit (RRM1) as a predictive factor in patients with cancer. Lancet Oncol. 2011;12:693–702. doi: 10.1016/S1470-2045(10)70244-8. [DOI] [PubMed] [Google Scholar]
- 68.Ceppi P., Volante M., Novello S., Rapa I., Danenberg K.D., Danenberg P.V., Cambieri A., Selvaggi G., Saviozzi S., Calogero R., et al. ERCC1 and RRM1 gene expressions but not EGFR are predictive of shorter survival in advanced non-small-cell lung cancer treated with cisplatin and gemcitabine. Ann. Oncol. 2006;17:1818–1825. doi: 10.1093/annonc/mdl300. [DOI] [PubMed] [Google Scholar]
- 69.Dong X., Hao Y., Wei Y., Yin Q., Du J., Zhao X. Response to first-line chemotherapy in patients with non-small cell lung cancer according to RRM1 expression. PLoS ONE. 2014;9:e92320. doi: 10.1371/journal.pone.0092320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bepler G., Sommers K.E., Cantor A., Li X., Sharma A., Williams C., Chiappori A., Haura E., Antonia S., Tanvetyanon T., et al. Clinical efficacy and predictive Mol.ecular markers of neoadjuvant gemcitabine and pemetrexed in resectable non-small cell lung cancer. J. Thorac. Oncol. 2008;3:1112–1118. doi: 10.1097/JTO.0b013e3181874936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jordheim L.P., Guittet O., Lepoivre M., Galmarini C.M., Dumontet C. Increased expression of the large subunit of ribonucleotide reductase is involved in resistance to gemcitabine in human mammary adenocarcinoma cells. Mol. Cancer Ther. 2005;4:1268–1276. doi: 10.1158/1535-7163.MCT-05-0121. [DOI] [PubMed] [Google Scholar]
- 72.Davidson J.D., Ma L., Flagella M., Geeganage S., Gelbert L.M., Slapak C.A. An increase in the expression of ribonucleotide reductase large subunit 1 is associated with gemcitabine resistance in non-small cell lung cancer cell lines. Cancer Res. 2004;64:3761–3766. doi: 10.1158/0008-5472.CAN-03-3363. [DOI] [PubMed] [Google Scholar]
- 73.Bergman A.M., Eijk P.P., Ruiz van Haperen V.W., Smid K., Veerman G., Hubeek I., van den Ijssel P., Ylstra B., Peters G.J. In vivo induction of resistance to gemcitabine results in increased expression of ribonucleotide reductase subunit M1 as the major determinant. Cancer Res. 2005;65:9510–9516. doi: 10.1158/0008-5472.CAN-05-0989. [DOI] [PubMed] [Google Scholar]
- 74.Bepler G., Kusmartseva I., Sharma S., Gautam A., Cantor A., Sharma A., Simon G. RRM1 modulated in vitro and in vivo efficacy of gemcitabine and platinum in non-small-cell lung cancer. J. Clin. Oncol. 2006;24:4731–4737. doi: 10.1200/JCO.2006.06.1101. [DOI] [PubMed] [Google Scholar]
- 75.Ferrandina G., Mey V., Nannizzi S., Ricciardi S., Petrillo M., Ferlini C., Danesi R., Scambia G., Del Tacca M. Expression of nucleoside transporters, deoxycitidine kinase, ribonucleotide reductase regulatory subunits, and gemcitabine catabolic enzymes in primary ovarian cancer. Cancer Chemother. Pharmacol. 2010;65:679–686. doi: 10.1007/s00280-009-1073-y. [DOI] [PubMed] [Google Scholar]
- 76.Itoi T., Sofuni A., Fukushima N., Itokawa F., Tsuchiya T., Kurihara T., Moriyasu F., Tsuchida A., Kasuya K. Ribonucleotide reductase subunit M2 mRNA expression in pretreatment biopsies obtained from unresectable pancreatic carcinomas. J. Gastroenterol. 2007;42:389–394. doi: 10.1007/s00535-007-2017-0. [DOI] [PubMed] [Google Scholar]
- 77.Avolio T.M., Lee Y., Feng N., Xiong K., Jin H., Wang M., Vassilakos A., Wright J., Young A. RNA interference targeting the R2 subunit of ribonucleotide reductase inhibits growth of tumor cells in vitro and in vivo. Anticancer Drugs. 2007;18:377–388. doi: 10.1097/CAD.0b013e328013c04f. [DOI] [PubMed] [Google Scholar]
- 78.Xu X., Page J.L., Surtees J.A., Liu H., Lagedrost S., Lu Y., Bronson R., Alani E., Nikitin A.Y., Weiss R.S. Broad overexpression of ribonucleotide reductase genes in mice specifically induces lung neoplasms. Cancer Res. 2008;68:2652–2660. doi: 10.1158/0008-5472.CAN-07-5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martin K.R., Barrett J.C. Reactive oxygen species as double-edged swords in cellular processes: Low-dose cell signaling versus high-dose toxicity. Hum. Exp. Toxicol. 2002;21:71–75. doi: 10.1191/0960327102ht213oa. [DOI] [PubMed] [Google Scholar]
- 80.Feig D.I., Reid T.M., Loeb L.A. Reactive oxygen species in tumorigenesis. Cancer Res. 1994;54:1890s–1894s. [PubMed] [Google Scholar]
- 81.Lee H.S., Lee N.C., Kouprina N., Kim J.H., Kagansky A., Bates S., Trepel J.B., Pommier Y., Sackett D., Larionov V. Effects of Anticancer Drugs on Chromosome Instability and New Clinical Implications for Tumor-Suppressing Therapies. Cancer Res. 2016;76:902–911. doi: 10.1158/0008-5472.CAN-15-1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pai C.C., Kearsey S.E. A Critical Balance: dNTPs and the Maintenance of Genome Stability. Genes. 2017;8:57. doi: 10.3390/genes8020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aye Y., Stubbe J. Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc. Natl. Acad. Sci. USA. 2011;108:9815–9820. doi: 10.1073/pnas.1013274108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Griffig J., Koob R., Blakley R.L. Mechanisms of inhibition of DNA synthesis by 2-chlorodeoxyadenosine in human lymphoblastic cells. Cancer Res. 1989;49:6923–6928. [PubMed] [Google Scholar]
- 85.Gandhi V., Plunkett W. Modulatory activity of 2′,2′-difluorodeoxycytidine on the phosphorylation and cytotoxicity of arabinosyl nucleosides. Cancer Res. 1990;50:3675–3680. [PubMed] [Google Scholar]
- 86.Stubbe J., van der Donk W.A. Ribonucleotide reductases: Radical enzymes with suicidal tendencies. Chem. Biol. 1995;2:793–801. doi: 10.1016/1074-5521(95)90084-5. [DOI] [PubMed] [Google Scholar]
- 87.Plunkett W., Huang P., Gandhi V. Preclinical characteristics of gemcitabine. Anticancer Drugs. 1995;6((Suppl. 6)):7–13. doi: 10.1097/00001813-199512006-00002. [DOI] [PubMed] [Google Scholar]
- 88.Lassmann G., Thelander L., Graslund A. EPR stopped-flow studies of the reaction of the tyrosyl radical of protein R2 from ribonucleotide reductase with hydroxyurea. Biochem. Biophys. Res. Commun. 1992;188:879–887. doi: 10.1016/0006-291X(92)91138-G. [DOI] [PubMed] [Google Scholar]
- 89.Nigovic B., Kujundzic N., Sankovic K. Electron transfer in N-hydroxyurea complexes with iron(III) Eur. J. Med. Chem. 2005;40:51–55. doi: 10.1016/j.ejmech.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 90.Agrawal K.C., Sartorelli A.C. The chemistry and biological activity of alpha-(N)-heterocyclic carboxaldehyde thiosemicarbazones. Prog. Med. Chem. 1978;15:321–356. doi: 10.1016/s0079-6468(08)70259-5. [DOI] [PubMed] [Google Scholar]
- 91.Cory J.G., Cory A.H., Rappa G., Lorico A., Liu M.C., Lin T.S., Sartorelli A.C. Structure-function relationships for a new series of pyridine-2-carboxaldehyde thiosemicarbazones on ribonucleotide reductase activity and tumor cell growth in culture and in vivo. Adv. Enzyme Regul. 1995;35:55–68. doi: 10.1016/0065-2571(94)00005-N. [DOI] [PubMed] [Google Scholar]
- 92.Cory J.G., Cory A.H., Rappa G., Lorico A., Liu M.C., Lin T.S., Sartorelli A.C. Inhibitors of ribonucleotide reductase. Comparative effects of amino- and hydroxy-substituted pyridine-2-carboxaldehyde thiosemicarbazones. Biochem. Pharmacol. 1994;48:335–344. doi: 10.1016/0006-2952(94)90105-8. [DOI] [PubMed] [Google Scholar]
- 93.Fritscher J., Artin E., Wnuk S., Bar G., Robblee J.H., Kacprzak S., Kaupp M., Griffin R.G., Bennati M., Stubbe J. Structure of the nitrogen-centered radical formed during inactivation of E. coli ribonucleotide reductase by 2′-azido-2′-deoxyuridine-5′-diphosphate: Trapping of the 3′-ketonucleotide. J. Am. Chem. Soc. 2005;127:7729–7738. doi: 10.1021/ja043111x. [DOI] [PubMed] [Google Scholar]
- 94.Keating M.J., Estey E., O’Brien S., Kantarjian H., Robertson L.E., Plunkett W. Clinical experience with fludarabine in leukaemia. Drugs. 1994;47((Suppl. 6)):39–49. doi: 10.2165/00003495-199400476-00007. [DOI] [PubMed] [Google Scholar]
- 95.Keating M.J., O’Brien S., Plunkett W., Robertson L.E., Gandhi V., Estey E., Dimopoulos M., Cabanillas F., Kemena A., Kantarjian H. Fludarabine phosphate: A new active agent in hematologic malignancies. Semin. Hematol. 1994;31:28–39. [PubMed] [Google Scholar]
- 96.Wright S.J., Robertson L.E., O’Brien S., Plunkett W., Keating M.J. The role of fludarabine in hematological malignancies. Blood Rev. 1994;8:125–134. doi: 10.1016/0268-960X(94)90072-P. [DOI] [PubMed] [Google Scholar]
- 97.Montefusco E., Fazi F., Cordone I., Ariola C., Nanni M., Spadea A., Spiriti M.A., Fenu S., Mandelli F., Petti M.C. Mol.ecular remission following high-dose hydroxyurea and fludarabine plus cytarabine in a patient with simultaneous acute myeloid leukemia and low-grade lymphoma. Leuk. Lymphoma. 2001;40:671–674. doi: 10.3109/10428190109097666. [DOI] [PubMed] [Google Scholar]
- 98.Huang P., Chubb S., Plunkett W. Termination of DNA synthesis by 9-beta-D-arabinofuranosyl-2-fluoroadenine. A mechanism for cytotoxicity. J. Biol. Chem. 1990;265:16617–16625. doi: 10.1016/S0021-9258(17)46267-3. [DOI] [PubMed] [Google Scholar]
- 99.Yang S.W., Huang P., Plunkett W., Becker F.F., Chan J.Y. Dual mode of inhibition of purified DNA ligase I from human cells by 9-beta-D-arabinofuranosyl-2-fluoroadenine triphosphate. J. Biol. Chem. 1992;267:2345–2349. doi: 10.1016/S0021-9258(18)45884-X. [DOI] [PubMed] [Google Scholar]
- 100.Gandhi V., Huang P., Plunkett W. Fludarabine inhibits DNA replication: A rationale for its use in the treatment of acute leukemias. Leuk. Lymphoma. 1994;14((Suppl. 2)):3–9. doi: 10.3109/10428199409052689. [DOI] [PubMed] [Google Scholar]
- 101.Chihara D., Arons E., Stetler-Stevenson M., Yuan C.M., Wang H.W., Zhou H., Raffeld M., Xi L., Steinberg S.M., Feurtado J., et al. Randomized Phase II Study of First-Line Cladribine With Concurrent or Delayed Rituximab in Patients With Hairy Cell Leukemia. J. Clin. Oncol. 2020;38:1527–1538. doi: 10.1200/JCO.19.02250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Qasrawi A., Bahaj W., Qasrawi L., Abughanimeh O., Foxworth J., Gaur R. Cladribine in the remission induction of adult acute myeloid leukemia: Where do we stand? Ann. Hematol. 2019;98:561–579. doi: 10.1007/s00277-018-3562-8. [DOI] [PubMed] [Google Scholar]
- 103.Fidias P., Chabner B.A., Grossbard M.L. Purine Analogs for the Treatment of Low-Grade Lymphoproliferative Disorders. Oncologist. 1996;1:125–139. doi: 10.1634/theoncologist.1-3-125. [DOI] [PubMed] [Google Scholar]
- 104.Benjamin R.C., Gill D.M. Poly(ADP-ribose) synthesis in vitro programmed by damaged DNA. A comparison of DNA Mol.ecules containing different types of strand breaks. J. Biol. Chem. 1980;255:10502–10508. doi: 10.1016/S0021-9258(19)70491-8. [DOI] [PubMed] [Google Scholar]
- 105.Wisitpitthaya S., Zhao Y., Long M.J., Li M., Fletcher E.A., Blessing W.A., Weiss R.S., Aye Y. Cladribine and Fludarabine Nucleotides Induce Distinct Hexamers Defining a Common Mode of Reversible RNR Inhibition. ACS Chem. Biol. 2016;11:2021–2032. doi: 10.1021/acschembio.6b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Faderl S., Gandhi V., Keating M.J., Jeha S., Plunkett W., Kantarjian H.M. The role of clofarabine in hematologic and solid malignancies--development of a next-generation nucleoside analog. Cancer. 2005;103:1985–1995. doi: 10.1002/cncr.21005. [DOI] [PubMed] [Google Scholar]
- 107.Pui C.H., Jeha S. Clofarabine. Nat. Rev. Drug Discov. 2005;4:369–370. doi: 10.1038/nrd1724. [DOI] [PubMed] [Google Scholar]
- 108.Kantarjian H.M., Jeha S., Gandhi V., Wess M., Faderl S. Clofarabine: Past, present, and future. Leuk. Lymphoma. 2007;48:1922–1930. doi: 10.1080/10428190701545644. [DOI] [PubMed] [Google Scholar]
- 109.Salzer W.L., Burke M.J., Devidas M., Chen S., Gore L., Larsen E.C., Borowitz M., Wood B., Heerema N.A., Carroll A.J., et al. Toxicity associated with intensive postinduction therapy incorporating clofarabine in the very high-risk stratum of patients with newly diagnosed high-risk B-lymphoblastic leukemia: A report from the Children’s Oncology Group study AALL1131. Cancer. 2018;124:1150–1159. doi: 10.1002/cncr.31099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Aye Y., Brignole E.J., Long M.J., Chittuluru J., Drennan C.L., Asturias F.J., Stubbe J. Clofarabine targets the large subunit (α) of human ribonucleotide reductase in live cells by assembly into persistent hexamers. Chem. Biol. 2012;19:799–805. doi: 10.1016/j.chembiol.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Genini D., Adachi S., Chao Q., Rose D.W., Carrera C.J., Cottam H.B., Carson D.A., Leoni L.M. Deoxyadenosine analogs induce programmed cell death in chronic lymphocytic leukemia cells by damaging the DNA and by directly affecting the mitochondria. Blood. 2000;96:3537–3543. doi: 10.1182/blood.V96.10.3537. [DOI] [PubMed] [Google Scholar]
- 112.Yamauchi T., Nowak B.J., Keating M.J., Plunkett W. DNA repair initiated in chronic lymphocytic leukemia lymphocytes by 4-hydroperoxycyclophosphamide is inhibited by fludarabine and clofarabine. Clin. Cancer Res. 2001;7:3580–3589. [PubMed] [Google Scholar]
- 113.Storniolo A.M., Enas N.H., Brown C.A., Voi M., Rothenberg M.L., Schilsky R. An investigational new drug treatment program for patients with gemcitabine: Results for over 3000 patients with pancreatic carcinoma. Cancer. 1999;85:1261–1268. doi: 10.1002/(SICI)1097-0142(19990315)85:6<1261::AID-CNCR7>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 114.Vallo S., Michaelis M., Rothweiler F., Bartsch G., Gust K.M., Limbart D.M., Rodel F., Wezel F., Haferkamp A., Cinatl J., Jr. Drug-Resistant Urothelial Cancer Cell Lines Display Diverse Sensitivity Profiles to Potential Second-Line Therapeutics. Transl. Oncol. 2015;8:210–216. doi: 10.1016/j.tranon.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scagliotti G.V., Parikh P., von Pawel J., Biesma B., Vansteenkiste J., Manegold C., Serwatowski P., Gatzemeier U., Digumarti R., Zukin M., et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 2008;26:3543–3551. doi: 10.1200/JCO.2007.15.0375. [DOI] [PubMed] [Google Scholar]
- 116.Pfisterer J., Vergote I., Du Bois A., Eisenhauer E., Ago O., Ncic C.T.G., Eortc G.C.G. Combination therapy with gemcitabine and carboplatin in recurrent ovarian cancer. Int. J. Gynecol. Cancer. 2005;15((Suppl. 1)):36–41. doi: 10.1136/ijgc-00009577-200505001-00007. [DOI] [PubMed] [Google Scholar]
- 117.Roy V., LaPlant B.R., Gross G.G., Bane C.L., Palmieri F.M., North Central Cancer Treatment G. Phase II trial of weekly nab (nanoparticle albumin-bound)-paclitaxel (nab-paclitaxel) (Abraxane) in combination with gemcitabine in patients with metastatic breast cancer (N0531) Ann. Oncol. 2009;20:449–453. doi: 10.1093/annonc/mdn661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xu H., Faber C., Uchiki T., Racca J., Dealwis C. Structures of eukaryotic ribonucleotide reductase I define gemcitabine diphosphate binding and subunit assembly. Proc. Natl. Acad. Sci. USA. 2006;103:4028–4033. doi: 10.1073/pnas.0600440103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang J., Lohman G.J., Stubbe J. Enhanced subunit interactions with gemcitabine-5′-diphosphate inhibit ribonucleotide reductases. Proc. Natl. Acad. Sci. USA. 2007;104:14324–14329. doi: 10.1073/pnas.0706803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lohman G.J., Gerfen G.J., Stubbe J. Inactivation of Lactobacillus leichmannii ribonucleotide reductase by 2′,2′-difluoro-2′-deoxycytidine 5′-triphosphate: Adenosylcobalamin destruction and formation of a nucleotide-based radical. Biochemistry. 2010;49:1396–1403. doi: 10.1021/bi9021318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ono H., Basson M.D., Ito H. P300 inhibition enhances gemcitabine-induced apoptosis of pancreatic cancer. Oncotarget. 2016;7:51301–51310. doi: 10.18632/oncotarget.10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Plunkett W., Huang P., Searcy C.E., Gandhi V. Gemcitabine: Preclinical pharmacology and mechanisms of action. Semin. Oncol. 1996;23:3–15. [PubMed] [Google Scholar]
- 123.Fryer R.A., Barlett B., Galustian C., Dalgleish A.G. Mechanisms underlying gemcitabine resistance in pancreatic cancer and sensitisation by the iMiD lenalidomide. Anticancer Res. 2011;31:3747–3756. [PubMed] [Google Scholar]
- 124.Kim M.P., Gallick G.E. Gemcitabine resistance in pancreatic cancer: Picking the key players. Clin. Cancer Res. 2008;14:1284–1285. doi: 10.1158/1078-0432.CCR-07-2247. [DOI] [PubMed] [Google Scholar]
- 125.Binenbaum Y., Na’ara S., Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updates. 2015;23:55–68. doi: 10.1016/j.drup.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 126.Wei L., Wen J.Y., Chen J., Ma X.K., Wu D.H., Chen Z.H., Huang J.L. Oncogenic ADAM28 induces gemcitabine resistance and predicts a poor prognosis in pancreatic cancer. World J. Gastroenterol. 2019;25:5590–5603. doi: 10.3748/wjg.v25.i37.5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yang G., Guan W., Cao Z., Guo W., Xiong G., Zhao F., Feng M., Qiu J., Liu Y., Zhang M.Q., et al. Integrative Genomic Analysis of Gemcitabine Resistance in Pancreatic Cancer by Patient-derived Xenograft Models. Clin. Cancer Res. 2021;27:3383–3396. doi: 10.1158/1078-0432.CCR-19-3975. [DOI] [PubMed] [Google Scholar]
- 128.Palam L.R., Gore J., Craven K.E., Wilson J.L., Korc M. Integrated stress response is critical for gemcitabine resistance in pancreatic ductal adenocarcinoma. Cell Death Dis. 2015;6:e1913. doi: 10.1038/cddis.2015.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Morimoto M., Matsuo Y., Koide S., Tsuboi K., Shamoto T., Sato T., Saito K., Takahashi H., Takeyama H. Enhancement of the CXCL12/CXCR4 axis due to acquisition of gemcitabine resistance in pancreatic cancer: Effect of CXCR4 antagonists. BMC Cancer. 2016;16:305. doi: 10.1186/s12885-016-2340-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Long J., Zhang Y., Yu X., Yang J., LeBrun D.G., Chen C., Yao Q., Li M. Overcoming drug resistance in pancreatic cancer. Expert Opin. Ther. Targets. 2011;15:817–828. doi: 10.1517/14728222.2011.566216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Gu J., Huang W., Wang X., Zhang J., Tao T., Zheng Y., Liu S., Yang J., Chen Z.S., Cai C.Y., et al. Hsa-miR-3178/RhoB/PI3K/Akt, a novel signaling pathway regulates ABC transporters to reverse gemcitabine resistance in pancreatic cancer. Mol. Cancer. 2022;21:112. doi: 10.1186/s12943-022-01587-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nimmakayala R.K., Leon F., Rachagani S., Rauth S., Nallasamy P., Marimuthu S., Shailendra G.K., Chhonker Y.S., Chugh S., Chirravuri R., et al. Metabolic programming of distinct cancer stem cells promotes metastasis of pancreatic ductal adenocarcinoma. Oncogene. 2021;40:215–231. doi: 10.1038/s41388-020-01518-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Qiu F., Chen J., Cao J., Diao F., Huang P. Low-intensity low-frequency ultrasound enhances the chemosensitivity of gemcitabine-resistant ASPC-1 cells via PI3K/AKT/NF-κB pathway-mediated ABC transporters. Oncol. Rep. 2020;44:1158–1168. doi: 10.3892/or.2020.7671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Okada Y., Takahashi N., Takayama T., Goel A. LAMC2 promotes cancer progression and gemcitabine resistance through modulation of EMT and ATP-binding cassette transporters in pancreatic ductal adenocarcinoma. Carcinogenesis. 2021;42:546–556. doi: 10.1093/carcin/bgab011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yang J., Sontag D., Gong Y., Minuk G.Y. Enhanced gemcitabine cytotoxicity with knockdown of multidrug resistance protein genes in human cholangiocarcinoma cell lines. J. Gastroenterol. Hepatol. 2021;36:1103–1109. doi: 10.1111/jgh.15289. [DOI] [PubMed] [Google Scholar]
- 136.Conroy T., Desseigne F., Ychou M., Bouche O., Guimbaud R., Becouarn Y., Adenis A., Raoul J.L., Gourgou-Bourgade S., de la Fouchardiere C., et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 137.Conroy T., Hammel P., Hebbar M., Ben Abdelghani M., Wei A.C., Raoul J.L., Chone L., Francois E., Artru P., Biagi J.J., et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018;379:2395–2406. doi: 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 138.Von Hoff D.D., Ervin T., Arena F.P., Chiorean E.G., Infante J., Moore M., Seay T., Tjulandin S.A., Ma W.W., Saleh M.N., et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hidalgo M. Pancreatic cancer. N. Engl. J. Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 140.Springfeld C., Jäger D., Büchler M.W., Strobel O., Hackert T., Palmer D.H., Neoptolemos J.P. Chemotherapy for pancreatic cancer. La Presse Med. 2019;48:e159–e174. doi: 10.1016/j.lpm.2019.02.025. [DOI] [PubMed] [Google Scholar]
- 141.Bao K., Li X., He X., Jian L. Pharmacoeconomic Evaluation of Erlotinib for the Treatment of Pancreatic Cancer. Clin. Ther. 2021;43:1107–1115. doi: 10.1016/j.clinthera.2021.04.012. [DOI] [PubMed] [Google Scholar]
- 142.Moore M.J., Goldstein D., Hamm J., Figer A., Hecht J.R., Gallinger S., Au H.J., Murawa P., Walde D., Wolff R.A., et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 143.Hammel P., Huguet F., van Laethem J.L., Goldstein D., Glimelius B., Artru P., Borbath I., Bouché O., Shannon J., André T., et al. Effect of Chemoradiotherapy vs Chemotherapy on Survival in Patients With Locally Advanced Pancreatic Cancer Controlled After 4 Months of Gemcitabine with or without Erlotinib: The LAP07 Randomized Clinical Trial. JAMA. 2016;315:1844–1853. doi: 10.1001/jama.2016.4324. [DOI] [PubMed] [Google Scholar]
- 144.Raymond E., Faivre S., Armand J.P. Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs. 2000;60((Suppl. 1)):15–23. doi: 10.2165/00003495-200060001-00002. discussion 41-12. [DOI] [PubMed] [Google Scholar]
- 145.Takahashi T., Nakashima A., Kanazawa J., Yamaguchi K., Akinaga S., Tamaoki T., Okabe M. Metabolism and ribonucleotide reductase inhibition of (E)-2′-deoxy-2′-(fluoromethylene)cytidine, MDL 101,731, in human cervical carcinoma HeLa S3 cells. Cancer Chemother. Pharmacol. 1998;41:268–274. doi: 10.1007/s002800050739. [DOI] [PubMed] [Google Scholar]
- 146.Skierski J.S., Koronkiewicz M., Grieb P. Effect of FMdC on the cell cycle of some leukemia cell lines. Cytometry. 1999;37:302–307. doi: 10.1002/(SICI)1097-0320(19991201)37:4<302::AID-CYTO7>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 147.Flaherty K.T., Stevenson J.P., Gallagher M., Giantonio B., Algazy K.M., Sun W., Haller D.G., O’Dwyer P.J. Dose escalation study of tezacitabine in combination with cisplatin in patients with advanced cancer. Cancer. 2003;97:1985–1990. doi: 10.1002/cncr.11273. [DOI] [PubMed] [Google Scholar]
- 148.Fernandes P.A., Ramos M.J. Theoretical studies on the mode of inhibition of ribonucleotide reductase by 2′-substituted substrate analogues. Chemistry. 2003;9:5916–5925. doi: 10.1002/chem.200304948. [DOI] [PubMed] [Google Scholar]
- 149.Bitonti A.J., Bush T.L., Lewis M.T., Sunkara P.S. Response of human colon and prostate tumor xenografts to (E)-2′-deoxy-2′-(fluoromethylene) cytidine, an inhibitor of ribonucleotide reductase. Anticancer Res. 1995;15:1179–1182. [PubMed] [Google Scholar]
- 150.Masuda N., Negoro S., Takeda K., Takifuji N., Hirashima T., Yana T., Kurata N., Kuwabara T., Kobayashi S., Kudoh S., et al. Phase I and pharmacologic study of oral (E)-2′-deoxy-2′-(fluoromethylene) cytidine: On a daily x 5-day schedule. Investig. New Drugs. 1998;16:245–254. doi: 10.1023/A:1006126212481. [DOI] [PubMed] [Google Scholar]
- 151.Seley K.L. Tezacitabine Hoechst Marion Roussel. Curr. Opin. Investig. Drugs. 2000;1:135–140. [PubMed] [Google Scholar]
- 152.Kanazawa J., Takahashi T., Akinaga S., Tamaoki T., Okabe M. The relationship between the antitumor activity and the ribonucleotide reductase inhibitory activity of (E)-2′-deoxy-2′-(fluoromethylene) cytidine, MDL 101,731. Anticancer Drugs. 1998;9:653–657. doi: 10.1097/00001813-199808000-00011. [DOI] [PubMed] [Google Scholar]
- 153.Zhou Y., Achanta G., Pelicano H., Gandhi V., Plunkett W., Huang P. Action of (E)-2′-deoxy-2′-(fluoromethylene)cytidine on DNA metabolism: Incorporation, excision, and cellular response. Mol. Pharmacol. 2002;61:222–229. doi: 10.1124/mol.61.1.222. [DOI] [PubMed] [Google Scholar]
- 154.Burtness B., Belker M., Stoltz M., Peccerillo K.M., Lamb L.A., Chmael S.E., McKeon A., Clark M.B., Winship J., Marsh J.C., et al. A phase I study of the antimetabolite (E)-2′-fluoromethylene-2′-deoxycytidine (MDL 101,731) administered as a twice-weekly infusion. Cancer J. 2000;6:309–315. [PubMed] [Google Scholar]
- 155.Rodriguez G.I., Jones R.E., Orenberg E.K., Stoltz M.L., Brooks D.J. Phase I clinical trials of tezacitabine [(E)-2′-deoxy-2′-(fluoromethylene)cytidine] in patients with refractory solid tumors. Clin. Cancer Res. 2002;8:2828–2834. [PubMed] [Google Scholar]
- 156.Bendell J.C., Eder J.P., Clark J.W., Fidias P., Lynch T.J., Seiden M.V., Ryan D.P. Phase I dose-escalation study of tezacitabine in combination with 5-fluorouracil in patients with advanced solid tumors. Cancer. 2005;103:1925–1931. doi: 10.1002/cncr.21002. [DOI] [PubMed] [Google Scholar]
- 157.Takenuki K., Matsuda A., Ueda T., Sasaki T., Fujii A., Yamagami K. Design, synthesis, and antineoplastic activity of 2′-deoxy-2′-methylidenecytidine. J. Med. Chem. 1988;31:1063–1064. doi: 10.1021/jm00401a001. [DOI] [PubMed] [Google Scholar]
- 158.Matsuda A., Takenuki K., Tanaka M., Sasaki T., Ueda T. Nucleosides and nucleotides. 97. Synthesis of new broad spectrum antineoplastic nucleosides, 2′-deoxy-2′-methylidenecytidine (DMDC) and its derivatives. J. Med. Chem. 1991;34:812–819. doi: 10.1021/jm00106a049. [DOI] [PubMed] [Google Scholar]
- 159.Miwa M., Eda H., Ura M., Ouchi K.F., Keith D.D., Foley L.H., Ishitsuka H. High susceptibility of human cancer xenografts with higher levels of cytidine deaminase to a 2′-deoxycytidine antimetabolite, 2′-deoxy-2′-methylidenecytidine. Clin. Cancer Res. 1998;4:493–497. [PubMed] [Google Scholar]
- 160.Brindley C.J., Morrison R., Gordon R.J., Devlin A.J., van der Gaast A., Verweij L., Funaki T. Clinical pharmacokinetics of 2′-deoxy-2′-methylidenecytidine (DMDC), a deoxycytidine analogue antineoplastic agent. Clin. Pharm. 2000;38:475–491. doi: 10.2165/00003088-200038060-00002. [DOI] [PubMed] [Google Scholar]
- 161.Eda H., Ura M., Kaori F.O., Tanaka Y., Miwa M., Ishitsuka H. The antiproliferative activity of DMDC is modulated by inhibition of cytidine deaminase. Cancer Res. 1998;58:1165–1169. [PubMed] [Google Scholar]
- 162.Gemma A., Kudoh S., Fukuoka M., Kurita Y., Hasegawa K., Harada M., Mori K., Ariyoshi Y., Kurihara M., Furuse K., et al. Phase I study on DMDC. Gan To Kagaku Ryoho. 1996;23:1799–1811. [PubMed] [Google Scholar]
- 163.Friberg L.E., Brindley C.J., Karlsson M.O., Devlin A.J. Models of schedule dependent haematological toxicity of 2′-deoxy-2′-methylidenecytidine (DMDC) Eur. J. Clin. Pharmacol. 2000;56:567–574. doi: 10.1007/s002280000181. [DOI] [PubMed] [Google Scholar]
- 164.Larsen I.K., Cornett C., Karlsson M., Sahlin M., Sjöberg B.M. Caracemide, a site-specific irreversible inhibitor of protein R1 of Escherichia coli ribonucleotide reductase. J. Biol. Chem. 1992;267:12627–12631. doi: 10.1016/S0021-9258(18)42323-X. [DOI] [PubMed] [Google Scholar]
- 165.Satyamoorthy K., Chitnis M.P., Advani S.H. In vitro cytotoxicity of caracemide alone and in combination with hydroxyurea or iron-chelating agents in human chronic myeloid leukemia cells and murine tumors. Neoplasma. 1988;35:27–35. [PubMed] [Google Scholar]
- 166.Newman R.A., Farquhar D., Lu K., Meyn R., Moore E.C., Massia S., Korp J.D., Wright J.A., McKinney M. Biochemical pharmacology of N-acetyl-N-(methylcarbamoyloxy)-N’-methylurea (caracemide; NSC-253272) Biochem. Pharmacol. 1986;35:2781–2787. doi: 10.1016/0006-2952(86)90190-5. [DOI] [PubMed] [Google Scholar]
- 167.Buccafusco J.J., Smith M.D. In vivo and in vitro cholinesterase inhibitor property of the antitumor agent caracemide. Res. Commun. Chem. Pathol. Pharmacol. 1990;67:219–227. [PubMed] [Google Scholar]
- 168.Pazdur R., Chabot G.G., Baker L.H. Phase I study and pharmacokinetics of caracemide (NSC-253272) administered as a short infusion. Investig. New Drugs. 1987;5:365–371. doi: 10.1007/BF00169976. [DOI] [PubMed] [Google Scholar]
- 169.Raber M.N., Adams F., Kavanagh J., Legha S., Dimery I., Krakoff I. Phase I trial of caracemide using bolus and infusion schedules. Cancer Treat. Rep. 1987;71:349–352. [PubMed] [Google Scholar]
- 170.Belani C.P., Eisenberger M., Van Echo D., Hiponia D., Aisner J. Phase II study of caracemide in advanced or recurrent non-small cell lung cancer. Cancer Treat. Rep. 1987;71:1099–1100. [PubMed] [Google Scholar]
- 171.Lad T., Schor J., Mullane M., Carroll R., Chernicoff D., Blough R., Weidner L. Phase II trial of caracemide (NSC 253272) in advanced unresectable non-small cell bronchogenic carcinoma. An Illinois Cancer Council study. Investig. New Drugs. 1992;10:27–28. doi: 10.1007/BF01275475. [DOI] [PubMed] [Google Scholar]
- 172.Witte R.S., Hsieh P., Elson P., Oken M.M., Trump D.L. A phase II trial of amonafide, caracemide, and homoharringtonine in the treatment of patients with advanced renal cell cancer. Investig. New Drugs. 1996;14:409–413. doi: 10.1007/BF00180819. [DOI] [PubMed] [Google Scholar]
- 173.Witte R.S., Lipsitz S., Goodman T.L., Asbury R.F., Wilding G., Strnad C.M., Smith T.J., Haller D.G. A phase II trial of homoharringtonine and caracemide in the treatment of patients with advanced large bowel cancer. Investig. New Drugs. 1999;17:173–177. doi: 10.1023/A:1006327418043. [DOI] [PubMed] [Google Scholar]
- 174.Donehower R.C. An overview of the clinical experience with hydroxyurea. Semin. Oncol. 1992;19:11–19. [PubMed] [Google Scholar]
- 175.Kennedy B.J. The evolution of hydroxyurea therapy in chronic myelogenous leukemia. Semin. Oncol. 1992;19:21–26. [PubMed] [Google Scholar]
- 176.Yarbro J.W. Mechanism of action of hydroxyurea. Semin. Oncol. 1992;19:1–10. [PubMed] [Google Scholar]
- 177.King S.B. Nitric oxide production from hydroxyurea. Free Radic. Biol. Med. 2004;37:737–744. doi: 10.1016/j.freeradbiomed.2004.02.073. [DOI] [PubMed] [Google Scholar]
- 178.Ho J.A., Pickens C.V., Gamcsik M.P., Colvin O.M., Ware R.E. In vitro induction of fetal hemoglobin in human erythroid progenitor cells. Exp. Hematol. 2003;31:586–591. doi: 10.1016/S0301-472X(03)00086-9. [DOI] [PubMed] [Google Scholar]
- 179.Baliga B.S., Pace B.S., Chen H.H., Shah A.K., Yang Y.M. Mechanism for fetal hemoglobin induction by hydroxyurea in sickle cell erythroid progenitors. Am. J. Hematol. 2000;65:227–233. doi: 10.1002/1096-8652(200011)65:3<227::AID-AJH9>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 180.Zhou B.S., Hsu N.Y., Pan B.C., Doroshow J.H., Yen Y. Overexpression of ribonucleotide reductase in transfected human KB cells increases their resistance to hydroxyurea: M2 but not M1 is sufficient to increase resistance to hydroxyurea in transfected cells. Cancer Res. 1995;55:1328–1333. [PubMed] [Google Scholar]
- 181.Zhou B., Mo X., Liu X., Qiu W., Yen Y. Human ribonucleotide reductase M2 subunit gene amplification and transcriptional regulation in a homogeneous staining chromosome region responsible for the mechanism of drug resistance. Cytogenet. Genome Res. 2001;95:34–42. doi: 10.1159/000057014. [DOI] [PubMed] [Google Scholar]
- 182.Desjardins A., Quinn J.A., Vredenburgh J.J., Sathornsumetee S., Friedman A.H., Herndon J.E., McLendon R.E., Provenzale J.M., Rich J.N., Sampson J.H., et al. Phase II study of imatinib mesylate and hydroxyurea for recurrent grade III malignant gliomas. J. Neurooncol. 2007;83:53–60. doi: 10.1007/s11060-006-9302-2. [DOI] [PubMed] [Google Scholar]
- 183.Swinnen L.J., Rankin C., Carraway H., Albain K.S., Townsend J.J., Budd G.T., Kish J.A., Rivkin S.E., Blumenthal D.T. A phase II study of cisplatin preceded by a 12-h continuous infusion of concurrent hydroxyurea and cytosine arabinoside (Ara-C) for adult patients with malignant gliomas (Southwest Oncology Group S9149) J. Neurooncol. 2008;86:353–358. doi: 10.1007/s11060-007-9483-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Dresemann G., Weller M., Rosenthal M.A., Wedding U., Wagner W., Engel E., Heinrich B., Mayer-Steinacker R., Karup-Hansen A., Fluge O., et al. Imatinib in combination with hydroxyurea versus hydroxyurea alone as oral therapy in patients with progressive pretreated glioblastoma resistant to standard dose temozolomide. J. Neurooncol. 2010;96:393–402. doi: 10.1007/s11060-009-9976-3. [DOI] [PubMed] [Google Scholar]
- 185.Kao J., Genden E.M., Gupta V., Policarpio E.L., Burri R.J., Rivera M., Gurudutt V., Som P.M., Teng M., Packer S.H. Phase 2 trial of concurrent 5-fluorouracil, hydroxyurea, cetuximab, and hyperfractionated intensity-modulated radiation therapy for locally advanced head and neck cancer. Cancer. 2011;117:318–326. doi: 10.1002/cncr.25374. [DOI] [PubMed] [Google Scholar]
- 186.Harrison C.N., Campbell P.J., Buck G., Wheatley K., East C.L., Bareford D., Wilkins B.S., van der Walt J.D., Reilly J.T., Grigg A.P., et al. Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N. Engl. J. Med. 2005;353:33–45. doi: 10.1056/NEJMoa043800. [DOI] [PubMed] [Google Scholar]
- 187.Charache S., Terrin M.L., Moore R.D., Dover G.J., McMahon R.P., Barton F.B., Waclawiw M., Eckert S.V. Design of the multicenter study of hydroxyurea in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea. Control. Clin. Trials. 1995;16:432–446. doi: 10.1016/S0197-2456(95)00098-4. [DOI] [PubMed] [Google Scholar]
- 188.Ma B., Goh B.C., Tan E.H., Lam K.C., Soo R., Leong S.S., Wang L.Z., Mo F., Chan A.T., Zee B., et al. A multicenter phase II trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) and gemcitabine in advanced non-small-cell lung cancer with pharmacokinetic evaluation using peripheral blood mononuclear cells. Investig. New Drugs. 2008;26:169–173. doi: 10.1007/s10637-007-9085-0. [DOI] [PubMed] [Google Scholar]
- 189.Shao J., Zhou B., Di Bilio A.J., Zhu L., Wang T., Qi C., Shih J., Yen Y. A Ferrous-Triapine complex mediates formation of reactive oxygen species that inactivate human ribonucleotide reductase. Mol. Cancer Ther. 2006;5:586–592. doi: 10.1158/1535-7163.MCT-05-0384. [DOI] [PubMed] [Google Scholar]
- 190.Aye Y., Long M.J.C., Stubbe J. Mechanistic studies of semicarbazone triapine targeting human ribonucleotide reductase in vitro and in mammalian cells: Tyrosyl radical quenching not involving reactive oxygen species. J. Biol. Chem. 2012;287:35768–35778. doi: 10.1074/jbc.M112.396911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Karp J.E., Giles F.J., Gojo I., Morris L., Greer J., Johnson B., Thein M., Sznol M., Low J. A phase I study of the novel ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) in combination with the nucleoside analog fludarabine for patients with refractory acute leukemias and aggressive myeloproliferative disorders. Leuk. Res. 2008;32:71–77. doi: 10.1016/j.leukres.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Giles F.J., Fracasso P.M., Kantarjian H.M., Cortes J.E., Brown R.A., Verstovsek S., Alvarado Y., Thomas D.A., Faderl S., Garcia-Manero G., et al. Phase I and pharmacodynamic study of Triapine, a novel ribonucleotide reductase inhibitor, in patients with advanced leukemia. Leuk. Res. 2003;27:1077–1083. doi: 10.1016/S0145-2126(03)00118-8. [DOI] [PubMed] [Google Scholar]
- 193.Gojo I., Tidwell M.L., Greer J., Takebe N., Seiter K., Pochron M.F., Johnson B., Sznol M., Karp J.E. Phase I and pharmacokinetic study of Triapine, a potent ribonucleotide reductase inhibitor, in adults with advanced hematologic malignancies. Leuk. Res. 2007;31:1165–1173. doi: 10.1016/j.leukres.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 194.Nutting C.M., van Herpen C.M., Miah A.B., Bhide S.A., Machiels J.P., Buter J., Kelly C., de Raucourt D., Harrington K.J. Phase II study of 3-AP Triapine in patients with recurrent or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2009;20:1275–1279. doi: 10.1093/annonc/mdn775. [DOI] [PubMed] [Google Scholar]
- 195.Knox J.J., Hotte S.J., Kollmannsberger C., Winquist E., Fisher B., Eisenhauer E.A. Phase II study of Triapine in patients with metastatic renal cell carcinoma: A trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC IND.161) Investig. New Drugs. 2007;25:471–477. doi: 10.1007/s10637-007-9044-9. [DOI] [PubMed] [Google Scholar]
- 196.Schelman W.R., Morgan-Meadows S., Marnocha R., Lee F., Eickhoff J., Huang W., Pomplun M., Jiang Z., Alberti D., Kolesar J.M., et al. A phase I study of Triapine in combination with doxorubicin in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2009;63:1147–1156. doi: 10.1007/s00280-008-0890-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Yen Y., Margolin K., Doroshow J., Fishman M., Johnson B., Clairmont C., Sullivan D., Sznol M. A phase I trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone in combination with gemcitabine for patients with advanced cancer. Cancer Chemother. Pharmacol. 2004;54:331–342. doi: 10.1007/s00280-004-0821-2. [DOI] [PubMed] [Google Scholar]
- 198.Wadler S., Makower D., Clairmont C., Lambert P., Fehn K., Sznol M. Phase I and pharmacokinetic study of the ribonucleotide reductase inhibitor, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, administered by 96-hour intravenous continuous infusion. J. Clin. Oncol. 2004;22:1553–1563. doi: 10.1200/JCO.2004.07.158. [DOI] [PubMed] [Google Scholar]
- 199.Li J., Zheng L.M., King I., Doyle T.W., Chen S.H. Syntheses and antitumor activities of potent inhibitors of ribonucleotide reductase: 3-amino-4-methylpyridine-2-carboxaldehyde-thiosemicarba-zone (3-AMP), 3-amino-pyridine-2-carboxaldehyde-thiosemicarbazone (3-AP) and its water-soluble prodrugs. Curr. Med. Chem. 2001;8:121–133. doi: 10.2174/0929867013373741. [DOI] [PubMed] [Google Scholar]
- 200.Zeidner J.F., Karp J.E., Blackford A.L., Smith B.D., Gojo I., Gore S.D., Levis M.J., Carraway H.E., Greer J.M., Ivy S.P., et al. A phase II trial of sequential ribonucleotide reductase inhibition in aggressive myeloproliferative neoplasms. Haematologica. 2014;99:672–678. doi: 10.3324/haematol.2013.097246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Elford H.L., Wampler G.L., van’t Riet B. New ribonucleotide reductase inhibitors with antineoplastic activity. Cancer Res. 1979;39:844–851. [PubMed] [Google Scholar]
- 202.Elford H.L., Van’t Riet B., Wampler G.L., Lin A.L., Elford R.M. Regulation of ribonucleotide reductase in mammalian cells by chemotherapeutic agents. Adv. Enzym. Regul. 1980;19:151–168. doi: 10.1016/0065-2571(81)90014-5. [DOI] [PubMed] [Google Scholar]
- 203.Tihan T., Elford H.L., Cory J.G. Studies on the mechanisms of inhibition of L1210 cell growth by 3,4-dihydroxybenzohydroxamic acid and 3,4-dihydroxybenzamidoxime. Adv. Enzym. Regul. 1991;31:71–83. doi: 10.1016/0065-2571(91)90009-B. [DOI] [PubMed] [Google Scholar]
- 204.Szekeres T., Gharehbaghi K., Fritzer M., Woody M., Srivastava A., van’t Riet B., Jayaram H.N., Elford H.L. Biochemical and antitumor activity of trimidox, a new inhibitor of ribonucleotide reductase. Cancer Chemother. Pharmacol. 1994;34:63–66. doi: 10.1007/BF00686113. [DOI] [PubMed] [Google Scholar]
- 205.Szekeres T., Vielnascher E., Novotny L., Vachalkova A., Fritzer M., Findenig G., Gobl R., Elford H.L., Goldenberg H. Iron binding capacity of trimidox (3,4,5-trihydroxybenzamidoxime), a new inhibitor of the enzyme ribonucleotide reductase. Eur. J. Clin. Chem. Clin. Biochem. 1995;33:785–789. doi: 10.1515/cclm.1995.33.11.785. [DOI] [PubMed] [Google Scholar]
- 206.Szekeres T., Fritzer M., Strobl H., Gharehbaghi K., Findenig G., Elford H.L., Lhotka C., Schoen H.J., Jayaram H.N. Synergistic growth inhibitory and differentiating effects of trimidox and tiazofurin in human promyelocytic leukemia HL-60 cells. Blood. 1994;84:4316–4321. doi: 10.1182/blood.V84.12.4316.bloodjournal84124316. [DOI] [PubMed] [Google Scholar]
- 207.Iyamu W.E., Adunyah S.E., Fasold H., Horiuchi K., Elford H.L., Asakura T., Turner E.A. Enhancement of hemoglobin and F-cell production by targeting growth inhibition and differentiation of K562 cells with ribonucleotide reductase inhibitors (didox and trimidox) in combination with streptozotocin. Am. J. Hematol. 2000;63:176–183. doi: 10.1002/(SICI)1096-8652(200004)63:4<176::AID-AJH3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 208.Horvath Z., Bauer W., Hoechtl T., Saiko P., Fritzer-Szekeres M., Tihan T., Szekeres T. Combination chemotherapy of BCNU and Didox acts synergystically in 9L glioma cells. Nucleosides Nucleotides Nucleic Acids. 2004;23:1531–1535. doi: 10.1081/NCN-200027746. [DOI] [PubMed] [Google Scholar]
- 209.Horvath Z., Hochtl T., Bauer W., Fritzer-Szekeres M., Elford H.L., Szekeres T., Tihan T. Synergistic cytotoxicity of the ribonucleotide reductase inhibitor didox (3,4-dihydroxy-benzohydroxamic acid) and the alkylating agent carmustine (BCNU) in 9L rat gliosarcoma cells and DAOY human medulloblastoma cells. Cancer Chemother. Pharmacol. 2004;54:139–145. doi: 10.1007/s00280-004-0795-0. [DOI] [PubMed] [Google Scholar]
- 210.Figul M., Soling A., Dong H.J., Chou T.C., Rainov N.G. Combined effects of temozolomide and the ribonucleotide reductase inhibitors didox and trimidox in malignant brain tumor cells. Cancer Chemother. Pharmacol. 2003;52:41–46. doi: 10.1007/s00280-003-0611-2. [DOI] [PubMed] [Google Scholar]
- 211.Wakisaka N., Yoshizaki T., Raab-Traub N., Pagano J.S. Ribonucleotide reductase inhibitors enhance cidofovir-induced apoptosis in EBV-positive nasopharyngeal carcinoma xenografts. Int. J. Cancer. 2005;116:640–645. doi: 10.1002/ijc.21096. [DOI] [PubMed] [Google Scholar]
- 212.Veale D., Carmichael J., Cantwell B.M., Elford H.L., Blackie R., Kerr D.J., Kaye S.B., Harris A.L. A phase 1 and pharmacokinetic study of didox: A ribonucleotide reductase inhibitor. Br. J. Cancer. 1988;58:70–72. doi: 10.1038/bjc.1988.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Carmichael J., Cantwell B.M., Mannix K.A., Veale D., Elford H.L., Blackie R., Kerr D.J., Kaye S.B., Harris A.L. A phase I and pharmacokinetic study of didox administered by 36 hour infusion. The Cancer Research Campaign Phase I/II Clinical Trials Committee. Br. J. Cancer. 1990;61:447–450. doi: 10.1038/bjc.1990.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Rubens R.D., Kaye S.B., Soukop M., Williams C.J., Brampton M.H., Harris A.L. Phase II trial of didox in advanced breast cancer. Cancer Research Campaign Phase I/II Clinical Trials Committee. Br. J. Cancer. 1991;64:1187–1188. doi: 10.1038/bjc.1991.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Inayat M.S., Chendil D., Mohiuddin M., Elford H.L., Gallicchio V.S., Ahmed M.M. Didox (a novel ribonucleotide reductase inhibitor) overcomes Bcl-2 mediated radiation resistance in prostate cancer cell line PC-3. Cancer Biol. Ther. 2002;1:539–545. doi: 10.4161/cbt.1.5.174. [DOI] [PubMed] [Google Scholar]
- 216.Fritzer-Szekeres M., Salamon A., Grusch M., Horvath Z., Hochtl T., Steinbrugger R., Jager W., Krupitza G., Elford H.L., Szekeres T. Trimidox, an inhibitor of ribonucleotide reductase, synergistically enhances the inhibition of colony formation by Ara-C in HL-60 human promyelocytic leukemia cells. Biochem. Pharmacol. 2002;64:481–485. doi: 10.1016/S0006-2952(02)01186-3. [DOI] [PubMed] [Google Scholar]
- 217.Novotny L., Rauko P., Liska J., Elford H.L., Szekeres T. Potentiation of the activity of cisplatin and cyclophosphamide by trimidox, a novel ribonucleotide reductase inhibitor, in leukemia-bearing mice. Cancer Lett. 2006;233:178–184. doi: 10.1016/j.canlet.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 218.Cooper C.E., Lynagh G.R., Hoyes K.P., Hider R.C., Cammack R., Porter J.B. The relationship of intracellular iron chelation to the inhibition and regeneration of human ribonucleotide reductase. J. Biol. Chem. 1996;271:20291–20299. doi: 10.1074/jbc.271.34.20291. [DOI] [PubMed] [Google Scholar]
- 219.Komoto K., Nomoto T., El Muttaqien S., Takemoto H., Matsui M., Miura Y., Nishiyama N. Iron chelation cancer therapy using hydrophilic block copolymers conjugated with deferoxamine. Cancer Sci. 2021;112:410–421. doi: 10.1111/cas.14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Wang Y., Liu Z., Lin T.M., Chanana S., Xiong M.P. Nanogel-DFO conjugates as a model to investigate pharmacokinetics, biodistribution, and iron chelation in vivo. Int. J. Pharm. 2018;538:79–86. doi: 10.1016/j.ijpharm.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dayani P.N., Bishop M.C., Black K., Zeltzer P.M. Desferoxamine (DFO)—mediated iron chelation: Rationale for a novel approach to therapy for brain cancer. J. Neurooncol. 2004;67:367–377. doi: 10.1023/B:NEON.0000024238.21349.37. [DOI] [PubMed] [Google Scholar]
- 222.Kalinowski D.S., Richardson D.R. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol. Rev. 2005;57:547–583. doi: 10.1124/pr.57.4.2. [DOI] [PubMed] [Google Scholar]
- 223.Merlot A.M., Kalinowski D.S., Richardson D.R. Novel chelators for cancer treatment: Where are we now? Antioxid. Redox Signal. 2013;18:973–1006. doi: 10.1089/ars.2012.4540. [DOI] [PubMed] [Google Scholar]
- 224.Richardson D.R., Kalinowski D.S., Lau S., Jansson P.J., Lovejoy D.B. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim. Biophys. Acta. 2009;1790:702–717. doi: 10.1016/j.bbagen.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 225.Fan L., Iyer J., Zhu S., Frick K.K., Wada R.K., Eskenazi A.E., Berg P.E., Ikegaki N., Kennett R.H., Frantz C.N. Inhibition of N-myc expression and induction of apoptosis by iron chelation in human neuroblastoma cells. Cancer Res. 2001;61:1073–1079. [PubMed] [Google Scholar]
- 226.Zhou T., Ma Y., Kong X., Hider R.C. Design of iron chelators with therapeutic application. Coord. Chem. Rev. 2012;41:6371–6389. doi: 10.1039/c2dt12159j. [DOI] [PubMed] [Google Scholar]
- 227.Donfrancesco A., Deb G., Dominici C., Pileggi D., Castello M.A., Helson L. Effects of a single course of deferoxamine in neuroblastoma patients. Cancer Res. 1990;50:4929–4930. [PubMed] [Google Scholar]
- 228.Blatt J. Deferoxamine in children with recurrent neuroblastoma. Anticancer Res. 1994;14:2109–2112. [PubMed] [Google Scholar]
- 229.Lang J., Zhao X., Wang X., Zhao Y., Li Y., Zhao R., Cheng K., Li Y., Han X., Zheng X., et al. Targeted Co-delivery of the Iron Chelator Deferoxamine and a HIF1alpha Inhibitor Impairs Pancreatic Tumor Growth. ACS Nano. 2019;13:2176–2189. doi: 10.1021/acsnano.8b08823. [DOI] [PubMed] [Google Scholar]
- 230.Krakoff I.H. Gallium nitrate in the treatment of cancer-related hypercalcemia. Semin. Oncol. 1991;18:3. [PubMed] [Google Scholar]
- 231.Higashi T., Wakao H., Yamaguchi M., Suga K. The relationship between Ga-67 accumulation and cell cycle in malignant tumor cells in vitro. Eur. J. Nucl. Med. 1988;14:155–158. doi: 10.1007/BF00293541. [DOI] [PubMed] [Google Scholar]
- 232.Chitambar C.R., Narasimhan J., Guy J., Sem D.S., O’Brien W.J. Inhibition of ribonucleotide reductase by gallium in murine leukemic L1210 cells. Cancer Res. 1991;51:6199–6201. [PubMed] [Google Scholar]
- 233.Bernstein L.R. Mechanisms of therapeutic activity for gallium. Pharmacol. Rev. 1998;50:665–682. [PubMed] [Google Scholar]
- 234.Chitambar C.R., Purpi D.P., Woodliff J., Yang M., Wereley J.P. Development of gallium compounds for treatment of lymphoma: Gallium maltolate, a novel hydroxypyrone gallium compound, induces apoptosis and circumvents lymphoma cell resistance to gallium nitrate. J. Pharmacol. Exp. Ther. 2007;322:1228–1236. doi: 10.1124/jpet.107.126342. [DOI] [PubMed] [Google Scholar]
- 235.Hashemy S.I., Ungerstedt J.S., Zahedi Avval F., Holmgren A. Motexafin gadolinium, a tumor-selective drug targeting thioredoxin reductase and ribonucleotide reductase. J. Biol. Chem. 2006;281:10691–10697. doi: 10.1074/jbc.M511373200. [DOI] [PubMed] [Google Scholar]
- 236.William W.N., Jr., Zinner R.G., Karp D.D., Oh Y.W., Glisson B.S., Phan S.C., Stewart D.J. Phase I trial of motexafin gadolinium in combination with docetaxel and cisplatin for the treatment of non-small cell lung cancer. J. Thorac. Oncol. 2007;2:745–750. doi: 10.1097/JTO.0b013e31811f4719. [DOI] [PubMed] [Google Scholar]
- 237.Edelman M.J., Otterson G., Leach J., Malpass T., Salgia R., Jones D., Mody T.D., Govindan R. Multicenter phase II trial of Motexafin gadolinium and pemetrexed for second-line treatment in patients with non-small cell lung cancer. J. Thorac. Oncol. 2011;6:786–789. doi: 10.1097/JTO.0b013e31820a443f. [DOI] [PubMed] [Google Scholar]
- 238.Evens A.M., Spies W.G., Helenowski I.B., Patton D., Spies S., Jovanovic B.D., Miyata S., Hamilton E., Variakojis D., Chen J., et al. The novel expanded porphyrin, motexafin gadolinium, combined with [90Y]ibritumomab tiuxetan for relapsed/refractory non-Hodgkin’s lymphoma: Preclinical findings and results of a phase I trial. Clin. Cancer Res. 2009;15:6462–6471. doi: 10.1158/1078-0432.CCR-09-0905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Brachman D.G., Pugh S.L., Ashby L.S., Thomas T.A., Dunbar E.M., Narayan S., Robins H.I., Bovi J.A., Rockhill J.K., Won M., et al. Phase 1/2 trials of Temozolomide, Motexafin Gadolinium, and 60-Gy fractionated radiation for newly diagnosed supratentorial glioblastoma multiforme: Final results of RTOG 0513. Int. J. Radiat. Oncol. Biol. Phys. 2015;91:961–967. doi: 10.1016/j.ijrobp.2014.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.McHaffie D.R., Chabot P., Dagnault A., Suh J.H., Fortin M.A., Chang E., Timmerman R., Souhami L., Grecula J., Nabid A., et al. Safety and feasibility of motexafin gadolinium administration with whole brain radiation therapy and stereotactic radiosurgery boost in the treatment of </= 6 brain metastases: A multi-institutional phase II trial. J. Neurooncol. 2011;105:301–308. doi: 10.1007/s11060-011-0590-9. [DOI] [PubMed] [Google Scholar]
- 241.Ford J.M., Seiferheld W., Alger J.R., Wu G., Endicott T.J., Mehta M., Curran W., Phan S.C. Results of the phase I dose-escalating study of motexafin gadolinium with standard radiotherapy in patients with glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2007;69:831–838. doi: 10.1016/j.ijrobp.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 242.Bradley K.A., Pollack I.F., Reid J.M., Adamson P.C., Ames M.M., Vezina G., Blaney S., Ivy P., Zhou T., Krailo M., et al. Motexafin gadolinium and involved field radiation therapy for intrinsic pontine glioma of childhood: A Children’s Oncology Group phase I study. Neuro-Oncology. 2008;10:752–758. doi: 10.1215/15228517-2008-043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Mehta M.P., Shapiro W.R., Phan S.C., Gervais R., Carrie C., Chabot P., Patchell R.A., Glantz M.J., Recht L., Langer C., et al. Motexafin gadolinium combined with prompt whole brain radiotherapy prolongs time to neurologic progression in non-small-cell lung cancer patients with brain metastases: Results of a phase III trial. Int. J. Radiat. Oncol. Biol. Phys. 2009;73:1069–1076. doi: 10.1016/j.ijrobp.2008.05.068. [DOI] [PubMed] [Google Scholar]
- 244.Meyers C.A., Smith J.A., Bezjak A., Mehta M.P., Liebmann J., Illidge T., Kunkler I., Caudrelier J.M., Eisenberg P.D., Meerwaldt J., et al. Neurocognitive function and progression in patients with brain metastases treated with whole-brain radiation and motexafin gadolinium: Results of a randomized phase III trial. J. Clin. Oncol. 2004;22:157–165. doi: 10.1200/JCO.2004.05.128. [DOI] [PubMed] [Google Scholar]
- 245.Lin T.S., Naumovski L., Lecane P.S., Lucas M.S., Moran M.E., Cheney C., Lucas D.M., Phan S.C., Miller R.A., Byrd J.C. Effects of motexafin gadolinium in a phase II trial in refractory chronic lymphocytic leukemia. Leuk. Lymphoma. 2009;50:1977–1982. doi: 10.3109/10428190903288464. [DOI] [PubMed] [Google Scholar]
- 246.Amato R.J., Jac J., Hernandez-McClain J. Motexafin gadolinium for the treatment of metastatic renal cell carcinoma: Phase II study results. Clin. Genitourin. Cancer. 2008;6:73–78. doi: 10.3816/CGC.2008.n.011. [DOI] [PubMed] [Google Scholar]
- 247.Zhou B., Su L., Hu S., Hu W., Yip M.L., Wu J., Gaur S., Smith D.L., Yuan Y.C., Synold T.W., et al. A small-Mol.ecule blocking ribonucleotide reductase holoenzyme formation inhibits cancer cell growth and overcomes drug resistance. Cancer Res. 2013;73:6484–6493. doi: 10.1158/0008-5472.CAN-13-1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Chen M.C., Zhou B., Zhang K., Yuan Y.C., Un F., Hu S., Chou C.M., Chen C.H., Wu J., Wang Y., et al. The Novel Ribonucleotide Reductase Inhibitor COH29 Inhibits DNA Repair In Vitro. Mol. Pharmacol. 2015;87:996–1005. doi: 10.1124/mol.114.094987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Orr R.M. GTI-2040. Lorus Therapeutics. Curr. Opin. Investig. Drugs. 2001;2:1462–1466. [PubMed] [Google Scholar]
- 250.Lee Y., Vassilakos A., Feng N., Lam V., Xie H., Wang M., Jin H., Xiong K., Liu C., Wright J., et al. GTI-2040, an antisense agent targeting the small subunit component (R2) of human ribonucleotide reductase, shows potent antitumor activity against a variety of tumors. Cancer Res. 2003;63:2802–2811. [PubMed] [Google Scholar]
- 251.Tu G.C., Tu X. GTI-2501. Lorus Therapeutics. Curr. Opin. Investig. Drugs. 2001;2:1467–1470. [PubMed] [Google Scholar]
- 252.Davis M.E., Zuckerman J.E., Choi C.H., Seligson D., Tolcher A., Alabi C.A., Yen Y., Heidel J.D., Ribas A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Cohen E.A., Gaudreau P., Brazeau P., Langelier Y. Specific inhibition of herpesvirus ribonucleotide reductase by a nonapeptide derived from the carboxy terminus of subunit 2. Nature. 1986;321:441–443. doi: 10.1038/321441a0. [DOI] [PubMed] [Google Scholar]
- 254.Dutia B.M., Frame M.C., Subak-Sharpe J.H., Clark W.N., Marsden H.S. Specific inhibition of herpesvirus ribonucleotide reductase by synthetic peptides. Nature. 1986;321:439–441. doi: 10.1038/321439a0. [DOI] [PubMed] [Google Scholar]
- 255.Climent I., Sjöberg B.M., Huang C.Y. Carboxyl-terminal peptides as probes for Escherichia coli ribonucleotide reductase subunit interaction: Kinetic analysis of inhibition studies. Biochemistry. 1991;30:5164–5171. doi: 10.1021/bi00235a008. [DOI] [PubMed] [Google Scholar]
- 256.Cosentino G., Lavallee P., Rakhit S., Plante R., Gaudette Y., Lawetz C., Whitehead P.W., Duceppe J.S., Lepine-Frenette C., Dansereau N., et al. Specific inhibition of ribonucleotide reductases by peptides corresponding to the C-terminal of their second subunit. Biochem. Cell Biol. 1991;69:79–83. doi: 10.1139/o91-011. [DOI] [PubMed] [Google Scholar]
- 257.Lycksell P.O., Ingemarson R., Davis R., Graslund A., Thelander L. 1H NMR studies of mouse ribonucleotide reductase: The R2 protein carboxyl-terminal tail, essential for subunit interaction, is highly flexible but becomes rigid in the presence of protein R1. Biochemistry. 1994;33:2838–2842. doi: 10.1021/bi00176a013. [DOI] [PubMed] [Google Scholar]
- 258.Yang F.D., Spanevello R.A., Celiker I., Hirschmann R., Rubin H., Cooperman B.S. The carboxyl terminus heptapeptide of the R2 subunit of mammalian ribonucleotide reductase inhibits enzyme activity and can be used to purify the R1 subunit. FEBS Lett. 1990;272:61–64. doi: 10.1016/0014-5793(90)80449-S. [DOI] [PubMed] [Google Scholar]
- 259.Wnuk S.F., Robins M.J. Ribonucleotide reductase inhibitors as anti-herpes agents. Antiviral. Res. 2006;71:122–126. doi: 10.1016/j.antiviral.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 260.Xu H., Fairman J.W., Wijerathna S.R., Kreischer N.R., LaMacchia J., Helmbrecht E., Cooperman B.S., Dealwis C. The structural basis for peptidomimetic inhibition of eukaryotic ribonucleotide reductase: A conformationally flexible pharmacophore. J. Med. Chem. 2008;51:4653–4659. doi: 10.1021/jm800350u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Lassmann G., Potsch S. Structure of transient radicals from cytostatic-active p-alkoxyphenols by continuous-flow EPR. Free Radic. Biol. Med. 1995;19:533–539. doi: 10.1016/0891-5849(95)00054-2. [DOI] [PubMed] [Google Scholar]
- 262.Potsch S., Drechsler H., Liermann B., Graslund A., Lassmann G. p-Alkoxyphenols, a new class of inhibitors of mammalian R2 ribonucleotide reductase: Possible candidates for antimelanotic drugs. Mol. Pharmacol. 1994;45:792–796. [PubMed] [Google Scholar]
- 263.Potsch S., Sahlin M., Langelier Y., Graslund A., Lassmann G. Reduction of the tyrosyl radical and the iron center in protein R2 of ribonucleotide reductase from mouse, herpes simplex virus and E. coli by p-alkoxyphenols. FEBS Lett. 1995;374:95–99. doi: 10.1016/0014-5793(95)01082-P. [DOI] [PubMed] [Google Scholar]
- 264.Wu X., Cooperman B.S. Synthesis and biological activity of a bivalent nucleotide inhibitor of ribonucleotide reductase. Bioorg. Med. Chem. Lett. 2000;10:2387–2389. doi: 10.1016/S0960-894X(00)00481-9. [DOI] [PubMed] [Google Scholar]
- 265.Petrelli R., Meli M., Vita P., Torquati I., Ferro A., Vodnala M., D’Alessandro N., Tolomeo M., Del Bello F., Kusumanchi P., et al. From the covalent linkage of drugs to novel inhibitors of ribonucleotide reductase: Synthesis and biological evaluation of valproic esters of 3′-C-methyladenosine. Bioorg. Med. Chem. Lett. 2014;24:5304–5309. doi: 10.1016/j.bmcl.2014.09.046. [DOI] [PubMed] [Google Scholar]
- 266.Ahmad M.F., Wan Q., Jha S., Motea E., Berdis A., Dealwis C. Evaluating the therapeutic potential of a non-natural nucleotide that inhibits human ribonucleotide reductase. Mol. Cancer Ther. 2012;11:2077–2086. doi: 10.1158/1535-7163.MCT-12-0199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Fan G.J., Liu X.D., Qian Y.P., Shang Y.J., Li X.Z., Dai F., Fang J.G., Jin X.L., Zhou B. 4,4′-Dihydroxy-trans-stilbene, a resveratrol analogue, exhibited enhanced antioxidant activity and cytotoxicity. Bioorg. Med. Chem. 2009;17:2360–2365. doi: 10.1016/j.bmc.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 268.Maccario C., Savio M., Ferraro D., Bianchi L., Pizzala R., Pretali L., Forti L., Stivala L.A. The resveratrol analog 4,4′-dihydroxy-trans-stilbene suppresses transformation in normal mouse fibroblasts and inhibits proliferation and invasion of human breast cancer cells. Carcinogenesis. 2012;33:2172–2180. doi: 10.1093/carcin/bgs244. [DOI] [PubMed] [Google Scholar]
- 269.Balan K.V., Wang Y., Chen S.W., Chen J.C., Zheng L.F., Yang L., Liu Z.L., Pantazis P., Wyche J.H., Han Z. Proteasome-independent down-regulation of estrogen receptor-alpha (ERalpha) in breast cancer cells treated with 4,4′-dihydroxy-trans-stilbene. Biochem. Pharmacol. 2006;72:573–581. doi: 10.1016/j.bcp.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 270.Kimura Y., Sumiyoshi M., Baba K. Antitumor activities of synthetic and natural stilbenes through antiangiogenic action. Cancer Sci. 2008;99:2083–2096. doi: 10.1111/j.1349-7006.2008.00948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Saha B., Patro B.S., Koli M., Pai G., Ray J., Bandyopadhyay S.K., Chattopadhyay S. trans-4,4′-Dihydroxystilbene (DHS) inhibits human neuroblastoma tumor growth and induces mitochondrial and lysosomal damages in neuroblastoma cell lines. Oncotarget. 2017;8:73905–73924. doi: 10.18632/oncotarget.17879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Savio M., Ferraro D., Maccario C., Vaccarone R., Jensen L.D., Corana F., Mannucci B., Bianchi L., Cao Y., Stivala L.A. Resveratrol analogue 4,4′-dihydroxy-trans-stilbene potently inhibits cancer invasion and metastasis. Sci. Rep. 2016;6:19973. doi: 10.1038/srep19973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Chen C.W., Li Y., Hu S., Zhou W., Meng Y., Li Z., Zhang Y., Sun J., Bo Z., DePamphilis M.L., et al. DHS (trans-4,4′-dihydroxystilbene) suppresses DNA replication and tumor growth by inhibiting RRM2 (ribonucleotide reductase regulatory subunit M2) Oncogene. 2019;38:2364–2379. doi: 10.1038/s41388-018-0584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Ahmad M.F., Huff S.E., Pink J., Alam I., Zhang A., Perry K., Harris M.E., Misko T., Porwal S.K., Oleinick N.L., et al. Identification of Non-nucleoside Human Ribonucleotide Reductase Modulators. J. Med. Chem. 2015;58:9498–9509. doi: 10.1021/acs.jmedchem.5b00929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Ahmad M.F., Alam I., Huff S.E., Pink J., Flanagan S.A., Shewach D., Misko T.A., Oleinick N.L., Harte W.E., Viswanathan R., et al. Potent competitive inhibition of human ribonucleotide reductase by a nonnucleoside small Mol.ecule. Proc. Natl. Acad. Sci. USA. 2017;114:8241–8246. doi: 10.1073/pnas.1620220114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Huff S.E., Mohammed F.A., Yang M., Agrawal P., Pink J., Harris M.E., Dealwis C.G., Viswanathan R. Structure-Guided Synthesis and Mechanistic Studies Reveal Sweetspots on Naphthyl Salicyl Hydrazone Scaffold as Non-Nucleosidic Competitive, Reversible Inhibitors of Human Ribonucleotide Reductase. J. Med. Chem. 2018;61:666–680. doi: 10.1021/acs.jmedchem.7b00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Misko T.A., Liu Y.T., Harris M.E., Oleinick N.L., Pink J., Lee H.Y., Dealwis C.G. Structure-guided design of anti-cancer ribonucleotide reductase inhibitors. J. Enzym. Inhib. Med. Chem. 2019;34:438–450. doi: 10.1080/14756366.2018.1545226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Tholander F., Sjöberg B.M. Discovery of antimicrobial ribonucleotide reductase inhibitors by screening in microwell format. Proc. Natl. Acad. Sci. USA. 2012;109:9798–9803. doi: 10.1073/pnas.1113051109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Berggren G., Sahlin M., Crona M., Tholander F., Sjoberg B.M. Compounds with capacity to quench the tyrosyl radical in Pseudomonas aeruginosa ribonucleotide reductase. J. Biol. Inorg. Chem. 2019;24:841–848. doi: 10.1007/s00775-019-01679-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Crona M., Codo P., Jonna V.R., Hofer A., Fernandes A.P., Tholander F. A ribonucleotide reductase inhibitor with deoxyribonucleoside-reversible cytotoxicity. Mol. Oncol. 2016;10:1375–1386. doi: 10.1016/j.molonc.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Cleaveland E.S., Monks A., Vaigro-Wolff A., Zaharevitz D.W., Paull K., Ardalan K., Cooney D.A., Ford H., Jr. Site of action of two novel pyrimidine biosynthesis inhibitors accurately predicted by the compare program. Biochem. Pharmacol. 1995;49:947–954. doi: 10.1016/0006-2952(95)00009-O. [DOI] [PubMed] [Google Scholar]
- 282.Knecht W., Loffler M. Redoxal as a new lead structure for dihydroorotate dehydrogenase inhibitors: A kinetic study of the inhibition mechanism. FEBS Lett. 2000;467:27–30. doi: 10.1016/S0014-5793(00)01117-0. [DOI] [PubMed] [Google Scholar]
- 283.Gaur K., Perez Otero S.C., Benjamin-Rivera J.A., Rodriguez I., Loza-Rosas S.A., Vazquez Salgado A.M., Akam E.A., Hernandez-Matias L., Sharma R.K., Alicea N., et al. Iron Chelator Transmetalative Approach to Inhibit Human Ribonucleotide Reductase. JACS Au. 2021;1:865–878. doi: 10.1021/jacsau.1c00078. [DOI] [PMC free article] [PubMed] [Google Scholar]